
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of hydrophilic block end groups and block junction on block copolymer self-assembly in solution†
Sungmin Ha and
Kyoung Taek Kim *
*
Department of Chemistry, Seoul National University, Seoul 08826, Republic of Korea. E-mail: ktkim72@snu.ac.kr
First published on 7th March 2022
Abstract
Recent research suggests that the end groups of polymers can affect their self-assembly. However, the effect of end groups on the self-assembly of block copolymers in solution remains unclear, and thus far, only micelle–vesicle transformations have been achieved via end-group modification. Herein, we report that hydrophilic block end groups and the junction between two blocks can affect the solution self-assembly of block copolymers, leading to the formation of different morphologies, including vesicles, cubosomes, and hexosomes. Poly(ethylene glycol)-b-polystyrene (PEG-b-PS) with hydroxyl, methoxy, azido, or amino groups at the PEG chain ends was synthesized and self-assembled in solution via the cosolvent method. As a result, the morphology of the block copolymers transformed from vesicles to hexosomes upon increasing the end-group hydrophobicity. In addition, a morphological transition from cubosomes to vesicles was observed upon changing the junction from a triazole to an amide, and the interaction between the solvent and end groups significantly affected the self-assembly behavior.
1. Introduction
Amphiphilic block copolymers (ABCPs) self-assemble in solution to form various morphologies such as micelles, vesicles,1–5 cubosomes,6–19 and hexosomes.10,11,20–28 The chemical structure, block ratio, and molecular weight distribution of ABCPs influence their self-assembly.4,18,29,30 In addition, recent studies have shown that the end groups of polymers, despite only accounting for small mass and volume fractions of an entire polymer, could affect or induce polymer self-assembly. The end group effect was first investigated in the self-assembly of hydrophilic homopolymers.31–33 Xu et al. synthesized hydrophilic homo- and copolymers with two hydrophobic pyrene or cholesterol groups at the ω-terminus.31 The polymers self-assembled to form polymer vesicles, and it was demonstrated that the two rigid hydrophobic end groups were responsible for the vesicle formation. O'Reilly and co-workers observed unusual micelle formation by the self-assembly of hydrophilic homopolymers with hydrophobic end groups at both ends.32 They synthesized a series of hydrophilic homopolymers with a pyrene and a dodecyl carbonotrithiocarbonate group at each end via reversible addition–fragmentation chain transfer (RAFT) polymerization. The homopolymers with the hydrophobic groups at both ends self-assembled in an acidic aqueous solution to form well-defined aggregated micelles that behaved similarly to ABA′ block copolymers. Liu et al. demonstrated that the terminal alkyne group of hydrophilic homopolymers could drive the polymer self-assembly into vesicles and flower-like complex particles.33 They prepared a series of differently end-functionalized hydrophilic homopolymers via atom transfer radical polymerization (ATRP). During self-assembly in aqueous solution, only the homopolymers with terminal alkyne groups formed various morphologies including spherical micelles, multicompartment vesicles, and flower-like complex particles. The authors suggest that intermolecular hydrogen bonding between the terminal protons of the alkyne groups and oxygen atoms of the carbonyl groups drives the self-assembly.The effect of end groups on block copolymer self-assembly has been investigated mostly based on bulk self-assembly34–37 or polymerization-induced self-assembly (PISA).38,39 Park and co-workers investigated the effect of the end groups of PEG blocks on the self-assembled morphology of PS-b-PEG along with other properties including conductivity and ion transport properties.34–37 They observed the formation of a disordered phase from hydroxyl-terminated PEG-b-PS while sulfonate-terminated PEG-b-PS formed lamellar, hexagonal, and gyroid phases depending on the concentration of lithium salt added during the self-assembly.34 Blackman et al. compared the photo- and thermally initiated PISA behaviors of poly(ethylene glycol)-b-(2-hydroxypropyl methacrylate).38 They found that photo-initiated PISA produced higher-order structures owing to the loss of the end group during the process. In addition, extended irradiation of light or heat could induce a worm-to-vesicle morphological transition. Davis and co-workers reported that vesicle formation could be induced from a micelle-forming PISA system by increasing the RAFT end-group hydrophobicity of the macromolecular chain transfer agent (macro-CTA).39 They prepared two hydrophilic macro-CTAs with a carboxylic acid or a methyl ester end group and compared the PISA morphologies after the RAFT aqueous emulsion polymerization of styrene. Consequently, the methyl-ester-terminated block copolymer formed vesicles, whereas the carboxyl-terminated one formed micelles. With regard to self-assembly using the film hydration method, Grzelakowski and Kita-Tokarczyk synthesized a series of ABA or BA block copolymers comprising poly(2-methyloxazoline) (PMOXA) and poly(dimethylsiloxane) (PDMS) with different functional groups at both ends.40 Notably, while the ABA block copolymer with hydroxyl groups at both ends formed micelles, the block copolymer self-assembled into vesicles after methacrylation of the hydroxyl groups despite a low conversion (ca. 5%) of the methacrylation. According to the authors, the morphological transition could be attributed to the difference between the hydroxyl and methacryl groups in terms of size, dipole moment, and polarizability.
Recently, Che et al. reported a morphological transition of azide-modified PEG-b-PS (N3-PEG-b-PS) vesicles into hexagonally packed hollow hoops (HHHs) induced by osmotic pressure during dialysis against NaCl solution.41 In contrast, vesicles of PEG-b-PS with a methoxy end group (mPEG-b-PS) deformed into stomatocytes. The authors suggest that azide coordination to water molecules increases the hydrodynamic volume of the PEG at the internal side of the vesicles, compared to the PEG volume at the external side, leading to the formation of HHHs.
Most studies on the effect of end groups on block copolymer self-assembly have focused on the morphological transition between micelles and vesicles. The effect of end groups on inverse-phase-forming block copolymers has rarely been studied. Herein, we investigated the influence of the end groups of the hydrophilic block on inverse-phase-forming block copolymer self-assembly in solution. Additionally, we report that the junction between two blocks can affect the solution self-assembly of block copolymers. First, branched PEG hydrophilic blocks with methoxy, hydroxyl, or azido groups at the PEG chain ends were synthesized and coupled with PSs via copper-catalyzed azide–alkyne cycloadditions (CuAAC) or amidation. PEG-b-PS with amino end groups was obtained via reduction of the PEG-PE with azido groups. Block copolymers with different end groups were self-assembled in solution via the cosolvent method to demonstrate the effect of the end groups on the self-assembled morphologies.4 In addition, the variation in morphological changes in response to a solvent change from dioxane to acetone was studied based on the block copolymers with different end groups.
2. Results and discussion
Synthesis of PEG-b-PS with different end groups
Starting from PEG monomethyl ether or PEG diol, we synthesized hydrophilic blocks with three PEG branches end-functionalized with methoxy, hydroxyl, and azido groups (Fig. S1†). Hydroxyl PEG block with an alkyne group at the other end was synthesized according to our previous study.13 Methoxy PEG blocks with an alkyne group or a carboxyl group were synthesized by tethering α-methoxy-ω-tosyl PEGs to a methyl gallate, which was followed by hydrolysis or reduction and subsequent etherification with a propargyl bromide to introduce carboxyl or alkyne group for conjugation with PS. Azido PEG block with a carboxyl group was prepared from α-azido-ω-tosyl PEG in the same way. Synthesis of the hydrophilic blocks was confirmed by MALDI-TOF mass spectrometry and NMR spectroscopy (Table S1,† Fig. 1 and S2–S5†).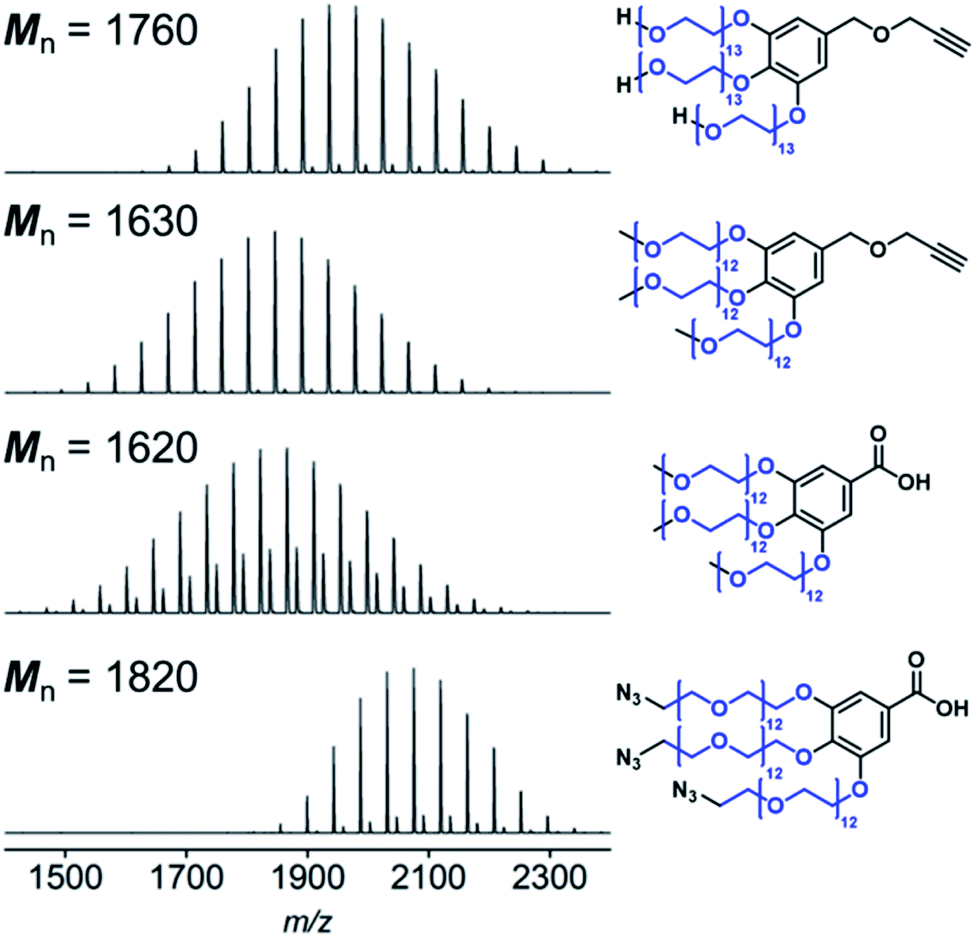 | ||
| Fig. 1 MALDI-TOF spectra of the PEG blocks. Mn of the PEG chains (shown in blue) was calculated from the spectra (Table S1†). | ||
Two different PSs, PS1 (Mn = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1) and PS2 (Mn = 15300 g mol−1), were obtained by ATRP. Their molecular weights were adjusted such that the block copolymers had a similar block ratio (wPEG), which was calculated as the ratio of the Mn of the PEG block to that of the PS block (Table 1 and S1†). The terminal bromo group of the PSs was converted into an azido group or an amino group for conjugation with the PEG blocks according to a previous study.42
900 g mol−1) and PS2 (Mn = 15300 g mol−1), were obtained by ATRP. Their molecular weights were adjusted such that the block copolymers had a similar block ratio (wPEG), which was calculated as the ratio of the Mn of the PEG block to that of the PS block (Table 1 and S1†). The terminal bromo group of the PSs was converted into an azido group or an amino group for conjugation with the PEG blocks according to a previous study.42
The PEG blocks and the PS were coupled via CuAAC (Fig. 2a) or amidation (Fig. 2b) to obtain hydroxyl, methoxy, and azido PEG-b-PSs with a triazole (denoted “trz”) or an amide bond (denoted “amd”) linking the two blocks. The azido PEG-b-PS was further reduced to obtain the amino PEG-b-PS, (H2N-PEG)3-amd-PS1 and the conversion was confirmed by the disappearance of the NMR proton peak for the carbon adjacent to the azido group (Fig. 3). As a result, we prepared five block copolymers with different end groups and junctions but similar block ratios, as confirmed by GPC and NMR analysis (Table 1, Fig. 4 and S6–S10†).
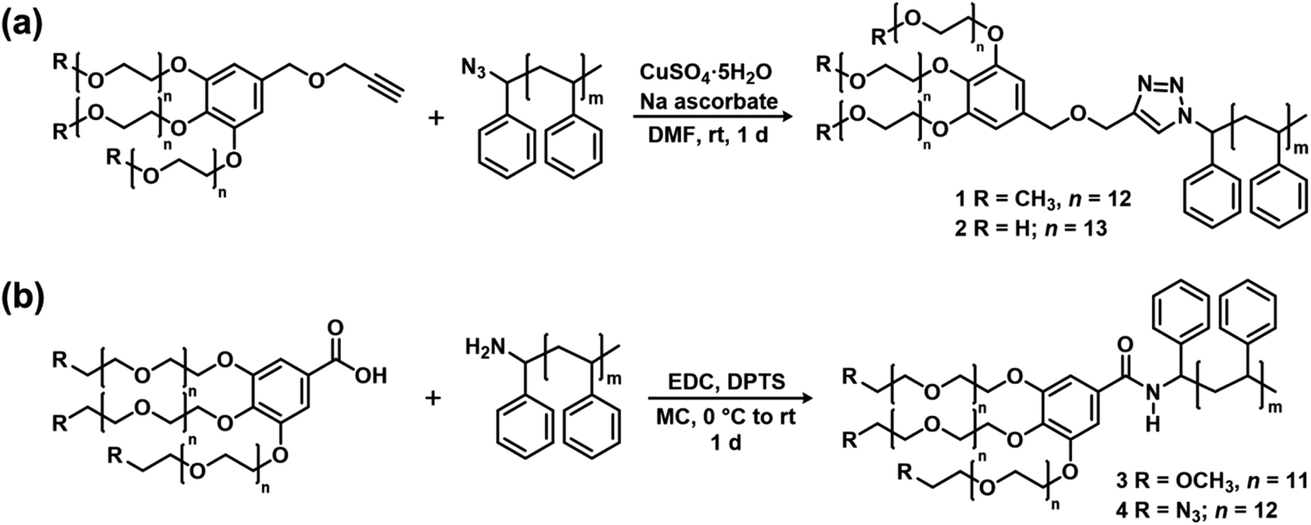 | ||
| Fig. 2 Synthesis of the block copolymers with different end groups and junctions via (a) CuAAC and (b) amidation. | ||
 | ||
| Fig. 3 (a) Reduction scheme of (N3-PEG)3-amd-PS1. (b) NMR spectra (right) of (N3-PEG)3-amd-PS1 (black) and (H2N-PEG)3-amd-PS1 (blue). | ||
| BCP | Mn (PEG)a (g mol−1) | Mn (PS)b (g mol−1) | Mn (BCP)b (g mol−1) | Đb (BCP) | wPEG (wt%) |
|---|---|---|---|---|---|
| a Mn (PEG) was calculated from the MALDI-TOF spectra of each PEG block.b Mn (PS) (Mn of PS), Mn (BCP) (Mn of a block copolymer), and Đ (BCP) (dispersity of a block copolymer) were determined by GPC (DMF, 35 °C, 1 mL min−1) using PS standards. | |||||
| (HO-PEG)3-trz-PS1 | 1760 | 16900 |
20200 |
1.09 | 10.4 |
| (MeO-PEG)3-trz-PS2 | 1630 | 15300 |
18700 |
1.09 | 10.7 |
| (MeO-PEG)3-amd-PS2 | 1620 | 15300 |
18900 |
1.05 | 10.6 |
| (N3-PEG)3-amd-PS1 | 1820 | 16900 |
20000 |
1.05 | 10.8 |
| (H2N-PEG)3-amd-PS1 | 1820 | 16900 |
19700 |
1.07 | 10.8 |
 | ||
| Fig. 4 GPC chromatograms (left) and chemical structures (right) of the BCPs. In the GPC chromatograms, PSs are shown in black and BCPs are shown in blue or red. | ||
Self-assembly of the block copolymers in solution
The block copolymers were self-assembled via the cosolvent method using acetone or dioxane, which are common solvents, as previously described.13 A block copolymer was dissolved in a common solvent and self-assembly was induced by slowly adding the same volume of water. We observed a wide range of morphologies from vesicles to hexosomes depending on the different end groups and junctions.(HO-PEG)3-trz-PS1 formed vesicles in dioxane (Fig. 5b), whereas a mixture of spongesomes and cubosomes was formed in acetone (Fig. 5a and S11c†). As previously reported, the PS chain is assumed to have a reduced critical chain length (lc) owing to its lower solubility in acetone than in dioxane, leading to the formation of morphologies with a higher packing parameter (p = V/a0lc).13 Under the same self-assembly conditions, (MeO-PEG)3-trz-PS2 self-assembled into cubosomes in both solvents (Fig. 5c, d, S11a and b†). Considering that the lc in dioxane is greater than in acetone, the hydrophilic headgroup area (a0) of (MeO-PEG)3-trz-PS2 should be smaller in dioxane than in acetone to compensate for the difference in lc. Moreover, it can be inferred that the a0 value is larger for (HO-PEG)3-trz-PS1 than for (MeO-PEG)3-trz-PS2 in both solvents, possibly owing to the higher hydrophilicity of the hydroxyl group compared to that of the methoxy group. These results are consistent with those of the previous study by Davis and co-workers that revealed micelle–vesicle transformations with an increasing packing parameter upon increasing the end-group hydrophobicity.39 The hydroxyl and methoxy end groups directly affect the BCP self-assembly by varying the a0 due to their different interactions with water and the common solvent.
 | ||
| Fig. 5 Representative SEM and TEM images of (a and b) (HO-PEG)3-trz-PS1 self-assembled in (a) acetone (cubosomes and spongesomes) and (b) dioxane (vesicles), and (c and d) (MeO-PEG)3-trz-PS2 in (c) acetone (cubosomes) and (d) dioxane (cubosomes). | ||
To further investigate the effect of azido and amino end groups, we compared the self-assembly of (MeO-PEG)3-amd-PS2, (N3-PEG)3-amd-PS1, and (H2N-PEG)3-amd-PS1 (Fig. 6 and 7). (MeO-PEG)3-amd-PS2 and (N3-PEG)3-amd-PS1 self-assembled in acetone to form a mixture of lamellae and vesicles or a mixture of sponges and vesicles, respectively (Fig. 6a and c). Interestingly, (N3-PEG)3-amd-PS1 self-assembled into hexosomes in acetone (Fig. 6d and S11d†) while (MeO-PEG)3-amd-PS2 only formed vesicles (Fig. 6b). This substantial difference in morphologies in terms of the packing parameter may arise from the higher hydrophobicity of azido PEGs. Hartwig and co-workers reported that the conversion of a hydrogen to an azide in cycloheximide and digoxigenin derivatives increased their distribution coefficient values.43 Therefore, the azido PEG should have higher hydrophobicity than that of the methoxy PEG.44–46 Specifically, (N3-PEG)3-amd-PS1 should possess a significantly smaller a0 value than that of vesicle-forming (MeO-PEG)3-amd-PS2, leading to high-packing-parameter hexosome formation in dioxane. Moreover, it can be inferred that (N3-PEG)3-amd-PS1 should possess a much smaller a0 value in dioxane than that in acetone to increase the packing parameter while the lc of PS decreases. Meanwhile, only vesicles were observed from the self-assembly of (H2N-PEG)3-amd-PS1 in both solvents owing to the high hydrophilicity of the amino groups.43
 | ||
| Fig. 6 Representative SEM and TEM images of (a and b) (MeO-PEG)3-amd-PS2 self-assembled in (a) acetone (vesicles and lamellae) and (b) dioxane (vesicles), (c and d) (N3-PEG)3-amd-PS1 in (c) acetone (vesicles and sponges) and (d) dioxane (hexosomes), and (e and f) (H2N-PEG)3-amd-PS1 in (e) acetone (vesicles) and (d) dioxane (vesicles). | ||
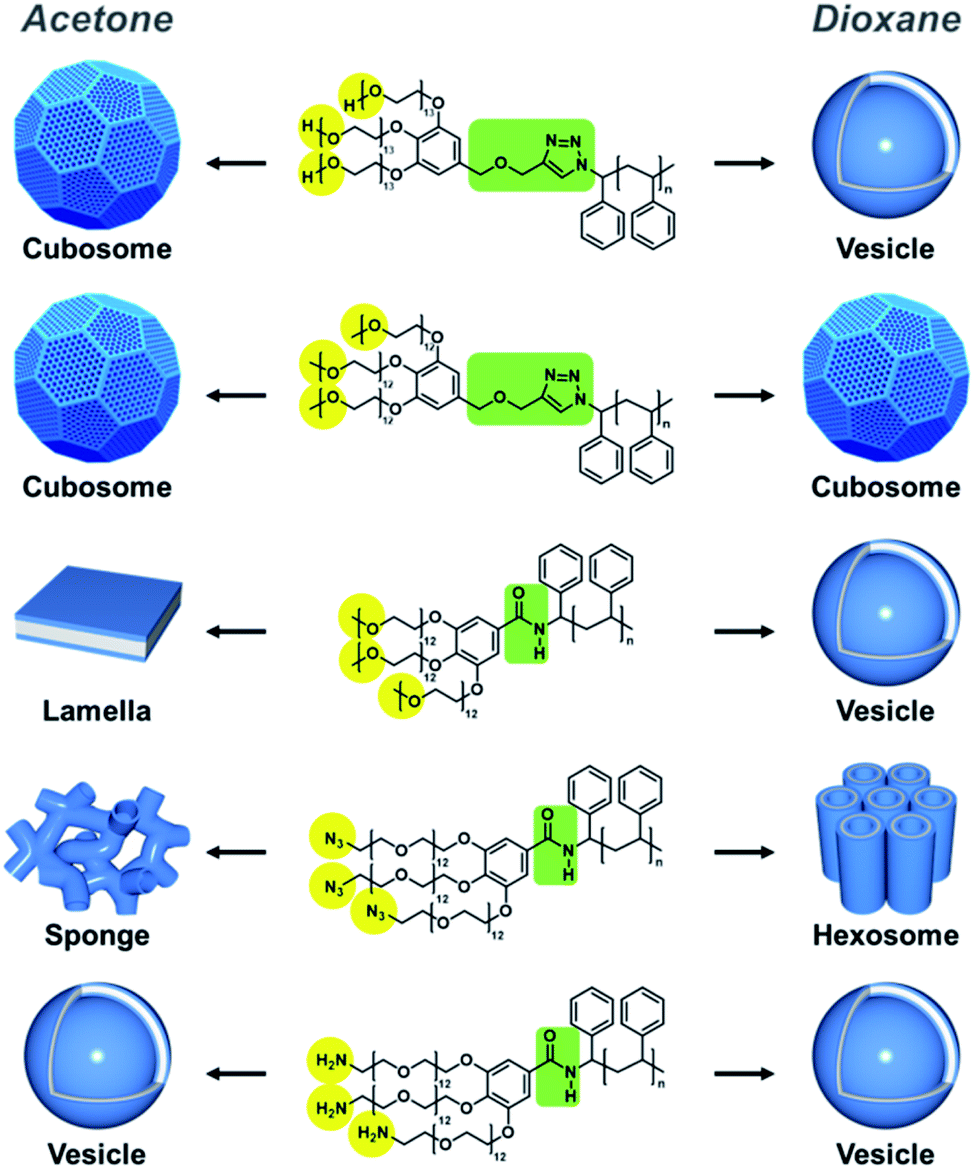 | ||
| Fig. 7 Schematic representation of BCP self-assembly showing major morphologies self-assembled in acetone (left) and dioxane (right). | ||
Remarkably, we found that the junction between the two blocks could also affect the BCP self-assembly. In both solvents, (MeO-PEG)3-trz-PS2 and (MeO-PEG)3-amd-PS2 self-assembled into cubosomes and mainly vesicles, respectively (Fig. 5c, d, S11a, b, S6a, and b†). Owing to the methoxy end groups, both BCPs exhibited small morphological changes upon varying the solvent from acetone to dioxane; this is due to the decrease in a0 offsetting the increase in lc. An amide unit can act both as a strong hydrogen-bonding donor and an acceptor, whereas a 1,2,3-triazole unit is a weak hydrogen bonding acceptor.47,48 Therefore, the hydrogen bonding between the amide bond in (MeO-PEG)3-amd-PS2 and other amide junctions or water molecules could affect the BCP conformation. The difference in the length of the junction between (MeO-PEG)3-trz-PS2 and (MeO-PEG)3-amd-PS2 could be another factor.
Based on the aforementioned reasons, (MeO-PEG)3-amd-PS2 may possess a larger a0 value than that of (MeO-PEG)3-trz-PS2, leading to vesicle formation, whereas (MeO-PEG)3-trz-PS2 forms cubosomes under the same self-assembly conditions. Moreover, the effect of hydrogen-bonding-forming units on the self-assembly process has been studied based on the supramolecular self-assembly of low-molecular-weight π-amphiphiles.49–53 Considering the differences in the self-assembly behaviors of (MeO-PEG)3-amd-PS2 and (MeO-PEG)3-trz-PS2, we suggest that a small difference in the junction could affect the self-assembly of high-molecular-weight BCPs. Regarding the effect of the junction, Hawker and co-workers previously demonstrated that the phase separation strength of a BCP could be increased by ionization of its triazole junction, which leads to electrostatic interactions between counterions of adjacent domain junctions.54 The self-assembly behavior with respect to the end groups and the junction is summarized in Table 2 and Fig. 7 for each common solvent.
| BCP | Self-assembled morphology | End group hydrophilicity | Junction | |
|---|---|---|---|---|
| Acetone | Dioxane | |||
| (HO-PEG)3-trz-PS1 | Cubosome spongesome | Vesicle | Hydrophilic | Triazole |
| (MeO-PEG)3-trz-PS2 | Cubosome | Cubosome | Hydrophobic | Triazole |
| (MeO-PEG)3-amd-PS2 | Lamella, vesicle | Vesicle | Hydrophobic | Amide |
| (N3-PEG)3-amd-PS1 | Sponge, vesicle | Hexosome | Hydrophobic | Amide |
| (H2N-PEG)3-amd-PS1 | Vesicle | Vesicle | Hydrophilic | Amide |
From the thermodynamic aspect, BCP self-assembly in solution relies on interfacial energy, which is determined by solvent affinity and block composition of the BCP.46,55 In the cosolvent method, the self-assembled morphology of a BCP depends on the affinity of the blocks to a solvent mixture of water and the common solvent. Considering the effect of the core-block hydrophobicity on BCP self-assembly,44–46 hydrophilicity of the corona block should also influence the self-assembled morphologies of the BCP. According to our findings, different end groups strongly affect the hydrophilicity of the corona block, thus leading to significant changes in self-assembled morphologies.
3. Conclusion
We have investigated the effect of different end groups on BCP self-assembly in solution. BCPs with hydroxyl, methoxy, azido, and amino groups at the end of the hydrophilic PEG block self-assembled into different morphologies ranging from vesicles to hexosomes under the same self-assembly conditions. Based on the packing parameter (p) analogy, we speculate that the interactions between the end groups and a common solvent (acetone and dioxane) or water could determine the headgroup area (a0) of the BCPs in solution, leading to the different self-assembly behaviors. Interestingly, different junctions between two blocks such as amide and triazole units also resulted in the self-assembly of different morphologies. This was attributed to the hydrogen bonding capability or the different lengths of the junctions. In summary, we have demonstrated that the end groups and junction, which account for a very small portion of an entire BCP, can significantly influence the BCP self-assembly. We envision that our findings will provide new insights to control the self-assembly of block copolymers.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge supports from the National Research Foundation (NRF) of Korea (2019R1A2C3007541), the Technology Innovation Program (or Industrial Strategic Technology Development Program-Alchemist Project) (20012390, 4D Molecular-Nano-Addressable Lithographic Self-Assembly (4D MONALISA)) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea), and Creative-Pioneering Researchers Programs (30520190050) by Seoul National University (SNU).Notes and references
- J. C. M. van Hest, D. A. P. Delnoye, M. W. P. L. Baars, M. H. P. van Genderen and E. W. Meijer, Science, 1995, 268, 1592–1595 CrossRef CAS PubMed
.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, Macromolecules, 2017, 50, 3439–3463 CrossRef CAS
- Y. La, C. Park, T. J. Shin, S. H. Joo, S. Kang and K. T. Kim, Nat. Chem., 2014, 6, 534–541 CrossRef CAS PubMed
- H. Yu, X. Qiu, S. P. Nunes and K.-V. Peinemann, Nat. Commun., 2014, 5, 4110 CrossRef CAS PubMed
- T. H. An, Y. La, A. Cho, M. G. Jeong, T. J. Shin, C. Park and K. T. Kim, ACS Nano, 2015, 9, 3084–3096 CrossRef CAS PubMed
- Y. La, T. H. An, T. J. Shin, C. Park and K. T. Kim, Angew. Chem., Int. Ed., 2015, 54, 10483–10487 CrossRef CAS PubMed
- Z. Lin, S. Liu, W. Mao, H. Tian, N. Wang, N. Zhang, F. Tian, L. Han, X. Feng and Y. Mai, Angew. Chem., Int. Ed., 2017, 56, 7135–7140 CrossRef CAS PubMed
- X. Lyu, A. Xiao, W. Zhang, P. Hou, K. Gu, Z. Tang, H. Pan, F. Wu, Z. Shen and X.-H. Fan, Angew. Chem., Int. Ed., 2018, 57, 10132–10136 CrossRef CAS PubMed
- Y. La, J. Song, M. G. Jeong, A. Cho, S.-M. Jin, E. Lee and K. T. Kim, Nat. Commun., 2018, 9, 5327 CrossRef CAS PubMed
- S. Ha and K. T. Kim, Polym. Chem., 2019, 10, 5805–5813 RSC
- J. Kim, M. Yoon, S.-M. Jin, J. Lee, Y. La, E. Lee and K. T. Kim, Polym. Chem., 2019, 10, 3778–3785 RSC
- H.-K. Liu, L.-J. Ren, H. Wu, Y.-L. Ma, S. Richter, M. Godehardt, C. Kübel and W. Wang, J. Am. Chem. Soc., 2019, 141, 831–839 CrossRef CAS PubMed
- X. Liu and I. Gitsov, Macromolecules, 2019, 52, 5563–5573 CrossRef CAS
- Y. Sun, J. Kim and K. T. Kim, RSC Adv., 2019, 9, 25423–25428 RSC
- S. Ha, Y. La and K. T. Kim, Acc. Chem. Res., 2020, 53, 620–631 CrossRef CAS PubMed
- H. Ma and K. T. Kim, Macromolecules, 2020, 53, 711–718 CrossRef CAS
- L. Zhang, C. Bartels, Y. Yu, H. Shen and A. Eisenberg, Phys. Rev. Lett., 1997, 79, 5034–5037 CrossRef CAS
- A. L. Parry, P. H. H. Bomans, S. J. Holder, N. A. J. M. Sommerdijk and S. C. G. Biagini, Angew. Chem., Int. Ed., 2008, 47, 8859–8862 CrossRef CAS PubMed
- B. E. McKenzie, F. Nudelman, P. H. H. Bomans, S. J. Holder and N. A. J. M. Sommerdijk, J. Am. Chem. Soc., 2010, 132, 10256–10259 CrossRef CAS PubMed
- S. J. Holder and N. A. J. M. Sommerdijk, Polym. Chem., 2011, 2, 1018–1028 RSC
- B. E. Mckenzie, S. J. Holder and N. A. J. M. Sommerdijk, Curr. Opin. Colloid Interface Sci., 2012, 17, 343–349 CrossRef
- B. E. McKenzie, J. F. de Visser, H. Friedrich, M. J. M. Wirix, P. H. H. Bomans, G. de With, S. J. Holder and N. A. J. M. Sommerdijk, Macromolecules, 2013, 46, 9845–9848 CrossRef CAS
- B. E. McKenzie, H. Friedrich, M. J. M. Wirix, J. F. de Visser, O. R. Monaghan, P. H. H. Bomans, F. Nudelman, S. J. Holder and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2015, 54, 2457–2461 CrossRef CAS PubMed
- B. E. McKenzie, J. F. de Visser, G. Portale, D. Hermida-Merino, H. Friedrich, P. H. H. Bomans, W. Bras, O. R. Monaghan, S. J. Holder and N. A. J. M. Sommerdijk, Soft Matter, 2016, 12, 4113–4122 RSC
- H. He, K. Rahimi, M. Zhong, A. Mourran, D. R. Luebke, H. B. Nulwala, M. Möller and K. Matyjaszewski, Nat. Commun., 2017, 8, 14057 CrossRef PubMed
- K. E. B. Doncom, L. D. Blackman, D. B. Wright, M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2017, 46, 4119–4134 RSC
- C. K. Wong, X. Qiang, A. H. E. Müller and A. H. Gröschel, Prog. Polym. Sci., 2020, 102, 101211 CrossRef CAS
- J. Xu, L. Tao, C. Boyer, A. B. Lowe and T. P. Davis, Macromolecules, 2011, 44, 299–312 CrossRef CAS
- J. Du, H. Willcock, J. P. Patterson, I. Portman and R. K. O'Reilly, Small, 2011, 7, 2070–2080 CrossRef CAS PubMed
- T. Liu, W. Tian, Y. Zhu, Y. Bai, H. Yan and J. Du, Polym. Chem., 2014, 5, 5077–5088 RSC
- G. Jo, H. Ahn and M. J. Park, ACS Macro Lett., 2013, 2, 990–995 CrossRef CAS
- G. Jo, O. Kim, H. Kim, U. Hyeok Choi, S.-B. Lee and M. Jeong Park, Polym. J., 2016, 48, 465–472 CrossRef CAS
- H. Y. Jung, P. Mandal, G. Jo, O. Kim, M. Kim, K. Kwak and M. J. Park, Macromolecules, 2017, 50, 3224–3233 CrossRef CAS
- J. Kim, H. Y. Jung and M. J. Park, Macromolecules, 2020, 53, 746–763 CrossRef CAS
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC
- S. Y. Khor, N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2017, 6, 1013–1019 CrossRef CAS
- M. Grzelakowski and K. Kita-Tokarczyk, Nanoscale, 2016, 8, 6674–6683 RSC
- H. Che, L. N. J. de Windt, J. Zhu, I. A. B. Pijpers, A. F. Mason, L. K. E. A. Abdelmohsen and J. C. M. van Hest, Chem. Commun., 2020, 56, 2127–2130 RSC
- K. Matyjaszewski, Y. Nakagawa and S. G. Gaynor, Macromol. Rapid Commun., 1997, 18, 1057–1066 CrossRef CAS
- R. R. Karimov, A. Sharma and J. F. Hartwig, ACS Cent. Sci., 2016, 2, 715–724 CrossRef CAS PubMed
- J. C. Foster, S. Varlas, B. Couturaud, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 15733–15737 CrossRef CAS PubMed
- S. Varlas, J. C. Foster, L. A. Arkinstall, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O'Reilly, ACS Macro Lett., 2019, 8, 466–472 CrossRef CAS PubMed
- J. C. Foster, I. Akar, M. C. Grocott, A. K. Pearce, R. T. Mathers and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 1700–1707 CrossRef CAS PubMed
- Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC
- D.-Y. Wang, L.-Y. You, J.-L. Wang, H. Wang, D.-W. Zhang and Z.-T. Li, Tetrahedron Lett., 2013, 54, 6967–6970 CrossRef CAS
- M. R. Molla and S. Ghosh, Chem.–Eur. J., 2012, 18, 9860–9869 CrossRef CAS PubMed
- P. Rajdev, M. R. Molla and S. Ghosh, Langmuir, 2014, 30, 1969–1976 CrossRef CAS PubMed
- A. Sikder, A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2015, 54, 6755–6760 CrossRef CAS PubMed
- A. Sikder, D. Ray, V. K. Aswal and S. Ghosh, Angew. Chem., Int. Ed., 2019, 58, 1606–1611 CrossRef CAS PubMed
- A. Sikder, Y. Xie, M. Thomas, M. J. Derry and R. K. O'Reilly, Nanoscale, 2021, 13, 20111–20118 RSC
- Y. Luo, D. Montarnal, N. J. Treat, P. D. Hustad, M. D. Christianson, E. J. Kramer, G. H. Fredrickson and C. J. Hawker, ACS Macro Lett., 2015, 4, 1332–1336 CrossRef CAS
- Q. Zhang, J. Lin, L. Wang and Z. Xu, Prog. Polym. Sci., 2017, 75, 1–30 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including synthetic procedures, characterization, and spectroscopy data. See DOI: 10.1039/d2ra00493c |
| This journal is © The Royal Society of Chemistry 2022 |