
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of carboxylic arylphosphines by hydrolysis of the trifluoromethyl group†
Daniel Herrera ,
Daniel Peral and
J. Carles Bayón*
,
Daniel Peral and
J. Carles Bayón*
Department of Chemistry, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallés, Spain. E-mail: joancarles.bayon@uab.cat
First published on 2nd March 2022
Abstract
Triarylphosphines substituted with carboxylic and trifluoromethlyl groups have been prepared by the hydrolysis of trifluoromethyl groups using fuming sulfuric acid and boric acid. The reaction has been studied in a set of homoleptic and heteroleptic trifluoromethylated triarylphosphines and offers a new synthetic procedure for the preparation of carboxylic phosphines with a relatively simple methodology. The degree of carboxylation is modulated by the reaction conditions and is sensitive to the substitution pattern of the starting trifluoromethylated phosphines. A pH-dependent procedure based on the amphiphilic character of these phosphines was developed for their separation and purification. The electronic properties of the synthesized carboxylic-trifluoromethylated phosphines have been analyzed by 31P NMR of the corresponding selenide derivatives. Finally, the structures of two palladium complexes, containing the para and meta carboxylic-trifluoromethylated phosphines are also described, showing different dimeric structures.
Introduction
Phosphines containing carboxylic groups have been widely reported in the literature. The amphiphilic behaviour that the carboxylic groups confer to the phosphines has been found of interest in several applications such as their use as water-soluble reducing agents of disulfide bonds1 or as ligands in transition metal-based catalysis, either by coordination through the phosphorus atom2 or acting as chelating ligands.3 Moreover, the carboxylic derivatives of triphenylphosphine have been used as building blocks in the preparation of porous coordination materials.4 In spite of the versatility of carboxylic phosphines, certain considerations should be born in mind in regard to their preparation. The most extended procedures for the general synthesis of phosphines, such as the reaction of chlorophosphines or phosphorus trichloride with Grignard reagents or organolithium derivatives, as well as the nucleophilic reaction between alkali phosphides and haloderivatives, are not compatible with the presence of acidic protons. In the last case, the incompatibility between the phosphide anions and the carboxylic acid can be overcome using strong basic conditions, although sometimes this is accompanied by solubility issues. In this regard, several other alternative strategies have been developed for the synthesis of carboxylic phosphines. Ravindar et al. reported the reaction of carbonation of organolithium derivatives to yield monocarboxylic triarylphosphines.5 The hydrolysis of the nitrile group has also been found to yield mono-, di- and tri-carboxylic triarylphosphines, and has also been applied to some alkyl phosphines.6 Finally, Pd(II)-catalysed P–C coupling has been also used for the preparation of carboxylic phosphines.7Carbonyl compounds can be prepared from a trifluoromethyl group under strong acidic conditions. In 1943, Simons and Ramler8 and Gilman and Blume9 reported the hydrolysis of the trifluoromethyl group, using sulfuric acid, as a methodology to establish the structure of different trifluoromethylbenzene derivatives. Later, Le Fave et al. reported the formation of benzoic acids from the hydrolysis of trifluoromethylbenzene and derivatives, using concentrated sulfuric acid, followed by the treatment of the mixture with water.10 They also proved that benzoate esters could be obtained when treating the reaction mixtures with alcohols instead of water.11 Furthermore, the same authors described the preparation of phthalic acids by hydrolysis of the corresponding bis(trifluoromethyl)benzenes in an equimolar mixture of sulfuric acid and chlorosulfuric acid.12 In spite of these early publications, the reaction of hydrolysis of the trifluoromethyl group has not attracted much interest and only a few more examples have been reported after the pioneering studies of Le Fave. Wen and co-workers applied the reaction of hydrolysis, using fuming sulfuric acid, in the preparation of cholesteryl p-perfluoroalkyl benzoates.13 Platonov and co-workers showed the formation of perfluorobenzoic acids from the reaction of the corresponding perfluorotoluene derivative with CF3CO2H/SbF5 and the subsequent aqueous treatment.14 Finally, Kethe et al. studied the hydrolysis of one trifluoromethyl group in 1,3,5-tris(trifluoromethyl)benzene with either triflic acid or fluorosulfuric acid.15
Despite the above mentioned applications of the hydrolysis of the trifluoromethyl group, to the best of our knowledge, no precedents are known about the application of this reaction in the synthesis of phosphines. We envisioned the use of fuming sulfuric acid in combination with boric acid to promote the hydrolysis of the trifluoromethyl group in trifluoromethylated triarylphosphines. Amongst the available superacid systems, fuming sulfuric acid is strong (−H0 up to 14.9 in 65 wt% SO3 oleum),16 non-expensive and, most importantly, with a relatively lower oxidation power compared to other stronger super acids such as magic acid (i.e. FSO3H·SbF5). Moreover, it has been previously studied in the sulfonation of phosphines.17 Herrmann and co-workers showed that the oxidizing power of fuming sulfuric acid could be reduced in combination with boric acid thus preventing the oxidation of the phosphines during the process of sulfonation.17a In this sense, the studies of Gillespie and co-workers showed that the addition of boric acid to sulfuric acid generates a strong superacid of the sulfuric acid system, H3SO4[B(OSO3H)4].18 More recently, Peral et al. reported the use of oleum and boric acid in the sulfonation of the strongly deactivated trifluoromethylated phosphines with minor yields of phosphine oxides.17b,c
Based on the previous examples, the reaction of hydrolysis of the trifluoromethyl group offers a new synthetic procedure to obtain carboxylic arylphosphines through a relatively simple procedure. In this methodology, the trifluoromethyl group will be used as a masked carboxylic acid, overcoming the difficulties that arise from the incompatibility of carboxylic acids with the reagents commonly used in the preparation of phosphines. Most interestingly, this methodology provides the preparation of arylphosphines containing both carboxylic and trifluoromethyl groups. On the one hand, the carboxylic group confers chemical versatility to phosphines, covering a wide variety of applications, including those derived from the amphiphilic character of the carboxylic group. On the other hand, the trifluoromethyl group confers greater π-acidity to the phosphines, which turns into a higher stability towards their oxidation.17c,19 Phosphines with π-acidic character have proven to increase the activity of certain transition metal-catalysed reactions such as some C–C couplings20 and carbonylations.17c,21
A few examples of carboxylic and trifluoromethylated arylphosphines have been previously described in the literature. Reek's group reported the synthesis of carboxylic and trifluoromethylated phosphines through a Pd catalysed P–C coupling.22 More recently, Starkov et al. described the synthesis of an ortho-carboxylic trifluoromethylated phosphine by a several-steps methodology in which the P–C bond is constructed through a Cu-mediated coupling with the carboxylic group protected as methyl ester.23
Here we report the preparation of carboxylic phosphines by the reaction of hydrolysis of the trifluoromethyl group in trifluoromethylated triarylphosphines, using fuming sulfuric acid and boric acid. The carboxylic-trifluoromethylated phosphines have been successfully separated and purified, and their electronic properties have been studied by 31P{1H} NMR using the selenium derivatives. Palladium complexes of the para and meta carboxylic and bis(trifluromethylated) triarylphosphine were also structurally characterized showing different supramolecular patterns, illustrating the capability of this family of ligands to build diverse intermolecular networks.
Results and discussion
The synthesis of carboxylic triarylphosphines was carried out from the hydrolysis of the trifluoromethyl group of a series of triarylphosphines with different substitution pattern, Scheme 1. The reaction was performed using a highly acidic media prepared with a mixture of boric acid and fuming sulfuric acid (oleum). The fine tuning of the reaction conditions allows the control of the yield and selectivity of the reaction. The protection towards the oxidation of the phosphines under these highly acidic conditions takes place through the protonation of the phosphorus atom.17a For this reason, complete solubility of the phosphines is necessary before the addition of fuming sulfuric acid. It is important to notice that water is formed when mixing H3BO3 and sulfuric acid, previous to the addition of the phosphines, eqn (1),17a,18 thus changing the polarity of the media and decreasing the acidity of the mixture. Therefore, sulfur trioxide from oleum is required to react with water shifting the equilibrium of eqn (1) towards the formation of the superacid.| H3BO3 + 2H2SO4 + 3SO3 ⇄ H3SO4+ + B(OSO3H)4− | (1) |
 | ||
| Scheme 1 Synthesis of carboxylic triarylphosphines from trifluoromethylated phosphines. (i) H3BO3/H2SO4/SO3. (ii) H2O. | ||
As it will be explained below, the superacid plays an important role, not only protecting the phosphines from oxidation, but also in the hydrolysis of the trifluoromethyl groups.
Synthesis of carboxylic phosphines from homoleptic trifluoromethylated phosphines
The hydrolysis of the trifluoromethyl groups of three homoleptic phosphines (SP1, SP2, SP3) were firstly studied.In a preliminary set of experiments, the para-substituted tris(4-trifluoromethylphenyl)phosphine (SP1) and the chosen amount of boric acid were dissolved in sulfuric acid before the addition of 65% SO3 fuming sulfuric acid. The evolution of the reaction was monitored by aliquoting the sulfuric mixture. After quenching the samples in cold water, the phosphines were extracted with diethyl ether and analysed by 31P{1H} NMR to quantify the distribution phosphorus compounds present in the mixture. Control analysis of the aqueous layers showed that all phosphorus compounds were completely extracted into the organic phase. Three carboxylic products were observed as result of the hydrolysis of one, two or all the trifluoromethyl groups.
The reaction was explored and optimized at different concentrations of phosphine, H3BO3 and SO3 (ESI Fig. S1†). The reaction rates of the hydrolysis of the trifluoromethyl group were observed to strongly depend on the concentration of H3BO3 and SO3. High concentrations of SO3 (above 12 M) gave faster reaction rates although led to the complete oxidation of the phosphine products at short reaction times. As expected, increasing H3BO3 concentration was found to decrease the rate of phosphine oxidation. However, and contrary to what was observed for the reactions of sulfonation of triarylphosphines,17b,c the use of H3BO3 showed a positive effect on the rate of the trifluoromethyl hydrolysis reaction. The most suitable conditions for the preparation of the monocarboxylic phosphine MC1 and the dicarboxylic phosphine DC1 were found at concentrations of SO3 and H3BO3 of 10.5 M and 0.8 M, respectively, with initial phosphine concentration of 0.15 M. Fig. 1 shows the graphical representation of the consumption of the trifluoromethylated starting phosphine SP1 together with the evolution of the monocarboxylic phosphine MC1, the dicarboxylic phosphine DC1 and the tricarboxylic phosphine TC1 under the optimized conditions. In addition to the molar fraction of the phosphines, the sum of phosphine oxides present in the mixture is also represented.
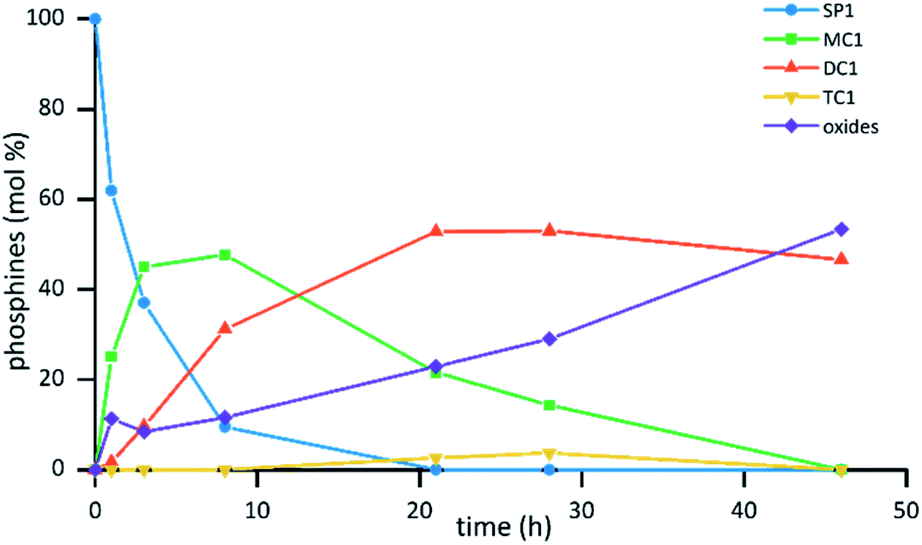 | ||
| Fig. 1 Evolution of the reaction of hydrolysis in phosphine SP1 in sulfuric acid with time. Reaction conditions [SP1]: = 0.15 M, [H3BO3] = 0.8 M, [SO3] = 10.5 M. | ||
In the optimized conditions, after 1 h, the reaction mixture showed 25% of the monocarboxylic compound MC1, together with 11% of phosphine oxides. After 3 h of reaction, the amount of MC1 increased to 45% and 10% of the dicarboxylic phosphine DC1 was detected, with no significant variation of the amount of phosphine oxides. When the reaction was analyzed after 21 h, the mixture showed complete consumption of the starting phosphine SP1, it being the dicarboxylic phosphine DC1 the major component of the mixture (53%) together with 22% of the monocarboxylic phosphine MC1 and 23% of oxides. Moreover, in this experiment, only traces of the tricarboxylic phosphine TC1 were detected (ca. 2%).
Fine tuning of the reaction conditions and reaction time allows the control of the composition of the reaction mixture. For the preparation of the monocarboxylic phosphine MC1, reaction times can be shortened to 4 hours at the optimized conditions described above, to get 50% of this carboxylic phosphine together with the unreacted phosphine SP1, which after proper separation, could be reused in a new reaction. If the desired product is the dicarboxylic phosphine DC1, the reaction mixture is quenched after 14 h to obtain a 50% mixture of MC1 and DC1, Table 1. Moreover, increasing the concentration of H3BO3 to 2.4 M, it is possible to obtain 46% of the tricarboxylic phosphine TC1, together with 36% of the dicarboxylic phosphine DC1 and below 25% of phosphine oxides after 20 hours. Although such conditions need fairly long reaction times, they open a new synthetic approach to the already known procedures for preparing this tricarboxylic phosphine.6
| Carboxylic phosphine | Starting phosphine | Reaction conditions | Hydrolysis yield (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| [SP] (M) | [SO3] (M) | [H3BO3] (M) | Time (h) | SP | MC | DC | TC | Oxides | ||
| MC1 | SP1 | 0.15 | 10.5 | 0.8 | 4 | 46 | 46 | 0 | 0 | 8 |
| DC1 | SP1 | 0.15 | 10.5 | 0.8 | 14 | 4 | 40 | 48 | 0 | 8 |
| MC2 | SP2 | 0.15 | 10.5 | 0.8 | 0.5 | 48 | 41 | 6 | 0 | 5 |
| DC2 | SP2 | 0.15 | 10.5 | 0.8 | 20 | 0 | 6 | 60 | 20 | 14 |
| MC4–DC4 | SP4 | 0.07 | 9 | 0.8 | 18 | 9 | 48 | 34 | — | 9 |
| MC5 | SP5 | 0.08 | 9 | 0.4 | 50 | 0 | 63 | — | — | 37 |
The synthesis of meta-carboxylic phosphines was also essayed from the homoleptic trifluoromethylated phosphine SP2 using the same optimized conditions than for the para-substituted phosphine SP1. The evolution of the reaction analysed by 31P NMR also showed the hydrolysis of one, two and three trifluoromethyl groups producing the monocarboxylic phosphine MC2, the dicarboxylic phosphine DC2 and the tricarboxylic phosphine TC2, respectively, Scheme 1. Interestingly, the rate of the reaction of hydrolysis of the trifluoromethyl groups in meta is much faster than that of the trifluoromethyl groups in para position (ESI Fig. S2†). For the synthesis of the monocarboxylic phosphine MC2, the reaction is quenched after 0.5 h to obtain a mixture of 48% of SP2 and 41% of MC2 with only traces of the dicarboxylic phosphine. For the preparation of the dicarboxylic phosphine DC2, the reaction was left for 20 h to obtain a mixture of 6% of MC2, 60% of DC2 and 20% of the tricarboxylic phosphine TC2, Table 1. As previously shown for SP1, with the meta-isomer SP2, it is also possible to tune the composition of the mixture before the separation process adjusting the reaction conditions.
Based on the faster reactivity of the trifluoromethyl groups in meta position with respect to the para substituted phosphines, the reaction of hydrolysis of the trifluoromethyl groups of the 3,5-substituted phosphine SP3 was tested, Chart 1. Unexpectedly, the hydrolysis in this doubly meta-substituted phosphine SP3 was much slower than for the hydrolysis of the single-substituted phosphine SP2. Using the optimized conditions for the preparation of the para- and meta-carboxylic phosphines, the progress of the reaction for SP3, analysed by NMR, showed considerable slower rates for the hydrolysis of the trifluoromethyl groups than in the case of SP1 or SP2, yielding phosphine oxides as main products. Mass spectrometry analysis of the reaction mixture after 20 h confirmed the presence of the monocarboxylic phosphine, the monocarboxylic phosphine oxide and a dicarboxylic phosphine oxide. However, the pattern of carboxylation in the dicarboxylic compound (i.e. in the same or different ring), could not be determined.
 | ||
| Chart 1 Trifluoromethylated phosphine SP3. | ||
Synthesis of carboxylic phosphines from heteroleptic trifluoromethylated phosphine
Given the feasibility of the synthesis of carboxylic phosphines from the hydrolysis of the para (SP1) and meta-substituted (SP2) trifluoromethylated phosphines and the poor reactivity of the trifluoromethyl groups in the 3,5-substituted phosphines (SP3), it was appealing to study the reaction of hydrolysis in heteroleptic phosphines containing both patterns of substitution, (SP4, SP5 in Scheme 1 and SP6 and SP7 in Chart 2). | ||
| Chart 2 Heteroleptic trifluoromethylated phosphines SP6 and SP7. | ||
As a general trend, low solubility of the heteroleptic phosphines in the H3BO3/H2SO4 mixture, previous to the addition of oleum, was observed yielding to high percentages of oxides at short reaction times. The solubility of the phosphines decreased when increasing the number of trifluoromethyl groups and this was more evident with the para-substituted heteroleptic phosphines (SP6 and SP7). Moreover, the hydrolysis of the trifluoromethyl group with the heteroleptic phosphines showed slower reaction rates for the para-isomers (SP6 and SP7) than for the meta-substituted phosphines (SP4 and SP5), a trend already observed for the homoleptic phosphines SP1 and SP2. Furthermore, hydrolysis of the trifluoromethyls at 3,5-positions was not detected. It is important to note that the reaction rates were found to be lower when increasing the number of trifluoromethyls in the phosphines (ESI Fig. S3†).
Optimized conditions were found for the synthesis of the meta-carboxylic phosphines MC4, DC4 and MC5, as shown in Table 1. Long reaction times were needed to obtain the carboxylic phosphines in moderate yields, due to the diluted phosphine concentrations required to minimize the initial oxidation of SP4 and SP5.
For the para-heteroleptic phosphines SP6 and SP7 the reaction was found to be very slow. The large amount of phosphine oxides and the slow reactivity of the para-trifluoromethyls jeopardize the preparation and isolation of the corresponding carboxylic phosphines. After several attempts to optimize the reactions, no suitable conditions were found for the preparation and isolation of the corresponding carboxylic phosphines. Therefore, other reported procedures would be more appropriate for the formation of these carboxylic phosphines.5,7,22,23
Separation and purification of the carboxylic phosphines
In all the synthetic procedures described above, a mixture of different phosphines is obtained after the reaction of the trifluoromethylated starting phosphines with H3BO3 and oleum. A general methodology for the separation and purification of the carboxylic-trifluoromethylated phosphines has been developed based on the pH-dependent solubility of carboxylic acids. The separation process has been optimized for a mixture containing all the possible phosphine compounds derived from the reaction of hydrolysis of trifluoromethylated phosphines, that is, starting phosphine (SP), monocarboxylic (MC), dicarboxylic (DC), tricarboxylic (TC) and phosphine oxides, to cover all possible scenarios. A scheme of the separation is presented in Fig. 2. | ||
| Fig. 2 Separation of the phosphines products of the reaction of hydrolysis of the trifluoromethyl group. | ||
After the hydrolysis reaction, the mixture was poured on ice and the phosphorus compounds were extracted from the aqueous sulfuric media using diethyl ether. Water was added to the previous Et2O solution, yielding a new biphasic organic-aqueous system. The aqueous phase was brought to pH 6.0 using an aqueous solution of NaOH to completely extract the tricarboxylated phosphine (TC) into the aqueous phase, together with its oxide and the oxide of DC, when present. After phase separation, water was added again to the ethereal phase and further basification to reach a pH of about 7.5 allowed the extraction of the dicarboxylic phosphine (DC) into the aqueous solution. Within this range of pH, also the oxide of the monocarboxylic compound (MC) is extracted. The acidification of each of the aqueous phases containing the sodium salts of the dicarboxylate and tricarboxylate phosphines to pH 2, allowed the re-extraction of the phosphines in the acidic form in diethyl ether.
The extraction of the monocarboxylic phosphine (MC) from the ethereal phase by further increasing the pH of the aqueous phase is not possible because of the formation of stable emulsions. Therefore, an alternative procedure was developed starting by vacuum evaporation of the diethyl ether. To the solid residue containing SP and MC, a solution of 10% NaOH in methanol was added dropwise. After complete deprotonation of the carboxylic group of MC, monitored by TLC (CH2Cl2), methanol was vacuum evaporated and cyclohexane was added to the solid residue to extract the unreacted starting phosphine (SP). Finally, phosphine MC was recovered again as carboxylic acid by acidification to pH 2 followed by extraction in diethyl ether.
With this procedure, it was possible to prepare a whole set of mono- and dicarboxylic trifluoromethylated triarylphosphines, Scheme 1.24,25 These phosphines are a proof concept of the application of the hydrolysis of the trifluoromethyl group in trifluoromethylated arylphosphines using sulfuric fuming acid and H3BO3, and open a new synthetic route in the synthesis of these sort of compounds.
Reaction mechanism
The nature of the highly acidic medium used for the hydrolysis of the trifluoromethyl groups hampers the study of the reaction mechanism. Nevertheless, with 31P{1H} and 19F{1H} NMR experiments it is possible to analyse in situ the progression of the reaction. Fig. 3 shows the evolution of the hydrolysis of the trifluoromethyl group in the p-tris(trifluoromethylphenyl)phosphine (SP1) monitored by 31P{1H} and 19F{1H} NMR. | ||
| Fig. 3 Evolution of the reaction of hydrolysis in phosphine SP1. Analysis of the crude reaction by 31P{1H} (101.27 MHz) (left) and 19F{1H} NMR (235.4 MHz) (right). Signals (intensity and position) relative to NH4PF6 used as internal standard (−144.45 ppm and −72.65 ppm, respectively). δ in ppm. In 31P{1H} NMR, only phosphine region is shown. p1, m1 and d1 refer to the precursors of phosphines SP1, MC1, and DC1 in sulfuric medium. Reaction conditions: [phosphine] = 0.16 M, [H3BO3] = 0.8 M, [SO3] = 11.5 M. | ||
Importantly, a singlet in the 19F{1H} spectra at 42.6 ppm, corresponding to the –OSO2F moiety26 was detected. The intensity of this signal increased as new phosphine signals were observed in the 31P{1H} NMR spectra. Besides the signal of the trifluoromethyl groups (around −63 ppm), no other fluorinated species were found in these experiments.
Based on the NMR experiments and the previously reported reactions of trifluoromethylated aryl compounds in acidic media,15,27 a mechanism for the hydrolysis of the trifluoromethyl group in trifluoromethylated triarylphosphines using fuming sulfuric acid and H3BO3 can be postulated, Scheme 2.
 | ||
| Scheme 2 Proposed reaction mechanism of the hydrolysis of the trifluoromethyl group in trifluoromethylated phosphines with sulfuric acid. | ||
In a first step, strong interaction between the acidic protons of the superacid and fluorine promotes the C–F bond cleavage releasing a HF molecule and generating the difluorobenzylic carbocation (A), in analogy with previously reported experimental and theoretical studies.27 Next, the nucleophilic attack of a molecule of H2SO4 on the carbocation A yields the formation of the hydrogensulfuric ester B. The reaction may be favoured by the highly electrophilic fluorinated carbocation. Following, the concerted elimination of HSO3F (ca. 40–42 ppm in 19F{1H}) would give the corresponding acyl fluoride (C) similarly to previously reported studies.15,27b Finally, the acyl fluoride (C) would react with sulfuric acid to yield the mixed sulfonic-carboxylic anhydride (D).28 Although the formation of mixed anhydrides by addition of SO3 to acyl fluorides has been reported in the literature,26 this option was discarded considering that such reaction requires the use of pure SO3.29 Moreover, the signal for the corresponding fluorinated mixed anhydride in 19F{1H} NMR (above 45 ppm) was not detected. After quenching the reaction in water, D is hydrolysed to form the carboxylic acid (E). It is worth mentioning that neither the signal for the fluorinated carbocation (A) (ca. 23 ppm)30 nor the corresponding signal for the acyl fluoride (C) (ca. 17.7 ppm)31 or any other intermediates were detected in the 19F{1H} NMR experiments. Therefore, the elimination of HF to form the carbocation (A) is likely to be the rate determining reaction step, in agreement with the mechanism proposed by Kethe et al. in the hydrolysis of the trifluoromethyl group in trifluoromethylbencenes using triflic acid or fluorosufuric acid.15
In the proposed mechanism, H3BO3 and SO3 would be involved in the generation of the superacid mixture. The 11B NMR spectra of the reaction mixtures containing phosphine, sulfuric acid, oleum and boric acid only showed the tetrahedral species B(OSO3H)4− (ESI Fig. S4†). Gillespie and co-workers showed that binary mixtures of sulfuric acid with either H3BO3 or SO3 give rise to an increase in the acidity of the media.16,18 In the case of the present work, the increase in the acidity produced by SO3 and H3BO3 would contribute to the C–F bond cleavage in the first step of the reaction.
Based on the proposed mechanism, the different reaction rates between phosphines with different substitution pattern can be explained through the resonant structures. Since phosphines are protonated under the reaction conditions, the formation of the difluorobenzylic carbocation (A) implies the generation of a second positive charge on the molecule. While in para-substituted phosphines the positive charge of the phosphonium can be delocalised at the adjacent atom to the carbocation, in meta-substituted phosphines this charge is never localised next to the other (ESI Fig. S5†). Therefore, the formation of the difluorobenzylic carbocation would be more favourable when the trifluoromethyl group is in meta-position than when located in para. Moreover, the extremely low reaction rates observed with those trifluoromethyls in 3,5-positions might be attributed to inductive effects.
Study of the σ-basicity of the trifluoromethylated-carboxylic phosphines: synthesis of the phosphine selenides
Phosphines have been widely studied as ligands in transition-metal catalysed reactions. The stereoelectronic properties of these ligands are determined by their substituents and play an essential role on the catalytic performance. Selenide derivatives have been used as a simple tool to study the σ-donation properties of phosphines through the 1JPSe coupling of the 77Se isotopomer, measured by 31P NMR.19,32,33 The selenide derivatives of the carboxylic-trifluoromethylated phosphines have been prepared following a previously reported methodology in trifluoromethylated triarylphosphines.19As previously observed in trifluoromethylated phosphines, the 1JPSe increases with the number of trifluoromethyl groups, thus decreasing the basicity of the ligands (ESI Table S1†). The hydrolysis of each trifluoromethyl group producing a new carboxylic group causes a moderate decrease, between 2 and 5 Hz, in the 1JPSe of the phosphine selenides, consistent with the decrease in the electron withdrawing power of the substituents. In previous works,19,33 it has been proven that the introduction of trifluoromethyl groups in triarylphosphines was cumulative and resulted in the increase of 12 Hz in the 1JPSe, regardless of the position of the group. Same observation was found in the case of the trifluoromethylated-carboxylic phosphines. Similarly, the introduction of carboxylic moieties is also cumulative and adds 9 Hz to the 1JPSe. Consequently, eqn (2) predicts the values of the 1JPSe in trifluoromethylated and/or carboxylic triarylphosphines with an error ≤3 Hz
| 1JPSe = 731 + 12n + 9m | (2) |
Trans-PdCl2L2 complexes of phosphines MC1 and MC2
The two monocarboxylic phosphines were used to synthesise the corresponding trans-PdCl2L2 (L = MC1 and MC2) complexes crystals structures of both complexes were solved showing the formation of different dimeric structures by hydrogen bonding.Both palladium complexes showed a slightly distorted square-planar geometry. The para-isomer PdCl2(MC1)2 was obtained as a dimeric structure where complexes are bridged by a carboxylic acid group of one of the phosphines of each of the monomers. The other carboxylic phosphine interacts through H-bonding with a molecule of acetone, Fig. 4. The meta-isomer PdCl2(MC2)2 also forms a dimeric structure. However, in this case, both carboxylic phosphines participate in the H-bond network, resulting in a cage type structure, Fig. 5. Inside this cage, badly disordered solvent molecules were observed, in the shape of residual peaks, fenced by the aromatic rings with the trifluoromethyl substituents.
 | ||
| Fig. 4 Stick representation of the dimeric structure of the complex of trans-[PdCl2(MC1)2]·acetone. Only one conformation of the disordered CF3 is shown. Hydrogen bond distances (Å): O1–O2, 2.612(3); O3–O1S, 2.708(5). | ||
 | ||
| Fig. 5 Stick representation of the dimeric structure of the complex of trans-[PdCl2(MC2)2]. Only one conformation of the disordered CF3 is shown. Hydrogen bond distances (Å): O1–O4, 2.673(9); O2–O3, 2.608(8). | ||
Conclusions
The reaction of hydrolysis of the trifluoromethyl group using fuming sulfuric acid and H3BO3 allows the preparation of carboxylic and trifluoromethylated triarylphosphines. The reactivity of the phosphines towards the hydrolysis of the trifluoromethyl group depends on the relative position of this group with respect to the phosphorus atom, with meta-position showing the fastest rate. The number of trifluoromethyl groups present in the phosphines also influences the rate of the reaction. Lower reactivity was found when increasing the number of trifluoromethyl groups. The reaction mechanism of the hydrolysis of the trifluoromethylated phosphines in the conditions described in the present work has been proposed based on in situ NMR experiments. The differences observed in the rate of the reaction between the para- and the meta-substituted phosphines are consistent with the formation of a difluorobenzyl carbocation in the rate determining step of the reaction.The separation and purification of the phosphines was carried out by a pH-dependent procedure, based on the amphiphilic character of the carboxylic moiety. The reaction has been successfully optimized to prepare seven carboxylic and trifluoromethylated phosphines in fair to good yields.
The σ-donation properties of the carboxylic-trifluoromethylated phosphines was studied by means of the 31P{1H} NMR spectra of the selenide derivatives. Both carboxylic and trifluoromethyl groups decrease the σ-basicity character of the phosphines although, as expected, trifluoromethyls have a larger influence. The correlation between the experimental values of 1JPSe and the number of carboxylic and trifluoromethyl groups was empirically studied allowing the prediction of the σ-donation properties of para- and meta-substituted carboxylic-trifluoromethylated triarylphosphines.
The trans-PdCl2(MC)2 complexes of carboxylic phosphines MC1 and MC2 were synthesised. Their crystal structures show the diversity of framework that complexes of these ligands can produce.
Experimental
General procedure and reagents
Unless otherwise mentioned, all the synthetic manipulations were performed under nitrogen atmosphere using standard Schlenk techniques.34 Solvents and liquid reagents were deoxygenated by bubbling nitrogen for 10–15 minutes. General chemical reagents and solvents were purchased from commercial sources and, otherwise specified were used without further purification. Anhydrous solvents and reagents were used when required and were dried by using different standard procedures.35 The NMR spectra were recorded in a Bruker Avance 250 and a Bruker Avance III 400 at the Servei de Ressonància Magnètica Nuclear at the UAB. Chemical shifts in 1H and 13C NMR are relative to the signal of the solvent.36 In 31P NMR chemical shifts are relative to 85% H3PO4 (0.0 ppm), used as external standard. For 19F NMR and 11B NMR experiments the signals are relative to flurobenzene (−113.15 ppm with respect to CFCl3) and BF3·Et2O (0.0 ppm), respectively. Unless otherwise stated, all the NMR experiments were performed at 298 K. High resolution mass-spectrometry analyses and elemental analyses (C and H) were performed by the Servei d'Anàlisi Química at UAB in a Bruker microTOF spectrometer coupled to an Apollo II electrospray source (ESI) and a Thermo Fisher Scientific Flash EA 2000 CHNS, respectively.Synthesis of the trifluoromethylated triarylphosphines SP1–SP7 was performed according to the previously reported methodologies in the literature.19,37
Synthesis of carboxylic phosphines38
3-((3,5-Bis(trifluoromethyl)phenyl)(3-(trifluoromethyl)phenyl)phosphanyl)benzoic acid (MC4) and 3,3′-((3,5-bis(trifluoromethyl)phenyl)-phosphanediyl)dibenzoic acid (DC4). The same procedure described for the hydrolysis of the trifluoromethyl groups in the homoleptic phosphines SP1 and SP2 was followed for the heteroleptic phosphine SP4. In a typical experiment, a solution of 2.5 g of phosphine SP4 (4.7 mmol) and 3.25 g of boric acid (52.6 mmol) in 30 ml of 98% sulfuric acid was cooled down to 0 °C in an ice bath. Following, 37 ml of 65% SO3 fuming sulfuric acid (0.6 mol of SO3) were added dropwise and the mixture was allowed to react for 18.5 h at room temperature. The mixture was then quenched on 180 g of deoxygenated ice and extracted three times with 100 ml of Et2O. Biphasic extractions at pH 6.7 allows the separation of the oxide of phosphine DC4 in the aqueous phase while the dicarboxylic phosphine DC4 is extracted pH 8.5. The monocarboxylic phosphine MC4 is purified as reported for phosphines MC1 and MC2. After full separation and purification, 0.67 g of DC4 (28% yield) and 0.70 g of MC4 30% yield) were obtained as white solids.
X-ray crystallography
Crystallographyc data for trans-[PdCl2(MC1)2]·acetone and trans-[PdCl2(MC2)2] are provided free of charge from the Cambridge Crystallographic Data Centre with deposition numbers 2097178 and 2106238, respectively.Crystallographyc details about the two structures can be found in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. H and D. P. thank Universitat Autònoma de Barcelona for their PIF grant.Notes and references
- (a) J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648–2650 CrossRef CAS; (b) E. B. Getz, M. Xiao, T. Chakrabarty, R. Cooke and P. R. Selvin, Anal. Biochem., 1999, 273, 73–80 CrossRef CAS PubMed.
- (a) A. Buhling, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Mol. Catal. A: Chem., 1995, 98, 69–80 CrossRef CAS; (b) A. Buhling, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. W. Elgersma, J. Mol. Catal. A: Chem., 1997, 116, 297–308 CrossRef CAS; (c) D. H. Chang, D. Y. Lee, B. S. Hong, J. H. Choi and C. H. Jun, J. Am. Chem. Soc., 2004, 126, 424–425 CrossRef CAS PubMed.
- (a) M. Peuckertt and W. Keim, Organometallics, 1983, 2, 594–597 CrossRef; (b) M. R. Ringenberg, D. L. Gray and T. B. Rauchfuss, Organometallics, 2011, 30, 2885–2888 CrossRef CAS; (c) M. M. Taqui Khan, M. K. Nazeeruddin and H. C. Bajaj, Inorg. Chim. Acta, 1988, 143, 177–184 CrossRef; (d) M. Guo, F. Jian and R. He, J. Fluorine Chem., 2006, 127, 177–181 CrossRef CAS.
- (a) A. J. Nuñez, L. N. Shear, N. Dahal, I. A. Ibarra, J. Yoon, Y. K. Hwang, J. S. Chang and S. M. Humphrey, Chem. Commun., 2011, 47, 11855–11857 RSC; (b) A. J. Nuñez, M. S. Chang, I. A. Ibarra and S. M. Humphrey, Inorg. Chem., 2014, 53, 282–288 CrossRef PubMed; (c) J. Václavík, M. Servalli, C. Lothschütz, J. Szlachetko, M. Ranocchiari and J. A. van Bokhoven, ChemCatChem, 2013, 5, 692–696 CrossRef; (d) A. A. Bezrukov, K. W. Törnroos and P. D. C. Dietzel, Cryst. Growth Des., 2017, 17, 3257–3266 CrossRef CAS.
- (a) V. Ravindar, H. Hemling, H. Schumann and J. Blum, Synth. Commun., 1992, 22, 841–851 CrossRef CAS; (b) V. Ravindar, H. Hemling, H. Schumann and J. Blum, Synth. Commun., 1992, 22, 1453–1459 CrossRef CAS.
- (a) M. M. Rauhut, I. Hechenbleikner, H. A. Currier, F. C. Schaefer and V. P. Wystrach, J. Am. Chem. Soc., 1959, 81, 1103–1107 CrossRef CAS; (b) J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648–2650 CrossRef CAS.
- O. Herd, A. Heßler, M. Hingst, M. Tepper and O. Stelzer, J. Organomet. Chem., 1996, 522, 69–76 CrossRef CAS.
- J. H. Simons and E. O. Ramler, J. Am. Chem. Soc., 1943, 65, 389–392 CrossRef CAS.
- H. Gilman and D. Blume, J. Am. Chem. Soc., 1943, 65, 2467–2468 CrossRef CAS.
- G. M. Le Fave, J. Am. Chem. Soc., 1949, 71, 4148–4149 CrossRef CAS.
- G. M. Le Fave and P. G. Scheurer, J. Am. Chem. Soc., 1950, 72, 2464–2465 CrossRef CAS.
- P. G. Scheurer and G. M. Le Fave, J. Am. Chem. Soc., 1950, 72, 3308–3309 CrossRef CAS.
- K. Wang, H. Li and J. Wen, J. Fluorine Chem., 2001, 109, 205–208 CrossRef CAS.
- Y. V. Zonov, V. M. Karpov and V. E. Platonov, J. Fluorine Chem., 2007, 128, 1058–1064 CrossRef CAS.
- A. Kethe, A. F. Tracy and D. A. Klumpp, Org. Biomol. Chem., 2011, 9, 4545–4549 RSC.
- R. J. Gillespie, T. E. Peel and E. A. Robinson, J. Am. Chem. Soc., 1971, 93, 5083–5087 CrossRef CAS.
- (a) W. A. Herrmann, G. P. Albanese, R. B. Manetsberger, P. Lappe and H. Bahrmann, Angew. Chem., Int. Ed. Engl., 1995, 34, 811–813 CrossRef CAS; (b) D. Peral Crespo, Triarilfosfines Sulfonades Trifluorometilades. Aplicacions en Processos Catalítics, PhD Thesis. Universitat Autònoma de Barcelona, 2013 Search PubMed; (c) D. Peral, D. Herrera, J. Real, T. Flor and J. C. Bayón, Catal. Sci. Technol., 2016, 6, 800–808 RSC.
- (a) S. J. Bass, R. H. Flowers, R. J. Gillespie, E. A. Robinson and C. Solomons, J. Chem. Soc., 1960, 4315–4320 RSC; (b) J. Barr, R. J. Gillespie and E. A. Robinson, Can. J. Chem., 1961, 39, 1266–1273 CrossRef CAS.
- D. Herrera, D. Peral, M. Cordón and J. C. Bayón, Eur. J. Inorg. Chem., 2021, 354–363 CrossRef CAS.
- (a) K. Itami, T. Kamei and J. I. Yoshida, J. Am. Chem. Soc., 2001, 123, 8773–8779 CrossRef CAS PubMed; (b) T. Vogler and A. Studer, Org. Lett., 2008, 10, 129–131 CrossRef CAS PubMed; (c) Y. Hirata, T. Yukawa, N. Kashihara, Y. Nakao and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 10964–10973 CrossRef CAS PubMed.
- (a) D. R. Palo and C. Erkey, Organometallics, 2000, 19, 81–86 CrossRef CAS; (b) S. I. Fujita, S. Fujisawa, D. M. Bhanage and M. Arai, Tetrahedron Lett., 2004, 45, 1307–1310 CrossRef CAS; (c) A. C. J. Koeken, M. C. A. van Vliet, L. J. P. van den Broeke, B.-J. Deelman and J. T. F. Keurentjes, Adv. Synth. Catal., 2008, 350, 179–188 CrossRef CAS.
- P. Dydio, R. J. Detz and J. N. H. Reek, J. Am. Chem. Soc., 2013, 135, 10817–10828 CrossRef CAS PubMed.
- P. Starkov, J. T. Moore, D. C. Duquette, B. M. Stoltz and I. Marek, J. Am. Chem. Soc., 2017, 139, 9615–9620 CrossRef CAS PubMed.
- D. Herrera Miranda, D. Peral Crespo, J. C. Bayon Rueda, Trifenilfosfinas Carboxílicas, ES2459790 (B1), Universitat Autònoma de Barcelona, 2012 Search PubMed.
- Phosphines MC1, DC1, MC2 and DC2 are currently part of the catalogue of Strem Chemicals company (branding name Miranphos and Miran2phos ligands) and are sold under license from the Universitat Autònoma de Barcelona for research purposes only.
- C. G. Krespan and D. C. England, J. Org. Chem., 1975, 40, 2937–2940 CrossRef CAS.
- (a) M. Aschi, B. Chiavarino, M. E. Crestoni and S. Fornarini, J. Phys. Chem., 1996, 100, 19859–19863 CrossRef CAS; (b) J. Zakzeski, I. S. Fan and A. T. Bell, Appl. Catal., A, 2009, 360, 33–37 CrossRef CAS; (c) F. Wang and J. Hu, Chin. J. Chem., 2009, 27, 93–98 CrossRef CAS.
- M. H. Karger and Y. Maztjr, J. Org. Chem., 1971, 36, 528–531 CrossRef.
- (a) C. G. Krespan and V. A. Petrov, Chem. Rev., 1996, 96, 3269–3301 CrossRef CAS PubMed; (b) V. A. Petrov, J. Org. Chem., 1998, 63, 2988–2992 CrossRef CAS.
- V. A. Petrov, A. Marchione and W. Marshall, J. Fluorine Chem., 2008, 129, 1011–1017 CrossRef CAS.
- P. S. Bhadury, M. Pandey and D. K. Jaiswal, J. Fluorine Chem., 1995, 73, 185–187 CrossRef CAS.
- J. Malito and E. C. Alyea, Phosphorus, Sulfur Silicon Relat. Elem., 1999, 54, 95–99 CrossRef.
- U. Beckmann, D. Süslüyan and P. C. Kunz, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 2061–2070 CrossRef CAS.
- D. A. Shriver, M. A. Drezdon, The Manipulation of Air-Sensitive Compounds, Wiley-Interscience, New York, 2nd edn, 1986 Search PubMed.
- W. L. F. Armarego, C. Chai, Purification of Laboratory Chemicals; Elsevier Science, 6th edn, 2009 Search PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- (a) G. R. Miller, A. W. Yankowsky and S. O. Grim, J. Chem. Phys., 1969, 51, 3185–3190 CrossRef CAS; (b) K. C. Eapen and C. Tamborski, J. Fluorine Chem., 1980, 15, 239–243 CrossRef CAS; (c) J. Jeschke, M. Korb, T. Rüffer, C. Gäbler and H. Lang, Adv. Synth. Catal., 2015, 357, 4069–4081 CrossRef CAS.
- Warning: In the preparation of carboxylic phosphines, highly corrosive fuming sulfuric acid is used. Appropriate protective clothing is required when handling it.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2097178 and 2106238. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ra00420h |
| This journal is © The Royal Society of Chemistry 2022 |