
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethyl nitrate energetic compounds based on bicyclic scaffolds of furazan–isofurazan (isoxazole): syntheses, crystal structures and detonation performances†
Qi Xueab,
Fuqiang Biab,
Yifen Luoab,
Jiarong Zhang ab,
Kaidi Yangab,
Bozhou Wang*ab and
Ganglin Xue*c
ab,
Kaidi Yangab,
Bozhou Wang*ab and
Ganglin Xue*c
aXi'an Modern Chemistry Research Institute, Xi'an, 710065, China. E-mail: wbz600@163.com
bState Key Laboratory of Fluorine & Nitrogen Chemicals, Xi'an, 710065, China
cCollege of Chemistry & Materials Science, Key Laboratory of Synthetic and Natural Functional Molecule Chemistry, Northwest University, Xi'an 710127, China. E-mail: xglin707@163.com
First published on 10th March 2022
Abstract
Two energetic bicyclic scaffolds (furazan–isoxazole and furazan–1,3,4-oxadiazole) were constructed via different cyclization reactions. Based on the energetic bicyclic scaffolds, the energetic compounds, 3-(4-nitraminofurazan-3-ly)-isoxazole-5-methylnitrate 1c and 5-(4-nitraminofurazan-3-ly)-1,3,4-oxadiazole-2-methylnitrate 2c, were designed and synthesized in good yields. Because of the acidity of nitramine, the corresponding energetic ionic salts, ammonium 3-(4-nitraminofurazan-3-ly)isoxazole-5-methylnitrate 1d and ammonium 5-(4-nitraminofurazan-3-ly)-1,3,4-oxadiazole-2-methylnitrate 2e, were also obtained and well characterized, their structures were further determined by X-ray single crystal diffraction. To have a better understanding of the structure–property relationships of furazan-bicyclic scaffolds and nitrate groups, their thermal behaviors, detonation performances and the sensitivities were investigated via differential scanning calorimetry (DSC), ESP analysis, Hirshfeld surfaces calculation, EXPLO5 program and BAM standard techniques. Compared with those of ammonium 5-(4-nitraminofurazan-3-ly)-1,2,4-oxadiazole-2-methylnitrate 3e, the results show that all these methyl nitrate energetic compounds based on bicyclic scaffolds of furazan–isofurazan exhibit good detonation performances and extraordinary insensitivities. As supported by experimental and theoretical data, the formation of energetic ionic salts causes an increase of the weak interactions, significantly improving the thermal performance over 110 °C.
Introduction
Since the discovery of the nitroglycerin (NG) by Ascanio Sobrero in 1874, nitrate ester compounds have been widespread and used in diversified fields, including medicines,1–4 fuel additives,5,6 and especially in military fields.7–10 On the one hand, the nitrate ester group could be easily synthesized in high yield by treatment of the corresponding alcohols with concentrated nitric acid or a nitrating mixture.11 On the other hand, introducing a nitrate ester group is an effective means to increase the energetic performance and oxygen balance of energetic molecules. Up to now, the aliphatic nitrate ester energetic compounds have been widely used as primary explosives, secondary explosives, and plasticizer ingredients for military purposes, such as nitroglycerin (NG),12 pentaerythritol tetranitrate (PETN),13 xylitol pentanitrate (XPN)14 (Fig. 1(a)). However, accompanied by high performances and great compatibility, these compounds often present high mechanical sensitivities and low thermal decomposition temperature, which greatly influence their handling safety.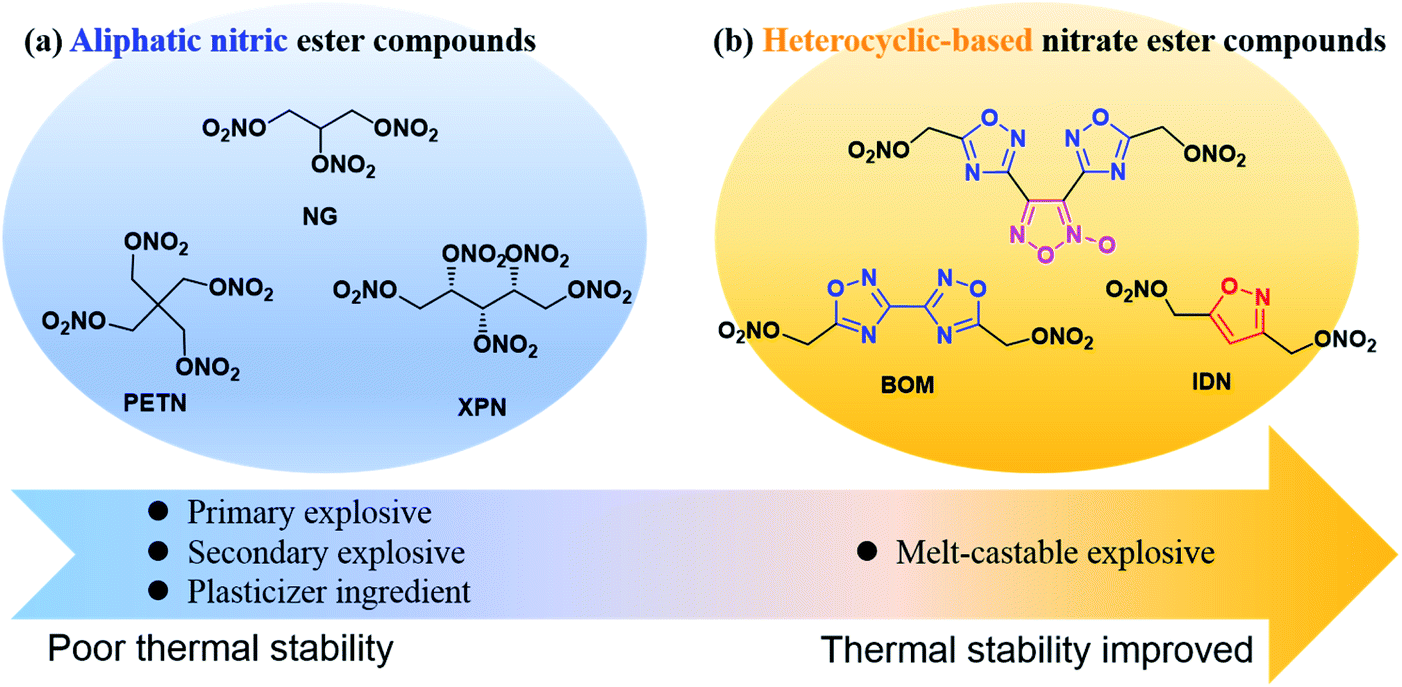 | ||
| Fig. 1 Representative examples: (a) typical aliphatic nitrate ester energetic compounds. (b) Typical heterocyclic-based nitrate ester compounds. | ||
In recent years, there are a few examples of heterocyclic-based nitrate ester compounds have been reported.15–19 In particular, the recently synthesized bis(1,2,4-oxadiazole)bis(methylene)dinitrate (BOM) displays calculated detonation pressure 50% higher than that of TNT and exhibits a relatively high decomposition temperature and lower sensitivities to impact and friction compared with RDX.19 Compared with aliphatic nitrate ester compounds, the thermal stability of heterocyclic-based nitrate ester compounds is significantly improved due to π–π stacking effect of heterocyclic skeleton and some of them can be used as melt-castable explosives (Fig. 1(b)). For example, with the same number of nitrate ester groups, 3,3′-biisoxazole-4,4′,5,5′-tetra(methylene)tetranitrate (BITN)16 and 4,5-bis(nitrooxy methylene)-1H-1,2,3-triazole-1-ethyl nitrate (PN3)20 have higher melting points, density thermal decomposition temperatures than those of PETN and NG (Scheme 1(a) and (b)), respectively. However, the mechanical sensitivity performance of heterocyclic-based nitrate ester compounds has not been markedly improved.
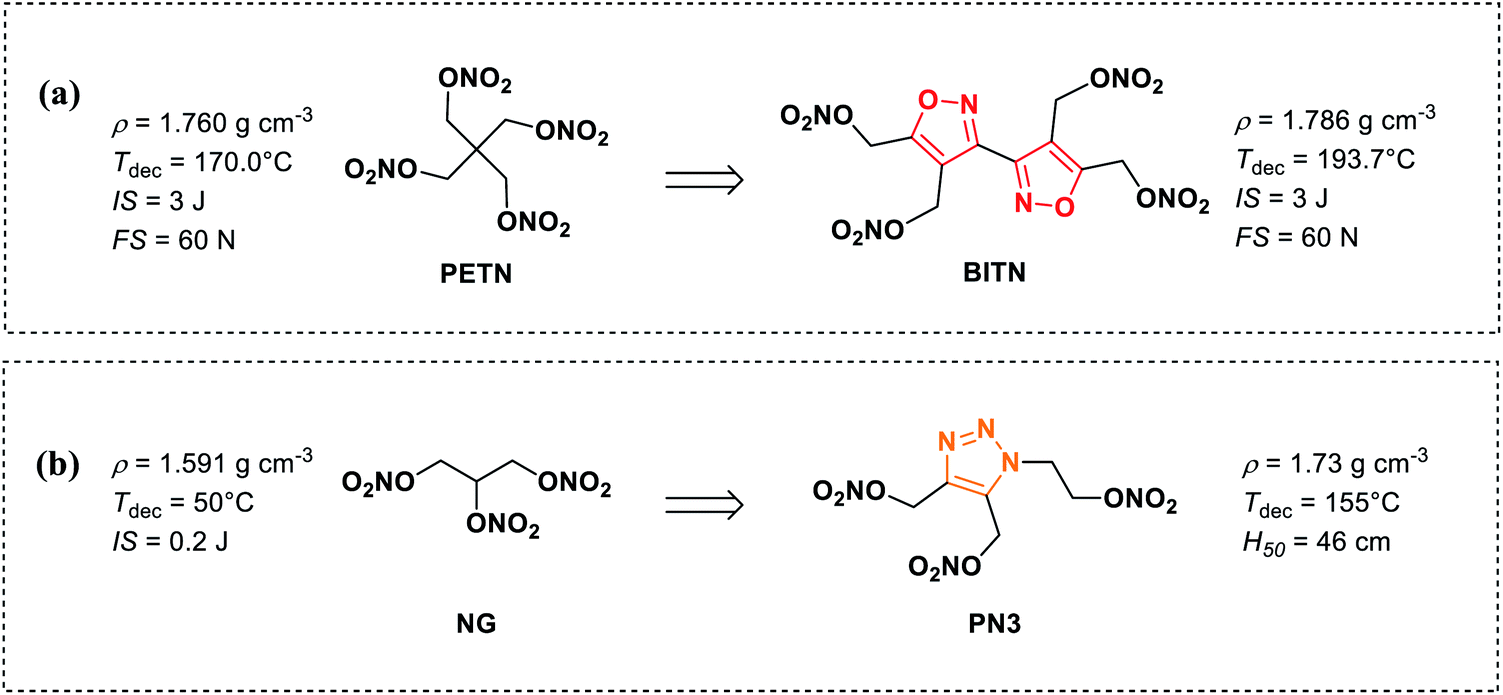 | ||
| Scheme 1 The performance comparison between (a) PETN and BITN (b) NG and PN3. | ||
The formation of energetic ionic salts has been proven to be a simple and effective way to improve the energy and safety of compounds.21–26 The cations, especially small cations like ammonium ions, which can functionalize as a bridge linking among molecules and layers to increase intra- and intermolecular forces by lots of hydrogen bonds. A typical example is dihydroxylammonium 5,5′-bis(tetrazolate-1N-oxide) (TKX-50). Yet, the synthesis of energetic salts based heterocyclic skeleton which incorporate nitrate ester group is rarely described and investigated.
In our previous work,27 a few of energetic compounds and its ionic salts based on furazan–1,2,4-oxadiazole backbone with nitrate ester and nitramine moieties were synthesized, which exhibit low insensitivity and good detonation performance. Unfortunately, single crystal structures of these ionic salts have not been obtained. To have a better understanding of these structure–property relationships, two new kinds of energetic bicyclic frameworks, including furazan–isoxazole and furazan–1,3,4-oxadiazole, are constructed through [3+2] cycloaddition, and condensation cyclization, respectively. Corresponding furazan–isofurazan (isoxazole) based nitrate ester-methyl neutral compounds and their ammonium ions salts are obtained. An intensive research and comparison on the crystal structures, thermal decomposition processes and energetic properties of three kinds of furazan–isofurazan (isoxazole) ammonium ions salts are presented in this work.
Results and discussion
Synthetic procedures
Synthetic pathways of target compounds are shown in Scheme 2. Synthesis of 3d and 3e was previously described by our research group.27 The starting material, compound 1a, was prepared according to the literature.28 The introduction of –ONO2 into the skeleton requires using fuming nitric acid, H2SO4/HNO3, 100% HNO3 or other reagents as nitration reagents. When compound 1b was treated with fuming nitric acid, only mixture products are obtained. To optimize the nitrating reaction, low temperature experiments were carried out in pure nitrate, and the required intermediate 1c was obtained without purification and with high yield (95%). Compound 1c, however, was unstable at room temperature in the absence of solvents. 1c could be converted to its ammonium salt by adding aqueous ammonia in methanol solution. A suitable single crystal of compound 1d was obtained by slow evaporation of the solvent at room temperature.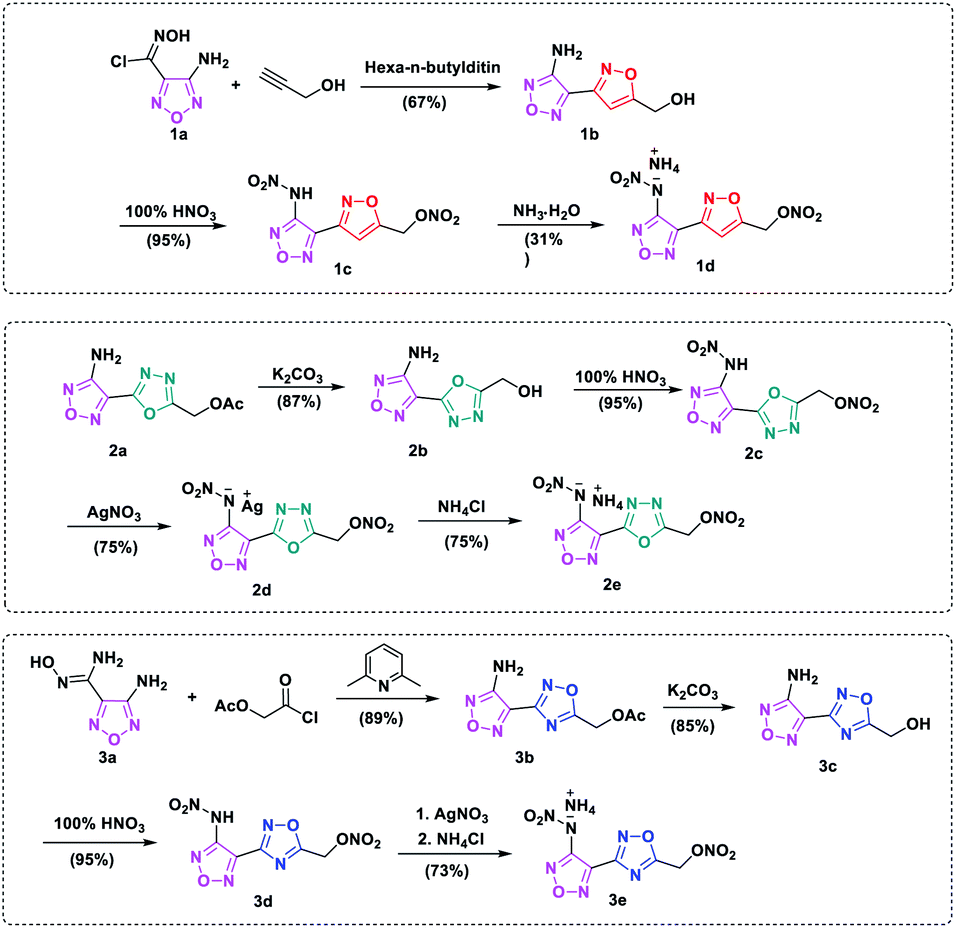 | ||
| Scheme 2 Synthetic routes of energetic compound 1c, 2c, 3d and their ionic salts 1d, 2e, 3e. The synthetic routes of 3d and 3e have been report on our previous work ref. 25. | ||
Compound 2a was synthesized from dehydrate diaroylhydrazine in weakly alkaline solution. Then through reaction of off-protecting and nitration, compound 2c could be obtained as light-yellow liquid, however, it was also found to be unstable at room temperature. The silver salt 2d was firstly prepared in high yield by treating 2c with AgNO3 in methyl alcohol, then ammonium salt 2e was synthesized by metathesis reaction in aqueous solution, as a colorless crystal solid.
All salts were characterized by IR, 1H NMR and 13C NMR spectroscopy. The 13C NMR spectra of 1d, 2e and 3e were shown in Fig. 2. The 13C spectrum for 1d was recorded in CD3OD as solvent, while the spectra for 2e and 3e were determined in D2O solution. All C-signals of each compound could be clearly observed. In contrast to 3e (δ = 55.40 ppm), the signals for CH2 group (C1) in 1d (δ = 63.52 ppm) and 2e (δ = 62.59 ppm) were shifted to higher field. For heterocyclic ring-connecting carbon atoms, the signals were observed in a typical range (105–180 ppm),16,29–31 depending on the electron-withdrawing or pushing character of atoms attached.
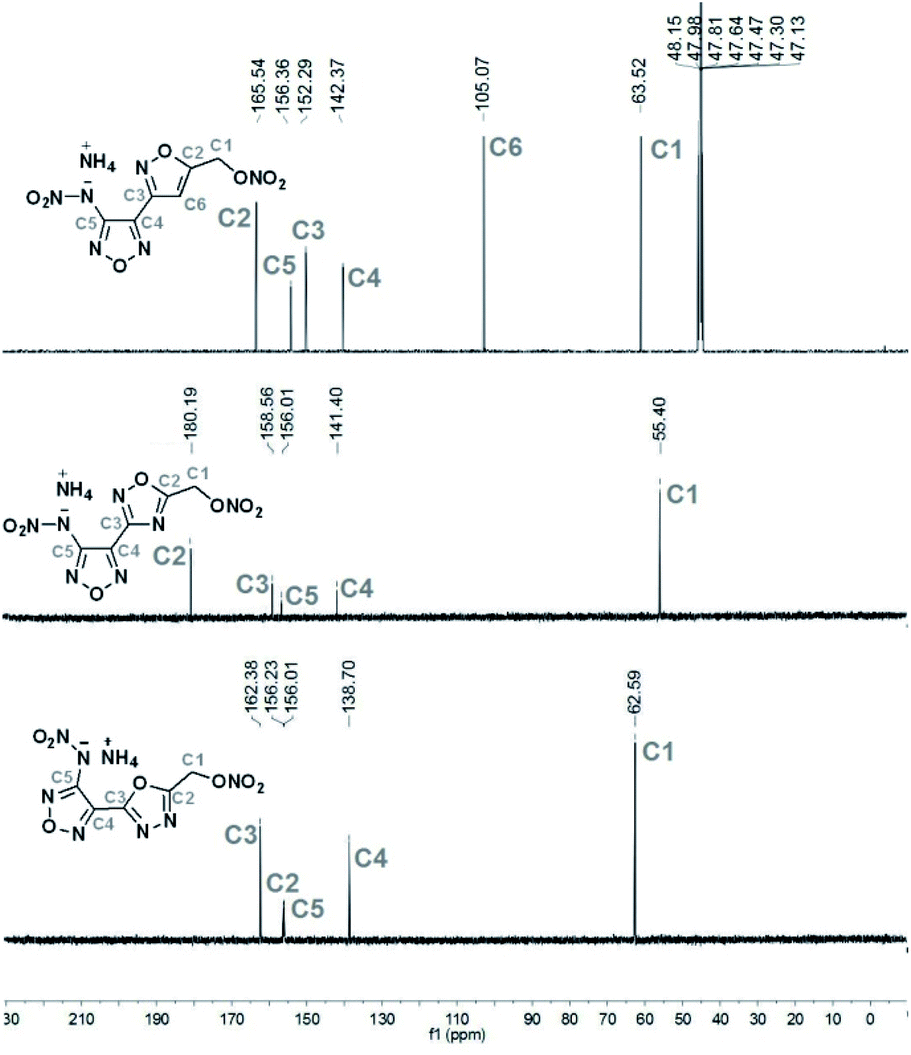 | ||
| Fig. 2 The experimental 13C NMR spectra of 1d in CD3OD, 2e and 3e in D2O, respectively. | ||
X-ray crystallography
Suitable crystals for 1d and 2e were obtained by slow evaporation of their saturated solutions in a methanol/H2O mixture, separately. The detailed crystal structure data (including selected bond lengths, angles, torsion angles and hydrogen bonds) were listed in Tables S1–S9.†Salt 1d crystallizes in the monoclinic space group P21, with two molecules per unit cell. The calculated density of 1.694 g cm−3 was determined from its X-ray crystal structure at 180(2) K. The atoms of diheterocycles and N–NO2 in the anion were in nearly coplanar, supported by the dihedral angles of C1–N2–N1–O1 (−176.933 (445)°) and C2–C1–N2–N1 (−173.473 (461)°) (Fig. 3(a)). The bond lengths of C1–N2 [1.373(7) Å] and N1–N2 [1.317(6) Å] are shorter than normal carbon–carbon or carbon–nitrogen single bond length (ca. 1.54 Å) markedly,32 suggesting that these atoms form a large π bond. However, the nitrate ester group and molecular skeleton formed a near right-angle (C4–C5–C6–O5 (105.398 (870)°)), and C6–O5 bond length [1.447(8) Å] was longer than that of a typical C–O bond (1.42 Å),32 indicating that this bond was relatively weak.
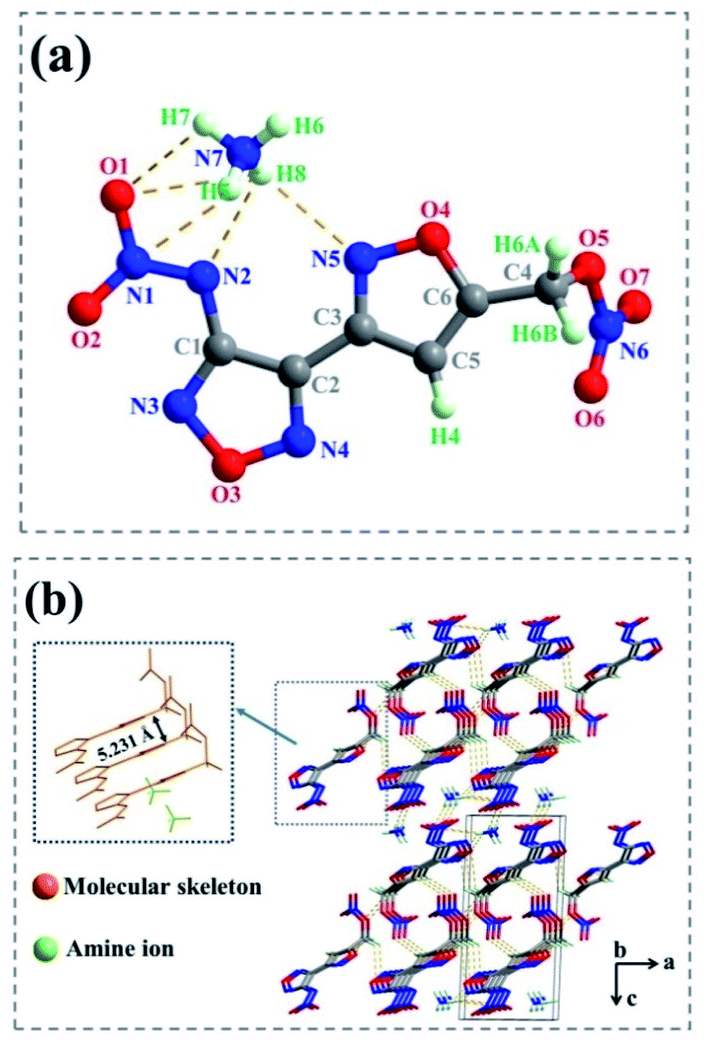 | ||
| Fig. 3 (a) Thermal ellipsoid plot (50%) and labelling scheme for 1d. (b) Packing diagram of 1d. | ||
Salt 2e crystallizes in the monoclinic P21/c space group with four formula units in the unit cell. 2e displayed a calculated density of 1.758 g cm−3 at 150 K. The nitramino group and bicyclic scaffolds in 2e were nearly coplanar and therefore form a conjugated system (Fig. 4(a)), as confirmed by the torsion angles of O4–C3–C4–C5 (170.362 (3)°) and C5–N6–N7–O6 (2.025 (6)°). The nitrate ester group and molecular skeleton formed an angle of nearly 90° (O4–C2–C1–O3 (72.788 (4)°) and N2–O2–C1–O3 (−111.138 (4)°), which were similar to the crystal structure of 1d.
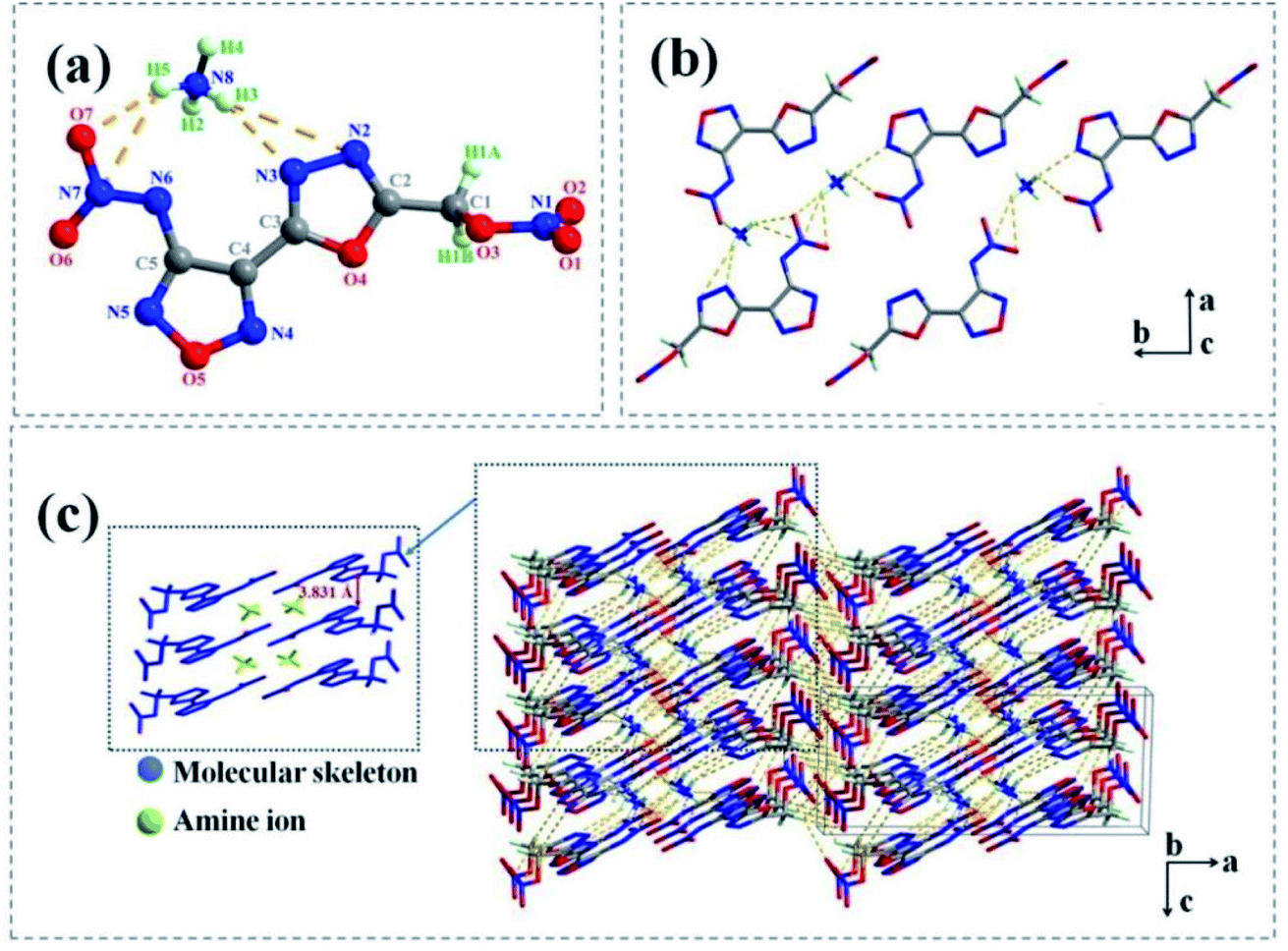 | ||
| Fig. 4 (a) Thermal ellipsoid plot (50%) and labelling scheme for 2e. (b) The 2D layer formed of 2e. Dashed lines indicate strong hydrogen bonding. (c) Packing diagram of 2e. | ||
As show in Fig. 4(b) and (c), the supermolecular structure of 2e was constructed by π–π stacking (the distance between the two neighbouring planes was 3.831 Å) and lots of hydrogen bonds among ammonium ions and atoms of energetic anions were formed. There were nine kinds of intermolecular and intermolecular strong hydrogen bonds in the independent unit (Table S9†). While in 1d (Fig. 3(b)), the ammonium ions were distributed on both sides of energetic anion layers and eleven kinds of strong hydrogen bonds were formed in the independent unit (Table S5†) (such as N7– H6⋯O3 [3.353 Å], N7–H6⋯N1 [3.051 Å], N7–H7⋯O4 [2.995 Å], N8–H6⋯N3 [2.281 Å] and so on).
To further investigating their weak inter-/intra molecular interaction, two-dimensional (2D)-fingerprint and relevant Hirshfeld surfaces of crystals 1d and 2e were fully studied. It was observed from the 2D fingerprint plot that O⋯H and N⋯H hydrogen bonds constitute 60% (1d) and 55% (2e) of the total weak interactions, respectively (Fig. 5(a) and (b)), which mainly concentrated in ammonium ions and nitro groups. This phenomenon demonstrates that introducing ammonium ions could greatly improve the ratios of intermolecular hydrogen bond interactions, and consequently to enhance the stability of molecules. In addition, compound 2e has 5% percentage of N⋯N, N⋯C and C⋯N interactions which denoted π–π stacking. However, because of the “head to head” packing method of nitrate ester groups, both 1d and 2e had a percentage (12% and 14%) of O⋯O interactions which might lead to a decrease in decomposition temperatures. Overall, strong hydrogen-bond interactions and interlayer π–π stacking interactions could improve thermal stability of energetic salts, which is consistent with the experimental decomposition temperature.
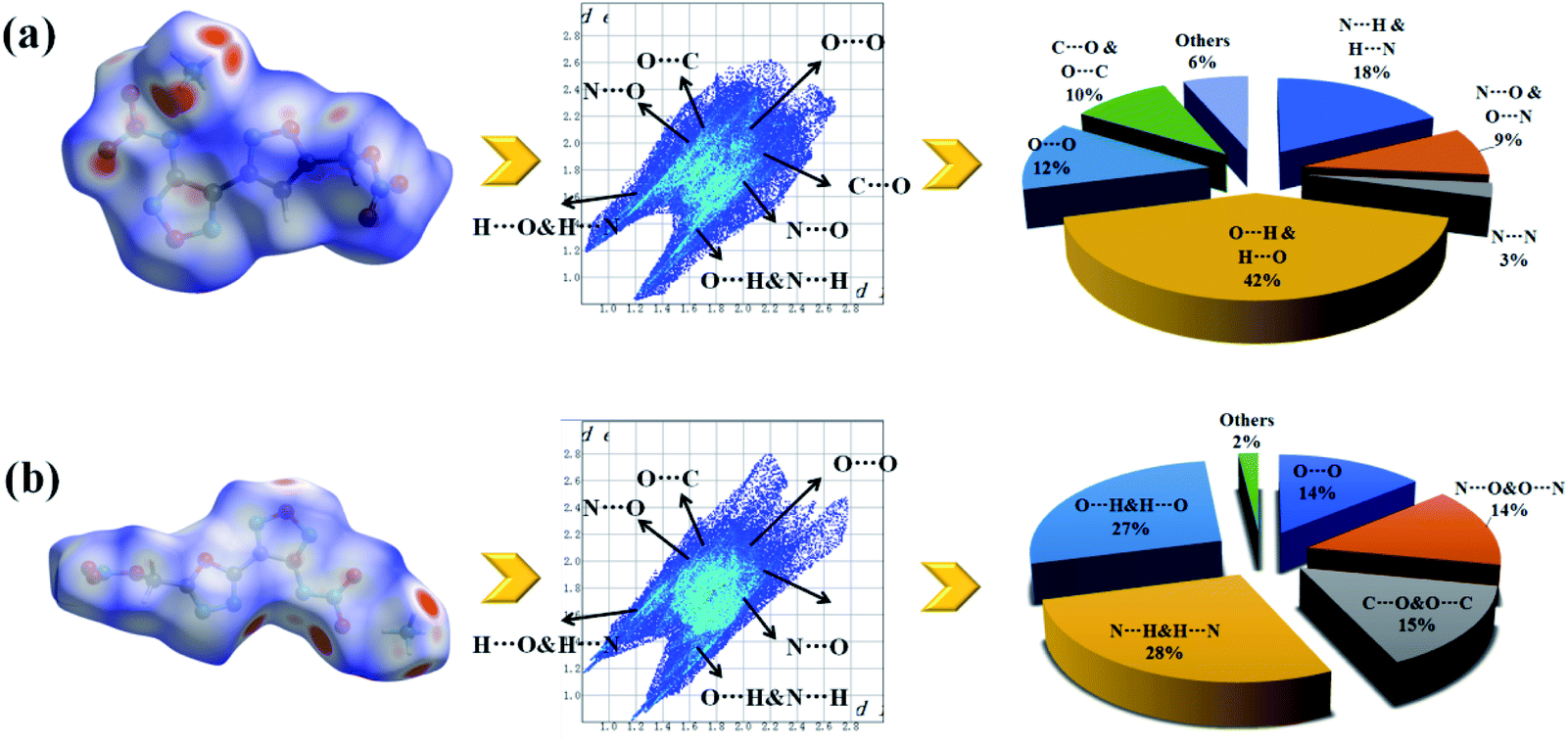 | ||
| Fig. 5 Hirshfeld surfaces calculation (white, distance d equals the van der Waals distance; blue, d exceeds the van der Waals distance; red, d is less than van der Waals distance) and two-dimensional fingerprint plots of (a) 1d and (b) 2e. | ||
ESP analysis and thermal behaviours
Thermal behaviors of 1d, 2e and 3e were investigated and compared by DSC under a heating rate of 10 °C min−1 in Ar atmosphere. Due to the presence of nitramino groups, molecule energetic compounds 1c and 2c were unstable at room temperature. Compared with the corresponding molecular compounds, the decomposition temperatures of the salts 1d, 2e and 3e increased by more than 110 °C (Fig. 7). Among them, 1d exhibits relatively good thermal stability (144.76 °C), which is consistent with the two-dimensional (2D)-fingerprint analysis result.To understand the influence of the energetic groups on the charge distribution of molecules, electrostatic potential energy surface (ESP) analysis of 1d, 2d and 2e was performed by using Multiwfn.33,34 The optimized structure of 2e was carried out based upon the B3LYP/6-31+G**35 method by Gaussian 09 software.36 As shown in Fig. 6, in contrast to compound 2d, salts 1d and 2e exhibited lower ESP maximum values and more negative charge accumulation in heterocyclic backbone, which might lead to relatively higher thermal stability.
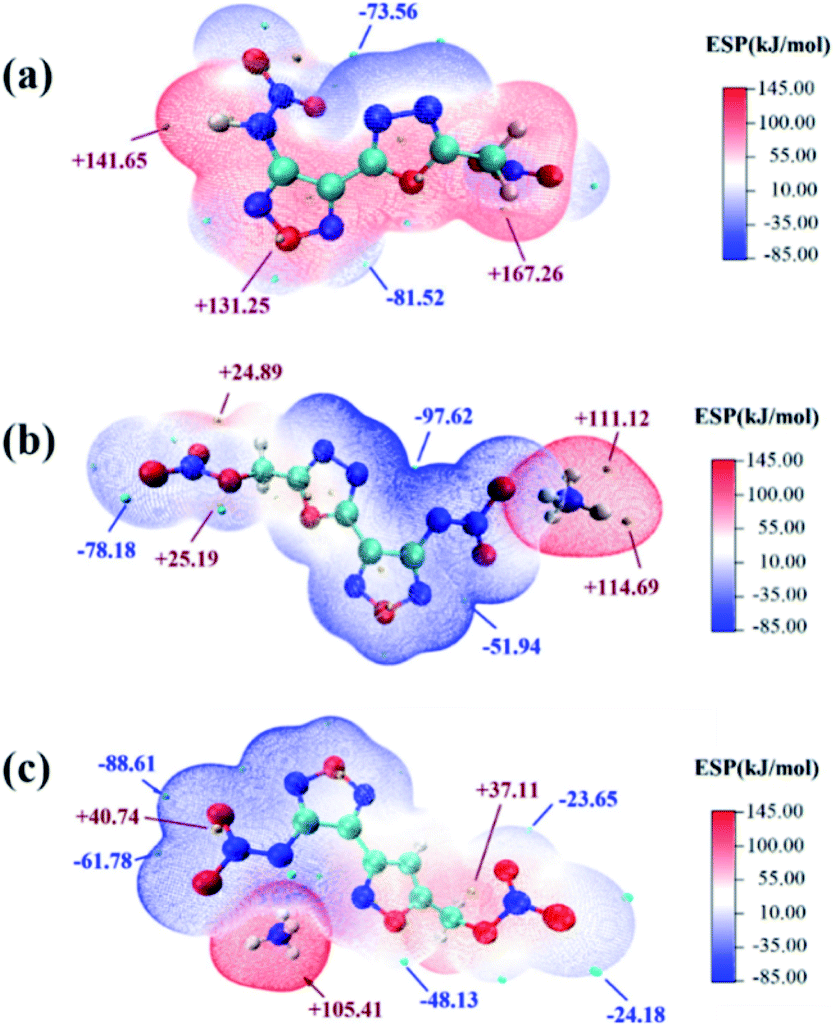 | ||
| Fig. 6 Electrostatic potential surfaces of compounds (a) 2d, (b) 2e and (c) 1d. The minimum and maximum of ESP are marked as blue and red points, respectively. The maximum value of ESP is labeled with red texts and units are in kcal mol−1. | ||
The thermal decomposition processes of 1d, 2e and 3e were further investigated using non-isothermal kinetic study and compared by Kissinger's method.37 In general, the kinetic parameters included the apparent activation energy (Ek), the per-exponential factor (A) and the linear coefficient (r) of the first exothermic decomposition process. The DSC traces of compounds were obtained at various heating rates (2.5, 5, 10 and 15 °C). As shown in Fig. 7, the linear fitting of 1d, 2e and 3e was straight lines with r = 0.997, 0.996 and 0.992, respectively, and it was demonstrated that Kissinger method is applied to calculate the thermal kinetic parameters. The apparent activation energy (Ek) of 1d, 2e and 3e were about 148.57 kJ mol−1, 186.91 kJ mol−1 and 148.98 kJ mol−1, respectively (Table S10†), and it was indicated that 2e displayed best stability at thermo-decomposition process.
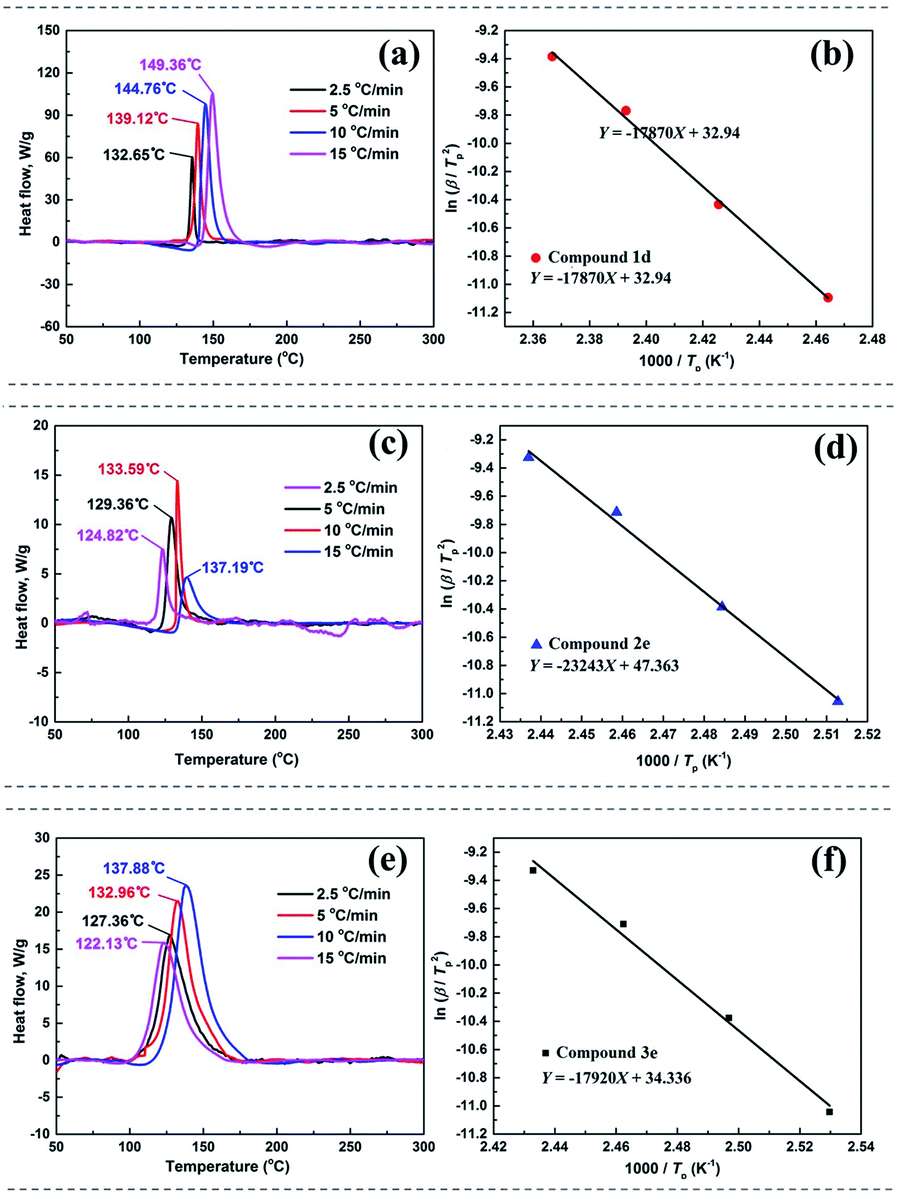 | ||
| Fig. 7 DSC traces of (a) 1d, (c) 2e and (e) 3e obtained at various heating rates. Kissinger's plots of (b) 1d, (d) 2e and (f) 3e. | ||
Computational energetic properties
The enthalpy of formation was one of the significant energetic characteristics of energetic compound. The heats of formation in solid phase (ΔHf) for 1d and 2e were calculated using the heat of formation in gas phase and heat of phase transition (lattice energy) according to the Born–Haber energy cycle.38 The gas-state enthalpies and energies of formation of molecule, cations, and anions were calculated using the quantum chemical CBS-4M calculations (HCBS-4M) method.39,40 1d, 2e and 3e had positive heats of formation of 265.1, 253.5 and 243.3 kJ mol−1, respectively (Table 1), which exceeded those of PETN (−538.6 kJ mol−1) and RDX (92.6 kJ mol−1).| Compound | Tda/°C | ρ/g cm−1 | OBc/% | ΔHfd/kJ mol−1 | Df/m s−1 | Pe/Gpa | IS/J |
|---|---|---|---|---|---|---|---|
| a Decomposition temperature (°C).b Densities obtained from X-ray measurements.c Oxygen balance (OB, %) for a compound with a molecular formula of CaHbNcOd without crystal water.d Standard enthalpy of formation (at 298 K, kJ mol−1).e Detonation pressure (GPa).f Detonation velocity (m s−1).g Impact sensitivity (J). | |||||||
| 1d | 144.8 | 1.674b | −47 | 265.1 | 7980 | 27 | 35g |
| 2e | 132.4 | 1.758b | −33 | 243.3 | 8361 | 31 | 33g |
| 3e19 | 135 | 1.79 | −33 | 253.5 | 8428 | 35 | 32 |
| RDX | 204 | 1.80 | −21 | 92.6 | 8795 | 35 | 7.5 |
| PETN9 | 172.9 | 1.76 | −10 | −538.6 | 8400 | 32 | 3 |
| BOM19 | 183.4 | 1.832 | −33 | −79.4 | 8180 | 29.4 | 8.7 |
The detonation performances of these salts were calculated using the EXPLO5 program (version 6.01)41 based on the calculated heats of formation and densities. The calculated detonation velocity (D) and pressure (P) values were shown in Table 1. Among these three kinds of salts based on bicyclic scaffolds of furazan–isofurazan (isoxazole), 1,2,4-oxadiazole-furazan based salt 3e displayed the best detonation performance (D: 8428 m s−1; P: 35 GPa), which is slightly better than PETN (D: 8400 m s−1; P: 32 GPa).
The impact sensitives of the salts 1d and 2e were determined by following BAM standard methods. Both salts showed excellent mechanical performance (IS > 30 J) due to strong noncovalent interactions, which was greatly superior to RDX and most reported nitrate ester energetic compounds, such as NG (IS < 0.2 J), PETN (IS = 3 J) and BOM (IS = 8.7 J).
Conclusions
In summary, two new heterocyclic based neutral compound nitrate ester compounds (1c and 2d) and its ammonium salts (1d and 2e) were synthesized and fully investigated. However, both neutral nitrate ester compounds were unstable at room temperature. By introducing ammonium ion, the decomposition temperatures of 1d, 2e and 3e were greatly improved over 110 °C. Among them, 1d exhibits relatively good thermal stability (144.8 °C) probably due to more strong hydrogen bond interactions besides π–π stacking in its crystal structure. Among these three kinds of salts based on bicyclic scaffolds of furazan–isofurazan (isoxazole), 1,2,4-oxadiazole-furazan based salt 3e exhibits good detonation performance and excellent mechanical performance, higher than most reported nitrate ester energetic compounds. The result indicated that introducing positive ion is a good strategy to decreasing mechanical sensitivity and thermal stability of nitrate ester compounds.Experimental
Reagents and sample preparation
Tris(hydroxymethyl)nitromethane was supplied by Xi'an Modern Chemistry Research Institute. All reagents and solvents were purchased from Aladdin Bio-Chem Technology CO., Ltd (Shanghai, China) and used without further purification unless otherwise indicated.Apparatus and measurements
13C and 1H NMR spectra were measured with AV 500 NMR spectrometer (Bruker, Switzerland). Infrared spectra were measured by an EQUINOX 55 Fourier transform Infrared spectrometer (Bruker, Germany). Elemental analyses were performed with the vario EL cube elemental analyzer (Elementar, Germany).The thermal analysis experiments were performed with a model TGA/DSC 1 instrument (METTLER, Switzerland). Single crystal X-ray experiment was carried out on a Bruker Apex II CCD diffractometer equipped with graphite monochromatized Ga and Cu Kα radiation using ω and φ scan mode. Structures were solved by the direct method using SHELXTL and refined by means of full-matrix least-squares procedures on F2 with the programs SHELXL-97. All nonhydrogen atoms were refined with anisotropic displacement parameters. The sensitivity towards impact (IS) was determined according to BAM standards.
DFT calculations
All quantum chemical calculations were carried out using the program package Gaussian 09. The geometry optimizations of the molecules and frequency analyses were accomplished by using the B3LYP with the 6-311+G** basis set. The ESP analysis of 1c was carried out based upon the B3LYP/6-31+G** method by Gaussian 09 software with optimized structure. The ESP analyses of 1d and 2e were carried out based on corresponding single crystal structures. The gas-state enthalpies and energies of formation of molecules were calculated using the quantum chemical CBS-4M calculations (HCBS-4M) method in order to obtain accurate values.Synthesis
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the financial support from National Natural Science Foundation of China (No. 22175139).Notes and references
- M. Smiljkovic, M. T. Matsoukas, E. Kritsi, U. Zelenko, S. G. Grdadolnik, R. C. Calhelha, I. C. F. R. Ferreira, S. Sankovic-Babic, J. Glamoclija, T. Fotopoulou, M. Koufaki, P. Zoumpoulakis and M. Sokovic, ChemMedChem, 2018, 13, 251–258 CrossRef CAS PubMed.
- S. Warren and G. Francis, Am. J. Med., 1978, 65, 53–62 CrossRef CAS PubMed.
- P. R. Sage, I. S. de la Lande, I. Stafford, C. L. Bennett, G. Phillipov, J. Stubberfield and J. D. Horowitz, Circulation, 2000, 102, 2810–2815 CrossRef CAS PubMed.
- J. T. Flaherty, P. Reid, D. Kelly, D. Taylor, M. Weisfeldt and B. Pitt, Circulation, 1975, 51, 132–139 CrossRef CAS PubMed.
- C. P. Zhang, X. M. Zhai, Y. J. Li and Z. G. Sun, Adv. Mater. Res., 2012, 354, 462–467 Search PubMed.
- E. R. Jonash, J. D. Wear and W. P. Cook, Effect of Fuel Additives on Carbon Deposition in a J33 Single Combustor: Nine Oxygen-bearing Compounds, National Advisory Committee for Aeronautics, 1956 Search PubMed.
- D. E. Chavez, M. A. Hiskey, D. L. Naud and D. Parrish, Angew. Chem., 2008, 120, 8431–8433 CrossRef.
- Z.-D. Sun, X. L. Fu, H. J. Yu, X. Z. Fan and X. H. Ju, J. Hazard. Mater., 2017, 339, 401–408 CrossRef CAS PubMed.
- J. J. Sabatini and E. C. Johnson, ACS Omega, 2021, 6, 11813–11821 CrossRef CAS PubMed.
- T. M. Klapötke, B. Krumm, T. Reith and C. Riedelsheimer, ChemistrySelect, 2021, 6, 8581–8586 CrossRef.
- J. Dong, Q. L. Yan, P. J. Liu, W. He, X. F. Qi and S. Zeman, J. Therm. Anal. Calorim., 2018, 131, 1391–1403 CrossRef CAS.
- A. T. Camp, H. S. Haiss and P. R. Mosher, Energetic Plasticizer and Improved Gas Producing Charges, Department of The Navy, Washington DC, 1993 Search PubMed.
- A. Booth and F. Llewellyn, J. Chem. Soc., 1947, 837–846 RSC.
- K. A. S. Stark, J. F. Alvino, K. P. Kirkbride, C. J. Sumby, G. F. Metha, C. E. Lenehan, M. Fitzgerald, C. Wall, M. Mitchell and C. Prior, Propellants, Explos., Pyrotech., 2019, 44, 541–549 CrossRef CAS.
- Y. Li, Y. Shu, B. Wang, S. Zhang and L. Zhai, RSC Adv., 2016, 6, 84760–84768 RSC.
- L. A. Wingard, E. C. Johnson, P. E. Guzmán, J. J. Sabatini, G. W. Drake, E. F. C. Byrd and R. C. Sausa, Eur. J. Org. Chem., 2017, 2017, 1765–1768 CrossRef CAS.
- J. Zhang, P. Yin, L. A. Mitchell, D. A. Parrish and J. n. M. Shreeve, J. Mater. Chem. A, 2016, 4, 7430–7436 RSC.
- L. Bauer, M. Benz, T. M. Klapötke, T. Lenz and J. Stierstorfer, J. Org. Chem., 2021, 86, 6371–6380 CrossRef CAS PubMed.
- E. C. Johnson, J. J. Sabatini, D. E. Chavez, R. C. Sausa, E. F. C. Byrd, L. A. Wingard and P. E. Guzmàn, Org. Process Res. Dev., 2018, 22, 736–740 CrossRef CAS.
- P. Gaur, S. Dev, S. Kumar, M. Kumar, A. A. Vargeese, P. Soni, P. F. Siril and S. Ghosh, ACS Omega, 2017, 2, 8227–8233 CrossRef CAS PubMed.
- Q. Wang, Y. Shao and M. Lu, Cryst. Growth Des., 2019, 20, 197–205 CrossRef.
- Y. Xu, L. Tian, D. Li, P. Wang and M. Lu, J. Mater. Chem. A, 2019, 7, 12468–12479 RSC.
- J. Ren, W. Zhang, T. Zhang, Y. Yin, L. Wang, Z. Li, Q. Zeng and T. Zhang, J. Energ. Mater., 2021, 39, 48–59 CrossRef CAS.
- C. He, G. H. Imler, D. A. Parrish and J. n. M. Shreeve, J. Mater. Chem. A, 2018, 6, 16833–16837 RSC.
- C. Lei, H. Yang and G. Cheng, Dalton Trans., 2020, 49, 1660–1667 RSC.
- P. Yang, H. Yang, Y. Zhao, J. Tang and G. Cheng, Dalton Trans., 2021, 50, 16499–16503 RSC.
- Q. Xue, F. q. Bi, J. l. Zhang, Z. j. Wang, L. j. Zhai, H. Huo, B. z. Wang and S. y. Zhang, Front. Chem., 2020, 7, 942 CrossRef PubMed.
- J. Zhang, L. A. Mitchell, D. A. Parrish and J. n. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 10532–10535 CrossRef CAS PubMed.
- L. L. Fershtat, A. S. Kulikov, I. V. Ananyev, M. I. Struchkova and N. N. Makhova, J. Heterocycl. Chem., 2016, 53, 102–108 CrossRef CAS.
- S. D. Shaposhnikov, N. V. Korobov, A. V. Sergievskii, S. V. Pirogov, S. F. Mel'nikova and I. V. Tselinskii, Russ. J. Org. Chem., 2002, 38, 1351–1355 CrossRef CAS.
- Z. Fu, R. Su, Y. Wang, Y. F. Wang, W. Zeng, N. Xiao, Y. Wu, Z. Zhou, J. Chen and F. X. Chen, Chem.–Eur. J., 2012, 18, 1886–1889 CrossRef CAS PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- S. Manzetti and T. Lu, J. Phys. Org. Chem., 2013, 26, 473–483 CrossRef CAS.
- J. L. Calais, Int. J. Quantum Chem., 1993, 47, 101 CrossRef.
- M. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian 09, Gaussian Inc., Wallingford, CT, 2009, vol. 200, p. 28 Search PubMed.
- R. L. Blaine and H. E. Kissinger, Thermochim. Acta, 2012, 540, 1–6 CrossRef CAS.
- H. D. B. Jenkins, D. Tudela and L. Glasser, Inorg. Chem., 2002, 41, 2364–2367 CrossRef CAS PubMed.
- J. W. Ochterski, G. A. Petersson and J. A. Montgomery Jr, J. Chem. Phys., 1996, 104, 2598–2619 CrossRef CAS.
- J. A. Montgomery Jr, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532–6542 CrossRef.
- M. Suceska, EXPLO5, version, Brodarski Institute, Zagreb, Croatia, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2131217 and 2131218. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ra00215a |
| This journal is © The Royal Society of Chemistry 2022 |