
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new core–shell magnetic mesoporous surface molecularly imprinted composite and its application as an MSPE sorbent for determination of phthalate esters†
Yuxin Liu‡
a,
Wei Songa,
Dianbing Zhoua,
Fang Hana,
Xiaoming Gongb and
Pan Pan‡ *c
*c
aTechnology Center of Hefei Customs, Hefei 230022, P. R. China
bTechnology Center of Jinan Customs, Jinan 250000, P. R. China
cSchool of Chemistry and Chemical Engineering, Hefei University of Technology, 193 Tunxi Road, Hefei 230009, Anhui, P. R. China. E-mail: etaion@mail.hfut.edu.cn; Fax: +86 551 62904405; Tel: +86 551 62904405
First published on 2nd March 2022
Abstract
In this study, a new core–shell magnetic mesoporous surface molecularly imprinted polymer (Fe3O4@SiO2@mSiO2-MIPs) which has specific adsorption and rapid adsorption rate for phthalate esters (PAEs) was prepared by a convenient method. Based on this composite as a magnetic solid phase extraction (MSPE) material, a rapid, efficient and sensitive matrix dispersion magnetic solid-phase extraction gas chromatography-mass spectrometry method (DMSPE-GC/MS) was developed for the determination of PAEs in multiple liquid samples. It is the first time that Fe3O4@SiO2@mSiO2-MIPs have been prepared by bonding amino groups on the surface of a double layer silicon substrate with diisononyl phthalate (DINP) as virtual template and 3-(2-aminoethyl)-aminopropyl trimethoxymethylsilane (TSD) as functional monomer. FT-IR, TEM, EDS, SEM, XRD, BET and VSM were used to characterize the composite. The adsorption isotherm and kinetics of Fe3O4@SiO2@mSiO2-MIPs showed that it possessed fast adsorption rates (approximately 5 min to reach equilibrium), high adsorption capacities (523.9 mg g−1) and good recognition of PAEs. The real samples were preconcentrated by Fe3O4@SiO2@mSiO2-MIPs, under the optimum DMSPE-GC/MS conditions. Validation experiments showed that the method presented good linearity (R2 > 0.9971), satisfactory precision (RSD < 5.7%) and high recovery (92.1–105.8%), and the limits of detection ranged from 1.17 ng L−1 to 3.03 ng L−1. The results indicated that the novel method had good sensitivity, high efficiency and wide sample application and was suitable for the determination of PAEs in liquid drink samples such as water, alcohol, beverages and so on.
Introduction
Phthalate esters (PAEs) are plasticizers widely used as polymer additives in the manufacture of food packaging, medical devices, plastics and other industries.1,2 It has found that PAEs have estrogen-like effects, which can interfere with the endocrine system, leading to human reproductive system lesions, and other potential health risks.3,4 There are no covalent bonds between PAEs and plastic molecules, so they easily migrate from packaging materials to food and the environment in the process of use and contact.5 Due to their extensive use for many years, they have become widespread pollutants throughout the world.6,7 Monitoring and detecting the content of PAEs in food samples is necessary to accurately assess exposure. However, due to the low content, it is very difficult to detect them by conventional methods.At present, a variety of GC-MS analytical methods have been applied to detect PAEs coupled with different pretreatments such as liquid–liquid extraction(LLE),8,9 solid phase extraction (SPE).10–14 However, the traditional LLE and SPE process has some disadvantages such as time-consuming, consumption of a large number of organic reagents, low mass transfer speed, non reusability and so on. In recent years, the magnetic solid phase extraction (MSPE) technology based on magnetic extraction materials has attracted extensive attention in sample pretreatment.15,16 Magnetic absorbent can be separated from the liquid phase under an external magnetic field to avoids tedious filtration or centrifugation procedures.17 Meanwhile, the magnetic adsorbent can be directly dispersed in the sample for extraction without column loading, which can greatly improves the extraction efficiency.18,19
Functional magnetic adsorbents have become the focus of research because of the development of MSPE.20,21 Magnetic mesoporous materials combined with molecularly imprinted technology (MIT) show good application prospects in the field of solid phase extraction.22–24 The Fe3O4 nanoparticles, commonly used as magnetic core due to their good magnetic properties and low toxicity, are functionalized by different materials to prepare magnetic composites.23,25–27 Wang synthesized magnetic graphitic carbon nitride nanocomposites for the separation and enrichment of PAEs in environmental water samples.28 Chang also reported a core–shell magnetic surface molecularly imprinted polymer nanosparticles synthesized by using unmodified SiO2 spheres as carriers, which are ideal material for the separation and preconcentration of bisphenol A in aqueous media.29
MIPs have shown potential applications as adsorbent materials for SPE.15,30–32 However, the MIPs prepared by traditional methods have some problems, such as uneven distribution of imprinted sites, too deep or tight embedding of imprinted molecules, poor dynamic performance and regeneration effect.33–35 Surface molecularly imprinted technology (SMIT) was used to modify the imprinted active sites on the surface of carriers to improve the selectivity and adsorption capacity of the materials for the target compounds. In recent years, kinds of surface molecularly imprinted materials have been synthesized and used for the detection of PAEs.13,28,36–40 Our team reported the yolk–shell type magnetic mesoporous carbon surface molecular imprinting composite for the detection of PAEs in water samples previously.22 The material has the characteristics of strong specific recognition ability and high saturated adsorption capacity, but the magnetic properties are greatly weakened (the saturation susceptibility is only 21.5 emu g−1) due to the large cavity between the mesoporous shell and the magnetic core, which makes it difficult to be separated by external magnetic field. And the adsorption equilibrium speed is slow and need to be improved.
In this paper, we prepared a new magnetic core–shell mesoporous surface molecularly imprinted composite (Fe3O4@SiO2@mSiO2-MIPs) as the adsorbent for selective extraction and determination of PAEs in multi matrix liquid food samples rapidly. The composite can significantly improve the detection efficiency of PAEs. Fe3O4@SiO2@mSiO2-MIPs were synthesized by using 3-(2-aminoethyl)-aminopropyl trimethoxymethylsilane (TSD) as amino functional group onto the magnetic core–shell composite and DINP as the virtual template. Based on this composite, we established a dispersion magnetic solid-phase extraction gas chromatography-mass spectrometry (DMSPE-GC/MS) method which has high extraction efficiency, special selectivity and accuracy for the determination of PAEs in multiple liquid food samples such as water, alcohol and beverage.
Experimental
Materials and chemicals
The standards of six phthalate esters, including dimethyl phthalate (DMP, 99%), diethyl phthalate (DEP, 99%), dibutyl phthalate (DBP, 99%), dipentyl phthalate (DPP, 99%), benzyl butyl phthalate (BBP, 98%) and diethylhexyl phthalate (DEHP, 99%) were obtained from Sigma-Aldrich LLC (Shanghai, China). Methacrylic acid (MAA, 98%) was purified by distillation under vacuum. Diisononyl phthalate (DINP), ethylene glycol dimethacrylate (EGDMA, 98%), 3-(2-aminoethyl)-aminopropyl trimethoxymethylsilane (TSD), tetraethoxysilane (TEOS, 99%), and ammonia solution (28%) were obtained from Aladdin Reagent Corp. (China). Azobisisobutyronitrile (AIBN), hydrochloric acid (HCl), sodium hydroxide(NaOH), ferric chloride hexahydrate (FeCl3·6H2O), ferrous chloride tetrahydrate (FeCl2·4H2O), anhydrous ethanol, methanol, chloroform, acetic acid, cetyltrimethyl ammonium bromide (CTAB), ammonium nitrate, dodecylbenzene (DB) and butyl benzoate (BB) were purchased from Sinopharm Chemical Corp. (China). Doubly distilled water obtained from a laboratory purification system was used throughout the experiments.Instrumental and analytical conditions
Nicolet iS10 Fourier transform (FT-IR) spectrometer (United States) was used to obtain IR spectra of the composites, with the resolution of 2 cm−1 and the spectral range of 4000–400 cm−1. Transmission electron microscopy (TEM) images and the Energy Dispersive Spectrometer (EDS) mapping were recorded on a high-resolution transmission electron Talos F200X (USA) operated at 200 kV. Scanning electron microscopy (SEM) images were obtained by Gemini 500 electron microscope (ZEISS, Germany) operated at 200 kV. Nitrogen sorption isotherms were measured at 77 K using a Micromeritics Tristar 3000 analyzer (Micromeritics, United States). The Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface areas. The X-ray diffraction (XRD) spectra were recorded using the Philips X'Pert PRO SUPER diffractometer (PANalytical, United Kingdom) with nickel-filtered Cu-Kα radiation (λ = 1.540598 Å) at 40 kV and 200 mA. The magnetic properties were analyzed with a super-conducting quantum interference device (SQUID) magnetometer (United States) at 300 K and the magnetic field intensity was ±3 T.Gas chromatography-mass spectrometry (GC-MS) was performed using an Agilent 5975C with a DB-5ms capillary column (30 m × 0.25 mm × 0.25 μm) and quadrupole mass spectrometers. The split/splitless injector was set to 250 °C, and 1 μL of the sample was injected without split. High purity helium (99.999%) was used as carrier gas (1.0 mL min−1). The oven temperature was programmed as follows: initial 60 °C, hold 1.0 min; rate 20 °C min−1 to 220 °C, hold 1.0 min; rate 5 °C min−1 to 280 °C and hold 1.0 min.
Synthesis of Fe3O4@SiO2@mSiO2-MIPs and Fe3O4@SiO2@mSiO2-NIPs
100 mL FeCl3·6H2O solution (0.2 mol L−1) and 100 mL FeCl2·4H2O solution (0.1 mol L−1) were put into a 500 mL three necked round-bottom flask, then added HCl aqueous solution (12.1 mol L−1, 0.85 mL). Next, at the stirring speed of 1200 rpm and T = 60 °C, the above solution was added dropwise into NaOH solution (1.5 mol L−1, 250 mL). After the addition, kept the temperature and the stirred for 30 min, then cooled to room temperature. All steps were carried out under N2. After the reaction, the precipitate was separated from the reaction medium by external magnet, and washed three times with 50 mL of deionized water and two times with 50 mL of anhydrous ethanol, dried overnight in vacuum at 60 °C to obtain Fe3O4 magnetic nanoparticles.The magnetic core–shell polymer composite was synthesized by the improved Stöber method. 1.0 g Fe3O4 magnetic nanoparticles were added to HCl solution (0.1 mol L−1, 10 mL) and ultrasonically dispersed for 5 min, then washed three times with 10 mL of deionised water, dispersed into a mixed solution of anhydrous ethanol (64 mL), deionized water (16 mL) and ammonia solution (28%, 2.0 mL), and then ultrasonically dispersed for 5 min. 5 mL tetraethoxysilane (TEOS) was added dropwise, and the mixture was stirred for 6 h. The particles were separated by external magnet and washed three times with 20 mL of deionised water and 20 mL of anhydrous ethanol. After dried in vacuum at 60 °C for 12 h, the composites coated with SiO2 layer were obtained (Fe3O4@SiO2). The particles were ultrasonically dispersed for 5 min in a mixture of anhydrous ethanol (40 mL), deionised water (50 mL) and ammonia solution (28%, 1.0 mL). Cetyltrimethylammonium bromide (CTAB, 2.0 g) was added as template and stirred for 30 min. TEOS (3.0 mL) was added dropwise and stirred at 50 °C for 6 h. The particles were separated by external magnetic field, washed three times with 20 mL of deionised water and 20 mL of anhydrous ethanol respectively, and then dried in vacuum at 60 °C for 6 h. The product was dispersed in ammonium nitrate ethanol solution (6.0 g L−1, 60 mL), then stirred at 60 °C for 2 h, repeated three times to remove CTAB. After that, the particles were separated by external magnet and washed with 20 mL of anhydrous ethanol for three times. The product was dried in vacuum at 60 °C for 6 h to obtain the nanocomposites (Fe3O4@SiO2@mSiO2).
1.0 g of the Fe3O4@SiO2@mSiO2 was added into a three-necked round-bottom flask. 60 mL of toluene, 6 mL of 3-(2-aminoethyl)-aminopropyl trimethoxymethylsilane (TSD) were slowly added, and then refluxed at 80 °C for 3 h. The particles were separated by external magnet. After drying in vacuum at 60 °C for 6 h the magnetic core–shell mesoporous composite modified by amino group (Fe3O4@SiO2@mSiO2-NH2) were obtained.
Fe3O4@SiO2@mSiO2-NH2 (0.5 g), diisononyl phthalate (DINP, 1 mmol) as virtual template, methacrylic acid (MAA, 4 mmol) were ultrasonically dispersed in 40 mL of chloroform for 15 min; then added 20 mmol of ethylene glycol dimethacrylate (EGDMA) and 50 mg of azodiisobutyronitrile (AIBN). The mixture was ultrasonically dispersed for 10 min and purged with nitrogen for 10 min, then stirred at 70 °C for 12 h. The particles were separated by external magnet and washed with 100 mL solution of methanol and acetic acid (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, V/V) in a Soxhlet apparatus for 60 h to remove the virtual template (DINP) and non-polymerized monomers. The particles were washed with 20 mL of methanol for three times. After drying in vacuum at 60 °C for 6 h, the magnetic core–shell mesoporous surface molecularly imprinted polymer (Fe3O4@SiO2@mSiO2-MIPs) was obtained. By the way, the synthesis method of non-imprinted polymer (Fe3O4@SiO2@mSiO2-NIPs) is similar to that mentioned above, except that the template molecule (DINP) was not added in the process.
:1, V/V) in a Soxhlet apparatus for 60 h to remove the virtual template (DINP) and non-polymerized monomers. The particles were washed with 20 mL of methanol for three times. After drying in vacuum at 60 °C for 6 h, the magnetic core–shell mesoporous surface molecularly imprinted polymer (Fe3O4@SiO2@mSiO2-MIPs) was obtained. By the way, the synthesis method of non-imprinted polymer (Fe3O4@SiO2@mSiO2-NIPs) is similar to that mentioned above, except that the template molecule (DINP) was not added in the process.
DINP was selected as the virtual template because no matter how thoroughly it was cleaned in the Soxhlet extraction stage, a small amount of residual template molecules could not be avoided to remain on the molecularly imprinted polymer. If the virtual template different from the target molecule is not used, the template molecules eluted in the subsequent detection process will affect the accuracy of the test results of the target molecule. This choice is made by referring to the reported literature, which is a more common method.
DMSPE-GC/MS procedure
DMSPE procedure for the extraction of PAEs was as follows: 10 mg of magnetic sorbents were added to 10 mL of sample solution in the tube with plug. After the tube was sealed and vibrated for 6 min, the magnetic sorbents were separated by external magnetic field. Then the magnetic sorbents were ultrasonicated for 10 min with 1.0 mL chloroform to desorb the PAEs, and then the solutions were dried with 0.5 g of anhydrous sodium sulfate. After the composite was separated by external magnetic field, the remaining solution was filtered through a 0.22 μm syringe-driven filter and put into a sample bottle for GC-MS analysis. The optimizations of MSPE dosage, eluting solvent, eluent dosage, elution time and pH value were provided in the ESI.†Collection and pre-treatment of actual samples
To demonstrate the suitability and application of the DMSPE-GC/MS method for actual liquid food samples, Five different kinds of plastic bottled drinks and two alcoholic samples were purchased from local supermarkets in Hefei, the different sample types are represented by pure water, apple juice, protein beverage, carbonated beverage, tea beverage, beer and white wine, which covered a wide variety of different substrates. These samples were filtered through 0.45 μm membrane to eliminate particulate and then analyzed under optimal DMSPE-GC/MS conditions. For the spiking experiments, standard analytes were added directly into the sample solutions.Results and discussion
Characterization of materials
Fig. 1(A) is the FT-IR spectra of Fe3O4 (a); Fe3O4@SiO2@mSiO2 (b); Fe3O4@SiO2@mSiO2-NH2 (c); Fe3O4@SiO2@mSiO2-MIPs (d). In the figure, the broad band of 3427 cm−1 is the anti stretching vibration of water on the surface of nanoparticles, 1633 cm−1 is the bending stretching vibration of water –OH, and 580 cm−1 is the stretching vibration of Fe–O. The strong and wide absorption peak at 1092 cm−1 is the antisymmetric stretching vibration absorption peak of Si–O–Si, and the absorption peak at 797 cm−1 is the symmetric stretching and bending vibration absorption peak of Si–O bond. The spectra of (c) and (d) are similar, and the characteristic absorption peak of N–H appears at 1546 cm−1, which indicates that amino groups have been successfully modified on the surface of magnetic composite microspheres. The absorption peak at 1382 cm−1 is the symmetric stretching and bending vibration absorption peak of Si–R bond.41 The characteristic absorption peak of N–H in (d) has lower intensity, which indicates that the template molecules have been completely removed. It is inferred that the MIP layer has been modified to the surface of mesoporous materials.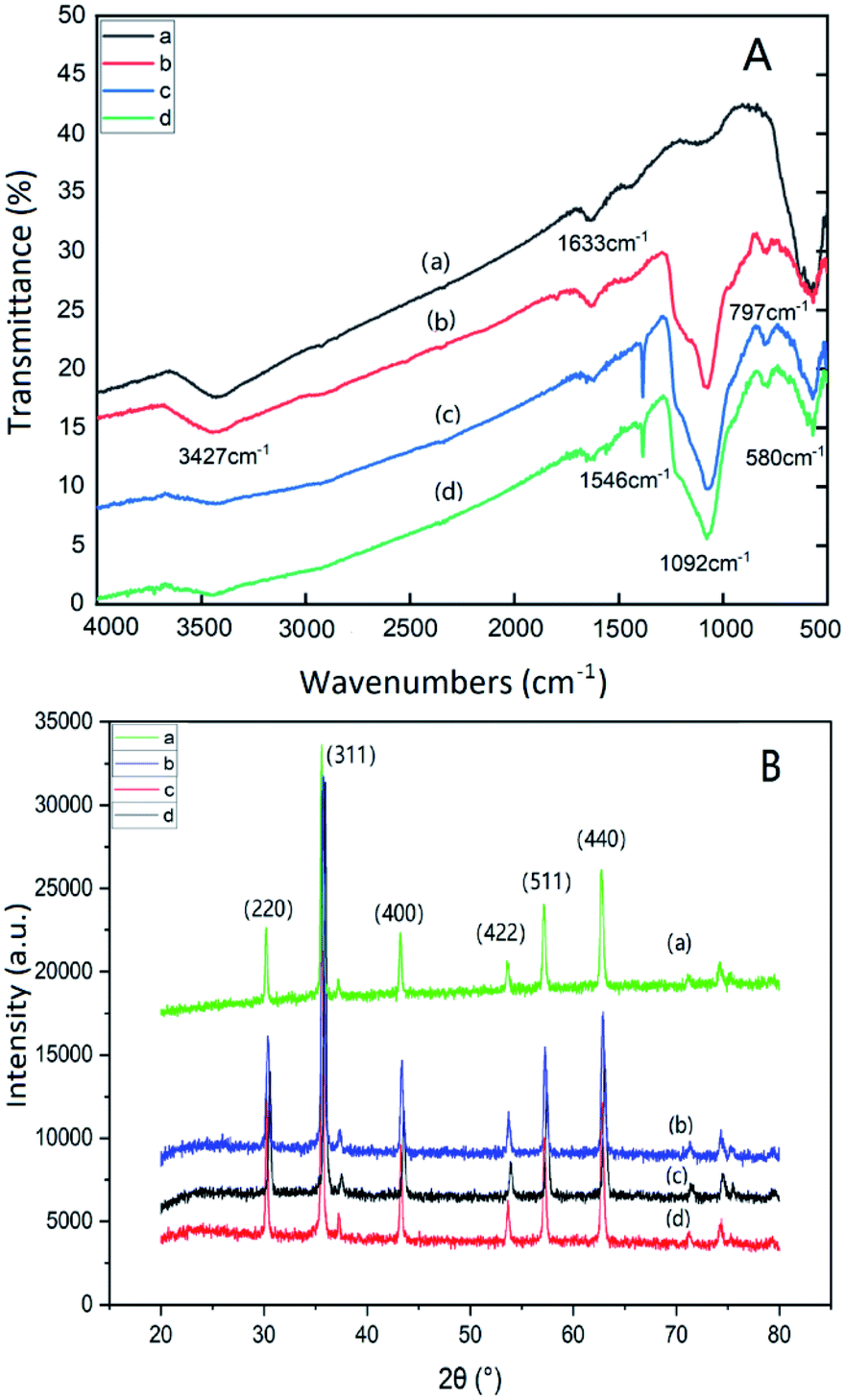 | ||
| Fig. 1 FT-IR spectra (A) and Wide-angle XRD patterns (B) of Fe3O4 (a), Fe3O4@SiO2@ mSiO2 (b), Fe3O4@SiO2@mSiO2-NH2 (c), Fe3O4@SiO2@mSiO2-MIPs (d). | ||
Fig. 1(B) shows the wide-angle X-ray diffraction patterns of Fe3O4 (a), Fe3O4@SiO2@mSiO2 (b), Fe3O4@SiO2@mSiO2-NH2 (c), Fe3O4@SiO2@mSiO2-MIPs (d). Sharp characteristic diffraction peaks appear at 30.32°, 35.62°, 43.32°, 53.76°, 57.22° and 62.78° respectively, corresponding to the characteristic diffraction peaks (220), (311), (400), (422), (511) and (440) of Fe3O4 nanoparticles respectively, which prove the existence of magnetic ions in all composite materials. The diffraction peaks of Fe3O4@SiO2@mSiO2 and Fe3O4@SiO2@mSiO2-NH2 did not change significantly compared with Fe3O4. It was found that the morphology of Fe3O4 was not changed by the introduction of amino groups and the SiO2 coated on the surface of Fe3O4 nanoparticles. The intensity of 311 peak was slightly weakened due to the SiO2 shell coating. There is no significant difference between Fe3O4 and Fe3O4@SiO2@mSiO2-MIPs, but the diffraction peak becomes narrow due to the increase of the crystal size of Fe3O4@SiO2@mSiO2-MIPs.
Fig. 2(A1) and (A2) are the comparison of SEM and TEM of Fe3O4@SiO2@mSiO2-MIPs, which shows that the surface of amino modified mesoporous material has been successfully modified with a thin layer of molecularly imprinted polymer. The regular pore structure can be observed in the outer layer and the overall distribution is spherical. EDS element mapping results of representative MIPs mesoporous nanospheres are shown in Fig. 2(B1)–(B6). From the distribution of elements, we can see the existence of Fe (B2), O (B3), Si (B4) and N (B5). Si is mainly distributed on the surface of the whole composite. The diameter of Si- and N-mapped spheres in Fig. 2 is much larger than those of Fe-mapped spheres, which also confirms the existence of Fe3O4 core and SiO2 outer layer, the composite has obvious core–shell structure. It also shows that the amino group and MIP layer have been successfully modified on the surface of the material.
 | ||
| Fig. 2 SEM and TEM images (A); scanning TEM (STEM) image (B1) of Fe3O4@SiO2@ mSiO2-MIPs; EDS elemental maps of Fe, O, Si and N respectively (B2–B5) and combined (B6). | ||
The nitrogen adsorption desorption isotherms are shown in Fig. 3(A). The results show that both of Fe3O4@SiO2@mSiO2-MIPs and Fe3O4@SiO2@mSiO2-NH2 are mesoporous materials with typical multilayered adsorption curves (type IV isothermal curve). In the region of high P/P0 (P/P0 = 0.5–0.8), the adsorbate condense in capillary, the isotherm obtained during desorption does not coincide with that obtained during adsorption, and the desorption hysteresis presents a hysteresis loop. The curve of mono-multilayer adsorption region (P/P0 = 0.1–0.5) is flat; when the relative pressure is close to 1 (P/P0 = 0.8–1.0), it adsorbs on the macropore, and the multilayer adsorption on the outer surface makes the curve rise rapidly. The BET surface area of Fe3O4@SiO2@ mSiO2-MIPs was calculated to be 327.6 m2 g−1, which make the composite has high adsorption capacity for the target analytes.
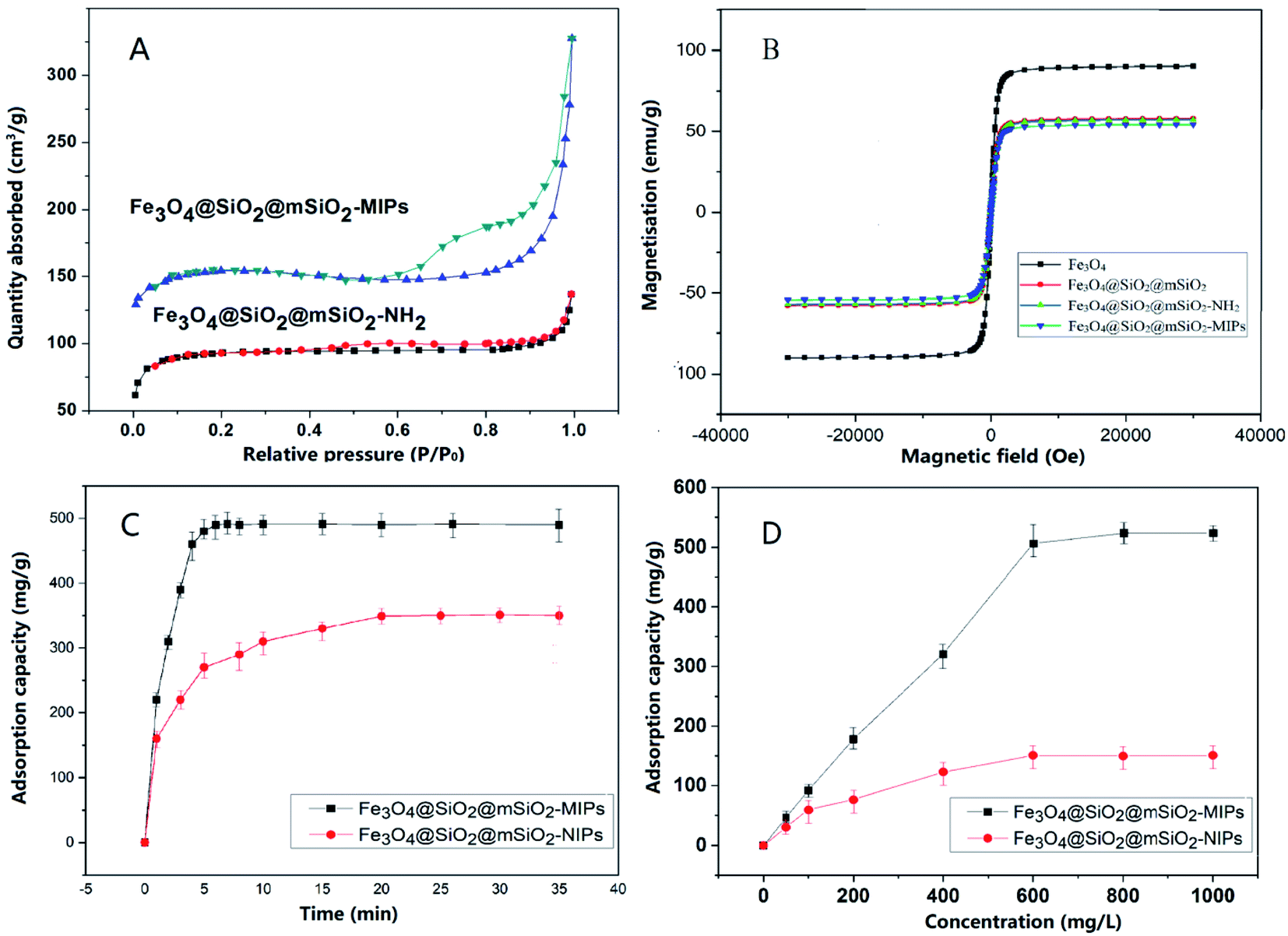 | ||
| Fig. 3 (A) Nitrogen adsorption/desorption isotherms of Fe3O4@SiO2@mSiO2-MIPs and Fe3O4@SiO2@mSiO2-NH2; (B) magnetization curves of Fe3O4, Fe3O4@SiO2@mSiO2, Fe3O4@SiO2@mSiO2-NH2 and Fe3O4@SiO2@mSiO2-MIPs; (C) adsorption kinetics and (D) adsorption isotherms of Fe3O4@SiO2@mSiO2-MIPs and Fe3O4@SiO2@mSiO2-NIPs. | ||
Fig. 3(B) is the magnetization curve of magnetic microspheres Fe3O4, Fe3O4@SiO2@mSiO2, Fe3O4@SiO2@mSiO2-NH2 and Fe3O4@SiO2@mSiO2-MIPs. As shown in the figure, the nanoparticles have no hysteresis loops and show good superparamagnetism. The saturation magnetization of Fe3O4 nanoparticles is 85.2 emu g−1 when Fe3O4 nanoparticles are coated with SiO2 and modified with amino groups, the saturation magnetization of Fe3O4 nanoparticles only decreases slightly. The saturation magnetization of Fe3O4@SiO2@mSiO2 and Fe3O4@SiO2@mSiO2-NH2 are still 55.9 emu g−1 and 55.8 emu g−1, respectively, showing strong magnetic properties. The saturation magnetization of the final product Fe3O4@SiO2@mSiO2-MIPs was exhibited to be 54.9 emu g−1, which was outstandingly higher than that of Fe3O4@void@C-MIPs (21.5 emu g−1),22 MGO@mSiO2-MIPs (39.1 emu g−1),37 and MAG-MIM (26 emu g−1).13 It can be separated easily under the action of external magnetic field.
Selective adsorption of Fe3O4@SiO2@mSiO2-MIPs
In order to evaluate the selective adsorption performance of Fe3O4@SiO2@mSiO2-MIPs sorbents, the adsorption properties of Fe3O4@SiO2@mSiO2-MIPs were compared with those of Fe3O4@SiO2@mSiO2-NIPs according to the process of section “DMSPE-GC/MS procedure”. The competitive samples were methanol solutions of the six PAEs and their analogues butyl benzoate (BB) and dodecylbenzene (DB) at 600 mg L−1, respectively. The results are shown in Table 1. The adsorption capacity of Fe3O4@SiO2@mSiO2-MIPs for the six PAEs is high with the maximum capacities ranging from 53.0 to 114.4 mg g−1, while the adsorption for two analogues is lower than 15 mg g−1, indicating that the imprinted sites on the Fe3O4@SiO2@mSiO2-MIPs lead to excellent adsorption performance and high adsorption capacity for the PAEs. There is no significant difference in the adsorption effect of non-molecularly imprinted composite on the target PAEs and the two analogues, The adsorption capacities of NIPs and MIPs for analogues DB and BB were similar, which indicated that the MIP layer on the surface of MIPs had no specificity for these analogues. The adsorption is mainly due to mesoporous physical adsorption, so it does not show selectivity. DEHP and DPP have larger adsorption capacity and imprinting factor (IF = QMIPs/QNIPs, Q is the equilibrium adsorption capacity) indicating that the higher the similarity with virtual template (DINP), the larger the adsorption capacity and imprinting factor. Taking the equilibrium adsorption capacity of MIPs minus the equilibrium adsorption capacity of NIPs as the specific adsorption capacity of MIPs (QMIPs − QNIPs), the selectivity factor (α) of MIPs to DINP is defined to 1, the selectivity factor (α) for other substrates is the ratio of the specific adsorption capacity of MIPs to DINP. α of six PAEs were between 0.36 and 0.91, indicating that MIPS have good selectivity for them. The Fe3O4@SiO2@mSiO2-MIPs is a good adsorbent for the separation of PAEs from complex matrices.| PAEs | QMIPs (mg g−1) | QNIPs (mg g−1) | IF (QMIP/QNIP) | α |
|---|---|---|---|---|
| DMP | 53.0 | 20.5 | 2.6 | 0.36 |
| DEP | 66.5 | 24.2 | 2.7 | 0.46 |
| DBP | 82.7 | 25.8 | 3.2 | 0.62 |
| DPP | 114.4 | 23.6 | 4.8 | 0.99 |
| BBP | 105.7 | 31.8 | 3.3 | 0.81 |
| DEHP | 107.6 | 24.7 | 4.4 | 0.91 |
| DINP (template) | 123.9 | 22.6 | 5.5 | 1.11 |
| BB | 14.2 | 14.7 | 0.9 | — |
| DB | 13.9 | 12.8 | 1.1 | — |
Adsorption kinetic studies
The adsorption capacity of Fe3O4@SiO2@mSiO2-MIPs for PAEs at different adsorption time was measured and the rate of reaching adsorption equilibrium was studied and evaluated. 10 mg of Fe3O4@SiO2@mSiO2-MIPs was added into 10 mL methanol solution of six PAEs at a concentration of 1000 mg L−1, respectively. The adsorption time of vortex agitation is 1 min, 3 min, 5 min, 8 min, 10 min, 15 min and 20 min respectively. The contents of PAEs were determined according to the “DMSPE-GC/MS procedure”. According to the curves in Fig. 3(C), the adsorption capacity of Fe3O4@SiO2@mSiO2-MIPs increased rapidly within 3 min, then, reached the adsorption equilibrium after 5 min. The adsorption capacity of Fe3O4@SiO2@mSiO2-NIPs increased in 10 min and reached equilibrium in 20 min. Compared with the previous MIPs material such us Fe3O4@void@C-MIPs (20 min) and MGO@mSiO2-MIPs (120 min),22,37 the adsorption equilibrium rate of Fe3O4@SiO2@ mSiO2-MIPs has been greatly improved, which is mainly due to the high specific surface area and the chemisorption force generated by the surface specific recognition sites, which reduces the mass transfer resistance and facilitates the rapid recognition and adsorption of PAEs targets.For studying the mass transfer mechanism of the adsorption process, the pseudo-first-order rate kinetic model and pseudo-second-order kinetic model were used to fit the kinetic data.
Pseudo-first-order model:
| ln(Qe − Qt) = lnQe − k1t |
Pseudo-second-order kinetic model:
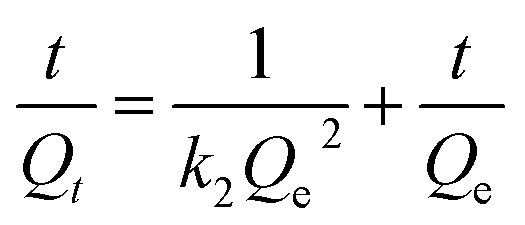
Adsorption isotherm studies
10 mg of Fe3O4@SiO2@mSiO2-MIPs or Fe3O4@SiO2@mSiO2-NIPs were added into 10 mL methanol solution of six PAEs in the concentration range of 0–1000 mg L−1 in methanol. The experimental steps were carried out according to the procedure in section “DMSPE-GC/MS procedure”. Fig. 3(D) shows the thermodynamic adsorption isotherms of Fe3O4@SiO2@mSiO2-MIPs and Fe3O4@SiO2@mSiO2-NIPs at 298 K. Qe (mg g−1) is the adsorption capacity, and the calculation formula is:where Ci and Cf are the concentrations of PAEs in the initial and final solution (mg L−1), V (mL) is the volume of bulk solution, and W (mg) is the weight of material.
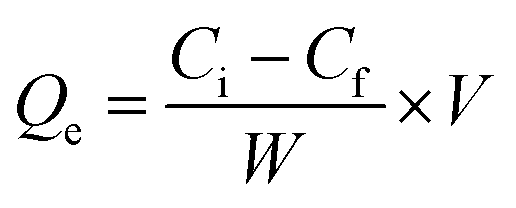
It showed that the adsorption capacities of MIPs and NIPs increase with the increasing concentrations of the mixture PAEs. The total maximum saturated equilibrium adsorption capacities of Fe3O4@SiO2@mSiO2-MIPs are 523.9 mg g−1, which are higher than those of Fe3O4@SiO2@mSiO2-NIPs (150.6 mg g−1). That's mainly due to the formation of a large number of molecularly imprinted active sites for the PAEs on the surface of Fe3O4@SiO2@mSiO2-MIPs.
We used Langmuir and Freundlich models to fit the experimental data to study the equilibrium adsorption process.
Langmuir equations:
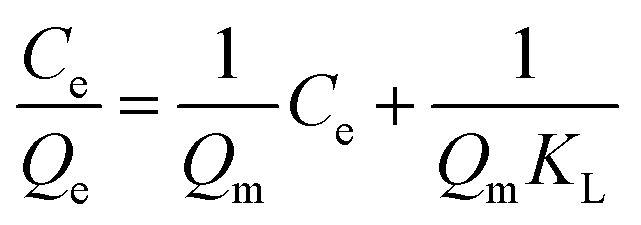
Freundlich equation:
| lnQe = mlnCe + lnKF |
Evaluation of Fe3O4@SiO2@mSiO2-MIPS as MSPE material
The enrichment effect of Fe3O4@SiO2@mSiO2-MIPs on PAEs was evaluated according to the process in section “DMSPE-GC/MS procedure”. 100 μg mL−1 solutions of the six PAEs in methanol were used as solution in method. As shown in Fig. 4(A), the recoveries of six PAEs by Fe3O4@SiO2@mSiO2-MIPs ranged from 92.6% to 103.8%. The composite can selectively separate and enrich trace PAEs in samples effectively.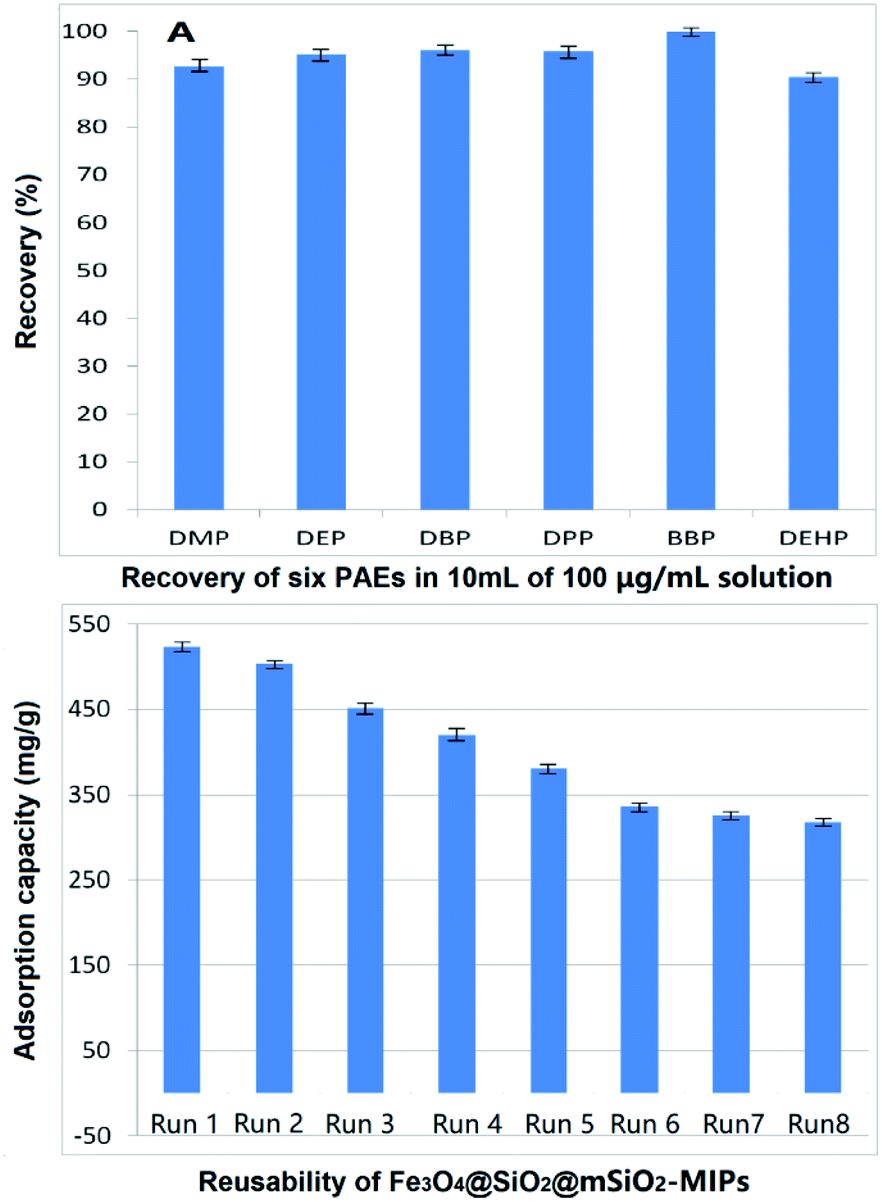 | ||
| Fig. 4 (A) Recovery of six PAEs in 10 mL 100 mg L−1 solution; (B) reusability of Fe3O4@SiO2@mSiO2-MIPs. | ||
Reusability of Fe3O4@SiO2@mSiO2-MIPS
From the perspective of environmental protection and economy, whether the material can be recycled is of great significance. Fig. 4(B) shows the trends of saturated total equilibrium adsorption capacities of the six PAEs with a concertation of 1000 mg L−1 in methanol during Fe3O4@SiO2@mSiO2-MIPs recycling for eight times. After each cycle, the Fe3O4@SiO2@mSiO2-MIPs was washed three times with 20 mL of methanol to regenerate the imprinted binding cavities. The results showed that the saturated equilibrium adsorption capacity of the composite for six PAEs was still 317.6 mg g−1 after eight cycles reused. The recoveries of PAEs were 87.1–93.3% for DMP, 89.8–95.2% for DEP, 90.9–96.2% for DBP, 90.1–96.1% for DPP, 92.2–97.3% for BBP and 85.5–92.8% for DEHP. Equilibrium adsorption capacities and recoveries were decreased slightly from the sixth cycle to the eighth cycle, indicating that the structure of composites became stable and almost has no change. The range of adsorption capacity is larger than the concentration range of the solution when investigating the recovery of PAEs. In this range, the recovery can still maintain a relatively stable result. Compared with the traditional disposable adsorption reagent, it has irreplaceable advantages on save the costs for the pre-treatment of samples.Evaluation of DMSPE-GC/MS method and application to real samples
| PAEs | Retention time (min) | Conventional regression equations | R2 | LOD (ng L−1) | RSD (%) |
|---|---|---|---|---|---|
| a y is the peak area; and x is the concentration of PAEs. | |||||
| DMP | 7.89 | y = 70623x + 1277.4a | 0.9990 | 2.89 | 3.5 |
| DEP | 8.76 | y = 67920x + 866.6 | 0.9986 | 2.36 | 4.3 |
| DBP | 11.31 | y = 109814x − 7614.3 |
0.9989 | 1.17 | 4.8 |
| DPP | 13.13 | y = 87702x − 12527 | 0.9977 | 2.06 | 3.9 |
| BBP | 15.46 | y = 18146x − 2666.3 | 0.9971 | 3.03 | 4.1 |
| DEHP | 17.85 | y = 22569x − 2417.8 | 0.9983 | 2.73 | 5.7 |
The recovery experiments were carried out to the real samples by the concentration of the known standard addition. According to the content level of the real samples, the samples were spiked with each target compound at the standard addition level of 5, 10, 20 μ g L−1 for plastic bottled drinks and 0.05, 1.0 and 5.0 mg L−1 for alcoholic samples. Recoveries for six PAEs at spiking levels in different matrices were satisfactory (between 92.1% and 105.8%), which show that the developed method is feasible in the detection of PAEs in real liquid drink samples.
Comparison of different extraction and determination methods of PAEs
To demonstrate the advantages of the proposed method, as shown in Table 3, the adsorption performance of Fe3O4@SiO2@mSiO2-MIPs and the analytical parameters of the DMSPE-GC/MS method were compared with other MSPE analytical methods reported in the previous literature. It is clear that the Fe3O4@SiO2@mSiO2-MIPs have wider range of sample matrixes, the shortest adsorption equilibrium time and larger adsorption capacity among the various MIP adsorbents. At the same time, the method proposed in this study was characterized by its lower LODs and satisfactory RSD, as well as higher recoveries than those reported in other literature, indicating that the developed DMSPE-GC/MS method is an effective and sensitive analysis method for the determination of trace PAEs in real liquid drink samples.| Sample matrix | Adsorbent | Adsorption equilibrium time (min) | Adsorption capacity (mg g−1) | Magnetism (emu g−1) | Linear range (μg L−1) | LOD (ng L−1) | RSD (%) | R (%) | Reference no |
|---|---|---|---|---|---|---|---|---|---|
| a Not mentioned in the reference; DMIPs: dummy template molecularly imprinted polymers; MWCNTs: multi-walled carbon nanotubes; MAG-MIM: magnetic dummy molecularly imprinted dispersive solid-phase extraction; Fe3O4@void@C-MIPs: molecularly imprinted polymers on the surface of yolk–shell magnetic mesoporous carbon; MGO@mSiO2-MIPs: magnetic graphene oxide modified with mesoporous silica molecularly imprinted polymers. | |||||||||
| Fruit and juice | DMIPs | 5 | 281 | —a | 5–1000 | 2–20 | <10.2 | 72–100.2 | 36 |
| Beverage and alcoholic | MWCNTs/SiO2 | 40 | — | — | 0.00002–1 | 6–30 | 2.57–4.11 | 68–115 | 38 |
| Plastic bottled beverages | MAG-MIM | 40 | — | 26 | 0.0040–0.4 | 530–1200 | 3.1–6.9 | 89.5–101.3 | 13 |
| Water and beverage | Fe3O4@void@C-MIPs | 15–20 | 569.2 | 21.5 | 0.035–12.2 | 1.5–5.3 | 4.1–6.7 | 82.7–104.4 | 22 |
| Water | MGO@mSiO2-MIPs | 120 | 4.8 | 39.1 | 1–50 | 10–50 | <3.8 | >92.9 | 37 |
| Water, beverage and alcoholic | Fe3O4@SiO2@mSiO2-MIPs | 5 | 523.9 | 54.9 | 0.010–10.0 | 1.17–3.03 | <5.7 | 92.1–105.8 | Present |
Conclusions
In this study, a convenient method was used to synthesize core–shell magnetic mesoporous surface molecularly imprinted polymer composite (Fe3O4@SiO2@mSiO2-MIPs). Based on the composite as magnetic solid phase extraction sorbent, a rapid, efficient and sensitive matrix dispersion magnetic solid phase extraction gas chromatography-mass spectrometry method (DMSPE-GC/MS) was developed for the determination of trace phthalate esters (PAEs) in liquid foods.The characterized results supported that the core–shell structure of the composite Fe3O4@SiO2@mSiO2-MIPs has high magnetisation and large specific surface area. Adsorption thermodynamics and kinetics studies showed that Fe3O4@SiO2@mSiO2-MIPs had excellent recognition performance, fast adsorption rate (5 min to reach equilibrium), high adsorption capacity (523.9 mg g−1) and good reusability for PAEs, which has a great improvement over the previous MIPs performance. Based on the results, Fe3O4@SiO2@mSiO2-MIPs could be used as a new solid phase extraction sorbent in magnetic solid phase extraction for the rapid enrichment, selective separation and preconcentration of trace PAEs in complicated liquid samples. After Fe3O4@SiO2@mSiO2-MIPs enrichment, matrix dispersion magnetic solid phase extraction and magnetic separation, the obtained DMSPE method combined with GC-MS method achieved superior extraction efficiency and recoveries and obtained LOD values in the low concentration range for six PAEs. The results indicate that the DMSPE combined with GC/MS method is a rapid, selective and efficient technique to determine PAEs residues in complicated liquid drinks such as water, alcohol and beverage.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Research projects of China General Administration of Customs (Item No. 2019HK092 and 2020HK197).References
- X. L. Cao, Compr. Rev. Food Sci. Food Saf., 2010, 9, 21 CrossRef CAS PubMed.
- A. Mousa, C. Basheer and A. R. Al-Arfaj, J. Sep. Sci., 2013, 36, 2003 CrossRef CAS PubMed.
- J. J. Rios, A. Morales and G. Marquez-Ruiz, Talanta, 2010, 80, 2076 CrossRef CAS PubMed.
- Y. Zhang, Y. Q. Jiao, Z. X. Li, Y. Tao and Y. Yang, Sci. Total Environ., 2021, 771, 145418 CrossRef CAS PubMed.
- E. Psillakis and N. Kalogerakis, J. Chromatogr., A, 2003, 999, 145 CrossRef CAS PubMed.
- U. Heudorf, V. Mersch-Sundermann and J. Angerer, Int. J. Hyg Environ. Health, 2007, 210, 623 CrossRef CAS PubMed.
- L. Arfaeinia, S. Dobaradaran, F. Nasrzadeh, S. Shamsi, Y. Poureshghd and H. Arfaeinia, Microchem. J., 2020, 155, 104719 CrossRef CAS.
- G. Cinelli, P. Avino, I. Notardonato, A. Centola and M. V. Russo, Anal. Chim. Acta, 2013, 769, 72 CrossRef CAS PubMed.
- A. Fabiani, C. Corzani and G. Arfelli, Talanta, 2010, 83, 281 CrossRef CAS PubMed.
- J. He, R. Lv, J. Zhu and K. Lu, Anal. Chim. Acta, 2010, 661, 215 CrossRef CAS PubMed.
- Y. K. Lv, L. M. Wang, L. Yang, C. X. Zhao and H. W. Sun, J. Chromatogr., A, 2012, 1227, 48 CrossRef CAS PubMed.
- S. Scorrano, L. Longo and G. Vasapollo, Anal. Chim. Acta, 2010, 659, 167 CrossRef CAS PubMed.
- J. D. Qiao, M. Y. Wang, H. Y. Yan and G. L. Yang, J. Agric. Food Chem., 2014, 62, 2782 CrossRef CAS PubMed.
- M. Moazzen, A. M. Khaneghah, N. Shariatifar, M. Ahmadloo, I. Es, A. N. Baghani, S. Yousefinejad, M. Alimohammadi, A. Azari, S. Dobaradaran, N. Rastkari, S. Nazmara, M. Delikhoon and G. J. Khaniki, Arab. J. Chem., 2019, 12, 476 CrossRef CAS.
- L. Sun, X. Sun, X. B. Du and Y. S. Yue, Anal. Chim. Acta, 2010, 665, 185 CrossRef CAS PubMed.
- E. Tahmasebi, Y. Yamini, M. Moradi and A. Esrafili, Anal. Chim. Acta, 2013, 770, 68 CrossRef CAS PubMed.
- J. Ding, Q. Gao, X. S. Li, W. Huang, Z. G. Shi and Y. Q. Feng, J. Sep. Sci., 2011, 34, 2498 CrossRef CAS PubMed.
- Y. Zhang, R. Liu, Y. Hu and G. Li, Anal. Chem., 2009, 81, 967 CrossRef CAS PubMed.
- J. Meng, J. Bu, C. Deng and X. Zhang, J. Chromatogr., A, 2011, 1218, 1585 CrossRef CAS PubMed.
- X. B. Zhang, H. W. Tong, S. M. Liu and G. P. Yong, J. Mater. Chem. A, 2013, 1, 7488 RSC.
- J. I. Abdelghani, R. S. Freihat and A. H. El-Sheikh, J. Environ. Chem. Eng., 2020, 8, 103527 CrossRef CAS.
- R. Yang, Y. X. Liu, X. Y. Yang and S. M. Liu, Talanta, 2016, 161, 114 CrossRef CAS PubMed.
- J. Li, X. B. Zhang, Y. X. Liu, H. W. Tong and S. M. Liu, Talanta, 2013, 117, 281 CrossRef CAS PubMed.
- C. Xu, X. Shen and L. Ye, J. Mater. Chem., 2012, 22, 7427 RSC.
- Q. H. Wu, Y. H. Song, Q. Q. Wang, W. H. Liu, L. Hao, Z. Wang and C. Wang, Food Chem., 2021, 339, 127855 CrossRef CAS PubMed.
- R. Yang, Y. X. Liu, X. Y. Yan and S. M. Liu, J. Mater. Chem. A, 2016, 4, 9807 RSC.
- M. X. Wei, X. Y. Yan, S. M. Liu and Y. X. Liu, J. Mater. Sci., 2018, 53, 4897 CrossRef CAS.
- M. Wang, X. D. Yang and W. T. Bi, J. Sep. Sci., 2015, 38, 445 CrossRef CAS PubMed.
- T. T. Chang, Y. X. Liu, X. Y. Yang and S. M. Liu, RSC Adv., 2016, 6, 66297 RSC.
- S. L. Wei, W. T. Liu, X. C. Huang and J. K. Ma, J. Sep. Sci., 2018, 41, 3806 CrossRef CAS PubMed.
- T. P. Cai, M. H. Ma, H. C. Liu, J. M. Li, J. Hou and B. L. Gong, J. Liq. Chromatogr. Relat. Technol., 2019, 42, 459 CrossRef CAS.
- C. X. Lu, Z. G. Tang, X. X. Gao, X. M. Ma and C. B. Liu, Microchim. Acta, 2018, 185, 373 CrossRef PubMed.
- L. X. Chen, S. F. Xu and J. H. Li, Chem. Soc. Rev., 2011, 40, 2922 RSC.
- Y. B. Fan, Y. M. Yin, W. B. Jiang, Y. P. Chen, J. W. Yang, J. Wu and M. X. Xie, Food Chem., 2014, 142, 170 CrossRef CAS PubMed.
- T. Zhu, D. Xu, Y. G. Wu and T. Li, J. Mater. Chem. B, 2013, 1, 6449 RSC.
- J. H. Hu, T. Feng, W. L. Li, H. Zhai and Y. Liu, J. Chromatogr., A, 2014, 1330, 6 CrossRef CAS PubMed.
- L. H. Guo, X. G. Ma, X. W. Xie, R. F. Huang, M. Y. Zhang, J. Li, G. L. Zeng and Y. M. Fan, Chem. Eng. J., 2019, 361, 245 CrossRef CAS.
- J. Li, Q. Su, K. Y. Li, C. F. Sun and W. B. Zhang, Food Chem., 2013, 141, 3714 CrossRef CAS PubMed.
- D. L. Deng, Y. N. He, M. Y. Li, L. D. Huang and J. Z. Zhang, Environ. Sci. Pollut. Res., 2021, 28, 5966 CrossRef CAS PubMed.
- Q. Ye, L. H. Liu, Z. B. Chen and L. M. Hong, J. Chromatogr., A, 2014, 1329, 24 CrossRef CAS PubMed.
- S. W. Jun, M. Shokouhimehr, D. J. Lee, Y. Jang, J. Park and T. Hyeon, Chem. Commun., 2013, 49, 7821–7823 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra09405j |
| ‡ Yuxin Liu and Pan Pan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |