
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExperimental and theoretical study of novel aminobenzamide–aminonaphthalimide fluorescent dyads with a FRET mechanism†
Ángel L. Garcíaa,
Adrián Ochoa-Terán *a,
Antonio Tirado-Guízara,
Jesús Jara-Cortés*b,
Georgina Pina-Luisa,
Hisila Santacruz Ortegac,
Victoria Labastida-Galvánd,
Mario Ordoñezd and
Jorge Peóne
*a,
Antonio Tirado-Guízara,
Jesús Jara-Cortés*b,
Georgina Pina-Luisa,
Hisila Santacruz Ortegac,
Victoria Labastida-Galvánd,
Mario Ordoñezd and
Jorge Peóne
aCentro de Graduados e Investigación en Química, Tecnológico Nacional de México/IT de Tijuana, Tijuana, Baja California, Mexico. E-mail: adrian.ochoa@tectijuana.edu.mx
bUnidad Académica de Ciencias Básicas e Ingenierías, Universidad Autónoma de Nayarit, Tepic, Nayarit, Mexico. E-mail: josejc@uan.edu.mx
cDepartamento de Investigación en Polímeros y Materiales, Universidad de Sonora, Hermosillo, Sonora, Mexico
dCentro de Investigaciones Químicas-(IICBA), Universidad Autónoma del Estado de Morelos, Cuernavaca, Morelos, Mexico
eInstituto de Química, Universidad Nacional Autónoma de México, Ciudad de México, Mexico
First published on 24th February 2022
Abstract
In this work, both experimental and theoretical methods were used to study the photophysical and metal ion binding properties of a series of new aminobenzamide–aminonaphthalimide (2ABZ–ANAPIM) fluorescent dyads. The 2-aminobenzamide (2ABZ) and 6-aminonaphthalimide (ANAPIM) fluorophores were linked through alkyl chains (C2 to C6) to obtain four fluorescent dyads. These dyads present a highly efficient (0.61 to 0.98) Förster Resonant Energy Transfer (FRET) from the 2ABZ to the ANAPIM due to the 2ABZ emission and ANAPIM excitation band overlap and the configurational stacking of both aromatic systems which allows the energy transfer. These dyads interact with Cu2+ and Hg2+ metal ions in solution inhibiting the FRET mechanism by the cooperative coordination of both 2ABZ and ANAPIM moieties. Both experimental and theoretical results are consistent and describe clearly the photophysical and coordination properties of these new dyads.
Introduction
Nowadays one of the main problems worldwide is population growth. It is known that the world population amounted to 2529 million in 1950 and it will be 9700 million in 2050.1 The population growth is intrinsically related to the consumption of natural resources, especially water. The data from the World Water Assessment Programme (WWAP) indicate that 2 million tons of garbage are dumped into the sea every day, and 70% of industrial waste is dumped without a prior treatment. Some of these pollutants are heavy metals, in particular Hg2+, Pb2+, Cu2+ and Co2+. The major risk of heavy metals is that they are not chemically or biologically degradable and cause health problems such as intoxication, blindness, amnesia and sometimes death, in relatively high concentrations.2Especially, Hg2+ ion is one of the most toxic and prevalent heavy metals, it is widely distributed by air, water and soil with cumulative and persistent characteristics, it causes damage in DNA, alterations in cell mitosis and defects in the nervous system.3 On the other hand, has been reported that a short time exposure to high concentrations of Cu2+ may result in anemic brain disorders, while a long-term exposure causes hepatic cirrhosis or perception of a nerve barrier.4 Therefore, more efficient, economical and viable techniques for detection and removal of heavy metals are desirable. A promising alternative for selective detection and extraction of heavy metals are the fluorescent chelating compounds, known as chemosensors, because they have several advantages such as simplicity, low-cost instrumentation, and the ability to be used for real-time monitoring.5,6
A common challenge in developing new fluorescent chemosensors is to find chemically stable fluorophores under the analysis conditions. The 1,8-naphthalimide (NAPIM) and 6-amino-1,8-naphthalimide (ANAPIM) fluorophores have been widely studied and applied in the design of fluorescent chemosensors due to its excellent photophysical properties such as high quantum yield, excitation and emission wavelengths at the visible spectrum, and high photo- and chemo-stability, among others.7–9
In literature there are reports regarding the design and study of naphthalimide based chemosensors for the detection of metal ions and anions as well as pH measurements. Anbu and coworkers developed a new naphthalimide–phenanthroimidazole fluorescent chemosensor for a turn-ON Cu2+ detection in living cancer cells,10 and a naphthalimide-aza 18-crown-6 ether ion-selective chemosensor for UO22+ ion was reported by Tsukahara.11 A ratiometric 4-amido-1,8-naphthalimide fluorescent sensor based on excimer-monomer emission for determination of pH and water content in organic solvents was reported by Bojinov and coworkers.12 Zhang and coworkers developed a novel naphthalimide–glutathione chemosensor for fluorescent detection of Fe3+ and Hg2+ in aqueous medium13 and the green synthesis of fluorescent N,O-chelating hydrazone Schiff base for multi-analyte sensing in Cu2+, F− and CN− ions was reported by Ervithayasuporn.14 Also, a highly Al3+-selective fluorescence probe based on naphthalimide-Schiff base and its application to practical water samples was reported by Whu and coworkers.15 Zhang and coworkers16 developed a selective chemosensor for Cu2+ ion, where ANAPIM is connected to a dihydroxibenzylimine receptor. This chemosensor displays an ON/OFF fluorescent response in a buffered aqueous media at pH 7.2. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 metal–ligand ratio was established for the supramolecular complex. Finally, they concluded that the quenching of fluorescence is due to the intrinsic strong paramagnetic effect of Cu2+ ion. Pachenko and coworkers17 designed a selective chemosensor for Ca2+ ion detection connecting the ANAPIM fluorophore to an aza-15-crown-5 ether in two different fashions to modulate the fluorescent response.
:2 metal–ligand ratio was established for the supramolecular complex. Finally, they concluded that the quenching of fluorescence is due to the intrinsic strong paramagnetic effect of Cu2+ ion. Pachenko and coworkers17 designed a selective chemosensor for Ca2+ ion detection connecting the ANAPIM fluorophore to an aza-15-crown-5 ether in two different fashions to modulate the fluorescent response.
A fluorescent dyad with an ICT-PET-FRET mechanism acting as a pH sensor was studied by Baoli and coworkers (Fig. 1a),18 using coumarin and ANAPIM chromophores as energy donor and acceptor to improve the FRET mechanism, connecting a morpholine group to ANAPIM inducing a PET process. Theoretical calculations showed that the HOMO of morpholine is in −5.4 eV and the HOMO of ANAPIM is −5.71 eV, which favors the PET from morpholine to ANAPIM. Coumarin and morpholine groups are sensible to pH and into acid environment the morpholine is protonated inhibiting the PET process allowing the FRET from coumarin to ANAPIM with an emission at 525 nm. On the contrary, in a basic medium the coumarin is deprotonated inducing an ICT and emitting at 467 nm. This work demonstrated the ability of ANAPIM chromophore as energy acceptor in a FRET process.
 | ||
| Fig. 1 Chemical structure of (a) coumarin–ANAPIM, (b) 2ABZ–NAPIM, (c) NAPIM–DANS and (d) 2ABZ–ANAPIM fluorescent dyads. | ||
Previously, we studied the photophysical properties and sensing behavior towards metal ions of a series of aminobenzamide–naphthalimide fluorescent dyads (2ABZ–NAPIM) with ICT-PET fluorescent mechanisms (Fig. 1b).19 In these dyads the exciting and emission wavelengths of both fluorophores are similar emitting around 400 nm. Interestingly, the fluorescence intensity of 2ABZ–NAPIM dyads was dependent on the spacer length and their quantum yield were lower than the individual fluorophores, which was attributed to a PET process. This PET was analyzed by theoretical methods finding that the HOMO of NAPIM and 2ABZ are at −6.98 eV and −5.47 eV respectively, indicating an electronic transfer from 2ABZ to NAPIM. Electrochemical analysis also confirmed the direction of this process. These dyads were sensitive towards two metal ions such as Hg2+ and Cu2+ inhibiting the PET in the shorter dyads and showing an OFF/ON fluorescent response by the increasing emission of NAPIM fluorophore and inhibition of an ICT in the 2ABZ. The NMR analysis demonstrated the interaction of the metal ions in the 2ABZ is through the amine and amide functional groups, as it was previously reported by other authors.20–22 Some fluorescent dyads containing a naphthalimide (NAPIM) and dansylamide (DANS) fluorophores linked by a polymethylene spacer were reported by Pischel (Fig. 1c).23 This work describes a singlet–singlet energy transfer (SSET) and PET processes between NAPIM and DANS. Both processes were affected by the presence of protons quenching DANS and enhancing NAPIM fluorescence. Ramaiah and collaborators24 found that the presence of Cu2+ and Zn2+ has the same quenching effect in the dyads with shorter spacing length due to the SSET and PET processes, instead a FRET process occurs in dyads with longer oligomethylene chain (six and eight methylenes).25
In this work new 2ABZ–ANAPIM dyads with an ICT-FRET mechanism were synthesized and evaluated against metal ions varying the distance between both fluorophores to optimize the resonant energy transfer as well as, to study the detection of metal ions (Fig. 1d). Additionally, the experimental results are complemented by excited state calculations in order to rationalize the photophysical mechanism of the dyads, as well as the changes in their optical response after complexation with metal ions.
Experimental section
Materials
All chemical reagents were purchased from a commercial supplier and were employed as received without further purification.Measurements
1H NMR spectra were recorded in a 400 MHz Brucker Advance III NMR spectrophotometer; the samples were dissolved in DMSO-d6 containing small amounts of TMS as the internal standard. FTIR spectra were recorded using a PerkinElmer spectrophotometer Spectrum 400 in the range from 4000 to 600 cm−1. MS (FAB+) HRMS analysis were recorded in an MStation JMS-700 JEOL. Optical properties of NDIs were analyzed by UV-Vis and fluorescence spectroscopy, recorded in a UV-Vis Spectrophotometer of double beam Shimadzu model UV2700 and a Varian Fluorescence Spectrophotometer model Eclipse Cary.Synthesis
1H NMR (DMSO-d6, 400 MHz): δ 8.68 (dd, J = 7.3, 1.2 Hz, 1H), 8.59 (dd, J = 8.5, 1.2 Hz, 1H), 8.44 (d, J = 7.9 Hz, 1H), 8.06 (d, J = 7.9 Hz, 1H), 7.87 (dd, J = 8.5, 7.3 Hz, 1H), 4.21–4.12 (m, 2H), 1.87–1.72 (m, 2H), 1.04 (t, J = 7.5 Hz, 3H). 13C NMR (DMSO-d6, 101 MHz): δ 162.8, 132.5, 131.4, 131.2, 130.9, 129.7, 129.0, 128.7, 128.2, 122.7, 121.9, 41.3, 20.7, 11.3. MS (FAB+) m/z: 318 [M + H+]. HRMS (FAB+): calculated for C15H13BrNO2: 318.0130; found: 318.0147.
6-((2-Aminoethyl)amino)-2-propyl-1H-benzo[de]isoquinoline-1,3(2H)-dione (2a). Yellow solid, 194.3 mg, 71% yield. 1H NMR (DMSO-d6, 400 MHz): δ 8.62 (d, J = 8.5 Hz, 1H), 8.44 (d, J = 7.2 Hz, 1H), 8.27 (d, J = 8.5 Hz, 1H), 8.23 (s, 1H), 7.79 (t, J = 4.6 Hz, 1H), 7.70 (t, J = 7.7 Hz, 1H), 6.85 (d, J = 8.6 Hz, 1H), 3.99 (t, J = 7.6 Hz, 2H), 3.46 (ddd, J = 13.6, 7.8, 6.7 Hz, 4H), 1.63 (hxt, J = 7.4 Hz, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (DMSO-d6, 101 MHz): δ 163.4, 162.4, 150.9, 134.6, 131.1, 129.8, 128.8, 124.8, 122.4, 120.6, 108.5, 104.2, 43.0, 41.2, 36.5, 21.4, 11.9. MS (FAB+) m/z: 298 [M + H+]. HRMS (FAB+): calculated for C17H20N3O2: 298.1556; found: 298.1562.
6-((3-Aminopropyl)amino)-2-propyl-1H-benzo[de]isoquinoline-1,3(2H)-dione (2b). Yellow solid, 246.2 mg, 86% yield. 1H NMR (DMSO-d6, 400 MHz): δ 8.69 (dd, J = 8.52, 0.84 Hz, 1H), 8.44 (dd, J = 7.28, 0.84 Hz, 1H), 8.28 (d, J = 8.52 Hz, 1H), 7.73 (t, J = 5.28 Hz, 1H), 7.69 (td, J = 8.32, 7.4 Hz, 1H), 6.80 (d, J = 8.6 Hz, 1H), 3.98 (t, J = 7.36 Hz, 2H), 3.41 (q, J = 6.64 Hz, 2H), 3.25 (q, J = 6.76 Hz, 2H), 1.87 (qnt, J = 6.96 Hz, 2H), 1.63 (m, 2H), 0.91 (t, J = 7.44 Hz, 3H). 13C NMR (DMSO-d6, 101 MHz): δ 163.4, 161.7, 151.0, 134.6, 131.1, 129.9, 128.9, 124.8, 122.4, 120.6, 108.2, 104.3, 41.2, 41.0, 35.6, 28.3, 21.4, 11.8. MS (FAB+) m/z: 312 [M + H+]. HRMS (FAB+): calculado para C18H22N3O2: 312.1712; found: 312.1724.
6-((4-Aminobutyl)amino)-2-propyl-1H-benzo[de]isoquinoline-1,3(2H)-dione (2c). Yellow solid, 122.2 mg, 41% yield. 1H NMR (DMSO-d6, 400 MHz): δ 8.70 (dd, J = 8.6, 1.2 Hz, 1H), 8.44 (dd, J = 7.4, 1.0 Hz, 1H), 8.27 (d, J = 8.5 Hz, 1H), 7.75 (t, J = 5.4 Hz, 1H), 7.68 (dd, J = 8.5, 7.3 Hz, 1H), 6.79 (d, J = 8.6 Hz, 1H), 4.98 (m, 2H), 3.40 (q, J = 6.6 Hz, 2H), 3.16 (m, 2H), 1.78–1.51 (m, 6H), 0.91 (t, J = 7.5 Hz, 3H). 13C NMR (DMSO-d6, 101 MHz): 163.4, 161.5, 151.1, 134.7, 131.1, 129.9, 129.0, 124.7, 122.4, 120.6, 108.0, 104.3, 42.9, 41.2, 37.3, 27.2, 25.7, 21.4, 11.8. MS (FAB+) m/z: 326 [M + H+]. HRMS (FAB+): calculated for C19H24N3O2: 326.1869; found: 326.1874.
6-((6-Aminohexyl)amino)-2-propyl-1H-benzo[de]isoquinoline-1,3(2H)-dione (2d). Yellow solid, 144.7 mg, 74% yield. 1H NMR (DMSO-d6, 400 MHz): δ 8.71 (d, J = 8.4 Hz, 1H), 8.44 (d, J = 7.3 Hz, 1H), 8.27 (d, J = 8.5 Hz, 1H), 7.73 (d, J = 6.0 Hz, 1H), 7.68 (t, J = 7.9 Hz, 1H), 6.78 (d, J = 8.6 Hz, 1H), 3.98 (dd, J = 8.5, 6.3 Hz, 2H), 3.45 (q, J = 7.0 Hz, 2H), 3.38 (q, J = 6.6 Hz, 2H), 1.70 (q, J = 7.1 Hz, 2H), 1.62 (qnt, J = 7.4 Hz, 2H), 1.40 (m, 6H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (DMSO-d6, 101 MHz): 163.4, 161.5, 151.1, 134.7, 131.09, 129.9, 129.0, 124.7, 122.3, 120.6, 108.0, 104.3, 42.9, 41.2, 37.3, 27.2, 25.7, 25.4, 21.4, 11.8. MS (FAB+) m/z: 354 [M + H+]. HRMS (FAB+): calculated for C21H28N3O2: 354.2182; found: 354.2202.
2-Amino-N-(2-((1,3-dioxo-2-propyl-2,3-dihydro-1H-benzo[de]isoquinolin-6-yl)amino)ethyl)benzamide (3a). Yellow solid, 111.5 mg, 77% yield. FTIR: 3459, 3349, 3062, 2956, 1677, 1615, 1631, 1578, 1537 cm−1. 1H NMR (DMSO-d6, 400 MHz): δ 8.65 (d, J = 8.6 Hz, 1H), 8.49 (t, J = 5.6 Hz, 1H), 8.45 (d, J = 7.2 Hz, 1H), 8.28 (d, J = 8.5 Hz, 1H), 7.94 (t, J = 8.5 Hz, 1H), 7.71 (t, J = 7.7 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.14 (t, J = 7.9 Hz, 1H), 6.93 (d, J = 8.6 Hz, 1H), 6.70 (d, J = 8.2 Hz, 1H), 6.51 (t, J = 7.5 Hz, 1H), 6.45 (s, 2H), 3.98 (t, J = 7.3 Hz, 2H), 3.55 (s, 4H), 1.63 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (DMSO-d6, 101 MHz): δ 170.0, 164.2, 163.4, 151.0, 150.2, 134.6, 132.3, 131.1, 129.83, 128.8, 128.6, 124.8, 122.3, 120.6, 116.9, 115.1, 114.9, 108.3, 104.2, 43.1, 41.2, 38.1, 21.4, 11.8. MS (FAB+) m/z: 417 [M + H+]. HRMS (FAB+): calculated for C24H25N4O3: 417.1927; found: 417.1947.
2-amino-N-(3-((1,3-dioxo-2-propyl-2,3-dihydro-1H benzo[de]isoquinolin-6-yl)amino)propyl)benzamide (3b). Yellow solid, 120.8 mg, 80% yield. FTIR: 3444, 3339, 3059, 2965, 1670, 1610, 1630, 1579, 1520 cm−1. 1H NMR (DMSO-d6, 400 MHz): δ 8.70 (dd, J = 8.6, 1.1 Hz, 1H), 8.44 (dd, J = 7.3, 1.04 Hz, 1H), 8.32 (t, J = 5.5 Hz, 1H) 8.26 (d, J = 8.6 Hz, 1H), 7.82 (t, J = 5.5 Hz, 1H), 7.69 (dd, J = 8.4, 8.5 Hz, 1H), 7.48 (d, J = 8.0, 1.6 Hz, 1H), 7.13 (ddd, J = 8.4, 7.1, 1.6 Hz, 1H), 6.81 (d, J = 8.7 Hz, 1H), 6.68 (dd, J = 8.2, 1.2 Hz, 1H), 6.51 (dd, J = 8.0, 1.2 Hz, 1H), 6.38 (s, 2H), 3.97 (t, J = 7.4 Hz, 2H), 3.36 (q, J = 8.8, 15 Hz, 2H), 1.95 (qnt, J = 6.7, 13.6 Hz, 2H), 1.62 (m, 2H), 0.90 (t, J = 7.5 Hz, 3H). 13C NMR (DMSO-d6, 101 MHz): δ 170.1, 164.3, 163.4, 151.1, 150.2, 134.6, 132.4, 131.2, 129.9, 128.9, 128.6, 124.9, 122.4, 120.6, 116.8, 115.0, 114.8, 108.2, 104.1, 43.0, 41.2, 38.1, 28.3, 21.4, 11.8. MS (FAB+) m/z: 431 [M + H+]. HRMS (FAB+): calculated for C25H27N4O3: 431.2083. Found: 431.2103.
2-Amino-N-(4-((1,3-dioxo-2-propyl-2,3-dihydro-1H benzo[de]isoquinolin-6-yl)amino)butyl)benzamida (3c). Yellow solid, 140.9 mg, 80% yield. FTIR: 3444, 3339, 3059, 2965, 1670, 1610, 1630, 1579, 1520 cm−1. 1H NMR (DMSO-d6, 400 MHz): δ 8.66 (d, J = 8.5 Hz, 1H), 8.38 (d, J = 7.2 Hz, 1H), 8.21 (s, 1H) 8.18 (d, J = 8.3 Hz, 1H), 7.87 (d, J = 7.9 Hz, 1H), 7.75 (t, J = 6.5, 1H), 7.40 (d, J = 7.7 Hz, 1H), 7.20 (t, J = 7.6 Hz, 1H), 6.76 (d, J = 8.6 Hz, 1H), 6.63 (d, J = 8.3 Hz, 1H), 6.44 (t, J = 7.4 Hz, 1H), 6.33 (s, 2H), 3.93 (t, J = 7.4 Hz, 2H), 3.4 (m, 2H), 3.28 (m, 2H), 1.64 (m, 6H), 0.90 (t, J = 7.5 Hz, 3H). MS (FAB+) m/z: 445 [M + H+]. HRMS (FAB+): calculated for C26H29N4O3: 445.2240. Found: 445.2297.
2-Amino-N-(6-((1,3-dioxo-2-propyl-2,3-dihydro-1H-benzo[de]isoquinolin-6-yl)amino)hexyl)benzamide (3d). Yellow solid, 68.5 mg, 68% yield. FTIR: 3471, 3334, 3075, 2931, 1679, 1620, 1631, 1573, 1541 cm−1. 1H NMR (DMSO-d6, 400 MHz): δ 8.71 (d, J = 8.5 Hz, 1H), 8.43 (d, J = 7.3 Hz, 1H), 8.26 (d, J = 8.6 Hz, 1H) 8.20 (t, J = 5.8 Hz, 1H), 7.79 (t, J = 5.6 Hz, 1H), 7.68 (t, J = 7.9, 1H), 7.44 (d, J = 7.9 Hz, 1H), 7.11 (t, J = 7.7 Hz, 1H), 6.78 (d, J = 8.6 Hz, 1H), 6.67 (d, J = 8.2 Hz, 1H), 6.49 (t, J = 7.4 Hz, 1H), 6.37 (s, 2H), 3.98 (t, J = 7.6 Hz, 2H), 3.36 (m, 2H), 3.21 (q, J = 6.7, 13.16 Hz, 2H), 1.71 (m, 2H), 1.63 (m, 2H), 1.53 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (DMSO-d6, 101 MHz): δ 169.3, 164.3, 163.4, 151.1, 149.9, 134.8, 131.9, 131.1, 129.9, 129.0, 128.4, 124.6, 122.2, 120.5, 116.7, 115.6, 115.1, 107.9, 104.2, 43.3, 41.2, 29.6, 28.3, 26.9, 26.8, 21.4, 11.8. MS (FAB+) m/z: 473 [M + H+]. HRMS (FAB+): calculated for C28H33N4O3: 473.2553. Found: 473.2528.
Determination of quantum yields
Fluorescence quantum yields were measured using N-propyl 6-aminonaphthalimide in acetonitrile as standard with a known ΦR of 0.58. Solutions of dyads 3a–d and pp-ANAPIM were prepared at 5 × 10−6 M in spectroscopic grade acetonitrile. Absorbance and fluorescence spectra were measured, and the data required for eqn (1) were obtained from these spectra.
 | (1) |
Metal ion interaction study by UV-vis titrations
The effect of metal ions upon the absorbance was examined by adding 6 μL of a 1 × 10−3 M perchlorate salt solution to a known volume (3 mL) of a 1 × 10−5 M dyad solution. The dyad and perchlorate salt stock solutions were prepared in spectroscopic grade acetonitrile. A quartz cuvette was filled with 3 mL of the dyad stock solution, then 6 μL aliquots of perchlorate salt stock solution were added successively with a calibrated micropipette. Thus, the ratio of the total concentration [salt]/[dyad] was in the desired range. The resulting complex solutions were stirred for 3 min, and the spectra were recorded. The absorption spectrum was confirmed to be unchanged with time at each measurement.Metal ion interaction study by fluorometric titrations
The effect of metal ions upon the fluorescence emission was examined by adding 6 μL of a 5 × 10−3 M perchlorate salt solution to a known volume (3 mL) of a 5 × 10−5 M dyad solution. The dyad and perchlorate salt stock solutions were prepared in spectroscopic grade acetonitrile. A quartz cuvette was filled with 3 mL of dyad stock solution, then 6 μL aliquots of perchlorate salt stock solution were added successively with a calibrated micropipette. Thus, the ratio of the total concentration [salt]/[dyad] was in the chosen range. The resulting complex solutions were stirred for 3 min, and the spectra were recorded. The emission spectrum was confirmed to be unchanged with time at each measurement.Computational methods
The calculations were carried out in two stages. First, a conformational search on the potential energy surfaces (PES) was done for dyads 3a–d using the basin hopping method, as it is implemented in the Tinker program.26 The Merck Molecular force field (MMFF) was used in this step,27 which shows a good performance in the conformational search for organic molecules.28,29 For the obtained structures, additional geometry optimizations were performed using the B3LYP density functional, including dispersion correction (D3BJ) and using the def2-SVP basis set.30–33 The implicit solvent effect was included through the polarizable continuum model using parameters corresponding to acetonitrile.34 To quantify the relative contribution of each one of the conformers, the average of some properties was obtained using the Boltzmann distribution, considering the Gibbs free energies (from thermochemical calculations at 298 K) to calculate the weight factors.35 The Time-Dependent Density Functional Theory (TDDFT) was employed to evaluate the electronic excited states.In the second stage additional calculations were achieved in order to rationalize the excitation energy transfer (EET). Although formally the EET for dyads 3a–d is an intramolecular process, the fragments 2ABZ and ANAPIM were analyzed individually as an acceptor and donor chromophores. For each relevant equilibrium geometry of the 3a–d compounds, a pair of 2ABZ and ANAPIM moieties were obtained by eliminating the alkyl chain spacer and capping the free valence of each unit with one hydrogen atom. The position of the included H atom was determined by a constrained geometry optimization. Later, the electronic coupling term (VDA) that appears in the expression of the EET rate was evaluated for the pairs 2ABZ–ANAPIM.36–38 All the electronic structure calculations were performed using the Gaussian 16 program,39 except the minimum energy conical intersections (MECI), which were obtained with the Q-Chem program.40
Results and discussion
Synthesis and characterization
The synthesis of 2ABZ–ANAPIM dyads (3a–d) is depicted in Scheme 1. | ||
| Scheme 1 Synthetic route for the synthesis of aminonapththalimide–aminobenzamide dyads. | ||
First, the N-propyl bromonaphthalimide 1 was prepared by a condensation reaction of 4-bromo-1,8-naphthalic anhydride with one molar equivalent of propylamine in refluxed ethanol with a 80% yield. Then, compound 1 was reacted with an excess of the corresponding alkyldiamine (a–d) in DMF obtaining the 2a–d intermediates in good yields (41 to 86%). Finally, the amines 2a–d were reacted with one molar equivalent of isatoic anhydride in dry THF obtaining 3a–d in 68 to 80% yield.
The intermediates 1, 2a–d and dyads 3a–d were characterized by 1H and 13C NMR, FTIR and MS (Fig. S1 to S30†). The FTIR spectrum of dyad 3a shows the characteristic stretching vibrations for a primary and secondary amine, as well as amide groups from 3459 to 3300 cm−1. The stretching vibrations of carbonyls are observed at 1677, 1631 and 1615 cm−1. The bending vibration of N–H groups is at 1527 cm−1. The spectra of 3b–3d dyads show similar vibrations (see Table S1†).
The 1H NMR spectrum of 1 obtained in DMSO-d6 (Fig. 2a) shows the napththalimide AB system corresponding to H4 and H5 at 8.27 and 8.06 ppm. The signals for H8 and H6 are present at 8.52 and 8.48 ppm, and H7 is observed at 7.95 ppm. The aliphatic hydrogens H3 are located at 3.98 ppm, H2 at 1.64 ppm and H1 at 0.92 ppm. The spectrum of 2a (Fig. 2b) shows the napththalimide AB system corresponding to H4 and H5 shifted to 8.24 and 6.85 ppm due to the protecting effect of the amino group over H5. The signals for H8 and H6 are observed at 8.62 and 8.44 ppm, respectively, and the signal for H7 is observed at 7.70 ppm. In addition to the dramatic chemical shift of H5 and H7, it is also significant the presence of new signals at 7.75 ppm and 3.47 ppm for the secondary amine and methylene hydrogens corresponding to the ethylenediamine chain, indicating a successful substitution of the bromine (Fig. S4†). The 1H NMR spectrum of 3a obtained in DMSO-d6 (Fig. 2c) shows the signals for both ANAPIM and 2ABZ aromatic systems, amine, amide and aliphatic hydrogens. The AB system appears at 8.28 ppm and 6.93 ppm for H4 and H5, respectively. The signals for H8 and H7 are observed at 8.65 ppm and 7.71 ppm, while H6 appears at 8.45 ppm. The secondary amine hydrogen is observed at 7.94 ppm, while the amide hydrogen appears at 8.49 ppm. The 2ABZ aromatic hydrogens signals were observed at 7.48 ppm for H12, 6.21 ppm for H11, 7.14 ppm for H10 and 6.70 for H9. Also, it is present the signal for the amino-aromatic hydrogens at 6.46 ppm.
 | ||
| Fig. 2 1H NMR of 1 (A), 2a (B) and 3a (C). | ||
The 13C NMR spectrum obtained in DMSO-d6 of 2a shows two signals at 163.4 and 162.4 ppm corresponding to the imide carbonyls (Fig. S5†). The ten signals of the ANAPIM system are observed from 151.0 to 104.0 ppm, the signals of the propyl group are at 43.0, 21.4 and 11.9 ppm, and the ethylene signals are at 41.2 and 36.6 ppm. The spectrum of 3a (Fig. S18†) shows three signals for carbonyls at 170.0 (amide), 164.2 and 163.4 ppm (imide). There are sixteen signals for both ANAPIM and 2ABZ aromatic systems from 151.0 to 104.0 ppm. The propyl signals are present at 43.1, 21.4 and 11.9 ppm and the ethylene carbons are observed at 41.2 and 38.0 ppm. The assignment of the NMR signals for dyads 3a–d are summarized in Tables S1 and S2.†
Optical properties of ANAPIM-2ABZ dyads
In order to study the photophysical properties of dyads 3a–d, the N-propyl 2-aminobenzamide (p-2ABZ) and 2-propyl-6-propylaminonaphthalimide (pp-ANAPIM) compounds were analyzed by UV-Vis and fluorescence spectroscopy. The UV-vis spectra of these compounds and dyads 3a–d were analyzed in acetonitrile. The p-2ABZ electronic absorption band shows a maximum absorbance at 325 nm associated to a π–π* transition (ε = 4370 L mol−1 cm−1), whilst the pp-ANAPIM band has three relative maximum peaks at 270, 296 and 420 nm, the latter also associated to a π–π* transition (ε = 11327 L mol−1 cm−1) (Fig. 3A). Both chromophores behave an ICT process where the non-bonding electrons localized in nitrogen atoms (donors) are delocalized through the aromatic system to reach a carbonyl group (acceptors). The absorbance band of 3a shows the characteristic peaks of the π–π* transition at 325 nm and 430 nm (ε = 20530 L mol−1 cm−1) corresponding to 2ABZ and ANAPIM chromophores, respectively. Interestingly, the absorbance of ANAPIM chromophore increases in 3a and a bathochromic shift (10 nm) is observed. The rest of dyads 3 have similar electronic absorption bands (Fig. S31†) and their ε values decrease as the spacer length increases (Table 1).
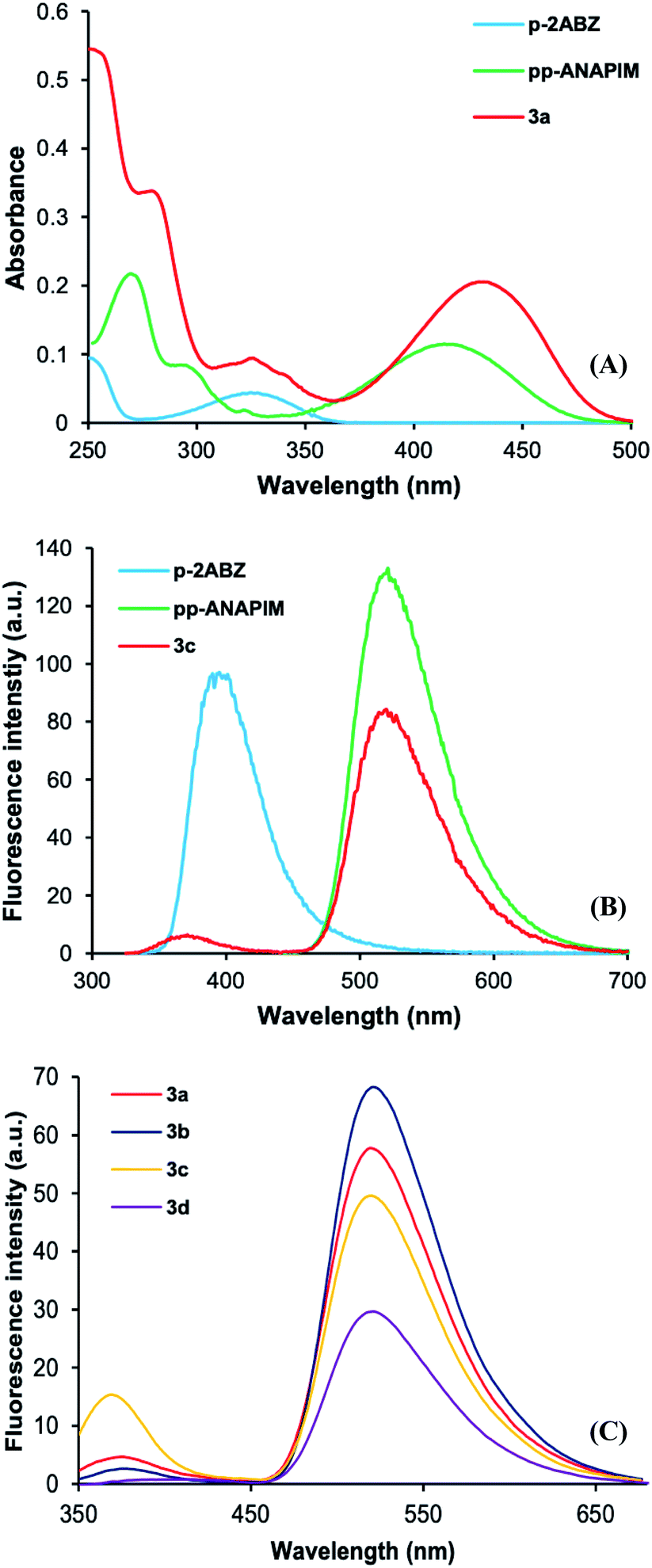 | ||
| Fig. 3 (A) Electronic absorption spectra of 3a–d obtained in acetonitrile at 1 × 10−5 M. (B) Emission spectra of p-2ABZ, pp-ANAPIM and 3a obtained in acetonitrile at 5 × 10−5 M. (C) Emission spectra of dyads 3a–3d (excited at 325 nm) obtained in acetonitrile at 5 × 10−5 M. | ||
| Compound | ε (L mol−1 cm−1) | ET | r (nm) | ΦF |
|---|---|---|---|---|
| 3a | 20530 |
0.87 | 2.3 | 0.61 |
| 3b | 18387 |
0.93 | 2.1 | 0.65 |
| 3c | 14943 |
0.61 | 2.9 | 0.46 |
| 3d | 14361 |
0.98 | 1.7 | 0.47 |
| p-2ABZ | 4370 | 0.19 | ||
| pp-ANAPIM | 11327 |
0.72 |
On the other hand, the fluorescence spectra of p-2ABZ have maximum excitation and emission at wavelengths of 325 nm and 391 nm, respectively. The pp-ANAPIM has a maximum excitation wavelength at 430 nm and the emission is at 517 nm. It is clearly observed that there is an overlap between the 2ABZ emission band and the ANAPIM excitation band. Interestingly, dyad 3a shows a low intensity emission peak at 370 nm and high intensity emission peak at 519 nm when the molecule is excited at 325 nm (p-2ABZ excitation wavelength) indicating an energy transfer in the excited state from the 2ABZ to ANAPIM fluorophore. The intensity of the emission band at 325 nm decreases as the alkyl spacer length increases in dyads 3a–d (Fig. 3C), indicating a more efficient energy transfer. In addition, the quantum yield (ΦF) of pp-ANAPIM is higher than those for p-2ABZ and dyads 3a–d (Table 1), the lower ΦF of dyads 3a–d may be related to the efficiency in the energy transfer process.
The energy transfer efficiency (ET) is related with the emission intensities of the donor–acceptor system (FDA) and the donor emission intensity (FD) in the eqn (2). Table 1 shows high ET values in dyads 3a–d (0.87 to 0.98) and there is a trend to increase as the alkyl spacer increases.
 | (2) |
The overlap integral was calculated from the emission spectrum of p-ABZ (energy donor) and electronic absorption spectrum of pp-ANAPIM (energy acceptor) applying the Simpson 1/3 method to obtain a function for each spectrum (Fig. 4). Thus, a value of J = 1.31 × 10−13 J cm3 L mol−1 was obtained from eqn (3).
 | (3) |
 | ||
| Fig. 4 Normalized emission spectrum of p-2ABZ (excited at 325 nm) and absorption spectrum of pp-ANAPIM obtained in acetonitrile at 5 × 10−5 M. | ||
The Förster radius (R0) is the distance between donor and acceptor, at which 50% of the energy transfer occurs, and it was calculated as 3.2 nm from eqn (4). The distance between both fluorophores in dyads 3a–d was calculated using eqn (5). As can be seen in Table 1, all the r values are in the 0.5R0 = 1.6 < r < 1.5R0 = 4.8 interval suggesting a favorable energy transfer process, and that the closer the fluorophores are to each other, the higher the energy transfer efficiency.
| R06 = 2.8 × 1017·k2·ΦD·εA·J | (4) |
 | (5) |
Interaction of ANAPIM–2ABZ dyads with metal ions
The supramolecular interaction of dyads 3a–d was studied experimentally by UV-vis spectrophotometric titrations in acetonitrile. In the experiments the concentration of dyads 3a–d solutions was 1 × 10−5 M and titrations were achieved keeping their concentration constant and varying the metal ion concentration by addition of aliquots of 1 × 10−3 M metal ion solutions.The UV-vis titrations were performed using monovalent and divalent cations such as Li+, Na+, K+, Mg2+, Ca2+, Hg2+, Cu2+, Cd2+, Co2+ and Pb2+. Interestingly, only Hg2+ and Cu2+ induced significant changes in the absorption band of these dyads. The titration of 3d with Hg2+ (Fig. 5A) induced a hypochromic change in the band at 325 nm with an isosbestic point at 290 nm, indicating the interaction of the metal ion with the 2ABZ, while Cu2+ has a hyperchromic effect over the band from 250 to 390 nm probably due to the absorbance contribution of the Cu2+-acetonitrile complex (Fig. S32†). However, there is a hypochromic change in the band at 430 nm with an isosbestic point at 395 nm. These changes are attributed to the inhibition of ICT processes by the coordination of Cu2+ with the amino groups of 2ABZ and ANAPIM chromophores. Besides the interaction with the cation induces a colorimetric change from green to colorless. The rest of dyads (3a, 3b, 3c) have the same behavior with Hg2+ and Cu2+ (Fig. S33 to S35†).
 | ||
| Fig. 5 Absorption spectra obtained by titration of dyad 3d with Hg2+ (A) and Cu2+ (B) in acetonitrile. [3d] = 1 × 10−5 M. | ||
Based on the previous results, the titrations by fluorescence were conducted only with Hg2+ and Cu2+ ions in acetonitrile. Fig. 6 shows the spectra obtained from the titration with Cu2+ (A and B) and Hg2+ (C and D) exciting at 315 nm and 435 nm, respectively. As can be seen, there is a quenching of fluorescence by the coordination with these metal ions through the 2ABZ fluorophore inhibiting the ICT and hence the energy transfer (FRET) to the ANAPIM fluorophore. Also, these results suggest a coordination of the ions with the ANAPIM fragment inducing a quenching of fluorescence when the molecule is excited at 435 nm. The rest of dyads (3a, 3b, 3c) have a similar behavior (Fig. S36 to S38†).
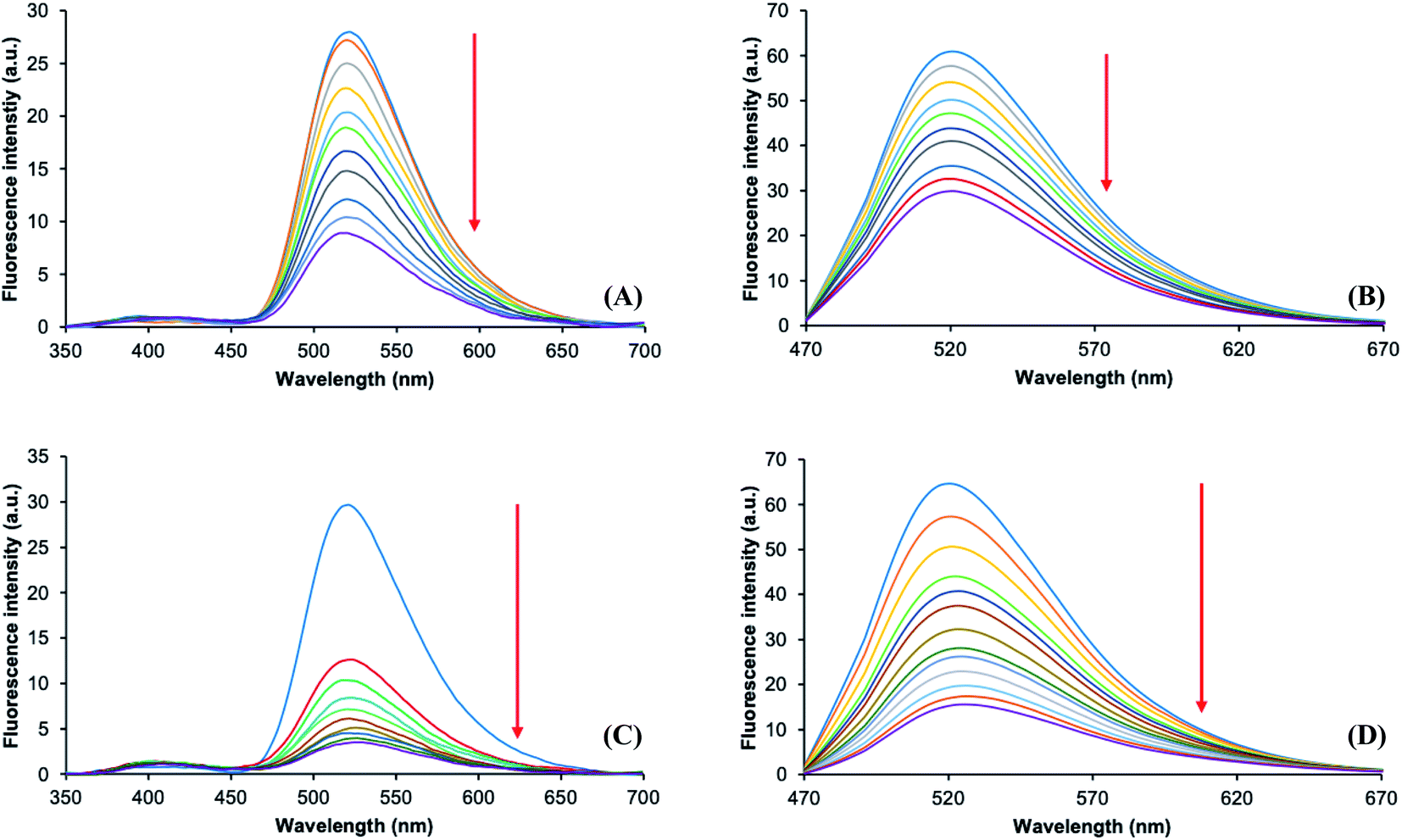 | ||
| Fig. 6 Emission spectra obtained by titration of dyad 3d with Cu2+ exciting at 315 nm (A) and at 435 nm (B) in acetonitrile. Emission spectra obtained by titration of dyad 3d with Hg2+ exciting at 315 nm (C) and at 435 nm (D) in acetonitrile. [3d] = 5 × 10−5 M. | ||
In order to corroborate the interaction of the ANAPIM moiety with the metal ions, the fluorescent behavior of pp-ANAPIM with Hg2+ and Cu2+ was analyzed (Fig. 6). The titration with Hg2+ shows a significant fluorescence quenching, while only a slight effect was observed with Cu2+ (Fig. S39†). This means that Cu2+ complexes with dyads 3 require the interaction of both 2ABZ and ANAPIM fragments to establish a cooperative coordination. Noteworthy, the quenching of fluorescence was more intense in dyad 3d, which has the longest alkyl chain between both fluorophores, while dyads 3a–c have a similar quenching behavior (Fig. S40 and Table S4†).
The Stern–Volmer constants (KSV) were determined for those complexes with Cu2+ and Hg2+ (Table 2) using eqn (6), where F0 is the free dyad fluorescence intensity, F is the fluorescence intensity in presence of the metal ion and [C] is the metal ion concentration.
 | (6) |
| Mn+ | 3a | 3b | 3c | 3d |
|---|---|---|---|---|
| Hg2+, λex: 315 nm | 3.79 × 104 M−1 (R2 = 0.99) | 3.30 × 104 M−1 (R2 = 0.99) | 3.13 × 104 M−1 (R2 = 0.99) | 5.51 × 104 M−1 (R2 = 0.95) |
| Cu2+, λex: 315 nm | 3.30 × 103 M−1 (R2 = 0.95) | 3.40 × 103 M−1 (R2 = 0.94) | 3.50 × 103 M−1 (R2 = 0.97) | 1.55 × 104 M−1 (R2 = 0.98) |
| KSV(Hg2+)/KSV(Cu2+) | 11.5 | 9.7 | 8.9 | 3.6 |
All the Stern–Volmer graphs were linear on the 0 to 150 μM interval of cation added, suggesting only one type of quenching (Fig. S41 to S44†). Considering that titrations by UV-Vis show the formation of a complex in the ground state, it is proposed a static quenching in these complexes. The KSV values corroborate the finding trend observed in the experimental study, showing more sensibility towards Hg2+ than Cu2+ in all days, however, the KSV(Hg2+)/KSV(Cu2+) ration indicates that the sensibility towards Cu2+ increases as the alkyl spacer are longer. Finally, the higher KSV were obtained for the complexes with dyad 3d.
Theoretical analysis of ANAPIM–2ABZ dyads photophysical properties
To rationalize the experimental findings corresponding to the photophysical behavior of dyads 3a–d, the starting point was to find the global minima of the PES for these compounds, as well as all the thermally accessible structures at 298 K, in accordance with the experimental conditions. Fig. 7 shows the structures obtained for the global minima of 3a–d, where the stacking of both fluorophores 2ABZ and ANAPIM is observed for all dyads. Additionally, at 298 K only a few conformations show a relevant contribution according to the calculated Boltzmann factors, and in almost the cases, for these structures we observed the stacking among the aromatic rings (Fig. S45†). | ||
| Fig. 7 Side and front views for the molecular structures corresponding to the global minima of the PES for 3a (A), 3b (B), 3c (C) and 3d (D). | ||
The ESI† includes the simulated electronic absorption and emission spectra for p-2ABZ and pp-ANAPIM (Fig. S46†), as well as the related dealkylated compounds (Fig. S47†). In general, there is a good match between the experimental and the simulated spectra, although the theoretical values are slightly shifted to lower wavelength. For example, for the S0 → S1 electronic transition of p-2ABZ and pp-ANAPIM the maximum of the bands are centered at 312 and 416 nm, against the experimental values of 326 and 420 nm, respectively. Also, the analysis of the TDDFT amplitudes reveals that for p-2ABZ and pp-ANAPIM the S1 state can be ascribed as 1ππ* nature, and also, the excited state of both molecules is well described by a single HOMO–LUMO configurational change.
The simulated electronic absorption spectra for dyads 3a–d are also similar to the experimental data (Fig. S48†). In these molecules the interaction between the chromophores causes a splitting of the S1 states of each individual fragment, giving rise to four singlet electronic states for the dyad. From these, two involve small oscillator forces and do not contribute significantly to the absorption spectra. A comparison of Fig. S46 and S48† suggests that the stacking interactions does not affect considerably the electronic transitions of chromophores 2ABZ and ANAPIM in dyads 3a–d because of the maximum absorbance wavelengths are nearly the same to those observed in p-2ABZ and pp-ANAPIM. In dyads 3a–d, the HOMO and HOMO−1 are constituted by the bonding and antibonding combination of the HOMOs of base chromophores, whilst the LUMO is centered at the ANAPIM chromophore and the LUMO+1 in 2ABZ (Fig. S49†). The lowest energy electronic transitions in 3a–d are described by significant amplitudes which involve the configurational changes between HOMO−1/HOMO and LUMO+1/LUMO, so is possible classify the electronic states as associated with the formation of an exciplex (π-complex).
The relative position of the electronic states, modulated by the conformation, has important consequences in the photophysical behavior of dyads 3a–d. For example, Fig. 8 shows the minimum energy path (MEP) for two different conformations of dyad 3a. In both cases, the photophysical pathway begins from an equilibrium geometry of S0 and the transition takes place towards the second bright electronic state. As can be seen, in the stacked conformation the MEP starts in S4, and the reaction profile proceeds from the Franck–Condon region towards the minimum of S1. Only a small barrier of 0.013 eV needs to be surpassed in order to access to an S3/S2 MECI. Since the initial absorption S0 → S4 occurs at 322 nm, while the S3 → S0 and S1 → S0 emissions from the respective minimums take place at 412 and 480 nm, conceptually this process may be interpreted as an energy transfer from the 2ABZ towards the ANAPIM system. Noteworthy, the PES crossings S4/S3, S3/S2 and S2/S1 take place along the MEP.
 | ||
| Fig. 8 Energy of the first singlet electronic states along the minimum energy path in the exciting state of stacked (A) and folded (B) conformations of 3a. The vertical dotted lines denote a vertical transition from/to S0. | ||
On the other hand, for the folded conformation the process starts at S0 → S3, and the MEP evolves to a minimum of S3. From this region is necessary to overcome a barrier of 0.25 eV to access to an S3/S2 MECI. Also, the emission S3 → S0 from the minimum of S3 is about 375 nm. Since this value is pretty similar to the maximum wavelength of the 2ABZ emission spectrum, it may be considered that it the folded conformation the energy is localized at the same chromophore. We also calculate the MEP for the global minima of dyad 3d and the same behavior as the stacked conformation of 3a was obtained (Fig. S50†). In other words, the stacked conformations favor the internal conversion process and in consequence, the EET. As the most stable conformations in dyads 3a–d involve stacked arrangements, the MEP data allow to rationalizing mechanistically the high energy transfer efficiencies observed experimentally.
With the purpose of explain the observed trend in the fluorescence quantum yields along the series of dyads 3a–d, the electronic couplings for the acceptor–donor pairs were calculated and are presented in Table 3. Briefly, the EET rate is given by kEET = πVDA2J/ℏ,38 where J corresponds to the overlap integral of the normalized emission and absorption spectra of the donor and acceptor, respectively. The coupling is given by the addition of all the coulombic contributions (Förster type), exchange–correlation (Dexter type), the overlap part and a term associated with the solvent, such that VDA = Vcoul + VXC + Vover + VPCM. Dyads 3a–d have the same J value, and hence, as higher is VDA higher is the energy transfer ratio. The R column in Table 3 denotes the distance between the geometric center of 2ABZ and the central carbon of ANAPIM system. In general, the values of VDA are high in all dyads and agree with the high energy transfer ratios as result of the closeness of chromophores in the stacked conformations. Also, the coulombic term makes the biggest contribution to the coupling. Although these values are consistent, they do not allow to explain the trend along the series, since according to experimental data a bigger magnitude of VDA for 3d would be expected, whilst is the smaller for 3c.
| Dyad | R | Vcoul | VXC | Vover | VPCM | VDA |
|---|---|---|---|---|---|---|
| 3a | 3.3847 | 0.0666 | −0.0006 | 0.0000 | −0.0154 | 0.0503 |
| 4.8670 | 0.0217 | 0.0001 | 0.0000 | −0.0065 | 0.0154 | |
| 5.1732 | 0.0264 | 0.0000 | 0.0000 | −0.0081 | 0.0184 | |
| 3b | 3.2382 | 0.0532 | −0.0001 | 0.0000 | −0.0133 | 0.0398 |
| 7.1545 | 0.0158 | 0.0000 | 0.0000 | −0.0055 | 0.0102 | |
| 7.3772 | 0.0182 | 0.0000 | 0.0000 | −0.0062 | 0.0119 | |
| 3c | 3.1503 | 0.0585 | −0.0003 | 0.0000 | −0.0146 | 0.0433 |
| 7.1578 | 0.0076 | 0.0000 | 0.0000 | −0.0027 | 0.0048 | |
| 7.2732 | 0.0100 | 0.0000 | 0.0000 | −0.0034 | 0.0066 | |
| 3d | 3.7338 | 0.0512 | −0.0004 | 0.0000 | −0.0126 | 0.0369 |
| 7.4223 | 0.0054 | 0.0000 | 0.0000 | −0.0020 | 0.0034 | |
| 7.8098 | 0.0074 | 0.0000 | 0.0000 | −0.0026 | 0.0047 |
Theoretical study of supramolecular complexes of dyads 3a–d with metal ions
The formation of supramolecular complexes of dyads 3a–d and metal ions induces significant changes in their structural and photophysical properties. In order to find the mechanisms responsible for these changes the complexes with Cu2+ and Hg2+ were theoretically studied. As can be seen in Fig. 9 and 10, an imide carbonyl of ANAPIM and the amino and amide carbonyl of 2ABZ interact with the metal ion (also see Fig. S51 and S52†) disrupting the stacking of the aromatic fragments observed in the free ligands. Although it was possible to find structures in which one of the NHR2 from the polymethylene bridge is bonded to the metal, the associated electronic energies (and also the free energies) are significantly higher with regard to the energy of the global minimum, so they are not relevant for the present analysis. Table 3 shows that the R values for the donor–acceptor fragments are higher in these metallic complexes. The simulated electronic absorption spectra for the complexes with Cu2+ and Hg2+ also agree with the experimental data, particularly, present absorbance band at positions similar to those for p-2ABZ and pp-ANAPIM. | ||
| Fig. 9 Lateral view of molecular structures corresponding to the global minimum for the complexes with Cu2+ and dyads 3a (A), 3b (B), 3c (C) and 3d (D). The ground state spin multiplicity corresponds to S = 1. | ||
 | ||
| Fig. 10 Lateral view of molecular structures corresponding to the global minimum for the complexes with Hg2+ and dyads 3a (A), 3b (B), 3c (C) and 3d (D). The ground state spin multiplicity corresponds to S = 0. | ||
However, for the supramolecular complexes the description of the electronic states is even more elaborated. Fig. S53† shows the frontier molecular orbitals (MO) for 3b-Hg2+ complex, this system presents a singlet spin multiplicity for the ground state. Comparing the MO of the complex and free ligand as well the amplitude information, it is possible to assign the nature of the electronic states for the complex. The configurational changes HOMO/LUMO and HOMO−1/LUMO+2 of 3b-Hg2+ are similar to the HOMO/LUMO of p-2ABZ and pp-ANAPIM. In fact, the S1 electronic state of 3b-Hg2+ is well described by the configurational change HOMO/LUMO and the transition S0 → S1 is localized in the ANAPIM fragment. This is also corroborated by analysis of the natural transition orbitals. Importantly, the electronic states of the complex described by the change HOMO−1/LUMO+1 and HOMO−1/LUMO+2 that would be involved in the excitation process localized in the 2ABZ system appear shifted to higher energy (¡S13 and S15!), so the respectively electronic transition from the ground state takes place approximately at 274 nm. This result agrees with the spectroscopic changes observed in the electronic absorption spectra of 3b-Hg2+ complex, where there is a hypochromic effect at 320 nm and hyperchromic change before 270 nm.
For 3b-Cu2+, the ground state spin multiplicity corresponds to a doublet. Unlike the complex with Hg2+ which has a filled d layer and where the d occupied orbitals are localized in the metal center, in 3d-Cu2+ exists a significant mixing of the molecular orbitals of the ANAPIM system and d-orbitals of the metal ion (Fig. S54†). In fact, the electronic state described by the configurational change HOMOα/LUMOα, which would be associated to the transition localized in ANAPIM system corresponds to S14. The analysis of the natural transition orbital reveals that the transitions toward lower energy states, which extend to the infrared zone, correspond to the metal–ligand excitations (Fig. S55†). Also, the electronic state where the excitation is localized in 2ABZ system is shifted to higher energy with regard to the free ligand. In general, the interaction of dyads 3a–d with metal ions considerably changes the electronic structure of these molecules and the mechanism of fluorescence quenching is different to the observed in the ligands. Due to the closeness of the electronic energies and the huge density of states in the supramolecular complexes, it is possible to consider the intersystem crossover towards S1(D0) may be favored according to the Kasha rule.41 The above was corroborated with geometry optimizations for 3b-Hg2+ and 3b-Cu2+ complexes. For example, in the searching of the minimum for S4 state (absorption at 324 nm) for 3b-Hg2+, the energy of the excited state decreases considerably, after seven or eight iterations changes to be S1. Interestingly, along this process the complexation with Hg2+ is disrupted releasing the metal ion and providing an unfolded dyad.
In summary, the data obtained by theoretical calculations suggest that in the complexes with Cu2+ and Hg2+ the excitation at 325 nm leads to an excited state different to the observed in free dyads. Very interestingly, the intensity decreases of the emission band at 530 nm by increasing the amount of metal ion in the solution, may be due to internal conversion toward inferior electronic states; in other words, the calculations suggest that both metal complexes should emit fluorescence in the near infrared.
Conclusions
The experimental and theoretical studies demonstrated a very efficient FRET process present in 2ABZ–ANAPIM fluorescent dyads. This is due to an overlap of the 2ABZ emission energy with the ANAPIM absorption energy, also the flexible interconnection between both fluorophores allows the stacking configuration, even with the shortest spacer, although with the longest the stacking is more favored. In mechanistic terms, the high FRET ratios showed by the dyads are consistent with the calculated barrierless MEP's, that lead directly from the state initially populated by light absorption toward the minimum of the first excited state, all through accessible crossings between the different potential energy surfaces. Additionally, the calculation of the EET couplings between the donor and acceptor chromophores are consistent with these results. The fluorescence quenching in the presence of Cu2+ and Hg2+ indicates the interaction with these metal ions inhibiting the FRET process between both fluorophores and their individual ICT processes, suggesting a cooperative interaction of both fluorophores. The optimization of the complexes geometry showed the interaction through the amine and carbonyl groups in 2ABZ and a carbonyl of ANAPIM, keeping the fluorophores away and changing their configuration and optical properties.Author contributions
A. L. G.: investigation. A. O.-T.: conceptualization, resources, formal analysis, funding acquisition, writing – original draft preparation. A. T.-G.: conceptualization, investigation. J. J.-C.: conceptualization, resources, formal analysis, writing – original draft preparation. G. P.-L.: conceptualization, writing-reviewing and editing. H. S. O.: investigation, resources. V. L.-G.: investigation, resources. M. O.: investigation, resources. J. P.: resources, writing-reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank to Consejo Nacional de Ciencia y Tecnología (CONACyT-México) (Grant no. SALUD-2015-1-261324) and Tecnológico Nacional de México (TECNM) (Grant no. 5366.19-P) for the financial support to this research. Ángel L. García thanks CONACyT for the postgraduate scholarship. Authors are grateful to Supramolecular Chemistry Thematic Network (Grant no. 294810) for their support. J. J.-C. and J. Peón thanks CONACyT-México grant Ciencia de Frontera 2019-51496 and DGTIC-UNAM for supercomputer resources (project LANCAD-UNAM-DGTIC-103). J. J-C. also thanks “Laboratorio Nacional de Supercómputo del Sureste de México” (project 202003047N) and to Patronato-UAN grant “Fortalecimiento a la Investigación”.Notes and references
- United Nations, Department of Economic and Social Affairs, Population Division, World Population Prospects 2019: Methodology of the United Nations population estimates and projections, 2019.
- United Nations Educational, Scientific and Cultural Organization (UNESCO), The United Nations World Water Development Report 2019, Leaving No One Behind.
- S. Atilgan, T. Ozdemir and E. U. Akkaya, Org. Lett., 2010, 12, 4792–4795 CrossRef CAS PubMed.
- S. Sarma, B. Devi, P. K. Bhattacharyya and D. K. Das, J. Fluoresc., 2020, 30, 1513–1521 CrossRef CAS PubMed.
- J. A. Cotruvo Jr, A. T. Aron, K. M. Ramos-Torresa and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4400–4414 RSC.
- X. Q. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS PubMed.
- J. H. Jung, J. H. Lee and S. Shinkai, Chem. Soc. Rev., 2011, 40, 4464–4474 RSC.
- M. Martinez-Quiroz, X. E. Aguilar-Martinez, M. T. Oropeza-Guzman, R. Valdez and E. A. Lopez-Maldonado, Molecules, 2019, 24, 1737 CrossRef CAS PubMed.
- T. Gunnlaugsson, P. E. Kruger, T.-C. Lee, R. Parkesh, F. M. Pfeffer and Y. M. Hussey, Tetrahedron Lett., 2003, 44, 6575–6578 CrossRef CAS.
- S. Anbu, A. Paul, K. Surendranath, A. Sidali and A. J. L. Pombeiro, J. Inorg. Biochem., 2021, 220, 111466 CrossRef CAS PubMed.
- M. S. Kim and T. Tsukahara, ACS Earth Space Chem., 2020, 4, 2270–2280 CrossRef CAS.
- N. I. Georgiev, P. V. Krasteva and V. B. Bojinov, J. Lumin., 2019, 212, 271–278 CrossRef CAS.
- L. Wang, Y.-Q. Fan, X.-W. Guan, W.-J. Qu, Q. Lin, H. Yao, T.-B. Wei and Y.-M. Zhang, Tetrahedron, 2018, 74, 4005–4012 CrossRef CAS.
- N. Saini, N. Prigyai, C. Wannasiri and V. Ervithayasuporn, J. Photochem. Photobiol., A, 2018, 358, 215–225 CrossRef CAS.
- K. Shen, S. Mao, X. Shi, F. Wang, Y. Xu, S. Opeyemi Aderinto and H. Wu, Luminiscence, 2017, 33, 54–63 CrossRef PubMed.
- H. Zhang, Y. Qu, K. Zhao, C. Wang, Y. Wu and H. A Wu, J. Chin. Chem. Soc., 2020, 67, 1062–1069 CrossRef CAS.
- P. A. Pachenko, Y. V. Fedorov, O. A. Fedorova and G. Jonusauskas, Dyes Pigm., 2013, 98, 347–357 CrossRef.
- B. Dong, X. Song, C. Wang, X. Kong, Y. Tang and W. Lin, Anal. Chem., 2016, 88, 4085–4091 CrossRef CAS PubMed.
- M. Landey-Álvarez, A. Ochoa-Terán, G. Pina-Luis, M. Martínez-Quiroz, M. Aguilar-Martínez, J. Elías-García, V. Miranda-Soto, J. Ramírez, L. Machi-Lara, V. Labastida-Galván and M. Ordoñez, Supramol. Chem., 2016, 28, 893–906 CrossRef.
- V. Ravindar, S. J. Swamy, S. Srihari and P. Lingaiah, Polyhedron, 1985, 4, 1511–1518 CrossRef CAS.
- J. Dharmaraja, T. Esakkidurai, P. Subbaraj and S. Shobana, Spectrochim. Acta, Part A, 2013, 114, 607–621 CrossRef CAS PubMed.
- J. Dharmaraja, P. Subbaraj, T. Esakkidurai and S. Shobana, Spectrochim. Acta, Part A, 2014, 132, 604–614 CrossRef CAS PubMed.
- S. Abad, M. Kluciar, M. A. Miranda and U. Pischel, J. Org. Chem., 2005, 70, 10565–10568 CrossRef CAS PubMed.
- B. H. Shankar and D. Ramaiah, J. Phys. Chem. B, 2011, 115, 13292–13299 CrossRef CAS PubMed.
- V. S. Jisha, A. J. Thomas and D. Ramaiah, J. Org. Chem., 2009, 74, 6667–6673 CrossRef CAS PubMed.
- J. A. Rackers, Z. Wang, C. Lu, M. L. Laury, L. Lagardère, M. J. Schnieders, J.-P. Piquemal, P. Ren and J. W. Ponder, J. Chem. Theory Comput., 2018, 14, 5273–5289 CrossRef CAS PubMed.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS.
- T. A. Halgren and R. B. Nachbar, J. Comput. Chem., 1996, 17, 587–615 CAS.
- T. Lewis-Atwell, A. Townsend Piers and N. Grayson Matthew, Tetrahedron, 2021, 79, 131865 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- G. Barone, D. Duca, A. Silvestri, L. Gomez-Paloma, R. Riccio and G. Bifulco, Chem.–Eur. J., 2002, 8, 3240–3245 CrossRef CAS.
- C. Curutchet and B. Mennucci, J. Am. Chem. Soc., 2005, 127, 16733–16744 CrossRef CAS PubMed.
- M. F. Iozzi, B. Mennucci, J. Tomasi and R. Cammi, J. Chem. Phys., 2004, 120, 7029–7040 CrossRef CAS PubMed.
- C. Azarias, R. Russo, L. Cupellini, B. Mennucci and D. Jacquemin, Phys. Chem. Chem. Phys., 2017, 19, 6443–6453 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresmany and D. J. Fox, Gaussian 16, Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. Min Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock III, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio Jr, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. Un Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Yeh Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Ying Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard III, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer III, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MS data; 1H NMR, 13C NMR, FTIR and electronic absorption and emission spectra of relevant compounds, as well as additional information from the calculations. See DOI: 10.1039/d1ra09278b |
| This journal is © The Royal Society of Chemistry 2022 |