
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrahigh oxygen evolution reaction activity in Au doped co-based nanosheets†
Chao Cai a,
Shaobo Hana,
Xiaotao Zhangb,
Jingxia Yua,
Xia Xianga,
Jack Yangc,
Liang Qiaoa,
Xiaotao Zu*ad,
Yuanzheng Chen*b and
Sean Li*c
a,
Shaobo Hana,
Xiaotao Zhangb,
Jingxia Yua,
Xia Xianga,
Jack Yangc,
Liang Qiaoa,
Xiaotao Zu*ad,
Yuanzheng Chen*b and
Sean Li*c
aYangtze Delta Region Institute (Huzhou), University of Electronic Science and Technology of China, Huzhou 313001, China. E-mail: xtzu@uestc.edu.cn
bSchool of Physical Science and Technology, Key Laboratory of Advanced Technology of Materials (Ministry of Education of China), Southwest Jiaotong University, Chengdu, Sichuan 610031, China. E-mail: cyz@swjtu.edu.cn
cSchool of Materials Science and Engineering, The University of New South Wales, Sydney 2052, Australia. E-mail: sean.li@unsw.edu.au
dInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, 610054, P. R. China
First published on 22nd February 2022
Abstract
Oxygen evolution reaction (OER) has attracted enormous interest as a key process for water electrolysis over the past years. The advance of this process relies on an effective catalyst. Herein, we employed single-atom Au doped Co-based nanosheets (NSs) to theoretically and experimentally evaluate the OER activity and also the interaction between Co and Au. We reveal that Au–Co(OH)2 NSs achieved a low overpotential of 0.26 V at 10 mA cm−2. This extraordinary phenomenon presents an overall superior performance greater than state-of-the-art Co-based catalysts in a sequence of α-Co(OH)2 < Co3O4 < CoOOH < Au–Co(OH)2. With ab initio calculations and analysis in the specific Au–Co(OH)2 configuration, we reveal that OER on highly active Au–Co(OH)2 originates from lattice oxygen, which is different from the conventional adsorbate evolution scheme. Explicitly, the configuration of Au–Co(OH)2 gives rise to oxygen non-bonding (ONB) states and oxygen holes, allowing direct O–O bond formation by a couple of oxidized oxygen with oxygen holes, offering a high OER activity. This study provides new insights for elucidating the origins of activity and synthesizing efficient OER electrocatalysts.
Introduction
The hydrogen economy is a sunrise industry that will provide a solution for the energy crisis and greenhouse emissions as well as stimulate the rapid growth of the economy.1,2 The core of research and development in the hydrogen economy is to search for cost-effective and ultra-efficient electrocatalysts for oxygen evolution reaction (OER) to realize high throughput hydrogen production on an industrial scale.3 Although the noble metal-based OER catalysts including Ru and Ir oxides exhibit high OER kinetics with a current density of 10 mA cm−2,4 high-cost and limited resources make these materials would not be sought after by the industry. Instead, earth-abundant metal-based nanomaterials have been refocused as alternatives to the noble metal-based OER catalysts. These include the first-row transition-metal (3d) oxides,5,6 nitrides,7 sulfides,8 and hydroxides.9Cobalt (Co)-based nanomaterials are the typical one because of their natural abundance and good OER performance.10,11 In particular, the multivalent nature of Co cations enables additional electrooxidation occurring in Co-based OER catalysts at high reaction potential [usually >1.23 V versus reversible hydrogen electrode (vs. RHE)]. The current efforts in developing efficient OER catalysts is to create the accelerated intrinsic active sites through manipulating the reaction conditions. Importantly, the formation of high-valance Co cations in the OER process can be facilitated through doping technology.12–14 For example, CoFe (oxy)hydroxides nanosheets (NSs) have high OER activity induced by the synergistic effects between Fe and Co, abundant oxygen and metal vacancies.15 It is reported that Au nanostructures can effectively enhance OER activity by positioning Au in a specific site to localize Au–Co interactions.16 Although many efforts have been put in to study the mechanism of Co-based materials for OER,11,17–20 as emerging hybrid electrocatalysts, the mechanism of the improved Co oxidation process is unclear. This is because the oxidation process of Co-based materials in the oxygen evolution reaction is too fast to be determined. It obstructs understanding the large activity gap between (oxy)hydroxides and precursors, impeding the development and rational design of high activity 3d metal-based electrocatalysts. Therefore, developing Co-based materials with a controllable oxidation state would be the first step to advance the highly active OER electrocatalysts.
Herein, we deposited ultrathin NSs of Co(OH)2-based catalysts on the surface of a highly electronegative Au single-atoms. By comparing non-modified Co-based NSs (e.g., Co(OH)2, Co3O4, CoOOH), we were able to explore their OER activity and interaction between Co and Au. We found that the OER activities of those NSs are IrO2 < α-Co(OH)2 < Co3O4 < CoOOH < Au–Co(OH)2 based on the geometric current density. The OER activity of Au–Co(OH)2 NSs is 30-fold of commercial IrO2. The surface chemistry of this particular material was analyzed using X-ray photoelectron spectroscopy (XPS). Ex situ scanning transmission electron microscopy (STEM) was used to characterize the preferential oxidation of Co atoms, which was coordinated with Au atoms, revealing the high structural stability of Au–Co(OH)2 NSs in an oxidation environment. Further theoretical calculations demonstrate that Au can improve the adsorption/desorption efficiency around the active Co sites during OER. It is believed that this is related to the decrease of *OH/*O free energy. The high OER activity of Au–Co(OH)2 NSs originates from the high intrinsic activity of Co as well as the strong synergistic effect between Au and Co, and the large efficacious active surface area.
Results and discussions
Synthesis and structural characterization of ultrathin Co-based NSs
The ultrathin two-dimension feature of NSs ensures high processability at the atomic scale, such as surficial and interfacial modification. Here we demonstrate that the Co-based NSs with modified electronic configurations exhibit superior OER activity. The growth procedure of Co-based NSs is schematically illustrated in Scheme 1. α-Co(OH)2 crystals are assembled to form NSs with CH3COO− adsorption, agreeing well with the reported Co-based NSs in ethylene glycol (EG).10,21 The manipulation of the oxidation state of Co was realized by processing α-Co(OH)2 NSs under operando conditions, forming Co3O4, CoOOH, and Au–Co(OH)2 NSs. The scanning electron microscopy (SEM) morphologies demonstrate the highly stable microstructure of Co-based NSs under operando conditions (Fig. S1†). As shown in Fig. S2,† peaks in the X-ray diffraction (XRD) pattern at 10.5°, 33.5° and 59.5° correspond to (003), (012) and (110) of α-Co(OH)2, respectively. Rhombohedral CoOOH was synthesized using the electrochemical oxidizing methodology from α-Co(OH)2 NSs. Subsequently, annealing α-Co(OH)2 formed Co3O4 NSs as shown in SEM morphology (Fig. S1† and XRD pattern in Fig. S2†). | ||
| Scheme 1 Growth procedure of ultrathin Co-based NSs. Co-Based NSs show high stability under operando conditions. | ||
The double-Cs corrected high angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was used to determine Au on NSs. The SEM image in Fig. 1a shows the NS morphology of Au–Co(OH)2 and the elemental distribution with Au homogeneously distributed on Co(OH)2 NSs was mapped using Energy Dispersive Spectra (EDS) as shown in Fig. 1b. The HAADF-STEM image demonstrates that the NSs are composed of four layers with a total thickness of about 1.2 nm (Fig. 1c), agreeing well with the reported Co/Co–O NSs.21 In the HAADF image (Fig. 1d), the bright dots correspond to Au atoms while the others are Co atoms because of the higher mass of Au atoms. It shows that Au atoms are isolated and attached to the surface of Co(OH)2.
 | ||
| Fig. 1 Structural characterization of Au–Co(OH)2. SEM image (a), EDS mapping (b), STEM image (c), and (d) HAADF image of Au–Co(OH)2. The newly formed Au–Co(OH)2 NSs maintain pure Co(OH)2 crystal features. NSs have four atomic layers with a thickness of 1.2 nm. Au attaches on NSs as single atoms, as indicated by white circles. | ||
Electrochemical characterization of Co-based NSs
The OER performance of Co-based NSs was evaluated with 1 M KOH (Fig. 2). The current density was counted on the geometry of the working electrode. Fig. 2a shows the polarization curves of Co-based NSs. For comparison, the commercial IrO2 presents a specific current density of 10 mA cm−2 at an overpotential of 320 mV (1.55 V vs. RHE), which is similar to the reported data.22,23 The OER activity of Co-based NSs shows a sequence of Au–Co(OH)2 (260 mV) > CoOOH (308 mV) > commercial IrO2 (320 mV) > Co3O4 (357 mV) > α-Co(OH)2 (410 mV) with a benchmark of the overpotential in a specific current density of 10 mA cm−2. In addition, it is noted that the achieved OER activity of Au–Co(OH)2 is higher than that of the state-of-the-art Au/Co-based catalysts (Table S1†) including CoSe2,24 NiCo–A (A = P, Se, O)6 and Au–CoSe2.25 Tafel plots of these samples summarized in Fig. 2b demonstrate that Au–Co(OH)2 NSs possess the highest charge transport efficiency around active sites with a slope of 52 mV dec−1, which is smaller than that of Co3O4 NSs (64 mV dec−1), commercial IrO2 (60 mV dec−1), α-Co(OH)2 NSs (104 mV dec−1), and CoOOH NSs (74 mV dec−1). The specific current density of Co-based NSs at 1.5 V vs. RHE is plotted in Fig. 2c. Au–Co(OH)2 NSs exhibit the highest OER activity, which is 30-fold higher than that of the commercial IrO2. Besides, the mass activity (MA) and turnover frequency (TOF) are calculated on basis of Fig. 2c.26,27 MA of Au–Co(OH)2 is 177 A g−1, which is much higher than 3.89 A g−1, 7 A g−1, 22.56 A g−1, and 5.66 A g−1 of Co(OH)2, Co3O4, CoOOH, and IrO2, respectively. The TOF of Au–Co(OH)2 is 27 s−1, which is much higher than 0.6 s−1, 1.07 s−1, 3.45 s−1, and 0.87 s−1 of Co(OH)2, Co3O4, CoOOH, and IrO2, respectively. These results demonstrate the high atomic utilization of Au–Co(OH)2 even better than that in IrO2. Fig. 2d shows the accelerating degradation of Co-based NSs at 1.49 V vs. RHE, showing a transformation from Co(OH)2 to CoOOH during the OER processing (Fig. S3†). It demonstrates that the stability of electrocatalysts may originate from CoOx(OH)y (e.g. CoOOH). | ||
| Fig. 2 Electrochemical properties of the series of Co-based NSs in 1 M KOH. Polarization curves (a), Tafel plots (b), a collection of specific current destiny at 1.5 V vs. RHE (c), accelerating degradation (d), Nyquist plots (e), and N2 adsorption/desorption isothermal (f) of as-prepared Co-based NSs. The loaded Au on Co(OH)2 can highly promote the OER activity (a–c). The data in (c) is normalized by the geometric area of the working electrode. Co-based NSs show high stability for OER in alkaline solution (d). Au–Co(OH)2 NSs have the highest mass transport efficiency (e) among those NSs. | ||
It is believed that the superior OER performance of Co-based NSs is related to the enhancement of electrical conductivity, which facilitates the relevant charge transfer between the support and catalysts.28,29 In this case, we used electrochemical impedance spectrometry (EIS) to characterize the electrode kinetics with different catalyst loading (Fig. 2e). Au–Co(OH)2 NSs demonstrate their highest charge transfer efficiency in OER at 1.49 V vs. RHE. To clarify that this phenomenon was not contributed by active sites number variations, we conducted the N2 adsorption/desorption isotherm test on Co-based NSs. Co(OH)2 NSs showed a higher surface area of 139.44 m2 g−1 than 131.07 m2 g−1 of Au–Co(OH)2 NSs (Fig. 2f). Even with the low active surface area of Au–Co(OH)2 NSs, which is 10-fold higher than that of pristine Co-based NSs. These results demonstrate that Au atoms on Co(OH)2 can efficiently increase the intrinsic activity of bonded Au–Co for OER. Therefore, we believe such exotic OER performance is associated with the highly efficient charge transfer and high intrinsic catalytic activity.
The oxidation state of Co-based NSs
High-valance Co (CoOx(OH)y) are usually considered the active sites for OER (Co3+/Co4+ group peaks located at 1.24–1.54 V vs. RHE).16 The OER activity of Co-based materials is highly sensitive to the oxygen vacancies surrounding Co cations. This is because the two electrons in the neighboring oxygen vacancies can be delocalized around Co3+/Co4+, ensuring that the low-coordinated Co sites are more active in adsorbing water molecules or active intermediate during OER.30 In our experiments, therefore, the core factors to analyze OER mechanisms are to clarify the oxidation states of species in Co(OH)2 NSs and Au–Co(OH)2 NSs. XPS was used to reveal the valence of cations and surface chemistry in NSs (Fig. 3). Spectra of Co, O, and Au as well as their deconvolution patterns are shown in Fig. 3a and b. It shows that the peaks at 781. 4 eV and 783.2 eV are assigned to Co3+ and Co2+ respectively.10,31 Co 2p3/2 has a red shift of 0.3 eV with the coated Au on Co(OH)2 NSs. The increased Co3+/Co2+ ratio in Au–Co(OH)2 NSs demonstrates that Au coating increases the Co valence in Co(OH)2, which was resulting from the low coordinated Co–O bonding because of the appearance of Au.12 Further study on O 1s shown in Fig. 3c indicates that spectra of O 1s can be deconvoluted into three peaks of Co–O bonding (529.1 eV), OH− in Co(OH)2 (530.1 eV), and adsorbed H2O (531.8 eV).32 The higher ratio Co–O peak supports the notion that Co valence in Au–Co(OH)2 NSs is increased by Au coating. Fig. 3d shows the Au 4f peak, where two peaks located at 84.0 eV and 88.2 eV are corresponding to Au 4f5/2 and 4f7/2, respectively.12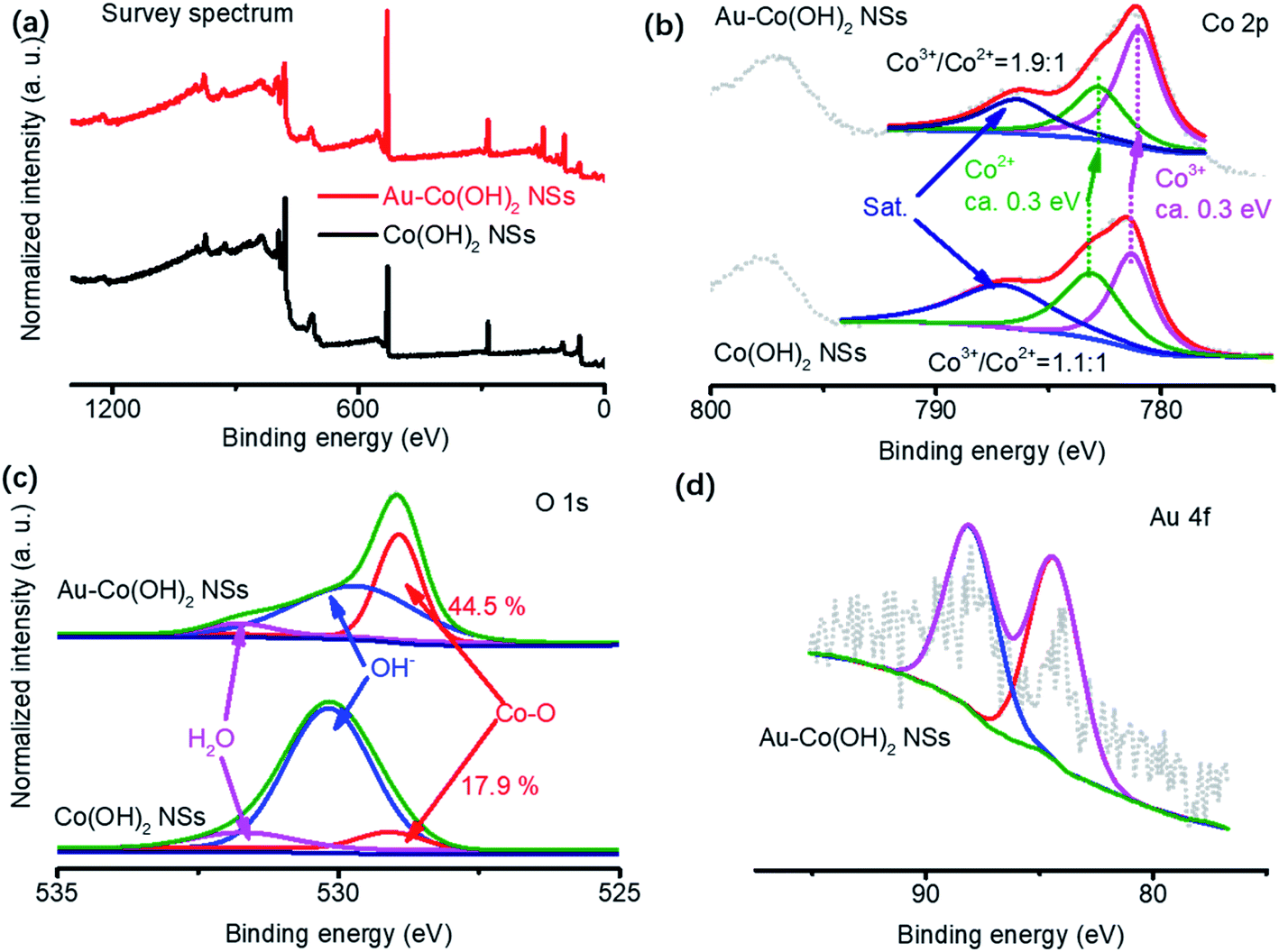 | ||
| Fig. 3 Surface chemistry characterization of Co(OH)2-based NSs. Survey (a), Co 2p spectrum (b), O 1s (c), and Au 4f spectrum (d). The extra Au can promote Co valence (c), and thus promotes the activity of high valence Co in OER. | ||
In general, the OER catalysts with high activity are usually accompanied by structural instability related to oxidation. In this work, we conducted ex situ STEM experiments, XPS, and XRD to characterize the stability of Au–Co(OH)2 NSs in the accelerating degradation process as shown in Fig. 4, S4 and S5.† XPS analysis demonstrates a very limited change in the ratio of Co2+/Co3+ (1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with the accelerating degradation process, which is similar to the value of 1.9:1 for the as-prepared Au–Co(OH)2 NSs. It suggests that Au dominated the selective electro-oxidation of Co in OER. The STEM image in Fig. 4b shows that the thickness of Au–Co(OH)2 NSs is about 1 nm, demonstrating the chemical stability of Au–Co(OH)2 NSs. Fig. 4c shows the EDS mapping of the individual elements in Au–Co(OH)2 NSs after the stability characterization. As shown in Fig. 4d, Au atoms still maintain the isolated feature of CoOx(OH)y. The high-resolution HAADF STEM image exhibits the co-existence of CoOOH and Co(OH)2 (Fig. 4d), indicating that only a part of Co2+ cations in NSs adapt further electrooxidation during OER. This finding is supported by the ex situ XRD characterization (Fig. S5†), where it shows the co-existence of CoOOH and Co(OH)2 phase. The individual Au atoms are surrounded by in situ formed CoOOH (Fig. 4d). Moreover, compared to the oxidation barriers of Co2+ in Co(OH)2 and Au–Co(OH)2, we found that Co2+ in Au–Co(OH)2 is easier to be further oxidized into high valence Co3+ (Fig. S8 and Table S2†). These results show that Co atoms near Au are preferentially oxidized in OER and Au–CoOOH is responsible for the high OER activity (Fig. 2).
:1) with the accelerating degradation process, which is similar to the value of 1.9:1 for the as-prepared Au–Co(OH)2 NSs. It suggests that Au dominated the selective electro-oxidation of Co in OER. The STEM image in Fig. 4b shows that the thickness of Au–Co(OH)2 NSs is about 1 nm, demonstrating the chemical stability of Au–Co(OH)2 NSs. Fig. 4c shows the EDS mapping of the individual elements in Au–Co(OH)2 NSs after the stability characterization. As shown in Fig. 4d, Au atoms still maintain the isolated feature of CoOx(OH)y. The high-resolution HAADF STEM image exhibits the co-existence of CoOOH and Co(OH)2 (Fig. 4d), indicating that only a part of Co2+ cations in NSs adapt further electrooxidation during OER. This finding is supported by the ex situ XRD characterization (Fig. S5†), where it shows the co-existence of CoOOH and Co(OH)2 phase. The individual Au atoms are surrounded by in situ formed CoOOH (Fig. 4d). Moreover, compared to the oxidation barriers of Co2+ in Co(OH)2 and Au–Co(OH)2, we found that Co2+ in Au–Co(OH)2 is easier to be further oxidized into high valence Co3+ (Fig. S8 and Table S2†). These results show that Co atoms near Au are preferentially oxidized in OER and Au–CoOOH is responsible for the high OER activity (Fig. 2).
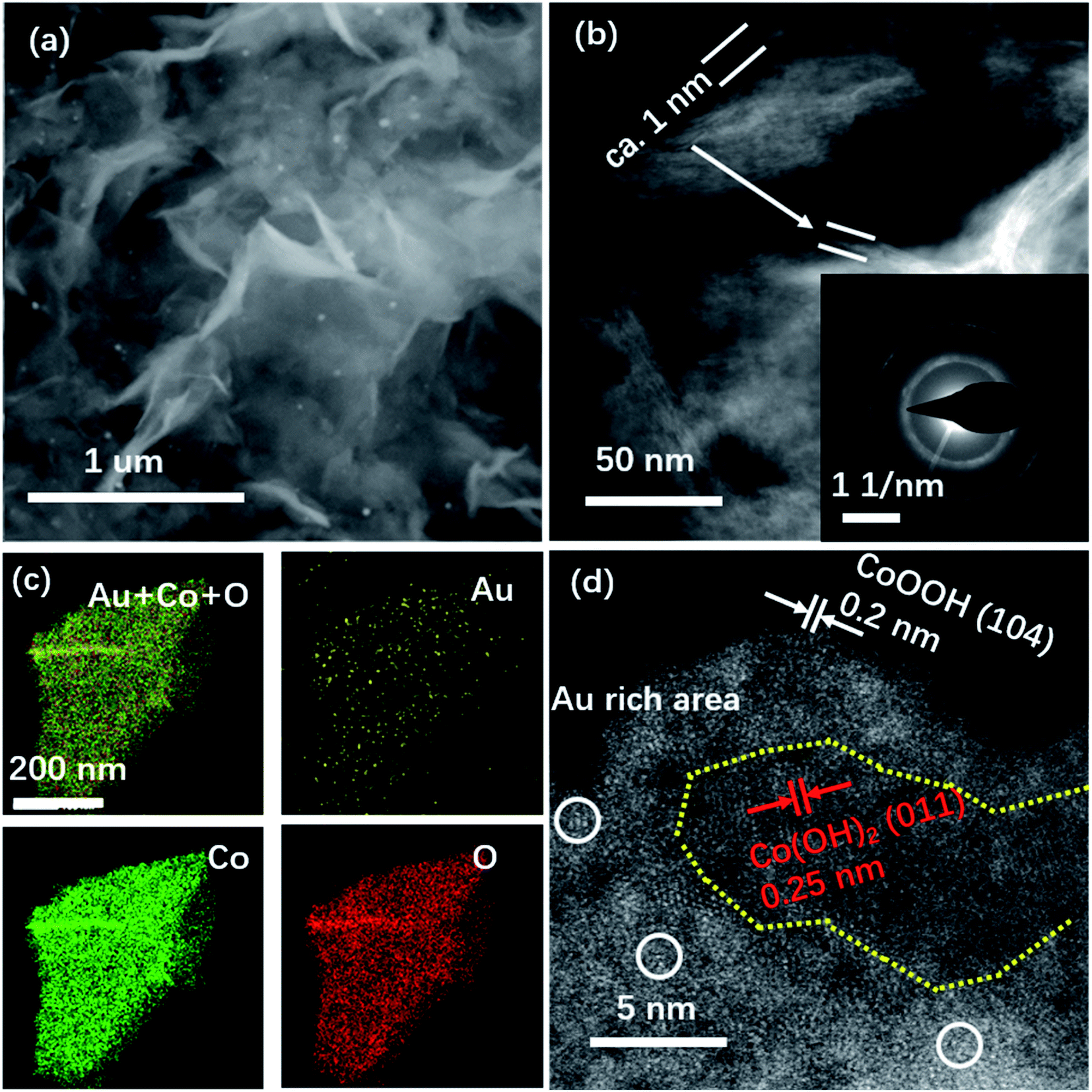 | ||
| Fig. 4 Structural characterization of Au–Co(OH)2 NSs after use. SEM image (a), HAADF image (b) (inset is SAED pattern), EDS mapping (c), and high magnification HAADF image of Au–Co(OH)2 NSs after use (d). Au–CoOx(OH)y NSs have high structural stability in OER (a and b). Co2+ ions around the Au atoms are preferentially activated and changed into Co3+, because of the accelerated OER process around Au–Co couples. Yellow dots mark the pure Co(OH)2 in NSs after use. | ||
Lattice-oxygen oxidation mechanism on Au–Co(OH)2
In general, the OER activity of Co-based materials is sensitive to the species loading amount. To clarify this, we synthesize samples with different Au loading amounts on Co(OH)2 NSs for OER. Fig. S6† shows the polarization curves and Tafel plots of Au–Co(OH)2 NSs. NSs with different Au doping levels show similar onset potential (1.43 V vs. RHE), demonstrating that the existence of Au in Co(OH)2 NSs to form Au–CoOOH is responsible for the high OER activity. Such a phenomenon may be attributed to the electron affinity difference between Au (223 kJ mol−1) and Co (64 kJ mol−1), where the higher electron affinity of Au enables the high concentration of negatively charged species (OH−, OOH−, and O2−) surrounding Au near Co cations. In this case, the improved activity of Au–Co(OH)2 NSs is dealt with the change of OH− species around active sites on the surface, where it shows a difference between the adsorption free energy (X–OH) and the reduced O (X–O).33 Meanwhile, because of the electron affinity difference and activated Au with a low excited barrier, the addition of Au may enhance the efficiency of the reversible absorption/adsorption process.34 This procedure can facilitate the formation of adsorbed OH− and reduced O intermediates on the Co3+ closed to the oxygen vacancies (agreeing well with Fig. S4†), enhancing the oxidation process from Co3+ to Co4+. This is also verified by the cyclic voltammetry method (Fig. S7†), where the integrated area of oxidation peak for the processes from Co2+ to Co3+ (1.1 V vs. RHE)35 and Co3+ to Co4+ (1.4 V vs. RHE) is increased 20-fold with individual Au atom deposition. However, further increasing the Au use in catalysts, the OER activity of Au–Co(OH)2 NSs decreases. It is believed that the excessive substitution is unfavorable to refill the oxygen vacancies through the lattice-oxygen oxidation mechanism (LOM).36 Consequently, Au excessive substitution in Au–Co(OH)2 would not only lower the overall activity but also lead to severe degradation of catalyst because of the significant loss of oxygen. Therefore, optimizing the Au doping level in Au–Co(OH)2 NSs can efficiently enhance the catalytic activity in the OER processing.In order to understand the reaction mechanism of the OER process of Au–Co(OH)2 nanosheets, we used the traditional adsorbate evolution mechanism to study the Gibbs free energy of possible adsorbates on their surfaces. In general, the basic OER process involves four fundamental electrochemical processes and one of the following desorption processes, as shown in Fig. 5a and S9.† The computed ΔG has no obvious difference between the Au–Co(OH)2 and Co(OH)2 (Fig. 5b). Their theoretical overpotential, which is equal to |ΔGMAX − 1.23|, is nearly unchanged (1.11 eV for the Au–Co(OH)2, 1.01 eV for Co(OH)2). It demonstrates that traditional AEM cannot explain the observed high catalytic performance in the OER for Au–Co(OH)2s.
 | ||
| Fig. 5 (a) The reaction pathways of OER for Au–Co(OH)2 in adsorbate evolution mechanism (AEM); (b) the free energy profiles of each reaction stage for Au–Co(OH)2 (denoted as Au–Co) and the pristine Co(OH)2 (denoted as Co) in AEM, and the ΔGMAX are labeled and the overpotential value is marked in parentheses; (c) the projected DOS for the Au–Co(OH)2 in comparison with that of Co(OH)2; (d) the schematic energy bands for the Au–Co(OH)2 and Co(OH)2, in which the antibonding band is split into one empty upper-Hubbard band (UHB) and one filled lower-Hubbard band (LHB). The oxygen non-bonding state (ONB) occurs above the Fermi level in the energy bands of Au–Co(OH)2; (e) the reaction pathways of OER in lattice oxygen reaction (LOM), in which the key step difference from AEM is labeled by the dotted line; (f) the free energy profiles of each reaction stage for Au–Co(OH)2 and Co(OH)2 in LOM. | ||
Recent studies on OER demonstrated that the doping of Co(OH)2 by heterogeneous atoms can oxidize oxygen atoms in lattice to generate oxygen with LOM. In this mechanism, the oxygen generated in the OER process is not only originating from the decomposition of water molecules, but also the oxidation of lattice oxygen atoms and adsorbed oxygen atoms. It is believed that this process improves the OER performance of the materials significantly. Considering the strong Coulomb effect of the d–d orbits between the heterogeneous atoms and the weak bonding formed between Au and Co, a similar ONB state is formed between Au and Co(OH)2. In this case, Au and Co(OH)2 would adsorb some unbonded oxygen. Therefore, it is inevitable that LOM states are formed on the surfaces of Au and Co(OH)2. In Au–Co(OH)2, as shown in Fig. 5c and S10,† the electric charge of Au along its C-axis region decreases, and subsequently transfers to the coordinated O atoms (Au–O bond direction). This forms the unoccupied ONB state above the Fermi level (EF). Moreover, the experiment indicates that the doped Au leads to the oxygen holes formation in Au–Co(OH)2. These experimental results reveal that the oxidized oxygen nearby oxygen holes in Au–Co(OH)2 is preferentially transformed into O–O. Herein, we proposed the likely reaction schemes of LOM for Au–Co(OH)2 involving the oxidation of lattice oxygen (Fig. 5e and S11†), though other possibilities may exist. This scheme mainly consists of two key chemical steps (L1 and L3), which first creates an O–O bond and subsequently an oxygen molecule from lattice oxygen sites via the formation of oxygen vacancies. As shown in Fig. 5f, ΔG calculations show that the overpotential decreases from 1.07 eV in Co(OH)2 to 0.78 eV in Au–Co(OH)2, suggesting that the OER of Au–Co(OH)2 along the LOM pathway is more thermodynamically favorable. It enables better performance in accordance with the experiment. Compared to AEM, the LOM mechanism can well explain the experimental observation in the highly efficient electrochemical performance of Au–Co(OH)2, which is related to the creation of ONB states and oxygen holes of the specific Au–Co(OH)2 configuration.
Conclusions
In this work, we synthesized Co-based NSs as OER catalysts. These Co-based NSs show a controllable oxidation state, leading to the OER activity change. Au–Co(OH)2 shows the best OER activity, which is 30-fold higher than the commercial IrO2. The existence of Au resulted in the OER activity adjustment by accelerating the oxidation process of high valence Co. This facilitates the adsorption/desorption efficiency on the activated Co center, enhancing the effective and active surface areas. The synergistic effect between Au and Co is highly sensitive to the d orbits of Au atoms, accelerating the ONB formation and OER activity. The excited barriers in Au–Co(OH)2 are lowered, accelerating the oxidation process of Co during OER. The high intrinsic activity of Co is further enhanced by the individual Au atoms.Experimental
Chemicals
Co(acac)3 (>95%), ethylene glycol (EG) (98%), KOH (>99.999%), ethanol (>97%), HAuCl4·4H2O (metal tracing, >42%), and isopropanol (>98%) were purchased from Aladdin. 5 wt% Nafion was purchased from Sigma.Preparation of α-Co(OH)2 NSs
In a typical synthesis of α-Co(OH)2 NSs, 200 mg of cobalt acetylacetonate (Co(acac)3) was dissolved in 40 mL of EG. The EG solution containing Co(acac)3 was mixed with 8 mL deionized water, heated at 160 °C for 48 h, and then cooled down to room temperature. The product was washed with a mixture of ethanol and deionized water several times, collected by centrifugation (10000 rpm, 5 min), and then dried in a vacuum oven at 70 °C, overnight.
Preparation of CoOOH NSs
20 mg fresh α-Co(OH)2 NSs were cast onto silicon support, and then a potential of 1.5 V vs. RHE was applied on the silicon support using chronoamperometry in 1 M KOH. After 10 h, the samples were sonicated and collected by centrifugation, washed with water and ethanol several times.Preparation of Co3O4 NSs
The as-prepared α-Co(OH)2 NSs were annealed at 400 °C for 5 min in the air (heating rate is ca. 10 °C min−1), and then cooled to room temperature. The black powders were obtained.Preparation of Au–Co(OH)2 NSs
According to the reports,37 α-Co(OH)2 NSs (40 mg) and required amounts of Au (2, 4, 10, 20, and 40 μL HAuCl4·4H2O (20 mg mL−1)) were dispersed in 10 mL ethanol under vigorous stirring, overnight. 1 mL NaBH4 solution (2 mg mL−1) was added to the solution dropwise. After stirring for 1 h, Au–Co(OH)2 NSs were collected and washed with water and ethanol, dried in a vacuum oven at 70 °C, overnight.Structure characterization
The morphology and microstructure characterizations of the as-prepared samples were investigated using scanning electron microscopy (SEM) (FEI, Helios Nanolab 600i, 2 kV), transmission electron microscopy (TEM) and selected area electron diffraction (SAED) (FEI, ETEM, G2, 200 kV), scanning transmission electron microscopy with an energy-dispersive X-ray spectroscopy attachment (STEM-EDS, FEI Titan Themis, 300 kV). X-ray diffraction (XRD) (Bruker ECO D8 power X-ray diffractometer with Cu Kα radiation) was utilized to determine the crystal structure of the samples.Electrochemical characterization
All experiments were carried out using the CHI760e in a three-electrode system. Rotate glassy disk carbon electrode (GCE) (3 mm) was the support of active materials to be used as the working electrode. Ag/AgCl (saturated with 4 M KCl) and a graphite rod were used as counter electrode and reference electrode, respectively. The electrolyte was 1 M KOH saturated with O2 (99.999)%. The working electrode for Co-based NSs was prepared by dropping 4 μL dispersion (catalyst ink (2 mg mL−1) with isopropanol/water (1:3) containing 10 μL 5 wt% Nafion solution) in a GCE. Cycling voltammetry (CV), linear sweep voltammetry (LSV), chronoamperometry and Tafel plot were measured at 298 K, 1600 rpm, and 5 mV s−1 on ALS-RRDE. The prepared working electrode was soaked in 1 M KOH solution for 2 h to form a Na+ Nafion before electrochemistry was conducted.
Computational models and methods
Vienna Ab initio Simulation Package was used for all density functional theory (DFT) calculations. The projector-augmented plane-wave method was performed. Exchange–correlation functional was treated in the Perdew–Burke–Ernzerhof (PBE). Fully relaxed calculations were simulated with 2 × 2 × 1 supercells. We used a slab cut along the (001) direction of Co(OH)2 as a model for the terminated surface. Au atom was introduced afterward in the model and then was optimized to the lowest energy position. The constructed supercell was isolated with an 18 Å vacuum space in the z-direction. The Brillouin zones of all systems were sampled with gamma-centred grids. The 10 × 10 × 1 gamma k-point setups were used for all structure optimization. The force and energy convergence criteria were set to 0.02 eV Å−1 and 10−5 eV, respectively. The Gibbs free energy calculations considered the zero-point energy (ZPE) in AEM and LOM pathways.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSAF Joint Foundation of China (U1630126, U1230124), the National Natural Science Foundation of China (No. 11604270) and Australian Research Council (DP190103661 and DP220103229).Notes and references
- M. W. Kanan and D. G. Nocera, In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+, Science, 2008, 321(5892), 1072–1075 CrossRef CAS PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction, Science, 2016, 353(6303), 1011–1014 CrossRef CAS PubMed.
- J. H. K. Pfisterer, Y. Liang, O. Schneider and A. S. Bandarenka, Direct instrumental identification of catalytically active surface sites, Nature, 2017, 549(7670), 74–77 CrossRef CAS PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, Synthesis and Activities of Rutile IrO2 and RuO2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions, J. Phys. Chem. Lett., 2012, 3(3), 399–404 CrossRef CAS PubMed.
- D. Zhou, S. Wang, Y. Jia, X. Xiong, H. Yang, S. Liu, J. Tang, J. Zhang, D. Liu, L. Zheng, Y. Kuang, X. Sun and B. Liu, NiFe Hydroxide Lattice Tensile Strain: Enhancement of Adsorption of Oxygenated Intermediates for Efficient Water Oxidation Catalysis, Angew. Chem., Int. Ed., 2019, 58(3), 736–740 CrossRef CAS PubMed.
- Z. Fang, L. Peng, Y. Qian, X. Zhang, Y. Xie, J. J. Cha and G. Yu, Dual Tuning of Ni-Co-A (A = P, Se, O) Nanosheets by Anion Substitution and Holey Engineering for Efficient Hydrogen Evolution, J. Am. Chem. Soc., 2018, 140(15), 5241–5247 CrossRef CAS PubMed.
- B. Zhang, C. Xiao, S. Xie, J. Liang, X. Chen and Y. Tang, Iron–Nickel Nitride Nanostructures in Situ Grown on Surface-Redox-Etching Nickel Foam: Efficient and Ultrasustainable Electrocatalysts for Overall Water Splitting, Chem. Mater., 2016, 28(19), 6934–6941 CrossRef CAS.
- H.-F. Wang, C. Tang, B. Wang, B.-Q. Li and Q. Zhang, Bifunctional Transition Metal Hydroxysulfides: Room-Temperature Sulfurization and Their Applications in Zn-Air Batteries, Adv. Mater., 2017, 29(35), 1702327 CrossRef PubMed.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, CoOOH Nanosheets with High Mass Activity for Water Oxidation, Angew. Chem., Int. Ed., 2015, 54(30), 8722–8727 CrossRef CAS PubMed.
- Y. Sun, S. Gao, F. Lei, J. Liu, L. Liang and Y. Xie, Atomically-thin non-layered cobalt oxide porous sheets for highly efficient oxygen-evolving electrocatalysts, Chem. Sci., 2014, 5(10), 3976 RSC.
- C. Cai, M. Wang, S. Han, Q. Wang, Q. Zhang, Y. Zhu, X. Yang, D. Wu, X. Zu, G. E. Sterbinsky, Z. Feng and M. Gu, Ultrahigh Oxygen Evolution Reaction Activity Achieved Using Ir Single Atoms on Amorphous CoOx Nanosheets, ACS Catal., 2021, 11(1), 123–130 CrossRef CAS.
- Z. Zhuang, W. Sheng and Y. Yan, Synthesis of Monodisperse Au@Co3O4 Core-Shell Nanocrystals and Their Enhanced Catalytic Activity for Oxygen Evolution Reaction, Adv. Mater., 2014, 26(23), 3950–3955 CrossRef CAS PubMed.
- F. Song and X. Hu, Ultrathin cobalt-manganese layered double hydroxide is an efficient oxygen evolution catalyst, J. Am. Chem. Soc., 2014, 136(47), 16481–16484 CrossRef CAS PubMed.
- J. Fester, A. Makoveev, D. Grumelli, R. Gutzler, Z. Sun, J. Rodriguez-Fernandez, K. Kern and J. V. Lauritsen, The Structure of the Cobalt Oxide/Au Catalyst Interface in Electrochemical Water Splitting, Angew. Chem., Int. Ed., 2018, 57(37), 11893–11897 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, Z. Liu, C. Xie, S. Feng, D. Liu, M. Shao and S. Wang, Layered Double Hydroxide Nanosheets with Multiple Vacancies Obtained by Dry Exfoliation as Highly Efficient Oxygen Evolution Electrocatalysts, Angew. Chem., Int. Ed., 2017, 56(21), 5867–5871 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, Enhanced Activity of Gold-Supported Cobalt Oxide for the Electrochemical Evolution of Oxygen, J. Am. Chem. Soc., 2011, 133(14), 5587–5593 CrossRef CAS PubMed.
- B. Sidhureddy, A. R. Thiruppathi and A. Chen, Au nanoparticle incorporated Co(OH)(2) hybrid thin film with high electrocatalytic activity and stability for overall water splitting, J. Electroanal. Chem., 2017, 794, 28–35 CrossRef CAS.
- M. Abu Sayeed, T. Herd and A. P. O'Mullane, Direct electrochemical formation of nanostructured amorphous Co(OH)(2) on gold electrodes with enhanced activity for the oxygen evolution reaction, J. Mater. Chem. A, 2016, 4(3), 991–999 RSC.
- R. Frydendal, M. Busch, N. B. Halck, E. A. Paoli, P. Krtil, I. Chorkendorff and J. Rossmeisl, Enhancing Activity for the Oxygen Evolution Reaction: The Beneficial Interaction of Gold with Manganese and Cobalt Oxides, Chemcatchem, 2015, 7(1), 149–154 CrossRef CAS.
- A. Moysiadou, S. Lee, C.-S. Hsu, H. M. Chen and X. Hu, Mechanism of Oxygen Evolution Catalyzed by Cobalt Oxyhydroxides: Cobalt Superoxide Species as a Key Intermediate and Dioxygen Release as a Rate-Determining Step, J. Am. Chem. Soc., 2020, 142(27), 11901–11914 CrossRef CAS PubMed.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel, Nature, 2016, 529(7584), 68–71 CrossRef CAS PubMed.
- C. Cai, S. Han, W. Caiyang, R. Zhong, Y. Tang, M. J. Lawrence, Q. Wang, L. Huang, Y. Liang and M. Gu, Tracing the Origin of Visible Light Enhanced Oxygen Evolution Reaction, Adv. Mater. Interfaces, 2018, 1801543 Search PubMed.
- C. Cai, Y. Mi, S. Han, Q. Wang, W. Liu, X. Wu, Z. Zheng, X. Xia, L. Qiao, W. Zhou and X. Zu, Engineering ordered dendrite-like nickel selenide as electrocatalyst, Electrochim. Acta, 2019, 295, 92–98 CrossRef CAS.
- I. H. Kwak, H. S. Im, D. M. Jang, Y. W. Kim, K. Park, Y. R. Lim, E. H. Cha and J. Park, CoSe(2) and NiSe(2) Nanocrystals as Superior Bifunctional Catalysts for Electrochemical and Photoelectrochemical Water Splitting, ACS Appl. Mater. Interfaces, 2016, 8(8), 5327–5334 CrossRef CAS PubMed.
- X. Zhao, P. Gao, Y. Yan, X. Li, Y. Xing, H. Li, Z. Peng, J. Yang and J. Zeng, Gold atom-decorated CoSe2 nanobelts with engineered active sites for enhanced oxygen evolution, J. Mater. Chem. A, 2017, 5(38), 20202–20207 RSC.
- J. Wang, C. Cai, Y. Wang, X. Yang, D. Wu, Y. Zhu, M. Li, M. Gu and M. Shao, Electrocatalytic Reduction of Nitrate to Ammonia on Low-Cost Ultrathin CoOx Nanosheets, ACS Catal., 2021, 11(24), 15135–15140 CrossRef CAS.
- C. Cai, K. Liu, Y. Zhu, P. Li, Q. Wang, B. Liu, S. Chen, H. Li, L. Zhu, H. Li, J. Fu, Y. Chen, E. Pensa, J. Hu, Y.-R. Lu, T.-S. Chan, E. Cortes and M. Liu, Optimizing Hydrogen Binding on Ru Sites with RuCo Alloy Nanosheets for Efficient Alkaline Hydrogen Evolution, Angew. Chem., Int. Ed., 2021, e202113664 Search PubMed.
- H. Zhou, F. Yu, J. Sun, R. He, S. Chen, C. W. Chu and Z. Ren, Highly active catalyst derived from a 3D foam of Fe(PO3)2/Ni2P for extremely efficient water oxidation, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(22), 5607–5611 CrossRef CAS PubMed.
- J. Zhang, Q. Zhang and X. Feng, Support and Interface Effects in Water-Splitting Electrocatalysts, Adv. Mater., 2019, e1808167 CrossRef PubMed.
- J. Bao, X. D. Zhang, B. Fan, J. J. Zhang, M. Zhou, W. L. Yang, X. Hu, H. Wang, B. C. Pan and Y. Xie, Ultrathin Spinel-Structured Nanosheets Rich in Oxygen Deficiencies for Enhanced Electrocatalytic Water Oxidation, Angew. Chem., Int. Ed., 2015, 54(25), 7399–7404 CrossRef CAS PubMed.
- Y. Liu, Z. Jin, P. Li, X. Tian, X. Chen and D. Xiao, Boron- and Iron-Incorporated α-Co(OH)2 Ultrathin Nanosheets as an Efficient Oxygen Evolution Catalyst, ChemElectroChem, 2018, 5(4), 593–597 CrossRef CAS.
- H. Zhang, J. Zhang, Y. Li, H. Jiang, H. Jiang and C. Li, Continuous oxygen vacancy engineering of the Co3O4 layer for an enhanced alkaline electrocatalytic hydrogen evolution reaction, J. Mater. Chem. A, 2019, 13506 RSC.
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Ultrathin Iron-Cobalt Oxide Nanosheets with Abundant Oxygen Vacancies for the Oxygen Evolution Reaction, Adv. Mater., 2017, 29(17), 1606793 CrossRef PubMed.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, Single-Atom Au/NiFe Layered Double Hydroxide Electrocatalyst: Probing the Origin of Activity for Oxygen Evolution Reaction, J. Am. Chem. Soc., 2018, 140(11), 3876–3879 CrossRef CAS PubMed.
- Y. Y. Liang, H. L. Wang, J. G. Zhou, Y. G. Li, J. Wang, T. Regier and H. J. Dai, Covalent Hybrid of Spinel Manganese-Cobalt Oxide and Graphene as Advanced Oxygen Reduction Electrocatalysts, J. Am. Chem. Soc., 2012, 134(7), 3517–3523 CrossRef CAS PubMed.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts, Nat. Energy, 2019, 4(4), 329–338 CrossRef CAS.
- M. Shariq, P. Majeric, B. Friedrich, B. Budic, D. Jenko, A. R. Dixit and R. Rudolf, Application of Gold(III) Acetate as a New Precursor for the Synthesis of Gold Nanoparticles in PEG Through Ultrasonic Spray Pyrolysis, J. Cluster Sci., 2017, 28(3), 1647–1665 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra09094a |
| This journal is © The Royal Society of Chemistry 2022 |