
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) P bond in phosphanylphosphaalkenes (CP–P bond system) in the reaction with nucleophilic reagents: MeLi, nBuLi and tBuLi†
P bond in phosphanylphosphaalkenes (CP–P bond system) in the reaction with nucleophilic reagents: MeLi, nBuLi and tBuLi†
Aleksandra Ziółkowska ,
Natalia Szynkiewicz and
Łukasz Ponikiewski*
,
Natalia Szynkiewicz and
Łukasz Ponikiewski*
Department of Inorganic Chemistry, Faculty of Chemistry, University of Technology, Gabriela Narutowicza Str. 11/12, 80-233 Gdansk, Poland. E-mail: lukasz.ponikiewski@pg.edu.pl
First published on 7th April 2022
Abstract
Three reactions of phosphanylphosphaalkene (1) with nucleophiles were performed to activate the diphosphorus monomer. We observed similar results in the reactions with MeLi and nBuLi, in which the P–P bond is cleavaged and triphosphorus systems [P(Me)2–CH(biph)–CH(biph)–P–(PtBu2)]− (1a′′) and [P(nBu)2–CH(biph)–CH(biph)–P–(PtBu2)]− (1b′′), respectively, are formed depending on the nucleophilic reagent (biph = biphenyl). In the case of tBuLi, the P–P bond remains intact; on the phosphorus atom, only one -tBu group is substituted, and as a result, [(biph)(H)C–P(tBu)–PtBu2]− (1c) is generated as a stable carbanion. We additionally investigated the effect of substitution in the phenyl ring in the course of these reactions by involving two other phosphanylphosphaalkenes (3 and 4). All initial reactions were conducted in tetrahydrofuran (THF) solution at ambient temperature.
Introduction
Compounds containing a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P bond, namely, phosphaalkenes, are well investigated in organophosphorus chemistry. The various methods of synthesis of these systems are widely described.1–4 However, not only the synthesis is of interest to many scientists, but also the use of their properties which are due to the presence of a reactive PC bond. One important area is the synthesis and engagement of phosphaalkene complexes in catalysis.5 The most studied metal complexes originate from nickel and copper groups. Alcohol allylation, cyclodehydration, hydroamination, hydroamidation and dehydrogenative silylation of ketones are some of the examples where platinum or palladium complexes supported with PC ligands are employed as catalysts.6–10 Copper complexes exhibit activity in CO2 hydrogenation and hydrosilylation, while gold-containing phosphaalkene complex was used in alkoxycyclization of 1,6-enyne intramolecular cyclizations to afford heterocyclic structures or in hydration reactions of terminal acetylenes.11,12 Phosphaalkene species are used in many organic syntheses, inter alia, in Diels–Alder reaction with dienes or via dimerization for e.g. 1-phosphabutadienes.13 Cycloaddition of phosphaalkenes to an arene ring (N-bound), the effect of which was bicyclic product may also by consider as specific application. Additionally, the authors demonstrated the reversibility of the process and the establishing equilibrium between the phosphaalkene and the phosphabicyclic product.14 A significant aspect is research on phosphaalkene polymerization and the importance of phosphorus polymers. In the literature, anion-, cation- and radical-initiated processes have been reported.15–19 In the first type of reaction, MeLi or nBuLi was most often used, while in the second type, triflic acid or Lewis acids (MCl3: M = Al, Ga, In) was mainly employed. In the third case, the most frequent choice was VAZO or TEMPO. The main starting monomer thus far has included MesPCPh2 compound and m-XylPCPh2 in a lesser extended, due to their moderate stability.20–23 Although, it should be emphasized that Mes*PCH2 was employed as monomer in cationic polymerization and AlCl3 was used as the initiator. However this led to intramolecular C–H activation of the Mes* substituent.19 Gates and coworkers demonstrated anionic polymerization of MesPC(Fc)Ph, which is a C-ferrocenyl-substituted phosphaalkene.24 However, no phosphaalkene with an aldehyde derivative was used for this purpose. In the following years, studies reported the polymerization of phosphaalkenes with the use of other substituents on carbon (C-aryl), including 1-naphthyl and 9-phenanthryl moieties.25 Another example is phosphaalkene bearing a C-pyrenyl substituent, MesPC(Ph)Pyr, which is subjected to anionic polymerization.26 Changing the substituent on the phosphorus atom with a phosphanyl group leads to the formation of phosphanylphosphaalkene (CP–P).27–32 These diphosphorus analogs are a considerable and lesser known group of compounds, and their reactivity is practically unknown. Our main goal was to perform the first step of anionic polymerization in solution (tetrahydrofuran, THF) at room temperature; for this purpose, we chose (biph)(H)CP–PtBu2 (1), (3,5-tBu2C6H3)(H)CP–PtBu2 (3) and (p-Me2N–C6H4)(H)CP–PtBu2 (4) containing an aldehyde derivative. We conducted reactions with three initiators (MeLi, nBuLi, tBuLi) and presented the results of our research.
P bond, namely, phosphaalkenes, are well investigated in organophosphorus chemistry. The various methods of synthesis of these systems are widely described.1–4 However, not only the synthesis is of interest to many scientists, but also the use of their properties which are due to the presence of a reactive PC bond. One important area is the synthesis and engagement of phosphaalkene complexes in catalysis.5 The most studied metal complexes originate from nickel and copper groups. Alcohol allylation, cyclodehydration, hydroamination, hydroamidation and dehydrogenative silylation of ketones are some of the examples where platinum or palladium complexes supported with PC ligands are employed as catalysts.6–10 Copper complexes exhibit activity in CO2 hydrogenation and hydrosilylation, while gold-containing phosphaalkene complex was used in alkoxycyclization of 1,6-enyne intramolecular cyclizations to afford heterocyclic structures or in hydration reactions of terminal acetylenes.11,12 Phosphaalkene species are used in many organic syntheses, inter alia, in Diels–Alder reaction with dienes or via dimerization for e.g. 1-phosphabutadienes.13 Cycloaddition of phosphaalkenes to an arene ring (N-bound), the effect of which was bicyclic product may also by consider as specific application. Additionally, the authors demonstrated the reversibility of the process and the establishing equilibrium between the phosphaalkene and the phosphabicyclic product.14 A significant aspect is research on phosphaalkene polymerization and the importance of phosphorus polymers. In the literature, anion-, cation- and radical-initiated processes have been reported.15–19 In the first type of reaction, MeLi or nBuLi was most often used, while in the second type, triflic acid or Lewis acids (MCl3: M = Al, Ga, In) was mainly employed. In the third case, the most frequent choice was VAZO or TEMPO. The main starting monomer thus far has included MesPCPh2 compound and m-XylPCPh2 in a lesser extended, due to their moderate stability.20–23 Although, it should be emphasized that Mes*PCH2 was employed as monomer in cationic polymerization and AlCl3 was used as the initiator. However this led to intramolecular C–H activation of the Mes* substituent.19 Gates and coworkers demonstrated anionic polymerization of MesPC(Fc)Ph, which is a C-ferrocenyl-substituted phosphaalkene.24 However, no phosphaalkene with an aldehyde derivative was used for this purpose. In the following years, studies reported the polymerization of phosphaalkenes with the use of other substituents on carbon (C-aryl), including 1-naphthyl and 9-phenanthryl moieties.25 Another example is phosphaalkene bearing a C-pyrenyl substituent, MesPC(Ph)Pyr, which is subjected to anionic polymerization.26 Changing the substituent on the phosphorus atom with a phosphanyl group leads to the formation of phosphanylphosphaalkene (CP–P).27–32 These diphosphorus analogs are a considerable and lesser known group of compounds, and their reactivity is practically unknown. Our main goal was to perform the first step of anionic polymerization in solution (tetrahydrofuran, THF) at room temperature; for this purpose, we chose (biph)(H)CP–PtBu2 (1), (3,5-tBu2C6H3)(H)CP–PtBu2 (3) and (p-Me2N–C6H4)(H)CP–PtBu2 (4) containing an aldehyde derivative. We conducted reactions with three initiators (MeLi, nBuLi, tBuLi) and presented the results of our research.
Results and discussion
Initially, we mixed MeLi with (biph)(H)CP-PtBu2 in THF-d8. The 31P{1H} nuclear magnetic resonance (NMR) spectrum obtained 24 hours later revealed that the signals of phosphanylphosphaalkene disappeared, whereas new signals were visible: 54.20 ppm (d, JPP = 325.2 Hz), −48.52 ppm (d, JPP = 108.4 Hz), and −56.33 ppm (dd, JPP = 325.2 Hz, JPP = 108.4 Hz) for one of the new compounds, 47.12 ppm and −51.41 ppm (d, JPP = 313.2 Hz) for another new compound and an additional two singlets at 40.28 ppm and −62.31 ppm (see ESI, Fig. S4 and S5†). The 31P{1H}/1H HMQC NMR spectrum shows that the phosphorus atom observed at 47.12 ppm correlates with protons at 1.21 ppm (tBu groups) and with protons at 2.91 ppm (weak correlation), while the phosphorus atom observed at −51.41 ppm indicates a strong correlation with protons at 2.91 ppm and a very weak correlation with protons observed at 1.03 ppm (see ESI, Fig. S11 and S12†). The integration revealed that at 2.91 ppm 1H, at 1.21 ppm 18H and at 1.03 ppm 3H protons are located. The obtained result shows that in the reaction, the anion [(biph)(H)C–P(Me)–PtBu2]− (1a) is formed, and the PC bond is activated. Similar CP bond activation by using nucleophilic reagents was described previously in the literature as the initiation step of the anionic polymerization of phosphaalkenes. Gates and coworkers presented the reactions of (Ph)2CPMes with MeLi and nBuLi, where the ionic compounds (Ph)2C–P(Mes)(Me) and (Ph)2C–P(Mes)(nBu), respectively, were generated.20–22 The other signals in the 31P{1H} NMR spectrum (especially singlets at 40.28 ppm and −62.31 ppm) may indicate that in the reaction of (biph)(H)CP–PtBu2 with MeLi, the P–P bond is not stable, and monophosphorus compounds are formed. In the 31P{1H}/1H HMBC spectrum, the phosphorus atom observed at 40.28 ppm correlates only with the doublet at 1.33 ppm (JPH = 10.1 Hz) from tBu groups (see ESI, Fig. S11†). The obtained result may suggest the formation of tBu2PLi, which confirms the cleavage of the P–P bond. For comparison, the signals of tBu2PLi (s) found in the literature lie in the range from 38.51 ppm (Li4 unit)33 to 46.6 ppm.34 Another compound observed in the 31P{1H} NMR spectrum is a singlet (−62.31 ppm) correlated with protons at 2.61 ppm (JPH = 5.4 Hz), 0.98 ppm (JPH = 2.4 Hz) and 0.89 ppm (JPH = 2.9 Hz) (see ESI, Fig. S11†). The obtained results reveal further reaction of 1a with MeLi remaining in the reaction solution and attachment of the second methyl group to the phosphorus atom. Substitution of the second methyl group leads to P–P bond cleavage and the formation of ionic compound with the formula [Li(solvent)n]+[(biph)(H)C–P(Me2)]− (1a′) and tBu2PLi (integration for compound 1a′ is shown in ESI Fig. S10†). All reactions of (biph)(H)CP–PtBu2 with RLi (R = Me, nBu, tBu) observed in the reaction mixture are presented in Scheme 1.
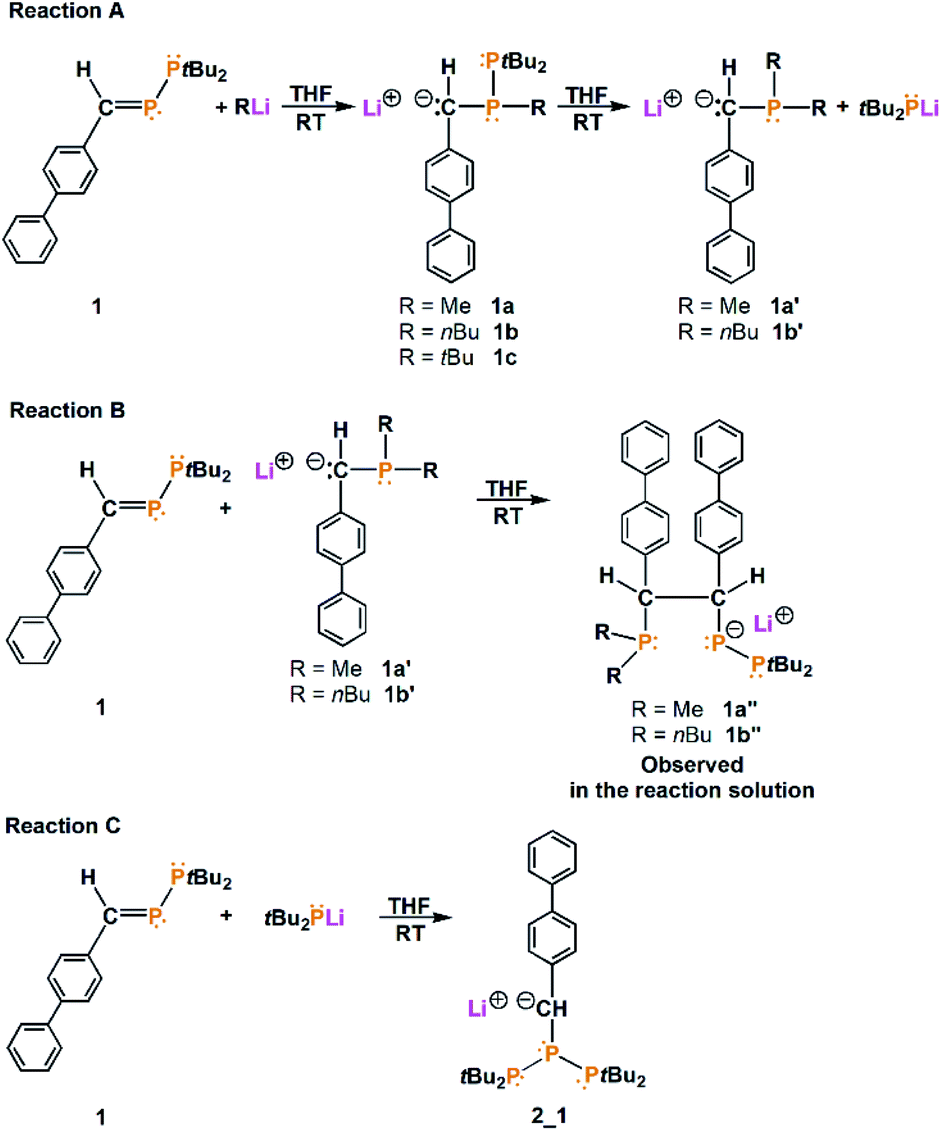 | ||
| Scheme 1 Reactions of phosphanylphosphaalkene (1) with RLi in THF solution (R = Me, nBu, tBu). | ||
Furthermore, the signals observed at 54.20 ppm (d, JPP = 325.2 Hz), −48.52 ppm (d, JPP = 108.4 Hz), and −56.33 ppm (dd, JPP = 325.2 Hz, JPP = 108.4 Hz) may suggest the formation of compounds with three different phosphorus atoms in solution. Repeating this reaction in THF and crystallization from dimethoxyethane (DME) allowed us to obtain good-quality crystals for X-ray measurements. Interestingly, we isolated two types of crystals from this reaction solution: deep red blocks and bright red needles. X-ray analysis of the red block crystals revealed an ionic compound with the formula [Li(DME)3]+[{P(Me)2–CH(biph)–CH(biph)–P–(PtBu2)}2Li]− (1a*). 1a* crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules in the unit cell. The independent part of the unit cell contains a total of 1a*; therefore, four stereocenters (C9, C22, C45, C58) can be distinguished. In one organic ligand, the carbon stereocenter atoms have the same configurations, C9, C22 – R, R and C45, C58 – S, S. The molecular structure of the anion of 1a* is presented in Fig. 1.
with two molecules in the unit cell. The independent part of the unit cell contains a total of 1a*; therefore, four stereocenters (C9, C22, C45, C58) can be distinguished. In one organic ligand, the carbon stereocenter atoms have the same configurations, C9, C22 – R, R and C45, C58 – S, S. The molecular structure of the anion of 1a* is presented in Fig. 1.
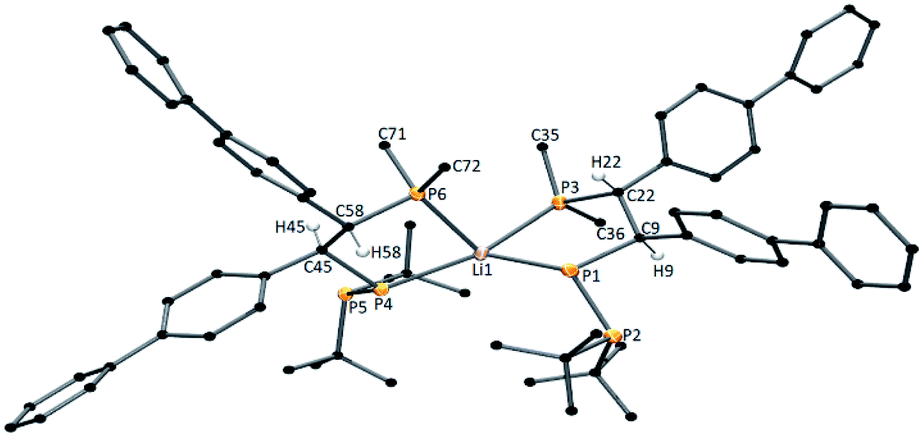 | ||
| Fig. 1 Molecular structure of the anion of 1a* (ellipsoids 30%, the H atoms have been omitted for clarity, except H9, H22, H45 and H58). Important distances (Å) and angles (deg): Li1–P1 2.479(7), Li1–P3 2.556(6), Li1–P4 2.517(6), Li1–P6 2.531(7), P1–P2 2.1585(15), P4–P5 2.1685(15), C9–C22 1.531(5), C45–C58 1.548(5). | ||
The X-ray structure of the anionic part of 1a* indicates a four-coordinate lithium compound between tetrahedral and square-planar geometry at the metal center (τ4 = 0.57(3), τ′4 = 0.54(6)).35,36 The P–P interatomic distances in 1a* differ slightly from one another (2.1585(15) Å and 2.1685(15) Å) and lie slightly below the lower limit of a single P–P bond (the range given by Corbridge for a P–P single bond is 2.17–2.24 Å).37 The geometries around P1, P2, P4 and P5 in 1a* are more pyramidal (ΣP1 = 338.31(12)°, ΣP2 = 316.03(16)°, ΣP4 = 325.65(12)° and ΣP5 = 315.81(11)°) and finally confirmed the single bond character between two phosphorus atoms (P1–P2 and P4–P5). The Li–P distances in 1a* are also differ from each other (2.479(7) Å, 2.556(6) Å, 2.517(6) Å, 2.531(1) Å). Similar Li–P distances are found in the following: [(Dipp)2P]Li(THF)3, 2.482(3) Å;38 iPrP{C6H4-2-CH(C6H4-2-CH2NMe2)NMe2}Li(THF)2, 2.509(6) Å;39 tBu(Ph)P–P(SiMe3)Li(THF)3, 2.518(17) Å;40 and [(Et2O)2Li{(Ph2P)2CH}], 2.581(4) Å and 2.583(4) Å.41 The 31P{1H} NMR spectrum conducted from the 1a* crystals revealed signals from six different phosphorus atoms: 29.07 ppm (JPP = 247.7 Hz, JPP = 17.2 Hz), −44.57 ppm (JPP = 73.4 Hz, JPP = 17.2 Hz), −67.56 ppm (JPP = 247.7 Hz, JPP = 73.4 Hz), 29.45 ppm (JPP = 239.3 Hz), −44.92 ppm (JPP = 36.5 Hz) −82.62 ppm (JPP = 239.3 Hz, JPP = 36.5 Hz) (see ESI, Fig. S13 and S14†). The Mulliken population analysis calculated for 1a* revealed different partial atomic charges on the phosphorus atoms: [{P(2.225)(Me)2–CH(biph)-CH(biph)–P(−0.583)–(P(−0.414)tBu2)}2]− and second ligand [{P(−0.025)(Me)2–CH(biph)–CH(biph)–P(−0.030)–(P(−0.255)tBu2)}2]. It shows that electron density on corresponding P atoms is not distributed equally after complexation (see ESI, Fig. S57 and S58†). Despite the seemingly analogous chemical environment, derived results indicate different coordination of the organophosphorus ligand to the lithium cation. Moreover it justifies presence of separate signal for each P atom in the 31P{1H} NMR spectrum of dissolved compound 1a*.
The second type of crystals isolated in the reaction as bright red needles were characterized as a symmetrical triphosphane with the formula [Li(DME)3]+[tBu2P–P–{C(H)–(biph)}–PtBu2]− (2_1) (the molecular structure of 2_1 is shown in ESI Fig. S2†). Furthermore, the reaction conducted at high concentrations of adducts led only to a mixture of 1a, 1a′ and 1a′′, while compound 2_1 was observed only at lower concentrations of adducts. The collected observations allowed us to eliminate compound 2_1 from the reaction by selecting the appropriate concentration of the reagents.
The reaction of (biph)(H)CP–PtBu2 with nBuLi was also conducted in THF-d8 and investigated by NMR technique. The 31P{1H} NMR spectrum revealed the formation of analogous compounds, as in the reaction with MeLi: [{(biph)(H)C–P(nBu)–PtBu2}]− (1b, d, 39.22 ppm, −37.43 ppm, JPP = 326.3 Hz), [{(biph)(H)C–P(nBu)2}]− (1b′, s, −41.56 ppm) and [{P(nBu)2–CH(biph)–CH(biph)–P–(PtBu2)}2]− (1b′′, d, 54.91 ppm, JPP = 326.0 Hz; d, −24.16 ppm, JPP = 102.4 Hz and dd, −59.24 ppm, JPP = 102.4 Hz, JPP = 326.0 Hz) (all NMR spectra of the reaction mixture are shown in ESI Fig. S25–S39†). To isolate compound 1b′′, the reaction was repeated in nondeuterated THF, and then, crystallization was conducted from organic solvents (DME, THF and toluene). Unfortunately, all attempts to isolate 1b′′ failed, and we isolated only a small amount of 2_1.
Because the reactions of (biph)(H)CP–PtBu2 with MeLi and nBuLi lead to substitution of two alkyl groups on the phosphorus atom and, in turn, P–P bond cleavage, we decided to examine another nucleophilic reagent – tBuLi. The reaction of (biph)(H)CP–PtBu2 with tBuLi was investigated by NMR spectroscopy and revealed the formation of only one compound. The 31P{1H} NMR spectrum obtained from the reaction mixture shows only two doublets at 30.08 ppm and −4.05 ppm, with a coupling constant of 333.3 Hz. The 31P{1H}/1H HMBC NMR spectrum revealed a correlation of the phosphorus atom observed at 30.08 ppm with only protons at 1.31 ppm (two doublets, JPH = 13.2 Hz and 13.7 Hz) from the tBu groups. Interestingly, the signal at −4.05 ppm correlates with a doublet of doublets at 3.15 ppm (JPH = 7.6 Hz, JPH = 1.3 Hz) and with a doublet at 1.14 ppm (JPH = 12.7 Hz) (all NMR spectra of the reaction mixture are shown in ESI Fig. S30–S32†). The obtained results evidently point out substitution of the phosphorus atom with only one tBu group and formation of a new carbanion-containing compound with the formula [(biph)(H)C–P(tBu)–PtBu2]− (1c). Stopping the reaction at this stage is probably associated with steric hindrance of the tBu groups. Despite all efforts, it was not possible to isolate the compound 1c in a crystalline form.
In order to investigate the influence of substituents on the phenyl ring, we synthesized a new phosphanylphosphaalkene with the formula (3,5-tBu2C6H3)(H)CP–PtBu2 (3) and we used previously synthesized phosphanylphosphaalkene (p-Me2N–C6H4)(H)CP–PtBu2 (4) additionally. In comparison to compound 1, in which the phenyl group was substituted with phenyl in the para position, compounds 3 and 4 constitute respectively examples where the phenyl is substituted with tBu groups (meta positions) and NMe2 (para position).
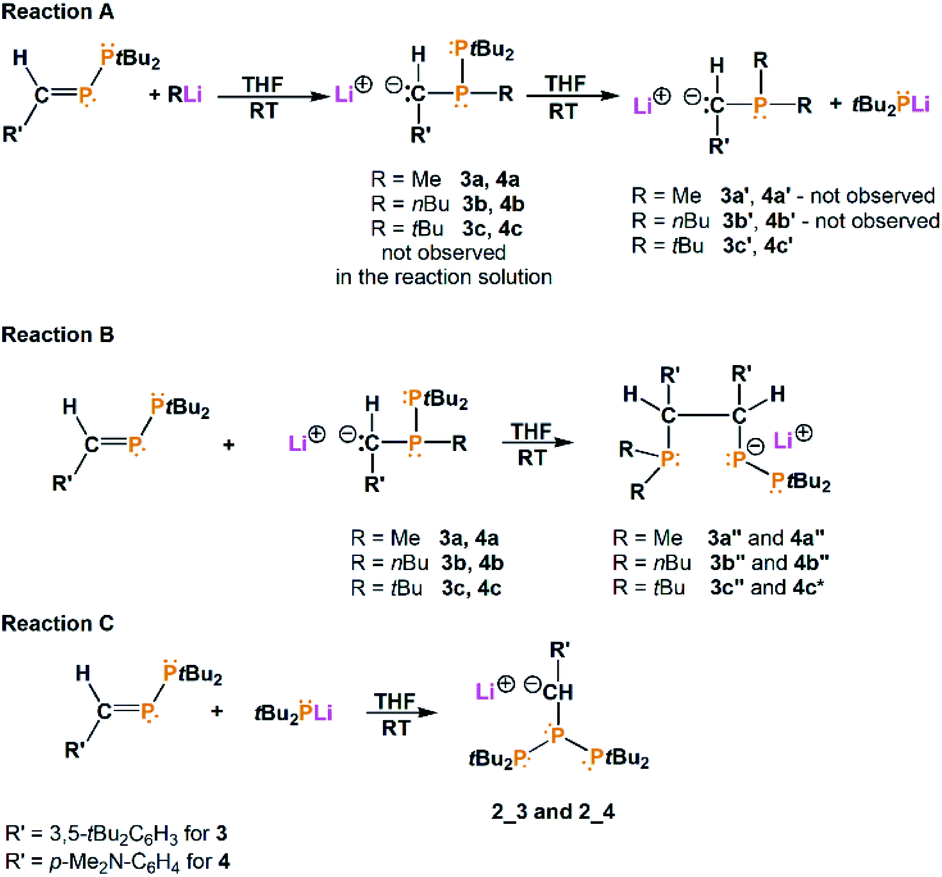 | ||
| Scheme 2 Reactions of phosphanylphosphaalkenes (3,5-tBu2C6H3)(H)CP–PtBu2 (3) and (p-Me2N–C6H4)(H)CP–PtBu2 (4) with RLi in THF solution (R = Me, nBu, tBu). | ||
All reactions were performed in THF-d8 and stirred for 24 hours before NMR measurements. The obtained results show that independently of the substituents in the phenyl ring and used lithium compounds, these reactions do not stop at the mono-substitution stage (compounds 3a–c and 4a–c were not observed in the reaction solutions) (Scheme 2). 31P{1H} NMR spectra of the reaction solution revealed that in each of these reactions appropriate triphosphane is formed: [tBu2P–P–{C(H)(3,5-tBu2C6H3)}–PtBu2]− (2_3, 66.12 ppm, JPP = 347.5 Hz, JPP = 62.5 Hz; 57.98 ppm, JPP = 231.7 Hz, JPP = 62.5 Hz; −67.30 ppm, JPP = 347.5 Hz, JPP = 231.7 Hz) and [tBu2P–P–{C(H)(p-Me2N–C6H4)}–PtBu2]− (2_4, 64.62 ppm, JPP = 348.2 Hz, JPP = 62.6 Hz; 56.85 ppm, JPP = 239.6 Hz, JPP = 62.6 Hz; −69.64 ppm, JPP = 348.2 Hz, JPP = 239.6 Hz). Additionally, the 31P{1H} NMR spectra show the compounds formation with a new C–C bond (3a′′–3c′′ and 4a′′–4c′′) and with three different phosphorus atoms, analogical to 1a′′ and 1b′′ compounds (see ESI Fig. S34–S47†). Importantly, the 4c′′ compound occurs in the reaction solution as a system of two three-phosphorus molecules coordinated to the lithium ion (31P{1H} NMR: 51.56 ppm, dd, JPP = 326.9 Hz, JPP = 5.3 Hz; 49.51 ppm, dd, JPP = 281.7 Hz, JPP = 51.5 Hz; 41.07 ppm, dd, JPP = 139.5 Hz, JPP = 5.3 Hz; 3.81 ppm, dd, JPP = 111.3 Hz, JPP = 51.5 Hz; −55.98 ppm, dd, JPP = 281.7 Hz, JPP = 111.3 Hz; −57.56 ppm, dd, JPP = 326.9 Hz, JPP = 139.5 Hz, the spectra see ESI, Fig. S44–S47†). For comparison, an analogous compound 1a* was obtained in the reaction of 1 with MeLi, however, was not observed in the reaction solution, but it revealed in the crystallization process.
Experimental
Materials and methods
All synthetic procedures were performed under inert gas (Ar) and were carried out using mBraun glovebox and standard Schlenk techniques. THF, DME, toluene and pentane were dried over Na/Ph2CO and distilled under argon atmosphere. All spectra in solution were recorded on Bruker AV400 MHz spectrometer (external standard tetramethylsilane for 1H, 13C; 85% H3PO4 for 31P). Elemental analysis for solid and liquid samples were recorded on Elementary Vario El Cube CHNS. (biph)(H)CP–PtBu2 and (p-Me2N–C6H4)(H)CP–PtBu2 were prepared according to procedure in the literature.32 MeLi, nBuLi and tBuLi were commercially purchased.
Synthesis of (3,5-tBu2C6H3)(H)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) P–PtBu2 (3)
P–PtBu2 (3)
To a solution of tBu2PP(SiMe3)Li·3THF (0.500 g; 1.058 mmol) in 8 mL of toluene, a solution of 3,5-di-tert-butylbenzaldehyde (0.231 g; 1.058 mmol) in 5 mL of toluene was added. Almost immediately solution turned yellow-orange. After 1 hour the solvent was evaporated, and the solid residue was treated with 20 mL of pentane. The mixture was filtered and concentrated more than half its volume. The solution was stored at +4 °C for 24 hours. The desired compound (3,5-tBu2C6H3)(H)CP–PtBu2 was obtained in the form of yellow crystals (0.320 g; 80%). Anal. calcd for C23H40P2: C, 72.98; H, 10.65%. Found: C, 72.89; H, 10.72%.
Reactions of (biph)(H)CP–PtBu2 with MeLi, nBuLi, tBuLi respectively
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) P–PtBu2 with MeLi. To a solution of (biph)(H)CP–PtBu2 (0.200 g; 0.584 mmol) in 4–5 mL of THF a MeLi (0.365 mL; 0.584 mmol; 1.6 M) was dropwise added. The color immediately changed from orange to dark red. After 24 hours of stirred, the solvent was evaporated, and 10 mL of DME was added. The solution was slowly concentrated to about half then allowed to crystallize at −23 °C. After 5–6 hours, the red block appeared which were characterized as [Li(DME)3]+[{P(Me)2–CH(biph)–CH(biph)–P–(PtBu2)}c2Li]− (1a*) (M = 1423.48 g mol−1; 0.105 g, 13%). Anal. calcd for C84H118Li2O6P6: C, 70.87; H, 8.36%. Found: C, 71.03; H, 8.45%. The crystals were isolated and concentrated one half more, and the solution was left at −23 °C again to afforded bright red needles, X-ray-quality crystals of [Li(DME)3]+[tBu2P–P–{C(H)(biph)}–PtBu2]− (2_1) (M = 764.86 g mol−1, 0.045 g, 10%). Anal. calcd for C41H76Li1O6P3: C, 64.38; H, 10.01%. Found: C, 64.25; H, 10.15%.
P–PtBu2 with MeLi. To a solution of (biph)(H)CP–PtBu2 (0.200 g; 0.584 mmol) in 4–5 mL of THF a MeLi (0.365 mL; 0.584 mmol; 1.6 M) was dropwise added. The color immediately changed from orange to dark red. After 24 hours of stirred, the solvent was evaporated, and 10 mL of DME was added. The solution was slowly concentrated to about half then allowed to crystallize at −23 °C. After 5–6 hours, the red block appeared which were characterized as [Li(DME)3]+[{P(Me)2–CH(biph)–CH(biph)–P–(PtBu2)}c2Li]− (1a*) (M = 1423.48 g mol−1; 0.105 g, 13%). Anal. calcd for C84H118Li2O6P6: C, 70.87; H, 8.36%. Found: C, 71.03; H, 8.45%. The crystals were isolated and concentrated one half more, and the solution was left at −23 °C again to afforded bright red needles, X-ray-quality crystals of [Li(DME)3]+[tBu2P–P–{C(H)(biph)}–PtBu2]− (2_1) (M = 764.86 g mol−1, 0.045 g, 10%). Anal. calcd for C41H76Li1O6P3: C, 64.38; H, 10.01%. Found: C, 64.25; H, 10.15%.
NMR data of isolated 1a* in THF-d8. 1H NMR (400 MHz, C6D6, 298 K) δ: 7.5–6.98 (aromatic protons), 3.85 (m, 1H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 3.67 (m, 1H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 3.61 (small amount of THF from THF-d8), 3.46 (DME protons), 3.44 (1H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 3.41 (m, 1H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 3.30 (DME protons), 1.76 (small amount of THF from THF-d8), 1.40 (d, JPH = 11.2 Hz, 9H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 1.39 (d, JPH = 11.0 Hz, 9H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 1.17 (d, JPH = 11.2 Hz, 9H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 1.11 (d, JPH = 4.5 Hz, 3H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 1.01 (d, JPH = 11.6 Hz, 9H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 0.73 (d, JPH = 3.9 Hz, 3H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 0.73 (d, JPH = 4.3 Hz, 3H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 0.66 (d, JPH = 4.0 Hz, 3H, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−) ppm.
13C{1H} NMR (100.6 MHz, C6D6, 298 K) δ: 142.04–125.43 (aromatic C atoms), 71.79 (DME), 66.42 (THF-d8), 57.93 (DME), 52.79 (d, JPC = 19.4 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 52.72 (d, JPC = 17.8 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 44.67 (d, JPC = 16.2 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 42.87 (d, JPC = 18.7 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 31.54 (dd, JPC = 28.8 Hz, JPC = 1.7 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(P{C(CH3)3}2)]−), 31.51 (dd, JPC = 28.7 Hz, JPC = 3.7 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(P{C(CH3)3}2)]−), 30.61 (dd, JPC = 15.0 Hz, JPC = 1.6 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(P{C(CH3)3}2)]−), 30.52 (dd, JPC = 14.0 Hz, JPC = 0.8 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(P{C(CH3)3}2)]−), 30.10 (dd, JPC = 13.1 Hz, JPC = 8.6 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(P{C(CH3)3}2)]−), 30.08 (dd, JPC = 14.3 Hz, JPC = 1.2 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(P{C(CH3)3}2)]−), 24.34 (THF-d8), 13.42 (d, JPC = 17.5 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 13.03 (d, JPC = 15.5 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 10.45 (d, JPC = 18.7 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−), 9.92 (d, JPC = 19.5 Hz, [Li(DME)3]+[P(Me)2–CH(biph)–CH(biph)–P(PtBu2)]−) ppm.
31P{1H} NMR (162 MHz, C6D6, 298 K) δ: 29.45 (d, JPP = 239.3 Hz, [Li(DME)3]+[P(Me2)–CH(biph)–CH(biph)–P(PtBu2)]−), 29.07 (dd, JPP = 245.7 Hz, JPP = 17.2 Hz, [Li(DME)3]+[P(Me2)–CH(biph)–CH(biph)–P(PtBu2)]−), −44.57 (dd, JPP = 73.4 Hz, JPP = 17.2 Hz, [Li(DME)3]+[P(Me2)–CH(biph)–CH(biph)–P(PtBu2)]−), −44.92 (d, JPP = 36.5 Hz, [Li(DME)3]+[P(Me2)–CH(biph)–CH(biph)–P(PtBu2)]−), −67.56 (dd, JPP = 245.7 Hz, JPP = 73.4 Hz, [Li(DME)3]+[P(Me2)–CH(biph)–CH(biph)–P(PtBu2)]−), −82.62 ppm, (dd, JPP = 239.3 Hz, JPP = 36.5 Hz, [Li(DMe)3]+[P(Me2)–CH(biph)–CH(biph)–P(PtBu2)]−) ppm.
NMR data of isolated 2_1 in THF-d8. 1H NMR (400 MHz, C6D6, 298 K) δ: 7.68–6.07 (aromatic protons), 3.46 (s, DME protons), 3.30 (s, DME protons), 3.22 (broad d, JPH = 5.3 Hz, 1H, [Li(DME)3]+[tBu2P–P–{C(H)–Ph–Ph}–PtBu2]−), 1.32 (broad s, 36H, [Li(DME)3]+[tBu2P–P–{C(H)–Ph–Ph}–PtBu2]−) ppm.
13C{1H} NMR (100.6 MHz, C6D6, 298 K) δ: 143.38–112.13 (aromatic C atoms), 71.79 (s, DME), 57.92 (s, DME), 54.80 (dt, JPC = 17.4 Hz, JPC = 3.9 Hz, [Li(DME)3]+[tBu2P–P–{C(H)–Ph–Ph}–PtBu2]−), 31.85 (broad dd, JPC = 15.0 Hz, JPC = 7.0 Hz, [Li(DME)3]+[{(H3C)3C}2P–P–{C(H)–Ph–Ph}–P{C(CH3)3}2]−), 31.24 (dd, JPC = 24.9 Hz, JPC = 2.3 Hz, [Li(DME)3]+[{(H3C)3C}2P–P–{C(H)–Ph–Ph}–P{C(CH3)3}2]−), 31.09 (dd, JPC = 28.2 Hz, JPC = 2.4 Hz, [Li(DME)3]+[{(H3C)3C}2P–P–{C(H)–Ph–Ph}–P{C(CH3)3}2]−) ppm.
31P{1H} NMR (162 MHz, C6D6, 298 K) δ: 40.11 (d, JPP = 285.4 Hz, [Li(DME)3]+[tBu2P–P–{C(H)(biph)}–PtBu2]−), 40.10 (d, JPP = 284.8 Hz, [Li(DME)3]+[tBu2P–P–{C(H)(biph)}–PtBu2]−), −54.71 (dd, JPP = 285.4 Hz, JPP = 284.8 Hz, [Li(DME)3]+[tBu2P–P–{C(H)(biph)}–PtBu2]−) ppm.
P–PtBu2 with nBuLi. To a solution of (biph)(H)CP–PtBu2 (0.08 g; 0.234 mmol) in 1 mL of THF-d8 a nBuLi (0.093 mL; 0.234 mmol; 2.5 M) was dropwise added. The color immediately changed from orange to dark red, and after 24 hours the solution was transferred to a NMR-tube and measurements were performed.1H NMR (400 MHz, C6D6, 298 K) δ: 7.55–6.57 (aromatic protons), 3.80 (broad m, 1H, [P(nBu)2–CH(biph)–CH(biph)–P–(PtBu2)]−, 1b′′), 3.52 (dd, JPH = 11.5 Hz, JPH = 3.7 Hz, 1H, [P(nBu)2–CH(biph)–CH(biph)–P–(PtBu2)]−, 1b′′), 2.95 (dd, JPH = 7.8 Hz, JPH = 1.5 Hz, 1H, [(biph)(H)C–P(nBu)–PtBu2]−, 1b), 2.73 (d, JPH = 5.3 Hz, [(biph)(H)C–P(nBu)2]−, 1b′), 1.65 (broad m, [P(nBu)2–CH(biph)–CH(biph)–P–(PtBu2)]−, 1b′′), 1.57 (broad m, [P(nBu)2–CH(biph)–CH(biph)–P–(PtBu2)]−, 1b′′), 1.56 (broad m, [(biph)(H)C–P(nBu)–PtBu2]−, 1b), 1.48 (broad m, [(biph)(H)C–P(nBu)2]−, 1b′), 1.45 (broad m, [P(nBu)2–CH(biph)–CH(biph)–P–(PtBu2)]−, 1b′′), 1.42 (broad m, [(biph)(H)C–P(nBu)2]−, 1b′), 1.34 (d, JPH = 12.2 Hz, 18H, [P(nBu2)–CH(biph)–CH(biph)–P–(PtBu2)]−, 1b′′), 1.32 (d, JPH = 12.2 Hz, 18H, [(biph)(H)C–P(nBu)–PtBu2]−, 1b), 1.32 (d, JPH = 10.9 Hz, 18H, tBu2PLi), 1.32 (DME protons), 1.15 (broad m, [P(nBu2)–CH(biph)–CH(biph)–P–(PtBu2)]−, 1b′′), 0.96 (d, JPH = 11.6 Hz, 18H, [P(nBu2)–CH(biph)–CH(biph)–P–(PtBu2)]−, 1b′′), 0.92 (DME protons) ppm.
31P{1H} NMR (162 MHz, C6D6, 298 K) δ: 54.91 (d, JPP = 326.0 Hz, [P(nBu)2–CH(biph)–CH(biph)–P–(PtBu2)]−, 1b′′), 41.80 (broad singlet, tBu2PLi), 39.22 (d, JPP = 326.3 Hz, [(biph)(H)C–P(nBu)–PtBu2]−, 1b), −24.16 (d, JPP = 102.4 Hz, [P(nBu)2–CH(biph)–CH(biph)–P–(PtBu2)]−, 1b′′), −37.43 (d, JPP = 326.3 Hz, [(biph)(H)C–P(nBu)–PtBu2]−, 1b), −41.56 (s, [(biph)(H)C–P(nBu)2]−, 1b′), −59.24 (dd, JPP = 102.4 Hz, JPP = 326.0 Hz, [P(nBu)2–CH(biph)–CH(biph)–P–(PtBu2)]−, 1b′′) ppm.
P–PtBu2 with tBuLi. To a solution of (biph)(H)CP–PtBu2 (0.08 g; 0.234 mmol) in 1 mL of THF-d8 a tBuLi (0.123 mL; 0.234 mmol; 1.9 M) was dropwise added. The color immediately changed from orange to very dark red, and after 24 hours the solution was transferred to a NMR tube and measurements were performed.1H NMR (400 MHz, C6D6, 298 K) δ: 7.58–6.07 (aromatic protons), 3.15 (dd, JPH = 7.6 Hz, JPH = 1.3 Hz, 1H, [(biph)(H)C–P(tBu)–PtBu2]−, 1c), 3.10 (d, JPH = 198.4 Hz, tBu2PH), 1.36 and 1.34 (hexane protons from 2.5 M solution of tBuLi in hexane), 1.31 (d, JPH = 13.2 Hz, [(biph)(H)C–P(tBu)–PtBu2]−, 1c), 1.31 (JPH = 13.7 Hz, [(biph)(H)C–P(tBu)–PtBu2]−, 1c), 1.14 (d, JPH = 12.7 Hz, [(biph)(H)C–P(tBu)–PtBu2]−, 1c), 1.09 (d, JPH = 12.3 Hz, tBu2PH), ppm.
31P{1H} NMR (162 MHz, C6D6, 298 K) δ: 30.08 (d, JPP = 333.28 Hz, [(biph)(H)C–P(tBu)–PtBu2]−, 1c), 19.87 (s, tBu2PH), −4.05 (d, JPP = 333.28 Hz, [(biph)(H)C–P(tBu)–PtBu2]−, 1c) ppm.
Reactions of (3,5-tBu2C6H3)(H)CP–PtBu2 (3) with MeLi, nBuLi, tBuLi respectively
Phosphanylphosphaalkene (80 mg, 0.223 mmol) was dissolved in 0.7 mL of THF-d8 and lithium compound was dropwise added. After 24 hours solution was transferred to NMR-tube and appropriate measurements were performed.
31P{1H} NMR (162 MHz, C6D6, 298 K) δ: 66.12 (dd, JPP = 347.5 Hz, JPP = 62.5 Hz, [tBu2P–P–{C(H)(3,5-tBu2C6H3)}–PtBu2]−), 57.98 (dd, JPP = 231.7 Hz, JPP = 62.5 Hz, [tBu2P–P–{C(H)(3,5-tBu2C6H3)}–PtBu2]−), 53.01 (d, JPP = 324.1 Hz, [P(Me)2–CH(3,5-tBu2C6H3)–CH(3,5-tBu2C6H3)–P–(PtBu2)]−), 39.78 (s, tBu2PLi), 19.81 (s, tBu2PH), −48.80 (d, JPP = 97.6 Hz, [P(Me)2–CH(3,5-tBu2C6H3)–CH(3,5-tBu2C6H3)–P–(PtBu2)]−), −56.70 (broad dd, JPP = 324.1 Hz, JPP = 97.6 Hz [P(Me)2–CH(3,5-tBu2C6H3)–CH(3,5-tBu2C6H3)–P–(PtBu2)]−), −67.30 (dd, JPP = 347.5 Hz, JPP = 231.7 Hz, [tBu2P–P–{C(H)(3,5-tBu2C6H3)}–PtBu2]−) ppm.
31P{1H} NMR (162 MHz, C6D6, 298 K) δ: 66.05 (dd, JPP = 347.5 Hz, JPP = 62.5 Hz, [tBu2P–P–{C(H)(3,5-tBu2C6H3)}–PtBu2]−), 57.88 (dd, JPP = 231.7 Hz, JPP = 62.5 Hz, [tBu2P–P–{C(H)(3,5-tBu2C6H3)}–PtBu2]−), 54.07 (dd, JPP = 325.2 Hz, JPP = 2.6 Hz, [P(nBu)2–CH(3,5-tBu2C6H3)–CH(3,5-tBu2C6H3)–P–(PtBu2)]−), −24.07 (dd, JPP = 96.8 Hz, JPP = 2.6 Hz, [P(nBu)2–CH(3,5-tBu2C6H3)–CH(3,5-tBu2C6H3)–P–(PtBu2)]−), −60.36 (dd, JPP = 325.2 Hz, JPP = 96.8 Hz, [P(nBu)2–CH(3,5-tBu2C6H3)–CH(3,5-tBu2C6H3)–P–(PtBu2)]−), −67.42 (dd, JPP = 347.5 Hz, JPP = 231.7 Hz, [tBu2P–P–{C(H)(3,5-tBu2C6H3)}–PtBu2]−) ppm.
31P{1H} NMR (162 MHz, C6D6, 298 K) δ: 65.94 (dd, JPP = 347.5 Hz, JPP = 62.5 Hz, [tBu2P–P–{C(H)(3,5-tBu2C6H3)}–PtBu2]−), 57.96 (dd, JPP = 231.7 Hz, JPP = 62.5 Hz, [tBu2P–P–{C(H)(3,5-tBu2C6H3)}–PtBu2]−), 48.79 (dd, JPP = 314.6 Hz, JPP = 35.3 Hz, [P(tBu)2–CH(3,5-tBu2C6H3)–CH(3,5-tBu2C6H3)–P–(PtBu2)]−), 39.78 (s, tBu2PLi), 19.84 (s, tBu2PH), 8.18 (dd, JPP = 114.7 Hz, JPP = 35.3 Hz, [P(tBu)2–CH(3,5-tBu2C6H3)–CH(3,5-tBu2C6H3)–P–(PtBu2)]−), −67.49 (dd, JPP = 347.5 Hz, JPP = 231.7 Hz, [tBu2P–P–{C(H)(3,5-tBu2C6H3)}–PtBu2]−), −67.54 (dd, JPP = 314.6 Hz, JPP = 114.7 Hz, [P(tBu)2–CH(3,5-tBu2C6H3)–CH(3,5-tBu2C6H3)–P–(PtBu2)]−) ppm.
Reactions of (p-Me2N–C6H4)(H)CP–PtBu2 (4) with MeLi, nBuLi, tBuLi respectively
Phosphanylphosphaalkene (80 mg, 0.257 mmol) was dissolved in 0.7 mL of THF-d8 and lithium compound was dropwise added. After 24 hours solution was transferred to NMR-tube and appropriate measurements were performed.
31P{1H} NMR (162 MHz, C6D6, 298 K) δ: 64.62 (dd, JPP = 348.2 Hz, JPP = 62.6 Hz, [tBu2P–P–{C(H)(p-Me2N–C6H4)}–PtBu2]−), 56.85 (dd, JPP = 239.6 Hz, JPP = 62.6 Hz, [tBu2P–P–{C(H) (p-Me2N–C6H4)}–PtBu2]−), 54.44 (d, JPP = 324.7 Hz, [P(Me)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)]−), 39.77 (s, tBu2PLi), 19.84 (s, tBu2PH), −47.08 (d, JPP = 111.51 Hz, [P(Me)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)]−), −64.84 (dd, JPP = 324.7 Hz, JPP = 111.51 Hz, [P(Me)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)]−), −69.64 (dd, JPP = 348.2 Hz, JPP = 239.6 Hz, [tBu2P–P–{C(H) (p-Me2N–C6H4)}–PtBu2]−) ppm.
31P{1H} NMR (162 MHz, C6D6, 298 K) δ: 64.73 (dd, JPP = 348.2 Hz, JPP = 62.6 Hz, [tBu2P–P–{C(H)(p-Me2N–C6H4)}–PtBu2]−), 57.05 (dd, JPP = 237.7 Hz, JPP = 62.6 Hz, [tBu2P–P–{C(H) (p-Me2N–C6H4)}–PtBu2]−), 54.84 (d, JPP = 328.6 Hz, [P(nBu)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)]−), 39.77 (s, tBu2PLi), 19.81 (s, tBu2PH), −22.45 (d, JPP = 109.6 Hz, [P(nBu)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)]−), −67.41 (dd, JPP = 328.6 Hz, JPP = 109.6 Hz, [P(nBu)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)]−), −69.52 (dd, JPP = 348.2 Hz, JPP = 237.7 Hz, [tBu2P–P–{C(H) (p-Me2N–C6H4)}–PtBu2]−) ppm.
31P{1H} NMR (162 MHz, C6D6, 298 K) δ: 64.65 (dd, JPP = 348.2 Hz, JPP = 62.6 Hz, [tBu2P–P–{C(H)(p-Me2N–C6H4)}–PtBu2]−), 57.03 (dd, JPP = 237.7 Hz, JPP = 62.6 Hz, [tBu2P–P–{C(H) (p-Me2N–C6H4)}–PtBu2]−), 51.56 (dd, JPP = 326.9 Hz, JPP = 5.3 Hz, [{P(tBu)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)}2Li]−), 49.51 (dd, JPP = 281.7 Hz, JPP = 51.5 Hz, [{P(tBu)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)}2Li]−), 41.07 (dd, JPP = 139.5 Hz, JPP = 5.3 Hz, [{P(tBu)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)}2Li]−), 31.44 (s, [(p-Me2N–C6H4)(H)C–P(tBu2)2]−), 3.81 (dd, JPP = 111.3 Hz, JPP = 51.5 Hz, [{P(tBu)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)}2Li]−), −55.98 (dd, JPP = 281.7 Hz, JPP = 111.3 Hz, [{P(tBu)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)}2Li]−), −57.56 (dd, JPP = 326.9 Hz, JPP = 139.5 Hz, [{P(tBu)2–CH(p-Me2N–C6H4)–CH(p-Me2N–C6H4)–P–(PtBu2)}2Li]−), −69.59 (dd, JPP = 348.2 Hz, JPP = 237.7 Hz, [tBu2P–P–{C(H) (p-Me2N–C6H4)}–PtBu2]−) ppm.
Conclusions
The performed experiments undoubtedly display that CP bond activation in the phosphanylphosphaalkene (biph)(H)CP–PtBu2 occurs with each of the three selected reagents. However, the reactions with MeLi and nBuLi ultimately lead to substitution of two organic groups (Me and nBu) on the phosphorus atoms and, consequently, to cleavage of the P–P bond. From the point of view of further research related to the polymerization of phosphanylphosphaalkenes, both MeLi and nBuLi are not suitable reagents for anionic activation. The research shows that the best reagent for anionic activation is tBuLi, because only one tBu group is substituted on the phosphorus atom. Additionally, in this substitution the new item (Scheme 2) is given in the text reaction of the second group (tBu), cleavage of the P–P bond was not observed. Moreover, replacement the –Ph group in the para position, with tBu groups (meta position) and NMe2 group (para position) does not improve the stability of the products after the first stage of the reaction – i.e. the activation of the PC bond consisting in the mono-substitution of the Me, nBu or tBu groups to the phosphorus atom. The obtained result indicates that (biph)(H)CP–PtBu2 (1) is the best phosphanylphosphaalkene to activate the PC bond as proved by its reaction with tBuLi.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. Z. thanks for financial support of these studies from Gdańsk University of Technology by the DEC-8/2020/IDUB/I.3.3 grant under the Argentum – ‘Excellence Initiative – Research University’ program is gratefully acknowledged. N. S. thanks the TASK Computational Centre and PLGrid Infrastructure for access to computational resources. Ł. P. thanks for financial support of these studies from Gdańsk University of Technology by the DEC-2/2021/IDUB/V.6/Si grant under the SILICIUM SUPPORTING CORE R&D FACILITIES – ‘Excellence Initiative – Research University’ program is gratefully acknowledged.Notes and references
- T. L. Breen and D. W. Stephan, J. Am. Chem. Soc., 1995, 117, 11914–11921 CrossRef CAS.
- A. Marinetti, S. Bauer, L. Ricard and F. Mathey, Organometallics, 1990, 9, 793–798 CrossRef CAS.
- M. Yam, J. H. Chong, C.-W. Tsang, B. O. Patrick, A. E. Lam and D. P. Gates, Inorg. Chem., 2006, 45, 5225–5234 CrossRef CAS PubMed.
- M. Yam, C.-W. Tsang and D. P. Gates, Inorg. Chem., 2004, 43, 3719–3723 CrossRef CAS PubMed.
- P. L. Floch, Coord. Chem. Rev., 2006, 250, 627–681 CrossRef.
- H. Murakami, Y. Matsui, F. Ozawa and M. Yoshifuji, J. Organomet. Chem., 2006, 691, 3151–3156 CrossRef CAS.
- F. Ozawa, H. Okamoto, S. Kawagishi, S. Yamamoto, T. Minami and M. Yoshifuji, J. Am. Chem. Soc., 2002, 124, 10968–10969 CrossRef CAS PubMed.
- R. S. Jensen, K. Umeda, M. Okazaki, F. Ozawa and M. Yoshifuji, J. Organomet. Chem., 2007, 692, 286–294 CrossRef CAS.
- T. Minami, H. Okamoto, S. Ikeda, R. Tanaka, F. Ozawa and M. Yoshifuji, Angew. Chem., Int. Ed., 2001, 40, 4501–4503 CrossRef CAS.
- O. Fumiyuki, Y. Shogo, K. Seiji, H. Masatomi, I. Shintaro, M. Tatsuya, I. Shigekazu and Y. Masaaki, Chem. Lett., 2001, 30, 972–973 CrossRef.
- S. Ito, M. Nanko and K. Mikami, ChemCatChem, 2014, 6, 2292–2297 CrossRef CAS.
- K. Takeuchi, Y. Tanaka, I. Tanigawa, F. Ozawa and J.-C. Choi, Dalton Trans., 2020, 49, 3630–3637 RSC.
- K. Ohtsuki, H. T. G. Walsgrove, Y. Hayashi, S. Kawauchi, B. O. Patrick, D. P. Gates and S. Ito, Chem. Commun., 2020, 56, 774–777 RSC.
- L. L. Liu, J. Zhou, L. L. Cao, Y. Kim and D. W. Stephan, J. Am. Chem. Soc., 2019, 141, 8083–8087 CrossRef CAS PubMed.
- P. W. Siu, S. C. Serin, I. Krummenacher, T. W. Hey and D. P. Gates, Angew. Chem., 2013, 125, 7105–7108 CrossRef.
- C.-W. Tsang, B. Baharloo, D. Riendl, M. Yam and D. P. Gates, Angew. Chem., 2004, 116, 5800–5803 CrossRef.
- C.-W. Tsang, M. Yam and D. P. Gates, J. Am. Chem. Soc., 2003, 125, 1480–1481 CrossRef CAS PubMed.
- C.-W. Tsang, C. A. Rohrick, T. S. Saini, B. O. Patrick and D. P. Gates, Organometallics, 2004, 23, 5913–5923 CrossRef CAS.
- C.-W. Tsang, C. A. Rohrick, T. S. Saini, B. O. Patrick and D. P. Gates, Organometallics, 2002, 21, 1008–1010 CrossRef CAS.
- K. J. T. Noonan and D. P. Gates, Angew. Chem., Int. Ed., 2006, 45, 7271–7274 CrossRef CAS.
- K. J. T. Noonan and D. P. Gates, Macromolecules, 2008, 41, 1961–1965 CrossRef CAS.
- B. H. Gillon, K. J. Noonan, B. Feldscher, J. M. Wissenz, Z. M. Kam, T. Hsieh, J. J. Kingsley, J. I. Bates and D. P. Gates, Can. J. Chem., 2007, 85, 1045–1052 CrossRef CAS.
- B. W. Rawe, A. M. Priegert, S. Wang, C. Schiller, S. Gerke and D. P. Gates, Macromolecules, 2018, 51, 2621–2629 CrossRef CAS.
- K. J. T. Noonan, B. O. Patrick and D. P. Gates, Chem. Commun., 2007, 3658–3660 RSC.
- B. W. Rawe, C. P. Chun and D. P. Gates, Chem. Sci., 2014, 5, 4928–4938 RSC.
- B. W. Rawe, C. M. Brown, M. R. MacKinnon, B. O. Patrick, G. J. Bodwell and D. P. Gates, Organometallics, 2017, 36, 2520–2526 CrossRef CAS.
- M. Balmer, H. Gottschling and C. von Hänisch, Chem. Commun., 2018, 54, 2659–2661 RSC.
- J. Mahnke, A. Zanin, W.-W. du Mont, F. Ruthe and P. G. Jones, Z. Anorg. Allg. Chem., 1998, 624, 1447–1454 CrossRef CAS.
- A. Ziółkowska, N. Szynkiewicz, J. Pikies and Ł. Ponikiewski, Dalton Trans., 2020, 49, 13635–13646 RSC.
- A. Ziółkowska, N. Szynkiewicz and Ł. Ponikiewski, Organometallics, 2019, 38, 2873–2877 CrossRef.
- A. Ziółkowska, N. Szynkiewicz and Ł. Ponikiewski, Dalton Trans., 2021, 50, 1390–1401 RSC.
- A. Ziółkowska, N. Szynkiewicz and Ł. Ponikiewski, Inorg. Chem. Front., 2021, 8, 3851–3862 RSC.
- R. A. Jones, A. L. Stuart and T. C. Wright, J. Am. Chem. Soc., 1983, 105, 7459–7460 CrossRef CAS.
- M. Xu, A. R. Jupp, Z.-W. Qu and D. W. Stephan, Angew. Chem., Int. Ed., 2018, 57, 11050–11054 CrossRef CAS PubMed.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- D. E. C. Corbridge, The structural chemistry of phosphorus, ed. E. C. Corbridge, Elsevier Scientific Pub. Co, Amsterdam, New York, 1974 Search PubMed.
- K. Izod, P. Evans and P. G. Waddell, Dalton Trans., 2017, 46, 13824–13834 RSC.
- K. Izod, P. O'Shaughness and W. Clegg, Organometallics, 2002, 21, 641–646 CrossRef CAS.
- A. Ziółkowska, N. Szynkiewicz, A. Wiśniewska, J. Pikies and Ł. Ponikiewski, Dalton Trans., 2018, 47, 9733–9741 RSC.
- J. Langer, V. K. Pálfi, B. Schowtka, H. Görls and M. Reiher, Inorg. Chem. Commun., 2013, 32, 28–31 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data and details, molecular structure of 1a* and 2_1 and 3. NMR spectra, DFT calculations details. CCDC 2114106, 2114107 and 2163558. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra09215d |
| This journal is © The Royal Society of Chemistry 2022 |