DOI:
10.1039/D1RA09060G
(Review Article)
RSC Adv., 2022,
12, 6214-6233
Recent advances in photochemical and electrochemically induced thiocyanation: a greener approach for SCN-containing compound formation
Received
14th December 2021
, Accepted 10th February 2022
First published on 22nd February 2022
Abstract
Techniques utilizing photo- and electrochemically induced reactions have been developed to accelerate organic processes. These techniques use light or electrical energy (electron transfer) as a direct energy source without using an initiator or reagent. Thiocyanates are found in biologically active and pharmacological compounds and can be converted into various functional groups. It is one of the most prominent organic scaffolds. Significant development in photo- and electro-chemically induced thiocyanation procedures has been made in recent years for the conception of carbon-sulfur bonds and synthesis of pharmaceutically important molecules. This review discusses different photo- and electro-chemically driven thiocyanation C(sp3)–SCN, C(sp2)–SCN, and C(sp)–SCN bond conception processes that may be useful to green organothiocyanate synthesis. We focus on the synthetic and mechanistic characteristics of organic photo- and electrochemically accelerated C–SCN bond formation thiocyanation reactions to highlight major advances in this novel green and sustainable research field.
1. Introduction
Synthetic organic chemistry relies heavily on organo-thiocyanates.1 In chemical synthesis and medicinal chemistry, the formation of C–SCN bonds is a common method for producing organothiocyanates,1a which are commonly used as synthetic precursors for a range of sulfur heterocycles and sulfur-containing compounds, such as sulfides, disulfides, thiols, thioethers, isothiocyanates, trifluoromethylthiolates, and thiocarbamates.2 These organothiocyanates contain many biological features that are useful in various sectors, such as medicine, pesticides, and materials.3 Organic synthetic chemists are fascinated by their remarkable biological activities and possible uses.
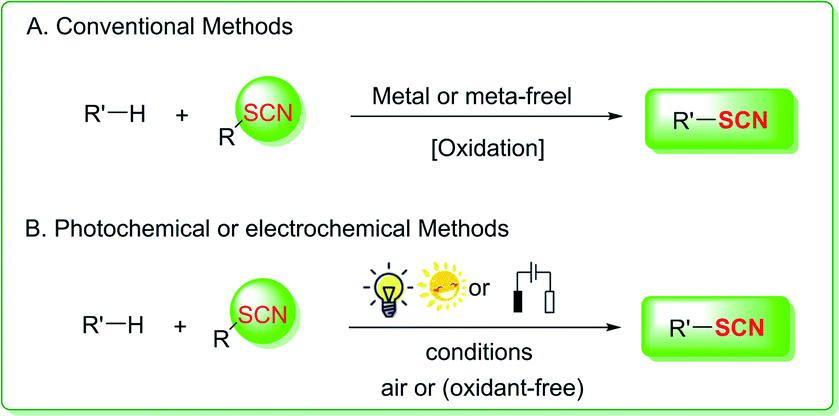
Organic chemists have concentrated their efforts into exploring thiocyanation reactions and applications, which mostly involve nucleophilic substitution, electrophilic substitution, and free radical thiocyanation (Fig. 1A) catalyzed by transition metals or nonmetals through a traditional technique or with the use of stoichiometric oxidants.4,5 As a result, a number of well-established and highly successful methods for generating C–SCN bonds have been developed and are already well-reviewed, which is critical to the advancement of organic thiocyanate research.1e
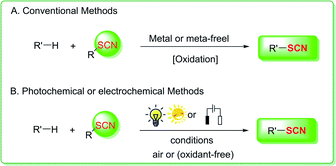 |
| | Fig. 1 Strategies for C–SCN bond formation through thiocyanation. | |
The development of green, sustainable, efficient, and extremely atomic-economical C–S bond conception procedure is one of the most noteworthy research subjects in synthetic organic chemistry.6 In this context, photoredox catalysis has lately appeared as a promising avenue for organic synthesis because it utilizes seemingly endless resources, such as visible light, air, and energy. Photocatalysis and electrocatalysis use light and electrical energy (electron transfer) as direct energy sources, respectively, and are thus in line with green chemistry principles. Photocatalytic approaches using air as a green antioxidant to perform thiocyanation reactions have been devised in recent years. Electrochemical synthesis is a novel and ecologically beneficial technology that uses continuous electron gain and loss rather than standard chemical redox reagents. Organic chemists have effectively leveraged this characteristic to construct a variety of metal-free and oxidant-free thiocyanation methods (Fig. 1B). The essential ideas of photocatalysis and electrocatalysis-induced processes for C–C and C–S bond formation have been thoroughly examined.7 Nevertheless, novel photo- and electrochemically promoted strategies for generating carbon-thiocyano (C–SCN) bonds have recently been described. The purpose of this review is to describe the latest advances in this rapidly expanding field of photo- or electrocatalysis. This review article presents a concise summary of the most important photocatalysis- and electrocatalysis-driven thiocyanation strategies for the creation of C(sp3)–SCN, C(sp2)–SCN, and C(sp)–SCN bonds to increase understanding of the evolution of green and sustainable thiocyanation progression.
2. Photochemical-induced thiocyanation reactions
2.1 Photochemical-induced C(sp3)–SCN bond formation through thiocyanation reaction
The direct thiocyanation of a stable C–H bond is one of the most advantageous routes because it facilitates the synthesis of organic thiocyanates with readily accessible C–H bond donors. Over the last decade, many catalytic techniques for the direct thiocyanation of stable C(sp2)–H and C(sp3)–H bonds have been satisfactorily established, allowing quick access to a wide range of thiocyanated molecules. The considerable activation energy required to activate inert C(sp3)–H bonds increases the difficulty and challenge of C(sp3)–H bond activation functionalization. Photocatalytic oxidative coupling of C(sp3)–H thiocyanation reaction is an effective way to achieve C(sp3)–H participation in thiocyanation reactions as compared to traditional C(sp3)–H thiocyanation reactions processes.
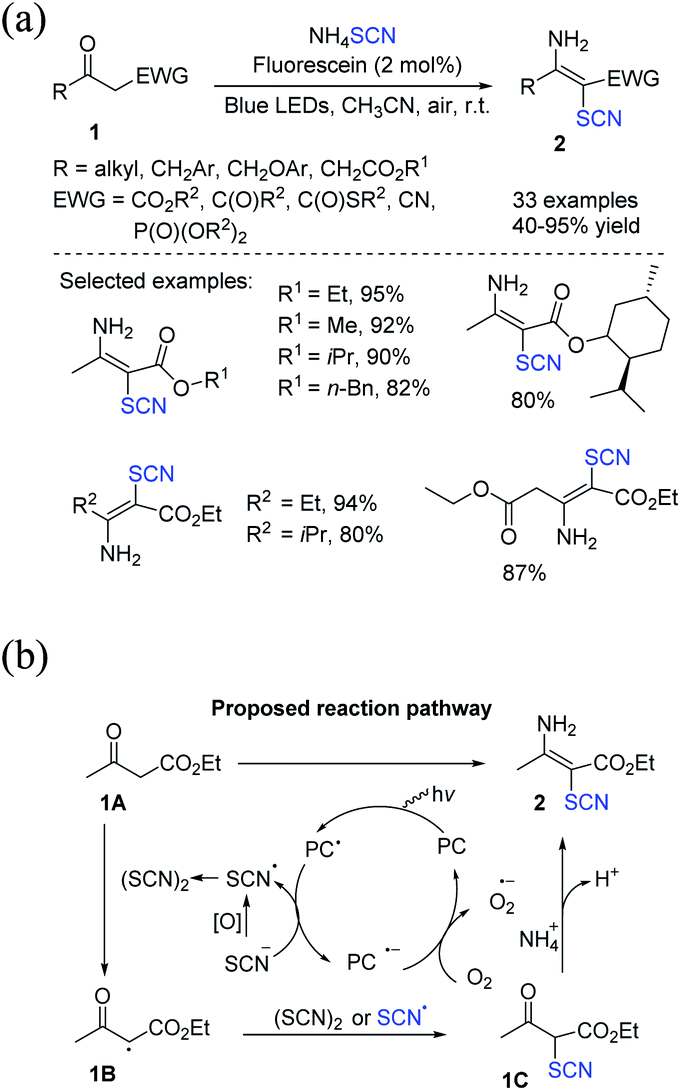
To produce multi-substituted olefins 2, Yuan et al. invented a strategy that uses atmospheric oxygen as the terminating oxidant for the metal-free photochemical-induced amino-thiocyanation of activated ketones 1.8 Ammonium thiocyanate has been used as the sole source of thiocyanate and amine, and water is the only secondary substance. This green approach may effectively and rapidly produce a range of multi-substituted olefins in high yields (Scheme 1a). The process starts with the SET oxidation of NH4SCN by the incited photocatalyst fluorescein*, which produces ˙SCN and the decreased form fluorescein˙−. In superoxide production, the photocatalyst can be regenerated by oxidizing fluorescein˙− with dioxygen. The ˙SCN radical then takes a hydrogen atom from 1A. This process results in a carbon-centered radical and thiocyanic acid (HSCN). ˙SCN or (SCN)2 captures the α-carbonyl radical 1B to produce a bridging precursor 1C, which contains an electron-poor carbonyl group and may be conveniently targeted through nucleophilic NH3. Eventually, the desired product 2 is obtained by removing water from the adduct of 1C and NH3 (Scheme 1b).
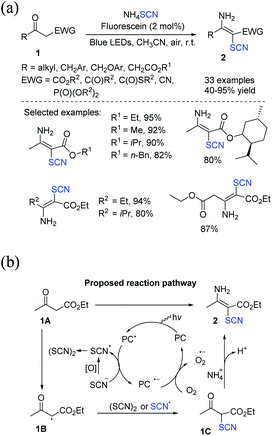 |
| | Scheme 1 a) Visible-light-induced amino-thiocyanation of activated ketones 1. (b) Proposed mechanism for visible-light-induced amino-thiocyanation of activated ketones 1. | |
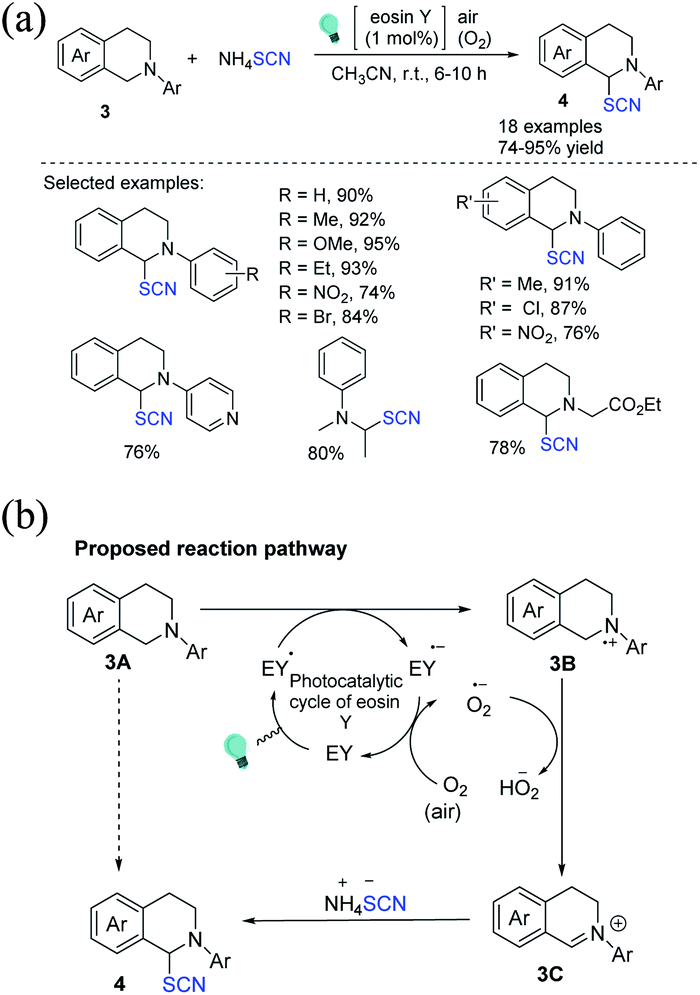
The first example of an organo-photoredox catalyst-driven C(sp3)–S bond formation through the thiocyanation reaction of tertiary amine 3 at ambient temperature was discovered by Yadav et al., who used ammonium thiocyanate as the source of thiocyanate.9 The reaction is gentle, and oxygen and green light are environmentally friendly and cost-effective reagents; only a small amount of eosin Y (EY; 1 mol%) is required (Scheme 2a). When light is absorbed, EY transforms into its excited state (EY*). Compound 3B and EY˙− are formed by a single electron transfer between 3A and EY*, and the photoredox cycle of eosin Y occurs through the aerobic oxidation of EY˙− by O2 to its ground state (EY). The superoxide radical anion (O2−˙) produced in situ removes a proton from the α-position of 3B to produce iminium ion 3C, which is subsequently attacked by the SCN nucleophile to provide the final product 4 (Scheme 2b).
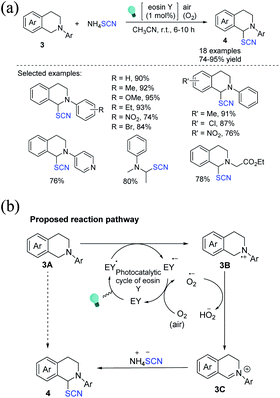 |
| | Scheme 2 a) Photoredox catalyst promoted α-C(sp3)–H thiocyanation of tertiary amines 3. (b) Proposed reaction pathway for the α-C(sp3)–H thiocyanation of tertiary amines 3. | |
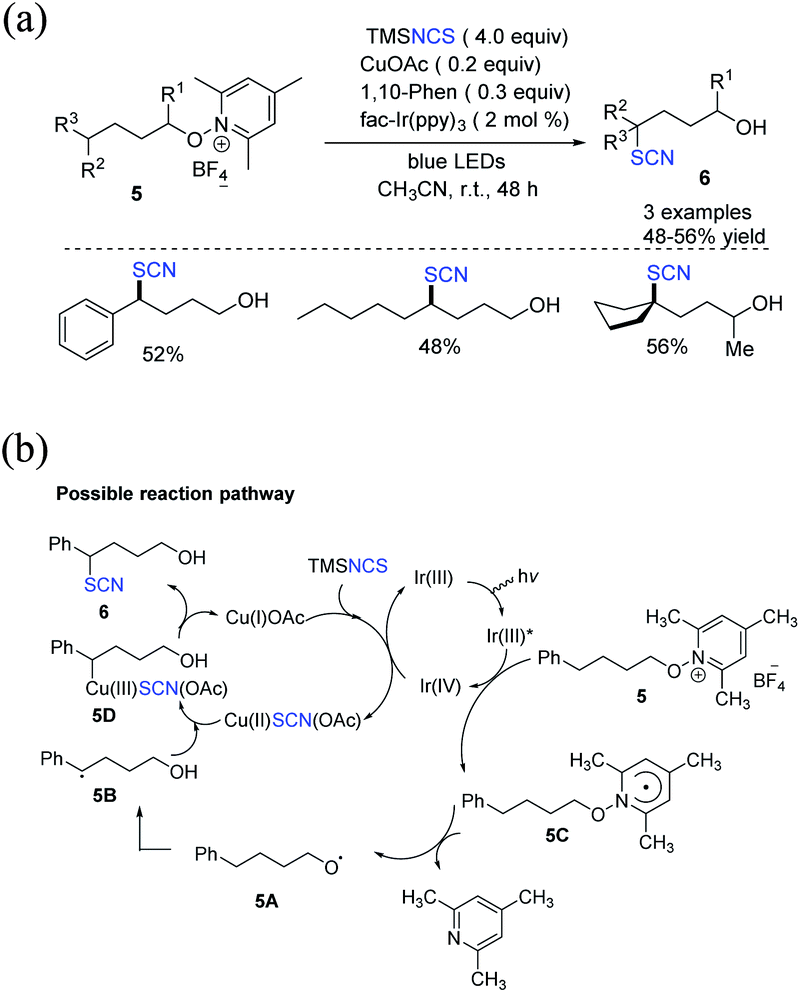
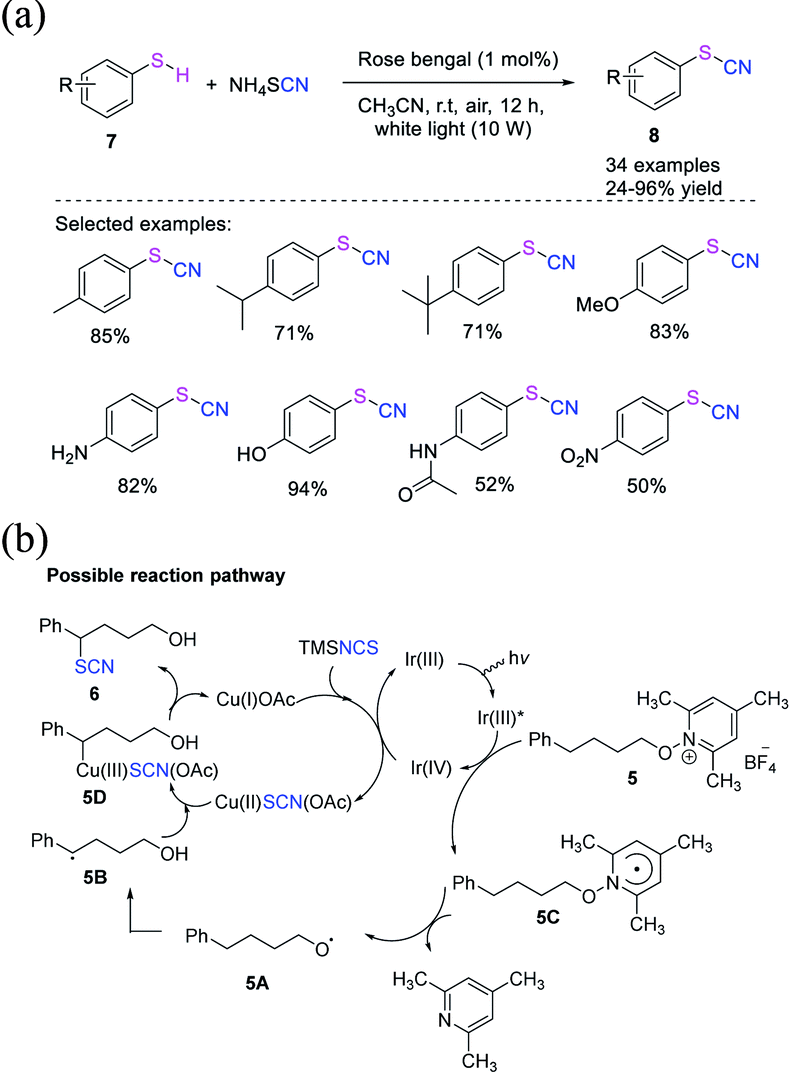
Zhu et al. created a distinctive approach for constructing δ-thiocyanato alcohols 6 in good to moderate yields. In this method, the reaction between N-alkoxypyridinium salts 5 and silyl isothiocyanate is catalyzed through dual photoredox or by copper.10 A domino reaction includes alkoxy radical production, 1,5-hydrogen atom transfer (1,5-HAT), and copper-catalyzed thiocyanation of the ensuing C-centered radical (Scheme 3a). Three alternative approaches for functionalizing benzylic, aliphatic, and tertiary C(sp3)H are demonstrated. The authors claim that Ir(III)* excited species reduce substrates 5 in the production of radical 5C and Ir(IV). In the presence of TMSNCS, Cu(I) is oxidized and produces Cu(II)SCN(OAc) salt. The process results in the regeneration of the Ir(III) species. After 1,5-HAT, radical 5C fragments become alkoxy radical 5A, which then fragments to form radical 5B. Cu(II)SCN(OAc) forms the Cu(III) species 5D when it interacts with Cu(III) species 5D. With the release of Cu(I) salt, reductive elimination forms the thiocyanate molecule 6 (Scheme 3b).
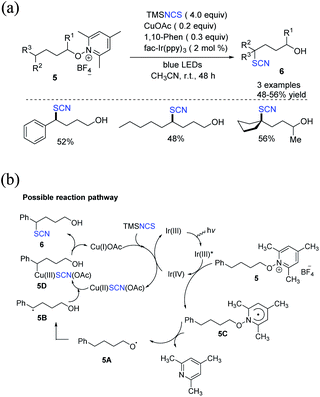 |
| | Scheme 3 a) Photoredox or copper-catalysis to access δ-thiocyanato alcohols 6. (b) Proposed mechanism for photoredox or copper-catalysis to access δ-thiocyanato alcohols 6. | |
2.2 C–S bond formation photochemical induced indirect thiocyanation reaction
Fan et al. proposed a direct photocatalyst-induced cyanation technique for the construction of thiocyanates 8 from thiols 7.11 This technique uses ammonium thiocyanate as an inorganic salt, which is a cheap and harmless and is a green cyano source and provides a wide range of thiocyanate products 8 in good to high yields (Scheme 4a). Under visible light, Rose Bengal (RB) can be used as a photocatalyst and air as an oxidant. RB is first stimulated to its excited state species (RB*) when released to visible light. Then, through energy transfer, RB* interacts with O2 to produce 1O2 and regenerates its ground state (RB). Under visible light irradiation, RB appears to be a better 1O2-generating photosensitizer than other photocatalysts. The produced 1O2 then captures a hydrogen atom from thiol, yielding the thioyl radical 7A. Through radical coupling, radical 7A is converted into intermediate 7B. Owing to the instability of disulfides under light irradiation, the homolytic dissociation of 7B produces radical 7A. The intermediate 7C is formed when thioyl radical 7A combines with SCN−. The intermediate 7D is propagated and formed when the intermediate 7C is trapped by another thiol. Finally, 7D is transformed into the desired product 8 with the release of HS− (Scheme 4b).
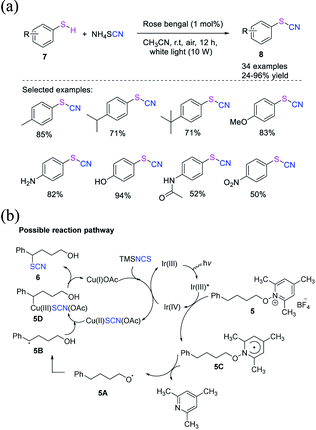 |
| | Scheme 4 a) Photocatalyst-promoted synthesis of thiocyanates 8 from thiols 7. (b) Possible reaction pathway for synthesis of thiocyanates 8 from thiols 7. | |
2.3 Photochemical induced C(sp2)–SCN bond formation thiocyanation reaction
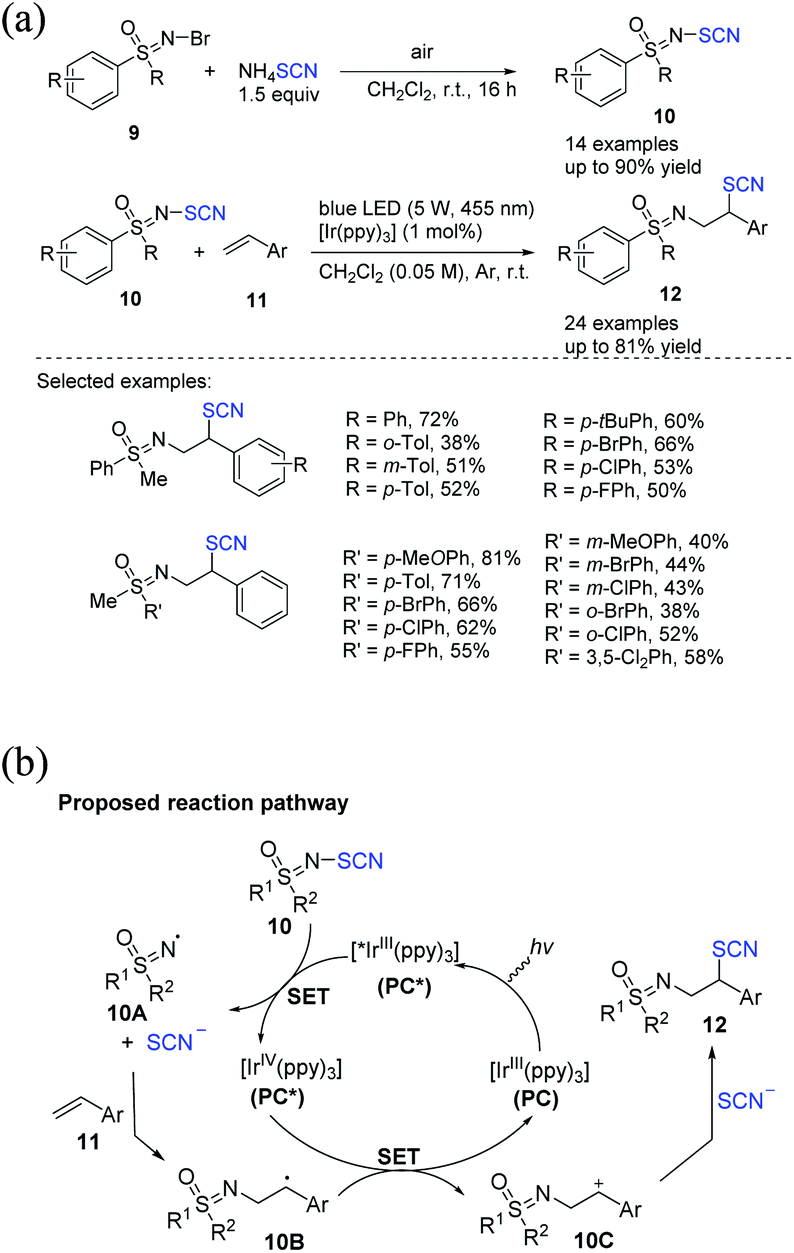
2.3.1 C(sp2)–SCN bond formation thiocyanation reaction of olefins and arenes. Bolm et al. proposed a method for synthesizing novel sulfur reagents as N-thiocyanato sulfoximines 10 from N–Br sulfoximines 9 and ammonium thiocyanate, which they then used in the photocatalytic difunctionalizations of styrenes 11 to obtain a variety of 1,2 difunctionalized compounds 12 carrying with a thiocyanato functions and a sulfoximine functions on vicinal positions in high to excellent yields (Scheme 5a).12 According to the proposed reaction pathway, the photocatalyst fac-[Ir(ppy)3] (PC) can efficiently receive a photon from poor fluorescent light. The process results in the incited state fac-[*Ir(ppy)3] (PC*). PC+ is formed through a single electron transfer (SET) from PC* to N-SCN sulfoximine 10. A thiocyanate anion and the sulfoximidoyl radical 10A are constructed. A novel radical, 10B, is created, which is oxidized through PC+ to produce the benzyle cation 10C when 11 is added to 10A. The photocatalyst then returns to its initial oxidation state (PC) and can initiate the original catalytic circle. When 10C interacts in the midst of SCN−, product 12 is produced (Scheme 5b).
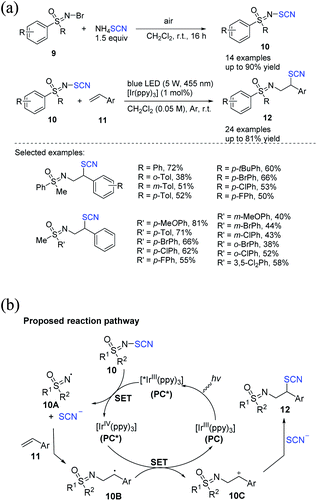 |
| | Scheme 5 a) Photocatalytic difunctionalization of alkenes 11 with N-SCN sulfoximines 10. (b) Proposed mechanism for photocatalytic difunctionalization of alkenes 11 with N-SCN sulfoximines 10. | |
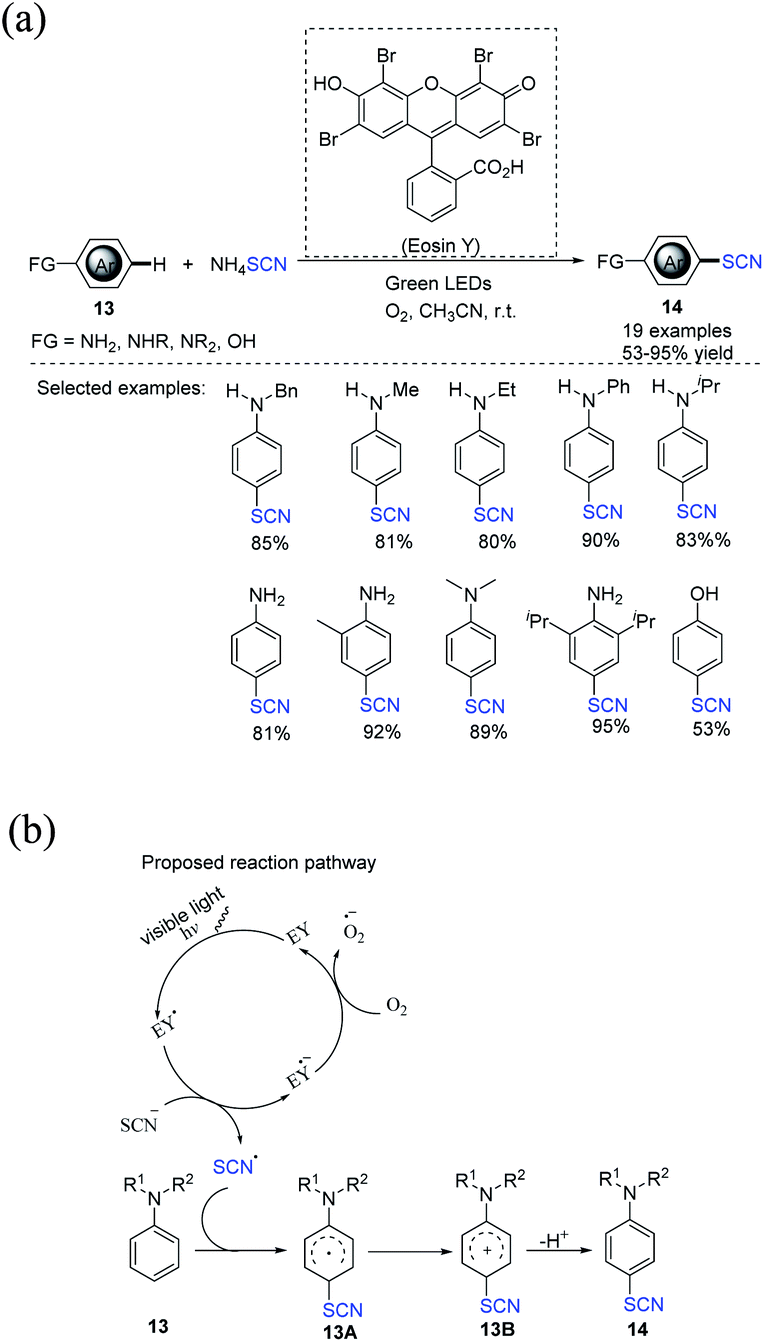
In 2020, Tan et al. developed a visible-light-assisted regioselective aerobic oxidative C(sp2)–H thiocyanation process for aromatic substances 13.13 The process is ecologically benign, effective, and simple to operate. EY, which is an useful photocatalyst, and oxygen, which is a green terminal oxidant, were used to produce the required compounds 14 in good to high yields. A wide variety of aromatic amines, including primary, secondary, and tertiary amines, can be used in this process (Scheme 6a). In the author's opinion, a workable mechanism has been offered. After being photo-excited in the presence of green LED light, EY is converted to its excited state EY*, which is then oxidized to radical SCN˙ through single-electron transfer (SET). Subsequently, the SCN˙ radical attacks substrate 13, promoting the formation of another radical intermediate 13A, which is then swiftly oxidized to produce the cationic intermediate 13B. The deprotonation of the cationic intermediate 13B results in the formation of the desired product 14. Furthermore, oxygen may be important to the completion of the photoredox cycle by oxidizing the EY˙− radical anion abaft to its ground state (EY), which then enters the next catalytic cycle, as previously stated (Scheme 6b).
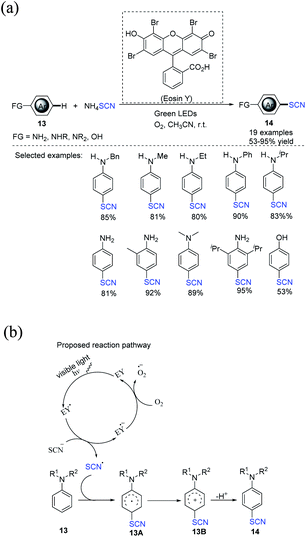 |
| | Scheme 6 a) Visible-light-assisted regioselective aerobic oxidative C(sp2)–H thiocyanation of aromatic compounds 13. (b) Proposed mechanism for visible-light-assisted regioselective aerobic oxidative C(sp2)–H thiocyanation of aromatic compounds 13. | |
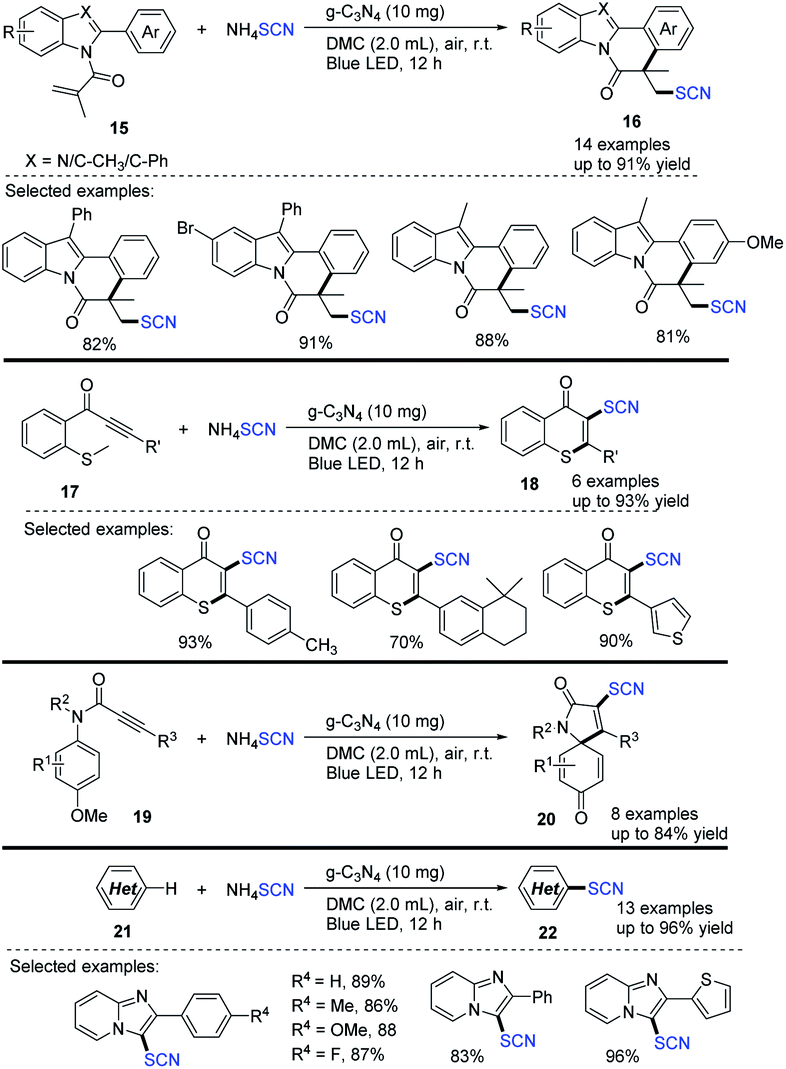
Yu et al. devised a metal-free and visible-light-mediated scheme for synthesizing thiocyanated heterocycles in green solvent, such as dimethyl carbonate (DMC), under an air atmosphere.14 They used ammonium thiocyanate and graphitic carbon nitride (g-C3N4), which are inexpensive, readily available, and reusable semiconductor photocatalysts, in designing and synthesizing various types of thiocyanated heterocycles, such as indolo/benzimidazo[2,1-a]isoquinolin-6(5H)-ones 16, thioflavones 18, azaspiro[4,5]trienones 20, and imidazo[1,2-a]pyridines 22 (up to 96%). The reusable g-C3N4 catalyst was reused many times before losing significant activity. They demonstrated the positive outcome of the protocol. They were able to use a reusable catalyst and a green solvent with a wide application without any metal or external oxidant (Scheme 7).
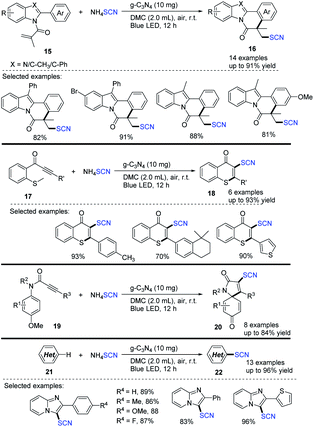 |
| | Scheme 7 Photocatalytic technique for the synthesis of thiocyanated heterocycles 16. | |
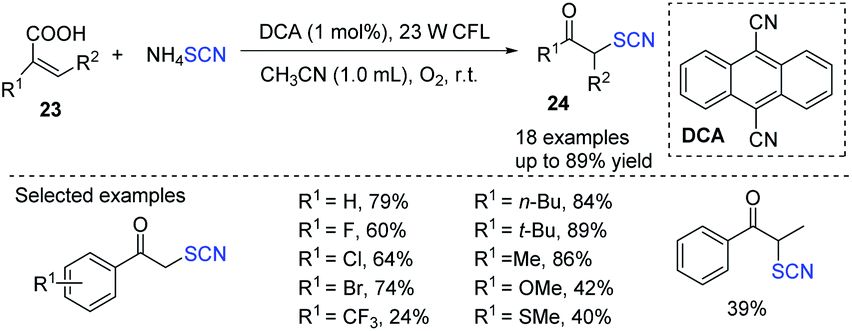
Guan and coworkers established an environmentally friendly photocatalytic additive-free decarboxylative reaction of acrylic acids 23 for the synthesis of a wide range of α-thiocyanate ketones 24.15 The author used a photocatalyst as an organic dye 9,10-dicyanoanthracene(DCA), instead of other photocatalysts. The ammonium thiocyanate was used as both a sulfur source and a reductant, and molecular dioxygen serves as both an oxygen supply and an oxidant which is inexpensive, and environmentally benign. This approach has a lot of possibilities in organic and medicinal chemistry because of the ease with which the product α-thiocyanate ketones 24 can be transformed (Scheme 8).
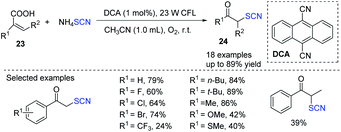 |
| | Scheme 8 Visible-light-mediated additive-free decarboxylative reaction of acrylic acid 23. | |
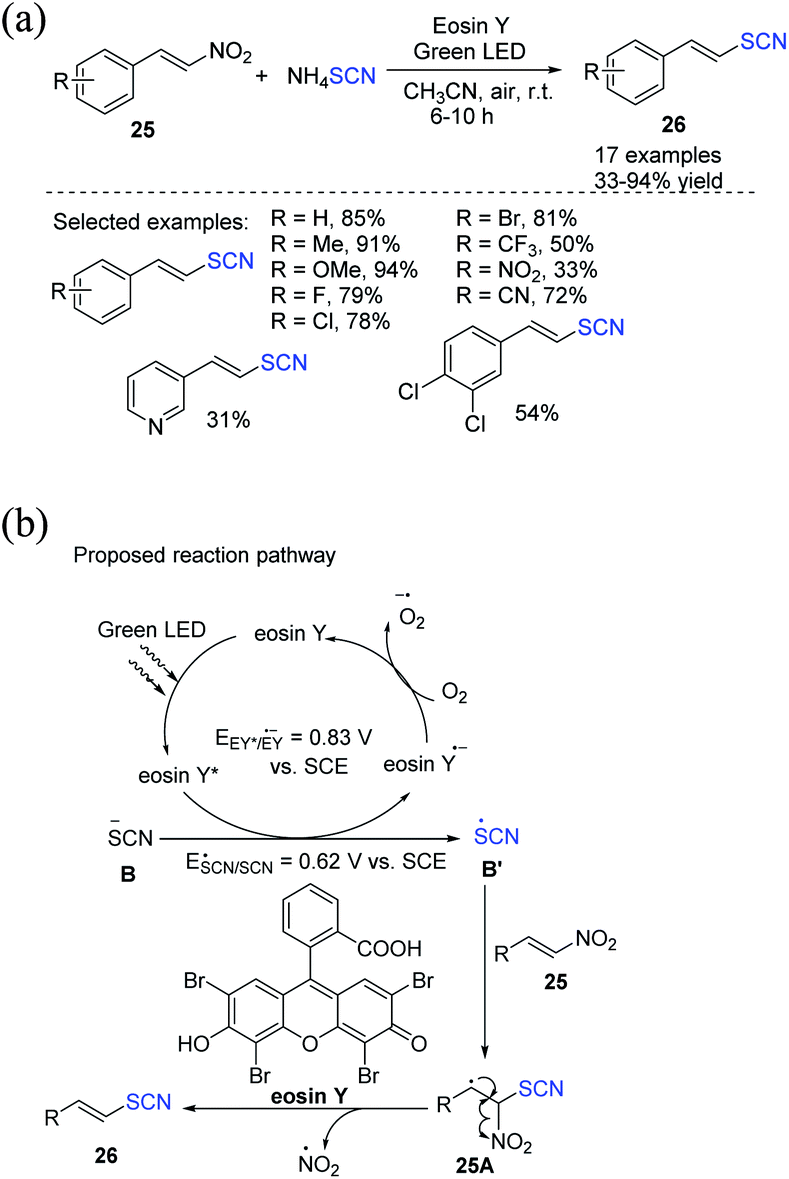
Kapoor et al. demonstrated the photocatalytic denitrative thiocyanation of β-nitrostyrenes 25.16 They employed EY as a photocatalyst and ammonium thiocyanate as a thiocyanating agent in acetonitrile as an organic solvent in open air under visible light to create (E)-vinyl thiocyanates 26 in excellent to outstanding yields (Scheme 9a). They proposed that visible light stimulates EY, which then uses the SET mechanism to oxidize thiocyanate anion B to SCN radical B′ through a reductive quenching circle. The carbon-centered radical intermediate 25A is formed when the generated thiocyanate radical B′ attacks the double bond of 25. 25A is subjected to denitration for the production of the desired product 26. The involvement of aerobic oxygen in fulfilling the photoredox catalytic process is likely essential. It returns the EY radical anion to its ground state by oxidizing it (Scheme 9b).
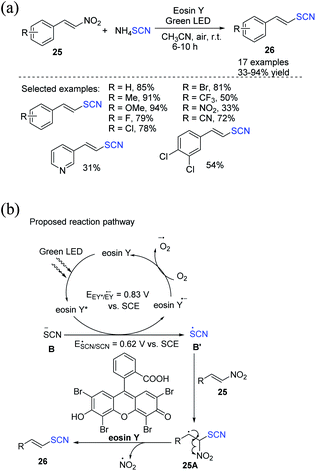 |
| | Scheme 9 a) Visible-light-induced denitrative thiocyanation of β-nitrostyrenes 25. (b) Proposed mechanism for visible-light-induced denitrative thiocyanation of β-nitrostyrenes 25. | |
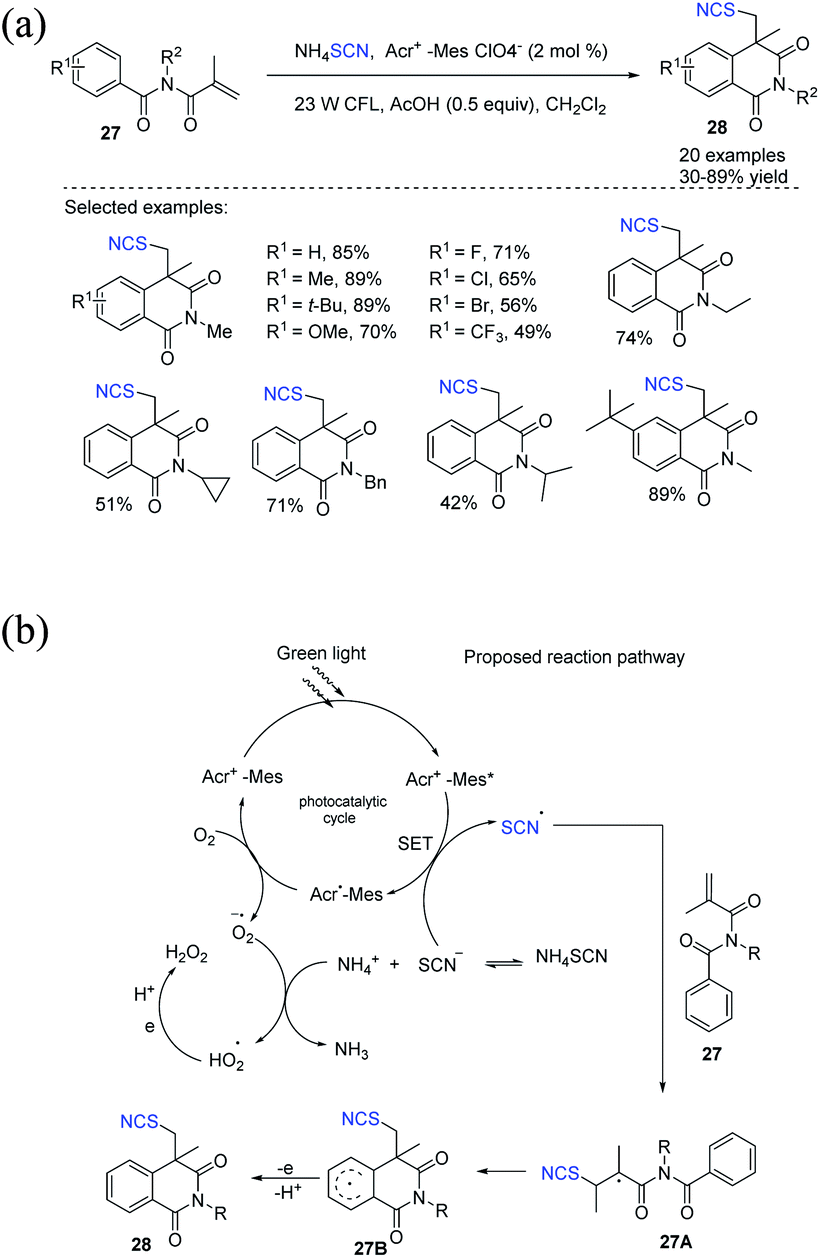
Guan et al. disclosed a new Acr+-MesClO4−-catalyzed thiocyanate radical addition or intramolecular cyclization cascade reaction from N-alkyl-N-methacryloylbenzamides 27 to obtain thiocyanato-containing isoquinolinediones 28 under gentle visible light photocatalytic conditions.17 They used ammonium thiocyanate as a reagent to synthesize thiocyanate radicals through a SET pathway. They also used molecular oxygen as the sole oblational reagent. They obtained moderate to good yields of several new compounds 28 (Scheme 10a). Acr+-Mes is stimulated to Acr+-Mes* when exposed to visible light, which removes an electron from the thiocyanate anion and generates a thiocyanate radical while producing Acr˙-Mes. To complete the photocatalysis cycle, molecular oxygen oxidizes Acr˙-Mes back to Acr+-Mes, releasing superoxide anion radical (O2˙−), which accepts a proton from NH4+ and forms a hydroperoxyl radical (HO2˙). When the thiocyanate radical is added to 27, it forms a C–S bond, which results in alkyl radical intermediate 27A. Intermediate 27A undergoes cyclization on the aromatic ring, yielding the cyclic radical intermediate 27B, which can then be oxidized and deprotonated by HO2˙ to generate the product 28 (Scheme 10b).
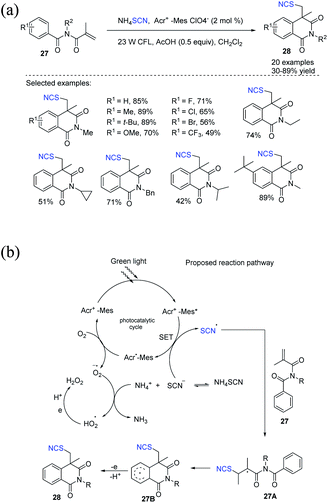 |
| | Scheme 10 a) Synthesis of isoquinoline-1,3-dione derivatives 28 through radical addition or cyclization cascade. (b) Proposed mechanism for the synthesis of isoquinoline-1,3-dione derivatives 28 through the radical addition or cyclization cascade. | |
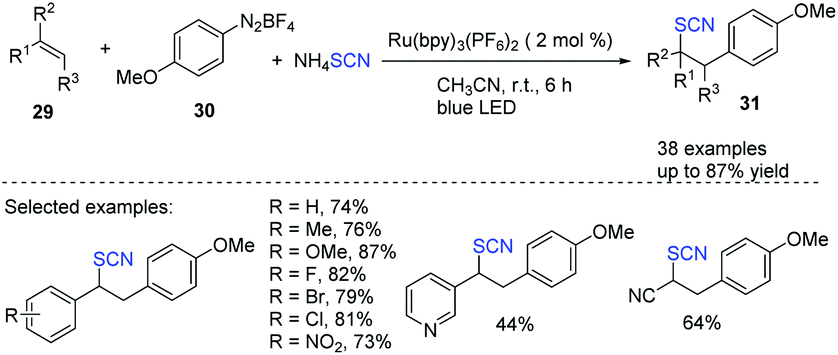
Using diazonium salts 30 as an arylating agent and ammonium thiocyanate as a thiocyanate source, Hoque et al. established a highly effective photoredox-catalyzed process for the intermolecular arylthiocyanation of alkenes 29.18 The process was found to be environmentally friendly and extremely efficient. Styrenes and acrylates produce diverse aryl-substituted alkylthiocyanates 31 good to excellent yields and in a variety of configurations (Scheme 11).
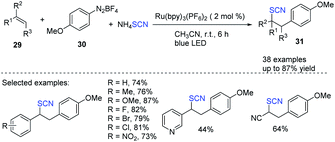 |
| | Scheme 11 Photoredox-catalyzed intermolecular radical arylthiocyanation of alkenes 29. | |
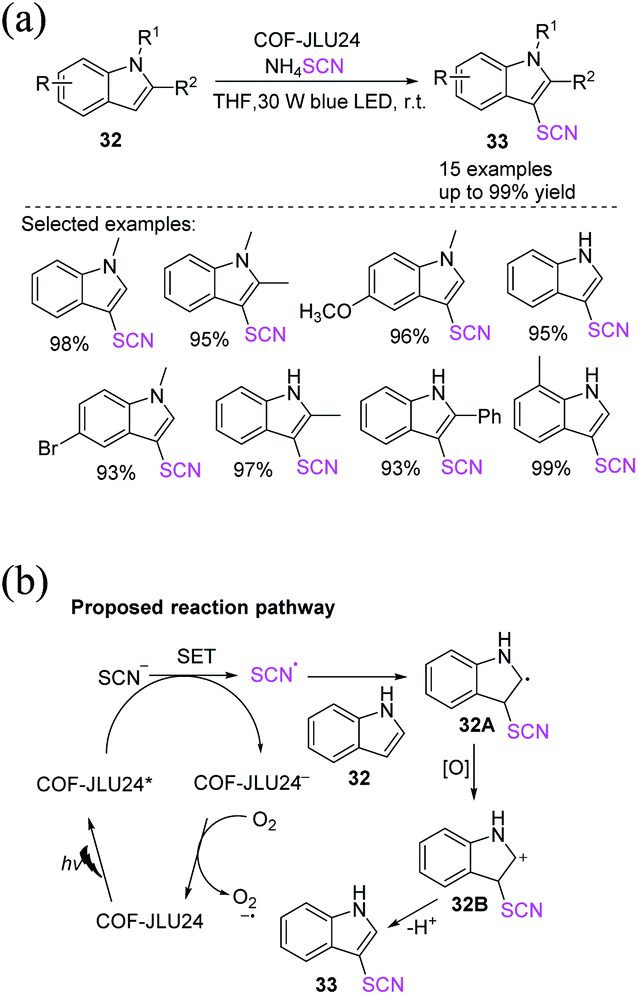
2.3.2 Photochemical-induced C(sp2)–SCN thiocyanation of aromatic heterocyclic compounds. Liu et al. identified an interesting approach for developing high-performance photocatalysts and novel isomorphic pyrene-based 2D-COFs (COF-JLU24) for the C-3 functionalization of indoles 32 with ammonium thiocyanate in tetrahydrofuran (THF) at room temperature (Scheme 12a).19 One of the possible mechanisms is the conversion of COF-JLU24 to its excited state COF-JLU24* by light irradiation. ˙SCN and COF-JLU24˙− radicals are generated through SET from the thiocyanate anion SCN to COF-JLU24*. The produced COF-JLU24˙− subsequently reacts with molecular oxygen to form the superoxide anion O2˙− and the ground state COF-JLU24. In the meantime, the newly formed radical ˙SCN attacks the substrate indole 32, generating intermediate 32A, which is subsequently oxidized to form cation intermediate 32B, which in turn is deprotonated to form the final 3-thiocyanoindole molecule 33 (Scheme 12b).
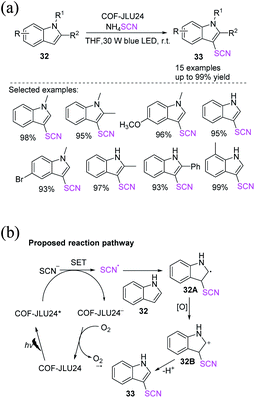 |
| | Scheme 12 a) C-3 functionalization reaction of indoles 32 with ammonium thiocyanate. (b) Proposed mechanism for C-3 functionalization reaction of indoles 32 with ammonium thiocyanate. | |
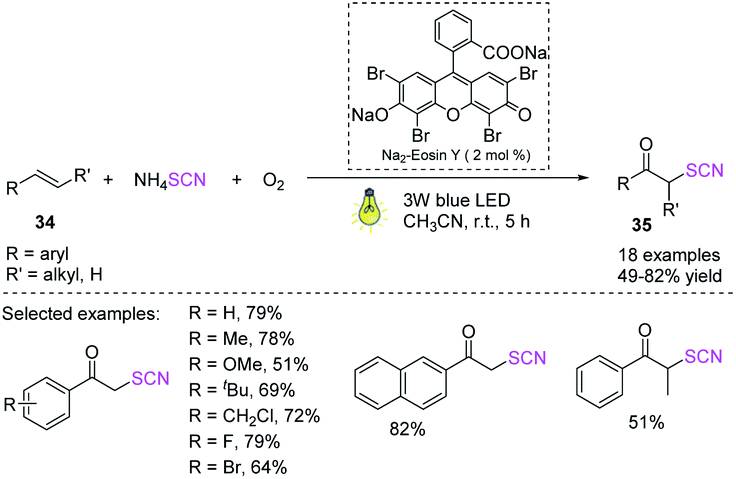
Yue et al. devised a fast and efficient visible-light-induced difunctionalization process for alkenes 34 at ambient temperature.20 The process uses ammonium thiocyanate and dioxygen. Owing to the generated carbon-sulfur and carbon–oxygen bonds and by using a nontoxic and cheap Na2-EY photocatalyst, a range of α-thiocyanato ketones 35 can be rapidly and effectively synthesized in moderate to high yields. This green technology offers an attractive alternative to obtaining different α-thiocyanato ketones 35 owing to its high functional flexibility, moderate reaction conditions, ecologically friendly energy source, and inexpensive and nontoxic catalyst (Scheme 13).
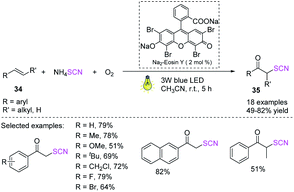 |
| | Scheme 13 Visible-light-enabled method for the formation of α-thiocyanato ketones 35. | |
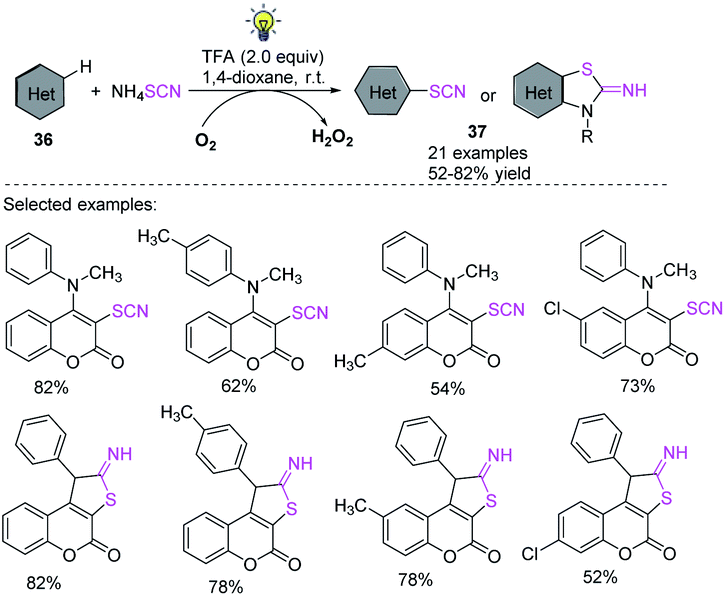
Using visible light and the widely available ammonium thiocyanate, Yang et al. found a novel photocatalyst-free oxidative C(sp2)–H thiocyanation process for electron-rich heteroarenes and arenes 36.21 According to mechanistic research, singlet oxygen is created when light is irradiated and can be a vital stimulant for the reaction to continue. The most appealing aspect of the reaction is that it allows simple bench-top processes because it only requires simple and widely affordable compounds 37 and does not necessitate the use of a transition metal catalysts, photocatalysts, bases, or strong oxidants. In a nutshell, the approach offers new options for C(sp2)–H thiocyanation reactions for medicinal and synthetic organic chemistry purposes (Scheme 14).
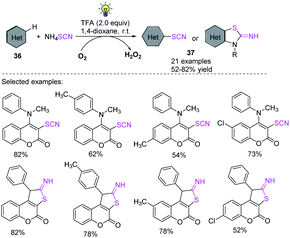 |
| | Scheme 14 Photocatalyst-free oxidative C(sp2)–H thiocyanation of heteroarenes and arenes 36. | |
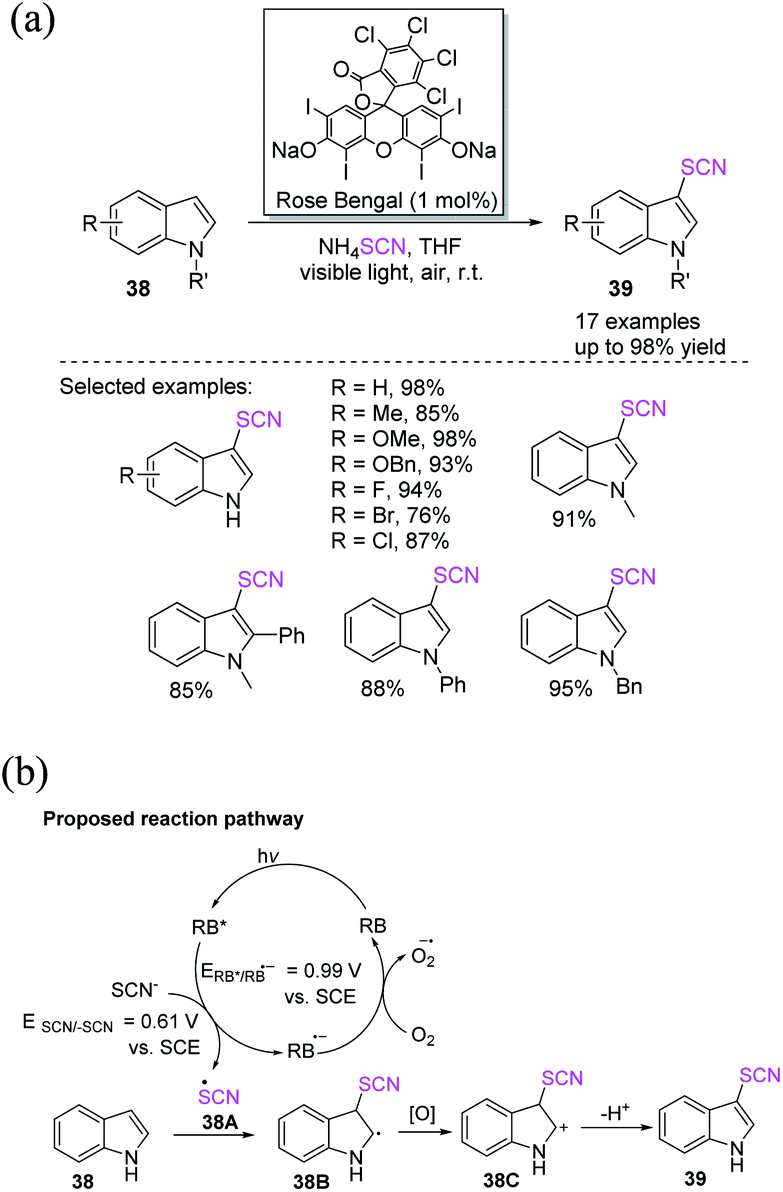
In recent years, chemists have designed energy-efficient photocatalytic thiocyanation processes that can directly use air as an oxidant in contrast to traditional free radical thiocyanation reactions and are thus more environmentally friendly. Li et al. invented a nontoxic and easy C-3 thiocyanation method for indoles 38. The author utilized RB, an reasonably priced and readily accessible organic dye, as the photocatalyst, and air serves as the terminal oxidant (Scheme 15a).22 When stimulated by visible light, RB transforms into excited RB*. A SET between –SCN and RB* produces the SCN radical 38A and an RB˙− radical anion. The photoredox cycle is completed when RB˙− is oxidized to its ground state. Intermediate 38B is formed after the electrophilic addition of radical 38A to 38. The oxidation of intermediate 38B results in the formation of cation intermediate 38C. Through the re-aromatization of intermediate 38C with a proton loss, the final 3-thiocyanoindole product 39 is formed (Scheme 15b).
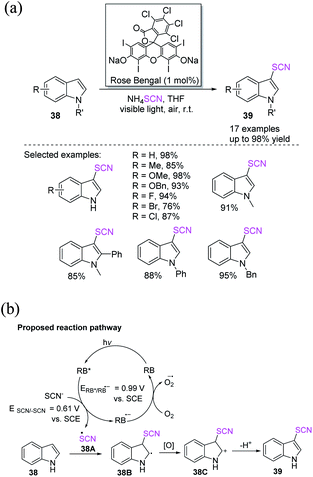 |
| | Scheme 15 a Photocatalyzed C-3 thiocyanation of indoles 38. (b) Proposed reaction pathway for photocatalyzed C-3 thiocyanation of indoles 38. | |
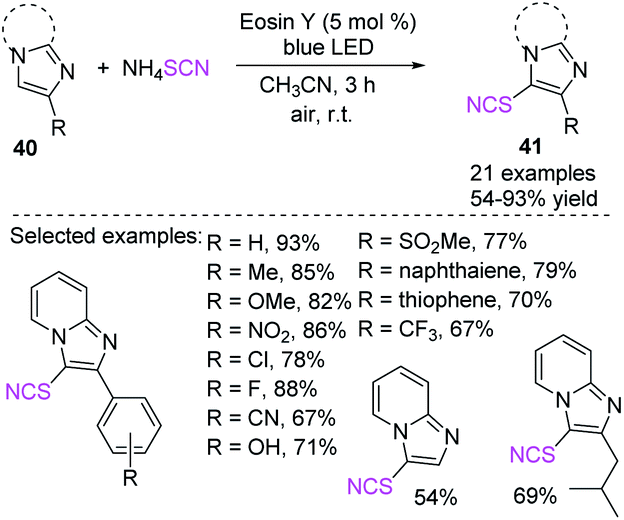
Using a green oxidant, Hajra et al. discovered an EY-catalyzed and visible-light-triggered C-3 thiocyanation of imidazoheterocycles 40 at room temperature.23 The new strategy is potent and cost effective, can use an extensive series of substrates, and can accommodate a large number of functional groups. This technique may offer substantial contributions to the fields of chemical synthesis, medicinal chemistry, and material science (Scheme 16).
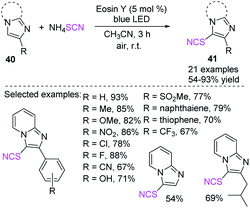 |
| | Scheme 16 Eosin Y-catalyzed C-3 thiocyanation of imidazoheterocycles 40. | |
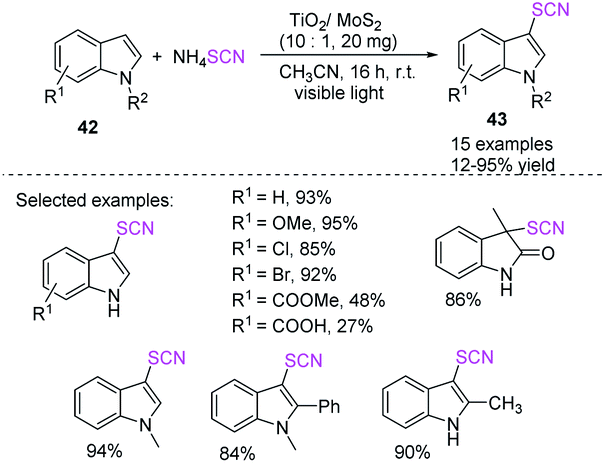
Ming-yang He et al. established a simple and nontoxic visible-light-assisted photocatalytic system for indole 42 thiocyanation.24 The system uses a TiO2/MoS2 nanocomposite. MoS2 is an effective photosensitizer, and its hybridization structure has a synergistic effect that boosts photocatalytic activity under visible light. This procedure is a practical and environmentally friendly choice to established techniques because of its high isolated yield and substrate compatibility and simple workup route and recyclability of the catalyst (Scheme 17).
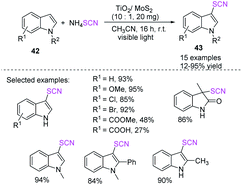 |
| | Scheme 17 Visible-light-mediated photocatalytic thiocyanation for indole 42. | |
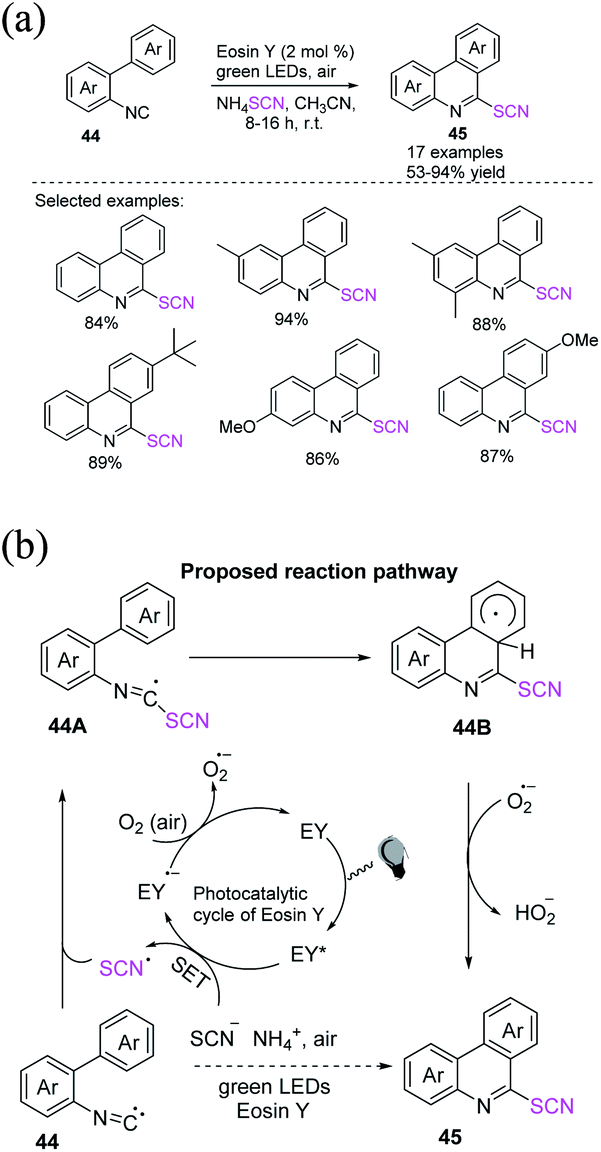
Singh et al. reported that the eosin Y-catalyzed sequential radical cyclization and aromatization of 2-isocyanobiphenyls 44 with ammonium thiocyanate produces 6-thiocyanatophenanthridines 45 through a simple, rapid, and metal-free mechanism.25 They used inexpensive, efficient, and sustainable natural resources, such as visible light and oxygen, at room temperature in this one-pot technique, which is more favorable for the process (Scheme 18a). When visible light is absorbed, the excited state of EY is generated. A single-electron transition between SCN− and EY* allows SCN and EY˙− to exist. The production of a superoxide radical anion (O2˙−) by the aerial oxidation of EY˙− to ground state EY˙− completes the photoredox cycle of EY. The imidoyl radical 44A is formed when the thiocyanate radical generated in situ combines with 2-isocyanobiphenyl 44. The imidoyl radical undergoes intramolecular radical aromatic substitution and generates intermediate 44B. O2˙− aromatize intermediate 44B by removing a hydrogen atom, resulting in the necessary 6-thiocyanato phenanthridine 45 (Scheme 18b).
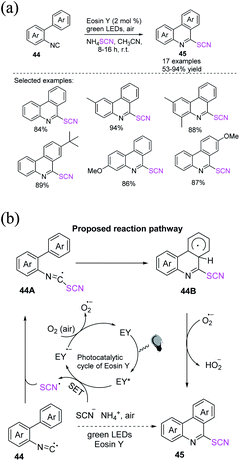 |
| | Scheme 18 a) Radical cyclization of 2-isocyanobiphenyls 44. (b) Proposed mechanism for the radical cyclization of 2-isocyanobiphenyls 44. | |
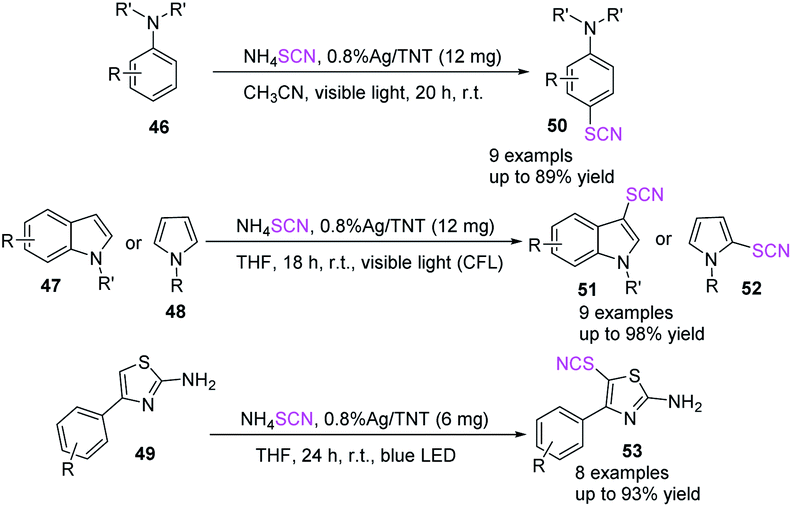
Sarvari et al. promoted the thiocyanation of anilines 46, indoles 47, pyrroles 48, and 2-amino thiazoles 49 treated with ammonium thiocyanate at room temperature by utilizing Ag/TNT as a photoredox catalyst.26 The produced thiocyano compounds had moderate to excellent yields under visible light. The convenience of usage, high to outstanding yields, gentle reaction conditions, and recyclability of the photoredox catalyst are the key benefits of this innovative approach (Scheme 19).
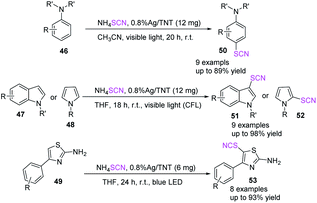 |
| | Scheme 19 Visible-light-promoted thiocyanation of aromatic and heteroaromatic compounds (46–49). | |
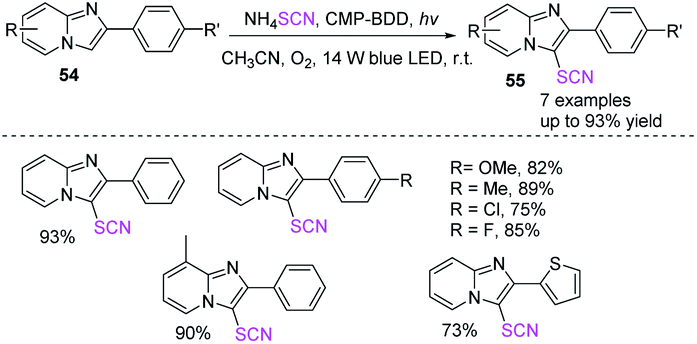
Zhang et al. exploited a commercially available molecule termed benzo[1,2-b:4,5-b′] dithiophene-4,8-dione (BDD) as a starting material to create a task-specific heterogeneous photocatalyst with a two-step process. The resulting BDD-based conjugated microporous polymer demonstrates effective visible light absorption as a result of the molecular engineering technique using an electron-output BDD because of the inclusion of a BDD moiety and an extended π-conjugated structure. The conjugated microporous polymer (CMP)-BDD complex displayed heterogeneous catalytic performance for common oxidative chemical reactions in the presence of light and oxygen, particularly in the selective thiocyanation of imidazoheterocycles 54 (Scheme 20).27
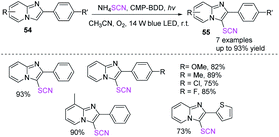 |
| | Scheme 20 Thiocyanation of imidazoheterocycles 54. | |
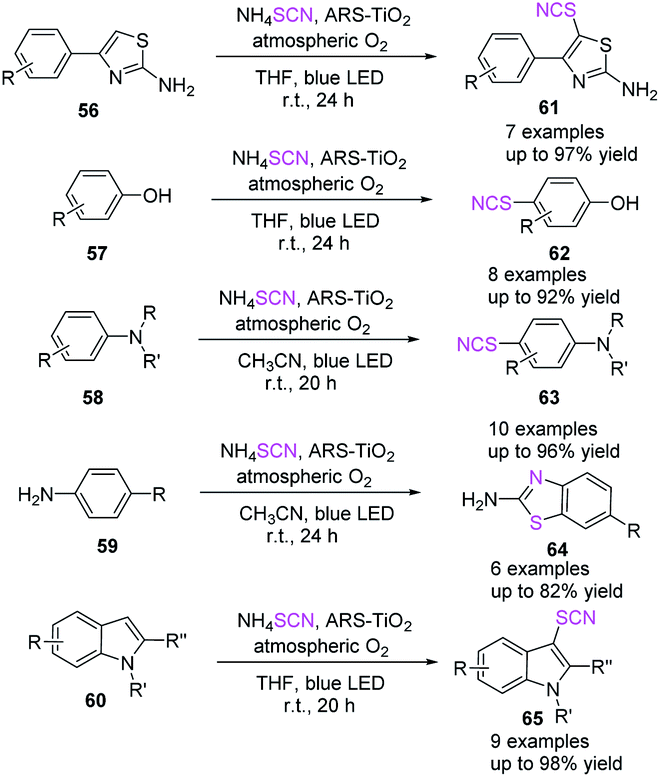
On the route to thiocyanation and cyclization processes, Koohgard et al. discovered an ARS-TiO2 photocatalyst that promotes the straightforward C–H functionalization of sp2 C–H bridges.28 They demonstrated the first visible-light-mediated phenol thiocyanation and synthesis of 2-aminobenzothiazole derivatives 64. They also reported a method for treating various aromatic and heteroaromatic frameworks (2-phenylamino-thiazole 56, phenol 57, aniline 58, pyrrole/indole 60 derivatives) with SCN− at room temperature (Scheme 21).
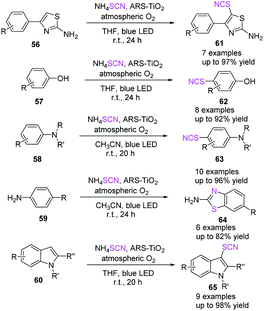 |
| | Scheme 21 Direct C–H functionalization of sp2 C–H bonds on the way to thiocyanation and cyclization reactions. | |
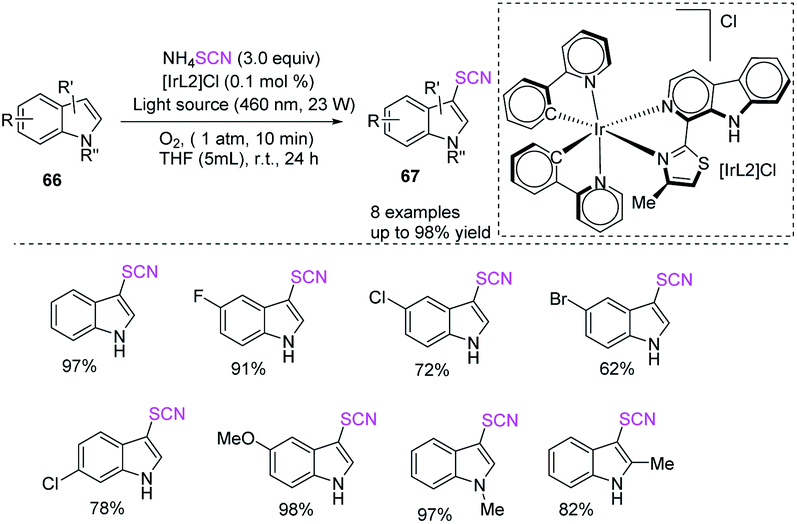
Espino et al. outlined the synthesis of innovative Ir(III) bis-cyclometallated photosensitizers with β-carboline ligands and a facile and unique photocatalytic one-pot two-step process for the chemoselective and C-3 regioselective oxidative thiocyanation of indolines 66 and production of 3-thiocyanatoindoles 67 in good to high yields.29 They used ammonium thiocyanate, molecular oxygen as a green oxidant, and Ir(III) photocatalysts in THF at room temperature in the presence of blue light as the energy source (Scheme 22).
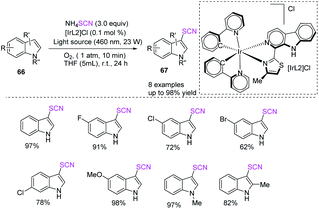 |
| | Scheme 22 Oxidative thiocyanation of indulines 66. | |
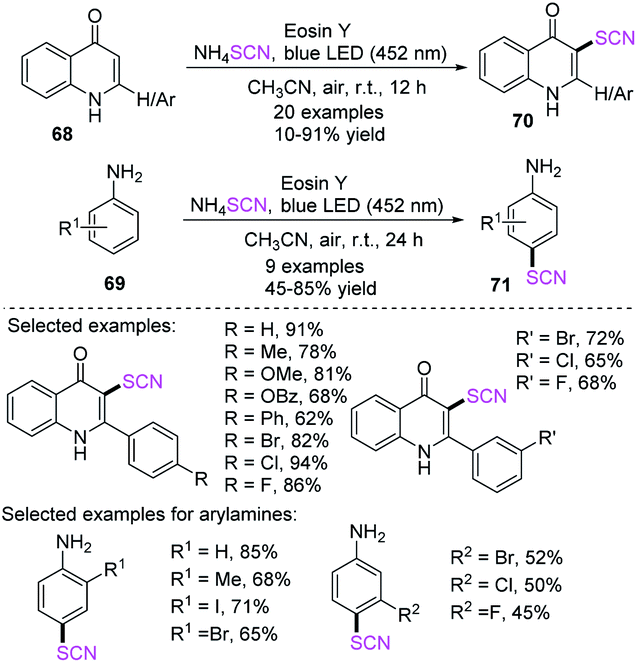
Jain et al. established an effective, easy, and metal-free regioselective C-3 thiocyanation process for 2-arylquinolin-4-ones 68 and a para-thiocyanation process for arylamines 69. The processes resulted in good to outstanding yields.30 They used eosin Y (EY) and air as the terminal oxidant in this photoredox process. The 3-thiocyanated quinolone derivatives 70 have many biological activities and can offer benefits to the field of pharmaceutical chemistry (Scheme 23).
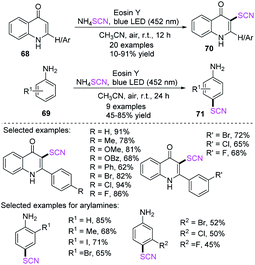 |
| | Scheme 23 Visible-light-promoted thiocyanation of heteroaromatics 68. | |
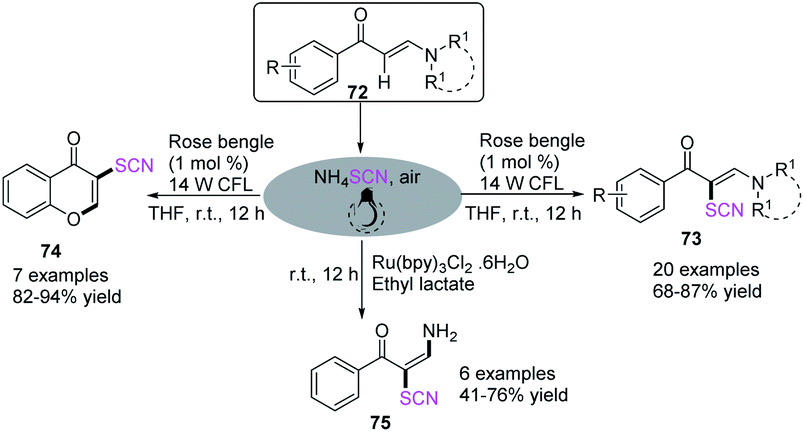
Wan et al. described the metal-free photocatalyst-induced C–H bond thiocyanation of tertiary enaminones 72. The process produced thiocyanated alkene derivatives 73 and chromones 74 with ammonium thiocyanate as the thiocyano source in an air atmosphere.31 They used ammonium thiocyanate and Ru (bpy)3Cl2·6H2O as a photocatalyst to transform the reaction path and produce NH2-functionalized thiocyanated enamines 75 through a difunctionalization route involving C–H bond thiocyanation and vinyl C–N bond trans-amination (Scheme 24).
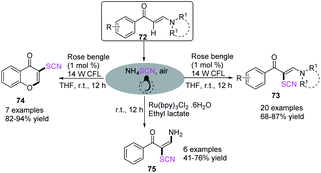 |
| | Scheme 24 Visible-light-induced thiocyanation of α-site of tertiary enaminones 72. | |
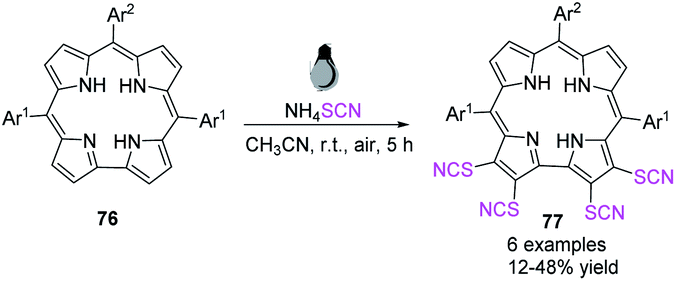
Kar et al. succeeded in developing a novel synthetic process for the simple synthesis of a novel class of corrole ligands 77 with four thiocyanate groups at the four surrounding β-positions in the corrole framework.32 They used an SCN radical created in situ to activate corrole macrocycles 76 at ambient temperature and fabricate tetrathio-cyanatocorroles 77 in high to excellent yields. The absorption properties of the modified corrole derivatives are strikingly similar to those of chlorophyll-a macrocycle. These compounds can be used as chlorophyll-a pigment spectroscopic model systems (Scheme 25).
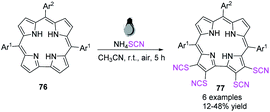 |
| | Scheme 25 Method for triggering corrole macrocycles 76. | |
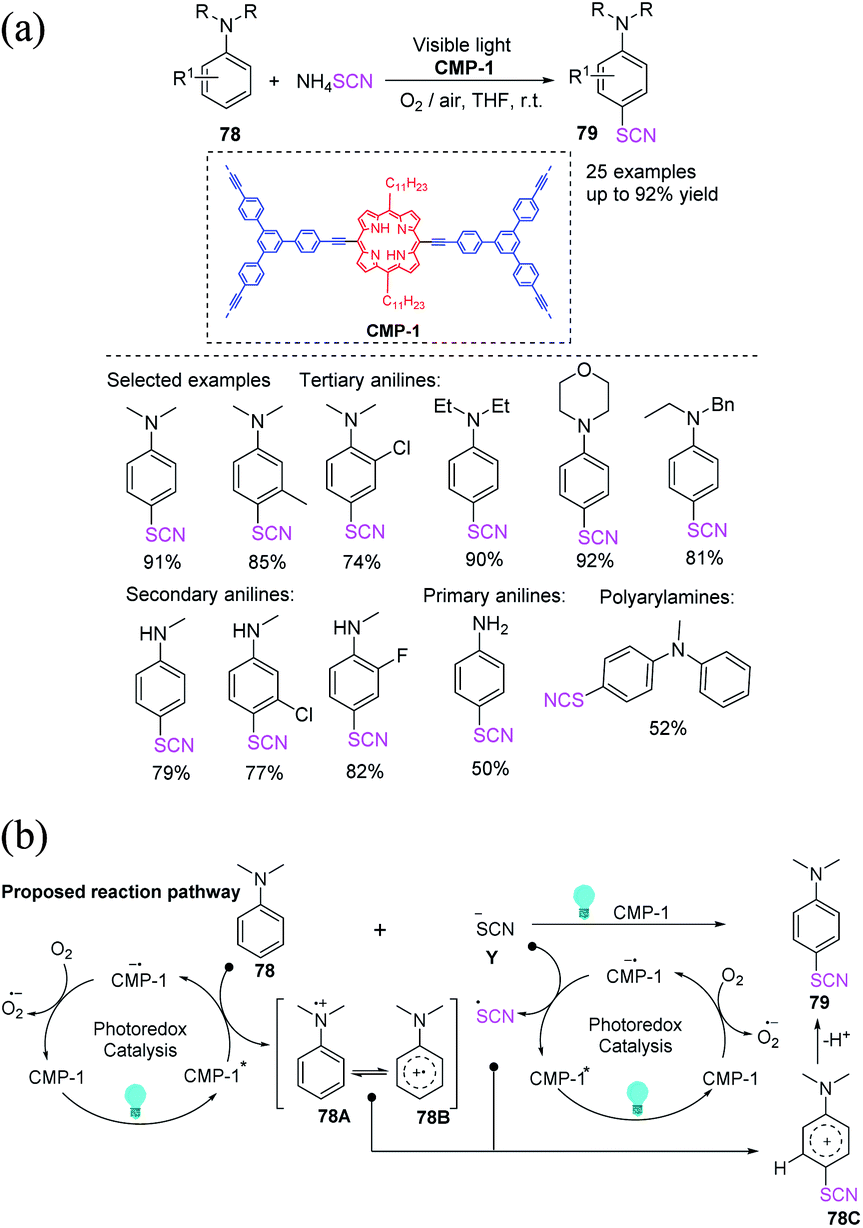
Mai et al. proposed a template-free procedure for the synthesis of porphyrin-based CMP tubes (CMP-1), which are generated by the scrolling and shutting of ribbon-like structures. The author used CMP-1 as a metal-free photocatalyst that demonstrates outstanding photocatalytic efficiency for the visible-light-driven chemoselective thiocyanation of anilines 78, obtaining excellent to high yields 79 with respect to the transformation of various types of anilines under ambient conditions.33 The photocatalyst can be used multiple times. CMP-1 plays two functions in photocatalytic thiocyanation according to the reaction mechanism. CMP tubes can be used as reusable carriers for single-atom catalysts in a variety of catalytic processes (Scheme 26a). Visible light is used to convert CMP-1 to CMP-1*. CMP-1* triggers two separate SET reactions: one that oxidizes an aromatic amine (78) to a radical cation, including intermediate 78A and its resonance structure (78B), and another that transform the –SCN anion (Y) to a ˙SCN free radical. The excited CMP-1* is then transformed into the radical anion CMP-1˙−. CMP-1˙− is a molecular oxygen oxidized to the initial state of CMP-1, completing the photoredox cycle. Additionally, the ˙−SCN free radical forms a cationic intermediate 78C when it connect to the radical cation 78B. The para-thiocyanated product 79 is generated through the rearomatization of the intermediate 78C by losing a proton (Scheme 26b).
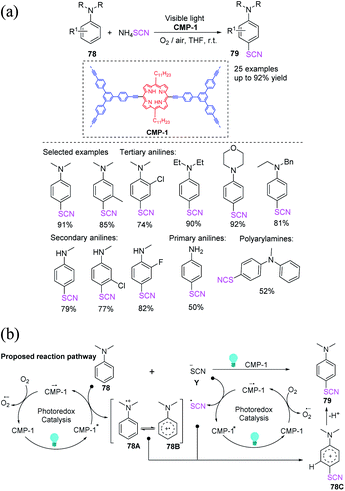 |
| | Scheme 26 a) Visible-light mediated thiocyanation of anilines 78. (b) Proposed mechanism for the visible-light-mediated thiocyanation of anilines 78. | |
2.4 Photochemical-induced C(sp)–SCN bond formation through thiocyanation reaction
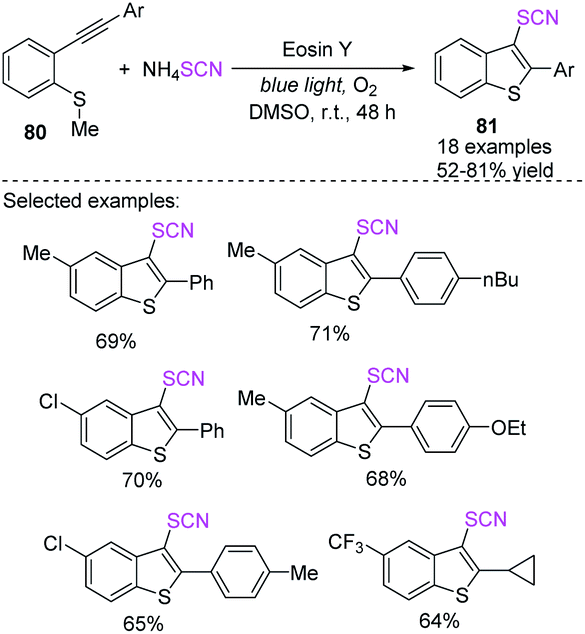
Kshirsagar et al. designed a photoredox-catalyzed simple green synthetic technique that uses visible light as a green source in the production of 3-thiocyanatobenzothiophenes 81 with an extensive substrate capacity in good to high yields.34 In the photoredox-catalyzed anion oxidation of the ammonium salt of a thiocyanate, they used EY and moleculer oxygen as a terminal oxidant, which incorporates the radical annulation of 2-alkynylthioanisoles 80 at room temperature (Scheme 27).
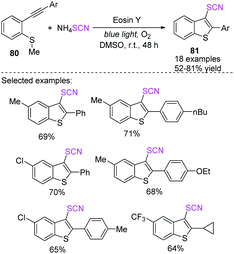 |
| | Scheme 27 Photoredox-catalyzed method for the synthesis of 3-thiocyanatobenothiophene 81. | |
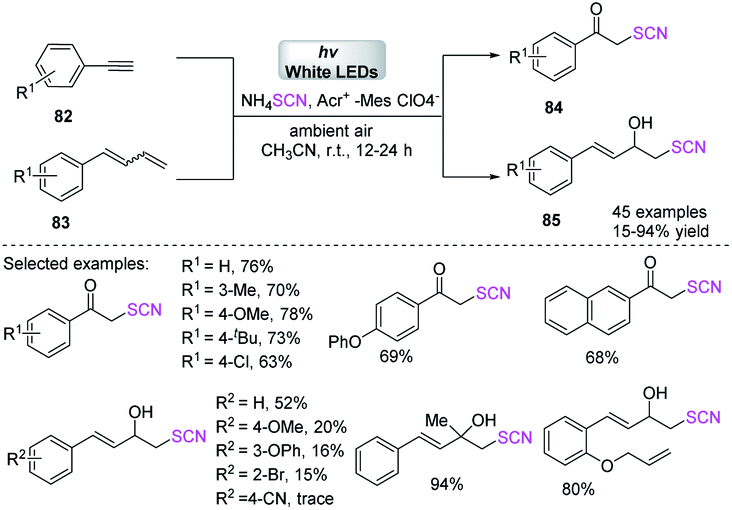
Gullapalli et al. reported a regio- and stereo-selective photocatalytic visible-light-mediated process for obtaining aryl α-thiocyanoketones 84 and α-thiocyanoalcohols 85 with excellent to high yields as of initiated aryl ultimate alkynes 82 and aryl conjugated dienes 83.35 They used ammonium thiocyanate as a thiocyanate source and sub-stoichiometric photocatalyst in ambient air. This technique is also suitable for the rapid one-pot synthesis of 2-hydroxy 4-substituted arylthiazoles, which is a potentially medicinal compound (Scheme 28).
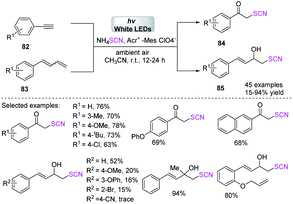 |
| | Scheme 28 Visible light promoted synthesis of aryl α-thiocyano ketones 84/thiocyano alcohols 85. | |
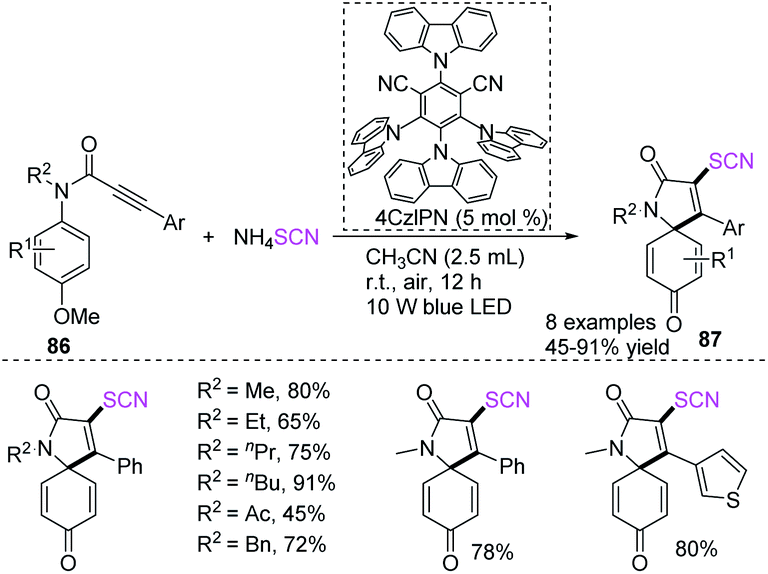
Yu and coworkers successfully created visible light mediated photocatalytic techniques for the synthesis of 3-thiocyanato azaspiro[4,5]trienones 87 with a wide reaction scope and high functional group acceptance using a radical-initiated cascade annulation reaction of N-arylpropiolamides 86 with ammonium thiocyanate and 2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN) as an photocatalyst (Scheme 29).36
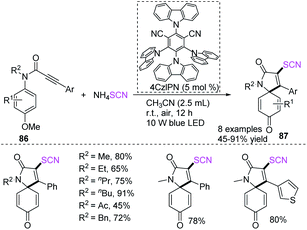 |
| | Scheme 29 Synthesis of 3-thiocyanato azaspiro [4,5] trienones 87. | |
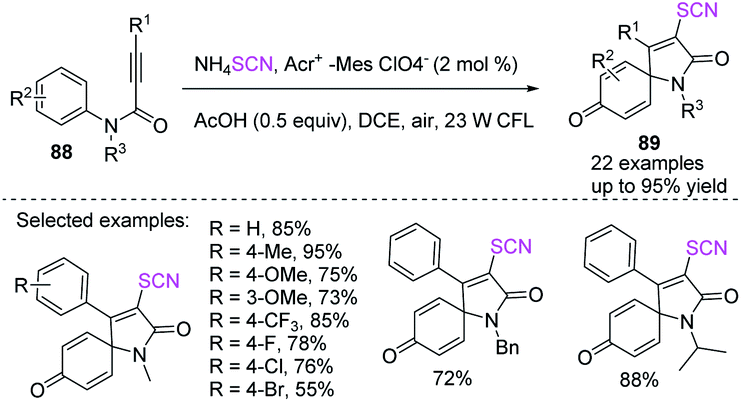
He et al. employed a visible-light-mediated metal-free thiocyanate radical addition/ipso-cyclization/oxidation cascade process to synthesize thiocyanato-containing azaspirotrienediones 89 in excellent to high yields.37 They used N-phenylpropanamide 88 with ammonium thiocyanate as a precursor to the thiocyanate free radical, DCE (1,2-dichloroethane) as a solvent, and recyclable air oxygen as the only protective reagent and oxygen source (Scheme 30).
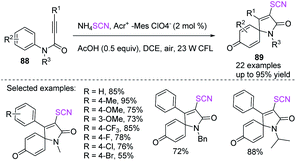 |
| | Scheme 30 Synthesis of thiocyanato-containing azaspirotrienediones 89. | |
3. Electrochemical-induced C–SCN bond formation through thiocyanation reaction
Oxidative cross-coupling has emerged as one of the most basic strategies for forming novel chemical bonds in the last decade.38 Without pre-functionalization, conveniently accessible starting materials are selected as substrates, significantly reducing the figure of synthetic processes. Nevertheless, an additional oblational oxidant is frequently needed to assist the oxidative cross-coupling procedure, resulting in significant material consumption and reduction in the atom economy of a complete transformation. Furthermore, chemical oxidants may have undesirable side effects, such as over-oxidation. As a result, achieving oxidative cross-coupling in the absence of chemical oxidants is extremely desirable.
Therefore, oxidative coupling approaches that do not require the use of chemical oxidants are urgently needed. Organic synthetic chemists have been paying particular attention to electrochemical organic synthesis methods that are effective, green, and environmentally benign.6,38 The exploit of electrons as mass-free reagents eliminates the need for a stoichiometric amount of chemical oxidant, resulting in no waste formation. Numerous electrochemical oxidation cross-coupling reactions have been exploited for the C–H bond activation through thiocyanation for the construction of C–S bonds in recent years.38n
3.1 Electrochemical-induced C(sp3)–SCN bond formation through thiocyanation reaction
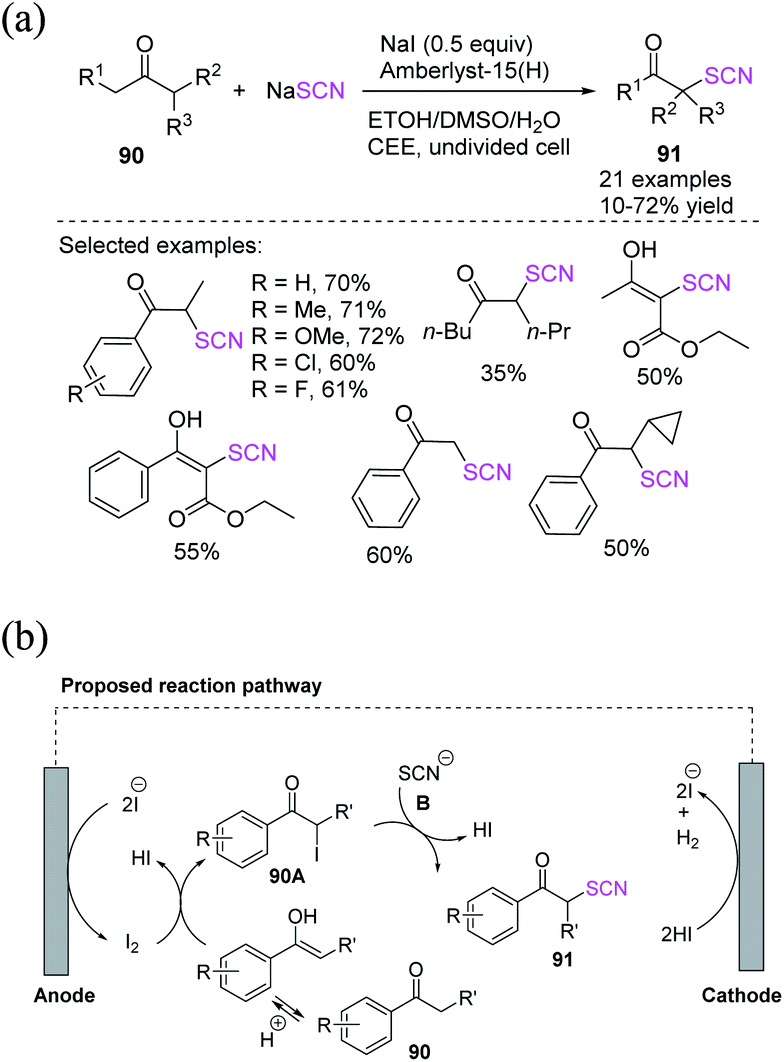
Liang et al. used NaI (sodium iodide) as a redox catalyst, sodium thiocyanate as a thiocyano source, and heterogeneous solid salt Amberlyst-15 (H) as a proton catalyst in a simple undivided cell to achieve the effective electrochemically oxidative α-C–H thiocyanation of ketones under constant current without the use of external conductive salts.39 They demonstrated that solid salt can be reused up to five times without showing decrease in effectiveness in this electrochemical oxidation thiocyanation process, which does not require the addition of an additional conductive salt (Scheme 31a). The process occurs with the anodic oxidation of the iodide ion to produce molecular iodine, which then reacts with the enol isomer of ketone 90 to produce α-iodo ketone 90A, as well as one molecule of HI. After the essential intermediate 90A is generated, it undergoes a nucleophilic substitution reaction with NaSCN or heterocyclic thiols. This process results in the final α-thiocyanation product 91, which is followed by the formation of a second molecule of HI. Consequently, the HI produced in situ is reduced to H2 on the cathode surface. The iodide ion is replenished during the reaction from stages of the α-iodination of ketone 90, resulting in 90A. After the nucleophilic substitution of 90A to 91, the iodide ion rejoins the redox catalyst process, requiring just a sub-stoichiometric or catalytic quantity of iodide ion (Scheme 31b).
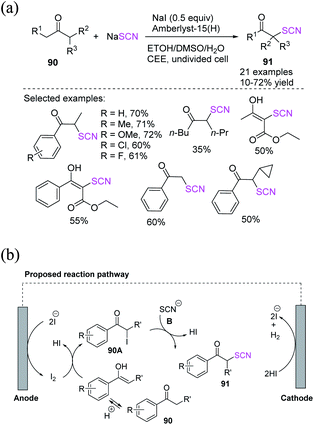 |
| | Scheme 31 a) Electrochemical oxidative α-thiocyanation of various ketones 90. (b) Proposed mechanism for electrochemical oxidative α-thiocyanation of various ketones 90. | |
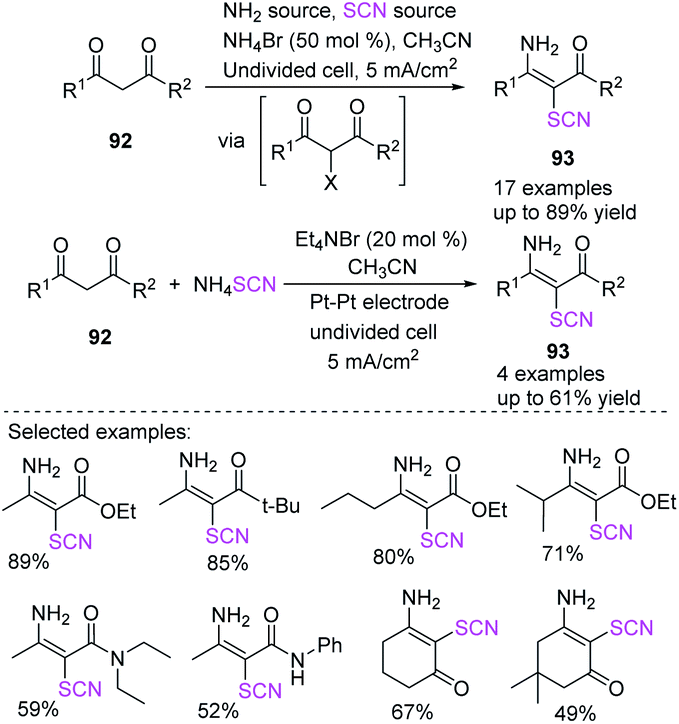
Kang et al. developed an electrochemically induced technique for the synthesis of 3-amino-2-thiocyanato-α,β-unsaturated carbonyl derivatives 93, using a single reagent or a mixture of ammonium acetate and potassium isocyanate without the use of any other conducting salt and a sub-stoichiometric amount of ammonium bromide.40 Additionally, the procedure can be performed with catalytic concentrations of bromide salt. When ammonium thiocyanate was used as a redox catalyst and 1,3-dicarbonyl compounds 92 were combined in the presence of Et4NBr (tetraethylammonium bromide), relevant adducts in moderate to good yields were obtained (Scheme 32).
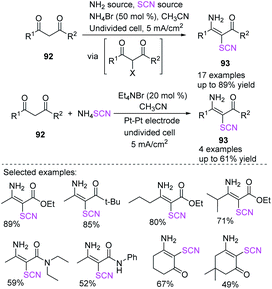 |
| | Scheme 32 Construction of precious 3-amino-2-thiocyanato-α,β-unsaturated carbonyl derivatives 93 by electro-synthesis. | |
3.2 Electrochemical-induced C(sp2)–SCN bond formation through thiocyanation reaction
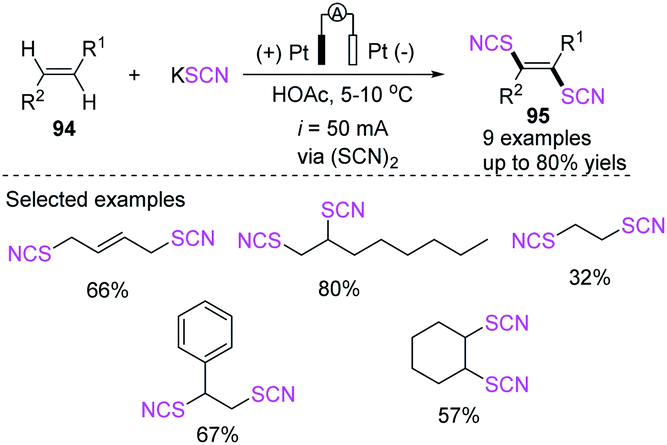
3.2.1 Electrochemical-induced thiocyanation of olefins. Electrocatalytic thiocyanation of olefins has been studied. In 1973, De Klein generated thiocyanate through the electro-oxidation of thiocyanate and generated dithiocyanation products 95 through the addition reaction of olefins 94.41 The authors stated that electrochemical thiocyanogen generation has several advantages over synthetic thiocyanogen sources in the thiocyanation of double bonds: initially, no process for the halogenation of the double bond is viable (this is particularly beneficial when a double bond responds slowly to thiocyanogen but reacts quickly with Cl or Br). Second, the oxidation potential may be easily manipulated either through controlled potential electrolysis or by performing at a set current density and keeping the reactant concentration over a certain point, reducing the number of side products produced. A series of novel dithiocyanate products 95 were obtained in 16–80% yield through constant current electrolysis at 5–10 °C in an acetic acid environment, which laid the theoretical foundation for the study of electrocatalytic thiocyanation reactions (Scheme 33).
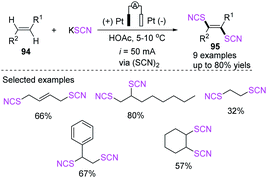 |
| | Scheme 33 Electrocatalytic dithiocyanation of olefins 94. | |
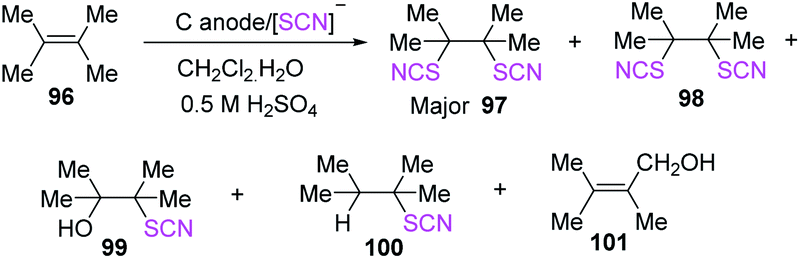
Becker et al. employed “the anodic one-pot approach” in a two-phase acidic environment of water and dichloromethane for the thiocyanation and isothiocyanation of polysubstituted olefins 96 in 2015.42 They evaluated the effects of various acids, anodes, current densities, charge consumption, and electrolysis procedures on the reactions, using tetramethyl ethylene as the template substrate. The two main products (major 97 and minor 98) were produced in moderate yields in two-phase media. Other alkyl-substituted olefins had similar addition products, but the two SCN addition products were the most important. The products of styrene and ninhydrin substrate reactions in a homogenous water-acetonitrile media were mostly SCN/OH adducts (Scheme 34).
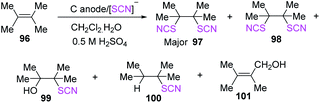 |
| | Scheme 34 Thiocyanation of polysubstituted olefins 96. | |
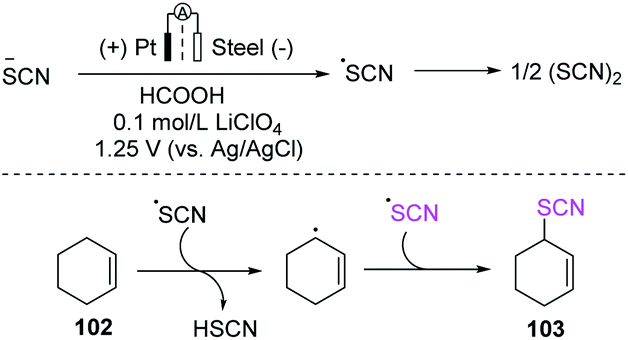
Becker et al. devised a two-step anodic oxidation approach for the thiocyanation of olefins 102 in 2016.43 First, ammonium thiocyanate was utilized as a source of thiocyanic acid in a separation cell with formic acid as a solvent and lithium perchlorate (LiClO4), and thiocyanogen (SCN)2 was formed through anodic oxidation in a constant voltage mode at 1.25 V. They used a platinum sheet as the anode, a steel sheet as the counter electrode, and Ag/AgCl as the reference electrode. Olefin derivatives were introduced and oxidized by free radicals. The desired product 103 was produced by a radical reaction pathway by using olefin derivatives (Scheme 35).
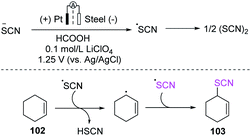 |
| | Scheme 35 Electrocatalytic thiocyanation of olefins 102. | |
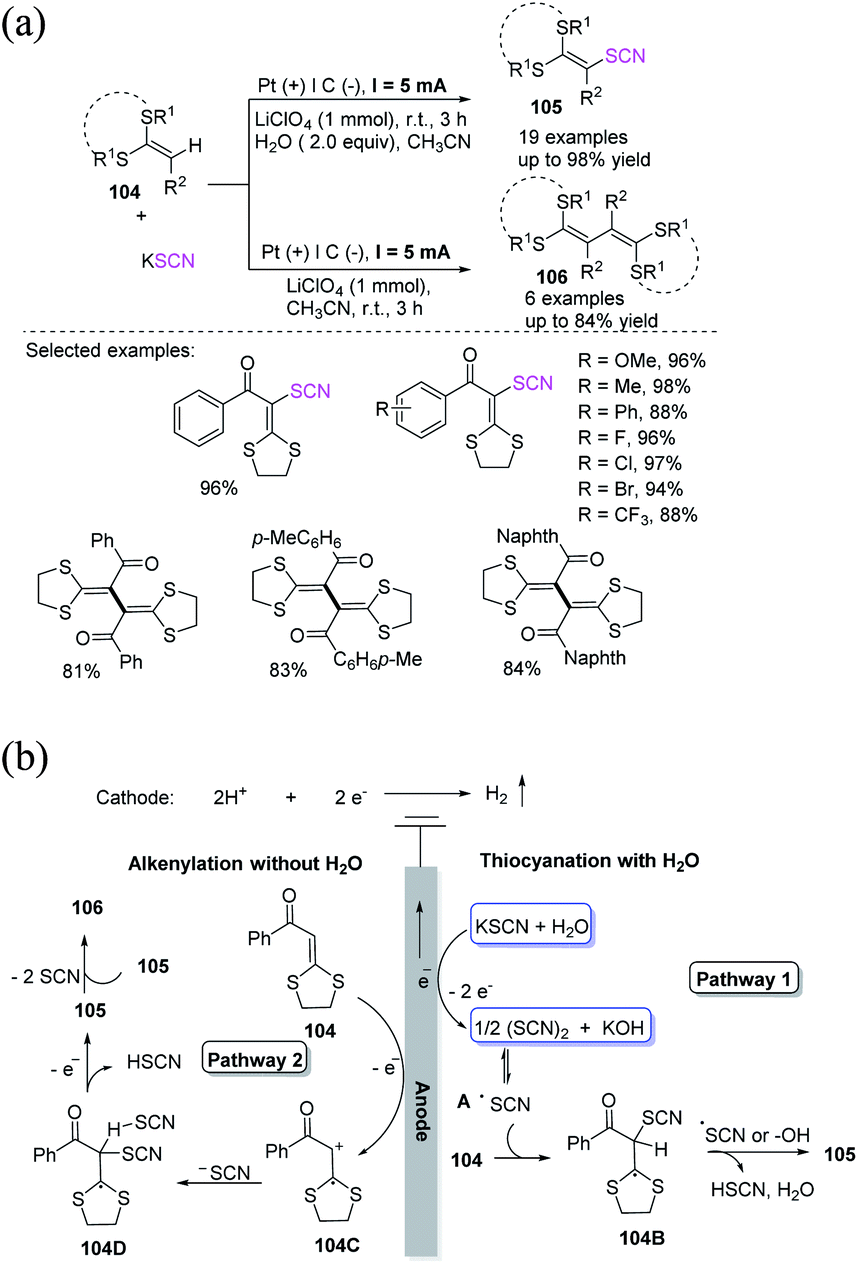
Under electrochemical oxidation, Wen et al. established an extremely effective and H2O-controlled thiocyanation as well as alkenylation of ketene dithioacetals 104 with potassium thiocyanate in good to high yields and exceptional lenience to functional groups.44 In contrast to previously disclosed strategies for fabricating internal alkenes, this electrochemical-oxidation-driven synthetic technique circumvents the use of transition metals and extrinsic oxidizing agents. This H2O-controlled anodic oxidation synthesis approach is green and can scalable, which are useful feature for a variety of pharmaceutical purposes (Scheme 36a). In the presence of water, potassium thiocyanate might be oxidized to (SCN)2 and is subsequently transformed into thiocyanate radical A through electrochemical anodic oxidation. The radical intermediate 104B is then obtained through the radical insertion of thiocyanate radical A to ketene dithioacetal 104, from which a proton can be extracted by another thiocyanate radical A. The product 105 is finally produced (pathway 1). In the absence of water, ketene dithioacetal 104 is oxidized on the anode's surface and generates a radical cation intermediate 104C, which is then proceeded with the basic electrophile potassium thiocyanate to create intermediate 104D, which loses an electron and a proton to create intermediate 105. After the removal of the pseudohalogen group SCN, the home-coupling product 106 is produced (pathway 2; Scheme 36b).
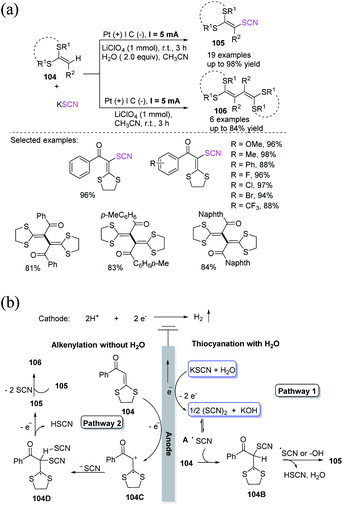 |
| | Scheme 36 a) Thiocyanation and alkenylation of ketene dithioacetals 104. (b) Proposed mechanism for thiocyanation and alkenylation of alkenes 104. | |
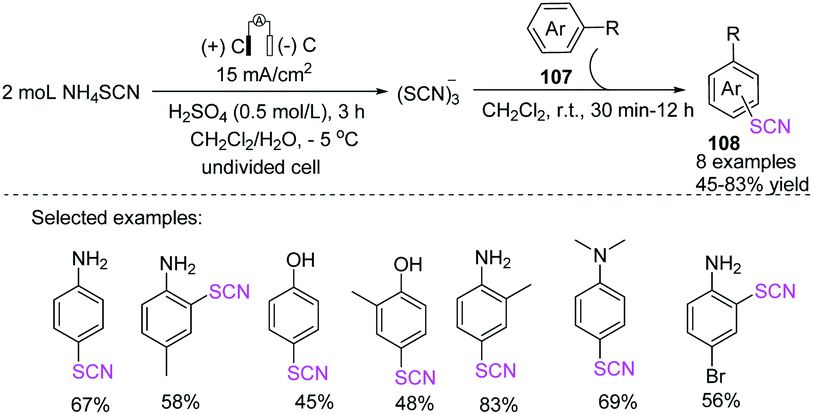
3.2.2 Electrochemical-induced C(sp2)–SCN bond formation through the thiocyanation of aromatic compounds. Thiocyanate derivatives are useful organic synthesis intermediates, especially in the creation of heterocyclic molecules. Gurjar et al. succeeded in selectively thiocyanating aniline and phenol derivatives 108 using a two-phase electrolysis of ammonium thiocyanate to generate (SCN)3− thiocyanate in 1992 (Scheme 37).45 Furthermore, they explored the effects of experimental parameters, such as electrolysis temperature, electrolysis time, non-aqueous solvent nature, and non-aqueous phase volume fraction on current efficiency. Cyclic voltammetry studies revealed that (SCN)3− was generated in the aqueous phase and then carried into the non-aqueous phase to participate in the current.46
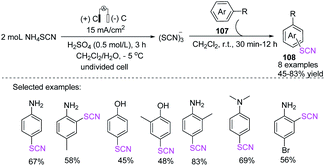 |
| | Scheme 37 Thiocyanation of aniline and phenol derivatives 107. | |
Kokarekin et al. evaluated the development of metal-free techniques for electro-oxidative C–H thiocyanation of a wide spectrum of high- and low-reactive (hetero) arenes (109, 110) in an undivided cell with GC electrodes. The desired aryl thiocyanates (111, 112) were obtained in moderate yields.47 The proposed technique for forming carbon-sulfur bonds has the advantages of having simple reaction conditions and being easily accessible and environmentally sustainable materials. Additionally, the technique does not require chemical oxidants and metal-containing catalysts (Scheme 38).
 |
| | Scheme 38 Electrochemical-induced synthesis of aryl thiocyanates 111, 112. | |
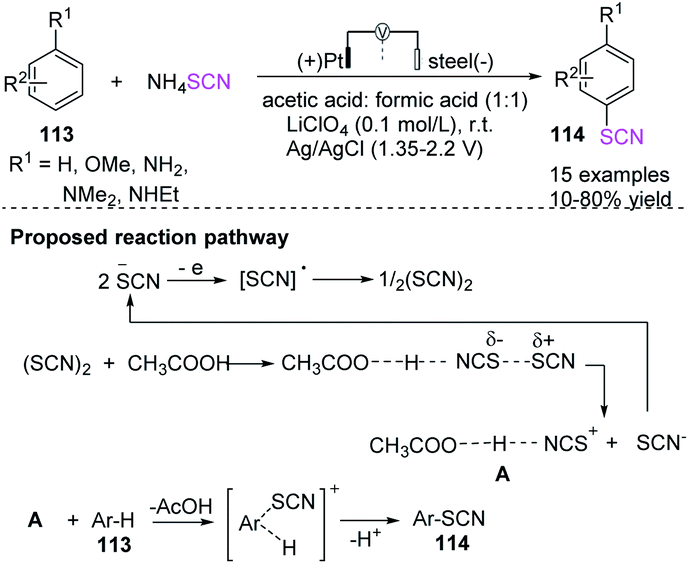
Becker et al. explored an environmentally sustainable thiocyanation of aromatic compounds 113 containing different derivatives of anisole and aniline to obtain aromatic thiocyanates 114 in the presence of organic acids.48 The initial electrochemical process is the anodic oxidation of the thiocyanate anion to its radical (SCN), followed by dimerization to thiocyanogen (SCN)2. The acidic solvent polarizes the latter, leading to the thiocyanate derivative and attacking the aromatic nucleus of the substrate. Mono- and disubstituted aromatics have excellent regioselectivity and high isomer-selectivity in their single thiocyanate derivatives (Scheme 39).
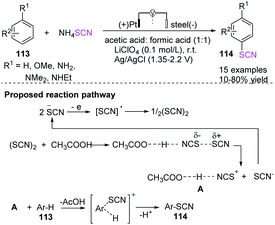 |
| | Scheme 39 Electrochemical thiocyanation of aromatic compounds 113. | |
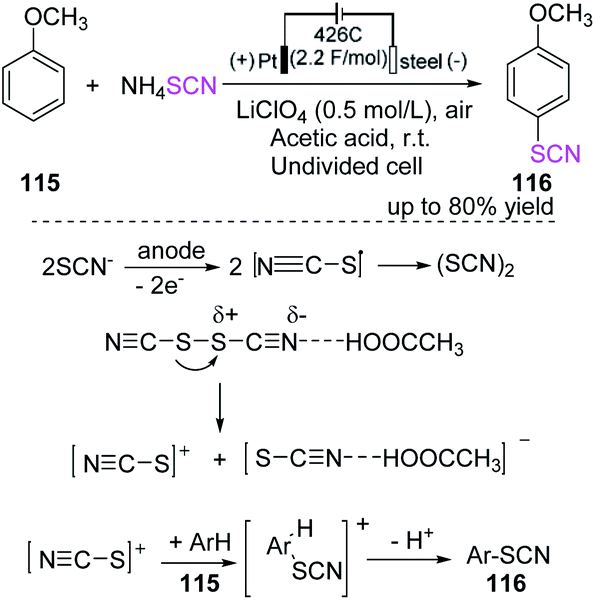
Gitkis et al. examined the straightforward one-pot electrochemical-induced thiocyanation of methoxy benzene (anisole) 115, which is highly regioselective in glacial acetic acid for the construction of 1-methoxy-4-thiocyanatobenzene 116.49 The yield of the product increases to over 80% at room temperature under the specified optimum conditions, and no detectable para-thiocyanogen owing to thiocyanogen polymerization is observed. Furthermore, controlled potential electrolysis is more effective than constant-current electrolysis in the synthesis of 1-methoxy-4-thiocyanatobenzene 116 (Scheme 40).
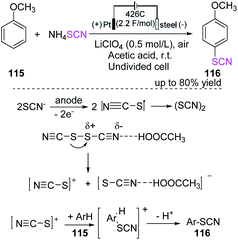 |
| | Scheme 40 One-pot electrochemical thiocyanation of methoxybenzene 115. | |
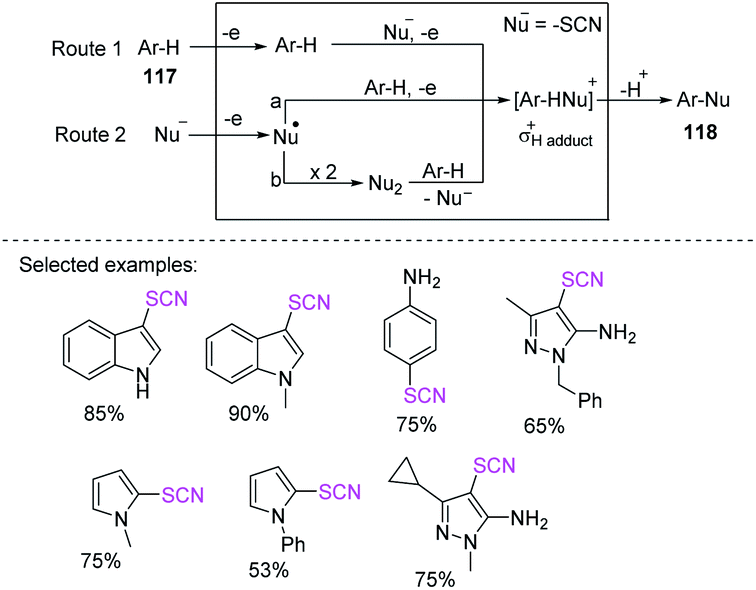
3.2.3 Electrochemical-induced C(sp2)–SCN bond formation thiocyanation of heteroaromatic heterocycles. In 2014, Petrosyan et al. successfully achieved the thiocyanation of N-heterocyclic derivatives 117, such as indole, N-methylpyrrole, pyrazole, and aniline through the anodic oxidation of thiocyanate to produce thiocyanate heterocycles 118 and nucleophilic substitution in aromatic compounds with yields of up to 95%.51 They utilized ammonium thiocyanate and acetonitrile as a solvent at 25 °C with a Pt anode in an undivided cell (Scheme 41).50a Notably, this method has a high yield (75%) for the construction of para-thiocyanoaniline compounds through a process that is more selective than that described by Krishnan et al.45
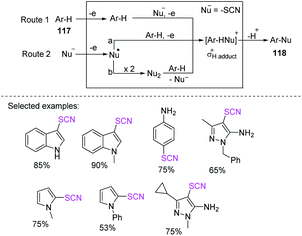 |
| | Scheme 41 Thiocyanation of heteroarenes 117 by anodic oxidation of ammonium thiocyanate. | |
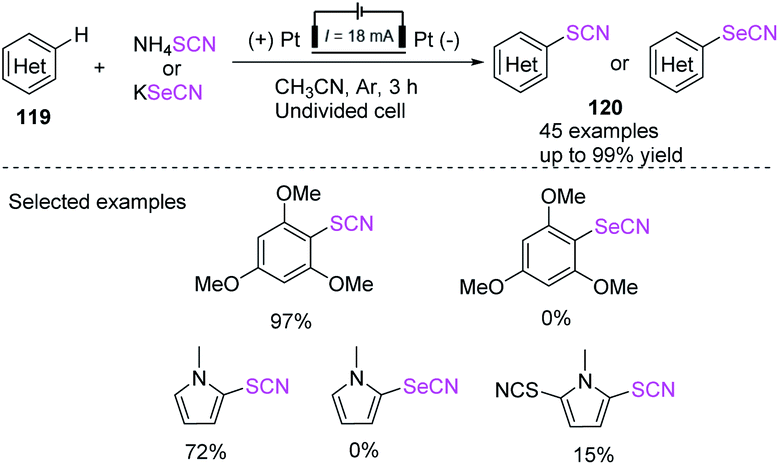
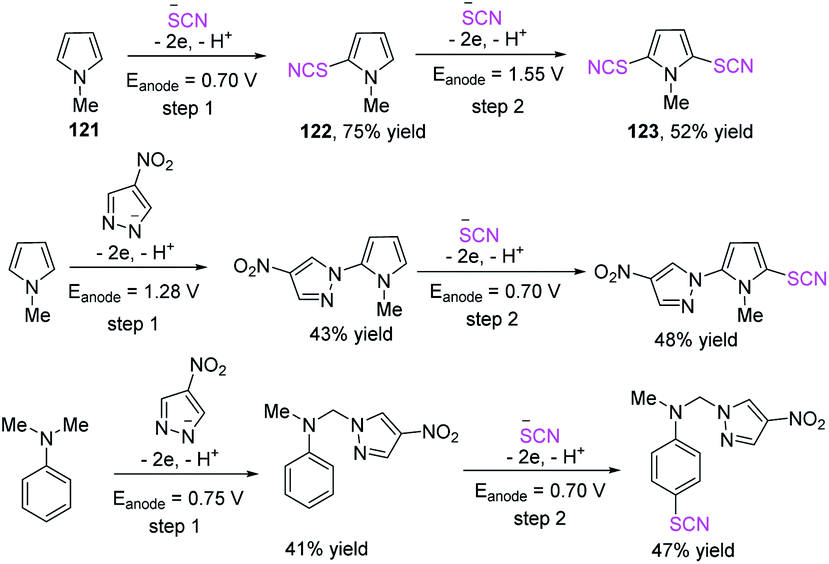
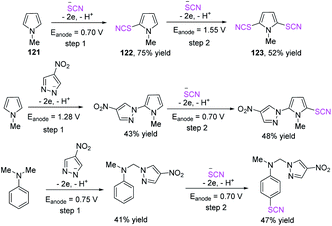
In 2018, Linhao Sun et al. achieved a low-cost and efficient technique for the thiocyanation and selenocyanation of electron-rich aromatics 119.50b This method does not require the use of any catalyst or oxidant in the thiocyanation and selenocyanation reactions of diverse hetero-aromatic compounds, such as the derivatives of indole, aniline, pyrrole, and anisole, to construct thio-and selenocyanated aromatic heterocyclic compounds 120 with various substituents in 52–99% yields. Unfortunately, this method cannot generate the 1,3,5-trimethoxy-2-selenocyanatobenzene product and the selenocyanated N-methylpyrrole product. It produces 1-methyl-2,5-dithiocyanato-1H-pyrrole product but in low yields (Scheme 42). In 2019, Yaubasarova et al. proposed a two-step controlled voltage electrolysis strategy based on this method and successfully achieved the double C–H bond activation thiocyanation of N-methylpyrrole 121 in 52% yield 123.51 They demonstrated that the procedure broadens the scope for the production of promising therapeutic drugs through direct C–S and C–N coupling, in which anode acts as a green oxidizing agent and contributes to the construction of polyfunctional hybrid molecules (Scheme 43).
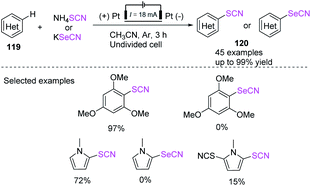 |
| | Scheme 42 Thiocyanation and seleno-cyanation techniques for electron-rich arenes 119. | |
 |
| | Scheme 43 Anodic C–H functionalization of N-methyl-1Hpyrrole 121 and N,N-dimethylaniline. | |
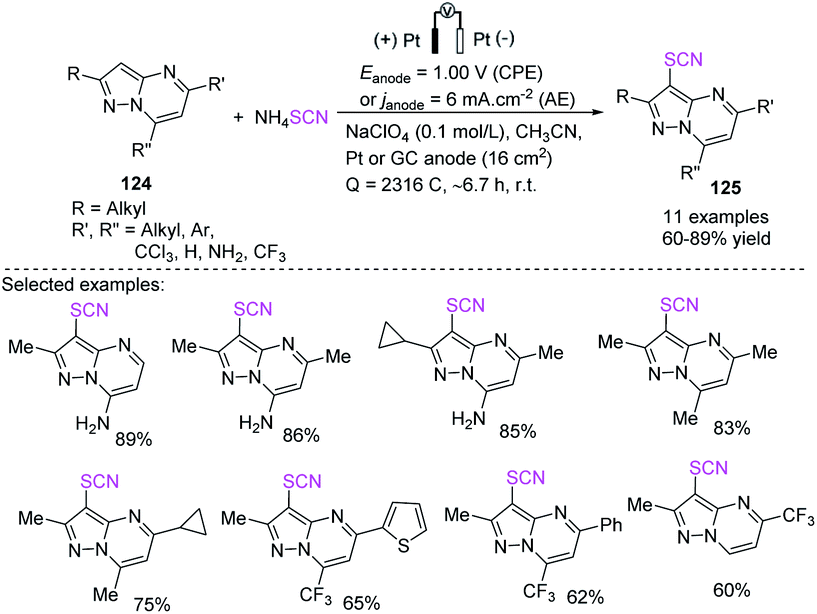
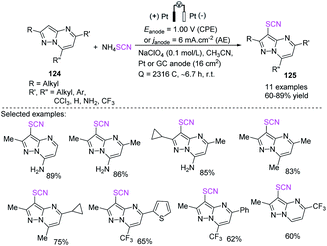
Kokorekin et al. established successful electrooxidative C–H thiocyanation of pyrazolo[1,5-a]pyrimidines 124 that have high and low levels of reactivity and utilize ammonium thiocyanate or potassium thiocyanate as a thiocyanating agent at room temperature.52 Owing to the moderate reaction conditions and excellent carbon-sulfur (C–S) bond-building efficiency, this method, which is based on the electrooxidative C–H thiocyanation of pyrazolo[1,5-a]pyrimidines 124, is expected to offer benefits to future applications. The development of the thiocyanation procedure, conversion of aryl thiocyanates to new thiols, and the considerable improvement in the antifungal activity of arenes when the thiocyano group is introduced are extremely important (Scheme 44).
 |
| | Scheme 44 Electrooxidative C–H thiocyanation of pyrazolo[1,5-a]pyrimidines 124. | |
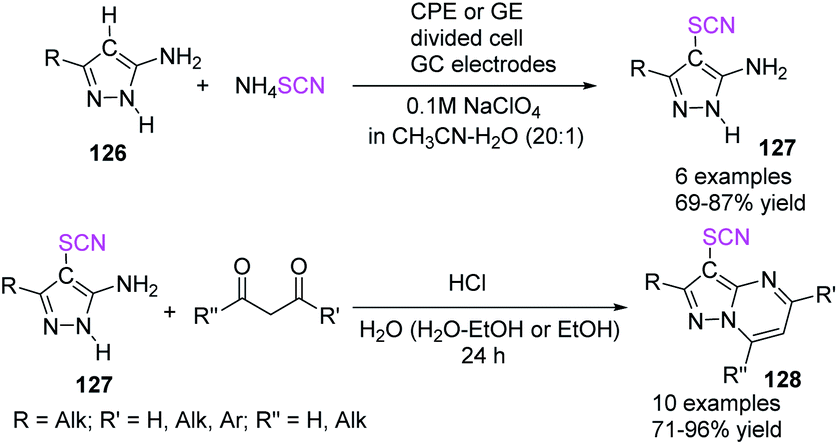
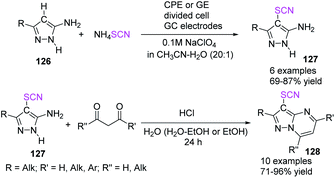
In 2020, Kokarekin et al. presented a novel facile metal-free electrochemical-mediated (anodic) C–H thiocyanation of 5-aminopyrazoles 126 for the construction of 3-thiocyanatopyrazolo [1,5-a]pyrimidines 128 in good to high yields.53 The author used ammonium thiocyanate as a thiocyanate ion source, obtaining 4-thiocyanato-5-aminopyrazoles 127 and then conducted chemical condensation with 1,3-dicarbonyl compounds or their derivatives. This innovative strategy for the two-stage synthesis of 3-thiocyanatopyrazolo[1,5-a]pyrimidines 128 is appealing because it combines electrochemical and chemical methodologies, has simple reaction conditions, uses widely accessible materials and solvents, is flexible, and conveniently isolates desired products (Scheme 45).
 |
| | Scheme 45 Synthesis of 3-thiocyanatopyrazolo [1,5-a]pyrimidines 128. | |
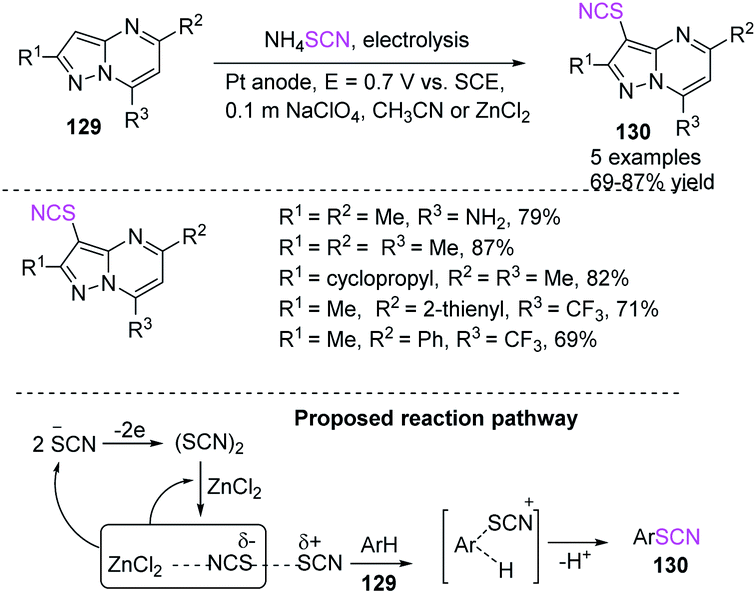
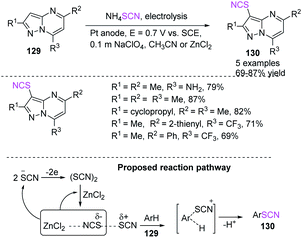
Petrosyan et al. established a method for the effective electrochemical-induced thiocyanation of pyrazolo[1,5-α]pyrimidines 129 in good to high yields.54 They used ammonium thiocyanate as a source of thiocyanogen, which is electrochemically generated on a Pt anode in the presence of acetonitrile as a solvent. The oxidation potentiality of a substrate determines system performance, which can be controlled using Lewis acid additives (Scheme 46).
 |
| | Scheme 46 Thiocyanation of pyrazolo[1,5-a]pyrimidines 129. | |
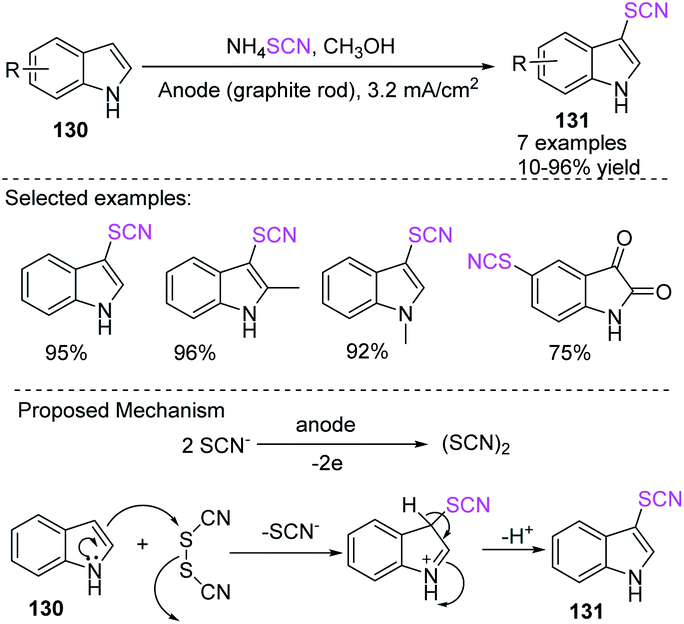
Nikoofar et al. discovered an easy and simple one-pot process for the electrochemical-mediated anodic thiocyanation of nitrogen-containing (hetero) aromatic compounds 130 in good to excellent yields 131 in methanol at ambient temperature.55 They studied the thiocyanation of many aromatic compounds in the presence of ammonium thiocyanate in situ in an undivided cell, which is electrochemically generated thiocyanogen (SCN)2 at a graphite electrode (anode) without the use of any other supporting electrolyte, toxic material, or harmful reagent. According to the possible mechanism for this process, SCN is oxidized to a radical through a one-electron oxidation process, which then dimerizes at the electrode surface for the production of thiocyanogen. The product is formed when the electrogenerated precursor thiocyanogen (SCN)2 interacts with substrates 130 (Scheme 47).
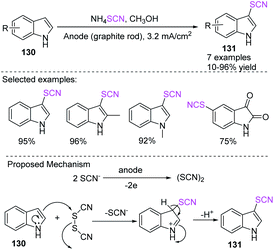 |
| | Scheme 47 Electrochemical-induced thiocyanation of indoles 130. | |
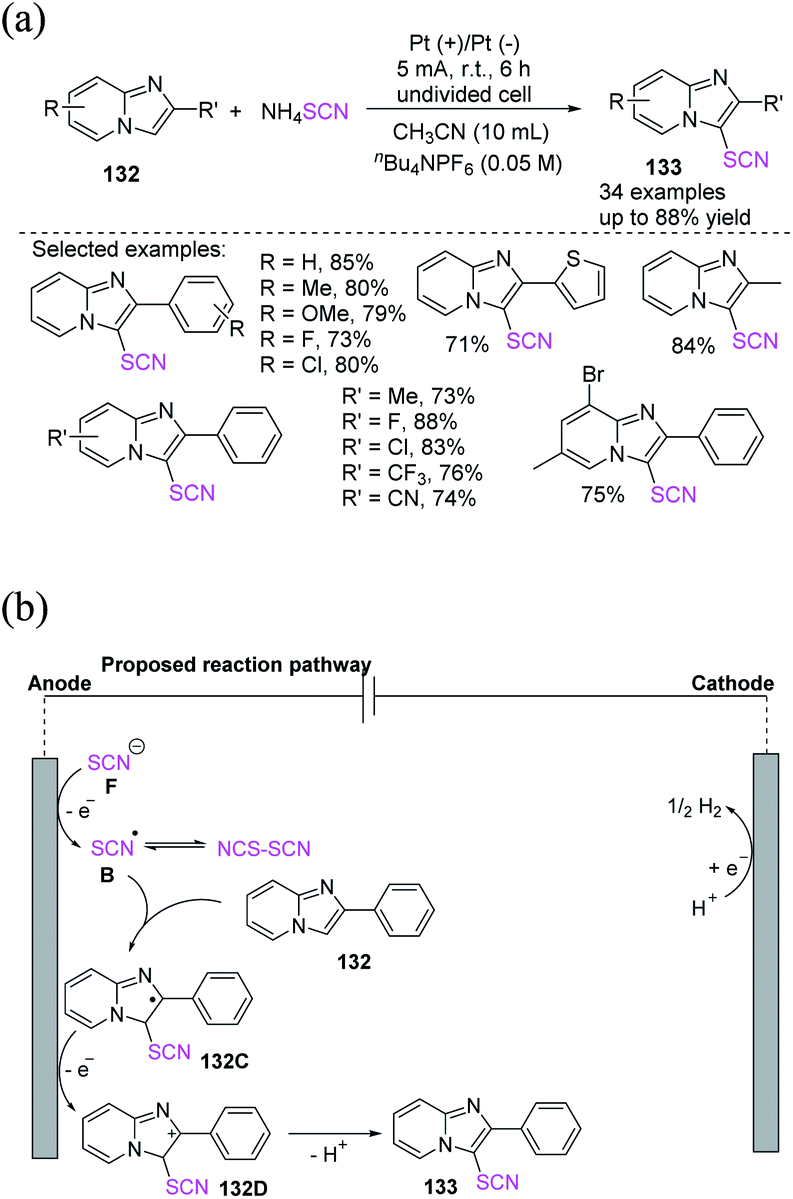
Cui et al. proposed an electrochemical approach for the oxidative C–H thiocyanation of imidazopyridines and electron-rich arenes 132, utilizing ammonium thiocyanate as a source of (thiocyanate) SCN for the formation of thiocyanated products 133 in moderate to excellent yields with a wide variety of substrates and functional groups.56 This convenient approach has great advantages in terms of thiocyanated imidazopyridine production in the field of medicinal chemistry (Scheme 48a). In the postulated reaction pathway, the thiocyanate radical intermediate B is generated through the anodic oxidation of the thiocyanate anion. Imidazopyridine 132 traps radical B, yielding the radical intermediate 132C, which is oxidized at the anode through SET to produce the cation intermediate 132D. Finally, the deprotonation of cation 132D produces 133 (Scheme 48b).
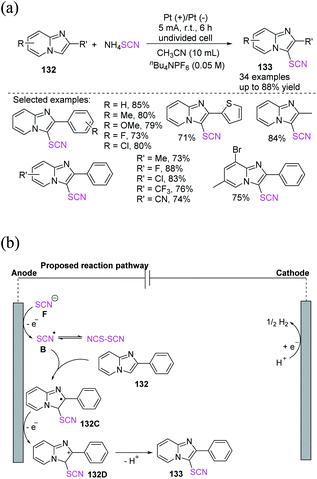 |
| | Scheme 48 a) Oxidative C–H thiocyanation of imidazopyridines 132. (b) Proposed mechanism of oxidative C–H thiocyanation of imidazopyridines 132. | |
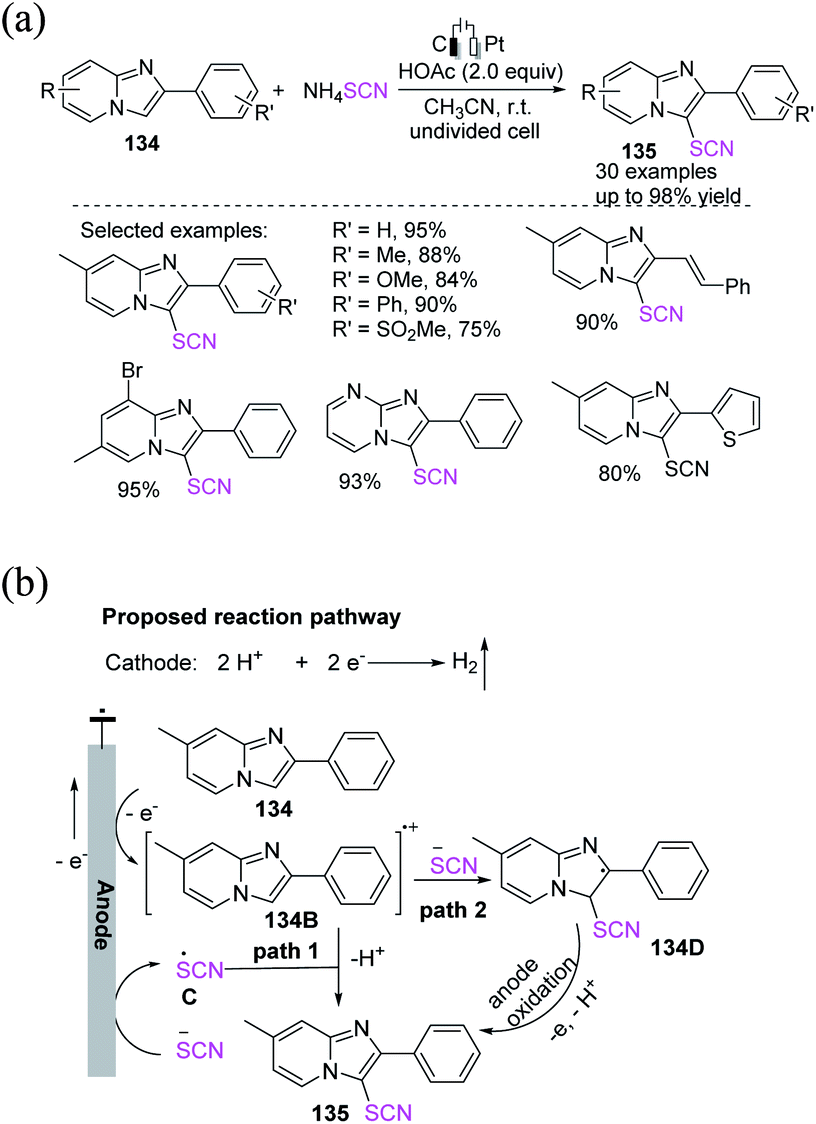
Chen et al. successfully formulated a simple and efficient electrochemical-induced oxidative thiocyanation process for imidazopyridines and imidazo[2,1-b] thiazoles 134 in a wide range of substrates.57 The process has great functional group tolerance and moderate to outstanding yields 135 because of anodic oxidation and cathode hydrogen evolution processes that do not require the use of a catalyst or an oxidant (Scheme 49a). In the proposed reaction pathways, the anodic oxidation of 7-methyl-2-phenylimidazo [1,2-a] pyridine 134 produces the radical cation 134B. Meanwhile, thiocyanate anion can be oxidized to thiocyanate radical C on the anode surface, which interacts with 134B. The intermediate is promptly deprotonated to generate 135 (path 1). Furthermore, the radical cation intermediate 134B has a low SOMO and a larger HOMO than the thiocyanate anion. The production of a radical intermediate 134D, which will then swiftly gives an electron to the anode and is deprotonated to yield the product 135, is expected to come from a substantial SOMO–HOMO interaction between the two species (path 2). Hydrogen gas is released as a result of the concurrent cathodic reduction of H+ (Scheme 49b).
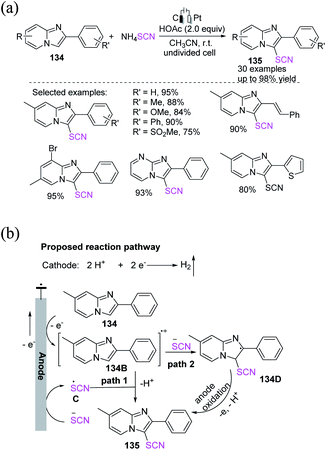 |
| | Scheme 49 a) Electrochemical oxidative thiocyanation of imidazopyridines 134. (b) Proposed mechanism of electrochemical oxidative thiocyanation of imidazopyridines 134. | |
3.3 Electrochemical-induced C–SCN bond formation through the thiocyanation of enaminones
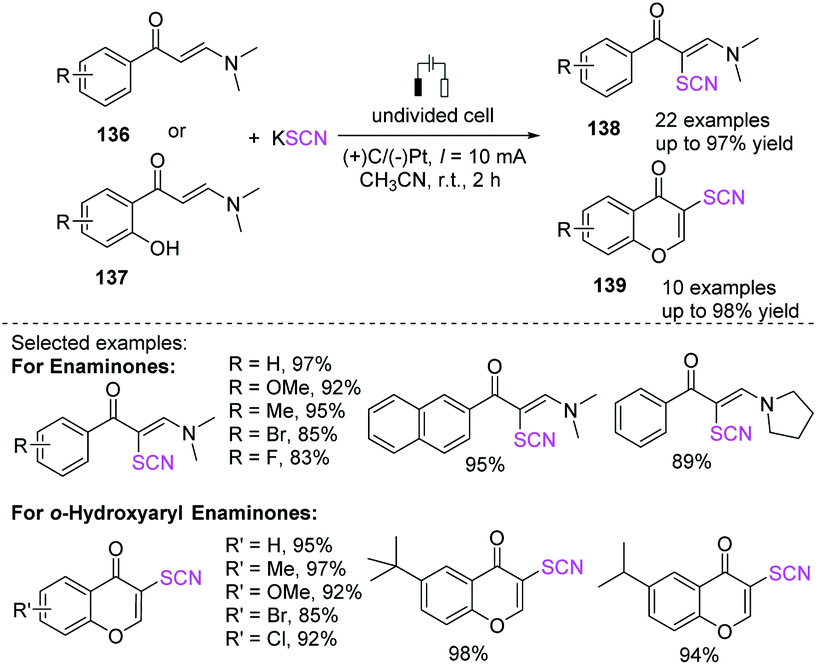
Under undivided electrolytic conditions, Zhou et al. produced a successful electrochemical-mediated thiocyanation of enaminones 136 or 137 by employing potassium thiocyanate as a thiocyanate source with a broad substrate scope and good to outstanding yields.58 This protocol produces a catalyst-free and external oxidant-free procedure for the preparation of polyfunctionalized alkenes 138 and chromones 139, in contrast to traditional methodologies for the thiocyanation of internal alkenes (Scheme 50).
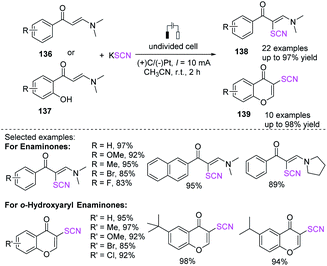 |
| | Scheme 50 Electrochemical-induced thiocyanation of enaminones 136 or 137. | |
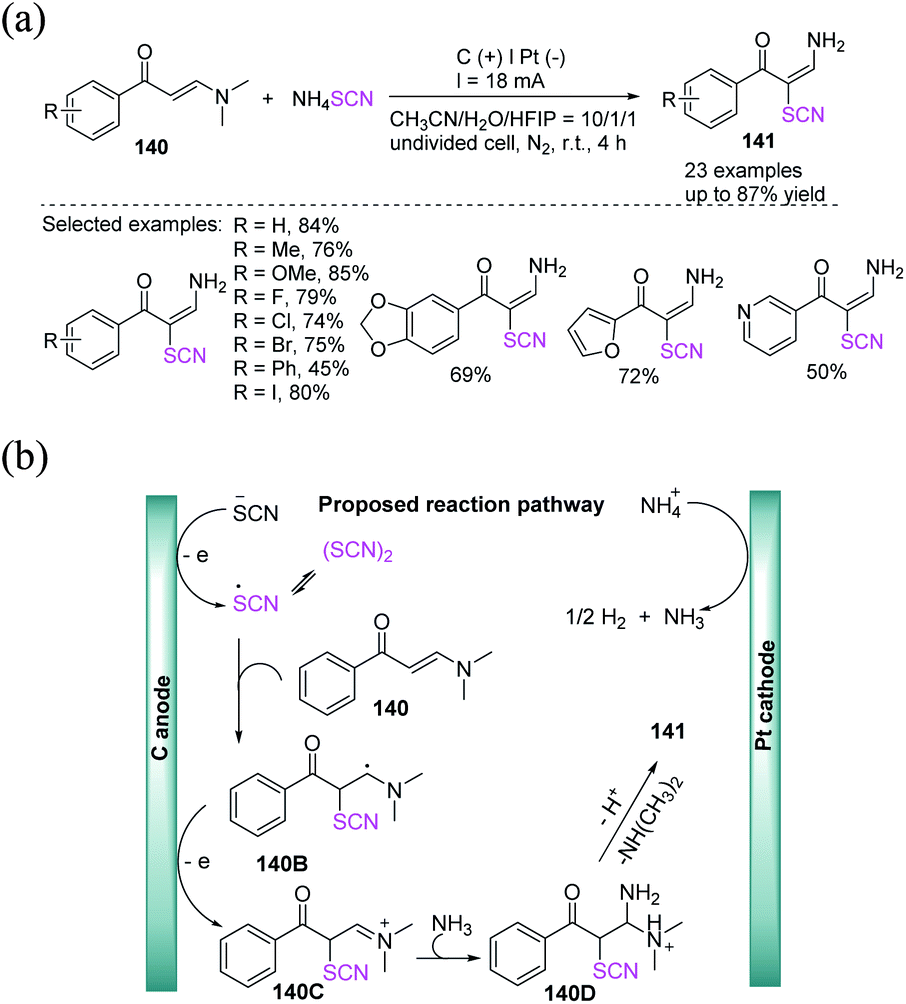
Lei et al. accomplished a highly stereoselective electrochemical technique for the production of thiocyanated enaminones 141in good to high yields. Interestingly, ammonium thiocyanate has been employed as an ammino and a thiocyanate source that comprises C–H bond thiocyanation and vinyl C–N bond trans-amination without the use of an extra oxidizing agent and a catalyst made of transition metal (Scheme 51a).59 They depicted a probable pathway for this electrochemical thiocyanation. SCN− is first anodically oxidized to the thiocyanate radical, which is rapidly dimerized to form a disulfide. The thiocyanate radical is then added to substrate 140 to produce intermediate 140B, which then undertakes single electron anodic oxidation to produce cation intermediate 140C, which is easily attacked by nucleophilic NH3 to generate 140D. Consequently, N, N-dimethylamine and proton are removed from intermediate 140D to produce the desired product 141. During the reaction, ammonium ion can be reduced at the cathode, releasing ammonia gas and hydrogen gas (Scheme 51b).
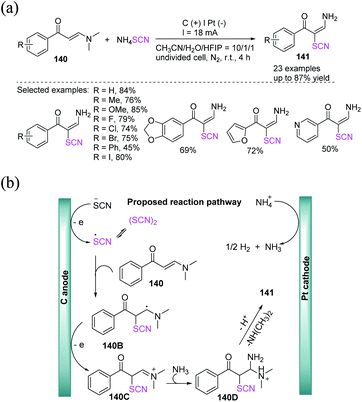 |
| | Scheme 51 a) Stereoselective electrochemical-induced synthesis of thiocyanated enaminones 141. (b) Proposed mechanism of stereoselective electrochemical-induced synthesis of thiocyanated enaminones 141. | |
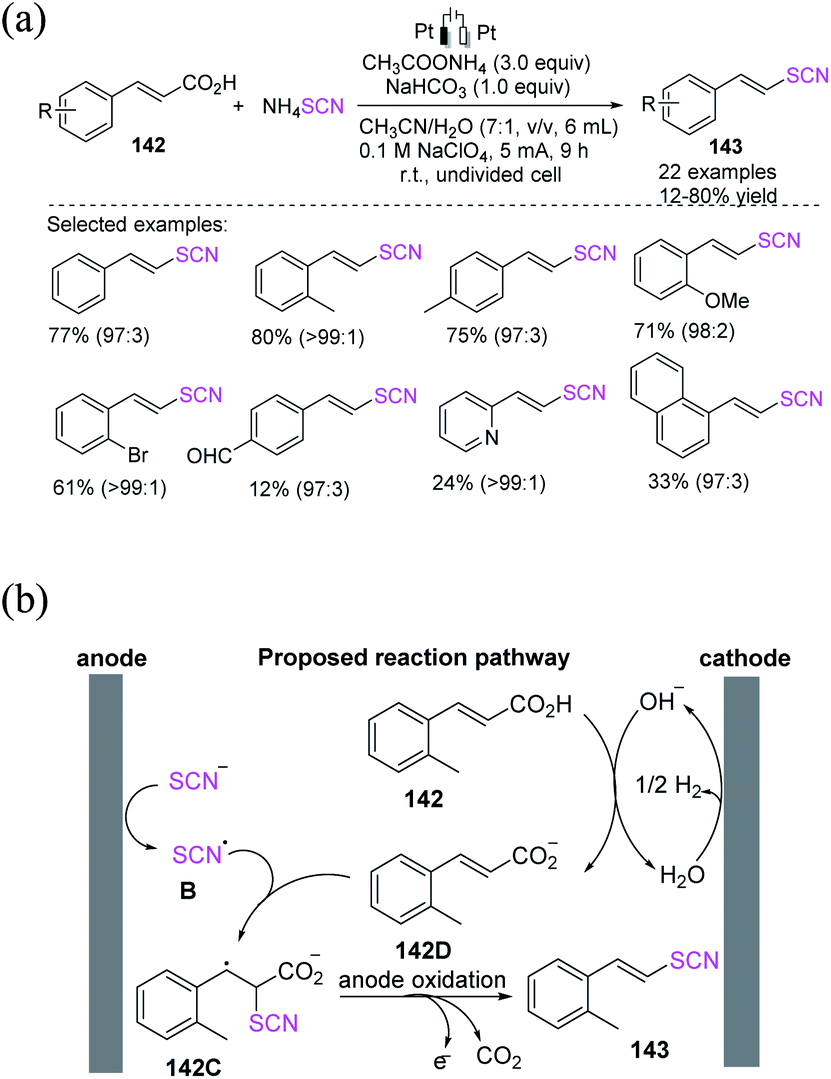
3.3.1 Electrochemical-induced C–SCN bond formation through the thiocyanation of cinnamic and boronic acid compounds. Yang et al. proposed a simple and convenient electrochemical-induced decarboxylation process for cinnamic acids by utilizing ammonium thiocyanate as a thiocyanate source for the construction of vinyl thiocyanates in moderate to high yields (Scheme 52a).60 The anodic electrode creates a thiocyanate radical intermediate B first when the thiocyanate anion is oxidized. By combining B and 142D, the intermediate 142C is produced. 142C is finally decarboxylated to yield 143, a vinyl thiocyanate. Anodic oxidation provides (SCN)2, which then participates in the electrophilic addition to 142, accompanied by the decarboxylation stage (Scheme 52b).
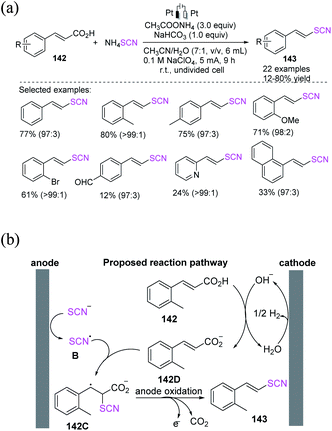 |
| | Scheme 52 a) Electrochemical decarboxylation of cinnamic acids 142. (b) Proposed mechanism of electrochemical decarboxylation of cinnamic acids 142. | |
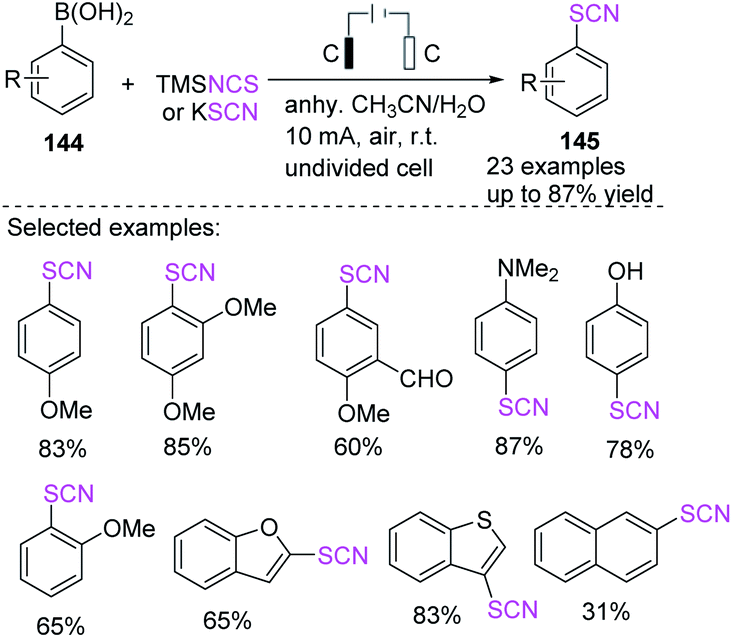
Cai et al. demonstrated green and simple electrochemical deborylative thiocyanation process for aryl boronic acids 144 to construct resulting aryl thiocyanates 145 in good to outstanding yields and with a broad range of functional groups.61 This electrochemical deborylative thiocyanation system requires electricity, and a variety of aryl boronic acids 144 can be engaged. This straightforward and metal-free technique uses potassium thiocyanate or TMSNCS (trimethylsilyl)isothiocyanate as a thiocyanate source in an air atmosphere with no adverse chemicals (Scheme 53).
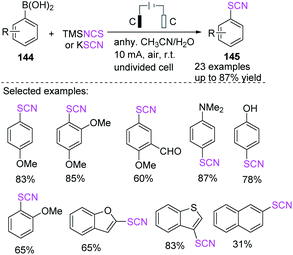 |
| | Scheme 53 Electrochemical deborylative thiocyanation of arylboronic acids 144. | |
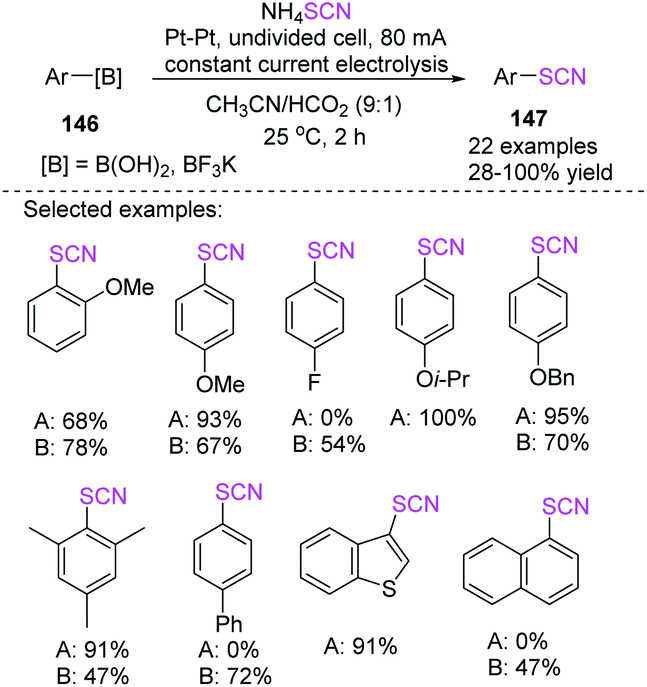
Without the use of any transition metals, Gooben et al. established a facile electrochemical technique for the ipso-thiocyanation of arylboronic acids with ammonium thiocyanate for the fabrication of aryl thiocyanates through excellent to high yields.62 According to the mechanistic suggestion for the regio-specific thiocyanation of arylboron compounds, thiocyanate (SCN) is oxidized to pseudohalogen (SCN)2, which then combines with the arylboron compound for the production of aryl thiocyanate. This reaction can be assisted by the coupling of SCN to boronic acid, which produces an anion, increasing the concentrations of reagents at the anode and activating the boron-based releasing group (Scheme 54).
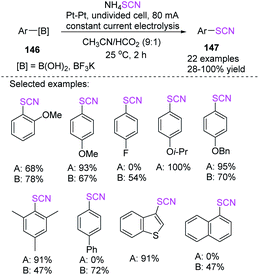 |
| | Scheme 54 Electrochemical induced synthesis of aryl thiocyanates 147. | |
4. Conclusion & viewpoint
Thiocyanates (SCN) are important organic intermediates in the advanced phases of bioactive molecule functionalization. Photo- or electrochemical-induced thiocyanation methods, which are novel methods for the environmentally sustainable, efficient, and safe synthesis of pharmaceuticals and functional materials, have garnered considerable interest in recent years. We have observed through previous discussions and published articles that photo- or electrochemical-induced synthesis in this field is sustainable and green and safer for the environment, thereby offering more significant contributions than previous conventional synthesis processes. In terms of future research, we can state that scientists will be able to conduct this type of photo- or electrochemical-triggered synthesis mode in a green and sustainable manner, opening new doors for the future where no toxic metals, oxidants, or other external catalysts are required. We have summarized entire research on photochemical- and electrochemical-induced thiocyanation techniques in this review, which is divided into three sections: (1) C(sp3)–SCN bond formation, (2) C(sp2)–SCN bond formation, and (3) C(sp)–SCN bond formation. We have discussed photochemically induced indirect thiocyanation of thiol compounds and electrochemically induced C–SCN bond formation of enaminones, cinnamic acid, and boronic acid compounds through thiocyanation. The construction of C–SCN bonds is finally described. The majority of photo- or electrochemical-mediated thiocyanation techniques seem to cover the conception of C(sp2)–SCN and C(sp3)–SCN bonds. Therefore, a wide range of thiocyanation approaches are available for C(sp2)–H and C(sp3)–H bonds. These strategies include undivided cell and metal-free (organic dye or EDA complex based) photocatalytic and electrocatalytic techniques. Photo- or electrochemistry-mediated C–SCN bond formation approaches are still limited. Organophoto- and electrocatalysis have attracted interest from academic and pharmaceutical laboratories. The development of potent and effective photo- and electrocatalysts ensures that the topic will remain an important subject of interest for many decades. Hence, C–SCN bond forming processes will be increasingly examined in the future.
Conflicts of interest
“There are no conflicts to declare”.
Acknowledgements
Financial support from the School-level Scientific Research Project, Neijiang Normal University (2020wj04), Directional Financial Transfer Payment Innovation and Development Technology Project of Neijiang National High Tech Zone (2018GZYZF0048), Special Fund Project of Municipal Science and Technology Incubation and Achievement Transformation (2018KJFH016), the National Natural Science Foundation of China (NO. 21777130 and 21277109) are gratefully acknowledged.
Notes and references
- For selected review see:
(a) T. Castanheiro, J. Suffert, M. Donnard and M. Gulea, Chem. Soc. Rev., 2016, 45, 494–505 RSC;
(b) R. H. Vekariya and H. D. Patel, Synth. Commun., 2017, 47, 87–104 CrossRef CAS;
(c) X. Qin, L.-Y. Zhang, G.-F. Feng and C.-A. Jin, Chin. J. Org. Chem., 2019, 39, 287–300 CrossRef;
(d) S. Majedi, L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31 Search PubMed;
(e) M. Gulea and M. Donnard, Curr. Green Chem., 2020, 7, 201–216 CrossRef CAS;
(f) M. Gao, M. Vuagnat, M.-Y. Chen, X. Pannecoucke, P. Jubault and T. Besset, Chem.–Eur. J., 2021, 27, 6145–6160 CrossRef PubMed.
-
(a) X.-Y. Duan, X.-J. Liu, X.-D. Cuan, L. Wang, K. Liu, H.-Y. Zhou, X. Chen, H.-M. Li and J.-Q. Wang, J. Org. Chem., 2019, 84, 12366–12376 CrossRef CAS PubMed;
(b) D. Wu, Y.-J. Duan, K. Liang, H.-Q. Yin and F.-X. Chen, Chem. Commun., 2021, 57, 9938–9941 RSC.
-
(a) R. G. Guy, The Chemistry of Functional Groups, Syntheses and Preparative Applications of Thiocyanates, in Interscience, ed. S. Patai, John Wiley, New York, 1977, ch. 18, p. 833 Search PubMed;
(b) V. A. Kokorekin, A. O. Terent’ev, G. V. Ramenskaya, N. E. Grammatikova, G. M. Rodionava and A. I. llovaiskii, Pharm. Chem. J., 2013, 47, 422–425 CrossRef CAS.
- For selected examples:
(a) C. Mudithanapelli and M.-H. Kim, Org. Biomol. Chem., 2020, 18, 450–464 RSC;
(b) G.-F. Zhang, L. Xuan, Y.-Y. Zhao and C.-R. Ding, Synlett, 2020, 31, 1413–1417 CrossRef CAS;
(c) L. Qi, S. Liu and L.-X. Xiao, RSC Adv., 2020, 10, 33450–33454 RSC;
(d) X. Zeng, B. Chen, Z. Lu, G. B. Hammond and B. Xu, Org. Lett., 2019, 21, 2772–2776 CrossRef CAS PubMed;
(e) A. Dey and A. Hajra, Adv. Synth. Catal., 2019, 361, 842–849 CrossRef CAS;
(f) M. Noikham and S. Yotphan, Eur. J. Org. Chem., 2019, 2019, 2759–2766 CrossRef CAS;
(g) J.-S. Qiu, D. Wu, L. Yuan, P.-L. Long, H.-Q. Yin and F.-X. Chen, J. Org. Chem., 2019, 84, 7917–7926 CrossRef CAS PubMed;
(h) J.-A. Xiao, X.-L. Cheng, R.-F. Meng, X.-S. Qin, H. Peng, J.-W. Ren, Z.-Z. Xie, J.-G. Cui and Y.-M. Huang, Synthesis, 2021, 53, 954–960 CrossRef CAS;
(i) N.-N. Yi, M.-J. Ouyang, H.-M. Liu, H. M. Yan, X.-Y. Wen, Y. Xiong and B. Yi, J. Chem. Res., 2021, 45, 21–27 CrossRef CAS;
(j) Q. A. Yadav, R. Kant and C. B. Tripathi, J. Org. Chem., 2020, 85, 2814–2822 CrossRef PubMed;
(k) M. Gao, M.-Y. Chen, X. Pannecoucke, P. Jubault and T. Besset, Chem.–Eur. J., 2020, 26, 15497–15500 CrossRef CAS PubMed;
(l) G.-W. Hu, P. Li, Z.-Q. Zhou, F. Yang, S. Xu, H. Fan, X.-C. Zhao and X.-X. Zhang, New J. Chem., 2021, 45, 3828–3832 RSC;
(m) C.-Q. Li, P.-L. Long, H.-P. Wu, H.-Q. Yin and F.-X. Chen, Org. Biomol. Chem., 2019, 17, 7131–7134 RSC;
(n) F. Meng, H.-L. Zhang, H. He, N. Xu, Q. Fang, K. Guo, S.-J. Cao, Y. Shi and Y.-G. Zhu, Adv. Synth. Catal., 2020, 362, 248–254 CrossRef CAS;
(o) P. G. Karmaker, J.-S. Qiu, D. Wu, H.-Q. Yin and F.-X. Chen, Synlett, 2018, 29, 954–958 CrossRef CAS;
(p) H.-P. Wu, C.-K. Shao, D. Wu, L. Jiang, H.-Q. Yin and F.-X. Chen, J. Org. Chem., 2021, 86, 5327–5335 CrossRef CAS PubMed.
- For selected examples see :
(a) T. Kittikool and S. Yotphan, Eur. J. Org. Chem., 2020, 8, 961–970 CrossRef;
(b) X.-K. Mao, J.-B. Ni, B. Xu and C.-Y. Ding, Org. Chem. Front., 2020, 7, 350–354 RSC;
(c) D. Wu, J.-S. Qiu, P. G. Karmaker, H.-Q. Yin and F.-X. Chen, J. Org. Chem., 2018, 83, 1576–1583 CrossRef CAS PubMed.
- For selected examples:
(a) E. O. L. Filho and I. Malvestiti, ACS Omega, 2020, 5, 33329–33339 CrossRef PubMed;
(b) Y.-S. Zhu, Y. Xue, W.-N. Liu, X.-J. Zhu, X.-Q. Hao and M.-P. Song, J. Org. Chem., 2020, 85, 9106–9116 CrossRef CAS PubMed;
(c) H. Mei, Z. Yin, J. Liu, H. Sun and J. Han, Chin. J. Chem., 2019, 37, 292–301 CAS;
(d) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef CAS;
(e) Y. Liu, J. Xiong and L. Wei, Chin. J. Org. Chem., 2017, 37, 1667–1680 CrossRef CAS;
(f) C. Shen, P. Zhang, Q. Sun, S. Bai, T. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC;
(g) Y. Xi, B. Dong, E. J. McClain, Q. Wang, T. L. Gregg, N. G. Akhmedov, J. L. Petersen and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 4657–4661 CrossRef CAS PubMed;
(h) Q. Lu, J. Zhang, G. Zhao, Y. Qi, H. Wang and A. Lei, J. Am. Chem. Soc., 2013, 135, 11481–11484 CrossRef CAS PubMed;
(i) Q. Lu, J. Zhang, F. Wei, Y. Qi, H. Wang, Z. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7156–7159 CrossRef CAS PubMed.
-
(a) S. P. Blum, K. Hofman, G. Manolikakes and S. R. Waldvogel, Chem. Commun., 2021, 57, 8236–8249 RSC;
(b) M. K. Bogdos, E. Pinard and J. A. Murphy, Beilstein J. Org. Chem., 2018, 14, 2035–2064 CrossRef CAS PubMed;
(c) N. P. Radhika, R. Selvin, R. Kakkar and A. Umar, Arabian J. Chem., 2019, 12, 4550–4578 CrossRef CAS.
- P.-F. Yuan, Q.-B. Zhang, X.-L. Jin, W.-L. Lei, L.-Z. Wu and Q. Liu, Green Chem., 2018, 20, 5464–5468 RSC.
- A. K. Yadav and L. D. S. Yadav, Tetrahedron Lett., 2015, 56, 6696–6699 CrossRef CAS.
- X. Bao, Q. Wang and J. Zhu, Angew. Chem., Int. Ed. Engl., 2019, 58, 2139–2143 CrossRef CAS PubMed.
- W. Guo, W. Tan, M. Zhao, L. Zheng, K. Tao, D. Chen and X. Fan, J. Org. Chem., 2018, 83, 6580–6588 CrossRef CAS PubMed.
- D. Zhang, H. Wang and C. Bolm, Chem. Commun., 2018, 54, 5772–5775 RSC.
- B. Yi, X.-Y. Wen, Z. Yi, Y. Xie, Q. Wang and J.-P. Tan, Tetrahedron Lett., 2020, 61(1–5), 152628 CrossRef CAS.
- F.-L. Zeng, H.-L. Zhu, X.-L. Chen, L.-B. Qu and B. Yu, Green Chem., 2021, 23, 3677–3682 RSC.
- Z.-L. Wang, J. Chen, Y.-H. He and Z. Guan, J. Org. Chem., 2021, 86(5), 3741–3749 CrossRef CAS PubMed.
- R. Kapoor, R. Chawla and L. D. S. Yadav, Tetrahedron Lett., 2020, 61, 152505–152508 CrossRef CAS.
- Y.-J. Chen, Y.-H. He and Z. Guan, Tetrahedron, 2019, 75, 3053–3061 CrossRef CAS.
- I. U. Hoque, S. R. Chowdhury and S. Maity, J. Org. Chem., 2019, 84, 3025–3035 CrossRef CAS PubMed.
- Z. Li, S. Han, C. Li, P.-P. Shao, H. Xia, H. Li, X. Chen, X. Feng and X.-M. Liu, J. Mater. Chem. A, 2020, 8, 8706–8715 RSC.
- G.-M. Nan and H.-L. Yue, Synlett, 2018, 29, 1340–1345 CrossRef CAS.
- G.-Q. Li, Q.-L. Yan, X.-Q. Gong, X.-M. Dou and D.-S. Yang, ACS Sustainable Chem. Eng., 2019, 7, 14009–14015 CrossRef CAS.
- W.-G. Fan, Q. Yang, F.-S. Xu and P.-X. Li, J. Org. Chem., 2014, 79, 10588–10592 CrossRef CAS PubMed.
- S. Mitra, M. Ghosh, S. Mishra and A. Hajra, J. Org. Chem., 2015, 80, 8275–8281 CrossRef CAS PubMed.
- L. Wang, C.-C. Wang, W.-J. Liu, Q. Chen and M.-Y. He, Tetrahedron Lett., 2016, 57, 1771–1774 CrossRef CAS.
- M. Singh, A. K. Yadav, L. D. S. Yadav and R. K. P. Singh, Synlett, 2018, 29, 176–180 CrossRef CAS.
- M. H. Sarvari, Z. Hosseinpour and M. Koohgard, New J. Chem., 2018, 42, 19237–19244 RSC.
- H. Zhang, Q. Huang, W.-J. Zhang, C.-Y. Pan, J. Wang, C.-X. Ai, J.-T. Tang and G.-P. Yu, ChemPhotoChem, 2019, 3, 645–651 CAS.
- M. Koohgard, Z. Hosseinpoura, A. M. Sarvestani and M. H. Sarvari, Catal. Sci. Technol., 2020, 10, 1401–1407 RSC.
- J. S. Villafruela, C. M. Alonso, I. Echevarría, M. Vaquero, A. Carbayo, J. Fidalgo, A. M. Rodríguez, J. V. C. Vicario, J. C. Lima, A. J. Moro, B. R. Manzano, F. A. Jalón and G. Espino, Inorg. Chem. Front., 2021, 8, 1253–1270 RSC.
- P. Chauhan, P. Ritu, S. Kumar and N. Jain, Eur. J. Org. Chem., 2019, 27, 4334–4340 CrossRef.
- Y. Gao, Y.-Y. Liu and J.-P. Wan, J. Org. Chem., 2019, 84, 2243–2251 CrossRef CAS PubMed.
- K. Sahu, S. Mondal, S. M. Mobin and S. Kar, J. Org. Chem., 2021, 86, 3324–3333 CrossRef CAS PubMed.
- P.-F. Zhang, Y. Yin, Z.-X. Wang, C.-Y. Yu, Y.-Z. Zhu, D. Yan, W.-M. Liu and Y.-Y. Mai, Macromolecules, 2021, 54(7), 3543–3553 CrossRef CAS.
- S. D. Tambe, M. S. Jadhav, R. S. Rohokale and U. A. Kshirsagar, Eur. J. Org. Chem., 2018, 35, 4867–4873 CrossRef.
- K. Gullapalli and S. Vijaykumar, Org. Biomol. Chem., 2019, 17, 2232–2241 RSC.
- F.-L. Zeng, X.-L. Chen, K. Sun, H.-L. Zhu, X.-Y. Yuan, Y. Liu, L.-B. Qu, Y.-F. Zhao and B. Yu, Org. Chem. Front., 2021, 8, 760–766 RSC.
- Y. Chen, Y.-J. Chen, Z. Guan and Y.-H. He, Tetrahedron, 2019, 75(1–17), 130763 CrossRef.
-
(a) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170–1214 CrossRef CAS PubMed;
(b) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed;
(c) M. Chen, X. Zheng, W. Li, J. He and A. Lei, J. Am. Chem. Soc., 2010, 132, 4101–4103 CrossRef CAS PubMed;
(d) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed;
(e) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC;
(f) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed;
(g) C. He, S. Guo, J. Ke, J. Hao, H. Xu, H. Chen and A. Lei, J. Am. Chem. Soc., 2012, 134, 5766–5769 CrossRef CAS PubMed;
(h) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed;
(i) C. Liu, D. Liu and A. Lei, Acc. Chem. Res., 2014, 47, 3459–3470 CrossRef CAS PubMed;
(j) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed;
(k) G. Zhang, C. Liu, H. Yi, Q. Meng, C. Bian, H. Chen, J.-X. Jian, L.-Z. Wu and A. Lei, J. Am. Chem. Soc., 2015, 137, 9273–9280 CrossRef CAS PubMed;
(l) C. Song, H. Yi, B. Dou, Y. Li, A. K. Singh and A. Lei, Chem. Commun., 2017, 53, 3689–3692 RSC;
(m) C. Song, X. Dong, H. Yi, C.-W. Chiang and A. Lei, ACS Catal., 2018, 8, 2195–2199 CrossRef CAS;
(n) C. Song, K. Liu, X. Dong, C.-W. Chiang and A. Lei, Synlett, 2019, 30, 1149–1163 CrossRef CAS.
- S. Liang, C.-C. Zeng, H.-Y. Tian, B.-G. Sun, X.-G. Luo and F.-Z. Ren, Adv. Synth. Catal., 2018, 360, 1444–1452 CrossRef CAS.
- L.-S. Kang, M.-H. Luo, C. M. Lam, L.-M. Hu, R. D. Little and C.-C. Zeng, Green Chem., 2016, 18, 3767–3774 RSC.
- W. J. De Klein, Electrochim. Acta, 1973, 18, 413–416 CrossRef CAS.
- A. Levy and J. Y. Becker, Electrochim. Acta, 2015, 178, 294–302 CrossRef CAS.
- A. Gitkis and J. Y. Becker, Electroanalysis, 2016, 28, 2802–2807 CrossRef CAS.
- J.-W. Wen, L.-F. Zhang, X.-T. Yang, C. Niu, S.-F. Wang, W. Wei, X.-J. Sun, J.-J. Yang and H. Wang, Green Chem., 2019, 21, 3597–3601 RSC.
- P. Krishnan and V. G. Gurjar, Synth. Commun., 1992, 22, 2741–2744 CrossRef CAS.
- P. Krishnan and V. G. Gurjar, J. Appl. Electrochem., 1995, 25, 792–796 CrossRef CAS.
- V. A. Kokorekin, E. I. Meĺnikova, R. R. Yaubasarova, N. V. Gorpinchenko and V. A. Petrosyan, Russ. Chem. Bull. Int. Ed., 2019, 68, 2140–2141 CrossRef CAS.
- A. Gitkis and J. Y. Becker, Electrochim. Acta, 2010, 55, 5854–5859 CrossRef CAS.
- A. Gitkis and J. Y. Becker, J. Electroanal. Chem., 2006, 593, 29–33 CrossRef CAS.
-
(a) V. A. Kokorekin, V. L. Sigacheva and V. A. Petrosyan, Tetrahedron Lett., 2014, 55, 4306–4309 CrossRef CAS;
(b) X. Zhang, C.-G. Wang, H. Jiang and L.-H. Sun, RSC Adv., 2018, 8, 22042–22045 RSC.
- R. R. Yaubasarova, V. A. Kokorekin, G. V. Ramenskaya and V. A. Petrosyan, Mendeleev Commun., 2019, 29, 334–336 CrossRef CAS.
- V. A. Kokorekin, R. R. Yaubasarova, S. V. Neverov and V. A. Petrosyan, Eur. J. Org. Chem., 2019, 26, 4233–4238 CrossRef.
- V. A. Kokorekin, S. V. Neverov, V. N. Kuzina and V. A. Petrosyan, Molecules, 2020, 25(1–13), 4169 CrossRef CAS PubMed.
- V. A. Kokorekin, R. R. Yaubasarova, S. V. Neverov and V. A. Petrosyan, Mendeleev Commun., 2016, 26, 413–414 CrossRef CAS.
- L. Fotouhi and K. Nikoofar, Tetrahedron Lett., 2013, 54, 2903–2905 CrossRef CAS.
- T. Cui, X.-F. Zhang, J. Lin, Z. Zhu, P. Liu and P.-P. Sun, Synlett, 2021, 32, 267–272 CrossRef CAS.
- J.-F. Chen, H.-J. Yang, M.-F. Zhang, H. Chen, J. Liu, K. Yin, S.-S. Chen and A.-L. Shao, Tetrahedron Lett., 2021, 65, 152755–152759 CrossRef CAS.
- Z. Yang, Y.-H. Wang, L.-P. Hu, J.-J. Yu, A. Li, L.-J. Li, T. Yang and C. Zhou, Synthesis, 2020, 52, 711–718 CrossRef CAS.
- F.-L. Lu, K. Zhang, Y.-X. Yao, Y. Yin, J.-F. Chen, X.-W. Zhang, Y. Wang, L.-L. Lu, Z.-W. Gao and A.-W. Lei, Green Chem., 2021, 23, 763–766 RSC.
- S.-M. Yang, T.-J. He, D.-Z. Lin and J.-M. Huang, Org. Lett., 2019, 21, 1958–1962 CrossRef CAS PubMed.
- D.-D. He, J.-J. Yao, B.-L. Ma, J.-H. Wei, G.-G. Hao, X. Tuo, S.-M. Guo, Z.-J. Fu and H. Cai, Green Chem., 2020, 22, 1559–1564 RSC.
- M. Dyga, D. Hayrapetyan, R. K. Rit and L. J. Gooßen, Adv. Synth. Catal., 2019, 361, 3548–3553 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Feng Huo
b and
Feng Huo