DOI:
10.1039/D1RA09084D
(Paper)
RSC Adv., 2022,
12, 7922-7934
Synthesis and fabrication of gelatin-based elastomeric hydrogels through cosolvent-induced polymer restructuring†
Received
15th December 2021
, Accepted 22nd February 2022
First published on 10th March 2022
Abstract
Hydrogels have a wide range of applications in tissue engineering, drug delivery, device fabrication for biological studies and stretchable electronics. For biomedical applications, natural polymeric hydrogels have general advantages such as biodegradability and non-toxic by products as well as biocompatibility. However, applications of nature derived hydrogels have been severely limited by their poor mechanical properties. For example, most of the protein derived hydrogels do not exhibit high stretchability like methacrylated gelatin hydrogel has ∼11% failure strain when stretched. Moreover, protein derived elastomeric hydrogels that are fabricated from low molecular weight synthetic peptides require a laborious process of synthesis and purification. Biopolymers like gelatin, produced in bulk for pharma and the food industry can provide an alternative for the development of elastomeric hydrogels. Here, we report the synthesis of ureidopyrimidinone (Upy) functionalized gelatin and its fabrication into soft elastomeric hydrogels through supramolecular interactions that could exhibit high failure strain (318.73 ± 44.35%). The hydrogels were fabricated through a novel method involving co-solvent optimization and structural transformation with 70% water content. It is anticipated that the hydrogel fabrication method involves the formation of hydrophobic cores of ureidopyrimidinone groups inside the hydrogel which introduced elastomeric properties to the resulting hydrogel.
1. Introduction
Elastomeric hydrogels have been gaining huge attention by researchers due to their application in stretchable electronics, biomedical implants, and device fabrication.1–4 For elastomeric hydrogel fabrication, synthetic polymers have been used due to ease of tailoring of physical, chemical, and structural properties.5 However natural polymers have the general advantages of being naturally biodegradable and non-toxic by products as well as biocompatible.6,7 Among natural polymers, gelatin is a protein-based biopolymer which is supportive of cell growth, biodegradable and biocompatible and is widely used in the biomedical and food industries.8 Gelatin is a collagen derivative and manufactured through acid/base hydrolysis of collagen present in bones and hides of bovine/pork. During hydrolysis process, most of the covalent and non-covalent crosslinks get lost which would result into a gelatin with random coil structure and it undergoes renaturation during gelation takes place below sol–gel transition temperature (20–25 °C).9 After gelation, gelatin forms a weak and brittle hydrogel with limited elastomeric properties. Gelatin based elastomeric hydrogels have been developed by researchers through blending with synthetic elastomeric polymers like polyacrylamide10 and polycitrate.11 However, they are unable to enhance the elastomeric properties of gelatin intrinsically. In a study by Feng and coworkers, hydrophobic groups 1-adamantyl isothiocyanate/phenyl isothiocyanate were modified over gelatin chain and also introduced as free molecules inside the hydrogel to enhance the elastomeric properties but fails to maintain ins integrity upon removal of free hydrophobic molecules.12
Supramolecular systems have been developed and introduced in various synthetic polymeric systems through various strategies. In supramolecular systems, non-covalent interactions play a vital role in gel structuring to attain a crosslinked network with reversible physical deformation.13–15 For elastomeric gel fabrication, numerous non-covalent systems have been investigated including inclusion complexes, host–guest complexes, and self-dimerized systems.16–19 In the last decade, biopolymers, protein-based biopolymers (PBB) and synthetic peptides have been explored by researchers utilizing blending or modification with self-assembled moieties for elastomeric gel fabrication.20 In this regards, Meijer and co-workers have developed ureidopyrimidinone (Upy), a hydrogen bonding based self-dimerized moiety with dimerization constant Kdim > 106 M−1 in chloroform.21 Due to its hydrophobicity and strong self-dimerization, Upy was conjugated to natural and synthetic polymers for hydrogel fabrication via copolymerization,22 and side chain/group modification.23–25 However, among the given methods only copolymerization of Upy with a monomer was able to establish the formation of an elastomeric hydrogel and the rest has formed soft hydrogels with limited elastomeric properties. Copolymerization of polyethylene glycol with Upy has been reported to form an elastomeric hydrogel with >500% strain at break.22 In case of natural polymers, Upy was substituted on functional groups present on subunits of polymer. End-group modification has resulted in the formation of soft visco-elastic hydrogels with limited elastomeric properties of synthetic and natural polymers. In gelatin biopolymer, end-group modification with Upy was observed to form a hydrogel with 296 MPa and 11.57%, ultimate tensile strength and strain at break, respectively.26
The major challenge reported by the researchers with Upy derivatives is dissolution of Upy substituted polymers caused by solvent incompatibility.26–28 Hence, film fabrication methods like solvent casting may be challenging because of molecular level segregation resulted in the formation of hydrogels with limited mechanical properties. Here, we present novel soft hydrogel/organo-hydrogels with excellent stretchability. These elastomeric organo-hydro gels and hydrogels were fabricated via restructuring of polymers through a cosolvent method. Upy has been studied to be present in a dimeric form and is soluble in given solvents including toluene, chloroform, and tetrahydrofuran (THF).29 Among these solvents, only THF is water miscible at macroscale and causes micro phase separation.30–32 The given micro-phase separation has been used by researchers as a template for synthesis of nano-capsules.31 In the present work, we synthesized Upy-functionalized gelatin derivatives through a two-step control method. Further, gelatin and Upy-functionalized gelatin derivatives were studied for their behaviour in water–THF for cosolvent optimization. Organo/hydro hybrid gels and hydrogel films were fabricated by the cosolvent method and characterized for their mechanical and structural properties. The structural transformation of Upy-functionalized gelatin biopolymer in the cosolvent system was further investigated in terms of mechanism.
2. Experimental
2.1 Materials and reagents
Gelatin A (300 bloom), 2-amino-4-hydroxyl-6 methyl pyrimidine, 1, 6-diisocyanatohexane, pyridine, hexane, tetrahydrofuran, ethanol, deuterated chloroform, deuterium oxide and deuterated dimethyl sulfoxide were procured from Sigma-Aldrich. Deionized (DI) water was used throughout the work.
2.2 Synthesis of Upy modified gelatin
2 grams of Gelatin A (300 bloom) was dried in vacuum at 60 °C to remove the moisture and was mixed in an anhydrous 70 ml DMSO at 60 °C for 48 hours with continuous stirring to get clear gelatin solution. Synthesis of Upy substituted gelatin involves two steps. In the first step, Upy (0.10, 0.30 and 0.50 grams of Upy/2 g gelatin) was added to the solution and the mixture was continuously stirred at 60 °C in N2 atmosphere for 24 hours. In the second step, dibutyltin dilaureate was added to the mixture as a catalyst and was stirred for another 24 hours at 60 °C in N2 atmosphere. After 48 hours of reaction, the solution was kept at room temperature to cool down and was precipitated with isopropanol. The precipitated sample was centrifuged and washed with isopropanol. The pellet was suspended in water and then freeze dried to obtain the Upy substituted gelatin synthesized by two-step method (GEUPY).
2.3 Solubility analysis of gelatin and Upy substituted gelatin derivatives in water and water–THF cosolvent
Transmittance analysis was carried out to test the solubility analysis of the gelatin/ureidopyrimidinone substituted gelatin derivatives in distilled water or cosolvent system (water–THF/ethanol) using Agilent UV-Vis NIR Spectrophotometer (Cary 5000). 0.10, 1.00 and 10.00 mg of gelatin/ureidopyrimidinone substituted gelatin derivatives were mixed in the desired 1 ml of solvent (distilled water/cosolvent) and was continuously stirred to get a homogeneous solution or suspension. The solution was then loaded in 36 well plates for transmittance at 600 nm using plate reader. For in situ analysis, 20 mg of gelatin and Upy substituted gelatin derivatives were mixed in 1 ml distilled water to make 20 mg ml−1 concentration for 6 hours at 60 °C with continuous stirring to get a homogeneous initial aqueous solution/suspension. Further, the samples were diluted with water/THF/ethanol by adding 100 μl of water/THF 10 times stepwise till concentration reaches to 10 mg ml−1 and was mixed at each step for 5 minutes using pipette inside cuvette and transmittance at 600 nm was measured for transmittance analysis.
2.4 Structural analysis of gelatin and Upy substituted gelatin derivatives for structural transformation
Scanning electron microscopy was used to study the micro-structural analysis of gelatin when mixed in water and water–THF cosolvent system. For microstructure analysis, gelatin and Upy substituted gelatin derivative GEUPY(0.10), GEUPY(0.30) and GEUPY(0.50) were mixed in 1 ml distilled water/x% v/v THF (x% = (volume of THF/total volume) × 100) at 70 °C with continuous stirring using magnetic stirrer hot plate to get a homogeneous suspension/solution with 10 mg ml−1 concentrations. 200 μl of samples was transferred to vials and were kept in fridge at −80 °C for 10–12 hours and was transferred to freeze dryer to get dried samples. Further freeze–dried samples were then loaded on to the sample holder using two-sided carbon tape followed by gold sputtering for conductivity and was loaded in the sample holder of JSM-FESEM 7600F, Jeol, Japan. Sample's microstructures were observed at 5 kV at different magnifications.
2.5 Fourier transform infrared spectroscopy (ATR-FTIR)
Ureidopyrimidinone, Gelatin and ureidopyrimidinone substituted gelatin derivatives were characterized by ATR-FTIR spectroscopy for their chemical structure and purity of the samples using PerkinElmer Fourier transform infrared (FTIR) spectrometer (Frontier). For ureidopyrimidinone, pellet was made using potassium bromide (KBr), whereas freeze dried powders/film were mounted directly over the zinc selenide (ZnSe) crystal. This was then scanned multiple times (50 scans) by infrared rays with wavenumber in the range of 400–4000 cm−1 by the instrument to obtain the emission/absorption spectrum.
2.6 1H nuclear magnetic resonance spectroscopy (NMR)
Ureidopyrimidinone, gelatin and ureidopyrimidinone substituted gelatin derivatives were characterized by 1H NMR spectroscopy for chemical analysis using AVANCE I 400 MHz spectrometer. For 1H NMR spectral analysis of ureidopyrimidinone, sample was prepared by mixing 10 mg of ureidopyrimidinone powder in 700 μl deuterated chloroform. Gelatin and ureidopyrimidinone modified gelatin derivative samples were prepared by mixing 10 mg of powder sample in deuterated dimethyl sulfoxide (Cambridge Isotope laboratories, Inc). Further samples were loaded in NMR tubes to obtain 1H NMR spectrum. Samples were scanned from 0 ppm to 10 ppm at 400 MHz frequency.
2.7 Film fabrication
1 g of biopolymer (gelatin/ureidopyrimidinone substituted gelatin) was mixed in 20 ml of distilled water and was continuously stirred at 60–70 °C to form a homogeneous suspension. After obtaining a homogeneous suspension, 20 ml of tetrahydrofuran was added to the sample dropwise and was continuously stirred at room temperature until the colour of the suspension changed from milky white to clear. Further 60 ml of tetrahydrofuran was added slowly to the solution to make the final cosolvent volume to 100 ml and concentration of 10 mg ml−1. Addition of THF would change the colour of the solution from clear to milky white again. The obtained milky suspension was incubated at 37 °C for 24 hours. After 24 hours, the biopolymer was sedimented at the bottom to form elastomeric film and a clear supernatant at the top of the film. Supernatant was removed and the film was peeled off carefully from the container for further processing and studies.
2.8 Physical and structural analysis of elastomeric films
2.8.1 Optical microscopy. Elastomeric film obtained at 80% v/v THF was investigated for its structural analysis using optical microscope at 10× resolution using Nikon 80i eclipse upright microscope. Film was mounted on to the glass slide and directly observed for its structure under a light microscope. For cross-section view, film cross-section was cut using surgery blade and then mounted on to glass slide for observation using the microscope.
2.8.2 Scanning electron microscopy of elastomeric films. Gelatin and GEUPY(0.50) elastomeric films obtained at 80% v/v THF were observed in scanning electron microscope for its microstructural analysis. The films were cut into pieces with dimension ∼2 mm × 3 mm and then kept at −80 °C for freezing. Samples were further transferred to freeze dryer to get the dry film. For cross-section analysis, dried films were broken manually and then mounted on to half stub. As the samples were non-conductive, they were sputter coated with gold atoms using a sputter coater for 45 seconds at 40 milli ampere. Then, the samples were mounted over the sample holder using carbon tape to load the sample inside JEOL FESEM 7600F scanning electron microscope. Samples were analysed at 5 kV at 6.00 mm working distance at low and high magnification to get SEM images of the films. For quantification of pore size analysis in films, the SEM images were processed using IMAGEJ 1.52 software and the pore diameters were calculated and were plotted in Microsoft excel for statistical analysis.To elucidate the film fabrication mechanism, point before cloud point was taken, which was 43.8% v/v THF for gelatin and 50% v/v THF for GEUPY(0.50). 100 μl of gelatin and GEUPY(0.50) samples were prepared with 43.8%, 50% v/v THF (concentration – 10 mg ml−1), respectively. In case of gelatin, THF proportion was adjusted from 43.8% THF to 50% and 60% v/v THF by addition of THF. In the case of GEUPY(0.50), THF proportion was adjusted from 50% to 52.6% and 60% v/v THF. After THF proportion adjustment, samples were mixed for 30 seconds with pipette manually and were transferred to freezer at −80 °C for 10–12 hours followed by drying.
2.8.3 Solvent exchange from the elastomeric films. Films fabricated at 80% v/v THF were dried at 37 °C under vacuum for 48 hours to remove the cosolvent from the films. Further, air dried films were rehydrated with distilled water for 24 hours at room temperature to make hydrogel. TGA analysis was carried out to ensure the complete removal of residual solvents inside the films.
2.8.4 Analysis of mechanical properties. Elastomeric films were characterized for the mechanical properties using MTS mechanical tester Criterion Model 42. Wet films were cut into standard dumbbell shaped films using cutter. The samples had a gauge length of 9.53 mm and width at gauge length of 3.18 mm. After sample preparation, thickness of the samples was measured using micro-meter screw gauge. Further, samples were loaded on to the tensile fixtures with 50 N load and tested at a crosshead speed of 1 mm s−1 at room temperature.
3. Results
Upy-functionalized gelatin derivatives (GEUPY) were prepared in a stepwise manner by reacting gelatin biopolymer with Upy–synthon in varying stoichiometric proportion (0.10, 0.30 & 0.50 g/2 g gelatin) in dimethyl sulphoxide as shown in Table 1 (see Fig. 1 for complete synthetic scheme). The solvent dimethyl sulphoxide is a polar solvent which prevents Upy dimerization and favours the tautomeric transition of Upy synthon from dimeric form (4[1H]-pyrimidinone & pyrimidin-4-ol) to monomeric form (6[1H]-pyrimidinone), enhancing its solubility as shown in ESI.†33 The synthetic protocol allowed modification of Upy on gelatin side groups to enhance the stretchability of resultant hydrogels. In gelatin, primary amino groups (–NH2) are present in lysine and hydroxylysine, and hydroxyl groups (–OH) are present in hydroxyproline, hydroxylysine, and serine, would facilitate the modification of Upy.34 Upy modification was monitored by ATR-FTIR and 1H NMR spectroscopy in which disappearance of isocyanate peak in ATR-FTIR spectrum at 2267 cm−1 and presence of 1H NMR chemical shift at 5.76 ppm confirms the modification of Upy in gelatin with purity (Fig. S1–S4†).26,35 Modified gelatin polymers were washed and precipitated with isopropanol to remove the unreacted Upy synthons followed by drying for further characterization and processing.
Table 1 Stoichiometric ratio or reactant and reaction conditions for the synthesis of Upy modified gelatin derivatives
| GEUPY samples |
Gelatin (mg ml−1) |
Upy (g/2 g gelatin) |
Temperature (°C) |
Time (hours) |
Catalyst (per 2 g gelatin) |
| Step I |
Step II |
| GEUPY(0.10) |
28.57 |
0.10 |
60 |
24 |
24 |
500 μl |
| GEUPY(0.30) |
28.57 |
0.30 |
60 |
24 |
24 |
500 μl |
| GEUPY(0.50) |
28.57 |
0.50 |
60 |
24 |
24 |
500 μl |
 |
| | Fig. 1 Scheme for the synthesis of (A) ureidopyrimidinone synthon and (B) GEUPY (Upy substituted gelatin derivatives). | |
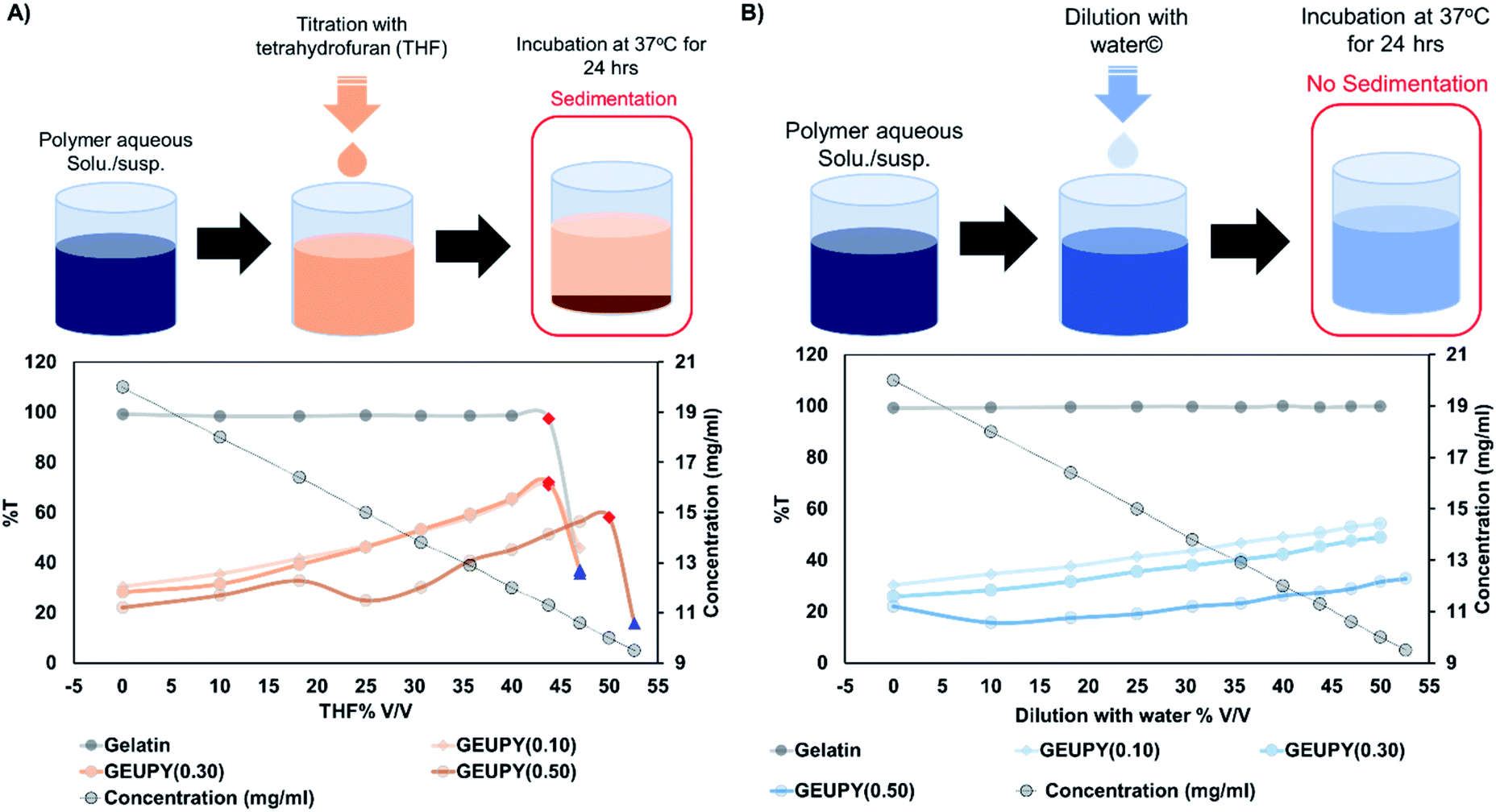
3.1 Cosolvent optimization
Gelatin is a hydrocolloid in nature due to its chemical structure which comprises of hydrophobic segments (tripeptide repeat, Glycine–X–Y) interspersed between hydrophilic segments.36 Presence of hydrophilic segments and charged functional groups are responsible for the stable dispersion of gelatin chains in aqueous medium.37 Modification of gelatin chains with hydrophobic groups has been observed to show poor aqueous dispersion or no dispersion of gelatin chains due to hydrophobic interactions.22,25,38 Similar effect was observed when gelatin chains were modified with Upy moiety due to the presence of hydrophobic groups (hexamethylene spacer and an isocytosine ring) in ureidopyrimidinone. To quantify these effects, aqueous solution/suspension of gelatin and GEUPY samples in different concentrations (0.10, 1.00, and 10.00 mg ml−1) were characterized for their transmittance at 600 nm. Gelatin formed clear solutions which had 89.65 ± 0.10 %T at all concentrations, whereas GEUPY samples formed milky suspensions with a significant decline in %T along with polymer concentration and Upy content due to hydrophobic interactions (HI) as shown in Fig. S5.† The HI has been known to be increased with an increase in the concentration of hydrophobic groups due to a decrease in ΔGHI (Gibbs free energy for hydrophobic interactions). It was observed that ΔGHI decreases linearly with an increase in the concentration of hydrophobic groups.39
These hydrophobic interactions lead to the formation of aggregates, which prevents Upy crosslinking via hydrogen bonding, required to form a supramolecularly reinforced hydrogel. Barzin and coworkers have reported the issue of insolubility and inability to form a hydrogel in aqueous environment for Upy-substituted gelatin derivatives.26 In the past few years, researchers have employed various strategies for the dissolution of Upy modified polymers in aqueous environment including heating,35 pH modulation,23 and cosolvent.40 Gelatin is a protein-based biopolymer which is heat as well as pH-sensitive and may cause denaturation/degradation due to which heating/pH modulation-based strategies cannot be employed for hydrogel fabrication. Apart from these, Upy is a non-polar moiety which is soluble in non-polar solvents like toluene, chloroform, tetrahydrofuran (THF), and dimethyl sulphoxide (DMSO). Among these solvents, DMSO is the only solvent which is compatible with both Upy and gelatin and was employed for the synthesis of Upy-substituted gelatin derivatives. However, Upy has been reported to undergo tautomerization in DMSO to form monomeric 6-[1H]-pyrimidinone and cannot undergo self-dimerization for crosslinking to form a hydrogel.33 Cosolvent systems have been employed to modulate the polarity of the solvents and have been proven useful in case of Upy-substituted peptides. Meijer et al. employed water–THF (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) for the dissolution of synthetic peptides modified with Upy to fabricate fibres through electrospinning.41 Due to this, we optimized the water–THF proportion required for the dissolution of Upy-modified gelatin derivatives.
:10) for the dissolution of synthetic peptides modified with Upy to fabricate fibres through electrospinning.41 Due to this, we optimized the water–THF proportion required for the dissolution of Upy-modified gelatin derivatives.
For cosolvent optimization, aqueous polymer solutions/suspensions were titrated with THF and %T was measured at 600 nm after every aliquot using Nanodrop till the cosolvent proportion reached 50% v/v THF (as the concentration decreased from 20 to 10 mg ml−1). Furthermore, samples were incubated at 37 °C for 24 hours to study the stability of the polymer solution/suspension in the given cosolvent proportion. A rise in %T was observed with an increase in THF proportion till 43.75% v/v THF in case of gelatin, GEUPY(0.10) and GEUPY(0.30), whereas till 50% v/v THF for GEUPY(0.50), termed as ‘clear point’ as shown in Fig. 2. In gelatin, GEUPY(0.10), and GEUPY(0.30) samples, adjustment of THF proportion to 47% v/v has resulted in a significant drop in %T, termed as ‘cloud point’, due to phase separation which cleared out after an overnight incubation at 37 °C due to sedimentation of aggregates. An initial increase in %T was due to the solvent compatibility of Upy with THF, which may have partially destroyed the hydrophobic interactions present in aqueous GEUPY solutions/suspensions.42,43 However, a further increase in THF proportion has resulted in phase separation due to incompatibility of gelatin in water–THF cosolvent and has been observed earlier in case of water–ethanol cosolvent, due to solvophobic interactions between gelatin and free THF molecules.15,23 Incubation of samples at 50% v/v THF caused sedimentation, resulting in a clear supernatant and a sedimented film. This happened perhaps due to coalescence of coacervates/aggregates present in water–THF cosolvent confirmed by SEM analysis of freeze–dried samples shown in the next section. However, GEUPY(0.50) did not show any phase separation till 50% v/v THF but underwent phase separation with a further increase in THF proportion to 52.6% v/v THF (shown in Fig. 1A).
 |
| | Fig. 2 Transmittance analysis of gelatin, GEUPY(0.10), GEUPY(0.30) and GEUPY(0.50) (A) titration with THF and (B) dilution with water (♦ – clear point (point of maximum transmittance during titration), ▲ – cloud point (point of phase separation)). | |
3.2 Cosolvent-induced structural transformation
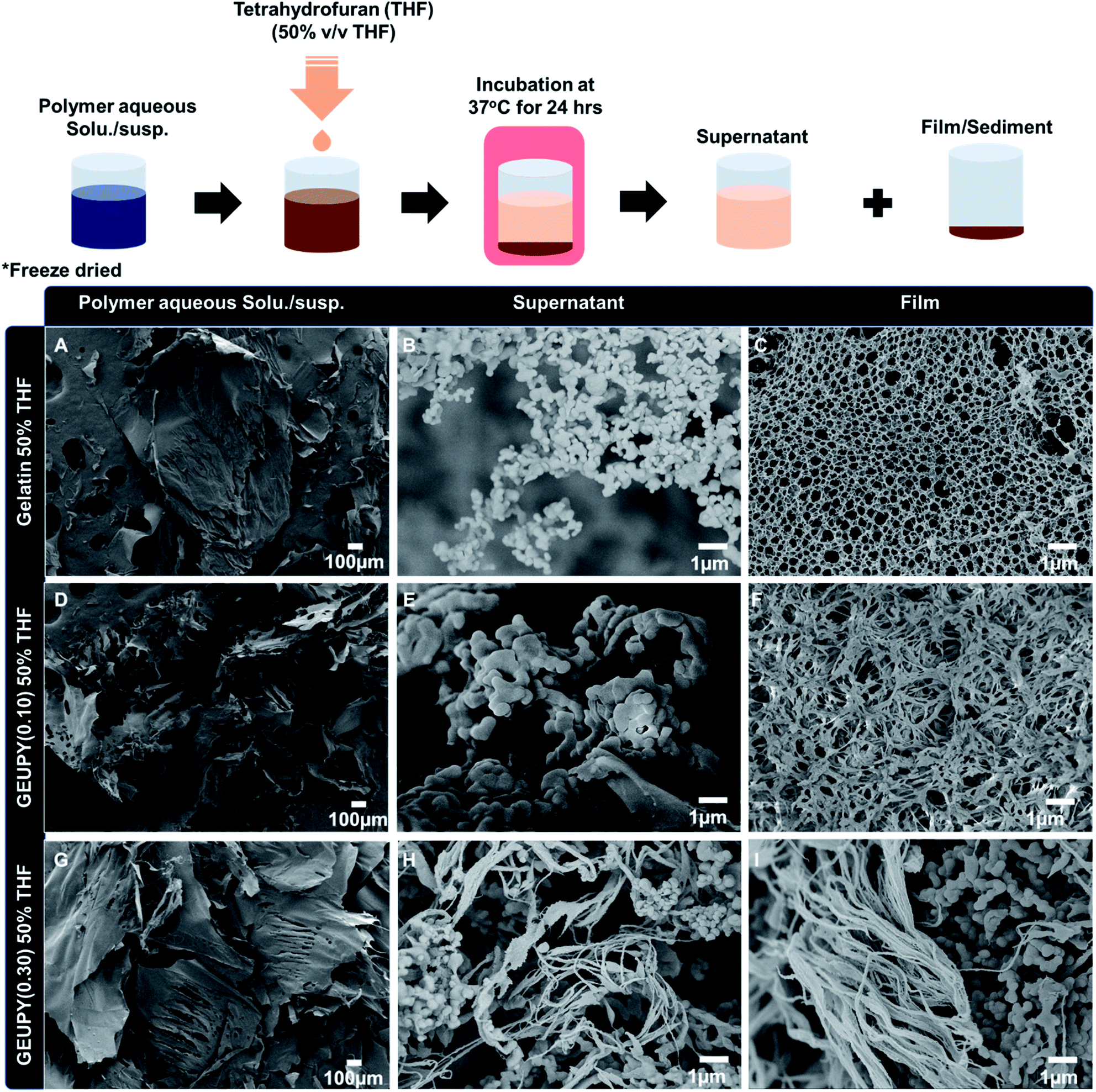
Amphiphilic polymers have been observed to undergo structural transformation when exposed to THF molecules. Zhou et al. have demonstrated the formation of spherical micellar structures and rod-like structure of poly[poly(ethylene glycol) monomethyl ether methacrylate] in water–THF cosolvent.44 Studies have shown that THF demonstrates good solubility for Upy and insolubility for gelatin biopolymer.45 To study the THF-induced structural transformation in gelatin and Upy-substituted gelatin derivatives, samples were characterized in liquid and freeze–dried state using microscopic techniques. Transmittance studies have demonstrated that %T of GEUPY samples increases with an increase in THF proportion, which implicates a decrease in size of aggregates. %T reached its maximum at 43.8% v/v THF and 50% v/v THF (clear point) in gelatin and GEUPY(0.50) suspension, respectively. Phase separation took place with a further increase in THF proportion to 50% v/v and 52.6% v/v THF in gelatin and GEUPY(0.50), respectively. Fig. S6A and B† present the optical microscopic images of GEUPY(0.50) in water and 50% v/v THF, respectively. It can be observed that the size of aggregates is decreased due to addition of THF, which could be due to a decrease in hydrophobic interactions responsible for the formation of aggregates and the increase in solvent compatibility. Further increase in THF proportion has resulted an increase in turbidity and were not visible through optical microscope at 10 mg ml−1. To observe the aggregation effect, concentration of polymer was decreased from 10 mg ml−1 to 1 mg ml−1, followed by THF proportion adjustment to 60% THF and all samples were stained with Nile red dye for CLSM analysis. CLSM images of GEUPY (0.50) at 60% THF demonstrates the aggregation of particles in wet state as well as individual particles present in the liquid phase (shown in Fig. S7†). Due to limited resolution of optical microscopy and turbidity, SEM analysis was carried out for freeze dried samples to identify the shape and size. Fig. 3A–D represents SEM images of gelatin and GEUPY(0.50) samples in water and water–THF phase at clear point and at point of phase separation. Spherical aggregates (Fig. 3B and D) of almost similar size can be clearly observed at clear point (gelatin – 43.8% v/v THF, GEUPY(0.50) – 50% v/v THF). Structural transformations with a further rise in THF proportion was observed by addition of THF aliquots to adjust the THF proportion to 50/60% v/v from clear point for gelatin and 52.6/60% v/v THF for GEUPY(0.50) followed by immediate freezing of samples after mixing for 30 seconds. SEM images of gelatin samples at 50 and 60% v/v THF are shown in Fig. 3(B1) and (B2) which demonstrates the appearance of larger spherical aggregates with 4.4 ± 1.2 μm size in addition to smaller aggregates present at clear point. Similar transformation in structure was observed in 52.6% and 60% THF. Water and THF are miscible at macroscale and immiscible at microscale due to which microphase separation has been observed in water–THF system in the form of THF micro/nano-droplet.46 Introduction of an amphiphilic polymer has been observed to form micellar structures or polymerosomes due to assembly of polymer chains around THF droplet.47,48 In the given studies an increase in THF proportion increased the size of aggregates due to fusion of THF.
 |
| | Fig. 3 THF-induced structural transformation in gelatin (A, B, B1, and B2) and GEUPY(0.50) (C, D, D1, and D2) at different THF proportions. | |
Structural transformation at 50% v/v THF of other samples including gelatin, GEUPY(0.10), and GEUPY(0.30) was also characterized via SEM analysis. At point of phase separation or cloud point (50% v/v THF), samples were incubated for 24 hours at 37 °C. Incubation of samples resulted in sedimentation of polymer aggregates to form a film at the bottom of the container with a clear supernatant above it. For structural analysis, both the supernatant and sedimented film were separated and freeze dried independently. In gelatin samples at 50% v/v THF, sedimentation formed a microporous film shown in Fig. 3C and it demonstrates the formation of a uniform film with pore diameter 425 ± 119.6 nm while spherical aggregates/coacervates of 314.8 ± 138.9 nm size were present in supernatant. However, GEUPY(0.10) formed a non-continuous film of interconnected micro-structures, and spherical aggregates of 430 ± 80 nm diameter were present in supernatant as shown in Fig. 4E and F. In case of GEUPY(0.30), tubular structures of 93 ± 20 nm diameter and 358 ± 11 nm diameter spherical structures were present both in the sedimented film and the supernatant, respectively. In case of gelatin, GEUPY(0.10), and GEUPY(0.30) at 50% v/v THF, a fraction of the biopolymer underwent phase separation, and subsequently sedimented to form a film at the bottom of the container when THF was adjusted from 43.8% v/v THF (xTHF (mole fraction of THF) − 0.15) to 50% v/v THF (xTHF − 0.26). Presence of free THF molecules at 50% v/v THF (0.26, xTHF) could be responsible for the phase separation and eventually sedimentation. Wu et al. has reported that microgels underwent swelling in pure water and pure THF, whereas they shrank in the range of 0.05 < xTHF < 0.15 due to intermolecular interactions between water and THF. A further increase in xTHF resulted in swelling of the microgels due to the presence of free THF molecules, which were not hydrogen-bonded to water molecules.49 One THF molecule was reported to form hydrogen bond with 3 water molecules, due to this the free THF molecules formed microdroplets inside the continuous phase of water–THF system.49,50 Fusion of the coacervates at point of phase separation to form a film was due to solvent incompatibility in case of gelatin. However, incomplete fusion and no fusion of aggregates took place in GEUPY(0.10) and GEUPY(0.30) respectively due to better solvent compatibility with suspended polymer. Phasing out of Upy-substituted gelatin derivates in case of GEUPY(0.30) could be due to hydrophobic effects implicated by free THF molecules on hydrophilic segments of gelatin. Bhosale et al. has reported the formation of spherical aggregates at 85% v/v THF and formation of fused structures with an increase in THF proportion in AIE active tetraphenylethylene (TPE) derivatives.51 Similar spherical aggregates formation has been reported in water THF systems for polymeric systems also.52–54 From the above studies, it can be concluded that THF could decrease the hydrophobic aggregation in case of Upy substituted gelatin derivatives till 43.8% v/v THF due to better solvent compatibility in gelatin, GEUPY(0.10), and GEUPY(0.30). However, an increase in THF proportion to 50% v/v THF, would implicate the solvophobic interactions by free THF molecules on the hydrophilic segment of gelatin chain and may cause phasing out of the structures through fusion of aggregates to form a film.
 |
| | Fig. 4 SEM images of freeze–dried samples of gelatin, GEUPY(0.10) and GEUPY(0.30) in water (A, D, and G) and at 50% v/v THF after 24 hours incubation at 37 °C (supernatant (B, E, and H), film (C, F, and I)). | |
3.3 Fabrication of elastomeric films and their structural characterization
Phenomenon of phase inversion has been employed by researchers in which a non-solvent induces phase separation before reaching the dried state.55,56 In this process, a concentrated polymer solution is exposed to non-solvent followed by phase separation due to solvent-non-solvent exchange. GEUPY(0.50) was chosen for film fabrication due to higher Upy substitution in comparison to other GEUPY samples. In the present study, THF is a good solvent for Upy and a non-solvent for gelatin, which caused the decrease of hydrophobic aggregation with an increase in THF proportion till 50% v/v in GEUPY(0.50) followed by a phase separation at a higher THF proportion. The polymer-rich phase formed a porous film and polymer-poor phase formed pores inside the film. For organo/hydro hybrid gel film fabrication, polymers were mixed in DI water followed by its THF proportion adjustment to clear point (50% v/v THF) with continuous stirring in a glass container shown in Fig. 5A. After complete mixing, THF proportion was adjusted to 80% v/v THF and was incubated overnight at 37 °C. Gelatin organo/hydro hybrid gel films were also fabricated by following a similar method as control. The film's structural characteristics were observed via optical microscopy and scanning electron microscopy. For optical microscopy, the cross-section of both the films were taken and then observed under an optical microscope at a 10× resolution as shown in Fig. 6C and F. Two different phases were observed in the films (a) polymer-rich phase (b) cosolvent-rich phase. In gelatin organo/hydro hybrid gel film at 80% v/v THF, the cosolvent rich phase was present uniformly throughout the gel due to phase separation by THF. However, GEUPY(0.50) organo/hydro hybrid gel had less cosolvent-rich phase in comparison to gelatin film and was present on one edge of the film. Similar porous structures have been observed by researchers in the polymeric films prepared by phase separation. Dufresne et al. have observed similar structures in gellan gum, alginate, and polydimethylacrylamide (PDMA) gels.57 Freeze–dried films were further characterized for their structural characteristic using SEM. The surface characteristic, pore size, and pore distribution of the films differed significantly. The surface of freeze dried GEUPY(0.50) organo/hydro hybrid gel film was solid and did not have any pores as shown in Fig. 6D, whereas pores with 0.51–1 μm diameter were present in cross-section SEM images. In gelatin organo/hydro hybrid gel film, pores with 10–65 μm diameter were observed on the surface as well as the cross section of the film as shown in the SEM images in Fig. 6A and B. The cross-section SEM images of gelatin and GEUPY(0.50) organo/hydro hybrid gel films were further analysed using ImageJ software for pore size analysis (shown in Fig. S8†). It was observed that the GEUPY(0.50) organo/hydro hybrid gel films film pores (0.51–1 μm) were ∼30 times smaller than gelatin film pores and were majorly present on one edge of the film, whereas gelatin film pores (10–25 μm) were uniformly distributed throughout the film. Fewer numbers of pores and smaller pore size in GEUPY(0.50) organo/hydro hybrid gel films could be due to fusion of coacervates in the presence of Upy to form an intact film in comparison to gelatin.
 |
| | Fig. 5 Schematics to demonstrate the fabrication of (A) organo/hydro hybrid gel films and (B) hydrogel films. | |
 |
| | Fig. 6 SEM images of freeze dried gelatin (A–C), GEUPY(0.50) organo/hydro hybrid gel films (D–F) and rehydrated GEYPY(0.050) hydrogel films (G–I). Top view (A, D and G), cross-section view (B, E, and H) and optical microscopic images of respective films cross-section (C, F, and I). | |
For hydrogel film fabrication, cosolvent from the Gelatin and GEUPY(0.50) organo/hydro hybrid gel films were removed by vacuum drying at 37 °C for 48 hours followed by rehydration (RH) with distilled water shown in Fig. 5B. Gelatin organo/hydro hybrid gel film disintegrated during rehydration process whereas GEUPY(0.50) organo/hydro hybrid gel films maintained their structural integrity and formed GEUPY(0.50) hydrogel films. SEM and optical microscopic images of GEUPY(0.50) hydrogel films demonstrate that rehydration led to appearance of pores and enlargement of pores within the films as shown in Fig. 6H and I.
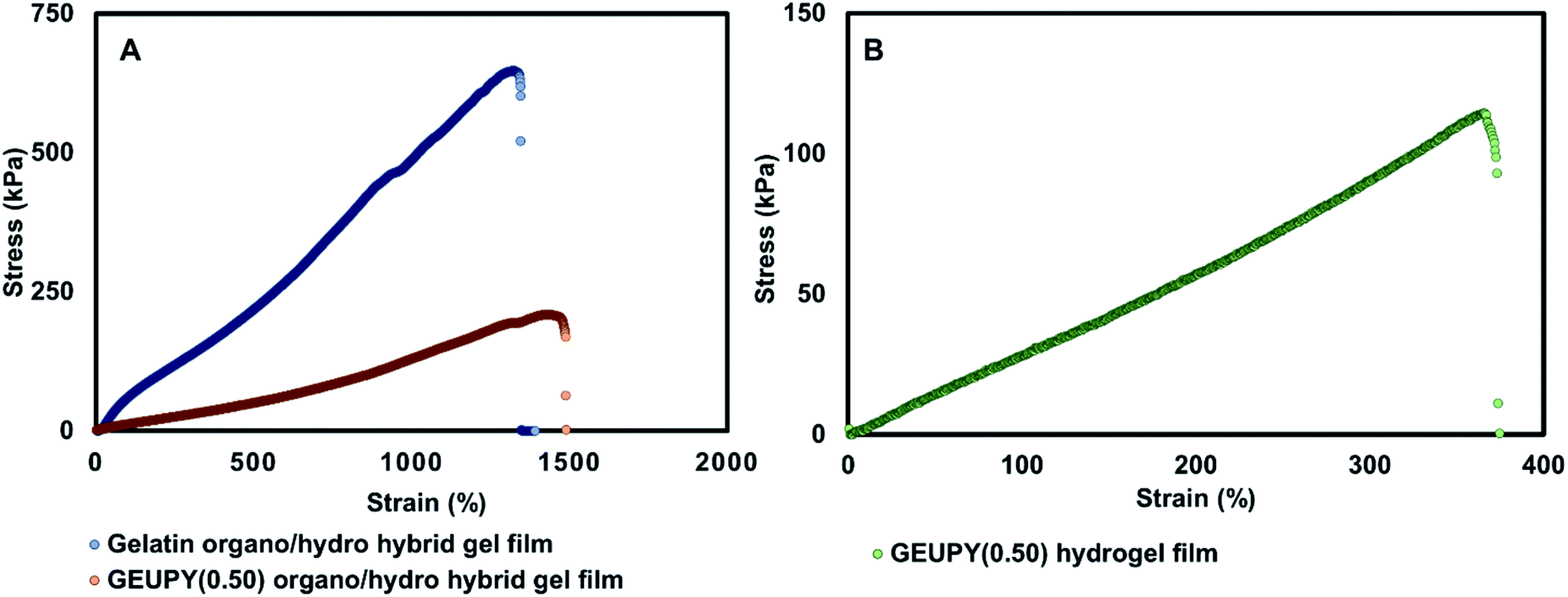
3.4 Tensile properties of elastomeric films
Both gelatin and GEUPY(0.50) organo/hydro hybrid gel films were fabricated by the process mentioned above and further processed to remove water–THF from the film before mechanical characterization. GEUPY(0.50) organo/hydro hybrid gel films displayed higher stretchability and lower Young's modulus in comparison to gelatin organo/hydro hybrid gel film as shown in Table 2. Higher stretchability and lower modulus of GEUPY(0.50) organo/hydro hybrid gel films could be due to Upy associations and solvent compatibility in comparison to gelatin organo/hydro hybrid gel film which has 7 times higher Young's modulus attributed by solvophobic interaction by free THF molecules.
Table 2 Mechanical properties of organo/hydro hybrid and hydrogel films
| Mechanical properties |
Gelatin organo/hydro hybrid gel films |
GEUPY(0.50) organo/hydro hybrid gel films |
GEUPY(0.50) hydrogel films |
| Strain at break (%) |
1269 ± 8 |
1405.9 ± 47.9 |
318.7 ± 44.4 |
| Young's modulus (kPa) |
78.71 ± 0.5 |
10.13 ± 1.3 |
27.35 ± 2.7 |
| Ultimate tensile strength (kPa) |
516.7 ± 106.3 |
207.96 ± 4.8 |
88.57 ± 50.3 |
In gelatin organo/hydro hybrid gel films, rehydration (RH) of vacuum-dried films led to loss of its integrity during handling. This could be due to lack of crosslinking, and consequently they were not characterized for their mechanical properties. However, GEUPY(0.50) hydrogel films remained stable and were characterized for their mechanical properties. A stress vs. strain curve for GEUPY(0.50) organo/hydro hybrid gel films and GEUPY(0.50) hydrogel films has been shown in Fig. 7A and B. Replacement of cosolvent water–THF with water has reduced the stretchability and has enhanced the Young's modulus of the GEUPY(0.50) elastomeric films (shown in Table 6.1†). The strain at break has reduced from 1405.9 ± 47.9% for GEUPY(0.50) organo/hydro hybrid gel films to 318.73 ± 44.4% for GEUPY(0.50) hydrogel films, respectively. However, Young's modulus has increased from 10.13 ± 1.3 kPa for GEUPY organo/hydro hybrid gel films to 27.35 ± 2.7 kPa for GEUPY(0.50) hydrogel film, respectively. Exchange of water–THF cosolvent with water would have increased the hydrophobic interactions in the films due to hydrophobic nature of Upy moieties and should be responsible for the increase in Young's modulus as well as decrease in stretchability. Solvent content of the films was quantified using TGA analysis. The solvent content of GEUPY(0.50) organo/hydro hybrid gel films gelatin organo/hydro hybrid gel films and GEUPY(0.50) hydrogel films was ∼48%, 36.9% and 67.8% respectively (Fig. S11a and 11b†).
 |
| | Fig. 7 Stress strain curve for (A) gelatin organo/hydro hybrid gel films, GEUPY(0.50) organo/hydro hybrid gel films and (B) GEUPY(0.50) hydrogel films. | |
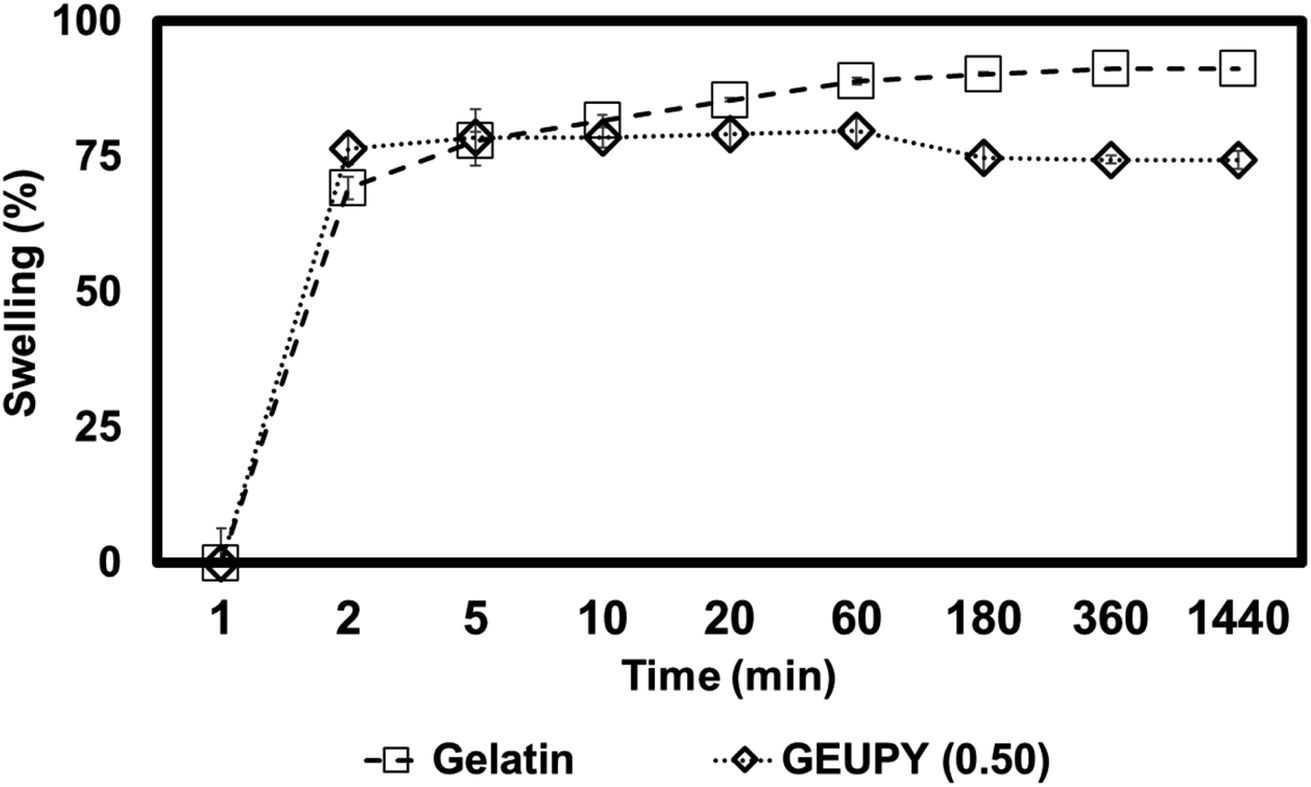
3.5 Swelling studies
Gelatin and GEUPY(0.50) organo/hydro hybrid gel films were dried in vacuum oven at 37 °C for 48 hours to analyse the swelling behaviour of the films. In both gelatin and GEUPY(0.50) air dried films, the swelling reached to >70% water content in less than 2 minutes when rehydrated in distilled water at room temperature as shown in Fig. 8. In case of gelatin it is due to large porous structure, whereas in GEUPY(0.50) film swelling of film could be due to hydrophilic cavities that allow fast swelling. Despite the hydrophobic Upy modification, the swelling of GEUPY(0.50) films were quite fast, and this could be due to hydrophilic pores present in the films. After 3 hours, the water content was stabilized in the GEUPY(0.50) film, whereas for gelatin films, equilibrium was reached after 48 hours at a water content of 91.17 ± 0.2% and started to disintegrate due to the absence of crosslinking. GEUPY(0.50) film reached saturation at 70.98 ± 1.3% and was stable. The lower swelling ability in the latter is expected due to Upy crosslinking as well as hydrophobic nature of the Upy present.
 |
| | Fig. 8 Swelling curve for air-dried gelatin and GEUPY(0.50) films at room temperature. | |
3.6 Mechanism of film fabrication
Cosolvent systems have been utilized by researchers for the fabrication of gelatin nanoparticles. It has been observed that incompatibility with the solvent compels gelatin polymer to reduce its spatial expansion into an aggregate of charged gelatin chains. In the given studies, gelatin has formed spherical aggregates at 43.8% v/v THF and 43.8% ethanol as shown in Fig. 8. Spherical aggregates formed in water–ethanol cosolvent tended to remain in a stable state when ethanol proportion was increased to 80% ethanol, whereas in water–THF spherical particles/aggregates fused with other particles to form an elastomeric film (Fig. S10†). In water–THF cosolvent, microdroplets of pure THF molecules have been known to form and were used as a template for the synthesis of polymeric nanoparticles.31,58,59 This significant variation in cosolvent behaviour and its effect on the polymers are demonstrated in Fig. 9. At stage I, pure water molecules are present with intermolecular hydrogen bonding among them. In pure water, gelatin has been reported to form hydrogen bonding with water molecules and have the affinity to form helical structures from random coils, whereas GEUPY samples had formed aggregates due to hydrophobicity attributed by modified Upy shown above.60 Addition of THF molecules to water would result in the formation of intermediate water–THF complexes and break hydrogen bonding among water molecules (stage II). At stage II, gelatin polymer chains remain stable (>90 %T) due to engagement of free water molecules with THF molecules, whereas GEUPY aggregates will extend due to compatibility with THF. However, a further increase in THF molecules would form THF nano/micro droplets of free THF molecules (stage III). At stage III, gelatin and GEUPY chains will orient themselves at water–THF droplet interface due to the amphiphilic nature of gelatin and Upy in GEUPY samples which led to the formation of spherical aggregates at clear point. Further addition of THF molecules would result in an increase of THF micro-droplets which could be either due to an increase in THF proportion or fusion of micro-droplets (stage IV) and eventually lead to the formation of an elastomeric film (stage V). Gelatin has been known to form triple helix structures in aqueous environment when incubated below sol–gel transition temperature. To study the effect of THF and Upy functionalization, gelatin, GUEPY(0.10) and GEUPY(0.30) were characterized for secondary structure through wide angle X-ray scattering (WAXS). Three transitions at (q ∼0.5(I), ∼1.25(II) and ∼3.25 nm (III)) were observed in pure gelatin and similar were observed in case of GEUPY(0.10) at 50% THF (shown in Fig. S11†). However, a single transition was observed at d ∼1.25 nm in case of in GEUPY (0.30) at 50% THF. In collagen, similar 3 transitions have been reported where the transition I corresponds to intermolecular lateral packing, transition II corresponds to isotropic amorphous phase of gelatin and transition III to axial periodicity of amino acid residues in chain.61 Loss of axial periodicity and lateral packaging (triple helix) took place upon Upy substitution.
 |
| | Fig. 9 Schematics demonstrating cosolvent composition and mechanism of film fabrication. | |
The elastomeric organo/hydro gel films of GEUPY(0.50) fabricated by adjustment of cosolvent proportion from 50% v/v THF to 80% v/v THF exhibited 1405.9 ± 47.9% strain at break, whereas gelatin organo/hydro gel films exhibited 1269 ± 8%. Elastomeric properties of the gelatin films could be due to solvophobic interactions exerted by the THF molecules over hydrophilic segments of gelatin chains. Higher stretchability of GEUPY(0.50) organo/hydro hybrid gel films in comparison to gelatin organo/hydro gel films could be attributed by Upy crosslinking in addition to solvophobic interactions. However, removal of cosolvent and rehydration of films has resulted in disintegration of gelatin hydrogel films due to absence of solvophobic interactions imposed by THF molecules. Lower stretchability of GEUPY(0.50) hydrogel films were attributed by an increase in the crosslinking by hydrophobic interactions imposed by water molecules on Upy moieties as well as due to lack of solvophobic interactions in the absence of THF molecules.
4. Conclusion
An elastomeric hydrogel from natural materials was successfully synthesized through Upy-modified gelatin polymers. It was anticipated that the elastomeric behaviour could be due to the potential of Upy to form supramolecular crosslinking through hydrophobic interactions and Upy dimerization. In this paper, the hydrophobic aggregation of GEUPY polymers in aqueous environments segregated by cosolvent optimization and cosolvent-induced structural transformations were characterized by microscopic techniques at various cosolvent compositions. A novel hydrogel fabrication method was optimized which would be able to induce structural transformation in ureidopyrimidinone-modified gelatin polymer and would form an elastomeric hydrogel. THF microdroplet phase of water–THF cosolvent system was utilized for the dissolution of polymers followed by fusion of polymer-rich THF microdroplets to form an elastomeric film. This film was able to form a stable elastomeric hydrogel with 70% water content with 318.7 ± 44.4% stretchability. The available conventional methods for hydrogel fabrication are not applicable when employed on supramolecular biomaterials which require a specific set of conditions for crosslinking and require a novel method to be devised. Using this novel method, and the supramolecular biomaterials, a natural hydrogel with elastomeric property was developed which is otherwise difficult to fabricate due to intra-chain crosslinking that prevents inter-chain crosslinking.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the financial support provided by Ministry of Education, Singapore, Singapore Centre for 3D Printing (SC3DP), A*STAR (Singapore Food Story R&D Programme), and School of Material Sciences and Engineering, Nanyang Technological University, Singapore.
Notes and references
- Q. Huang and Y. Zhu, Adv. Mater. Technol., 2019, 4, 1800546 CrossRef.
- M. Hao, L. Li, S. Wang, F. Sun, Y. Bai, Z. Cao, C. Qu and T. Zhang, Microsyst. Nanoeng., 2019, 5, 1–10 CrossRef CAS PubMed.
- R. Carta, P. Jourand, B. Hermans, J. Thoné, D. Brosteaux, T. Vervust, F. Bossuyt, F. Axisa, J. Vanfleteren and R. Puers, Sens. Actuators, A, 2009, 156, 79–87 CrossRef CAS.
- R. Garifullin and M. O. Guler, Mater. Today Bio, 2021, 100099 CrossRef CAS.
- A. Panwar and L. P. Tan, Molecules, 2016, 21, 685 CrossRef.
- M. M. Sk, P. Das, A. Panwar and L. P. Tan, Mater. Sci. Eng. C, 2021, 111694 CrossRef CAS PubMed.
- P. Das, M. D. DiVito, J. A. Wertheim and L. P. Tan, Mater. Sci. Eng. C, 2020, 111, 110723 CrossRef CAS.
- B. H. Lee, H. Shirahama, M. H. Kim, J. H. Lee, N.-J. Cho and L. P. Tan, NPG Asia Mater., 2017, 9, e412 CrossRef CAS.
- B. H. Lee, N. Lum, L. Y. Seow, P. Q. Lim and L. P. Tan, Materials, 2016, 9, 797 CrossRef.
- X. Yan, Q. Chen, L. Zhu, H. Chen, D. Wei, F. Chen, Z. Tang, J. Yang and J. Zheng, J. Mater. Chem. B, 2017, 5, 7683–7691 RSC.
- C. Feng, Y. Du, Y. Li and B. Lei, Mater. Sci. Eng. C, 2017, 75, 1339–1342 CrossRef CAS.
- Q. Feng, K. Wei, K. Zhang, B. Yang, F. Tian, G. Wang and L. Bian, NPG Asia Mater., 2018, 10, e455 CrossRef.
- S. Seiffert and J. Sprakel, Chem. Soc. Rev., 2012, 41, 909–930 RSC.
- H. Li, H. Wang, D. Zhang, Z. Xu and W. Liu, Polymer, 2018, 153, 193–200 CrossRef CAS.
- Q. Wang, Z. Liu, C. Tang, H. Sun, L. Zhu, Z. Liu, K. Li, J. Yang, G. Qin and G. Sun, ACS Appl. Mater. Interfaces, 2021, 123617 Search PubMed.
- K. Miyamae, M. Nakahata, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2015, 54, 8984–8987 CrossRef CAS PubMed.
- T. Kakuta, Y. Takashima and A. Harada, Macromolecules, 2013, 46, 4575–4579 CrossRef CAS.
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, eaaf3627 CrossRef PubMed.
- H. Yan, Q. Jiang, J. Wang, S. Cao, Y. Qiu, H. Wang, Y. Liao and X. Xie, Polymer, 2021, 123617 CrossRef CAS.
- M. S. Tjin, P. Low and E. Fong, Polym. J., 2014, 46, 444–451 CrossRef CAS.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. Dankers and E. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS.
- R. E. Kieltyka, A. Pape, L. Albertazzi, Y. Nakano, M. M. Bastings, I. K. Voets, P. Y. Dankers and E. Meijer, J. Am. Chem. Soc., 2013, 135, 11159–11164 CrossRef CAS.
- C. L. Lewis and M. Anthamatten, Soft Matter, 2013, 9, 4058–4066 RSC.
- H. Chen, Y. Li, G. Tao, L. Wang and S. Zhou, Polym. Chem., 2016, 7, 6637–6644 RSC.
- R. Z. Alavijeh, P. Shokrollahi and J. Barzin, J. Mater. Chem. B, 2017, 5, 2302–2314 RSC.
- T. V. Chirila, H. H. Lee, M. Oddon, M. M. Nieuwenhuizen, I. Blakey and T. M. Nicholson, J. Appl. Polym. Sci., 2014, 131 Search PubMed.
- X. Zhao, Y. Liang, Y. Huang, J. He, Y. Han and B. Guo, Adv. Funct. Mater., 2020, 30, 1910748 CrossRef CAS.
- K. Yamauchi, J. R. Lizotte and T. E. Long, Macromolecules, 2003, 36, 1083–1088 CrossRef CAS.
- J. N. Nayak, M. I. Aralaguppi, B. V. Kumar Naidu and T. M. Aminabhavi, J. Chem. Eng. Data, 2004, 49, 468–474 CrossRef CAS.
- Y.-Z. Ni, W.-F. Jiang, G.-S. Tong, J.-X. Chen, J. Wang, H.-M. Li, C.-Y. Yu, X.-h. Huang and Y.-F. Zhou, Org. Biomol. Chem., 2015, 13, 686–690 RSC.
- M. D. Smith, B. Mostofian, L. Petridis, X. Cheng and J. C. Smith, J. Phys. Chem. B, 2016, 120, 740–747 CrossRef CAS PubMed.
- A.-M. Alexander, M. Bria, G. Brunklaus, S. Caldwell, G. Cooke, J. F. Garety, S. G. Hewage, Y. Hocquel, N. McDonald and G. Rabani, Chem. Commun., 2007, 2246–2248 RSC.
- A. Duconseille, T. Astruc, N. Quintana, F. Meersman and V. Sante-Lhoutellier, Food Hydrocolloids, 2015, 43, 360–376 CrossRef CAS.
- S. Hou, X. Wang, S. Park, X. Jin and P. X. Ma, Adv. Healthcare Mater., 2015, 4, 1491–1495 CrossRef CAS PubMed.
- A. A. Karim and R. Bhat, Trends Food Sci. Technol., 2008, 19, 644–656 CrossRef CAS.
- E. Hoch, T. Hirth, G. E. Tovar and K. Borchers, J. Mater. Chem. B, 2013, 1, 5675–5685 RSC.
- K. Yoshizawa and T. Taguchi, Int. J. Mol. Sci., 2014, 15, 2142–2156 CrossRef.
- A. Ben-Naim, in Hydrophobic Interactions, Springer, 1980, pp. 49–115 Search PubMed.
- P. Y. W. Dankers, M. J. A. van Luyn, A. Huizinga-van der Vlag, G. M. L. van Gemert, A. H. Petersen, E. W. Meijer, H. M. Janssen, A. W. Bosman and E. R. Popa, Biomaterials, 2012, 33, 5144–5155 CrossRef CAS.
- P. Y. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. Van Luyn and E. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS.
- Y.-H. Lin, W. Nie, H. Tsai, X. Li, G. Gupta, A. D. Mohite and R. Verduzco, RSC Adv., 2016, 6, 51562–51568 RSC.
- P. Y. W. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. A. Van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS.
- W. Lin, C. Zheng, X. Wan, D. Liang and Q. Zhou, Macromolecules, 2010, 43, 5405–5410 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- T. Takamuku, A. Yamaguchi, D. Matsuo, M. Tabata, M. Kumamoto, J. Nishimoto, K. Yoshida, T. Yamaguchi, M. Nagao and T. Otomo, J. Phys. Chem. B, 2001, 105, 6236–6245 CrossRef CAS.
- V. Bütün, Ş. Sönmez, S. Yarligan, F. F. Taktak, A. Atay and S. Bütün, Polymer, 2008, 49, 4057–4065 CrossRef.
- J. áde Visser, Soft Matter, 2016, 12, 4113–4122 RSC.
- C. Yang, W. Li and C. Wu, J. Phys. Chem. B, 2004, 108, 11866–11870 CrossRef CAS.
- M. J. Shultz and T. H. Vu, J. Phys. Chem. B, 2015, 119, 9167–9172 CrossRef CAS.
- M. Salimimarand, D. D. La, M. Al Kobaisi and S. V. Bhosale, Sci. Rep., 2017, 7, 42898 CrossRef CAS PubMed.
- X. Li, H. Shan, M. Cao and B. Li, J. Membr. Sci., 2018, 555, 237–249 CrossRef CAS.
- T. Higuchi, A. Tajima, H. Yabu and M. Shimomura, Soft Matter, 2008, 4, 1302–1305 RSC.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS PubMed.
- K.-V. Peinemann, V. Abetz and P. F. Simon, Nat. Mater., 2007, 6, 992–996 CrossRef CAS PubMed.
- P. Molla-Abbasi, Polym. Adv. Technol., 2021, 32, 391–401 CrossRef CAS.
- R. W. Style, T. Sai, N. Fanelli, M. Ijavi, K. Smith-Mannschott, Q. Xu, L. A. Wilen and E. R. Dufresne, Phys. Rev. X, 2018, 8, 011028 CAS.
- C. K. Wong, A. F. Mason, M. H. Stenzel and P. Thordarson, Nat. Commun., 2017, 8, 1–10 CrossRef CAS.
- T. Takamuku, A. Nakamizo, M. Tabata, K. Yoshida, T. Yamaguchi and T. Otomo, J. Mol. Liq., 2003, 103, 143–159 CrossRef.
- L. Nuvoli, P. Conte, C. Fadda, J. A. R. Ruiz, J. M. García, S. Baldino and A. Mannu, Polymer, 2021, 214, 123244 CrossRef CAS.
- G. Tronci, A. Doyle, S. J. Russell and D. J. Wood, J. Mater. Chem. B, 2013, 1, 5478–5488 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra09084d |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Md Moniruzzaman Ska,
Bae Hoon Leec and
Lay Poh Tan
*ab,
Md Moniruzzaman Ska,
Bae Hoon Leec and
Lay Poh Tan