DOI:
10.1039/D1RA08552B
(Paper)
RSC Adv., 2022,
12, 2436-2442
Reduction of imines with a reusable bimetallic PdCo–Fe3O4 catalyst at room temperature under atmospheric pressure of H2†
Received
22nd November 2021
, Accepted 27th December 2021
First published on 18th January 2022
Abstract
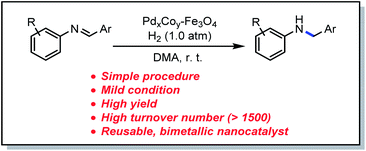
Bimetallic nanocatalysts have been used for the development of organic reactions, owing to the synergistic effect between the transition metals. A new procedure for synthesizing amines by the reduction of imines with H2 at atmospheric pressure and room temperature in the presence of PdCo–Fe3O4 nanoparticles is reported. The straightforward procedure, mild reaction conditions, high turnover number, and recyclability extend the scope of this reaction to practical applications.
Introduction
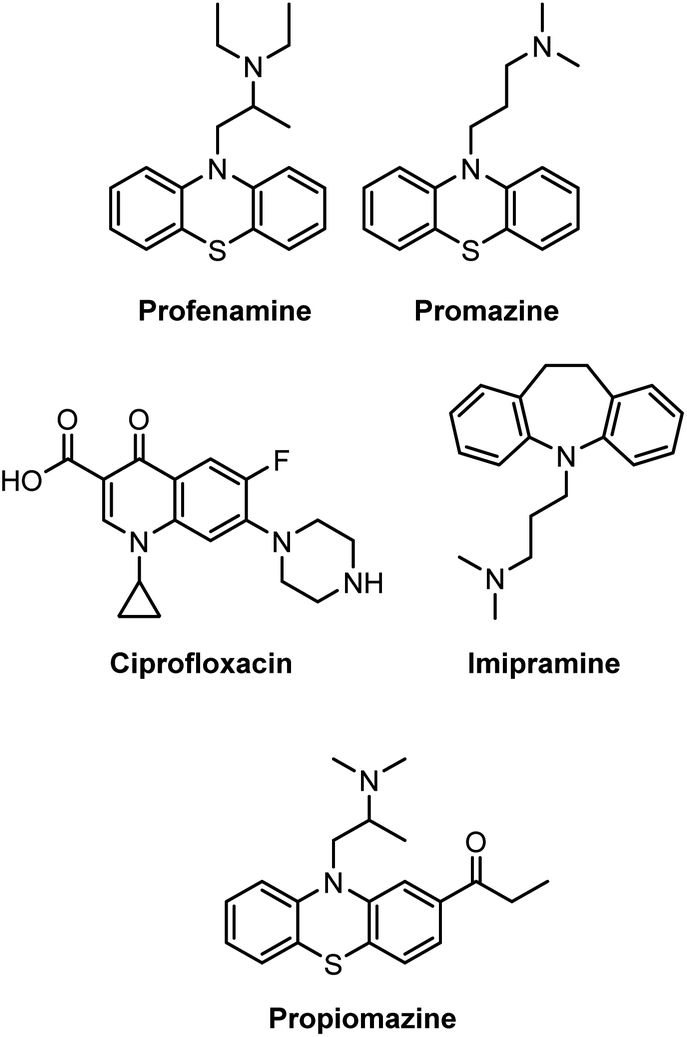
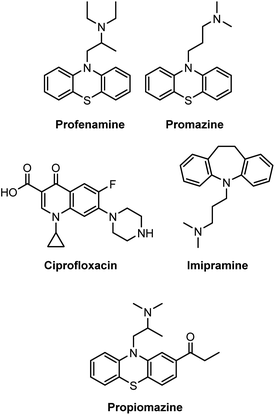
Amines are versatile organic molecules found in various natural products,1 drug molecules,2 and dyes.3 Of particular interest is a large number of pharmaceuticals, such as profenamine (antidyskinetic),4 promazine (antiemetic),5 ciprofloxacin (antibiotic),6 imipramine (antidepressant),7 and propiomazine (antipsychotic agent),8 which all contain the amine functionality (Scheme 1). In this context, research to develop continually mild and convenient synthetic procedures for synthesizing amines from various precursors remains highly relevant to the present pharmaceutical industry.9
 |
| | Scheme 1 Amines in some commercially available drugs. | |
Among many established procedures, the reduction of imines is one of the most straightforward methods, owing to the fact that imines can be readily produced from precursor amines and carbonyl compounds.10 Although the reduction of imines can be facilitated by adding stoichiometric hydride reagents, such as NaBH4,11 NaBH3CN,12 or NaBH(OAc)3,13 catalytic processes are preferred for a large-scale production. Various homogeneous transition metal catalysts such as Pd,14a Ir,15a Ru,16a,b Mn,17a and Mo18 have been employed in the reduction of imines using H2. Drawbacks associated with reactions on these catalysts include high temperature and high H2 pressure required to achieve high turnover numbers (TON). In the meantime, heterogeneous catalysis employing transition metals have been gaining increasing attention as an alternative to homogeneous catalysis, owing to the usability and recyclability.19 For the purpose of overcoming the shortcomings mentioned above, efforts have been concentrated on recycling the catalyst, thereby allowing repeated applications after the hydrogenation process.14b–e,15b,c,16c,17b Therefore, we envisioned that further development of the catalytic reaction reduction of imines under mild reaction conditions, i.e., room temperature and at atmospheric pressure of H2, with a high turnover number, and good catalyst reusability is still in need (Table 1).
Table 1 Representative reports on the reduction of imines with various transition metal catalysts in the presence of H2
| Entry |
Metal |
Condition |
TON |
Note |
Ref. |
| HM = homogeneous catalysis. HT = heterogeneous catalysis. |
| 1 |
Pd |
r. t., 40 atm H2 |
20 |
HMa |
14a |
| 2 |
Ir |
80 °C, 120 atm H2 |
3960 |
HM |
15a |
| 3 |
Ru |
40 °C, 50 atm H2 |
31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
HM |
16a |
| 4 |
Ru |
70 °C, 5.0 atm H2 |
1000 |
HM |
16b |
| 5 |
Mn |
50 °C, 50 atm H2 |
49 |
HM |
17a |
| 6 |
Mo |
r. t., 30 atm H2 |
333 |
HM |
18a |
| 7 |
Mo |
140 °C, 60 atm H2 |
800 |
HM |
18b |
| 8 |
Pd |
r. t., 1.0 atm H2 |
404 |
HTb |
14c |
| 9 |
Pd |
r. t., 1.0 atm H2 |
8 |
HT |
14e |
| 10 |
Ir |
50 °C, 100 atm H2 |
12400 |
HT |
15b |
| 11 |
Ir |
r. t., 10 atm H2 |
1188 |
HT |
15c |
| 12 |
Ru |
90 °C, 30 atm H2 |
1 |
HT |
16c |
| 13 |
Mn |
r. t., 5.0 atm H2 |
19 |
HT |
17b |
| 14 |
PdCo |
r. t., 1.0 atm H2 |
1583 |
HT |
This work |
There has been increasing interest in bimetallic heterogeneous catalysts because they often exhibit a synergistic effect between two types of metals, which is not observed in metals of single type.20 A series of bimetallic heterogeneous catalysts, supported on iron oxide (Fe3O4) nanoparticles (NPs), have recently been developed for application in the development of various organic reactions. This included the development of PdPt–Fe3O4 NPs that have been employed in nitroarene reduction,21 arylsilylation,22 as well as secondary amine synthesis from benzonitriles and nitroarenes.23 Additionally, AuPd–Fe3O4 NP catalysts have been developed for reductive amination,24 oxidation of furan-2,5-dimethylcarboxylate,25 and N-formylation of secondary amines.26 However, all these catalysts employed expensive, precious metals (Pd and Pt, Au and Pd) among the transition metal element group. Further research therefore focused on the development of a bimetallic nanocatalyst that incorporated at least one inexpensive transition metal. Accordingly, PdCo bimetallic catalysts were identified as a promising alternative, with applications in various organic transformations.27 These include the development of a PdCo catalyst for the transfer hydrogenation of carbonyl compounds by Kumar et al.,27a while Wang group employed a PdCo electrocatalyst for the reduction of oxygen and the oxidation of ethanol.27b Chen group also successfully employed a PdCo alloy for oxygen reduction,27c while Salmeron group investigated the effect of a PdCo nanocatalyst in CO oxidation.27d A PdCo bimetallic catalyst has, however, not yet been employed in imine reduction. In the present study, we have developed PdCo–Fe3O4 as a recyclable bimetallic nanocatalyst for the reduction of imines (Scheme 2) under very mild conditions.
 |
| | Scheme 2 Synthesis of amines from imines using reusable PdCo–Fe3O4 bimetallic catalyst under mild condition. | |
Results and discussion
Catalyst characterization
Here, bimetallic PdCo–Fe3O4 NPs were synthesized through the modification of an existing synthetic procedure for AuPd–Fe3O4.24 Sequentially adding palladium(II) chloride (PdCl2) in ethylene glycol (EG), cobalt(II) chloride hexahydrate (CoCl2·6H2O) in water, and aqueous sodium borohydride solution dropwise to a Fe3O4 solution in water followed by stirring under 60 °C for 24 h afforded PdCo–Fe3O4 NPs. The synthesized nanocatalyst was characterized employing various techniques. Scanning electron microscopy (SEM) images (Fig. 1a and S1†) were obtained using a JSM-7800F Prime microscope (JEOL Ltd., Tokyo, Japan). High-resolution transmission electron microscopy (HR-TEM) images (Fig. 1b, S4 and S6†) were obtained using a JEM-3010 microscope (JEOL Ltd., Tokyo, Japan), indicating successful deposition of Pd and Co atoms on the Fe3O4 support. Scanning transmission electron microscopy images (STEM, Fig. 2 and S5†) were obtained using a JEM-ARM200F microscope (JEOL Ltd., Tokyo, Japan), while energy-dispersive X-ray spectroscopy (EDS) images (Fig. S2 and S3†) were obtained using a JSM-7800F Prime Field Emission Scanning Electron Microscope (JEOL Ltd., Tokyo, Japan), showing that Pd and Co were randomly distributed. The electron energy loss spectroscopy (EELS) was performed using a Themis Z STEM (ThermoFisher, MA, USA), where the high-energy loss spectrum images indicated that the Co lines are located at 780–800 eV (Fig. S7†). In addition, the X-ray diffraction (XRD) data obtained using a D8 Advance XRD (Bruker, Billerica, MA, USA) indicated that the diffraction peaks at ∼40° and 46–47° 2θ were shifted. This suggested that PdCo–Fe3O4 is made up with Pd–Co bimetallic alloy (Fig. S8†).27h X-ray photoelectron spectroscopy (XPS) data were obtained using a SIGMA PROBE XPS (ThermoFisher Scientific, UK) and provided information on the electronic states of the catalyst (Fig. S9†). Here, first major peaks at the Pd 3d5/2 and Pd 3d3/2 energy levels were observed at 335.35 and 340.61 eV, respectively, which correspond to metallic Pd(0) species.28 In addition, first major peaks at the Co 2p3/2 and Co 2p1/2 energy levels were observed at 779.84 and 795.64 eV, respectively, corresponding to metallic Co(0) species.29 Finally, an OPTIMA 8300 inductively coupled plasma-atomic emission spectrometer (ICP-AES) (PerkinElmer, Waltham, MA, USA), was employed to determine that the PdCo–Fe3O4 NPs consisted of 7.49 wt% Pd and 3.89 wt% Co, at a molar ratio of 0.99:1.00 (Fig. S11†). The synthesized PdCo–Fe3O4 NPs were subsequently utilized to optimize the imine reduction reaction.
 |
| | Fig. 1 (a) SEM and (b) HR-TEM image of PdCo–Fe3O4 catalyst. | |
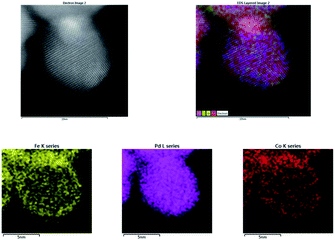 |
| | Fig. 2 STEM-EDS image of PdCo–Fe3O4 catalyst. | |
Reaction optimization
For the initial screening study, N-benzylideneaniline (1a) was selected as a representative substrate and N,N-dimethylacetamide (DMA) as the solvent. Various reaction conditions were examined, with the results presented in Tables 2 and S1–S4.† When the reaction was carried out with only Fe3O4 as a catalyst under 1.0 atm of H2, the reaction did not proceed and 1a remained unchanged (entry 1). Although the reaction resulted in hydrogenolysis side products when Pd/C was employed as a catalyst, none of the desired product was produced (entry 2). However, employing 4.0 mol% of Pd–Fe3O4 resulted in a 60% yield of the desired product 2a, while catalysis employing Co–Fe3O4 alone did not produce any 2a (entries 3 and 4, respectively). Finally, employing the PdCo–Fe3O4 bimetallic catalyst resulted in a 95% yield, indicating an outstanding synergistic effect between Pd and Co (entry 5). To confirm this synergy, the reaction yields of several substrates from the reactions employing monometallic catalysts were compared (Table S6†). Here, the reaction yields using PdCo–Fe3O4 were higher than those obtained when Pd–Fe3O4 or Co–Fe3O4 was used independently. The initial kinetics associated with employing either monometallic catalysts or PdCo–Fe3O4 catalyst were also investigated (Fig. S17†). An enhanced reaction rate was observed for the reaction when the PdCo–Fe3O4 catalyst was employed, demonstrating an additional benefit associated with the use of the bimetallic catalyst. The effect of other solvents was also evaluated. Here, employing methanol and N,N-dimethylformamide (DMF) (entries 6 and 7), resulted in lower yields when compared to yields obtained with DMA. Reactions with other reducing agents such as phenylsilane (entry 8), ammonia borane (entry 9), and sodium borohydride (entry 10) also resulted in lower product yields. Ultimately, approximately quantitative yields were obtained (entries 11 and 12, respectively) even when the catalyst loading was reduced from 2.0 mol% to 1.0 or 0.50 mol%, respectively. Furthermore, the turnover number (TON) of the catalyst could be higher than 1500 even when the catalyst loading was low, demonstrating the efficiency of the catalyst to facilitate imine reduction (entry 13).
Table 2 Screening data of the synthesis of amine from imine reductiona

|
| Entry |
Catalyst |
Reductant |
Solvent |
Yieldb |
TONc |
| Reaction conditions: 1a (0.20 mmol), catalyst (2.0 mol%), H2 (1.0 atm), solvent (1.0 mL), r. t., 18 h. Determined from 1H NMR spectral analysis through the use of anisole as an internal standard. Turnover number (TON) = mmol of product/mmol of total metal except Fe. N. D. = not detected. Pd/C (4.0 mol%) was used as a catalyst. Pd–Fe3O4 (4.0 mol%) was used as a catalyst. Co–Fe3O4 (4.0 mol%) was used as a catalyst. Yield of isolated product. Result with 3.0 equiv. of reductant instead of 1.0 atm of H2. PdCo–Fe3O4 (1.0 mol%) was used as a catalyst. PdCo–Fe3O4 (0.50 mol%) was used as a catalyst. Result with 1a (3.3 mmol), PdCo–Fe3O4 (0.03 mol%), 78 h. |
| 1 |
Fe3O4 |
H2 |
DMA |
N. D.d |
— |
| 2e |
Pd/C |
H2 |
DMA |
N. D. |
— |
| 3f |
Pd–Fe3O4 |
H2 |
DMA |
60 |
15 |
| 4g |
Co–Fe3O4 |
H2 |
DMA |
N. D. |
— |
| 5 |
PdCo–Fe3O4 |
H2 |
DMA |
95 (91h) |
24 |
| 6 |
PdCo–Fe3O4 |
H2 |
MeOH |
71 |
18 |
| 7 |
PdCo–Fe3O4 |
H2 |
DMF |
83 |
21 |
| 8i |
PdCo–Fe3O4 |
PhSiH3 |
DMA |
2 |
0.50 |
| 9i |
PdCo–Fe3O4 |
BH3NH3 |
DMA |
47 |
12 |
| 10i |
PdCo–Fe3O4 |
NaBH4 |
DMA |
39 |
10 |
| 11j |
PdCo–Fe3O4 |
H2 |
DMA |
>99 |
50 |
| 12k |
PdCo–Fe3O4 |
H2 |
DMA |
98 (97h) |
98 |
| 13l |
PdCo–Fe3O4 |
H2 |
DMA |
95 |
1583 |
Substrate scope
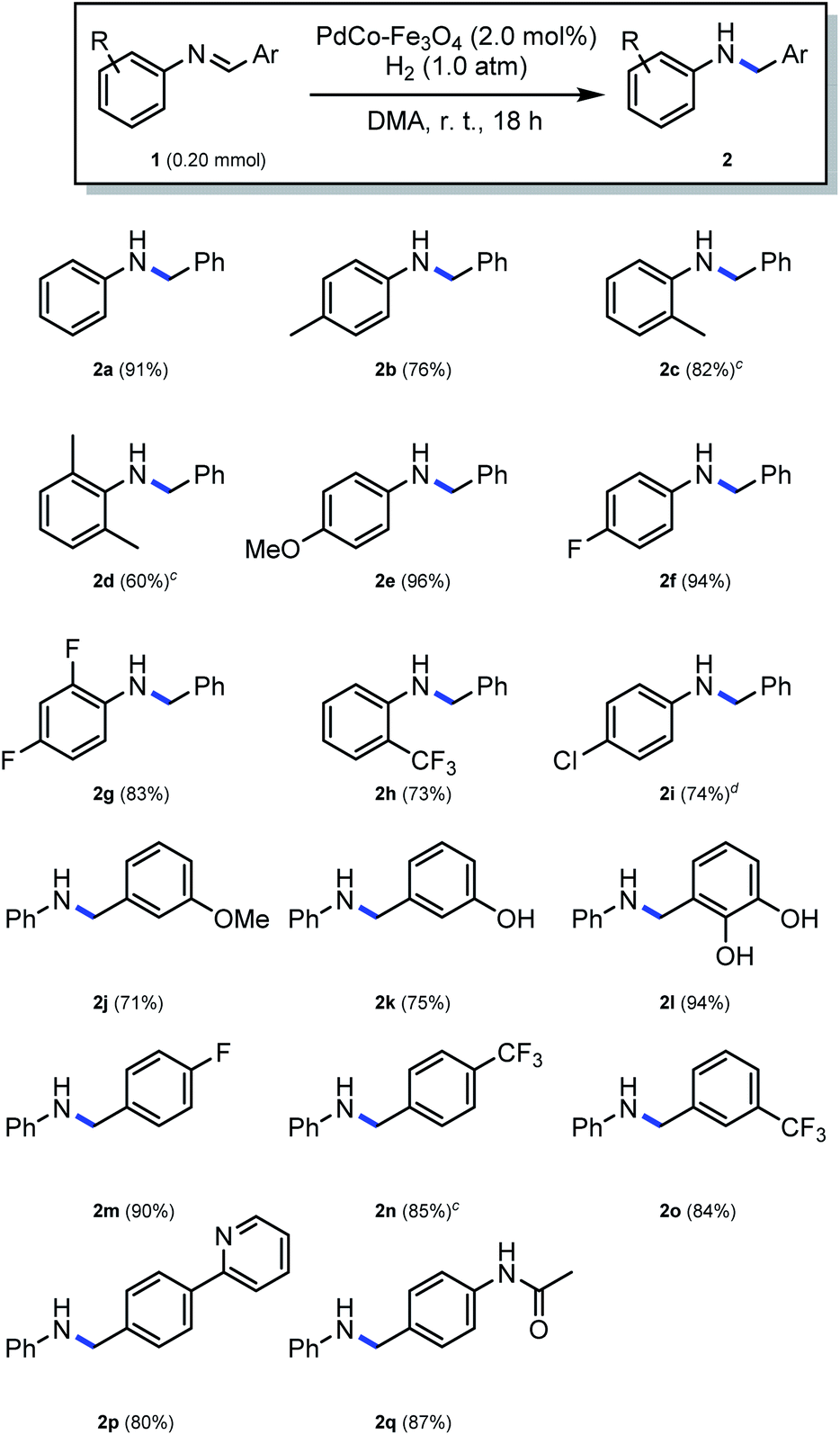
The reduction of several imines with various substituents on the aromatic ring was subsequently evaluated at the optimized conditions, employing a 0.20 M substrate concentration for a period of 18 h. Here, as shown in Scheme 3, the reduction of N-benzylideneaniline, with no substituent on the aniline or benzene ring, proceeded smoothly, and a 91% yield of the desired product (2a) could be isolated. Substrates with a methyl group on the aniline moiety resulted in moderate to good yields (76% and 82% for 2b and 2c, respectively), while the reaction of a substrate with a single methyl group at position 2 of the aromatic ring did not have an impact on the yield (2c). However, substitution with two methyl groups at positions 2 and 6 resulted in a low yield (2d), demonstrating the influence of steric hindrance. Strong electron-donating methoxy group on the aromatic ring was well tolerated (2e). The reduction of substrates with strong electron-withdrawing groups, such as fluorine, were associated with high yields (94% and 83% for 2f and 2g, respectively). The reduction of an imine containing a relatively bulky trifluoromethyl group at position 2 of the aniline ring provided a 73% yield (2h). For the reduction of substrates containing a halogen such as Cl, Br, and I, the use of PdCo–Fe3O4 was not appropriate, because substrate dehalogenation occurred.30 It was therefore hypothesized that reducing the content of Pd relative to that of Co to a minimum, may suppress dehalogenation while maintaining the synergy between Pd and Co. This was confirmed as the reaction with Pd0.05Co1–Fe3O4 proceeded successfully without knocking out the chloride group at position 4 of the aniline ring, resulting in a 74% yield obtained within 30 h (2i). Various substrates with substituents on the benzyl ring were further tested. Here, the reaction of a substrate containing a methoxy group at position 3 provided a 71% yield (2j), while a good yield (75%, 2k) was also obtained using a 3-hydroxy substituted substrate. The reaction of a substrate with multiple hydroxyl groups at positions 2 and 3 on the benzyl ring also resulted in a high yield (94%, 2l). Similarly, when a substrate possessing fluoro or trifluoromethyl group on the benzyl ring was employed in the reaction, high yields (90%, 85% and 84% for 2m, 2n and 2o, respectively) could be obtained under similar conditions. Reduction of a substrate containing a heteroaromatic ring, such as pyridine was also well tolerated, resulting in an 80% yield (2p). Finally, an 87% yield was obtained when a substrate with an acetamide group on the benzyl ring was employed in the reaction. In this instance, the amide functionality was intact (2q).
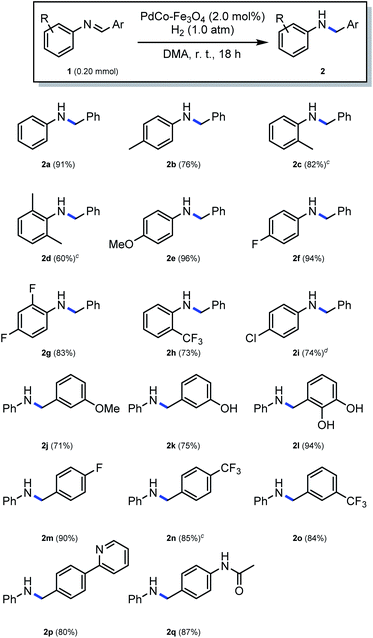 |
| | Scheme 3 Substrate scope of the synthesis of amines by the reduction of imines.a,b aResult with 1 (0.20 mmol), PdCo–Fe3O4 (2.0 mol%), H2 (1.0 atm), DMA (1.0 mL), r. t., 18 h. bYield of isolated product. cResult with 1 (0.50 mmol) in DMA (2.0 mL). dPd0.05Co1–Fe3O4 (0.10 mol%), r. t., 30 h. | |
Effect of Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Co ratio and various supports
:Co ratio and various supports
Based on the results from Table 2, the role of Co in the PdCo bimetallic catalyst appeared to accelerate hydrogenation since the bimetallic catalyst works more efficiently than either of the monometallic catalyst. Therefore, we were curious whether any differences in catalytic activity could be attributed to the composition of the two metals in the catalyst. Catalysts with various Pd:Co ratios were therefore prepared (Fig. S12†) and tested under the same reaction conditions (Table 3, entries 1–6). Here, the experimental data indicated that the yields were excellent regardless of the Pd:Co molar ratios. Reaction using the catalyst with 0.04:1 molar ratio of Pd:Co resulted in 91% yield in 18 h (Table 3, entry 1). In an attempt to evaluate the effect of the Fe3O4 support on the catalyst performance, PdCo catalysts with various supports were synthesized (Fig. S16 and Table S5†) and the reactions employing catalysts with new supports were tested (Table 3, entries 7–9). Although PdCo–CeO2 (entry 8) exhibited a similar reactivity to that of PdCo–Fe3O4, it was concluded that PdCo–Fe3O4 offered additional advantages owing to the simplicity associated with the recycling process. This will be discussed in the following section.
Table 3 Reactivity comparison of various PdCo catalystsa

|
| Entry |
Catalyst |
Yieldb |
| Reaction conditions: 1a (0.20 mmol), catalyst (2.0 mol%), H2 (1.0 atm), DMA (1.0 mL), r. t., 18 h. Determined from 1H NMR spectral analysis through the use of anisole as an internal standard. |
| 1 |
Pd0.04Co1–Fe3O4 |
91 |
| 2 |
Pd0.26Co1–Fe3O4 |
96 |
| 3 |
Pd0.46Co1–Fe3O4 |
96 |
| 4 |
Pd1Co1–Fe3O4 |
95 |
| 5 |
Pd1Co0.45–Fe3O4 |
93 |
| 6 |
Pd1Co0.28–Fe3O4 |
93 |
| 7 |
PdCo–TiO2 |
82 |
| 8 |
PdCo–CeO2 |
93 |
| 9 |
PdCo–C |
63 |
Recycling test
Considering that heterogeneous catalysts are often employed based on their convenient recovery and the possibility to reuse the catalyst, the recyclability of various PdxCoy–Fe3O4 NPs was tested under optimized conditions (Fig. 3 and S13†). PdCo–Fe3O4 NPs were used as a representative catalyst, after being recovered using a small magnet and washed with MeOH several times. In the recycling experiment with 1a, a consistent catalytic activity was observed for 12 cycles. However, starting from the 13th cycle, a reduction in reactivity was observed. Evaluation of the SEM and HR-TEM images of the catalyst after the 14th cycle indicated that the used PdCo particles exhibited a high degree of agglomeration as well as detachment of metal atoms from the Fe3O4 support (Fig. S14 and S15†). Additionally, ICP-AES analysis of the used catalyst showed a reduction in both Pd and Co contents, from 7.49 wt% to 1.69 wt% for Pd and from 3.89 wt% to 0.28 wt% for Co, respectively (Fig. S11†), reflecting the decreased reactivity after the repeated catalytic cycles.
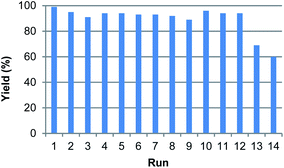 |
| | Fig. 3 Results of recycling test under PdCo–Fe3O4 NPs. | |
Gram scale reaction
The scalability of the reaction (Scheme 4) was further investigated, and at a 6.0 mmol scale, the reaction of 1a resulted in an 80% yield of 2a, demonstrating its potential suitability in organic synthesis applications.
 |
| | Scheme 4 A gram scale reaction. | |
Conclusions
In summary, a new catalytic system was developed for the synthesis of amines via the reduction of imines. Utilizing a PdCo bimetallic nanocatalyst, various imines could be reduced to amines at room temperature and under 1.0 atm of H2. This process therefore allowed the catalytic reduction of imines to amines without the use of toxic or polluting stoichiometric reducing agents. Furthermore, the catalytic performance of the bimetallic PdCo–Fe3O4 NPs was superior to that of monometallic NPs, such as Pd–Fe3O4 and Co–Fe3O4. This indicated the existence of a synergistic effect between Pd and Co. The application scope of the reaction was broad, and it could tolerate substrates with various functional groups. The reduction reaction was further shown to be independent of the Pd:Co ratio, while the Fe3O4 NPs were proven to be a more effective support material than other supporting materials such as TiO2, CeO2, and C. The PdCo–Fe3O4 NPs could be used in more than 10 cycles without a significant loss of catalytic activity. Moreover, it was possible to run the reaction on a gram scale. The high turnover number combined with the simple experimental procedure overcomes the limitations associated with previous approaches such as high reaction temperatures, high H2 pressures, as well as the use of expensive transition metal precursors. This catalyst will be investigated further to expand its potential applications in organic chemistry.
Experimental
Materials
All commercially available chemicals were purchased from Acros Organics (Pittsburgh, PA, USA), Sigma-Aldrich Aldrich (St. Louis, MO, USA), Alfa Aesar (Ward Hill, MA, USA), Tokyo Chemical Industry (Tokyo, Japan), and used without further purification. Imine substrates were synthesized by a known procedure.31
Synthesis of PdCo–Fe3O4 NPs
Initially, 88.7 mg (0.50 mmol) of PdCl2 and 1.00 g of PVP (Mw ∼ 10000 g mol−1, 0.10 mmol) were placed in 20 mL of ethylene glycol (EG) in a 100 mL round-bottom flask. This mixture was sonicated for 10 min and stirred for 1 h at 100 °C in an oil bath. In a separate 100 mL round-bottom flask, 119.0 mg (0.50 mmol) of CoCl2·6H2O and 500 mg of PVP (0.050 mmol) were added to 20 mL of water. This mixture was sonicated for 10 min and stirred for 30 min at 60 °C in an oil bath. Meanwhile, 500 mg of Fe3O4 NPs were added to 150 mL of water in a two-necked 500 mL round-bottom flask and then sonicated for 10 min. The prepared Pd precursor solution was then injected dropwise onto the Fe3O4 suspension with vigorous stirring. After 5 min, the Co precursor solution was added and 90 mg (2.38 mmol) of sodium borohydride in 20 mL of water was injected dropwise. The resulting mixture was stirred for 24 h at 60 °C. Subsequently, the PdCo alloy on Fe3O4 nanoparticles was retrieved via sonication and washing with ethanol (40 mL × 10 times) and dried on a rotary evaporator to give PdCo–Fe3O4 NPs (550 mg, 77% yield based on PdCl2).
Synthesis of Pd–Fe3O4 NPs
Initially, 177 mg of PdCl2 (1.0 mmol) and 2.00 g of PVP (0.20 mmol) were placed in 40 mL of EG in a 100 mL round-bottom flask. This mixture was sonicated for 10 min and stirred for 1 h at 100 °C. Meanwhile, 500 mg of Fe3O4 NPs was added to 150 mL of EG in a two-necked 500 mL round-bottom flask. The prepared precursor solution was then injected dropwise to Fe3O4 NPs in 150 mL of EG, and stirred at 100 °C for an additional 24 h. The resultant product was washed with ethanol (40 mL × 10 times) and dried on a rotary evaporator to give Pd–Fe3O4 NPs (440.0 mg, 36% yield based on PdCl2).
Synthesis of Co–Fe3O4 NPs
Initially, 23.8 mg of CoCl2·6H2O (0.30 mmol) and 200 mg of PVP (0.020 mmol) were placed in 4.0 mL of water in a 10 mL round-bottom flask. This solution was sonicated for 1 min and stirred for 30 min at 60 °C. Meanwhile, 100 mg of Fe3O4 NPs was added to 30 mL of water in a two-necked 100 mL round-bottom flask. The prepared precursor solution was then injected dropwise to Fe3O4 NPs in 30 mL of water, followed by dropwise addition of 30 mg of sodium borohydride (0.79 mmol) in 4.0 mL of water. The mixture was stirred at 60 °C for an additional 24 h. The resultant product was washed with ethanol (40 mL × 10 times) and dried on a rotary evaporator to give Co–Fe3O4 NPs (98.5 mg, 30% yield based on CoCl2·6H2O).
Synthesis of PdxCoy–Fe3O4 NPs
For PdxCoy–Fe3O4 synthesis, the same method used for the synthesis of PdCo–Fe3O4 NPs was employed, with different quantities of metals. To prepare Pd0.26Co1–Fe3O4 NPs, PdCl2 (6.6 mg, 0.038 mmol) with PVP (0.0060 mmol), CoCl2·6H2O (35.7 mg, 0.15 mmol) with PVP (0.010 mmol), and sodium borohydride (0.79 mmol) were used. For the synthesis of Pd0.46Co1–Fe3O4 NPs, PdCl2 (11.1 g, 0.063 mmol) with PVP (0.010 mmol), CoCl2·6H2O (35.7 mg, 0.15 mmol) together with PVP (0.010 mmol), and sodium borohydride (0.79 mmol) were used. In the case of Pd1Pd0.45–Fe3O4 NPs, PdCl2 (17.7 mg, 0.10 mmol) with PVP (0.020 mmol), CoCl2·6H2O (18.2 mg, 0.077 mmol) with PVP (0.0050 mmol), and sodium borohydride (0.79 mmol) were used. Finally, for the preparation of Pd1Co0.28–Fe3O4 NPs, PdCl2 (17.7 mg, 0.10 mmol) with PVP (0.020 mmol), CoCl2·6H2O (11.1 mg, 0.047 mmol) with PVP (0.0030 mmol), and sodium borohydride (0.79 mmol) were used. Meanwhile, 100 mg of Fe3O4 NPs were added to 30 mL of water in a two-necked 100 mL round-bottom flask and then sonicated for 10 min. The prepared Pd precursor solution in 4.0 mL of EG was then injected dropwise onto the Fe3O4 suspension with vigorous stirring. After 5 min, the Co precursor solution in 4.0 mL of water was added and sodium borohydride in 4.0 mL of water was injected dropwise. The resulting mixture was stirred for 24 h at 60 °C. Subsequently, the PdxCoy alloy on Fe3O4 nanoparticles was retrieved via sonication and washing with ethanol (40 mL × 10 times) and dried on a rotary evaporator to give Pd0.26Co1–Fe3O4 NPs (99.0 mg, 59% yield based on PdCl2), Pd0.46Co1–Fe3O4 NPs (96.0 mg, 57% yield based on PdCl2), Pd1Pd0.45–Fe3O4 NPs (98.0 mg, 42% yield based on CoCl2·6H2O), and Pd1Co0.28–Fe3O4 NPs (98.0 mg, 69% yield based on CoCl2·6H2O).
Catalyst characterization
SEM images were obtained using JSM-7800F Prime (JEOL Ltd., Tokyo, Japan) and MERLIN Compact (ZEISS, Oberkochen, Germany). HR-TEM images were obtained using JEM-3010 (JEOL Ltd., Tokyo, Japan). STEM images were obtained using JEM-ARM200F (JEOL Ltd., Tokyo, Japan). XPS data were obtained using SIGMA PROBE (ThermoVG, U.K). ICP-AES data were obtained using OPTIMA 8300 (PerkinElmer, Waltham, MA, USA). The machines mentioned above are installed at the National Center for Inter-University Research Facilities (NCIRF) at Seoul National University. EELS images were obtained using Themis Z (Thermo Fisher, MA, USA) installed at the Research Institute of Advanced Materials at Seoul National University.
The powder X-ray diffraction (XRD) was performed using a D8 Advance (Bruker, Billerica, MA, USA) installed at the National Instrumentation Center for Environmental Management (NICEM) at Seoul National University. Fourier-transform infrared spectroscopy images were obtained using Spectrum Two (PerkinElmer, Waltham, MA, USA) installed at Seoul National University.
General procedure of the synthesis of amines
A glass vial (10 mL) were charged with an imine (0.20 mmol), PdCo–Fe3O4 (2.0 mol%), and N,N-dimethylacetamide (1.0 mL). The mixture was sonicated for 1 min and stirred at room temperature. Next, the vial was purged with H2 using a balloon filled with H2 for 1 min and stirred for 18 h at room temperature. The reaction mixture was extracted by ethyl acetate and the organic layer was filtered through a layer of Celite® and magnesium sulfate. The crude product was purified by column chromatography.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
B. M. K. thanks the Nano Material Development Program (NRF-2012M3A7B4049644) and the Mid-career Researcher Program (NRF-2019R1A2C1004173) for an NRF grant funded by MEST.
Notes and references
- H. Luo, Y. Yang, B. Yang, Z. Xu and D. Wang, J. Chem. Res., 2021, 708–715 CrossRef CAS.
-
(a) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed;
(b) S. Nandi, P. Patel, N.-u. H. Khan, A. V. Biradar and R. I. Kureshy, Inorg. Chem. Front., 2018, 5, 806–813 RSC.
- H. M. Pinheiro, E. Touraud and O. Thomas, Dyes Pigm., 2004, 61, 121–139 CrossRef CAS.
- P. L. Goldschmidt, L. Savary and P. Simon, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 1984, 8, 257–261 CrossRef CAS PubMed.
- W. J. Cubała, K. J. Wojten, A. Burkiewicz and A. Wrońska, Psychiatr. Danubina, 2011, 23, 198–199 Search PubMed.
- C. M. Oliphant and G. M. Green, Am. Fam. Physician, 2002, 65, 455–464 Search PubMed.
- H. d'A. Heck, S. E. Buttrill Jr, N. W. Flynn, R. L. Dyer, M. Anbar, T. Cairns, S. Dighe and B. E. Cabana, J. Pharmacokinet. Pharmacodyn., 1979, 7, 233–248 CrossRef CAS PubMed.
- J. K. Aronson, Meyler's Side Effects of Drugs: The International Encyclopedia of Adverse Drug Reactions and Interactions, Elsevier, 2015, p. 985 Search PubMed.
-
(a) A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035–2052 CrossRef CAS PubMed;
(b) A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed.
-
(a) B. S. Takale, S. M. Tao, X. Q. Yu, X. J. Feng, T. Jin, M. Bao and Y. Yamamoto, Org. Lett., 2014, 16, 2558–2561 CrossRef CAS PubMed;
(b) J. B. Rivera, Y. Xu, M. Wills and V. K. Vyas, Org. Chem. Front., 2020, 7, 3312–3342 RSC.
-
(a) K. A. Schellenberg, J. Org. Chem., 1963, 28, 3259–3261 CrossRef CAS;
(b) R. P. Tripathi, S. S. Verma, J. Pandey and V. K. Tiwari, Curr. Org. Chem., 2008, 12, 1093–1115 CrossRef CAS;
(c) H. Alinezhad, H. Yavari and F. Salehian, Curr. Org. Chem., 2015, 19, 1021–1049 CrossRef CAS.
-
(a) R. F. Borch, M. D. Bernstein and H. D. Durst, J. Am. Chem. Soc., 1971, 93, 2897–2904 CrossRef CAS;
(b) C. F. Lane, Synthesis, 1975, 135–146 CrossRef CAS.
-
(a) G. W. Gribble and C. F. Nutaitis, Org. Prep. Proced. Int., 1985, 17, 317–384 CrossRef CAS;
(b) A. F. A. Magid, C. A. Mayanoff and K. G. Carson, Tetrahedron Lett., 1990, 31, 5595–5598 CrossRef;
(c) A. F. A. Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef PubMed;
(d) G. W. Gribble, Chem. Soc. Rev., 1998, 27, 395–404 RSC;
(e) A. F. A. Magid and S. J. Mehrman, Org. Process Res. Dev., 2006, 10, 971–1031 CrossRef;
(f) G. W. Gribble, Org. Process Res. Dev., 2006, 10, 1062–1075 CrossRef CAS.
-
(a) X. Y. Zhou, M. Bao and Y. G. Zhou, Adv. Synth. Catal., 2011, 353, 84–88 CrossRef CAS;
(b) H. P. Hemantha and V. V. Sureshbabu, Org. Biomol. Chem., 2011, 9, 2597–2601 RSC;
(c) S. M. Islam, A. S. Roy, P. Mondal and N. Salam, Appl. Organomet. Chem., 2012, 26, 625–634 CrossRef CAS;
(d) H. A. Patel, M. Rawat, A. L. Patel and A. V. Bedekar, Appl. Organomet. Chem., 2019, 33, e4767 CrossRef;
(e) X. Jv, S. Sun, Q. Zhang, M. Du, L. Wang and B. Wang, ACS Sustainable Chem. Eng., 2020, 8, 1618–1626 CrossRef CAS.
-
(a) D. Kong, M. Li, G. Zi, G. Hou and Y. He, J. Org. Chem., 2016, 81, 6640–6648 CrossRef CAS PubMed;
(b) H.-U. Blaser, B. Pugin, F. Spindler and A. Togni, C. R. Chim., 2002, 5, 379–385 CrossRef CAS;
(c) Y. Motoyama, M. Taguchi, N. Desmira, S.-H. Yoon, I. Mochida and H. Nagashima, Chem.–Asian J., 2014, 9, 71–74 CrossRef CAS PubMed.
-
(a) D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., Int. Ed., 2013, 52, 2538–2542 CrossRef CAS PubMed;
(b) M. H. Juárez, J. L. Serrano, P. Lara, J. P. M. Cerón, M. Vaquero, E. Álvarez, V. Salazar and A. Suárez, Chem.–Eur. J., 2015, 21, 7540–7555 CrossRef PubMed;
(c) D. Deng, Y. Kita, K. Kamata and M. Hara, ACS Sustainable Chem. Eng., 2019, 7, 4692–4698 CrossRef CAS.
-
(a) D. Wei, A. B. Voisine, D. A. Valyaev, N. Lugan and J.-B. Sortais, Chem. Commun., 2018, 54, 4302–4305 RSC;
(b) U. Chakraborty, E. R. Rodriguez, S. Demeshko, F. Meyer and A. J. von Wangelin, Angew. Chem., Int. Ed., 2018, 57, 4970–4975 CrossRef CAS PubMed.
-
(a) A. Dybov, O. Blacque and H. Berke, Eur. J. Inorg. Chem., 2011, 652–659 CrossRef CAS;
(b) S. Chakraborty, O. Blacque, T. Fox and H. Berke, Chem.–Asian J., 2014, 9, 328–337 CrossRef CAS PubMed.
-
(a) D. J. C. Hamilton, Science, 2003, 299, 1702–1706 CrossRef PubMed;
(b) R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed.
-
(a) H.-L. Jiang and Q. Xu, J. Mater. Chem., 2011, 21, 13705–13725 RSC;
(b) A. K. Singh and Q. Yu, ChemCatChem, 2013, 5, 652–676 CrossRef CAS.
- S. Byun, Y. Song and B. M. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 14637–14647 CrossRef CAS PubMed.
- J. Jang, S. Byun, B. M. Kim and S. Lee, Chem. Commun., 2018, 54, 3492–3495 RSC.
- J. H. Cho, S. Byun, A. Cho and B. M. Kim, Catal. Sci. Technol., 2020, 10, 4201–4209 RSC.
- A. Cho, S. Byun and B. M. Kim, Adv. Synth. Catal., 2018, 360, 1253–1261 CrossRef CAS.
- A. Cho, S. Byun, J. H. Cho and B. M. Kim, ChemSusChem, 2019, 12, 2310–2317 CrossRef CAS PubMed.
- S. Yang, A. Cho, J. H. Cho and B. M. Kim, Nanomaterials, 2021, 11, 2101–2115 CrossRef CAS PubMed.
-
(a) B. S. Kumar, P. Puthiaraj, A. J. Amali and K. Pitchumani, ACS Sustainable Chem. Eng., 2018, 6, 491–500 CrossRef;
(b) Z. Zhang, S. Liu, X. Tian, J. Wang, P. Xu, F. Xiao and S. Wang, J. Mater. Chem. A, 2017, 5, 10876–10884 RSC;
(c) G.-R. Xu, C.-C. Han, Y.-Y. Zhu, J.-H. Zeng, J.-X. Jiang and Y. Chen, Adv. Mater. Interfaces, 2018, 5, 1701322 CrossRef;
(d) C. H. Wu, C. Liu, D. Su, H. L. Xin, H.-T. Fang, B. Eren, S. Zhang, C. B. Murray and M. B. Salmeron, Nat. Catal., 2019, 2, 78–85 CrossRef CAS;
(e) H. Xue, J. Tang, H. Gong, H. Guo, X. Fan, T. Wang, J. He and Y. Yamauchi, ACS Appl. Mater. Interfaces, 2016, 8, 20766–20771 CrossRef CAS PubMed;
(f) Y. Chen, J. Mao, R. Shen, D. Wang, Q. Peng, Z. Yu, H. Guo and W. He, Nano Res., 2017, 10, 890–896 CrossRef CAS;
(g) M. A. Ehsan, A. S. Hakeem and A. Rehman, Sci. Rep., 2020, 10, 14469–14479 CrossRef CAS PubMed;
(h) E. C. Avila, E. J. R. Ruiz, A. H. Ramirez, F. J. R. Varela, M. D. M. Acosta and D. M. Acosta, Int. J. Hydrogen Energy, 2017, 42, 30349–30358 CrossRef.
-
(a) P. Yu, J. Ma, R. Zhang, J. Z. Zhang and G. G. Botte, ACS Appl. Energy Mater., 2018, 1, 267–272 CrossRef CAS;
(b) S. Sankar, G. M. Anilkumar, T. Tamaki and T. Yamaguchi, ACS Appl. Energy Mater., 2018, 1, 4140–4149 CrossRef CAS;
(c) D. Liu, Q. Guo, H. Hou, O. Niwa and T. You, ACS Catal., 2014, 4, 1825–1829 CrossRef CAS;
(d) C. Xu, Y. Liu, H. Zhang and H. Geng, Chem.–Asian J., 2013, 8, 2721–2728 CrossRef CAS PubMed;
(e) W. Ye, X. Shi, Y. Zhang, C. Hong, C. Wang, W. M. Budzianowski and D. Xue, ACS Appl. Mater. Interfaces, 2016, 8, 2994–3002 CrossRef CAS PubMed;
(f) S. Zhang, W. Wang, Y. Gao, S. Deng, L. Ding, H. Zhuo, Z. Bao, W. Ji, C. Qiu and J. Wang, Appl. Surf. Sci., 2021, 567, 150680 CrossRef CAS.
-
(a) T. Li, R. Wang, M. Yang, S. Zhao, Z. Li, J. Miao, Z.-D. Gao, Y. Gao and Y.-Y. Song, Sustainable Energy Fuels, 2020, 4, 380–386 RSC;
(b) S. Sobhani, H. Zarei and J. M. Sansano, Sci. Rep., 2021, 11, 17025–17045 CrossRef CAS PubMed;
(c) L. Zhang, L. Wan, Y. Ma, Y. Chen, Y. Zhou, Y. Tang and T. Lu, Appl. Catal., B, 2013, 138, 229–235 CrossRef;
(d) Y. Li, H. Zhou, T. Li, X. Jian, Z. Gao and Y.-Y. Song, J. Mater. Chem. B, 2021, 9, 2016–2024 RSC;
(e) Z.-Y. Liu, G.-T. Fu, L. Zhang, X.-Y. Yang, Z.-Q. Liu, D.-M. Sun, L. Xu and Y.-W. Tang, Sci. Rep., 2016, 6, 32402–32411 CrossRef CAS PubMed.
-
(a) T. Hara, T. Kaneta, K. Mori, T. Mitsudome, T. Mizugaki, K. Ebitani and K. Kaneda, Green Chem., 2007, 9, 1246–1251 RSC;
(b) C. A. Ohlin, Z. Béni, G. Laurenczy, N. Ruiz and A. M. Masdeu-Bulto, Appl. Organomet. Chem., 2007, 21, 156–160 CrossRef CAS;
(c) B. Y. Kara, M. Yazici, B. Kilbas and H. Goksu, Tetrahedron, 2016, 72, 5898–5902 CrossRef CAS;
(d) W.-L. Jiang, J.-C. Shen, Z. Peng, G.-Y. Wu, G.-Q. Yin, X. Shi and H.-B. Yang, J. Mater. Chem. A, 2020, 8, 12097–12105 RSC.
- A. K. Chakraborti, S. Bhagat and S. Rudrawar, Tetrahedron Lett., 2004, 45, 7641–7644 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst characterization, experimental details, and 1 H and 13C NMR spectra. See DOI: 10.1039/d1ra08552b |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*