
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModeling the syn-cycle in the light activated opening of the channelrhodopsin-2 ion channel†
Qi Xina,
Jie Chenga,
Hongwei Wangb,
Wenying Zhang*a,
Hong Lua,
Junpeng Zhoua,
Glenn V. Loc,
Yusheng Douc and
Shuai Yuan *a
*a
aChongqing Key Laboratory of Big Data for Bio Intelligence, Chongqing University of Posts and Telecommunications, Chongqing 40065, China. E-mail: zhangwenying@cqupt.edu.cn; yuanshuai@cqupt.edu.cn
bSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane St Lucia, QLD 4072, Australia
cDepartment of Chemistry and Physical Sciences, Nicholls State University, P.O. Box 2022, Thibodaux, LA 70310, USA
First published on 24th February 2022
Abstract
The ion channel of channelrhodopsin-2 (ChR2) is activated by absorbing light. The light stimulates retinal to isomerize to start the photocycle. There are two pathways for photocycles, which are caused by isomerization of the retinal from all-trans, 15-anti to 13-cis, 15-anti in the dark-adapted state (anti-cycle) and from 13-cis, 15-syn to all-trans, 15-syn in the light-adapted state (syn-cycle). In this work, the structure of the syn-cycle intermediate and mechanism of channel opening were studied by molecular dynamics (MD) and steered molecular dynamics (SMD) simulations. Due to the lack of crystal structure of intermediates in the syn-cycle of ChR2, the intermediate models were constructed from the homologous intermediates in the anti-cycle. The isomerization of retinal was shown to cause the central gate (CG) hydrogen bond network to rearrange, cutting the link between TM2 and TM7. TM2 is moved by the intrahelical hydrogen bond of E90 and K93, and induced the intracellular gate (ICG) to expand. The ion penetration pathway between TM1, TM2, TM3 and TM7 in the P500* state was observed by MD simulations. However, this channel is not fully opened compared with the homologous P500 state in the anti-cycle. In addition, the protons on Schiff bases were found to be unable to form hydrogen bonds with the counter residues (E123 and D253) in the P500* state, preventing an evolution of the P500* state to a P390-like state in the syn-cycle.
Introduction
Channelrhodopsin-2 (ChR2) is a nonselective cation channel protein and can be expressed on mammalian neurons, non-invasively stimulate neurons and activate nerve cells by adjusting light.1–5 It is a good system to explore in the field of optogenetics. Although ChR2 is widely used in the field of neuroscience, the detailed mechanism of channel activation and the complete light cycle are not clear.In wild type (WT) ChR2, the chromophore retinal, which is covalently linked to K257 and generates a protonated Schiff base (RSBH+), is in the all-trans conformation. Absorption of blue light converts the all-trans retinal to the 13-cis conformation, and then triggers the photocycle reaction, which leads to a series of complex reactions and conformational transformations. Nagel et al.6 established a three-state photocycle model based on the recorded photocurrent, including a closed ground state (C), an open state (O) and a closed desensitized state (D), as shown in Fig. 1a. This model qualitatively reproduces the relative amplitude of the peak photocurrent and the overall dynamics in ChR2. However, the photocurrent of ChR2 was later found, experimentally, to decay in a double exponential manner under constant illumination, which means that the decay process has two independent time constants.7,8 Therefore, a four-state model was proposed to describe the photocycle of ChR1 and ChR2.7–12 This model involves two closed states (C1 and C2) and two open states (O1 and O2) as illustrated in Fig. 1b. The main structural difference between C1 and C2 is that the retinal is in the all-trans, 15-anti conformation in C1 and is in the 13-cis, 15-syn conformation in C2.11,13–17 Zamani et al.18 found the C1 state evolves to a highly conductive O1 state under light irradiation, while the C2 state evolves to the O2 state. This finding suggested that C1 → O1 and C2 → O2 were two independent kinetic processes.7,8,18 In the fast C1 → O1 transition, the peak current is proportional to the amount of O1.19
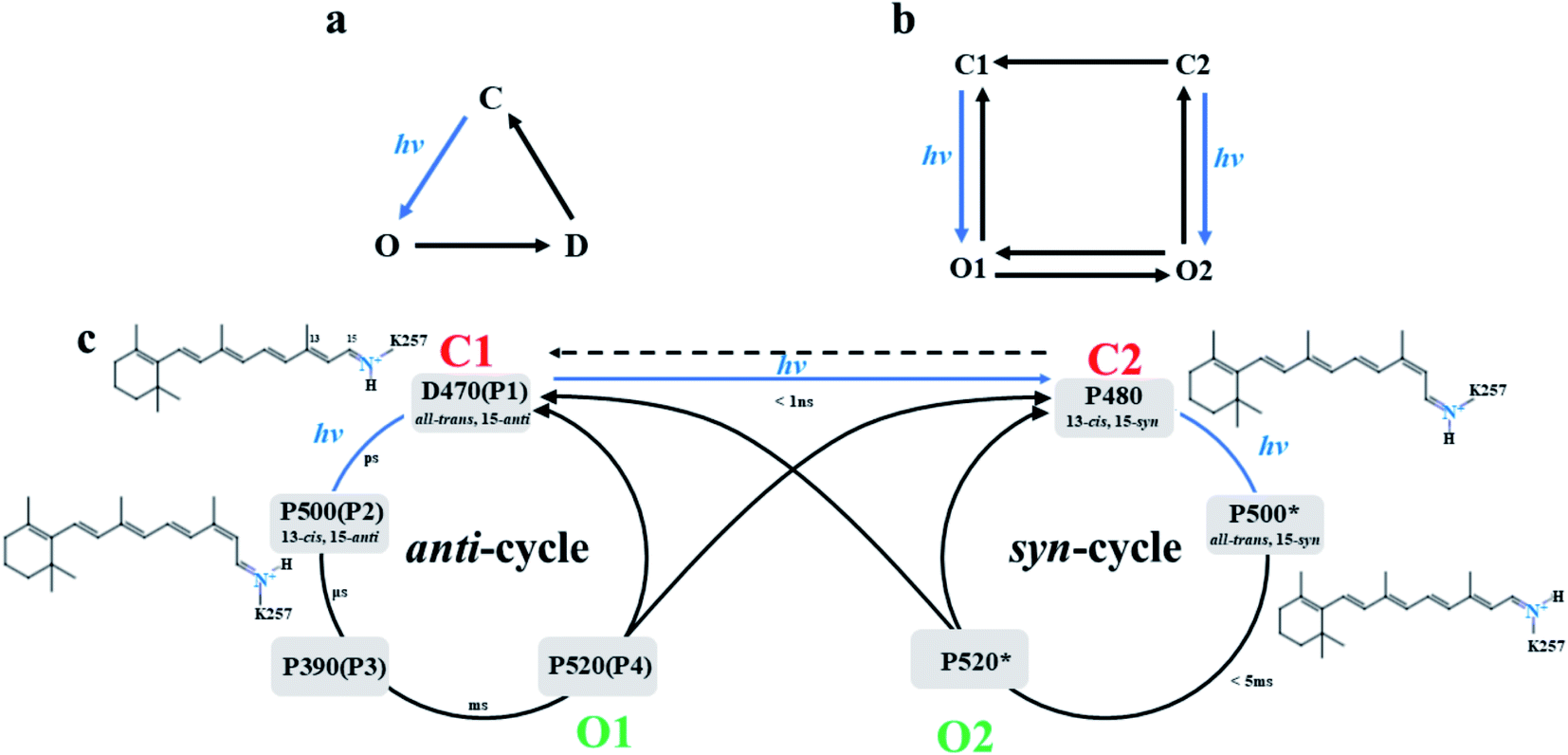 | ||
| Fig. 1 (a) The three-state model and (b) the four-state model of photocurrent in ChR2, and (c) proposed double photocycle model of ChR2. | ||
Bamann et al.20 activated ChR2 by using a short laser pulse (480 nm, 10 ns) and the results revealed four relaxation processes with different time constants during the photocycle of ChR2. This suggested four different intermediates (designated as P1, P2, P3, and P4) were formed in the photocycle induced by photoisomerization of retinal. Conversion of P4 to P1 was found6,13–18 under longer irradiation time. This led to a modified photocycle model, which involves anti- and syn-branches according to the conformation of 15-RSBH+ of retinal as summarized in Fig. 1c. The D470 state (P1) is the dark-adapted state of ChR2 with retinal conformation of the all-trans, 15-anti in the anti-cycle.21 The retinal molecule isomerizes to 13-cis, 15-anti conformation by absorbing light and leads to the P500 state (P2).21 Subsequent deprotonation of RSBH+ yields the P390 state (P3). It was reported that a small amount of cations had passed through the channel in late P390 state,11,13,22 so the P390 state was considered to be the early O1 state.13 Subsequently, the Schiff base is reprotonated to form the opened P520 state (O1 state),23–25 and a large number of cations enters the cell.11,20,22 The P520 state decays to the desensitized P480 state or returns to D470 state. The P480 state configuration, is 13-cis, 15-syn retinal, is consistent with a negative band near 1183 cm−1 of infrared absorption spectrum,13,26,27 and was assigned to the desensitized C2 state. The P480 links the two parallel cycles (anti- and syn-cycle) and is regarded as the starting point of syn-cycle. Ritter15 and Schneider28 showed that these two cycles both involve homologous photocycle intermediates. However, the existence of P390*, a state in the syn-cycle that would be homologous to the P390 state in anti-cycle, is still controversial. Kuhne et al.13 did not detect related band similar to the P390 state in syn-cycle. The P520* state is an opened state, and could be assumed to correspond to the O2 state in Fig. 1b. The O2 state is responsible for maintaining a steady-state photocurrent.8,9
The mechanism of direct isomerization from all-trans, 15-anti to 13-cis, 15-syn conformation has been well studied.13,14,18 It is suggested that C2 may also be directly excited from C1. Currently, structural information for intermediates in the photocycle cannot be obtained experimentally. Moreover, controversies still exist regarding the syn-cycle.13,17,29 The O2 state was detected with lower conductivity but stable photocurrent.13,17 However, the amount of O2 state increased after continuous illumination,8,29 and generated another peak photocurrent.29 The photocurrent dynamics of ChRs at a light-adapted state are considerably different compared to a dark-adapted state,18 which implied that the mechanism of the syn-cycle is significantly different from that of anti-cycle. Elucidating the mechanism of channel opening in syn-cycle is also very important.
In this paper, the model of P480 intermediate with a 13-cis, 15-syn retinal was built based on the structure of the D470 state. Then the retinal was manually rotated to obtain the all-trans, 15-syn conformation and establish P500* state. MD simulations were performed on the two intermediates in syn-cycle to examine the structural changes in the ion channel during the syn-cycle.
Methods
Setting up the early P480 and P500*intermediate models of syn-cycle
The initial structure of the D470 (monomer) state was obtained from the X-ray crystal structure of the A chain of ChR2 (PDBID: 6EID).25 MD simulation and the K-means algorithm in the MMTSB tool set30 were used to do a cluster analysis of the trajectory to obtain the equilibrium structure of D470 state. The 13-cis, 15-syn retinal was picked from bacteriorhodopsin (BR, PDBID: 1X0S)31 since the photoisomerization of retinal cannot be directly simulated by MD. Following Guo et al.,32,33 the main chain atoms and six-membered ring of retinal with the 13-cis, 15-syn conformation were superimposed (and replaced corresponding atoms) on the all-trans, 15-anti retinal in the D470 state to generate the initial P480 state. Taking into account the complex interactions between retinal and adjacent residues, the hybrid QM/MM method34 was used to minimize the energy of the Schiff base region in order to reduce the influence of the substituted retinal in the binding pocket. It has been reported that E123 and D253 have electrostatic interactions with the protonated RSBH+.36,37 Therefore, the QM region included E123, D253, K257 and retinal molecules and were calculated using the self-consistent charge density functional based on tight-binding (SCC-DFTB) method.38 The rest of the structures were to the MM region (as seen in Fig. S3†). In the QM/MM calculations, the SHAKE algorithm39 was used to freeze the bonds containing hydrogen atoms during the minimization. Particle-Mesh-Ewald (PME)40 was described by the long-distance static electricity with a cut-off radius of 12 Å. Then the SHAKE restriction on hydrogen bond was removed and a constant-temperature of 310 K simulation was run by QM/MM approach. The time step was 0.02 fs per step, and the trajectories were saved every 100 fs, and 1 ns of the production run was performed. All the QM/MM calculations are performed in the Amber18 package.41 Subsequently, a 300 ns MD simulation was performed on the optimized initial P480 state and a cluster analysis was performed to obtain the representative structure of P480 state. Following Kuhne et al.,35 the dihedral angle of C13 = C14 of the retinal in the representative conformation of P480 state was twisted with an interval of 20° to generate the all-trans, 15-syn retinal due to it has not been found in any rhodopsin crystal structure currently. For each torsion step, the retinal and K257 atoms are frozen to perform a 0.5 ns QM/MM simulation. After 9 steps, with a total dihedral angle change of 180°, the initial P500* state was produced. A similar MD simulation was performed to sample conformations and cluster analysis procedure was used to obtain the representative structure of the P500* intermediate.Both P480 and P500* states were described by CHARMM36 all-atom force field.42 The Membrane Builder43 of CHARMM-GUI44,45 was used to model the membrane and solvation. According to previous researches, the E90 was protonated in the D470 state, deprotonated in P480 and P500* state. D156 is protonated and other titratable residues are set to standard status in these intermediates.11,13,27,33 The intermediates of ChR2 are embedded in the center of the pre-equilibrated 16:0/18:1 c9-palmitoyloleyl phosphatidylcholine (POPC) lipid bilayer. Then, TIP3P water molecules were added on both sides of the lipid bilayer46 with 0.15 M NaCl. The complete simulation system is consisted of the ChR2 protein, 114 POPC molecules and 8561 TIP3P water molecules, 22 Na+ and 24 Cl− in a periodic box of size 70 × 70 × 90 Å with 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 103 atoms in total.
103 atoms in total.
MD simulation
NAMD 2.13 software47 was used to perform MD, SMD, and umbrella sampling (US) simulations48,49 for P480 and P500* intermediates. The PME method was used to describe the long-range electrostatic interaction, the grid spacing was set to be less than 1 Å, and the cut-off radius of short-range non-bonded interaction was set to 12 Å. For all the chemical bonds containing hydrogen atoms in ChR2, the SHAKE constraint algorithm was used to restrict and limit vibrations. The SHAKE step was set to 2 fs. The steepest descent method was used to simulate 10000 steps to minimize the energy of the system. Then the system is heated to 310 K in the NVT ensemble. The Langevin Piston algorithm was used in the heating process to maintain keep the constant pressure (1 bar), with the damping coefficient set to 1 ps−1. Then an unconstrained 300 ns production was run in the NPT ensemble (the system maintains 310 K and 1 bar) with 2 fs steps; and the trajectories were saved after every 2500 steps (i.e. 5 ps). The last 50 ns was selected for analysis. Each model has 5 parallel simulations in this work.
SMD and potential of mean force (PMF)
Since the P500* state is not an opened state, ions cannot freely pass through ChR2. SMD was used to simulate the process of Na+ passing through ion channel. It was assumed that a steering force is applied to Na+ to enable it to pass through the ion channel in the SMD simulation process. Then the coordinate information for the ion permeation process as well as relevant information for ion transmembrane conduction can be obtained by the analysis of structure and energy. However, the stretching may cause the energy to be quite high and inaccurate at some coordinates. Therefore, US was used to divide the reaction coordinates into separate “windows” and the equilibrium state determined for each window. Then, the weighted histogram analysis method (WHAM)50 was used to construct the PMF.The constant speed SMD simulation51 was ran in PLUMED plugin of the NAMD 2.13 software package. Initially, Na+ is placed at the port of the potential ion channel in the representative conformation. Na+ is stretched along the Z coordinate axis, which is parallel to the channel. The protein translates and rotates during SMD simulation. Therefore, a Hamilton limit of 5 kcal mol−1 Å−2 was added to the residues on the periphery of each helix to avoid protein structure change. The stretching speed is set to 0.2 Å ns−1. The spring coefficient k is set to 4 kcal mol−1 Å−2 (hard spring).32
The US simulation uses the same direction as SMD, and divides the “window” along the Z axis into 1 Å intervals. A harmonic potential with a force constant of 2.5 kcal mol−1 Å−1 was applied to ensure the gap of each window along the Z axis. In addition, the 0.1 Å translocation colvar was set to overlap adjacent windows in the WHAM method to form a smooth PMF curve.
Results and discussion
Structure analysis of intermediates based on clustering method
Both the P480 and P500* intermediates were studied with five independent MD simulations with timescale of 300 ns in the NPT ensemble respectively (i.e. total of 1.5 μs for each state). The root mean squared deviations (RMSD, as seen in Fig. S1†) of all trajectories indicated the transmembrane helix was stable. Cluster analysis method was used to get representative conformations of the P480 state and P500* state from last 50 ns of RMSD.The intracellular gate (ICG), and the extracellular gate (ECG) were found to change. The local conformations of P480 and P500* intermediates are shown in Fig. 2. In the P480 state (Fig. 2a), the extracellular gate (ECG) was opened and allowed a large number of water molecules to enter. A water molecule acts as a bridge to link R120 and E123 by hydrogen bonds. The protonated K93 formed hydrogen bonds with E90, E123 and D253 respectively, creating a tight local hydrogen bond network at central gate (CG). Furthermore, the proton in RSBH+ orients to D253 and formed a hydrogen bond with it. These hydrogen bonds networks blocked the ion channel indicating that the CG has not yet opened. Almost no water molecule entered the potential channel from CG to ICG. R268 and E82 formed two hydrogen bonds at ICG in the D470 state.32 However, one the twisting of R268, left only one (with E82) and led to the formation of a new one with E83 in the P480 state.
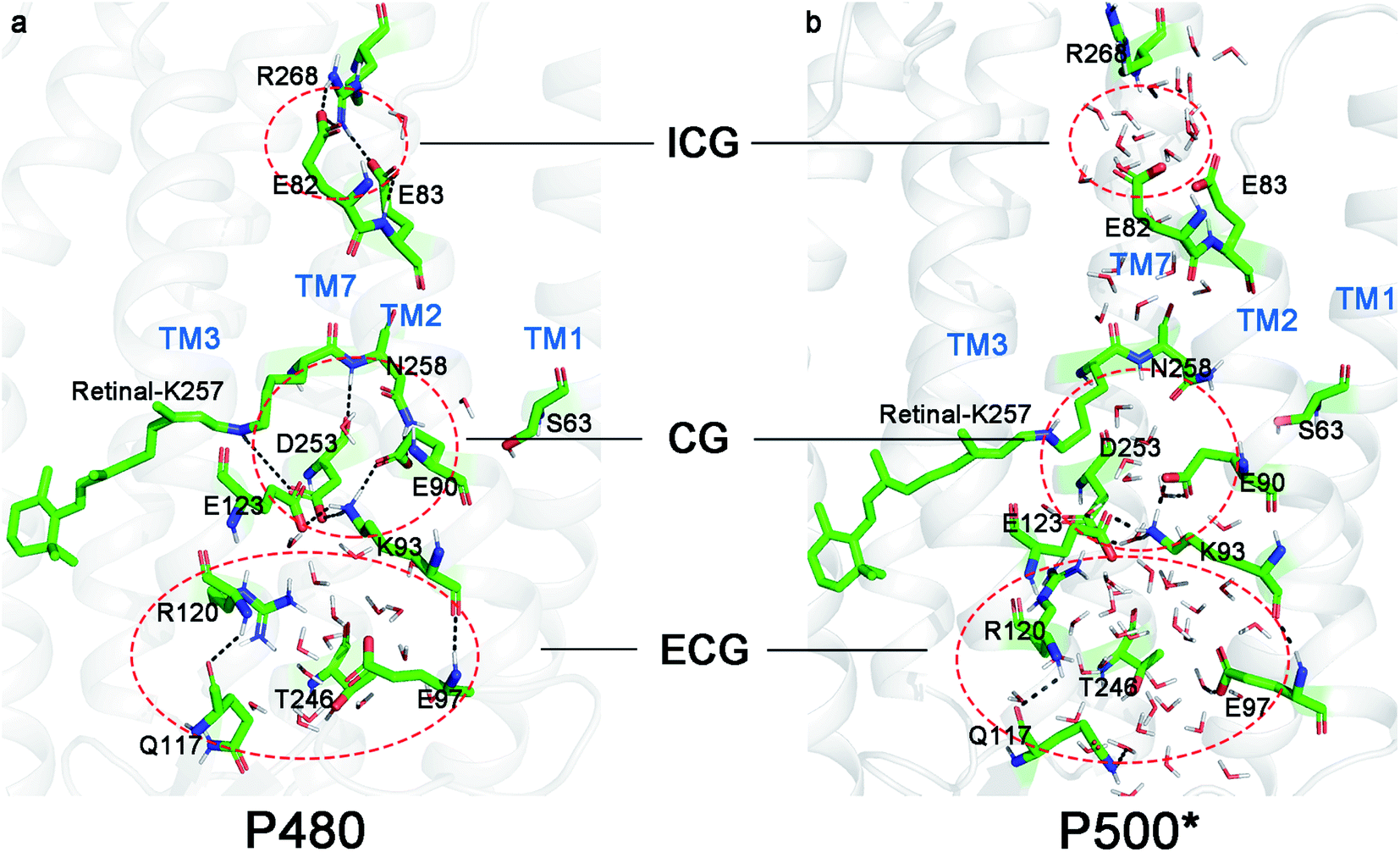 | ||
| Fig. 2 Representative structures in (a) P480 state and (b) P500* state. Areas enclosed by red dashed lines represent the ICG, CG and ECG in ChR2. | ||
It can be seen from Fig. 2b that there are significantly more water molecules in the ECG of P500* state than P480 state, which implies that ECG is more open. Previous studies24,35,52,53 have shown that the hydrogen bond between E90 and K93 in CG induced the movement of TM2 in the P500 state in the anti-cycle. Similar structural changes were found in the P500* state because it is homologous to the P500 state. The E90 and K93 also formed a hydrogen bond causing TM2 to be displaced. This caused the movement of K93 to break the hydrogen bond with D253, forming a cavity at the CG and water molecules entering in the P500* state. However, E123 and E90 still maintained the hydrogen bonds with K93. At the ICG, R268 flipped to the cytoplasm, breaking the hydrogen bonds with E82 and E83. However, the count of water molecules has not increased.
After the isomerization of retinal from 13-cis, 15-syn to all-trans, 15-syn in the P500* state, it is interesting that the proton of RSBH+ did not point to E123 or D253, which is expected to be proton acceptors for RSBH+. It suggests that the RSBH+ may not be able to transfer the proton and stays in protonated state. In a previous study of anti-cycle,11,22 the deprotonation of RSBH+ at the end of the P500 state was identified the key step in the formation of the subsequent P390 intermediate. But this evolution seems to be impossible in the syn-cycle. According to Jeffrey,54 a hydrogen bond can be formed when the distance between the proton and the proton acceptor is less than 4 Å and the angle of H⋯X⋯C (X is O or N atom) is greater than 120°. We calculated the possibility of hydrogen bond formation between RSBH+ and potential proton acceptors in the last 50 ns trajectories of D470, P480 and P500* states by counting the ration between hydrogen bond formation conformations and total conformations during last 50 ns. We found it obvious from Fig. 3 that the probability of hydrogen bond formation between RSBH+ and D253 is 45.16% in D470 state and 36% in P480 state respectively, which is much higher than that of E123. The distance between the proton in RSBH+ and the proton acceptor D253 were 3.8 Å and 3.4 Å, in the D470 and P480 states, respectively. However, the probability of RSBH+ forming hydrogen bonds with D253 was 0 in P500* state based on five independent simulation trajectories, and the probability for E123 was very low (Table S1†). The variations of distances of RSBH+ and D253 or E123 versus time in last 50 ns of five trajectories are shown in Fig. S2.† It shows that the distances of D253 and E123 were larger than 4 Å in most cases. Both Fig. 2b and 3 showed that the proton of RSBH+ point in the opposite direction of E123 and D253 in the P500* state, and the distances of 5.4 Å and 7.2 Å are too large for a hydrogen bond to form. Therefore, proton transfers from RSBH+ cannot occur. This result may explain why Kuhne et al.13 failed to detect the P390* state in syn-cycle.
 | ||
| Fig. 3 The probabilities of hydrogen bonding between RSBH+ and potential proton acceptors (E123 and D253) based on proton to proton acceptor distances in the D470, P480 and P500* states. | ||
Changes in the transmembrane helix of protein
The movements of the protein helices are the main conformational changes during anti-photocycle. Kuhne et al.24 has reported that the E90 flips down and deprotonates to rearrange the hydrogen bond network at CG and induce the tilt of TM2 after the isomerization of retinal, leading to further opening of the channel. Müller et al.52 also observed the movement of TM2 during the opening of the channel in ChR2. The displacements of TM6 and TM7 were also found during the process of photoactivation in ChR2.28,52,53,55We observed similar helix movements in the syn-cycle. As shown in Fig. 4a and b, the TM2 is tilted outward by 2.8 Å from P480 state to P500* state, and extracellular part moved with a significant displacement of 3.1 Å. The intrahelical hydrogen bond linked E90 and K93 induced the displacement of TM2 (Fig. 4c). Moreover, the E123 on TM3 also has a certain degree of twisting and displacement with the movement of TM2, which may have caused the extracellular part of TM3 to shift 2.5 Å. The outward tilts of TM2 and TM3 enlarged the pores in ECG, resulting in more water molecules to enter. At the same time, the movement of TM2 caused E82 and E83 to move outward, cutting off their hydrogen bonds with R268 on TM7. It was also observed that the hydrogen bond between N258 on TM7 and S63 on TM1 disappeared in P480 state. This broke the link between TM1 and TM7 and made the intracellular part of TM1 to shift outward 2.1 Å. The two changes in the hydrogen bond networks on TM7 broke the connections with TM1 and TM2, causing the intracellular part of TM7 to shift outward 2.4 Å. TM6 only shifted by 1.0 Å on the cytoplasmic part. Since the ECG was already open in the P480 state, it implies that the movements of the helix resulted from the changes in relevant residues at CG. The RMSF of K93 and E90 are obviously higher than other residues (Fig. 4d), which means that these two residues are most flexible during the simulation. In addition, the residue 257 (including K257 and covalently bonded retinal), D253 on TM7, and E123 on TM3 also have higher RMSF, suggesting that the CG undergoes significant conformational changes during the opening of the ion channel.
 | ||
| Fig. 4 (a) The front view of the superimposed structure of P480 state and P500* state, (b) the top view of the superimposed structure of P480 state and P500* state from the cytoplasm side, (c) the superimposed diagram of the key residues (E90, K93, E123, D253, K257) at CG, (d) RMSF of these residues, the no. 257 residue is the combination of K257 and retinal molecule. The P480 state is orange, and the P500* state is red. | ||
Water density distributions
The influx of water plays an important role in opening the ion channel of ChR2.56 Water molecules have been observed entering the cavity of the ECG in the D470 state.32 The subsequent movement of E90 leads to CG opening and allows more water molecules to flow into the interior of ChR2, which results in TM2 tilting outwards and further expansion of the channel.24 It means that water molecules are distributed along ion channel,24,28,56 which implies that water molecules have already occupied the cavities in the channel before ion penetration.The water distributes all over the ECG in the P480 state (Fig. 5a) since the ECG has already been opened in D470 state. However, some hydrophobic sites formed by the tight hydrogen bond networks composed of E90, K93, E123 and D253 at the CG prevent an even continuous distribution of water. In addition, this hydrogen bond networks tie TM2, TM3 and TM7 to block passage from the CG to the ICG. A similar hydrophobic cavity is observed in the ICG.
 | ||
| Fig. 5 The water density distributions in (a) P480 state and (b) P500* state. The red circles respectively represent the water density distribution at CG and ICG. | ||
The isomerization of retinal in the P500* state involved a rearrangement of the hydrogen bond networks at the CG, resulting in the outward displacement of TM2 that breaks the hydrogen bond between K93 and D253 and allows water molecules to enter CG (Fig. 5b). The sparse water distribution at the ICG is lower than that at the CG, which suggests the ICG is not fully opened in P500* state. At the same time, due to the movements of TM1, TM2, TM3 and TM7 around ion channel, the water distribution is also observed in the channel from the CG to the ICG.
Hydrogen bond analysis
The change in the hydrogen bond networks around the channel is a key factor in the opening of the ion channel. We calculated the number of hydrogen bonds in the residues at the ECG, CG and ICG in the P480 and P500* states.Fig. 6a show that the average total number of hydrogen bonds (including hydrogen bonds of residue–residue (R–R) and residue–water (R–W)) is 9–10 in the channel of P480 intermediate, 7–8 for the P500* intermediate. This suggests that the 13-cis to all-trans isomerization of retinal induced the rearrangement of the hydrogen bond network. The reduction in the number of hydrogen bonds lowers the barrier to permeate. However, the variations of R–W hydrogen bonds increased from 4–5 in the P480 state to 7–8 in the P500* state. This would be consistent with a large number of water molecules entering the channel, leading to more R–W hydrogen bonds in P500* state. The total number of hydrogen bonds is significantly larger than the number of R–W hydrogen bonds in the P480 state, which means that the R–R hydrogen bonds are the predominant types at this point. The interhelix R–R hydrogen bonds lock the helix and form rigid conformation to maintain the close relationship between the helices. These hydrogen bonds close the channel and hinder the entry of water molecules. The number of total hydrogen bonds is almost the same as the R–W hydrogen bonds in the P500* state, which would be consistent with the condition of the most the R–R hydrogen bonds being broken and the cavities among helices being filled with water molecules.
 | ||
| Fig. 6 (a) The quantity of the total hydrogen bonds of key residues (S63, E82, E83, E90, K93, E97, E101, Q117, R120, E123, T246, D253, N258, R268) around the ion channel, including the residue–residue (R–R) hydrogen bonds and residue–water (R–W) hydrogen bonds. (b) The quantity of residue–water (R–W) hydrogen bonds. (c) The quantity of residue–residue (R–R) hydrogen bonds. All the data were obtained from the last 50 ns of the trajectory (P480 to P500* state). | ||
Ion steering and PMF
The time scale for ion penetration of the channel is more than millisecond.16,56 This is an almost impossible task for the current MD simulation. Therefore, the SMD was used to simulate the process of the ionic transmembrane conduction and the US method was used to construct the PMF to determine the mechanism. The interaction between ions and related residues in three gates when passing through the ion channel, and the optimal path of ion penetration were deduced from the changes in tension and PMF.Fig. 7a illustrates the steering process of Na+ through the P500* state. The ionic transmembrane conduction involves movement of the ion from the extracellular channel entrance, followed by entry through the ECG, CG and ICG. With the ion approaching the ECG, the tensile force mostly stayed within ±200 pN (Fig. 7b). The tensile force reached a maximum of 400 pN at about Z = 8 Å. The PMF from the extracellular matrix to ECG with extremely low energy shows that this is barrierless and spontaneous process (Fig. 7c). The energy barrier of PMF with 6.45 kcal mol−1 near Z = 8 Å indicates that ions need to overcome the attraction of adjacent E97. As the Na+ moved towards the CG, the pulling force rose to 600 pN because the channel has not been fully opened yet. At the same time, the PMF has increased to 13.98 kcal mol−1. This increase in energy suggests that the Na+ needs to overcome a higher energy barrier due to the attraction of negatively charged residues such as E90 to leave CG. Therefore, the CG could be the major blocking point and Na+ binding site in the ion channel of ChR2, consistent with our previous results in the anti-cycle of ChR2.32 The PMF decreased after passing through the CG. When the ion approached the ICG, the tension exceeded 400 pN. At the same time, PMF also shows an energy barrier of 9.59 kcal mol−1. As Na+ leaves the ICG, the tensile force and PMF remained at high values due to the attraction of the two negatively charged residues E82 and E83. The ICG is another blocking point and Na+ binding site in the ion channel of ChR2.
 | ||
| Fig. 7 (a) A schematic diagram of the steering Na+ in ion channel, (b) the tension change during the steering process, and (c) the PMF constructed by US. | ||
As in our previous work, we found that there was no barrier at the ECG, and lower force and energy are needed to pass CG and ICG in P500 state of anti-cycle.32 However, a higher steering force and energy barriers were observed during ion steering in the P500* intermediate. This suggests that the ion channel of syn-cycle may be smaller than that of anti-cycle. SMD calculations revealed some key information about the ionic transmembrane conduction. Combined with the water density distribution, we found that the ion penetration pathway is the same as the previous study, i.e., located between TM1, TM2, TM3 and TM7 25. The CG and ICG are the main blocking points of in ion channel, and the electrostatic attraction of negative residues in three gates hinders the conduction of ions across the membrane. Ions may aggregate at the binding sites in the initial stage of transmembrane conduction, then a large amount of ions pass through the channel to form a stable photocurrent when the pore becomes sufficiently large. It is obvious that conformational changes occur in the process from the P500* state to the fully open P520* state. These structural changes may be focused on the residue clusters of the CG and ICG.
Conclusions
In this study we modelled the syn-cycle intermediates, P480 and P500* based on crystal structure of ChR2 and performed MD and SMD simulations to investigate mechanism of channel opening. This research found the structural evolution of intermediates of syn-cycle is different from that of anti-cycle. To start with, we drew differences between P480 and P500* structurally, including the change of hydrogen bond networks and movements in helices. What's more, the increased water molecules were discovered in ECG and ICG. Meanwhile, water density distribution proved a more open intermediate has formed, which is required for ion penetration. Last but not least, an overall situation within the channel was described by PMF, and revealed the constriction sites of ChR2 in syn-cycle.While ECG, CG and ICG still have certain degree of blockages in P500* state, the extent of blocking was found to be lower in the P500 state. The results suggest that anti-cycle is more suitable for ion channel opening, which explains why the P390* state not exist in syn-cycle. From the analysis of the representative conformation of the P500* state, we found that the distance between the proton of the RSBH+ and the potential proton acceptors (E123 and D253) are too large so that proton transfer cannot occur. Therefore, the evolution from P500* to P390* state is not available in syn-cycle.
Author contributions
S. Y. and W. Z. conceived the idea and designed the calculation strategy. Q. X., J. C., H. W. and H. L. performed the molecular dynamics simulations. J. C. and J. Z. performed the statistical analysis. W. Z. and J. C. prepared the original draft. G. L. and Y. D. reviewed the manuscript. Q. X. completed the revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Natural Science Foundation of Chongqing, China (cstc2020jcyj-msxmX0653) and Chongqing Education Commission Foundation (KJQN201900618).References
- M. Häusser and S. L. Smith, Nature, 2007, 446, 617–619 CrossRef PubMed.
- F. Zhang, L. P Wang, M. Brauner, J. F. Liewald, K. Kay, N. Watzke, P. G. Wood, E. Bamberg, G. Nagel, A. Gottschalk and K. Deisseroth, Nature, 2007, 446, 633–639 CrossRef CAS PubMed.
- A. W. Roe, M. M. Chernov, R. M. Friedman and G. Chen, Front. Neuroanat., 2015, 9, 135 Search PubMed.
- S. Jiang, Y. F. Liu, X. M. Wang, K. F. Liu, D. H. Zhang, Y. D. Li, A. P. Yu, X. H. Zhang, J. Y. Zhang, J. G. Xu and Y. D. Gu, Biomed. Opt. Express, 2016, 7, 3478–3490 CrossRef CAS PubMed.
- S. D. Klapper, A. S. wiersy, E. Bamberg and V. Busskamp, Front. Syst. Neurosci., 2016, 10, 74 Search PubMed.
- G. Nagel, T. Szellas, W. Huhn, S. Kateriya, N. Adeishvili, P. Berthold, D. Ollig, P. Hegemann and E. Bamberg, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13940–13945 CrossRef CAS PubMed.
- P. Hegemann, S. Ehlenbeck and D. Gradmann, Biophys. J., 2005, 89, 3911–3918 CrossRef CAS PubMed.
- K. Nikolic, N. Grossman, M. S. Grubb, J. Burrone, C. Toumazou and P. Degenaar, Photochem. Photobiol., 2009, 85, 400–411 CrossRef CAS PubMed.
- R. A. Stefanescu, R. G. Shivakeshavan, P. P. Khargonekar and S. S. Talathi, Bull. Math. Biol., 2013, 75, 2208–2240 CrossRef CAS PubMed.
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Chem. Rev., 2014, 114, 126–163 CrossRef CAS PubMed.
- V. A. Lórenz-Fonfría and J. Heberle, Biochim. Biophys. Acta, 2014, 1837, 626–642 CrossRef PubMed.
- K. Stehfest, E. Ritter, A. Berndt, F. Bartl and P. Hegemann, Mol. Biol., 2010, 398, 690–702 CrossRef CAS PubMed.
- J. Kuhne, J. Vierock, S. A. Tennigkeit, M.-A. Dreier, J. Wietek, D. Petersen, K. Gavriljuk, S. F. El-Mashtoly, P. Hegemann and K. Gerwert, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9380–9389 CrossRef CAS PubMed.
- S. Bruun, D. Stoeppler, A. Keidel, U. Kuhlmann, M. Luck, A. Diehl, M. A. Geiger, D. Woodmansee, D. Trauner, P. Hegemann and H. Oschkinat, Biochemistry, 2015, 54, 5389–5400 CrossRef CAS PubMed.
- E. Ritter, P. Piwowarski, P. Hegemann and F. J. Bartl, J. Biol. Chem., 2013, 288, 10451–10458 CrossRef CAS PubMed.
- M. Saita, F. P. Sellnau, T. Resler, R. Schlesinger, J. Heberle and V. A. Lorenz-Fonfria, J. Am. Chem. Soc., 2018, 140, 9899–9903 CrossRef CAS PubMed.
- V. A. Lórenz-Fonfría, B. J. Schultz, T. Resler, R. Schlesinger, C. Bamann, E. Bamberg and J. Heberle, J. Am. Chem. Soc., 2015, 137, 1850–1861 CrossRef PubMed.
- A. Zamani, S. Sakuragi, T. Ishizuka and H. Yawo, Biophys. Physicobiol., 2017, 14, 13–22 CrossRef CAS PubMed.
- C. Lavigne and P. Brumer, J. Chem. Phys., 2020, 153, 034303 CrossRef CAS PubMed.
- C. Bamann, T. Kirsch, G. Nagel and E. Bamberg, J. Mol. Biol., 2008, 375, 686–694 CrossRef CAS PubMed.
- M. K. Verhoefen, C. Bamann, R. Blöcher, U. Förster, E. Bamberg and J. Wachtveitl, ChemPhysChem, 2010, 11, 3113–3122 CrossRef CAS PubMed.
- T. Kottke, V. A. Lórenz-Fonfría and J. Heberle, J. Phys. Chem. B, 2017, 121, 335–350 CrossRef CAS PubMed.
- V. A. Lórenz-Fonfría, T. Resler, N. Krause, M. Nack, M. Gossing, G. Fischer von Mollard, C. Bamann, E. Bamberg, R. Schlesinger and J. Heberle, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E1273–E1281 CrossRef PubMed.
- J. Kuhne, K. Eisenhauer, E. Ritter, P. Hegemann, K. Gerwert and F. Bartl, Angew. Chem., Int. Ed., 2015, 54, 4953–4957 CrossRef CAS PubMed.
- O. Volkov, K. Kovalev, V. Polovinkin, V. Borshchevskiy, C. Bamann, R. Astashkin, E. Marin, A. Popov, T. Balandin, D. Willbold and G. Büldt, Science, 2017, 358, eaan8862 CrossRef PubMed.
- E. Ritter, K. Stehfest, A. Berndt, P. Hegemann and F. J. Bartl, J. Biol. Chem., 2008, 283, 35033–35041 CrossRef CAS PubMed.
- I. Radu, C. Bamann, M. Nack, G. Nagel, E. Bamberg and J. Heberle, J. Am. Chem. Soc., 2009, 131, 7313–7319 CrossRef CAS PubMed.
- F. Schneider, C. Grimm and P. Hegemann, Annu. Rev. Biophys., 2015, 44, 167–186 CrossRef CAS PubMed.
- M. Nack, I. Radu, C. Bamann, E. Bamberg and J. Heberle, FEBS Lett., 2009, 583, 3676–3680 CrossRef CAS PubMed.
- M. Feig, J. Karanicolas and C. L. Brooks, J. Mol. Graphics Modell., 2004, 22, 377–395 CrossRef CAS PubMed.
- T. Nishikawa, M. Murakami and T. Kouyama, J. Mol. Biol., 2005, 352, 319–328 CrossRef CAS PubMed.
- T. Yang, W. Zhang, J. Cheng, Y. Nie, Q. Xin, S. Yuan and Y. Dou, Int. J. Mol. Sci., 2019, 20, 3780 CrossRef CAS PubMed.
- Y. Guo, F. E. Beyle, B. M. Bold, H. C. Watanabe, A. Koslowski, W. Thiel, P. Hegemann, M. Marazzi and M. Elstner, Chem. Sci., 2016, 7, 3879–3891 RSC.
- R. C. Walker, M. F. Crowley and D. A. Case, J. Comput. Chem., 2008, 29, 1019–1031 CrossRef CAS PubMed.
- J. Kuhne, K. Eisenhauer, E. Ritter, P. Hegemann, K. Gerwert and F. Bartl, Angew. Chem., Int. Ed., 2015, 54, 4953–4957 CrossRef CAS PubMed.
- Y. Guo, F. E. Wolff, I. Schapiro, M. Elstner and M. Marazzi, Phys. Chem. Chem. Phys., 2018, 20, 27501–27509 RSC.
- I. Dokukina and O. Weingart, Phys. Chem. Chem. Phys., 2015, 17, 25142–25150 RSC.
- G. D. M. Seabra, R. C. Walker, M. Elstner, D. A. Case and A. E. Roitberg, J. Phys. Chem. A, 2007, 111, 5655–5664 CrossRef PubMed.
- K. D. Hammonds and J. P. Ryckaert, Comput. Phys. Commun., 1991, 62, 336–351 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, Y. Huang, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. SalomonFerrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, University of California, San Francisco, CA, 2018 Search PubMed.
- J. Huang and A. D. MacKerell Jr, J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed.
- E. L. Wu, X. Cheng, S. Jo, H. Rui, K. C. Song, E. M. Dávila-Contreras, Y. Qi, J. Lee, V. Monje-Galvan, R. M. Venable, J. B. Klauda and W. Im, J. Comput. Chem., 2014, 35, 1997–2004 CrossRef CAS PubMed.
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. Brooks 3rd, A. D. MacKerell Jr., J. B. Klauda and W. Im, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin, W. Jiang and R. McGreevy, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- J. Kästner, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2011, 1, 932–942 CrossRef.
- P. Akhshi and G. Wu, Phys. Chem. Chem. Phys., 2017, 19, 11017–11025 RSC.
- B. Shi, Y. K. Shin, A. A. Hassanali and S. J. Singer, J. Phys. Chem. B, 2015, 119, 11030–11040 CrossRef CAS PubMed.
- B. Isralewitz, M. Gao and K. Schulten, Curr. Opin. Struct. Biol., 2001, 11, 224–230 CrossRef CAS PubMed.
- M. Müller, C. Bamann, E. Bamberg and W. Kühlbrandt, J. Biol. Chem., 2015, 427, 341–349 Search PubMed.
- T. Sattig, C. Rickert, E. Bamberg, H. J. Steinhoff and C. Bamann, Angew. Chem., Int. Ed., 2013, 52, 9705–9708 CrossRef CAS PubMed.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York and Oxford, 1997 Search PubMed.
- M. Takemoto, H. E. Kato, M. Koyama, J. Ito, M. Kamiya, S. Hayashi, A. D. Maturana, K. Deisseroth, R. Ishitani and O. Nureki, PLoS One, 2015, 10, e0131094 CrossRef PubMed.
- A. Ardevol and G. Hummer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3557–3562 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08521b |
| This journal is © The Royal Society of Chemistry 2022 |