
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge-scale synthesis of ultrafine Fe3C nanoparticles embedded in mesoporous carbon nanosheets for high-rate lithium storage†
Ying Yu‡
abc,
Xuanli Wang‡a,
Hongkun Zhang *bc,
Zhiqin Caod,
Haoyang Wu*a,
Baorui Jiaa,
Jun Jun Yanga,
Xuanhui Quae and
Mingli Qin*ae
*bc,
Zhiqin Caod,
Haoyang Wu*a,
Baorui Jiaa,
Jun Jun Yanga,
Xuanhui Quae and
Mingli Qin*ae
aInstitute for Advanced Materials and Technology, University of Science and Technology Beijing, Beijing 100083, China. E-mail: wuhaoyang@ustb.edu.cn; qinml@mater.ustb.edu.cn
bChina United Test & Certification Co., Ltd, China. E-mail: zhk@cutc.net
cGRINM Group Corporation Limited, China
dCollege of Vanadium and Titanium, Panzhihua University, Panzhihua 617000, China
eBeijing Advanced Innovation Center of Materials Genome Engineering, University of Science and Technology Beijing, Beijing 100083, China
First published on 24th February 2022
Abstract
Fe3C modified by the incorporation of carbon materials offers excellent electrical conductivity and interfacial lithium storage, making it attractive as an anode material in lithium-ion batteries. In this work, we describe a time- and energy-saving approach for the large-scale preparation of Fe3C nanoparticles embedded in mesoporous carbon nanosheets (Fe3C-NPs@MCNSs) by solution combustion synthesis and subsequent carbothermal reduction. Fe3C nanoparticles with a diameter of ∼5 nm were highly crystallized and compactly dispersed in mesoporous carbon nanosheets with a pore-size distribution of 3–5 nm. Fe3C-NPs@MCNSs exhibited remarkable high-rate lithium storage performance with discharge specific capacities of 731, 647, 481, 402 and 363 mA h g−1 at current densities of 0.1, 1, 2, 5 and 10 A g−1, respectively, and when the current density reduced back to 0.1 A g−1 after 45 cycles, the discharge specific capacity could perfectly recover to 737 mA h g−1 without any loss. The unique structure could promote electron and Li-ion transfer, create highly accessible multi-channel reaction sites and buffer volume variation for enhanced cycling and good high-rate lithium storage performance.
1. Introduction
Transition metal carbides (TMCs) offer good electrical and thermal conductivity, excellent hardness and mechanical stability, which makes them promising candidates in the fields of dye-sensitized solar cells, Fischer–Tropsch synthesis, proton exchange membrane fuel cells and lithium ion batteries (LIBs).1–5 Among these, Fe3C displays high additional capacity and excellent electrochemical performance in LIBs owing to the reversible gel-like film and interfacial lithium storage on the surface.6–8 However, bulk Fe3C suffers from high resistance and volume expansion during the intercalation/deintercalation of Li ions, leading to capacity loss and poor cycling life. Nanostructure materials and hybrid composites with conducting matrices are widely used to enhance the lithium storage property.9–11 Due to their large specific surface area, high conductivity and mechanical flexibility, carbon materials are considered the most effective strategy to promote the electrochemical performance of Fe3C,5–8,12–17 for e.g., amorphous carbon,5,6 graphene,7,13 nanospindles,8 porous carbon nanosheets,12 porous carbon spheres14 and expanded graphite.17 Porous carbon materials not only provide a fast electron transport network but also create highly accessible multi-channel reaction sites in electrochemical reactions,18–21 as well as buffer volume expansion and act as a barrier against Fe3C aggregation.12,22,23Consequently, much effort has been devoted to preparing Fe3C/carbon composites. For instance, Su et al. obtained core–shell Fe@Fe3C/C nanocomposites by annealing a mixture of α-FeOOH nanorods, glucose and urea, which presented a reversible capacity of 500 mA h g−1 with no capacity loss after 30 cycles at a current density of 50 mA g−1.6 Tan and coworkers synthesized N-doped graphene/Fe–Fe3C nanocomposites through a one-pot pyrolysis process; these composites exhibited a high capacity of 607 mA h g−1 after 100 cycles at 1 A g−1.13 Chen et al. prepared porous carbon spheres doped with Fe3C by a hydrothermal reaction, which afforded 533.6 mA h g−1 after 250 cycles at 100 mA g−1.14 Additionally, chemical vapor deposition,5,7 pyrolysis,8,15 proline-derived in situ synthesis12 and thermal treatment23 have been chosen to fabricate Fe3C/carbon composites. Even though notable success has been achieved, most synthesis methods still have multiple steps, high energy consumption and low output. Thus, it is highly desirable to seek a time- and energy-saving approach for the large-scale preparation of TMC/carbon composites in an environmentally friendly manner, and solution combustion synthesis (SCS) is the most effective approach.24,25
Herein, we describe a facile strategy for the large-scale synthesis of Fe3C nanoparticles embedded in mesoporous carbon nanosheets (Fe3C-NPs@MCNSs) by SCS and subsequent carbothermal reduction (CTR).26,27 The Fe3C-NPs were highly crystallized and compactly dispersed in mesoporous carbon nanosheets and Fe3C-NPs@MCNSs exhibited a high reversible specific capacity of 706 mA h g−1 at a current density of 1 A g−1 and maintained its original morphology even after 300 cycles, which is much better than that of previously reported materials.6,8,13–17 More importantly, the unique mesoporous structure also facilitated a remarkable high-rate lithium storage property. The discharge specific capacities of Fe3C-NPs@MCNSs are 731, 647, 481, 402 and 363 mA h g−1 at current densities of 0.1, 1, 2, 5 and 10 A g−1, respectively, and it could perfectly recover to 737 mA h g−1 without any loss when the current density reduced back to 0.1 A g−1 after 45 cycles. The excellent cycling and high-rate performance indicate that Fe3C-NPs@MCNSs is a promising anode material in lithium ion batteries.
2. Experimental section
2.1 Material synthesis
The precursor was prepared from SCS using Fe(NO3)3·9H2O (11.8 g), NH2CH2COOH (6.26 g) and C6H12O6·H2O (5 g) as the oxidizer, fuel and carbon source, respectively. These raw materials were dissolved in 150 mL deionized water under stirring to obtain a homogeneous mixed solution. The mixture was poured into 1000 mL beakers, and heated to 300 °C in air on a temperature-controlled electrical furnace. The solution evaporated and a gelatinous mass was formed. In a few minutes, the gel suddenly swelled with the release of a lot of gas, and black powder was obtained. The carbonization of the precursor was performed by CTR in a tube furnace by heating at a constant rate of 10 °C min−1 up to 800 °C for 2 h under an argon flow (150 mL min−1). After heating, the product was allowed to cool to room temperature in the Ar flow.2.2 Material characterization
Phase analysis of the precursors and carbonized products was performed on an X-ray diffractometer (XRD, Ultima IV) equipped with graphite monochromatized Cu Kα radiation as the X-ray source. Thermogravimetric-differential scanning calorimetry (TG-DSC, TA Q600) were used for weight loss and gas release measurements, which were taken in the range of room temperature to 600 °C at a constant heating rate of 10 °C min−1 in air. The surface composition of the precursors was analyzed with X-ray photoelectron spectroscopy (XPS, Thermo EscaLab 250Xi) at a background pressure of about 10−9 Torr using Al Kα X-rays as the excitation source (1486.6 eV). Surface morphology, elemental analysis and the corresponding elemental mapping were examined using field emission scanning electron microscopy (FE-SEM, Quanta FEG-450) at 30 kV. Standard and high-resolution transmission electron microscopy (TEM and HRTEM, respectively) were performed using a Tecnai G2 F20 instrument operated at an accelerated voltage of 200 kV. Atomic force microscopy (AFM) was performed on a Dimension Icon-AFM under the tapping mode. The scanning rate was 1 Hz and the resonance vibration frequency was in the range of 310–330 kHz. The samples for AFM measurement were prepared by dropping an aqueous suspension on a silicon chip. The Raman spectra were recorded via a Raman spectrometer. The pore size and pore volume were determined using a Brunauer–Emmett–Teller (BET) apparatus, an automated surface area and pore size analyzer (QUADRASORB SI-MP, Quantachrome Instruments, Boynton Beach, FL). The surface area was determined by evaluating the isotherm of the physical adsorption of nitrogen and the specific surface area was determined by the BET method.2.3 Electrochemical characterization
The active materials (Fe3C-NPs@MCNSs), acetylene black (AB) and polyvinylidene fluoride (PVDF), with a standard weight ratio (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10), were dissolved in N-methyl-2-pyrrolidone (NMP) (solvent) and thoroughly ground in an agate mortar to obtain a homogeneous slurry. Then, the slurry was evenly spread on a copper foil 14 mm in diameter and dried at 120 °C in a vacuum oven overnight. This working electrode was assembled into 2032 coin-type cells in an argon-filled glove box. In the cells, polyethene served as a separator, lithium foil as a counter electrode, and 1 M LiPF6 in a mixture of 1:1 (vol%) ethylene carbonate (EC) and dimethyl carbonate (DMC) served as the electrolyte. The assembled cells were galvanostatically discharged and charged at current densities of 0.1 A g−1 and 1 A g−1, respectively, within the voltage range of 0.01–3.0 V. To evaluate the rate performance, the discharge and charge current densities were gradually increased from 0.1 A g−1 to 1, 2, 5 and 10 A g−1, and suddenly reduced back to 0.1 A g−1 after 45 cycles. Cyclic voltammetry (CV) was performed on an electrochemical workstation (CHI618D) at a scan rate of 0.5 mV s−1 in the range of 0.01–3.0 V.
:10:10), were dissolved in N-methyl-2-pyrrolidone (NMP) (solvent) and thoroughly ground in an agate mortar to obtain a homogeneous slurry. Then, the slurry was evenly spread on a copper foil 14 mm in diameter and dried at 120 °C in a vacuum oven overnight. This working electrode was assembled into 2032 coin-type cells in an argon-filled glove box. In the cells, polyethene served as a separator, lithium foil as a counter electrode, and 1 M LiPF6 in a mixture of 1:1 (vol%) ethylene carbonate (EC) and dimethyl carbonate (DMC) served as the electrolyte. The assembled cells were galvanostatically discharged and charged at current densities of 0.1 A g−1 and 1 A g−1, respectively, within the voltage range of 0.01–3.0 V. To evaluate the rate performance, the discharge and charge current densities were gradually increased from 0.1 A g−1 to 1, 2, 5 and 10 A g−1, and suddenly reduced back to 0.1 A g−1 after 45 cycles. Cyclic voltammetry (CV) was performed on an electrochemical workstation (CHI618D) at a scan rate of 0.5 mV s−1 in the range of 0.01–3.0 V.
3. Results and discussion
Fig. 1 displays the schematic diagram of the synthesis of TMC-NPs@MCNSs by solution combustion synthesis and subsequent carbothermal reduction. The transition metal salts, organic fuel and water-soluble carbon sources were dissolved in deionized water to obtain a homogeneous mixed solution and heated on a hotplate. Then, the solution was evaporated and concentrated to form a gelatinous mass. Meanwhile, a large amount of water vapor escaped from the gel. In this process, the gel gradually polymerized and formed numerous large bubbles due to the continuous liberation of water vapor. As the heating temperature increased, the gel swelled and the bubble walls gradually thinned and broke, and a sheet-like precursor of the transition metal oxide/carbon (TMO/C) composites was obtained. Afterwards, the precursor was carbonized in an inert atmosphere for several hours. During the CTR process, TMO particles reacted with the surrounding close-knit carbons to form TMC particles and consume parts of the unstable carbons to generate plenty of oxy carbide gases, leaving lots of mesopores in the sheets. Finally, TMC particles embedded in mesoporous carbon nanosheets were obtained, denoted as TMC-NPs@MCNSs.26,27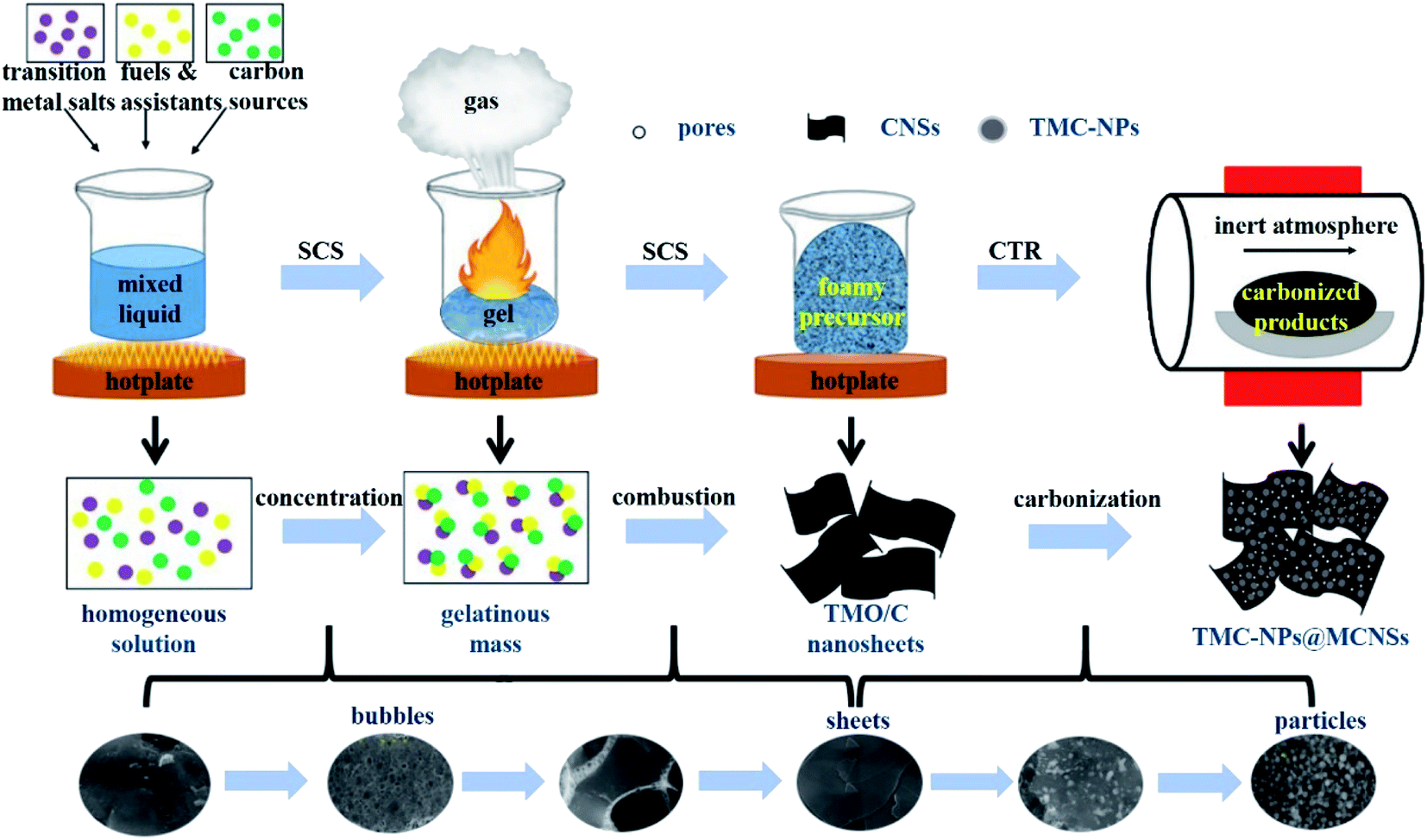 | ||
| Fig. 1 Schematic illustration of the synthesis of TMC-NPs@MCNSs. | ||
The crystal structure and phase composition of the SCS precursor of Fe3C-NPs@MCNSs are characterized by X-ray diffraction and X-ray photoelectron spectroscopy. As shown in Fig. 2a, there are no obvious diffraction peaks in the XRD pattern, indicating the amorphous structure of the precursor. This may be ascribed to the pyrolysis of glucose, which absorbs heat and decreases the reaction temperature in the combustion process; thus, the energy generated from the combustion reaction is not sufficient for the crystallization of FeOx (ESI Fig. S1†). From Fig. 2b, the characteristic peaks of C 1s, N 1s, O 1s and Fe 2p can be observed at about 285, 400, 530 and 712 eV, respectively, confirming the existence of these elementary components in the precursor. Moreover, in the partial XPS spectrum of Fe 2p (Fig. 2c), the peaks of Fe 2p3/2 and Fe 2p1/2 are observed at around 710 and 723 eV, respectively, confirming that Fe ions feature a tri-valence oxidation state. The Fe 2p3/2 peak is also associated with a satellite peak located approximately 7 eV higher than the main peak, which is a characteristic of hematite,28 indicating the formation of the α-Fe2O3 phase in the precursor.
 | ||
| Fig. 2 (a) XRD pattern; (b) survey XPS spectrum and (c) Fe 2p high-resolution XPS spectrum of the SCS precursor. | ||
Fig. 3a shows a typical field emission scanning electron microscopy image of the SCS precursor, which presents free-standing and ultrathin sheets with a smooth surface. The corresponding energy dispersive spectroscopy (EDS, Fig. 3b) elemental mapping results clearly reveal that the elements Fe, O and C are evenly distributed on the sheets, indicating that α-Fe2O3 and carbon are in intimate contact. Furthermore, a low magnification transmission electron microscopy image of the SCS precursor is shown in Fig. 3c, wherein the ultrathin sheet structure is in good agreement with the observation from the FE-SEM image. Meanwhile, the amorphous structure of the sheets can be further confirmed by selected area electron diffraction (SAED, the inset in Fig. 3c), and there is a typical amorphous ring diffraction pattern rather than speckles or distinct rings, which agrees well with the XRD pattern in Fig. 2a. In addition, the thickness of sheets can be estimated to be ∼100 nm according to atomic force microscopy (AFM, Fig. 3d). These experimental results indicate that the SCS precursor contains a homogeneous mixture of amorphous α-Fe2O3 and carbon nanosheets.
 | ||
| Fig. 3 (a) FE-SEM image; (b) EDS analysis; (c) TEM image (inset: SAED pattern) and (d) AFM image of the SCS precursor. | ||
As shown in Fig. 4a, the XRD pattern of the CTR product can be indexed to Fe3C (JCPDS 89-2867) according to the diffraction peak position and relative intensity.29 Furthermore, there are two distinguishable peaks from the Raman spectrum (Fig. 4b) at ∼1350 cm−1 (D-band) and ∼1580 cm−1 (G-band), in which the D-band corresponds to amorphous carbon and the G-band is related to crystalline graphite.30 The peak intensity ratio ID/IG is calculated to be ∼0.92, demonstrating that the carbon in Fe3C-NPs@MCNSs is almost in the amorphous form. Moreover, the nitrogen adsorption and desorption isotherms of the product show a remarkable type-IV isotherm with an H2-type hysteresis loop (Fig. 4c), indicating mesoporous structure with narrow pore-size distribution,31 which is centered at 3–5 nm. In addition, the Brunauer–Emmett–Teller (BET) surface area of the product is measured to be 297.9 m2 g−1. The TEM image in Fig. 4d shows that Fe3C-NPs@MCNSs has a sheet-like structure, which maintains the morphology of the SCS precursor. The magnified TEM image (Fig. 4e) reveals that well-dispersed Fe3C nanoparticles with a nearly spherical structure are embedded in the matrix of carbon sheets. Furthermore, it is clear from Fig. 4f that the Fe3C nanoparticles are highly crystallized and perfectly embedded in the amorphous carbon sheets, and the average diameter is ∼5 nm. Moreover, lattice fringes with a spacing of 0.2067 nm can be observed, which correspond to the (102) crystal plane of the orthorhombic Fe3C structure. The SAED patterns (the inset in Fig. 4f) confirm that the Fe3C nanoparticles are polycrystalline, which matches well with the XRD result in Fig. 4a. According to the AFM images (Fig. 4g and h), it can be clearly seen that the Fe3C nanoparticles have a relatively homogeneous particle size and the thickness of the carbon sheet can be estimated to be within the range of 80–100 nm.
 | ||
| Fig. 4 (a) XRD pattern; (b) Raman spectrum; (c) nitrogen adsorption and desorption isotherms (inset: corresponding BJH pore distribution); (d and e) TEM images under different magnifications; (f) HRTEM image (inset: SAED pattern); (g) 2D AFM image and (h) 3D AFM image of the CTR product. | ||
The electrochemical performance of Fe3C-NPs@MCNSs is presented in Fig. 5. As shown in Fig. 5a, an explicit reduction peak at ∼0.5 V (vs. Li+/Li) can be observed from the CV curves in initial discharge corresponding to the formation of the SEI film.6,13 The oxidation peaks between 0.3 V and 0.01 V are related to the intercalation/deintercalation of lithium ions in the carbon nanosheets.13 In the following CV curves, the reduction peak related to the SEI film increases to 0.8 V because of the decomposition of the SEI film under the catalytic effect of Fe3C NPs.13 In Fig. 5b, Fe3C-NPs@MCNSs exhibits a high initial discharge specific capacity of 1240 mA h g−1 and a relatively low initial charge specific capacity of 662 mA h g−1 at 0.1 A g−1. There is no obvious variation in the discharge and charge curves after the first cycle, which proves the enhanced lithium ion intercalation/deintercalation property.
 | ||
| Fig. 5 Electrochemical properties of the Fe3C-NPs@MCNSs anode in a lithium ion battery. (a) CV curves obtained in a potential range of 0.01 to 3.0 V (vs. Li+/Li) and at a scan rate of 0.5 mV s−1. (b) Galvanostatic discharge and charge voltage profiles for the 1st, 2nd, 3rd, 10th and 30th cycles at a current density of 0.1 A g−1. (c) Cycling performance and the corresponding coulombic efficiency at a current density of 1 A g−1 and (d) rate performance at various discharge and charge current densities from 0.1 to 10 A g−1. | ||
Compared to the commercial graphite anode (Fig. 5c), Fe3C-NPs@MCNSs displays a very high reversible specific capacity of 706 mA h g−1 after 300 cycles at a current density of 1 A g−1, which is much higher than that in related reports [ESI Table S1†], and it maintains its original morphology even after 300 cycles (Fig. 6). More importantly, it also exhibits a remarkable high-rate lithium storage property, as demonstrated in Fig. 5d. Specifically, the discharge specific capacities of the anode are 731, 647, 481, 402 and 363 mA h g−1 at current densities of 0.1, 1, 2, 5 and 10 A g−1, respectively. When the current density reduces back to 0.1 A g−1 after 45 cycles, the discharge specific capacity can perfectly recover to 737 mA h g−1 without any loss. These results clearly highlight the excellent electrochemical performance of Fe3C-NPs@MCNSs, which can be ascribed to the unique nanostructure.13,14 Firstly, the mesoporous carbon nanosheets with a large specific surface area enhance electrical conductivity, promote electron and lithium ion transfer, and create highly accessible multi-channel reaction sites for lithium storage.13,18,19 Secondly, the smaller particle size of Fe3C contributes to the deintercalation of lithium ions from the carbon sheets and the reversible formation/decomposition of the SEI film. Third, the mesoporous structure can accommodate the volume change and prevent the agglomeration of Fe3C, which maintains the structural integrity.21,32–34
 | ||
| Fig. 6 FE-SEM images of Fe3C-NPs@MCNSs under different magnification (a and c) before and (b and d) after 300 electrochemical cycles. | ||
4. Summary
We demonstrate a time- and energy-saving approach for the large-scale preparation of Fe3C nanoparticles embedded in mesoporous carbon nanosheets by solution combustion synthesis and subsequent carbothermal reduction. The obtained Fe3C nanoparticles are uniformly and compactly embedded in amorphous mesoporous carbon nanosheets. Fe3C-NPs@MCNSs displays a high reversible specific capacity of 706 mA h g−1 at a current density of 1 A g−1 after 300 cycles and a good high-rate lithium storage property with discharge specific capacities of 731, 647, 481, 402 and 363 mA h g−1 at current densities of 0.1, 1, 2, 5 and 10 A g−1, respectively. The unique structure facilitates lithium ion and electron transfer, creates highly accessible multi-channel reaction sites for lithium storage, prevents the agglomeration of Fe3C and maintains the structural integrity. Thus, this work provides a versatile and energy-efficient method to construct TMC-NPs@MCNSs nanocomposites for large-scale preparation and scale-up application in electrochemistry and other areas.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the National Natural Science Foundation Program of China (52131307, 52130407, 52071013, 52104359, 51774035, and 52174344), National Key Research and Development Program of China (2021YFB3701900), the Natural Science Foundation Program of Beijing (2202031, 2174079 and 2162027), S&T Program of Hebei (20311001D), the Fundamental Research Funds for the Central Universities (FRF-TP-19-003C2, FRF-IDRY-19-025, FRF-IDRY-20-022, FRF-TP-20-032A2, FRF-TP-20-100A1Z), Scientific and Technological Innovation Foundation of Foshan (BK21BE007), the Postdoctor Research Foundation of Shunde Graduate School of University of Science and Technology Beijing (2020BH014), and the Natural Science Foundation Program of Hunan (2021JJ30250).References
- Z. Kou, T. Meng, B. Guo, I. S. Amiinu, W. Li, J. Zhang and S. Mu, Adv. Funct. Mater., 2017, 27, 1604904 CrossRef.
- P. Vijayakumar, R. Govindaraj, N. Santhosh, M. S. Pandian, A. Pandikumar and P. Ramasamy, J. Mater. Sci., 2018, 53, 4444–4455 CrossRef CAS.
- J. G. Rivera De La Cruz, M. K. Sabbe and M. F. Reyniers, J. Phys. Chem. C, 2017, 121(45), 25052–25063 CrossRef CAS.
- L. Wei, H. Sun, T. Yang, S. Deng, M. Wu and Z. Li, Appl. Surf. Sci., 2018, 439, 439–446 CrossRef CAS.
- L. Hou, W. Yang, X. Xu, B. Deng, Z. Chen, S. Wang, J. Tian, F. Yang and Y. Li, Chem. Eng. J., 2019, 375, 122061 CrossRef CAS.
- L. Su, Z. Zhou and P. Shen, Electrochim. Acta, 2013, 87, 180–185 CrossRef CAS.
- Y. Yang, X. Fan, G. Gasillas, Z. Peng, G. Ruan, G. Wang, M. J. Yacaman and J. M. Tour, ACS Nano, 2014, 8, 3939–3946 CrossRef CAS PubMed.
- J. Zhang, K. Wang, Q. Xu, Y. Zhou, F. Cheng and S. Guo, ACS Nano, 2015, 9, 3369–3376 CrossRef CAS PubMed.
- H. Wang, J. Li, K. Li, Y. Lin, J. Chen, L. Gao, V. Nicolosi, X. Xiao and J.-M. Lee, Chem. Soc. Rev., 2021, 50, 1354–1390 RSC.
- P. Prabhu, V. Jose and J.-M. Lee, Matter, 2020, 2, 526–553 CrossRef.
- Y. Dong, Z. Zhang, Y. Xia, Y.-S. Chui, J.-M. Lee and J. A. Zapien, J. Mater. Chem. A, 2015, 3, 16206–16212 RSC.
- J. Zhang, L. Qi, X. Zhu, X. Yan, Y. Jia, L. Xu, D. Sun and Y. Tang, Nano Res., 2017, 10, 3164–3177 CrossRef CAS.
- Y. Tan, K. Zhu, D. Li, F. Bai, Y. Wei and P. Zhang, Chem. Eng. J., 2014, 258, 93–100 CrossRef CAS.
- S. Chen, J. Wu, R. Zhou, L. Zuo, P. Li, Y. Song and L. Wang, Electrochim. Acta, 2015, 180, 78–85 CrossRef CAS.
- X. Zhao, D. Xia, J. Yue and S. Liu, Electrochim. Acta, 2014, 116, 292–299 CrossRef CAS.
- C. Zhao, R. Fan, B. Murugesan, Y. Xu, J. Ma, J. Yao, I. Krucinska, Y. Cai and J. Gao, J. Alloys Compd., 2021, 881, 10661 CrossRef.
- Y. Huang, X. Lin, X. Zhang, Q. Pan, Z. Yan, H. Wang, J. Chen and Q. Li, Electrochim. Acta, 2015, 178, 468–475 CrossRef CAS.
- Y. Xing, Y. Wang, C. Zhou, S. Zhang and B. Fang, ACS Appl. Mater. Interfaces, 2014, 6, 2561–2567 CrossRef CAS PubMed.
- B. Fang, J. Kim, M.-S. Kim and J.-S. Yu, Acc. Chem. Res., 2013, 46, 1397–1406 CrossRef CAS PubMed.
- S. Yu, S. Song, R. Li and B. Fang, Nanoscale, 2020, 12, 19536–19556 RSC.
- Y. Zhang, X. Wang, F. Luo, Y. Tan, L. Zeng, B. Fang and A. Liu, Appl. Catal., B, 2019, 25, 117852 CrossRef.
- H. Fan, Y. Zhang, H. Yu, Y. Zheng, Y. Luo, Z. Dai, B. Li, Y. Zong and Q. Yan, Angew. Chem., Int. Ed., 2017, 56, 12566–12570 CrossRef CAS PubMed.
- Y. Liu, X. Xu, P. Sun and T. Chen, Int. J. Hydrogen Energy, 2015, 40, 4531–4539 CrossRef CAS.
- H. Wu, M. Qin, W. Wang, Z. Cao, Z. Liu, Q. Yu, C. Lao, D. Zhang, B. Jia, D. He, T. Liu, A. A. Volinsky, P. Cao and X. Qu, J. Mater. Chem. A, 2018, 6, 7053–7061 RSC.
- H. Wu, Q. Yu, C. Lao, M. Qin, W. Wang, Z. Liu, C. Man, L. Wang, B. Jia and X. Qu, Energy Storage Mater., 2019, 18, 43–50 CrossRef.
- A. Varma, A. S. Mukasyan, A. S. Rogachev and K. V. Manukyan, Chem. Rev., 2016, 116, 14493–14586 CrossRef CAS PubMed.
- F. Li, J. Ran, M. Jaroniec and S. Qiao, Nanoscale, 2015, 7, 17590–17610 RSC.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- G. Ren, X. Lu, Y. Li, Y. Zhu, L. Dai and L. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 4118–4125 CrossRef CAS PubMed.
- A. Antwi-Boasiako, D. Dunn, S. Dasary, Y. K. Jones, S. Barnes and A. Singh, J. Raman Spectrosc., 2017, 48, 1056–1064 CrossRef CAS PubMed.
- Y. Gu, Z. Xiong, W. A. Abdulla, G. Chen and X. S. Zhao, Chem. Commun., 2014, 50, 14824–14827 RSC.
- X. Zhang, J. Zhou, X. Chen and H. Song, ACS Appl. Energy Mater., 2018, 1, 48–55 CrossRef CAS.
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS.
- L. Zu, W. Zhang, L. Qu, L. Liu, W. Li, A. Yu and D. Zhao, Adv. Energy Mater., 2020, 10, 2002152 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08516f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |