
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochemical modifications for DNA/RNA oligonucleotides
Amirrasoul Tavakoli†
 and
Jung-Hyun Min
*
and
Jung-Hyun Min
*
Department of Chemistry & Biochemistry, Baylor University, Waco, TX 76706, USA. E-mail: JungHyun_Min@baylor.edu; Tel: +1-254-710-2095
First published on 24th February 2022
Abstract
Light-triggered chemical reactions can provide excellent tools to investigate the fundamental mechanisms important in biology. Light is easily applicable and orthogonal to most cellular events, and its dose and locality can be controlled in tissues and cells. Light-induced conversion of photochemical groups installed on small molecules, proteins, and oligonucleotides can alter their functional states and thus the ensuing biological events. Recently, photochemical control of DNA/RNA structure and function has garnered attention thanks to the rapidly expanding photochemistry used in diverse biological applications. Photoconvertible groups can be incorporated in the backbone, ribose, and nucleobase of an oligonucleotide to undergo various irreversible and reversible light-induced reactions such as cleavage, crosslinking, isomerization, and intramolecular cyclization reactions. In this review, we gather a list of photoconvertible groups used in oligonucleotides and summarize their reaction characteristics, impacts on DNA/RNA thermal stability and structure, as well as their biological applications.
 Amirrasoul Tavakoli | Dr Amirrasoul Tavakoli received his Bachelor's degree in Chemistry and Master's degree in Nanotechnology from the University of Tehran (Tehran, IRAN). He obtained his PhD degree in Biochemistry under the supervision of Prof. Jung-Hyun Min from Baylor University (Texas, USA) on light-induced modulation of DNA recognition by the Rad4/XPC damage sensor protein using photoreactive DNA. He is currently a postdoctoral fellow in Prof. Daniella Nicastro's lab at the University of Texas Southwestern Medical Center (Texas, USA), developing methods for localizing and identifying specific molecules inside cells at near-atomic resolution using cryo-electron tomography. |
 Jung-Hyun Min | Prof. Jung-Hyun Min is an Associate Professor of Chemistry and Biochemistry at Baylor University (Texas, USA). She received her Bachelor's degree in Chemistry from Seoul National University (Seoul, South Korea) and a PhD degree in Biochemistry from University of Washington (Washington, USA). She conducted postdoctoral research in Structural Biology at the Memorial Sloan-Kettering Cancer Center/Howard Hughes Medical Institute in New York, USA. She was also an Assistant/Associate Professor of Chemistry at the University of Illinois at Chicago before moving to Baylor. Her research focuses on determining the mechanism of the nucleotide excision repair process in eukaryotes using various structural, biochemical and biophysical tools including photoreactive oligonucleotides. |
Introduction
Optical control of chemical reactions has recently gained popularity.1–4 These controls rely on photoconvertible groups that undergo structural changes upon irradiation by light.5–10 Light can be readily applied to and removed from a reaction, and the wavelength, localization, and intensity of the light can be precisely controlled.11,12 Thus, light offers distinct advantages in triggering and controlling reactions, compared with other more common methods such as chemical inhibition, rapid mixing, temperature-, salt- or pH-jumps. In some reversible reactions, the forward or reverse reactions are promoted by distinct wavelengths of light, offering a unique advantage.13,14Photoconvertible modifications in small molecules,15,16 oligonucleotides,12,17,18 peptides,19 and proteins (mostly enzymes)20–24 have been applied to control and monitor biological events such as gene expression, enzyme activity, oligomerization states, cellular localization, and immune responses. Here, we have compiled a list of photoreactive modifications on DNA/RNA oligonucleotides and summarized the literature on their reaction characteristics, impacts on DNA/RNA thermal stability and structure, as well as their biological applications (Table 1).
Many photochemical groups entail ‘bulky’ modifications that alter the DNA/RNA structures in unique ways and some modifications can induce site-specific strand-breaks, thus mimicking cellular DNA damage. Thus, these applications may be applicable to studying various DNA damage repair and response mechanisms as well as in the more commonly used applications such as gene expression control. We hope this review will provide useful information for the community of researchers looking for ways to use light to study biochemical/molecular events.
Photochemical modifications for DNA/RNA oligonucleotides
Photochemical modifications are most commonly incorporated into oligonucleotides by solid-phase synthesis using phosphoramidite chemistry in which a phosphoramidite building block containing the desired photochemical group is first synthesized and subsequently incorporated to an oligonucleotide chain.25 Post-synthetic approaches have also been used in which site-specific chemical reactions were carried out directly on nucleic acids: this approach can bypass the need for specialized equipment such as DNA/RNA synthesizer.26–28 Most of the modifications in this review are incorporated into oligonucleotides via the phosphoramidite chemistry unless noted otherwise.The family of photochemical groups for oligonucleotides (1 to 12) is named after the parent molecule and can be grouped into four broad categories (I to IV) according to their reaction types as follows:
I. Photocleavage − irreversible:
(1) o-Nitrobenzyl
(2) p-Hydroxyphenacyl
(3) TEEP-OH
(4) Aryl sulfide
(5) Nitroindole
(6) Benzophenone/acetophenone
(7) Coumarin
II. Intermolecular photocrosslinking via [2 + 2] cycloaddition − reversible
(7) Coumarin
(8) Carbazole
(9) Vinyl derivatives
III. Cis–trans photoisomerization − reversible
(9) Vinyl derivative
(10) Azobenzene
IV. Intramolecular photocyclization − reversible
(11) Spiropyrans
(12) Diarylethene
1. o-Nitrobenzyl
ortho-Nitrobenzyl (oNB) group is the most extensively studied and applied photoremovable group. A wide variety of functional groups can be introduced into the oNB scaffold, and oNB derivatives have been used as a part of DNA, RNA, small molecules, and proteins.11 Photocleavage wavelengths are tunable (λ = 345–420 nm).29 In DNA/RNA, they can be incorporated in the backbone, ribose, or nucleobase. Initially, oNB derivatives have been employed as a part of the backbone linkers that can trigger light-induced strand breaks (Fig. 1).30–32 These cleavable oNB linkers have been used in various systems including circular antisense oligonucleotides,33 DNAzyme,34 negatively charged peptide nucleic acids,35 single-stranded circular RNAs as RNA interference (siRNA) precursors,36 single guide RNA (sgRNA) for CRISPR-Cas9-based gene editing,37–39 and splice-switching oligonucleotides.40 Caging the 2′-OH with oNB group41,42 has been applied to regulate DNAzymes by the Lu group (Fig. 1).43–46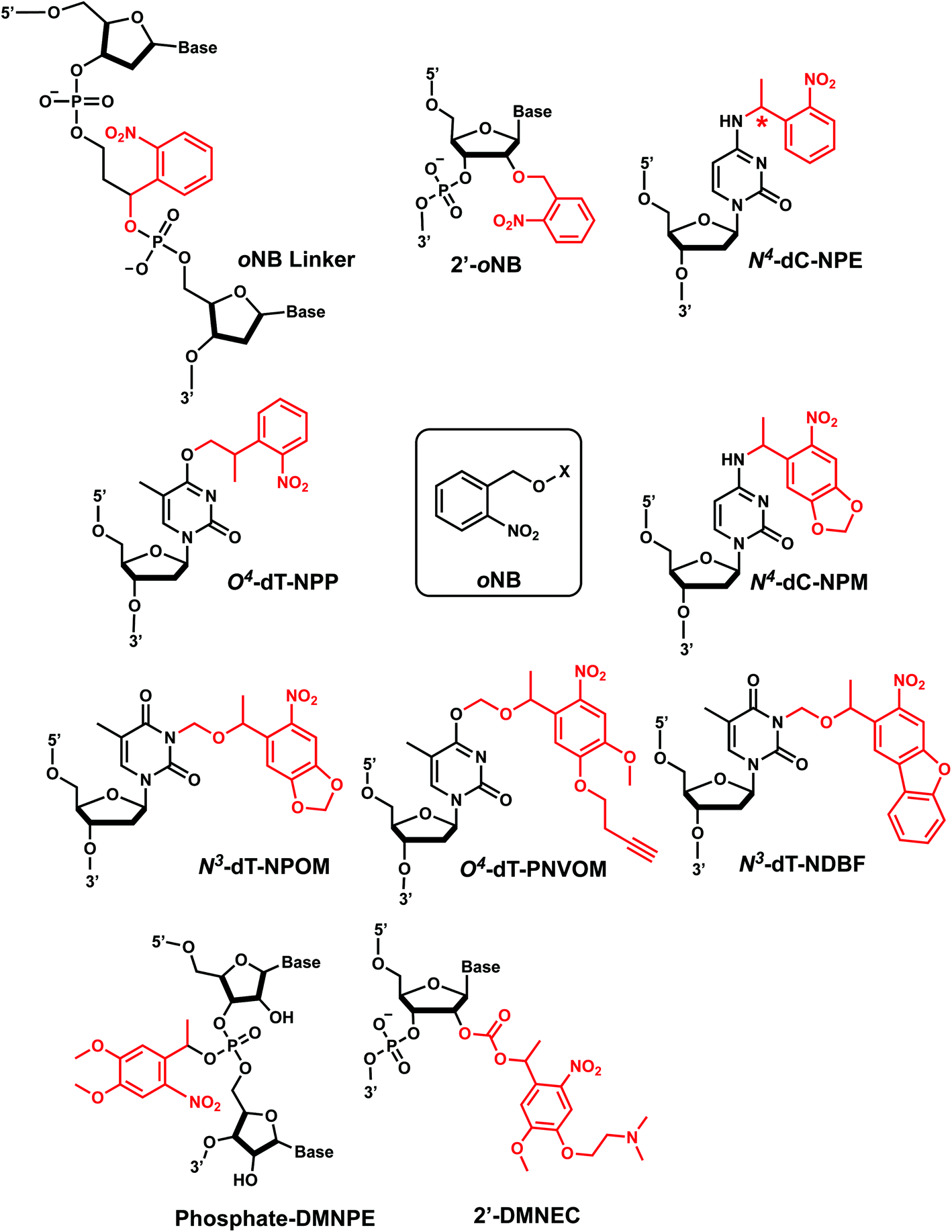 | ||
| Fig. 1 Representative structures of oNB modifications on oligonucleotides. oNB's X represents a caged substrate such as an oligonucleotide. Other types of oNB derivatives are also available but not shown for clarity. | ||
oNB and its derivatives have also been incorporated in nucleobases to make caged nucleobases for various applications (reviewed by Deiters30). 1-(ortho-Nitrophenyl)-ethyl (NPE) and 2-(ortho-nitrophenyl)-propyl (NPP) caged nucleotides were among the first oNB-modified nucleobase.30 Later, 6-nitropiperonyl methyl group (NPM) on N4-dC and its corresponding hydroxymethylene analogs (NPOM) on N3-dT, N3–U and N1-dG were developed; these groups offered longer photocleavage wavelengths (∼365 nm) than that used for oNB and better stability in an aqueous environment at various pHs.30 In particular, NPOM, developed by the Deiters group, has been extensively used for various in vitro and in vivo biological applications.9,40,47–51
Closely related propargyl-6-nitroveratryloxymethyl (PNVOM) modification contains an alkyne group available for post-synthetic click reaction.52 The nitrodibenzofuran (NDBF) group attached to N3-dT53 or N4-dC and N6-dA54 showed photocleavage with >400 nm wavelength.54 4,5-Dimethoxy-2-nitrophenylethyl (DMNPE) has been used to modify internal55 and termini56 phosphate in siRNA. 1-(4-(2-(Dimethylamino)ethoxy)-5-methoxy-2-nitrophenyl)ethyl carbonyl (DMNEC) moiety has been utilized for acylating 2′-hydroxyls of RNA.26,57
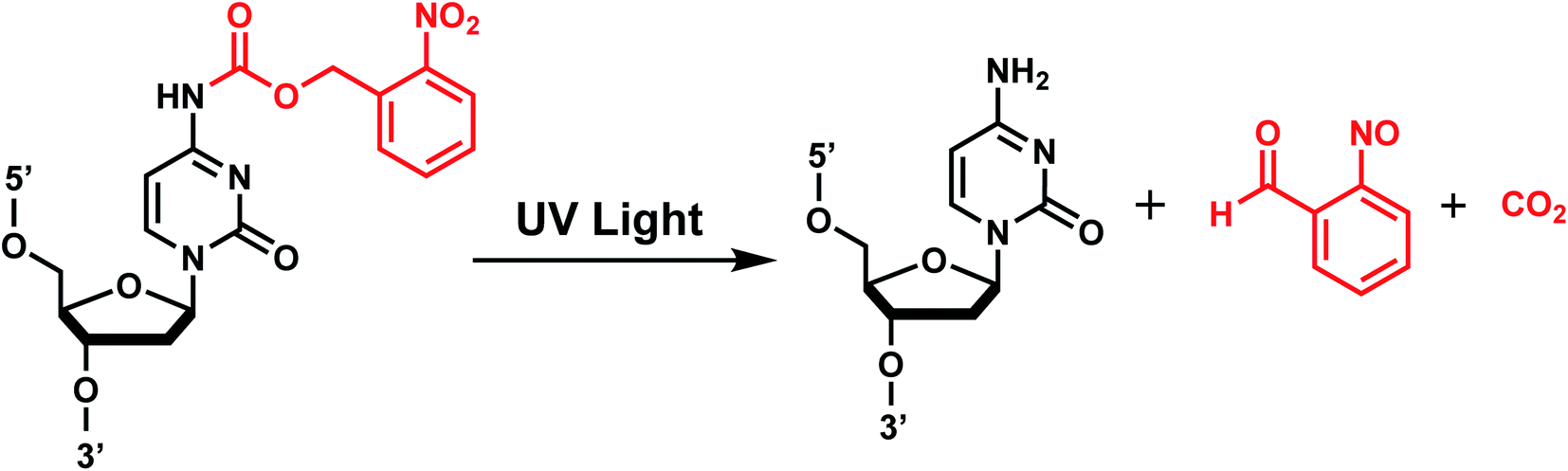 | ||
| Fig. 2 oNB photocleavage reaction. Upon irradiation with UV-A light, the bond between the oNB and the leaving group (e.g., dC nucleoside) is cleaved in a radical-mediated reaction. The byproduct (e.g., CO2) varies depending on the type of oNB photocage. | ||
oNB derivatives in oligonucleotides are shown to be removed in seconds to minutes range using a wide range of power mW–W.39,49,58,59 In a study by Stephanopoulos et al., 85% removal of NPOM-caged DNA occurred in 3 s using 18.2 W lamp.60
NPP and NPE modifications. NPP-dT and NPE-dT were used to block the binding of the MutS mismatch repair protein to a DNA bulge, which then could be removed by photoirradiation and enable the binding.63
NPP, NPE, and oNB linkers were also widely used as photocleavable linkers in the backbone of oligonucleotides to control gene expression and editing. For instance, NPE has been utilized in modulating siRNA activity.64 In a recent work, single-stranded RNA circularized via an oNB linker in the phosphodiester backbone was used as siRNA precursor, which could efficiently be activated to linear RNAs by 365 nm irradiation in vitro.36 Various types of oNB-based linkers were also used: e.g., as internal photocleavable linkers within single guide RNA (sgRNA) to inactivate Cas9 nuclease and attenuate genome editing by CRISPR-Cas9 within cells37,39 and as a way to control RNA-cleaving DNAzyme's activity.34
NPOM. The ability of NPOM to disrupt DNA and RNA hybridization has been used in various applications such as DNA nano-tweezer,60 DNA triplex nanostructures,65 DNA computation,66 as well as controlling antisense DNA agent activity,52 DNAzyme activity,50,67 restriction endonuclease,49 and polymerase chain reaction.68,69 NPOM was also used as a part of gene promoters, triplex-forming oligonucleotides, microRNA, siRNA47 as a tool to regulate gene transcription and translation. In more recent works, NPOM was applied to the CRISPR-Cas9 gene editing system. In one study, NPOM-caged guide RNAs (gRNAs) conferred complete suppression of gRNA:dsDNA-target hybridization, which could subsequently be restored with light irradiation.51 In another study, NPOM-modified gRNA hybridized with DNA and allowed Cas9 to bind DNA, but the gene cleavage was suppressed until light-induced activation.70 This approach, referred to as very fast CRISPR (vfCRISPR), also created double-strand breaks (DSBs) at a submicrometer scale within seconds, which could be used to track the recruitment of DSB repair proteins to the damaged sites.70 Min et al. also showed that NPOM-modified dT could be specifically bound by the Rad4/XPC DNA nucleotide excision repair protein and that such binding was abolished upon light-induced photocleavage of NPOM.58 Other biological applications of NPOM include modulating mRNA splicing (splice switching) in cells and zebrafish40 and controlling TLR9 in immune responses.18
DMNPE & DMNEC. DMNPE was used to control the activity of siRNA as a part of the internal backbone or its termini.56 For instance, the regioselective incorporation of DMNPE groups in the four phosphate termini of an siRNA duplex effectively limited the RNAi activity, which could be restored upon irradiation.56 On the other hand, the DMNEC modifications were used to post-synthetically acylate 2′-hydroxyls of RNA ribose. Hammerhead ribozyme activity could be photo-regulated using this method in which multiple DMNEC groups were incorporated along the RNA molecule.26 Later, Zhou et al. used DMNEC on gRNA to suppress CRISPR-Cas gene editing, which could be reversed by 365 nm light.57
2. p-Hydroxyphenacyl
p-Hydroxyphenacyl (pHP) modification has been first introduced by the Reese group.71 Currently, pHP modification and their photolysis reactions have been reported for base modifications on N3-dT,59 O4-dT,72 and O6-dG (Fig. 3).73 Addition of a benzothiazole to pHP as 2-(2′-hydroxyphenyl)benzothiazole (HBT) has also been introduced as fast decaging moiety on O6-dG (Fig. 3B).74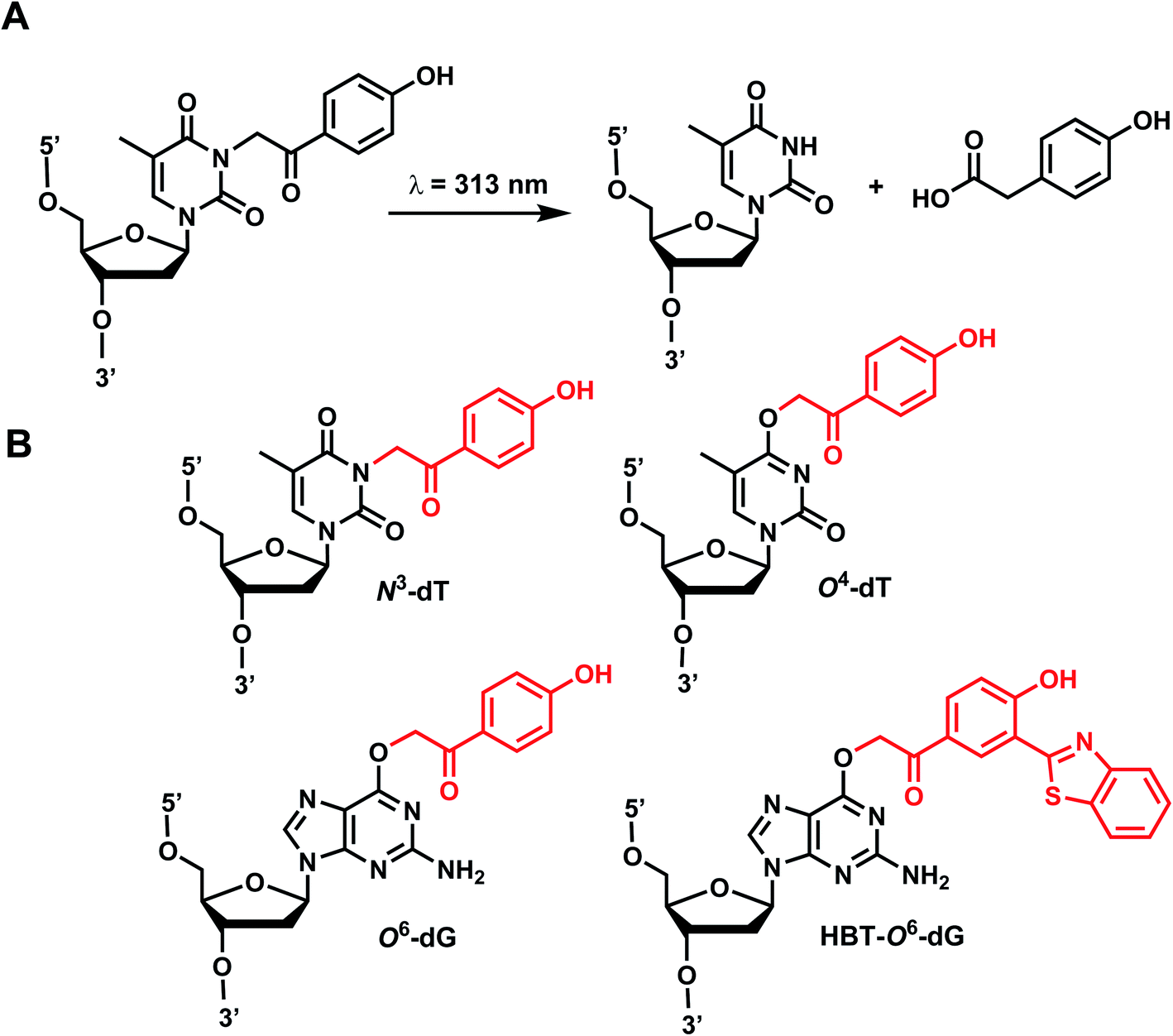 | ||
| Fig. 3 pHP modifications on oligonucleotides nucleobase. (A) Upon light irradiation, the pHP group undergoes skeletal rearrangements and is removed from the nucleobases of DNA and RNA, restoring the native structure. (B) pHP and pHP-derived modification on DNA nucleobases on N3-dT, O4-dT, O6-dG. | ||
The photocleavage reaction of pHP on N3-dT was slow (1 h using 313 nm),59 but the incorporation of pHP in O4-dT shortened the photodecaging, and complete photodecaging was achieved in 0.3 min.72 pHP at the O6-dG position was decaged with a time constant t1/2 of 17 s upon irradiation with 295 nm UV light.73 Later, Singh et al.75 improved the photodecaging reaction by synthesizing HBT. HBT has strong fluorescence and can use longer wavelength light (400 nm) for decaging. HBT-modification on O6-dG cleaved using blue light (405 nm) in a short pulse (≤30 ms) was used for rapidly initiating the folding and activation of twister ribozyme in a single molecule study.74
3. TEEP-OH (thioether-enol phosphate, phenol substituted)
Photolabile TEEP-OH (thioether-enol phosphate, phenol substituted) was inspired by the p-hydroxyphenacyl bromide group (pHP, Section 2)76 and can be incorporated via post-synthetic modification on the phosphodiester backbone of phosphorothioate DNA.28 Upon light irradiation, TEEP–OH is photocleaved and the phosphate backbone reverts to its native form (Fig. 4). This modification was used for photoregulation of an RNA-cleaving DNAzyme, a G-quadruplex peroxidase-mimicking DNAzyme, and a thrombin-binding aptamer.27,28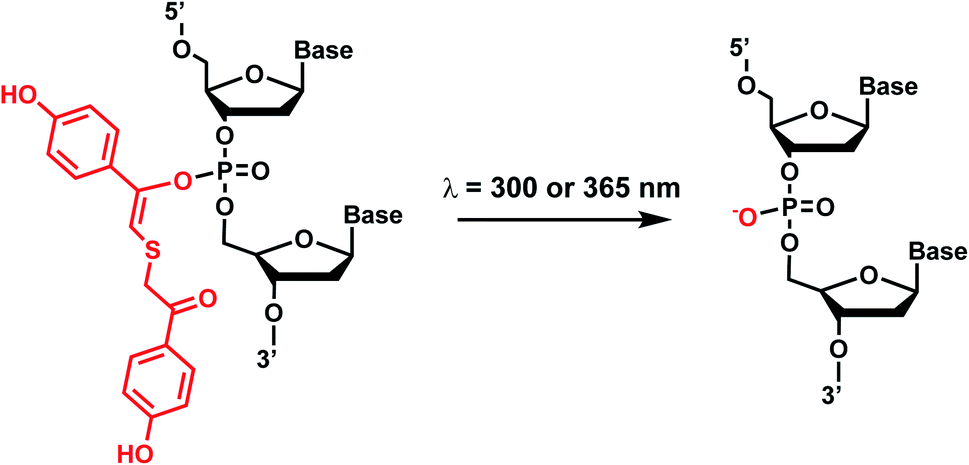 | ||
| Fig. 4 Light-induced photocleavage reaction of the TEEP-OH group from a phosphate backbone. Schematic of the photocleavage reaction. Upon light irradiation (λ = 300 or 365 nm), the TEEP-OH group is cleaved from the phosphate group in the DNA, restoring the DNA backbone. | ||
4. Aryl sulfide
Originally reported by the Greenberg group as a way to study nucleobase radical formation, electron-rich aryl sulfide (ArS, dimethoxythiophenyl) undergoes carbon–sulfur bond homolysis upon irradiation.77 ArS-modified nucleobase (e.g., on C5-methyluridine or C6-hydrothymidine) disrupts nucleic acid structure by perturbing base stacking.77,78 ArS on 5-methyluridine prevents RNA hairpin formation in short RNA as well as the folding of the preQ1 class I riboswitch.78,79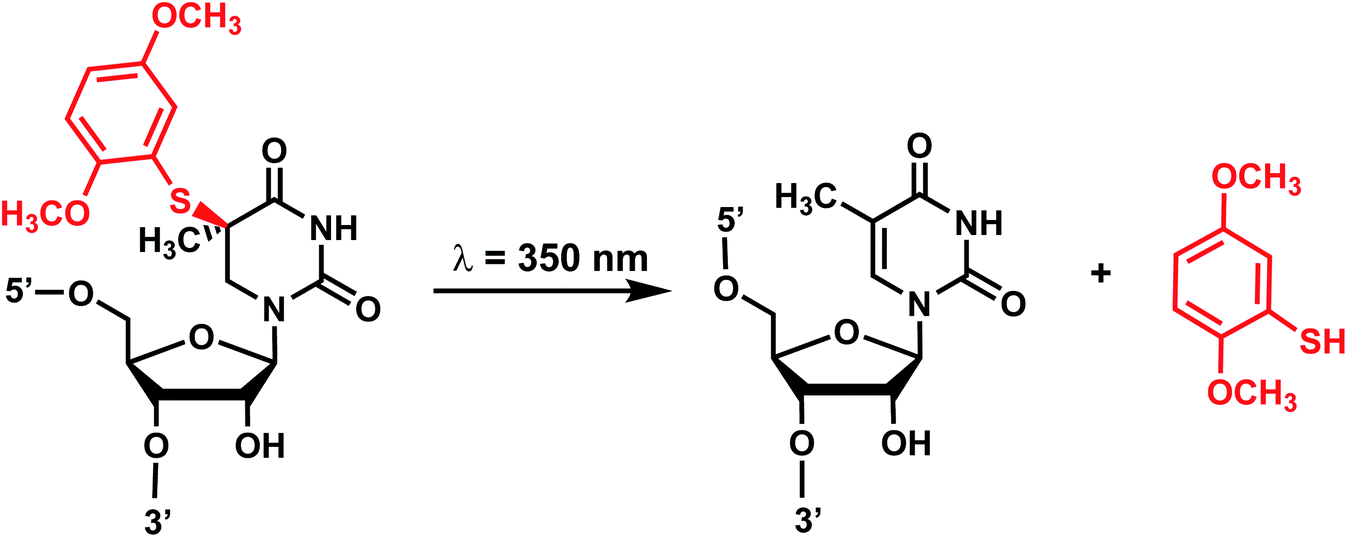 | ||
| Fig. 5 Light-induced photocleavage reaction of the ArS group from nucleobase. Schematic of the photocleavage reaction. Upon light irradiation (λ = 350 nm), the ArS group is cleaved, restoring 5-methyluridine in the RNA. | ||
5. Nitroindole group
Photocleavable nitroindole nucleoside was introduced by Lhomme and colleagues.80,81 Irradiation (λ = 350 nm) of oligonucleotides containing 7-nitroindole or 5-nitroindole triggers a radical process: the excited nitro group induces an intramolecular H1′ abstraction leading to the release of a nitrosoindole group while forming a highly labile deoxyribonolactone, an abasic site (Fig. 6).82 Subsequent mild basic or thermal treatment leads to cleavage of the DNA backbone via β- and δ-elimination at the abasic site.82 | ||
| Fig. 6 Nitroindole modification on oligonucleotides. (A) Upon light irradiation, the nitroindole group is cleaved from the ribose in the DNA and generating abasic lactone and nitrosoindole. (B) Nitroindole-derived modification on DNA deoxyribose 7-nitroindole (d-(7-Ni)), 5-nitroindole (d-(5-Ni)). | ||
6. Benzophenone and acetophenone
Benzophenones contain a carbonyl carbon that undergoes intersystem crossing in high yields, making it a robust triplet photosensitizer for use in organic and biological chemistry.84 While a wide range of applications have utilized its light-induced C–C photocrosslinking properties,85 the Rentmeister group reported that benzophenone modification at N7-G in the context of RNA oligonucleotides can undergo a photocleavage reaction (Fig. 7).86 Upon irradiation with 365 nm light, benzophenone is cleaved from nucleobase through hydrogen abstraction mechanism. The photocaged guanosines used as a 5′-cap blocked the RNA's interactions with the translation initiation factor eIF4E and the RNA decapping enzyme DcpS.86 Benzophenones and acetophenones also can form UV-induced (C–C) cross-links with protein amino acids.84 For example, terminal deoxynucleotidyl transferase enzyme could be crosslinked to 3′-tails of DNA containing benzophenones and acetophenones on N4-dC's using 365 nm light (Fig. 8)87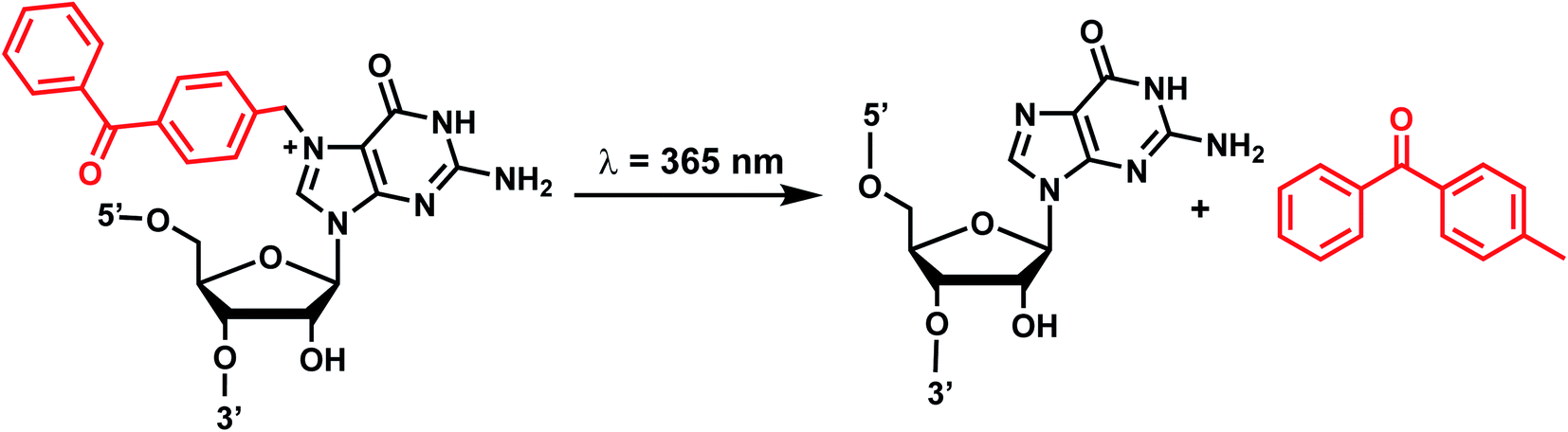 | ||
| Fig. 7 Schematic of benzophenone cleavage from N7-G in an RNA nucleoside upon light irradiation at λ = 365 nm. | ||
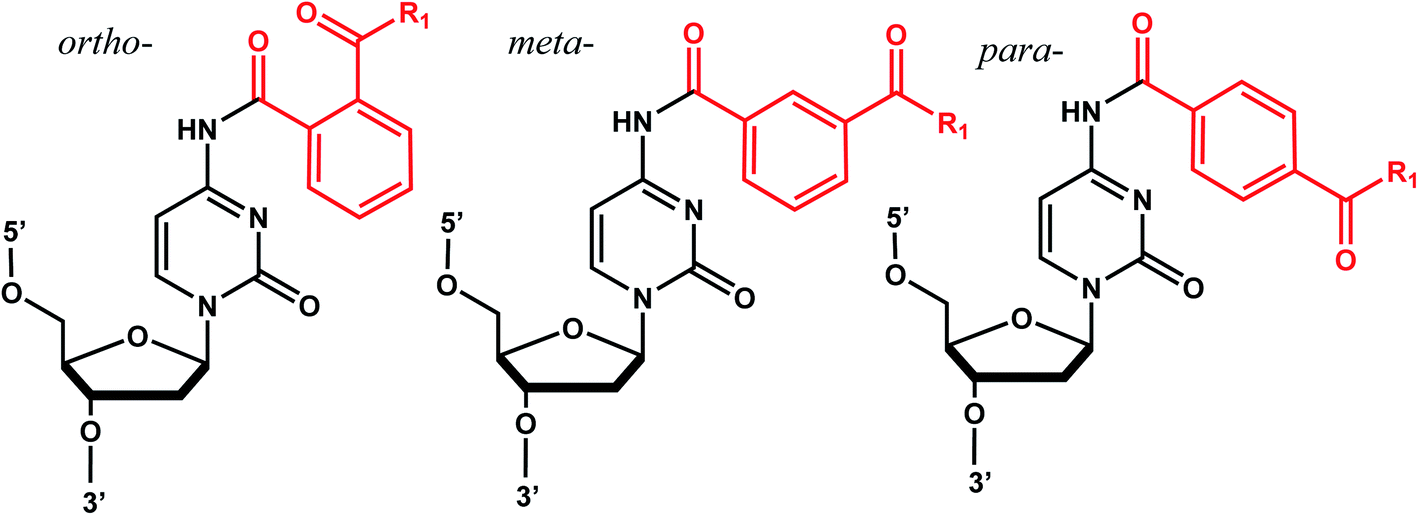 | ||
| Fig. 8 Acetophenone (R1= methyl) and benzophenone (R1= phenyl) groups as crosslinkers. Acetophenone and benzophenone modified N4-dC nucleosides. These modifications can be installed in ortho, meta, and para positions of the benzene ring. The C–C crosslinking occurs between the C of the carbonyl group in phenones and a Cα of the peptide bond. | ||
7. Coumarin
Coumarin-based groups are widely used photo-removable groups because of their large molar absorption coefficients at longer wavelengths, high release rates, and fluorescent properties. They are also capable of photo-crosslinking via [2 + 2] cycloaddition. One of the representative coumarin derivatives, (7-diethylaminocoumarin-4-yl)methyl (DEACM) was first introduced by Hagen et al.88 (Fig. 9). DEACM could be introduced in the backbone of the DNA or on N3-dT,72 O6-dG,89 and O4-dT (without and with a linker as in DEACM-O-Bn-dT)90,91 as well as on the γ-phosphate group of ATP (Fig. 9A).92 6-bromo-7-hydroxycoumarin-4-ylmethyl (Bhc) was first introduced by Tsien et al.93 Bhc has been employed in modifying C4-dC (as Bhcmoc or Bmcmoc),94 5′-position of the ribose in adenosine,95 and the phosphate backbone (Fig. 9B).96,97 The mechanism for photo-removal of these coumarin derivatives is through solvent-assisted photo-heterolysis (SN1 mechanism). Coumarin-modified oligonucleotides have been applied in various ways, for instance, in generating DNA strand breaks (e.g., by using DEACM linker in Fig. 9A) and in regulating DNA polymerization, translation and transcription. Inspired by Bhc, a series of photo-removable groups based on quinoline was reported by Guo et al. Among them, 8-bromo-2-diazomethyl-7-hydroxyquinolinyl (BHQ-diazo) showed the highest caging and restoration efficiency for the anti-thrombin aptamer HD1 (Fig. 9C).98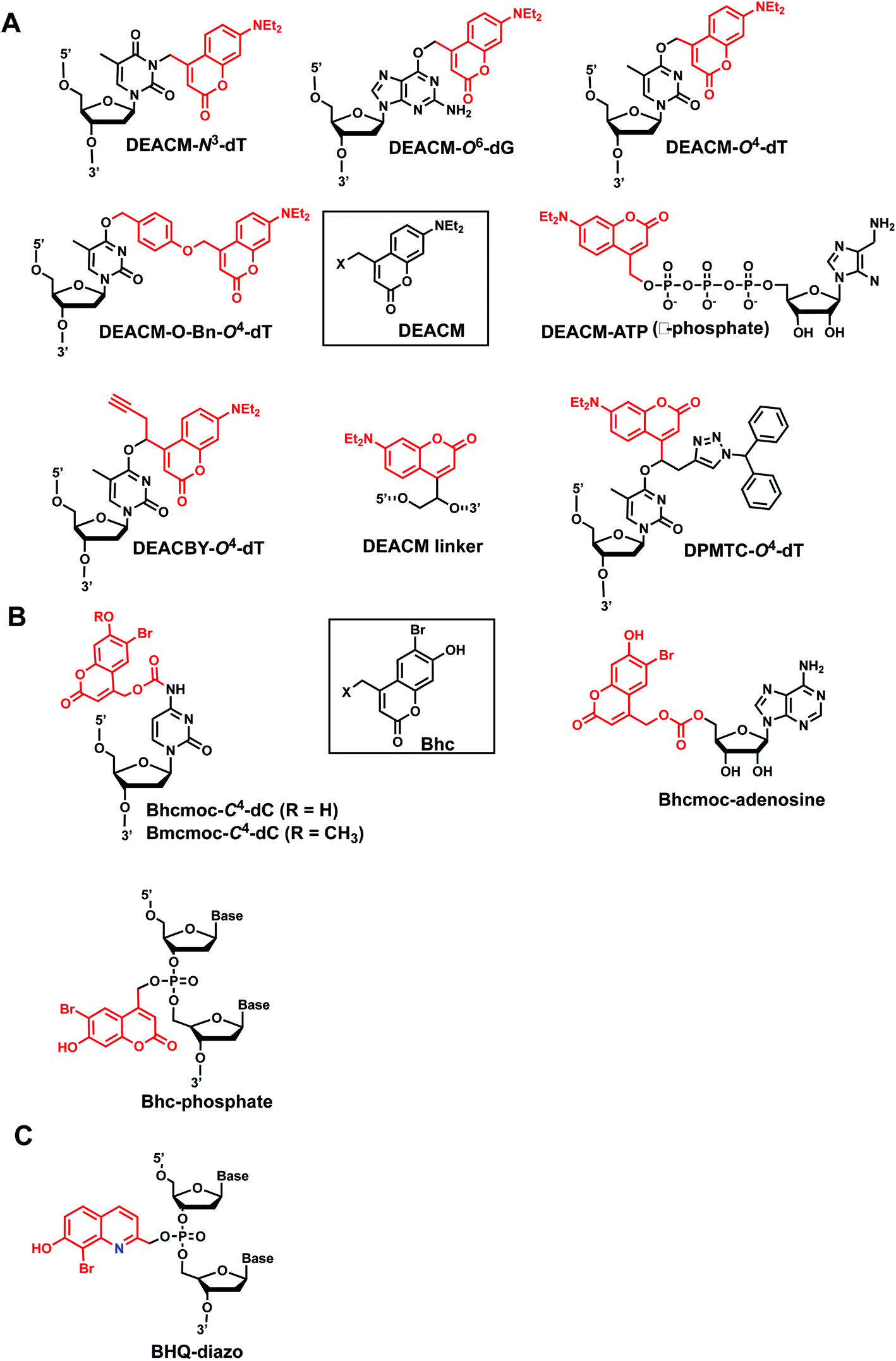 | ||
| Fig. 9 Representative coumarin-derived modification on oligonucleotides. (A) DEACM and (B) Bhc modifications in various positions of oligonucleotides. (C) Quinoline-based BHQ-diazo modification on the phosphodiester backbone. | ||
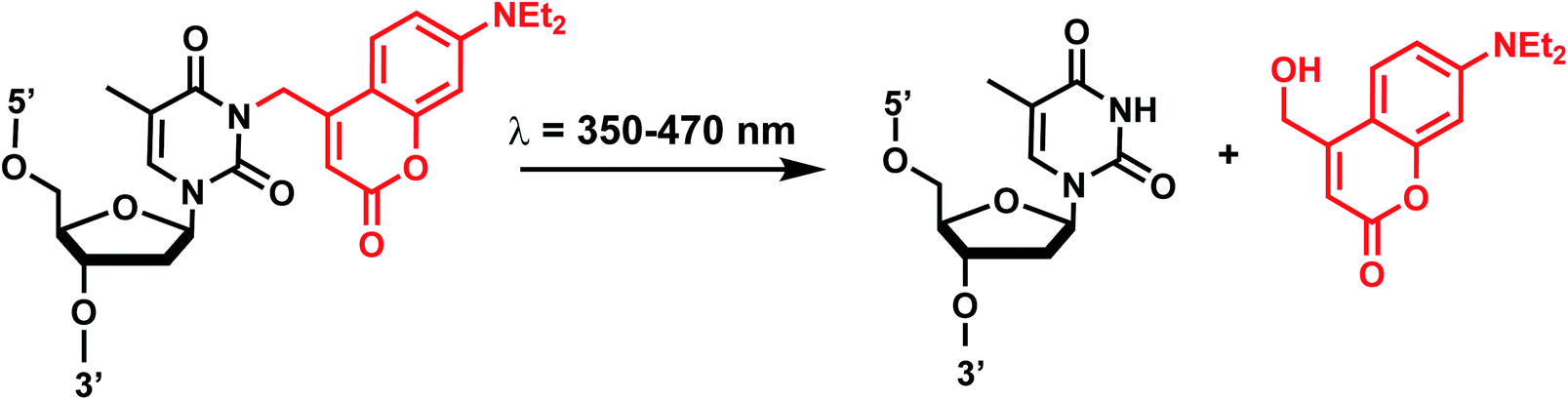 | ||
| Fig. 10 Coumarin photocleavage reaction. Upon irradiation with 350–470 nm light, the DEACM group is cleaved from the nucleobase dT, which restores the native structure. | ||
In addition to the photocleavage reaction, coumarin molecules can also undergo reversible photocrosslinking via a photo-induced [2 + 2] cycloaddition reaction similarly as psoralen (Fig. 11).101,102 The photocyclization reaction between coumarin and thymidine leads to fast and quantitative DNA interstrand crosslink (ICL) formation (>98%).101 The DNA crosslinks were generated by 350 nm irradiation whereas the reverse reaction, cyclo-reversion of the photo-adducts were achieved by 254 nm light (Fig. 11).101 ICL formation between a coumarin moiety containing a flexible two-carbon or longer chain and thymidine on the opposite strand completely quenches the fluorescence of coumarin, which allows for the monitoring of DNA crosslinking process over time via fluorescence spectroscopy.101 DNA crosslinking by coumarins shows a kinetic preference when flanked by an A:T base pair as opposed to a G:C pair.101
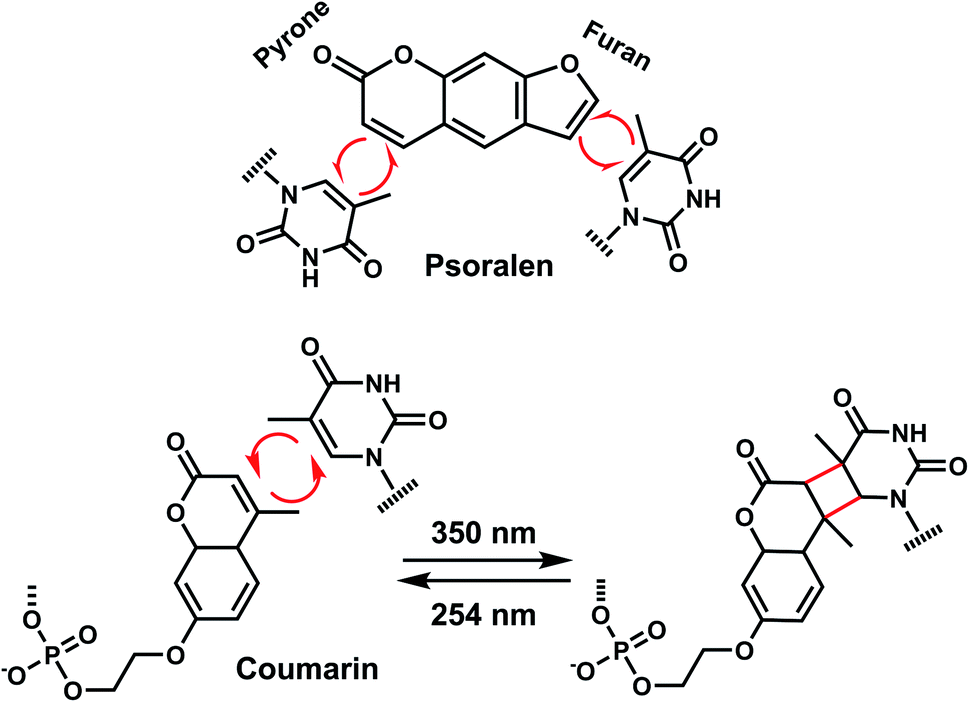 | ||
| Fig. 11 Light-induced reversible [2 + 2] cycloaddition of the Coumarin group. Schematic of the ICL formation upon light irradiation (λ = 350 nm), and reversible reaction (λ = 254 nm). | ||
8. Carbazole
The photo-crosslinking has been widely used to stabilize complexes with DNA by a covalent-bond formation.104,105 For instance, photo-crosslinkers such as psoralen (a member of furocoumarin family, related to coumarin, Fig. 11) can produce DNA inter-strand crosslinks via [2 + 2] cycloaddition reaction when irradiated by UV-A (365 nm) either via their furan or pyrone photoreactive site. The crosslinks can be reversed upon irradiation 254 nm shorter wavelength. Although psoralen is widely used as a thymine-selective photo-crosslinker in biological studies, there are limitations such as requiring a TpA step in the sequence and causing photodamage to DNA by forming pyrimidine photodimers upon cycloreversion that uses UV-C (254 nm).104,106 To alleviate these issues, Fujimoto et al., reported 3-cyanovinylcarbazole nucleoside (CNVK) as a reversible photo-crosslinker that can photo-crosslink to pyrimidine base located 5′ to the complementary base through [2 + 2] cycloaddition with 385 or 365 nm irradiation (Fig. 12).106 The resulting photo-adducts can be uncrosslinked by 312 nm irradiation without causing DNA damage.106 CNVK shows higher reactivity compared with psoralens, showing 97% yield with 1 s of longer wavelength UV-A light (366 nm).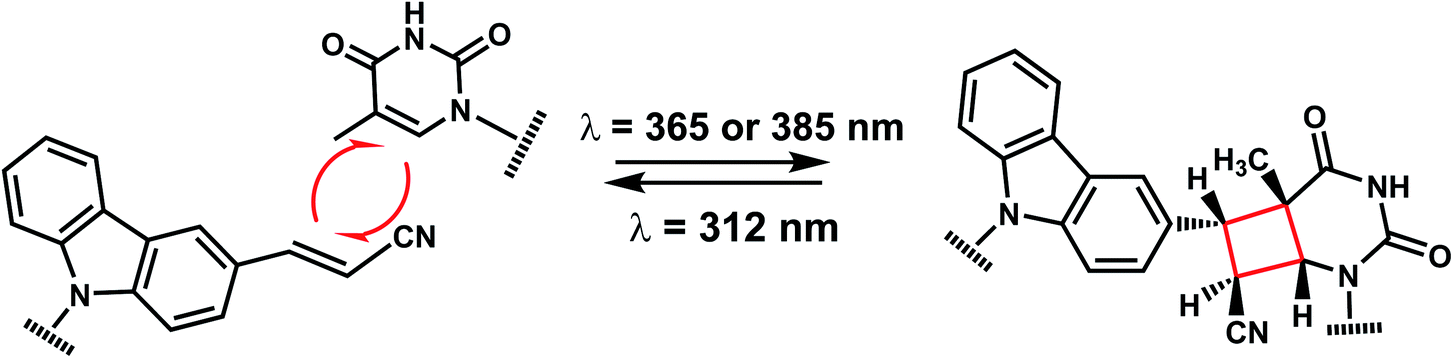 | ||
| Fig. 12 Reversible [2 + 2] reaction in CNVK. 3-cyanovinylcarbazole nucleoside (CNVK) can undergo rapid photo-crosslinking to the complementary strand at one wavelength. Rapid reversal of the crosslink is also possible at a second wavelength. | ||
CNVK nucleoside was further developed to improve photoreactivity. 3-Cyanovinylcarbazole-modified D-threoninol (CNVD) which has a flexible structure showed enhanced photoreactivity for the pyrimidine base at the −1 position in the complementary strand (Fig. 13): the photoreactivity of CNVD was 1.8- (for crosslinking with dT), 8- (with dC), and 2.8-fold (with U in RNA) greater than that of CNVK.107
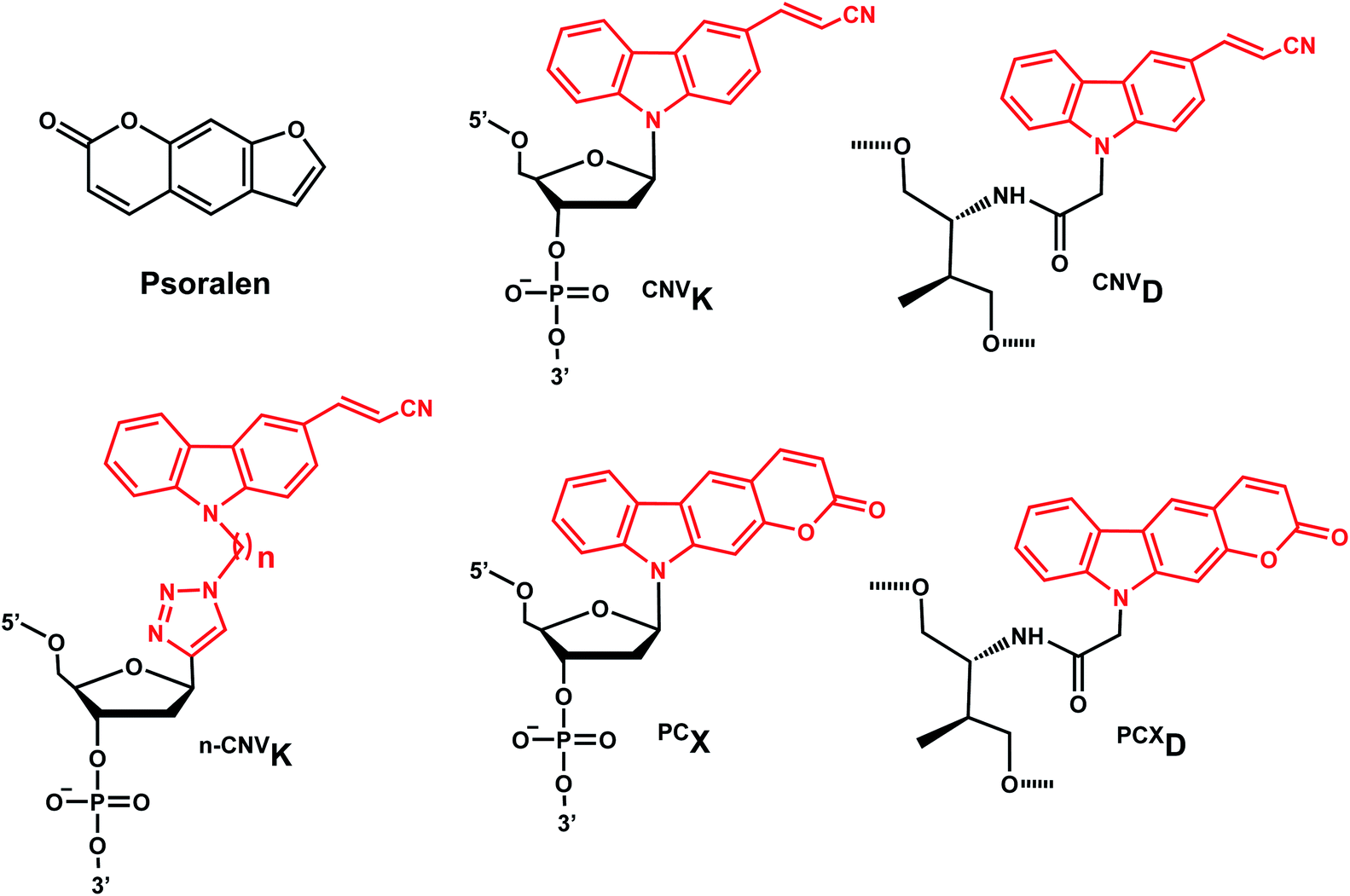 | ||
| Fig. 13 Carbazole derivatives. Both CNVK and PCX carbazole derivatives are inspired from psoralen. Either the vinyl in CNVK, CNVD, n-CNVK or the pyrone in PCX, and PCXD undergo reversible [2 + 2] photoreaction. | ||
PCX, pyranocarbazole nucleoside was developed to use visible light instead of UV-A, therefore less toxic and damaging (Fig. 13).14 Recently D-threoninol version of the PCX photo-crosslinker (PCXD) was reported, showing a higher photoreactivity than PCX (Fig. 13).8 In addition, n-CNVK with variable linker lengths (n = 2–5) was developed to use with click chemistry.108 This probe is capable of photo-crosslinking with pyrimidine bases at locations other than the −1 position (Fig. 13).108
Fujimoto et al. reported that the rate constant of the photo-crosslinking reaction of CNVD is 0.106 s−1 which is comparable to CNVK (0.059 s−1). Within the same sequence, psoralen showed a much slower photo-crosslinking rate (0.003 s−1) using the same wavelength (365 nm).110 The reaction rate constant of PCXD with cytosine is 4.3-fold larger than that of PCX.8
Fujimoto group reported the use of CNVK photocrosslinking in antisense DNA technology: photocrosslinkable antisense oligonucleotides containing CNVK can regulate GFP expression in a sequence-specific manner only after 10 s photocrosslinking with 365 nm light in HeLa cells. In a recent study, they investigated and compared the photo-crosslinking rate and its inhibitory effect including CNVD, CNVK, and psoralen on gene expression.110 The inhibitory effect on gene expression was the highest with CNVD (93%), while no inhibitory effects were observed with psoralen.
In another recent study, they regulated the DNAzyme activity by photoirradiation through the photochemically reversible formation of covalent bonds.109 While photo-crosslinking using 365 nm completely abolished the activity of the DNAzyme harboring CNVK, uncrosslinking using 312 nm irradiation restored the activity.109 CNVK also was shown to accelerate in vitro DNA strand displacement reactions, which may be employed for rapid-response DNA nano device technology using higher-order DNA structures.117
9. Vinyl derivatives
As with the vinyl groups in 3-cyanovinylcarbazole (CNV) modifications (Section 8), other DNA/RNA modifications containing vinyl group have been reported with photo-crosslinking properties (Fig. 14A). In addition, some vinyl-derivatives can also undergo cis- to trans-photoisomerization around the C–C double bond in the absence of a crosslinkable partner; this then alters the orientations of the attached moieties and subsequently the DNA/RNA structures (Fig. 15A).118 Here, we discuss the vinyl derivatives reported for these two reaction categories.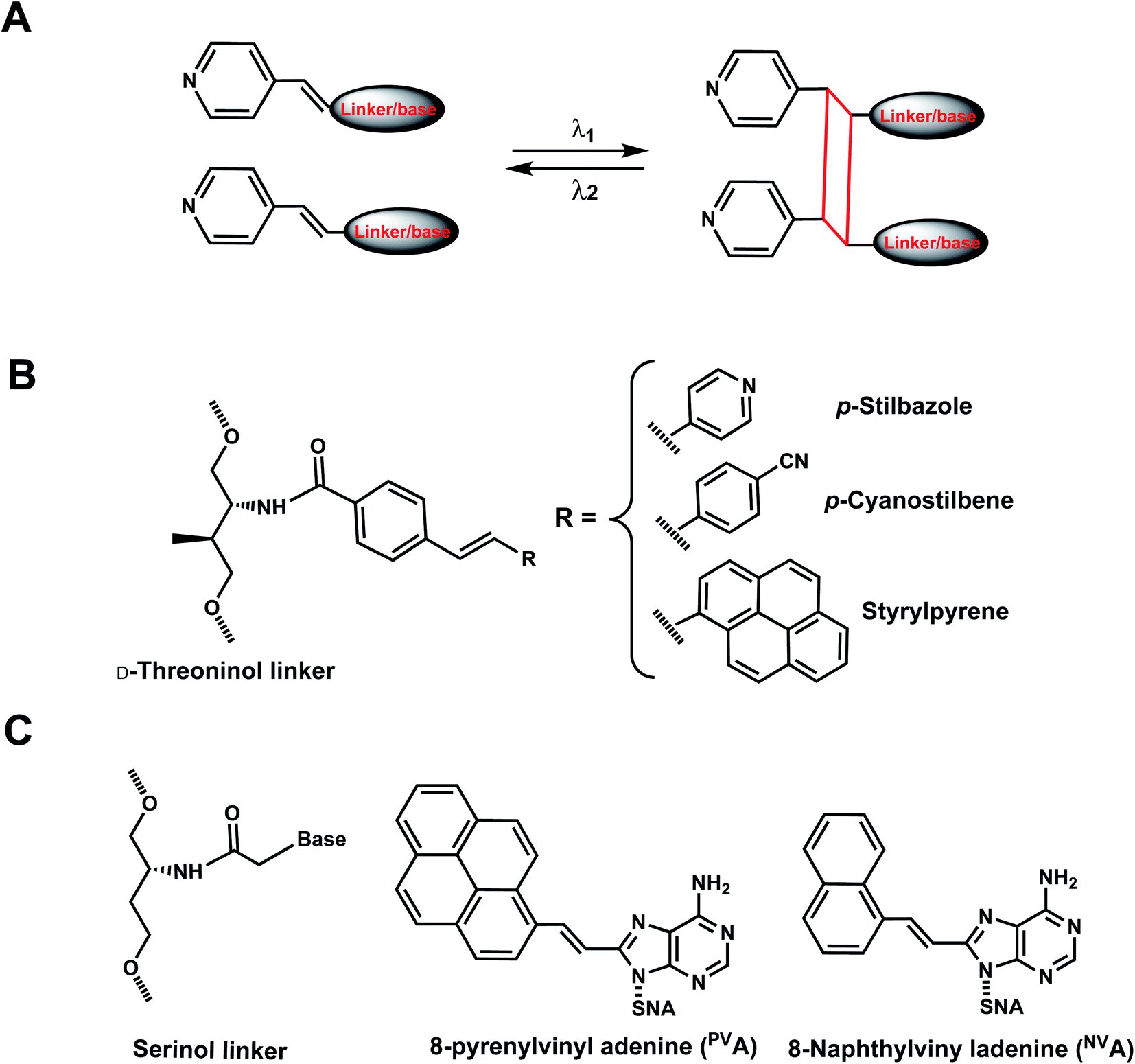 | ||
| Fig. 14 Vinyl derivatives for light-induced [2 + 2] cycloaddition. (A) Schematic of the [2 + 2] photo cycloaddition and its reverse reaction (cycloreversion) upon light irradiation in two different wavelengths. Usually, the wavelength for cycloaddition (crosslinking) is longer than that for the reverse reaction (un-crosslinking). (B) D-Threoninol linkers can be used with p-stilbazole, p-cyanostilbene, and styrylpyrene as a part of DNA backbone. (C) Serinol nucleic acid (SNA) linkers can also be used to introduce PVA and NVA to the oligonucleotides. | ||
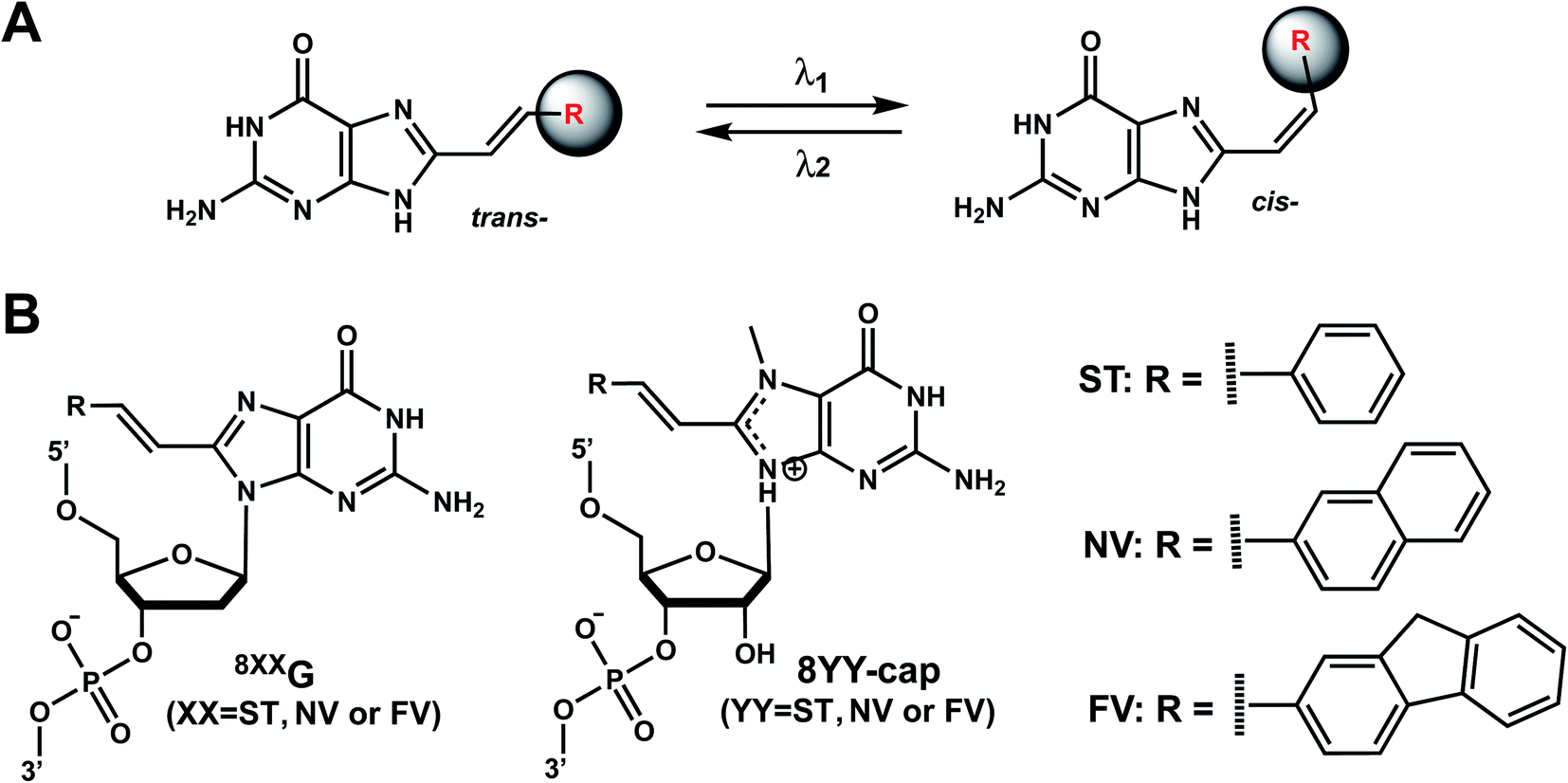 | ||
| Fig. 15 Vinyl group derivatives undergo reversible trans–cis photoisomerization. (A) trans-to-cis and cis-to-trans photoisomerization are catalyzed by two different wavelengths. (B) 8-styryl (8ST), 8-naphthylvinyl (8NV), and 8-fluorenylvinyl (8FV) modifications can be attached to C8-dG in DNA (“8xxG”) or to C8-methylG-cap in mRNA cap (“8YY-cap”) and can undergo trans to cis photoisomerization. | ||
More recently, 8-pyrenylvinyl adenine (PVA) was employed as a way to control duplexation between serinol nucleic acid (SNA) and RNA (Fig. 14C). When incorporated in SNA in adjacent positions, PVA could undergo intrastrand photodimerization by 455 nm light, which abolished the duplexation with a complementary RNA. However, the crosslinks could be reversed with cycloreversion catalyzed by 340 nm light.122 It is noteworthy that both the forward and reverse reactions could be carried out to completion at constant room temperature.122 8-Naphthylvinyladenine (NVA) is also used in SNA for crosslinking/uncrosslinking reaction, similarly as PVA, but uses a shorter wavelength of light than PVA: intrastrand crosslink by irradiation with 340–405 nm light and reverse reaction by ≤300 nm light. In an SNA strand with adjacent NVA and PVA residues, irradiation with 405–465 nm led to intrastrand crosslink, which was reversed by irradiation with ≤340 nm light.123 In all these cases, the intrastrand photo-crosslinking destabilize SNA/RNA duplexes, resulting in duplex dissociation while its cycloreversion led to duplex formation.123 With these NVA/NVA and NVA/PVA photo-switches, the hybridization states of SNA/RNA duplexes could be independently controlled by using light of varying wavelengths.123
Reaction characteristics. Strylpyrene pairs (Spa and Spb) introduced as a part of D-threoninol linker in the opposite strands of DNA duplex could also undergo [2 + 2] photocycloaddition using visible light (λ ≈ 455 nm), but gave two diastereomers as a result of the rotation of the styrylpyrene residues.121 The reaction progress and stacking of the Sp dimers could be monitored using UV-Vis absorption spectroscopy. Upon visible light irradiation of SPa/SPb at λ = 455 nm, the absorption band at λ ≈ 390 nm decreased with irradiation time and almost disappeared after 60 min of irradiation while new bands concurrently appeared at λ = 338 and 354 nm. The progress of photocycloaddition was apparent also due to changes in color and fluorescence of the solutions from colored to colorless.121
PVA-containing oligonucleotide features an absorption band at around 400 nm (characteristic of vinylpyrene) and upon irradiation with 455 nm light immediately decreased and almost disappeared after 2 min using 203 mW cm−2 power.122 Simultaneously, new bands appeared at 270 and 354 nm, which correspond to absorption bands of alkylpyrene, a product of crosslinking. Upon irradiation of the crosslinked product with 340 nm light, the initial absorption bands were restored, indicating the recovery of PVA monomers.122 The crosslinking and uncrosslinking reactions were rendered complete after 1 h (irradiation at 455 nm) and 15 min (340 nm), respectively.122
In the case of NVA, irradiation with 405 nm light for 60 s led to the disappearance of the absorption band around 360 nm, and irradiation with 300 nm light of this photo-adduct led to the cycloreversion: 61% of the initial absorption band was recovered within 120 s.123 Four hybridization states of two SNA/RNA duplexes containing either the NVA/NVA pair or NVA/PVA could be orthogonally controlled using different wavelengths of light.123
Thermodynamic or structural characteristics. Photocrosslinking between Sp groups is thermodynamically stabilizing for DNA duplexes, as expected. Melting measurements revealed that both diastereomer products after crosslinking had melting temperatures significantly higher (22–25 °C) than that of the uncrosslinked dimer, SPa/SPb. Melting measurements also indicated that the crosslink had been reversed upon cycloreversion.121
PVA slightly destabilized the duplexes when compared with the unmodified SNA/RNA duplex.122 On the other hand, NVA in SNA/RNA duplex slightly increased Tm compared with the control SNA/RNA123while NVA intrastrand photo-crosslink caused severe destabilization of a SNA/RNA duplex containing NVA or PVA: this resulted in the melting of the duplex to single strands. The reverse reaction, cycloreversion, led to the restoration of the duplexes.123
Biological applications. NVA and PVA were studied as a part of SNA/RNA as mentioned above as reversible crosslinkers that stabilize the SNA/RNA duplexes.121,123,124
Reaction & thermodynamic characteristics. In 12-bp duplexes containing 8STG, 8NVG, or 8FVG, the trans forms of the dG modifications were photoisomerized to the corresponding cis forms when irradiated for 5 min with 370, 410, and 420 nm light with 86%, 63%, and 77% conversion efficiencies. In addition, subsequent irradiation for 2 min at 254, 290, and 310 nm yielded the trans forms with 94%, 87%, and 77% conversion efficiencies, respectively.125
Thermal stability study of 12-bp duplexes containing 8STG, 8NVG, or 8FVG showed both the cis and trans isomers were thermally stable. 8STG-containing duplex showed the Tm of the trans form was 7.9 °C higher than that of the cis form. This is probably due to a difference in the steric hindrance of the benzene ring with its neighboring nucleobase and backbone. In contrast, the Tm of the trans forms of 8NVG- and 8FVG-containing duplexes were only 1.6 and 1.4 °C lower than the cis forms, respectively.125 This may indicate that the bulky substituents, naphthalene and fluorene in 8NVG and 8FVG, may cause serious steric hindrance with the backbone, even in the trans form.125
Biological applications. The trans to cis isomerization of vinyl derivatives can regulate oligonucleotide duplex hybridizations125 and has been applied in various biological applications.101,127 For example, The mRNA containing the 8NV-cap at the 5′-end could be switched between a translating (ON) state when in cis form and a non-translating (OFF) state when in the trans form in a reversible fashion by alternately irradiating with 410 nm or 310 nm light.118 In addition, 8ST-cap can reversibly regulate translation by controlling the interaction with eukaryotic translation initiation factor eIF4E through its cis–trans photoisomerization in living mammalian cells as shown in PC12 neuronal cell line through its neurite expansion and contraction.127 Furthermore, trans (E)-to-cis (Z) photoisomerization of the 8STG was utilized by Zhou et al. to reversibly switch between a B-form and Z-form DNA by alternately illuminating with monochromatic 254 nm and 365 nm light.128
10. Azobenzene
Azobenzenes (AzoB) are the most widely used reversible photoswitches in oligonucleotides due to their high quantum yields, fast switching, low rate of photobleaching, easy synthesis, high fatigue resistance (high repeatability of photoswitching), and good thermal stability.12 Irradiation with UV light converts planar trans-N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond with zero dipole to non-planar cis isomer (dipole moment of ∼3 D), which can be accelerated by heat (Fig. 16).129 The reverse cis to trans isomerization can be achieved by visible light.129 AzoB was first introduced in nucleic acids as a part of a flexible backbone linker based on propionic acid by Asanuma et al.130 and was used to regulate duplex131 and triplex5,132 DNA formation.
N bond with zero dipole to non-planar cis isomer (dipole moment of ∼3 D), which can be accelerated by heat (Fig. 16).129 The reverse cis to trans isomerization can be achieved by visible light.129 AzoB was first introduced in nucleic acids as a part of a flexible backbone linker based on propionic acid by Asanuma et al.130 and was used to regulate duplex131 and triplex5,132 DNA formation.
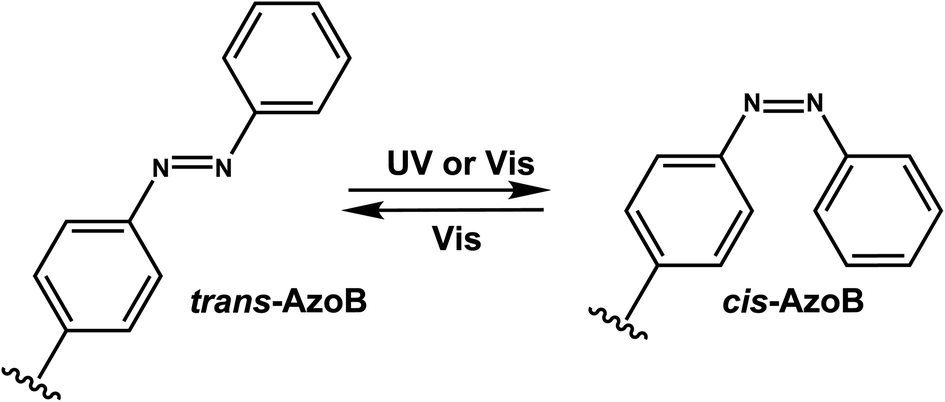 | ||
| Fig. 16 Schematic of reversible azobenzene trans–cis photoisomerization with different wavelengths. This conversion occurs through reversible rotation of planar trans NN to non-planar cis isomer. λ1 is often in the UV-A range, and λ2 in the visible range. | ||
To enatioselectively introduce AzoB into DNA or RNA, optically pure D-threoninol and L-threoninol linkers were employed. D-Threoninol-linked AzoB (D-tAzo)133 (Fig. 17A) induces larger changes in Tm between the trans and cis isomers than L-threoninol (L-tAzo) (Fig. 17B) and is now commercially available, making it one of the most commonly used forms of AzoB in DNA/RNA.12
 | ||
| Fig. 17 Azobenzene derivatives. Different azobenzene derivatives were developed with different linkers and base analogs and incorporated to different segments of oligonucleotides. | ||
Introducing methylthio-modification at para-position of azobenzene induced a bathochromic (red-) shift of absorption maximum, allowing trans-to-cis isomerization by 400 nm visible light (Fig. 17C). Additional methylation at the ortho-position of the distal benzene ring enhanced the stacking interaction of trans-azobenzene while further destabilizing cis-AzoB (Fig. 17C). This in turn raised the Tm of the trans-form and lowered the Tm of the cis-form, and the resulting large ΔTm enhanced photoregulatory efficiency. More recently, an AzoB modified with a highly branched secondary alkylthio group was incorporated into DNA via an L-threoninol scaffold for which the photoisomerization was carried out by visible light (λ = 400 nm for the trans-to-cis reaction with 58% efficiency and λ = 520 nm for the reverse reaction) (Fig. 17C). In contrast to other AzoB, these modifications also showed that the trans-form is duplex-destabilizing than the cis-form (Fig. 17C).13
 | ||
| Fig. 18 Spiropyran modification on oligonucleotides. (A) Spiropyran-derived DNA backbone linker can undergo reversible isomerization through ring opening/closing with light or heat. (B) DNA modifications synthesized by the Heckel group with different photophysical and thermodynamic properties. | ||
More recently, several modifications have been made to improve the modest photoisomerization efficiency of D-tAzo (e.g., 30% of cis-isomer at 37 °C with irradiation with 365 nm (ref. 134)) or L-tAzo.13 For instance, Liang et al., introduced AzoB through R-glycerol linker (R-gAzo) which has improved photoisomerization efficiency to 70–80% at room temperature (Fig. 17D).135 Later, Asanuma, Heckel, and co-workers developed p-Azo and m-Azo C-nucleosides photoswitches which exhibited complete photoisomerization at room temperature (Fig. 17E and F).136
The AzoB group was also incorporated as a part of the backbone in DNA/RNA: Tang et al. introduced 4′-bis(hydroxymethyl)-azobenzene to dumbbell hairpin antisense strand complementary to target RNA at the loop position to reversibly control the stability of the hairpin structure via UV or visible light (Fig. 17G).137
Desaulniers et al., developed photo-regulatable siRNAs with internal azobenzene derivative spacers (siRNAzos) (Fig. 17H).138–140 A related, tetra ortho-chlorinated azobenzene-containing siRNAs (Cl-siRNAzos) shifted the trans to cis conversion wavelength to 660 nm (red-shift) and was applied in cell culture gene inactivation studies (Fig. 17H).141
AzoB can also be introduced as a part of the purine ring as 2-phenyldiazenyl-substituted 2′-deoxyadenosine (dAAzo) and 2′-deoxyguanosine (dGAzo) (Fig. 17I and J).10 GAzo has been developed by Ogasawara and used as a photoresponsive 5′-cap of mRNA in vivo to control protein expression.142
N bond and the formation of the cis isomer. The transition associated with the absorption at 430 nm is related to the cis to trans isomerization. These properties can be influenced by the substitution of the azobenzene core structure and the choice of solvent.The impact of AzoB on the thermal stability of the DNA also depends on the stereochemical environment of the group. For instance, the trans-form of D-tAzoB is more stable than that of L-tAzoB because D-threoninol prefers a clockwise winding, as does the DNA double helix. Cis-form is also more destabilized in D-tAzoB than in L-tAzoB, resulting in a larger trans-to-cis stability difference (ΔTm) for D-tAzoB.
Newer applications include siRNAzos151 in gene silencing in cells and in vivo: siRNAzos use AzoB as internal spacers within the sense strand in HeLa cells.138 siRNAzos in the cis form would distort the siRNA helix, thus rendering it non-functional, but irradiation with UV light would make it functional and lead to gene silencing.138 siRNAzos also has been used in the 3′-end of the sense strand with improved nuclease resistance for gene silencing applications.139 Red-shifted Cl-siRNAzos were used in cell culture with reversibility.141 Additionally, dGAzo developed by Ogasawara was used as a photoresponsive 5′-cap demonstrating the impact of the distal aromatic ring on the dGAzo in the development of double-headed zebrafish by controlling the expression of squint protein.142
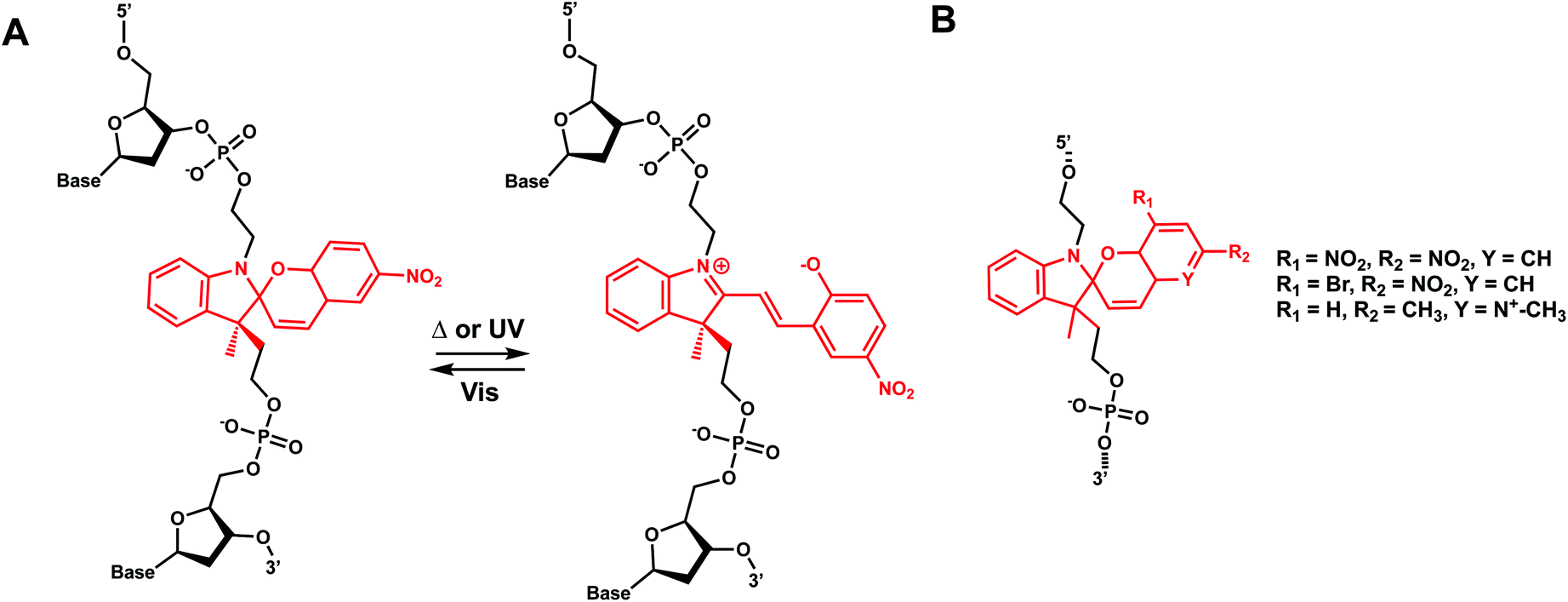
11. Spiropyran
Hirshberg and Fischer reported the first photochemical reactions and photochromic phenomena of spiropyrans.152 Spiropyrans are unique among the broad spectrum of photoswitches, due to the range of stimuli (e.g., temperature, visible light, mechanical forces, and solvent effects) able to induce its reversible isomerization.153 Spiropyran consists of orthogonally orientated indoline and chromene moieties, joined by a quaternary carbon atom and thus is largely nonplanar.Early work on spiropyran-modification on DNA oligonucleotides met with various obstacles including fast hydrolysis of spiropyrans in aqueous buffer solutions and the loss of the photoswitching ability in DNA.6,154 This problem was largely alleviated when Heckel et al. incorporated spiropyran as a part of the DNA backbone using phosphoramidite chemistry and solid-phase synthesis (Fig. 18).6 This photoswitch was also reported to work when incorporated at the 5′-end of homothymidine oligonucleotide in duplex DNA.153
 | ||
| Fig. 19 Light-induced reversible [2 + 2] cycloaddition of the diarylethene group. Schematic of the ring formation upon light irradiation at UV range, and reversible reaction ring opening upon light irradiation at visible range. | ||
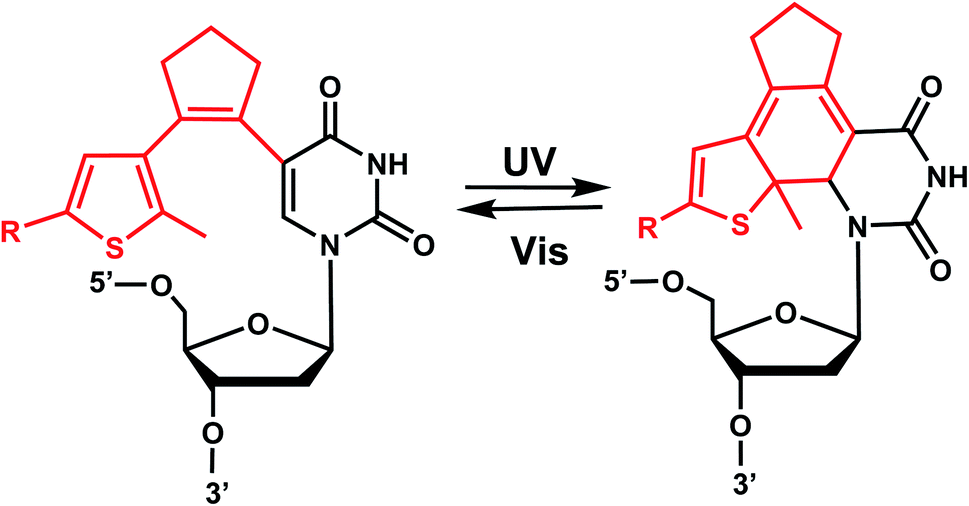
12. Diarylethene group
Diarylethenes (DAE) are known for excellent photochromic properties, such as negligible thermal relaxation, spectral tunability, and strong absorption bands upon photoconversion as well as high fatigue resistance against multiple photoswitching cycles.156,157 Diarylethenes containing thiophene moieties and a cyclopentene ring are a special class of stilbene-type structures in which the ortho-hydrogens are substituted to suppress irreversible oxidation after photocyclization of the cis isomer. Typically, the incorporated aryl rings are replaced by heterocycles to elongate the lifetime of the closed form, and the ethene moiety is often embedded in a small ring to prohibit cis–trans isomerization (Fig. 19).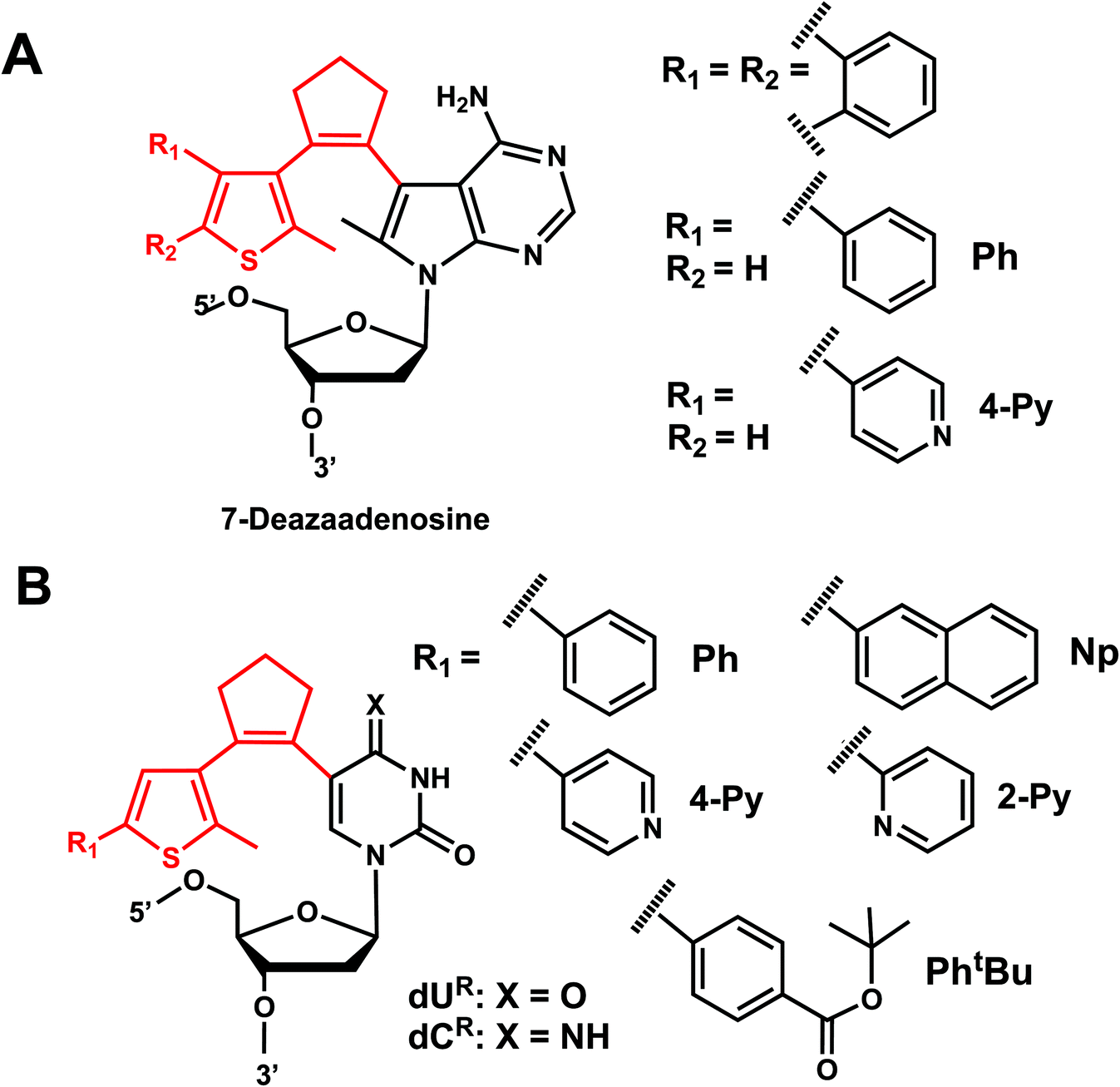 | ||
| Fig. 20 Diarylethene group modification on oligonucleotides. (A) Diarylethene-derived modifications on 7-deazaadenosine nucleoside analogue. (B) Diarylethene-derived modifications on pyrimidine nucleosides. | ||
Diarylethene derivatives were first introduced to oligonucleotides by Jäschke's group through 7-deazaadenosine.158 Diarylethene under the irradiation of different wavelengths (250–370 nm) undergoes an electrocyclic rearrangement, generating strongly colored closed-ring isomers, whereas visible light (>400 nm) triggers the cyclo-reversion to the colorless opened-ring form which is thermally stable (Fig. 19).158
Originally reported as a photoswitchable reaction in non-aqueous solvent,158 the relatively low efficiency of photoisomerization in aqueous solvents had to be optimized using various substituents on the thiophene ring on 7-deazaadenosine which resembles purine159,160 or on a (deoxy)uridine as a pyrimidine analog (Fig. 20).161,162 In comprehensive testing of 13 different substituents on dU and dC nucleosides, dU with 2-pyridyl (2Py) and tert-butylester-phenyl (PhtBu) were found to be the best in the photoisomerization efficiency and thermal and photochemical stability (Fig. 20).163 In particular, the photochromism (e.g., quantum yields, composition of the photostationary states, thermal and photochemical stability, and reversibility) of the modified dU with 2-Py or PhtBu was maintained in the environment of the single-stranded oligonucleotide, and for PhtBu even in the duplex. These modifications were also shown to be useful in controlling transcriptional activation.163
Concluding remarks
Photoconvertible groups offer a convenient way to alter molecular structures in a spatially and temporally controlled manner using light as the reaction initiator. Ideal photoreactive groups for biological applications would feature fast and complete photoconversions under mild, physiologically relevant conditions and would be capable of multiplexed, orthogonally controllable reactions (e.g., by choosing different wavelengths of trigger light). In recent years, significant strides have been made in the availability and applicability of photoreactive oligonucleotides. Here, we compiled a list of currently available photoreactive groups for oligonucleotides to regulate DNA/RNA structure and function for diverse biological applications (Table 1). The photoreactions are either irreversible (e.g., cleavage) or reversible (e.g., crosslink, isomerization, and intramolecular cyclization reactions), each with their own strengths but also limitations. For instance, reactions that use UV light may cause DNA or tissue damage and can interfere with the excitation/emission of fluorescent reporters used in in vitro/in vivo studies. Relatively moderate or low photoconversion yields (reaction completeness) of photoreactive groups also remain as hurdles. Expanding the array of available photoreactive modifications with enhanced photostability, biocompatibility and tunability would be exciting future directions.Continued research and development of light-convertible oligonucleotides promise to provide powerful tools for studying complex genetic mechanisms that uses DNA/RNA as their platforms including transcription, replication, and repair. For example, for DNA repair pathways such as NER where bulky lesions are recognized and repaired, o-nitrobenzyl, p-hydroxyphenacyl, and coumarin-related modifications may be used as a bulky DNA lesion surrogate which can be readily switched on and off for various structural and mechanistic studies. Further improvements of photoconvertible nucleic acids to achieve higher photo-reaction efficiency and tunability of light at longer wavelengths may expand the applicability of photoreactions for biological investigations and modulation.
| # | Photoreaction type | Photoreactive group | Structure | Reaction wavelength (nm) | Reversible?b | Tm effect | Photoreaction time scale | Nucleic acid position | Biological application |
|---|---|---|---|---|---|---|---|---|---|
| a Some derivatives (e.g., D-tAzo) are commercially available.b Wavelengths in parentheses indicate those for the reverse, uncrosslinking reactions.c Wavelengths for (un)crosslinking reaction.d Wavelengths for cis–trans/trans–cis isomerization.e N.B. All modifications can be incorporated in solid-phase oligonucleotide syntheses (e.g. via phosphoramidite chemistry) unless otherwise noted (e.g., DMNEC as a part of oNB, TEEP-OH, and DPMTC as a part of coumarin). | |||||||||
| 1 | (I) Photocleavage – irreversible | o-Nitrobenzyla (& NPE, NPM, NPOM, PNVOM, NDBF, etc) | 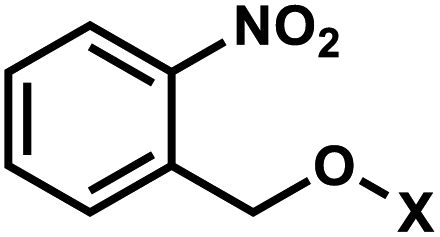 |
345–420 | No | ↓ | sec–min | Bases, backbone, ribose | Variety of biological systems (see text) |
| 2 | (I) Photocleavage – irreversible | p-Hydroxyphenacyl (& HBT) | 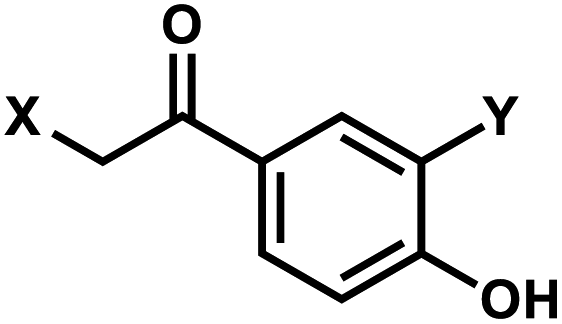 |
295–405 | No | ↓ | msec–sec | G, T bases | Control of antisense RNA annealing |
| 3 | (I) Photocleavage – irreversible | TEEP-OH | 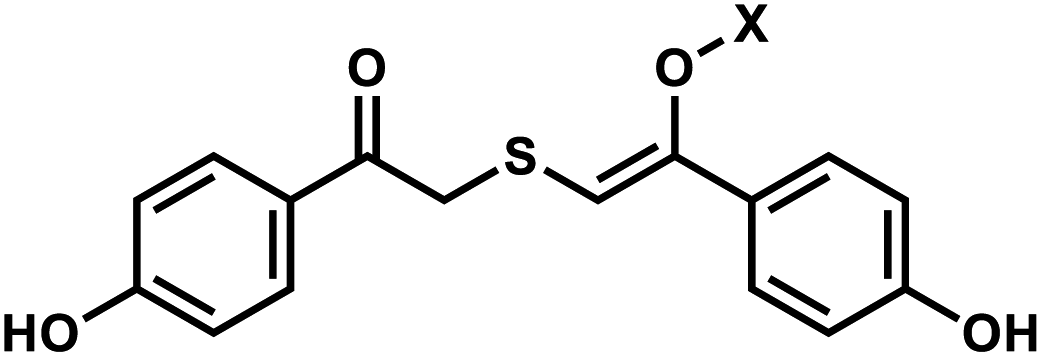 |
300–365 | No | ND | min | Phosphate backbone on DNA | Regulation of DNAzyme activity |
| 4 | (I) Photocleavage – irreversible | Aryl sulfide | 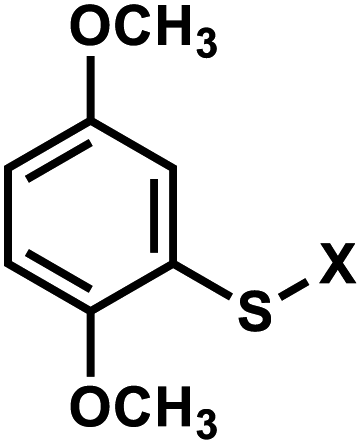 |
350 | No | ↓ | μsec–min | U, T bases | Control of RNA riboswitch folding |
| 5 | (I) Photocleavage – irreversible | Nitroindole | 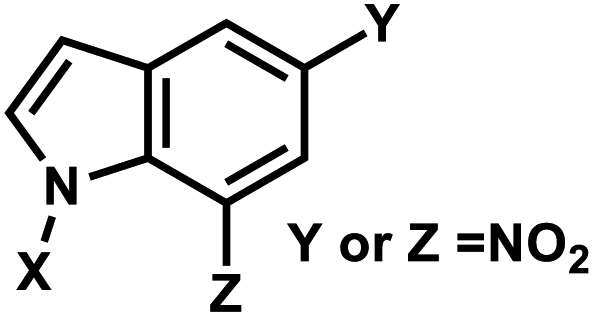 |
350 | No | ↓ | min | Deoxyribose as a purine analogue | Catch and Release DNA Decoys |
| 6 | (I) Photocleavage – irreversible | Benzophenone, acetophenone | 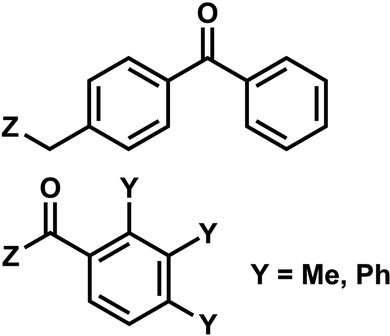 |
365 | No | ND | min | G, C bases | Regulation of mRNA translation; photocrosslink with bound proteins |
| 7 | (I) Photocleavage – irreversible, (II) Intermolecular photocrosslinking via [2 + 2] cycloaddition – reversible | Coumarin (& DEACM, Bhc; quinoline. Cf. psoralen) | 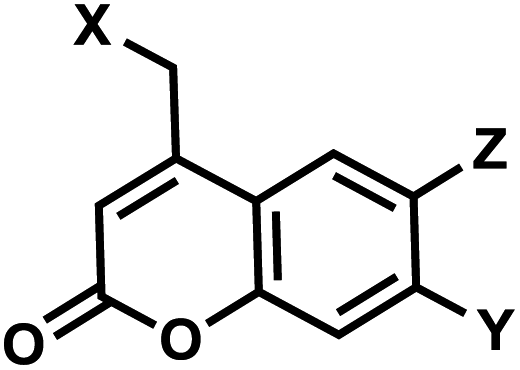 |
350–470 | No for cleavage; Yes for crosslinking (254) | ↓ | sec–min | G, T bases, phosphate, backbone linker | Catch-and-release DNA decoy, regulation of mRNA caging, transient DNA polymerization, aptamer |
| 8 | (II) Intermolecular photocrosslinking via [2 + 2] cycloaddition – reversible | Carbazolea (&CNVK, CNVD, PCX, and PCXD) | 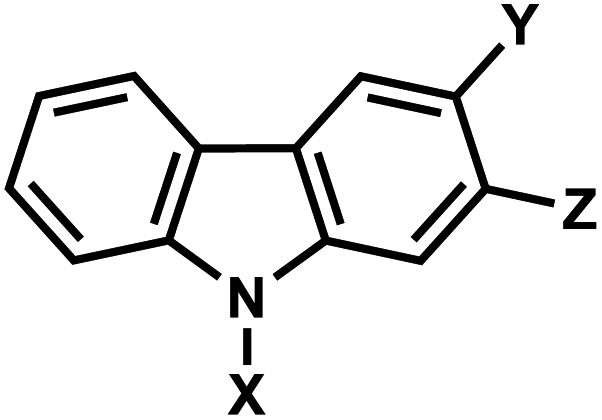 |
365–450 | Yes (312) | ↓↑ | sec–min | Nucleoside | RNA FISH, plasmid labeling, antisense DNA, regulation of DNAzyme activity |
| 9 | (II) Intermolecular photocrosslinking via [2 + 2] cycloaddition – reversible, (III). Cis–trans photoisomerization – reversible | Vinyl-derivative (& stilbazole, cyanostilbene, styrylpyrene; 8ST, 8NV and 8FV) | 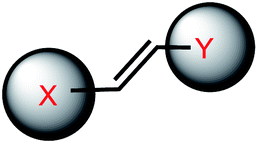 |
340–465c, 370–420d | Yes (≤300–340c) (254–310d) | ↓↑ | min–hour | Nucleoside, G base | DNA hybridization, regulation of gene expression |
| 10 | (III) Cis–trans photoisomerization – reversible | Azobenzenea | 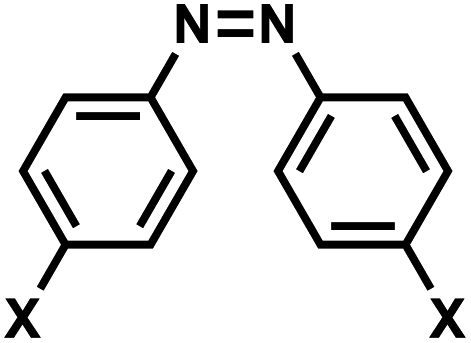 |
365 | Yes (400–420) | ↓↑ | sec–min | Nucleoside backbone linker, G, A bases | Variety of biological systems (see text) |
| 11 | (IV) Intramolecular photocyclization – reversible | Spiropyrans | 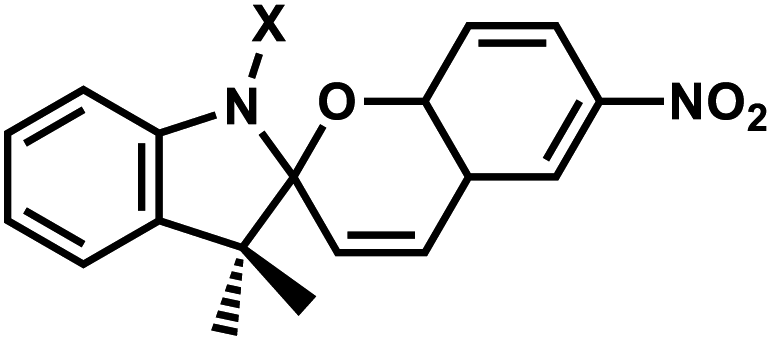 |
365 | Yes (400–520) | ND | min–hour | Backbone | ND |
| 12 | (IV) Intramolecular photocyclization – reversible | Diarylethene | 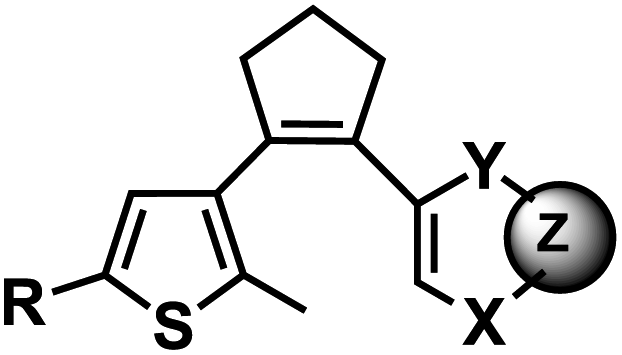 |
250–370 | Yes (>400) | ↓ | sec–min | U, C bases, A analogue (deazapurine) | ND |
Abbreviations
| NPE | 1-(ortho-Nitrophenyl)-ethyl |
| NPP | 2-(ortho-Nitrophenyl)-propyl |
| DMNEC | 1-(4-(2-(Dimethylamino)ethoxy)-5-methoxy-2-nitrophenyl)ethyl carbonyl |
| HBT | 2-(2′-Hydroxyphenyl) benzothiazole |
| dAAzo | 2-Phenyldiazenyl-substituted 2′-deoxyadenosine |
| dGAzo | 2-Phenyldiazenyl-substituted 2′-deoxyguanosine |
| 2Py | 2-Pyridyl |
| CNVK | 3-Cyanovinylcarbazole |
| CNVD | 3-Cyanovinylcarbazole modified D-threoninol |
| DMNPE | 4,5-Dimethoxy-2-nitrophenylethyl |
| Bhc | 6-Bromo-7-hydroxycoumarin-4-ylmethyl |
| NPOM | 6-Nitropiperonyl hydroxymethylene |
| DEACM | (7-Diethylaminocoumarin-4-yl)methyl |
| BHQ-diazo | 8-Bromo-2-diazomethyl-7-hydroxyquinolinyl |
| 8FV | 8-Fluorenylvinyl |
| 8NV | 8-Naphthylvinyl |
| NVA | 8-Naphthylvinyladenine |
| PVA | 8-Pyrenylvinyl adenine |
| 8ST | 8-Styryl |
| ATP | Adenosine triphosphate |
| ArS | Aryl sulfide |
| BP | Benzophenone |
| CRDDs | Catch and release DNA decoys |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| Cas9 | CRISPR-associated protein 9 |
| DNA | Deoxyribonucleic acid |
| DAE | Diarylethenes |
| DSBs | Double-strand breaks |
| D-tAzo | D-Threoninol-linked AzoB |
| PCXD | D-Threoninol version of the PCX |
| FISH | Fluorescence in situ hybridization |
| GFP | Green fluorescent protein |
| gRNA | Guide RNA |
| ICL | Interstrand crosslink |
| D-tAzo | L-Threoninol-linked AzoB |
| Tm | Melting temperature |
| mRNA | Messenger RNA |
| NDBF | Nitrodibenzofuran |
| NF-κB | Nuclear factor κB |
| NMR | Nuclear magnetic resonance |
| oNB | Ortho-Nitrobenzyl |
| PSS | Photostationary state |
| pHP | p-Hydroxyphenacyl |
| PNVOM | Propargyl-6-nitroveratryloxymethyl |
| PCX | Pyranocarbazole |
| R-gAzo | R-Glycerol-linked AzoB |
| RNA | Ribonucleic acid |
| SNA | Serinol nucleic acid |
| sgRNA | Single guide RNA |
| siRNA | Small interference RNA |
| SP | Spiropyran |
| SP | Styrylpyrene |
| PhtBu | tert-Butylester-phenyl |
| TFO | Triplex-forming oligonucleotides |
| TLR9 | Toll-like receptor 9 |
| TEEP-OH | Thioether-enol phosphate, phenol substituted |
| UV | Ultraviolet |
| vfCRISPR | Very fast CRISPR |
| Vis | Visible |
Conflicts of interest
There are no conflicts to declare.References
- D. Devasia, A. Das, V. Mohan and P. K. Jain, Annu. Rev. Phys. Chem., 2021, 72, 423–443 CrossRef CAS PubMed
.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS PubMed
- G. S. Kumar and Q. Lin, Chem. Rev., 2021, 121, 6991–7031 CrossRef CAS PubMed
- H. Lai, J. Zhang, F. Xing and P. Xiao, Chem. Soc. Rev., 2020, 49, 1867–1886 RSC
- H. Asanuma, X. Liang, T. Yoshida, A. Yamazawa and M. Komiyama, Angew. Chem., Int. Ed., 2000, 39, 1316–1318 CrossRef CAS PubMed
- C. Brieke and A. Heckel, Chem.–Eur. J., 2013, 19, 15726–15734 CrossRef CAS PubMed
- K. Fujimoto, M. Hashimoto, N. Watanabe and S. Nakamura, Bioorg. Med. Chem. Lett., 2019, 29, 2173–2177 CrossRef CAS PubMed
- K. Fujimoto, T. Yamaguchi, T. Inatsugi, M. Takamura, I. Ishimaru, A. Koto and S. Nakamura, RSC Adv., 2019, 9, 30693–30697 RSC
- J. M. Govan, M. O. Lively and A. Deiters, J. Am. Chem. Soc., 2011, 133, 13176–13182 CrossRef CAS PubMed
- N. Grebenovsky, V. Hermanns and A. Heckel, ChemPhotoChem, 2020, 4, 5245–5248 CrossRef CAS
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed
- A. S. Lubbe, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2017, 46, 1052–1079 RSC
- H. Asanuma, T. Ishikawa, Y. Yamano, K. Murayama and X. Liang, ChemPhotoChem, 2019, 3, 418–424 CrossRef CAS
- K. Fujimoto, S. Sasago, J. Mihara and S. Nakamura, Org. Lett., 2018, 20, 2802–2805 CrossRef CAS PubMed
- K. Aggarwal, M. Banik, B. Medellin and E. L. Que, Biochemistry, 2019, 58, 48–53 CrossRef CAS PubMed
- T. Tian, Y. Song, J. Wang, B. Fu, Z. He, X. Xu, A. Li, X. Zhou, S. Wang and X. Zhou, J. Am. Chem. Soc., 2016, 138, 955–961 CrossRef CAS PubMed
- Y. Wu, Z. Yang and Y. Lu, Curr. Opin. Chem. Biol., 2020, 57, 95–104 CrossRef CAS PubMed
- J. M. Govan, D. D. Young, M. O. Lively and A. Deiters, Tetrahedron Lett., 2015, 56, 3639–3642 CrossRef CAS PubMed
- T. Kakiyama, K. Usui, K.-y. Tomizaki, M. Mie, E. Kobatake and H. Mihara, Polym. J., 2013, 45, 535–539 CrossRef CAS
- T. M. Courtney and A. Deiters, Nat. Commun., 2019, 10, 4384 CrossRef PubMed
- G. M. Grotenbreg, N. R. Roan, E. Guillen, R. Meijers, J.-h. Wang, G. W. Bell, M. N. Starnbach and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3831 CrossRef CAS PubMed
- J. Hemphill, E. K. Borchardt, K. Brown, A. Asokan and A. Deiters, J. Am. Chem. Soc., 2015, 137, 5642–5645 CrossRef CAS PubMed
- J. Hemphill, C. Chou, J. W. Chin and A. Deiters, J. Am. Chem. Soc., 2013, 135, 13433–13439 CrossRef CAS PubMed
- S. Sankaran and A. del Campo, Adv. Biosyst., 2019, 3, 1800312 Search PubMed
- A. M. MacMillan and G. L. Verdine, J. Org. Chem., 1990, 55, 5931–5933 CrossRef CAS
- W. A. Velema, A. M. Kietrys and E. T. Kool, J. Am. Chem. Soc., 2018, 140, 3491–3495 CrossRef CAS PubMed
- M. Feng, Z. Ruan, J. Shang, L. Xiao, A. Tong and Y. Xiang, Bioconjugate Chem., 2017, 28, 549–555 CrossRef CAS PubMed
- X. Wang, M. Feng, L. Xiao, A. Tong and Y. Xiang, ACS Chem. Biol., 2016, 11, 444–451 CrossRef CAS PubMed
- M. J. Hansen, W. A. Velema, M. M. Lerch, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2015, 44, 3358–3377 RSC
- Q. Liu and A. Deiters, Acc. Chem. Res., 2014, 47, 45–55 CrossRef CAS PubMed
- P. Ordoukhanian and J.-S. Taylor, J. Am. Chem. Soc., 1995, 117, 9570–9571 CrossRef CAS
- A. Dussy, C. Meyer, E. Quennet, T. A. Bickle, B. Giese and A. Marx, ChemBioChem, 2002, 3, 54–60 CrossRef CAS PubMed
- X. Tang, M. Su, L. Yu, C. Lv, J. Wang and Z. Li, Nucleic Acids Res., 2010, 38, 3848–3855 CrossRef CAS PubMed
- J. L. Richards, G. K. Seward, Y.-H. Wang and I. J. Dmochowski, ChemBioChem, 2010, 11, 320–324 CrossRef CAS PubMed
- X. Tang, S. Maegawa, E. S. Weinberg and I. J. Dmochowski, J. Am. Chem. Soc., 2007, 129, 11000–11001 CrossRef CAS PubMed
- Y. Kimura, Z. Shu, M. Ito, N. Abe, K. Nakamoto, F. Tomoike, S. Shuto, Y. Ito and H. Abe, Chem. Commun., 2020, 56, 466–469 RSC
- J. Carlson-Stevermer, R. Kelso, A. Kadina, S. Joshi, N. Rossi, J. Walker, R. Stoner and T. Maures, Nat. Commun., 2020, 11, 5041 CrossRef CAS PubMed
- P. K. Jain, V. Ramanan, A. G. Schepers, N. S. Dalvie, A. Panda, H. E. Fleming and S. N. Bhatia, Angew. Chem., Int. Ed., 2016, 55, 12440–12444 CrossRef CAS PubMed
- R. S. Zou, Y. Liu, B. Wu and T. Ha, Mol. Cell, 2021, 81, 1553–1565.e1558 CrossRef CAS PubMed
- J. Hemphill, Q. Liu, R. Uprety, S. Samanta, M. Tsang, R. L. Juliano and A. Deiters, J. Am. Chem. Soc., 2015, 137, 3656–3662 CrossRef CAS PubMed
- S. G. Chaulk and A. M. MacMillan, Nucleic Acids Res., 1998, 26, 3173–3178 CrossRef CAS PubMed
- S. G. Chaulk and A. M. MacMillan, Nat. Protoc., 2007, 2, 1052–1058 CrossRef CAS PubMed
- E. M. McConnell, I. Cozma, Q. Mou, J. D. Brennan, Y. Lu and Y. Li, Chem. Soc. Rev., 2021, 50, 8954–8994 RSC
- Z. Yang, K. Y. Loh, Y.-T. Chu, R. Feng, N. S. R. Satyavolu, M. Xiong, S. M. Nakamata Huynh, K. Hwang, L. Li, H. Xing, X. Zhang, Y. R. Chemla, M. Gruebele and Y. Lu, J. Am. Chem. Soc., 2018, 140, 17656–17665 CrossRef CAS PubMed
- K. Hwang, Q. Mou, R. J. Lake, M. Xiong, B. Holland and Y. Lu, Inorg. Chem., 2019, 58, 13696–13708 CrossRef CAS PubMed
- K. Hwang, P. Wu, T. Kim, L. Lei, S. Tian, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 13798–13802 CrossRef CAS PubMed
- J. M. Govan, D. D. Young, H. Lusic, Q. Liu, M. O. Lively and A. Deiters, Nucleic Acids Res., 2013, 41, 10518–10528 CrossRef CAS PubMed
- J. Hemphill, J. Govan, R. Uprety, M. Tsang and A. Deiters, J. Am. Chem. Soc., 2014, 136, 7152–7158 CrossRef CAS PubMed
- D. D. Young, J. M. Govan, M. O. Lively and A. Deiters, ChemBioChem, 2009, 10, 1612–1616 CrossRef CAS PubMed
- D. D. Young, M. O. Lively and A. Deiters, J. Am. Chem. Soc., 2010, 132, 6183–6193 CrossRef CAS PubMed
- W. Zhou, W. Brown, A. Bardhan, M. Delaney, A. S. Ilk, R. R. Rauen, S. I. Kahn, M. Tsang and A. Deiters, Angew. Chem., Int. Ed., 2020, 59, 8998–9003 CrossRef CAS PubMed
- J. M. Govan, R. Uprety, M. Thomas, H. Lusic, M. O. Lively and A. Deiters, ACS Chem. Biol., 2013, 8, 2272–2282 CrossRef CAS PubMed
- H. Lusic, R. Uprety and A. Deiters, Org. Lett., 2010, 12, 916–919 CrossRef CAS PubMed
- F. Schäfer, K. B. Joshi, M. A. H. Fichte, T. Mack, J. Wachtveitl and A. Heckel, Org. Lett., 2011, 13, 1450–1453 CrossRef PubMed
- S. Shah, S. Rangarajan and S. H. Friedman, Angew. Chem., Int. Ed., 2005, 44, 1328–1332 CrossRef CAS PubMed
- A. Kala, P. K. Jain, D. Karunakaran, S. Shah and S. H. Friedman, Nat. Protoc., 2014, 9, 11–20 CrossRef CAS PubMed
- S. Wang, L. Wei, J.-Q. Wang, H. Ji, W. Xiong, J. Liu, P. Yin, T. Tian and X. Zhou, ACS Chem. Biol., 2020, 15, 1455–1463 CrossRef CAS PubMed
- A. Tavakoli, D. Paul, H. Mu, J. Kuchlyan, S. Baral, A. Ansari, S. Broyde and J.-H. Min, RSC Chem. Biol., 2021, 2, 523–536 RSC
- A. Rodrigues-Correia, X. M. M. Weyel and A. Heckel, Org. Lett., 2013, 15, 5500–5503 CrossRef CAS PubMed
- M. Liu, S. Jiang, O. Loza, N. E. Fahmi, P. Šulc and N. Stephanopoulos, Angew. Chem., Int. Ed., 2018, 57, 9341–9345 CrossRef CAS PubMed
- A. Rodrigues-Correia, M. B. Koeppel, F. Schäfer, K. B. Joshi, T. Mack and A. Heckel, Anal. Bioanal. Chem., 2011, 399, 441–447 CrossRef CAS PubMed
- H. S. Steinert, F. Schäfer, H. R. A. Jonker, A. Heckel and H. Schwalbe, Angew. Chem., Int. Ed., 2014, 53, 1072–1075 CrossRef CAS PubMed
- K. Seio, Y. Ohno, K. Ohno, L. Takeshita, T. Kanamori, Y. Masaki and M. Sekine, Bioorg. Med. Chem. Lett., 2016, 26, 4861–4863 CrossRef CAS PubMed
- L. Wu, F. Pei, J. Zhang, J. Wu, M. Feng, Y. Wang, H. Jin, L. Zhang and X. Tang, Chem.–Eur. J., 2014, 20, 12114–12122 CrossRef CAS PubMed
- Y. Yamagata, T. Emura, K. Hidaka, H. Sugiyama and M. Endo, Chem.–Eur. J., 2016, 22, 5494–5498 CrossRef CAS PubMed
- A. Prokup, J. Hemphill and A. Deiters, J. Am. Chem. Soc., 2012, 134, 3810–3815 CrossRef CAS PubMed
- H. Lusic, D. D. Young, M. O. Lively and A. Deiters, Org. Lett., 2007, 9, 1903–1906 CrossRef CAS PubMed
- D. D. Young, W. F. Edwards, H. Lusic, M. O. Lively and A. Deiters, Chem. Commun., 2008, 462–464 RSC
- D. D. Young, H. Lusic, M. O. Lively and A. Deiters, Nucleic Acids Res., 2009, 37, e58 CrossRef PubMed
- Y. Liu, R. S. Zou, S. He, Y. Nihongaki, X. Li, S. Razavi, B. Wu and T. Ha, Science, 2020, 368, 1265 CrossRef CAS PubMed
- J. C. Anderson and C. B. Reese, Tetrahedron Lett., 1962, 3, 1–4 CrossRef
- A. Rodrigues-Correia, D. Knapp-Bühle, J. W. Engels and A. Heckel, Org. Lett., 2014, 16, 5128–5131 CrossRef CAS PubMed
- S. Panja, R. Paul, M. M. Greenberg and S. A. Woodson, Angew. Chem., Int. Ed., 2015, 54, 7281–7284 CrossRef CAS PubMed
- A. Korman, H. Sun, B. Hua, H. Yang, J. N. Capilato, R. Paul, S. Panja, T. Ha, M. M. Greenberg and S. A. Woodson, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12080 CrossRef CAS PubMed
- S. Barman, S. K. Mukhopadhyay, S. Biswas, S. Nandi, M. Gangopadhyay, S. Dey, A. Anoop and N. D. Pradeep Singh, Angew. Chem., Int. Ed., 2016, 55, 4194–4198 CrossRef CAS PubMed
- A. Specht, S. Loudwig, L. Peng and M. Goeldner, Tetrahedron Lett., 2002, 43, 8947–8950 CrossRef CAS
- J. M. N. San Pedro and M. M. Greenberg, J. Am. Chem. Soc., 2014, 136, 3928–3936 CrossRef CAS PubMed
- M. J. E. Resendiz, A. Schön, E. Freire and M. M. Greenberg, J. Am. Chem. Soc., 2012, 134, 12478–12481 CrossRef CAS PubMed
- U. Rieder, K. Lang, C. Kreutz, N. Polacek and R. Micura, ChemBioChem, 2009, 10, 1141–1144 CrossRef CAS PubMed
- M. Kotera, A.-G. Bourdat, E. Defrancq and J. Lhomme, J. Am. Chem. Soc., 1998, 120, 11810–11811 CrossRef CAS
- M. Kotera, Y. Roupioz, E. Defrancq, A.-G. Bourdat, J. Garcia, C. Coulombeau and J. Lhomme, Chem.–Eur. J., 2000, 6, 4163–4169 CrossRef CAS
- C. Crey-Desbiolles, N. Berthet, M. Kotera and P. Dumy, Nucleic Acids Res., 2005, 33, 1532–1543 CrossRef CAS PubMed
- N. B. Struntz and D. A. Harki, ACS Chem. Biol., 2016, 11, 1631–1638 CrossRef CAS PubMed
- M. M. Hassan and O. O. Olaoye, Molecules, 2020, 25, 2285 CrossRef CAS PubMed
- G. Dormán, H. Nakamura, A. Pulsipher and G. D. Prestwich, Chem. Rev., 2016, 116, 15284–15398 CrossRef PubMed
- L. Anhäuser, N. Klöcker, F. Muttach, F. Mäsing, P. Špaček, A. Studer and A. Rentmeister, Angew. Chem., Int. Ed., 2020, 59, 3161–3165 CrossRef PubMed
- J. Jakubovska, D. Tauraitė and R. Meškys, Sci. Rep., 2018, 8, 16484 CrossRef PubMed
- V. Hagen, J. Bendig, S. Frings, T. Eckardt, S. Helm, D. Reuter and U. B. Kaupp, Angew. Chem., Int. Ed., 2001, 40, 1045–1048 CrossRef PubMed
- C. Menge and A. Heckel, Org. Lett., 2011, 13, 4620–4623 CrossRef CAS PubMed
- S. Tang, J. Cannon, K. Yang, M. F. Krummel, J. R. Baker and S. K. Choi, J. Org. Chem., 2020, 85, 2945–2955 CrossRef CAS PubMed
- P. Seyfried, L. Eiden, N. Grebenovsky, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2017, 56, 359–363 CrossRef CAS PubMed
- J. Deng, D. Bezold, H. J. Jessen and A. Walther, Angew. Chem., Int. Ed., 2020, 59, 12084–12092 CrossRef CAS PubMed
- T. Furuta, S. S. H. Wang, J. L. Dantzker, T. M. Dore, W. J. Bybee, E. M. Callaway, W. Denk and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1193 CrossRef CAS PubMed
- T. Furuta, T. Watanabe, S. Tanabe, J. Sakyo and C. Matsuba, Org. Lett., 2007, 9, 4717–4720 CrossRef CAS PubMed
- A. Z. Suzuki, T. Watanabe, M. Kawamoto, K. Nishiyama, H. Yamashita, M. Ishii, M. Iwamura and T. Furuta, Org. Lett., 2003, 5, 4867–4870 CrossRef CAS PubMed
- H. Ando, T. Furuta, R. Y. Tsien and H. Okamoto, Nat. Genet., 2001, 28, 317–325 CrossRef CAS PubMed
- Y. Li, J. Shi, Z. Luo, H. Jiang, X. Chen, F. Wang, X. Wu and Q. Guo, Bioorg. Med. Chem. Lett., 2009, 19, 5368–5371 CrossRef CAS PubMed
- Y. M. Li, J. Shi, R. Cai, X. Chen, Z. F. Luo and Q. X. Guo, J. Photochem. Photobiol., A, 2010, 211, 129–134 CrossRef CAS
- X. M. M. Weyel, M. A. H. Fichte and A. Heckel, ACS Chem. Biol., 2017, 12, 2183–2190 CrossRef CAS PubMed
- Y. Zhu, C. M. Pavlos, J. P. Toscano and T. M. Dore, J. Am. Chem. Soc., 2006, 128, 4267–4276 CrossRef CAS PubMed
- H. Sun, H. Fan and X. Peng, J. Org. Chem., 2014, 79, 11359–11369 CrossRef CAS PubMed
- S. R. Trenor, A. R. Shultz, B. J. Love and T. E. Long, Chem. Rev., 2004, 104, 3059–3078 CrossRef CAS PubMed
- P. Seyfried, M. Heinz, G. Pintér, D.-P. Klötzner, Y. Becker, M. Bolte, H. R. A. Jonker, L. S. Stelzl, G. Hummer, H. Schwalbe and A. Heckel, Chem.–Eur. J., 2018, 24, 17568–17576 CrossRef CAS PubMed
- J. Elskens and A. Madder, RSC Chem. Biol., 2021, 2, 410–422 RSC
- E. Gyssels, N. De Laet, E. Lumley and A. Madder, in Modified Nucleic Acids in Biology and Medicine, ed. S. Jurga, V. A. Erdmann and J. Barciszewski, Springer International Publishing, Cham, 2016, pp. 339–369, DOI:10.1007/978-3-319-34175-0_15
- Y. Yoshimura and K. Fujimoto, Org. Lett., 2008, 10, 3227–3230 CrossRef CAS PubMed
- T. Sakamoto, Y. Tanaka and K. Fujimoto, Org. Lett., 2015, 17, 936–939 CrossRef CAS PubMed
- K. Fujimoto, K. Ishida, L. Xue and S. Nakamura, Photochem. Photobiol. Sci., 2020, 19, 776–782 CrossRef CAS PubMed
- Y. Watanabe and K. Fujimoto, ChemBioChem, 2020, 21, 3244–3248 CrossRef CAS PubMed
- K. Fujimoto, H. Yang-Chun and S. Nakamura, Chem.–Asian J., 2019, 14, 1912–1916 CrossRef CAS PubMed
- K. Fujimoto, A. Yamada, Y. Yoshimura, T. Tsukaguchi and T. Sakamoto, J. Am. Chem. Soc., 2013, 135, 16161–16167 CrossRef CAS PubMed
- K. Fujimoto and N. Watanabe, ChemistrySelect, 2020, 5, 14670–14676 CrossRef CAS
- K. Fujimoto, K. Hiratsuka-Konishi, T. Sakamoto, T. Ohtake, K.-i. Shinohara and Y. Yoshimura, Mol. BioSyst., 2012, 8, 491–494 RSC
- T. Sakamoto, A. Shigeno, Y. Ohtaki and K. Fujimoto, Biomater. Sci., 2014, 2, 1154–1157 RSC
- A. Choudhary, D. P. Vanichkina, C. Ender, J. Crawford, G. J. Baillie, A. D. Calcino, K. Ru and R. J. Taft, RNA, 2018, 24, 597–608 CrossRef CAS PubMed
- K. Fujimoto, K. Toyosato, S. Nakamura and T. Sakamoto, Bioorg. Med. Chem. Lett., 2016, 26, 5312–5314 CrossRef CAS PubMed
- S. Nakamura, H. Hashimoto, S. Kobayashi and K. Fujimoto, ChemBioChem, 2017, 18, 1984–1989 CrossRef CAS PubMed
- S. Ogasawara and M. Maeda, Bioorg. Med. Chem. Lett., 2011, 21, 5457–5459 CrossRef CAS PubMed
- H. Kashida, T. Doi, T. Sakakibara, T. Hayashi and H. Asanuma, J. Am. Chem. Soc., 2013, 135, 7960–7966 CrossRef CAS PubMed
- Y. Kamiya, K. Iishiba, T. Doi, K. Tsuda, H. Kashida and H. Asanuma, Biomater. Sci., 2015, 3, 1534–1538 RSC
- T. Doi, H. Kawai, K. Murayama, H. Kashida and H. Asanuma, Chem.–Eur. J., 2016, 22, 10533–10538 CrossRef CAS PubMed
- K. Murayama, Y. Yamano and H. Asanuma, J. Am. Chem. Soc., 2019, 141, 9485–9489 CrossRef CAS PubMed
- Y. Yamano, K. Murayama and H. Asanuma, Chem.–Eur. J., 2021, 27, 4599–4604 CrossRef CAS PubMed
- Y. Nakasone, H. Ooi, Y. Kamiya, H. Asanuma and M. Terazima, J. Am. Chem. Soc., 2016, 138, 9001–9004 CrossRef CAS PubMed
- S. Ogasawara and M. Maeda, Angew. Chem., Int. Ed., 2008, 47, 8839–8842 CrossRef CAS PubMed
- S. Ogasawara and M. Maeda, Angew. Chem., Int. Ed., 2009, 48, 6671–6674 CrossRef CAS PubMed
- S. Ogasawara, ChemBioChem, 2014, 15, 2652–2655 CrossRef CAS PubMed
- Y. Liu, C. Zhong, Z. He, S. Rao, H. Su, F. Wu, B. Fu, J. Wu, Y. Song, Y. Du and X. Zhou, Sens. Actuators, B, 2018, 255, 2151–2154 CrossRef CAS
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC
- H. Asanuma, T. Ito and M. Komiyama, Tetrahedron Lett., 1998, 39, 9015–9018 CrossRef CAS
- H. Asanuma, T. Ito, T. Yoshida, X. Liang and M. Komiyama, Angew. Chem., Int. Ed., 1999, 38, 2393–2395 CrossRef CAS PubMed
- X. Liang, H. Asanuma and M. Komiyama, J. Am. Chem. Soc., 2002, 124, 1877–1883 CrossRef CAS PubMed
- H. Asanuma, T. Takarada, T. Yoshida, D. Tamaru, X. Liang and M. Komiyama, Angew. Chem., Int. Ed., 2001, 40, 2671–2673 CrossRef CAS PubMed
- M. Liu, H. Asanuma and M. Komiyama, J. Am. Chem. Soc., 2006, 128, 1009–1015 CrossRef CAS PubMed
- B. Kou, X. Guo, S.-J. Xiao and X. Liang, Small, 2013, 9, 3939–3943 CrossRef CAS PubMed
- T. Goldau, K. Murayama, C. Brieke, H. Asanuma and A. Heckel, Chem.–Eur. J., 2015, 21, 17870–17876 CrossRef CAS PubMed
- L. Wu, Y. He and X. Tang, Bioconjugate Chem., 2015, 26, 1070–1079 CrossRef CAS PubMed
- M. L. Hammill, C. Isaacs-Trépanier and J.-P. Desaulniers, ChemistrySelect, 2017, 2, 9810–9814 CrossRef CAS
- M. L. Hammill, A. Patel, M. A. Alla and J.-P. Desaulniers, Bioorg. Med. Chem. Lett., 2018, 28, 3613–3616 CrossRef CAS PubMed
- M. L. Hammill, G. Islam and J.-P. Desaulniers, Org. Biomol. Chem., 2020, 18, 41–46 RSC
- M. L. Hammill, G. Islam and J.-P. Desaulniers, ChemBioChem, 2020, 21, 2367–2372 CrossRef CAS PubMed
- S. Ogasawara, ACS Chem. Biol., 2017, 12, 351–356 CrossRef CAS PubMed
- H. Asanuma, X. Liang, H. Nishioka, D. Matsunaga, M. Liu and M. Komiyama, Nat. Protoc., 2007, 2, 203–212 CrossRef CAS PubMed
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed
- Z. Wu and L. Zhang, Biomater. Sci., 2019, 7, 4944–4962 RSC
- X. Liang, M. Zhou, K. Kato and H. Asanuma, ACS Synth. Biol., 2013, 2, 194–202 CrossRef CAS PubMed
- J. M. Govan and A. Deiters, in From Nucleic Acids Sequences to Molecular Medicine, ed. V. A. Erdmann and J. Barciszewski, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 275–291, DOI:10.1007/978-3-642-27426-8_11
- X. Wang and X. Liang, RSC Adv., 2016, 6, 93398–93402 RSC
- D. Y. Tam, X. Zhuang, S. W. Wong and P. K. Lo, Small, 2019, 15, 1805481 CrossRef PubMed
- Y. Kim, J. A. Phillips, H. Liu, H. Kang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6489 CrossRef CAS PubMed
- M. L. Hammill, G. Islam and J.-P. Desaulniers, Curr. Protoc. Nucleic Acid Chem., 2020, 83, e119 CAS
- E. Fischer and Y. Hirshberg, Journal, 1952, 4522–4524 CAS
- H. Asanuma, K. Shirasuka, T. Yoshida, T. Takarada, X. Liang and M. Komiyama, Chem. Lett., 2001, 30, 108–109 CrossRef
- C. Beyer and H.-A. Wagenknecht, Synlett, 2010, 2010, 1371–1376 CrossRef
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC
- M. Herder, B. M. Schmidt, L. Grubert, M. Pätzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS PubMed
- H.-h. Liu and Y. Chen, New J. Chem., 2012, 36, 2223–2227 RSC
- M. Singer and A. Jäschke, J. Am. Chem. Soc., 2010, 132, 8372–8377 CrossRef CAS PubMed
- T. Buckup, C. Sarter, H.-R. Volpp, A. Jäschke and M. Motzkus, J. Phys. Chem. Lett., 2015, 6, 4717–4721 CrossRef CAS PubMed
- H. Cahová and A. Jäschke, Angew. Chem., Int. Ed., 2013, 52, 3186–3190 CrossRef PubMed
- S. Barrois and H. A. Wagenknecht, Beilstein J. Org. Chem., 2012, 8, 905–914 CrossRef CAS PubMed
- C. Sarter, S. Dey and A. Jäschke, ACS Omega, 2019, 4, 12125–12129 CrossRef CAS PubMed
- T. Kolmar, S. M. Büllmann, C. Sarter, K. Höfer and A. Jäschke, Angew. Chem., Int. Ed., 2021, 60, 8164–8173 CrossRef CAS PubMed
Footnote |
| † Current address: Department of Cell Biology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA. |
| This journal is © The Royal Society of Chemistry 2022 |