
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrothermal synthesis of water-soluble Mn- and Cu-doped CdSe quantum dots with multi-shell structures and their photoluminescence properties†
Hisaaki Nishimura a,
Kazushi Enomotob,
Yong-Jin Pub and
DaeGwi Kim*a
a,
Kazushi Enomotob,
Yong-Jin Pub and
DaeGwi Kim*a
aDepartment of Applied Physics, Osaka City University, Osaka 558-8585, Japan. E-mail: tegi@a-phy.eng.osaka-cu.ac.jp
bRIKEN Center for Emergent Matter Science (CEMS), Saitama 351-0198, Japan
First published on 22nd February 2022
Abstract
Optical properties of semiconductor quantum dots (QDs) can be tuned by doping with transition metal ions. In this study, water-soluble CdSe/ZnS:Mn/ZnS QDs with the core/shell/shell structure were synthesized through a hydrothermal method, in which the surface of the CdSe core was coated with a ZnS:Mn shell and ZnS capping shell. Herein, the CdSe core QDs were prepared first and then doped with Mn2+; therefore, the QD size and doping level could be controlled independently and interference from the self-purifying effect could be avoided. When CdSe cores with diameters less than 1.9 nm were used, Mn-related photoluminescence (PL) was observed as the main PL band, whereas the band-edge PL was mainly observed when larger CdSe cores were used. Furthermore, using ZnS:Cu as the doping shell layer, CdSe/ZnS:Cu/ZnS and ZnSe/ZnS:Cu/ZnS nanoparticles were successfully synthesized, and Cu-related PL was clearly observed. These results indicate that the core/shell/shell QD structure with doping in the shell layer is a versatile method for synthesizing doped QDs.
Introduction
The optical properties of semiconductor quantum dots (QDs) can be controlled through particle size owing to the quantum size effect.1–5 Furthermore, new optical and magnetic properties can be imparted to QDs by doping with transition metal ions.6–9 Therefore, transition metal-doped quantum dots (doped QDs) have attracted attention in various fields, such as highly efficient photoluminescent materials,6,7,10–15 wavelength conversion materials,16–21 and spin-photonics.22–28 Focusing on the optical properties of doped QDs, their photoluminescence (PL) properties can be controlled by doping with Mn or Cu ions.2,6–14,29–33 In semiconductor QDs doped with Mn2+ ions as emission centres, excitation energy in the host QDs is transferred to Mn2+, resulting in orange emission (Mn PL) due to the intra 3d-shell 4T1 → 6A1 transition of Mn2+.2,6,7,34In contrast, upon host excitation in Cu-doped QDs, the photogenerated hole nonradiatively localizes from the valence band to the d-orbital levels of the Cu dopant, and radiative recombination of electron from the conduction band with the copper-localized hole can occur as Cu emission (Cu-related PL).35–37 In addition, some QDs doped with transition ions display interesting magnetic properties such as dilute magnetic semiconductors because of the giant excitonic Zeeman splitting arising from the exchange interaction between the host QDs and transition metal ions.38–41 Thus, the correlation between the intrinsic energy of exciton states of host QDs and the energy levels of dopant ions is significant in determining the optical and magnetic properties of doped QDs.2,37,42–44 For example, if the excitonic states of the host QDs are located above the dopant level, then energy transfer to the dopant ions become dominant.2,42,45,46 Therefore, to apply doped QDs as functional materials, it is essential to select the appropriate host semiconductor materials and control the QD size.
One difficulty in doping QDs with transition metal ions is the so-called self-purification effect, wherein impurities are removed during QD growth.6,47–49 This effect is useful for achieving high crystallinity when preparing non-doped QDs; however, it becomes a disadvantage when the intentional doping with transition metal ions is desired. Many studies have been conducted on the synthesis of Mn-doped QDs as model systems. In 1994, Bhargava et al. first reported the synthesis of colloidal ZnS:Mn QDs.50 Subsequently, Mn ions were doped into II–VI semiconductors including ZnSe,51–53 CdSe,42,45,46,54 CdS,55,56 and ZnTe57,58 QDs and perovskite QDs such as CsPbCl3.59–61 Their optical and magneto-optical properties have also been studied.23–28
We previously used a hydrothermal method to synthesize ZnSe/ZnS:Mn/ZnS core/shell/shell QDs with a PL quantum yield (PLQY) of 84%.21 Instead of directly doping the core, Mn2+ doping was carried out in a shell layer over non-doped ZnSe core QDs as the host. Because this method separates the host synthesis from the doping process, it could be applied to various types of doped QDs with different host materials and QD sizes.
In this study, Mn-doped CdSe QDs were synthesized with a core/shell/shell structure of CdSe/ZnS:Mn/ZnS. Because the Mn PL originates from energy transfer from exciton in the QD host to the Mn2+ ions, the energy gap between the 4T1 state of Mn2+ dopant and the host QD excitonic state (ΔE) has a significant impact on the PL properties. Furthermore, as shown in Fig. S1 in ESI,† because the host exciton energy can be controlled by the QD size, the ΔE value may be adjusted by varying the diameter of CdSe QDs. Beaulac et al. reported that when the diameter of the host CdSe QDs is less than 3.3 nm, exciton energy is transferred to Mn2+ and Mn PL is observed. For larger CdSe QDs, the host exciton and the 4T1 state of Mn2+ are reversed in their relative energies. Thus, energy transfer to Mn2+ ions does not occur, and the Mn PL is quenched.2,42
Although the energy gap, ΔE, is an essential factor in determining the PL properties, it is difficult to synthesize high-quality doped QDs with precise control. When a longer reaction time is required to increase the QD size, a stronger self-purification effect also occurs. In previous studies, high-quality doped QDs were prepared using various synthetic techniques such as the nucleation-doping method62 and high-temperature organometallic synthesis.51 Nevertheless, it is not easy to synthesize doped QDs with both strong Mn PL and controlled QD size. This problem could be overcome using our previously proposed method,21 in which the non-doped core QDs are prepared independently and then two shell layers including a doped one are deposited to form the core/shell/shell QDs. In other words, because the QD size of the host CdSe core (i.e. the exciton energy) and the Mn-doped shell layers (i.e. the Mn doping conditions) are determined separately, it is possible to find the optimal Mn doping level for each core QD size, which is a promising way to synthesize Mn-doped CdSe QDs with controlled ΔE values.
Herein, we used CdSe core QDs of different sizes (1.6, 1.7, 1.8, 1.9, 2.2, 2.4, and 2.7 nm) as the host, and investigated the synthesis conditions that maximise the Mn PL intensity for each core size. In addition, Cu was used as a dopant in place of Mn to prepare ZnSe/ZnS:Cu/ZnS and CdSe/ZnS:Cu/ZnS core/shell/shell QDs. These results confirm that the method to synthesize QDs with doped shell layers is versatile and applicable to various host materials and dopant ions.
Experimental section
Chemicals
Cd(ClO4)2·6H2O, Zn(ClO4)2·6H2O, and Mn(ClO4)2·6H2O were purchased from Fujifilm Wako Chemicals, and selenium was purchased from Aldrich. N-Acetyl-L-cysteine (NAC), Na2S·9H2O and CuCl2·2H2O were purchased from Kishida Chemical.Synthesis of CdSe and CdSe:Mn QDs
The CdSe precursor solution was prepared according to the report by Lee et al.63 Briefly, Cd(ClO4)2·6H2O (1.0 mmol) and NAC (3.0 mmol) as a ligand were dissolved in 50 mL of deionised (DI) water. The pH of the solution was adjusted to 8.5, and freshly prepared NaHSe solution (0.3 mmol) was added. Then, the pH was adjusted to 5.0 by adding diluted HCl.The CdSe:Mn precursor solution was prepared in the same way as the CdSe precursor solution, except that Mn(ClO4)2·6H2O (0.02 mmol) was added before the addition of NaHSe. The CdSe and CdSe:Mn precursor solutions (10 mL) were separately placed in reaction vessels and incubated at 200 °C for 3 min. The QD size was estimated as 1.6 nm from the absorption peak energy.64
Preparation of ZnS:Mn precursor solution
The ZnS:Mn precursor solution was prepared according to a previous report.21 First, Zn(ClO4)2·6H2O (1.2 mmol) and NAC (8.7 mmol) were dissolved in 20 mL of DI water. NaOH was added to adjust the pH to 4.0, and Na2S·9H2O (0.24 mmol) and Mn(ClO4)2·6H2O (0.18 mmol) were added. Finally, the pH of the ZnS:Mn precursor solution was adjusted to 10.5 using NaOH.Preparation of ZnS precursor solution
The ZnS precursor solution was prepared according to previous reports.21 First, NAC (7.2 mmol) was dissolved in 50 mL of DI water, and Zn(ClO4)2·6H2O (3.0 mmol) was added as a Zn2+ source. After adjusting the solution pH to 4.0 using NaOH, Na2S·9H2O (0.6 mmol) was added, and then the pH was adjusted to 4.5 using NaOH.Synthesis of CdSe/ZnS:Mn/ZnS core/shell/shell QDs
First, the CdSe core QDs were prepared using the hydrothermal method. The prepared CdSe precursor solution (described above) was placed in a reaction vessel and heated by microwave irradiation. CdSe core QDs of various sizes (1.6–2.7 nm) were prepared by heating for 5 min at different temperatures (100–180 °C). The ZnS:Mn precursor solution was then added, and CdSe/ZnS:Mn core/shell QDs were synthesized by microwave irradiation for 15 min at 80 °C using a single-mode CEM Discover system. The ratio between the two solutions was varied to determine the optimal amount of ZnS:Mn precursor. Finally, the ZnS precursor was added to the CdSe/ZnS:Mn core/shell QD solution to produce CdSe/ZnS:Mn/ZnS core/shell/shell QDs by microwave irradiation at 140 °C. Here, the total amount of added ZnS:Mn and ZnS precursors was fixed, and the ZnS precursor was added gradually in five portions and heated after each addition for 4, 4, 6, 6, and 10 min, respectively.Synthesis of CdSe/ZnS:Cu/ZnS QDs and ZnSe/ZnS:Cu/ZnS QDs
To prepare the ZnS:Cu precursor solution, Zn(ClO4)2·6H2O (0.4 mmol) and NAC (0.96 mmol) were dissolved in 20 mL of DI water. NaOH was added to adjust the pH to 4.0, and Na2S·9H2O (0.08 mmol) and CuCl2·2H2O (0.036 mmol) were added. Finally, the pH of the ZnS:Cu precursor solution was adjusted to 6.0.The ZnS precursor solution was prepared as follows. First, Zn(ClO4)2·6H2O (1.0 mmol) and NAC (2.4 mmol) were dissolved in 50 mL of DI water. NaOH was used to adjust the solution pH to 4.0, and Na2S·9H2O (0.2 mmol) was added. The pH of the ZnS precursor solution was also adjusted to 6.0.
To prepare the Cu-doped QDs, first the CdSe (or ZnSe) core solution and ZnS:Cu precursor solution were mixed in a [ZnS:Cu]/[CdSe (ZnSe)] ratio of 0.5 based on the molar ratio of cations in each solution. The mixed solution was heated at 120 °C for 2 min under microwave irradiation. Finally, the ZnS precursor was added to the CdSe/ZnS:Cu (ZnSe/ZnS:Cu) core/shell QD solution. After microwave irradiation, the CdSe/ZnS:Cu/ZnS (ZnSe/ZnS:Cu/ZnS) core/shell/shell QDs were synthesized.
Characterisation and spectroscopic measurements
High-resolution electron microscopy images were obtained using a transmission electron microscope (JEOL JEM-2100F/SP). Energy-dispersive X-ray spectroscopy (EDX) analysis was performed to estimate the actual doping amount of Mn2+. For the X-ray photoelectron spectroscopy (XPS) measurements, the monolayer structure of QDs was prepared according to the following procedure. First, Si substrates were cleaned by immersing them in fresh piranha solution (1/3 (v/v) mixture of 30% H2O2 and 98% H2SO4) for 20 min (caution: piranha solution reacts violently with organic materials). The substrates were then rinsed with water and used immediately after cleaning. At the beginning of the sample preparation, the adhesion layer of polyelectrolytes of positively charged poly(diallyldimethylammonium chloride) (PDDA) was deposited to enhance the binding of QDs. The monolayer structure of QDs was then deposited onto the PDDA layer. A Shimadzu ESCA-3400 with a Mg Kα (1.253 keV) source was used for the XPS measurements.The absorption spectra were obtained using a JASCO V-650 UV-Vis spectrophotometer with a spectral resolution of 0.5 nm. The PL spectra were measured with a JASCO FP-8300 spectrofluorometer at an excitation wavelength of 375 nm and spectral resolution of 0.5 nm. The PL intensity of each sample was normalized by the absorption intensity at the excitation wavelength. For the ZnSe/ZnS:Mn/ZnS and ZnSe/ZnS:Cu/ZnS core/shell/shell QDs, the excitation wavelength was 355 nm. The PLQY was measured using a JASCO ILF-835 100 mm integrating sphere system coupled with a spectrofluorometer. To measure the decay profiles of Mn PL, the excitation source was third-harmonic-generated light (355 nm) from a laser diode-pumped YAG laser with a pulse duration of 20 ns and repetition rate of 20 Hz, and the Mn PL decay profiles were observed using a 500 MHz digitising oscilloscope. To measure the decay profiles of Cu-related PL, a laser diode (375 nm, Hamamatsu PLP M10306-27) with a pulse duration of 50 ps and a repetition rate of 100 kHz was used as the excitation light, and the PL decay profiles were obtained using a time-correlated single-photon counting method.
Results and discussion
Before synthesizing the CdSe/ZnS:Mn/ZnS core/shell/shell QDs, we first tested the possibility of directly doping Mn ions into CdSe core QDs by using the CdSe:Mn precursor solution. Fig. 1a shows the absorption and PL spectra of the obtained CdSe:Mn QDs (solid curve) and non-doped CdSe QDs (dashed curve) as reference. Clearly, the two spectra are consistent with a peak at 2.90 eV. Donegá et al. reported that in CdSe QDs with diameter d, the lowest exciton energy is given as follows:64
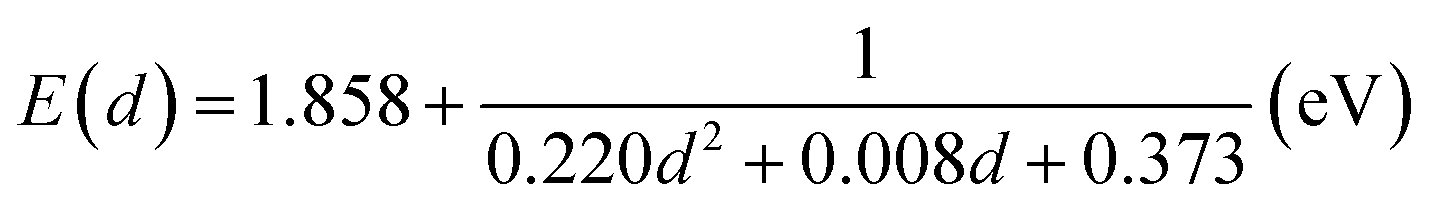 | (1) |
 | ||
| Fig. 1 (a) Absorption and PL spectra of non-doped CdSe and Mn-doped CdSe:Mn QDs with a diameter of 1.6 nm. (b) PL decay profiles detected at 1.9 eV in CdSe and CdSe:Mn QDs. | ||
Next, we applied our reported method for synthesizing doped QDs with multi-shell structures. First, CdSe core QDs with d = 1.6 nm were prepared by the hydrothermal method, and then the ZnS:Mn inner-shell precursor solution and ZnS outer-shell precursor solution were added step-by-step to the prepared core QD solution and heated by microwave irradiation to synthesize CdSe/ZnS:Mn/ZnS core/shell/shell QDs. This process was initially carried out under the optimum conditions previously reported for synthesizing the ZnSe/ZnS:Mn/ZnS core/shell/shell QDs.21 As shown in Fig. S2,† the prepared CdSe/ZnS:Mn/ZnS QDs clearly displayed Mn PL. However, there was a simultaneous strong band-edge PL, suggesting that a certain amount of QDs in the solution were not doped with Mn ions. Therefore, when CdSe cores are used in place of ZnSe cores, a higher Mn ion concentration is necessary to increase Mn doping in the ZnS:Mn shell.
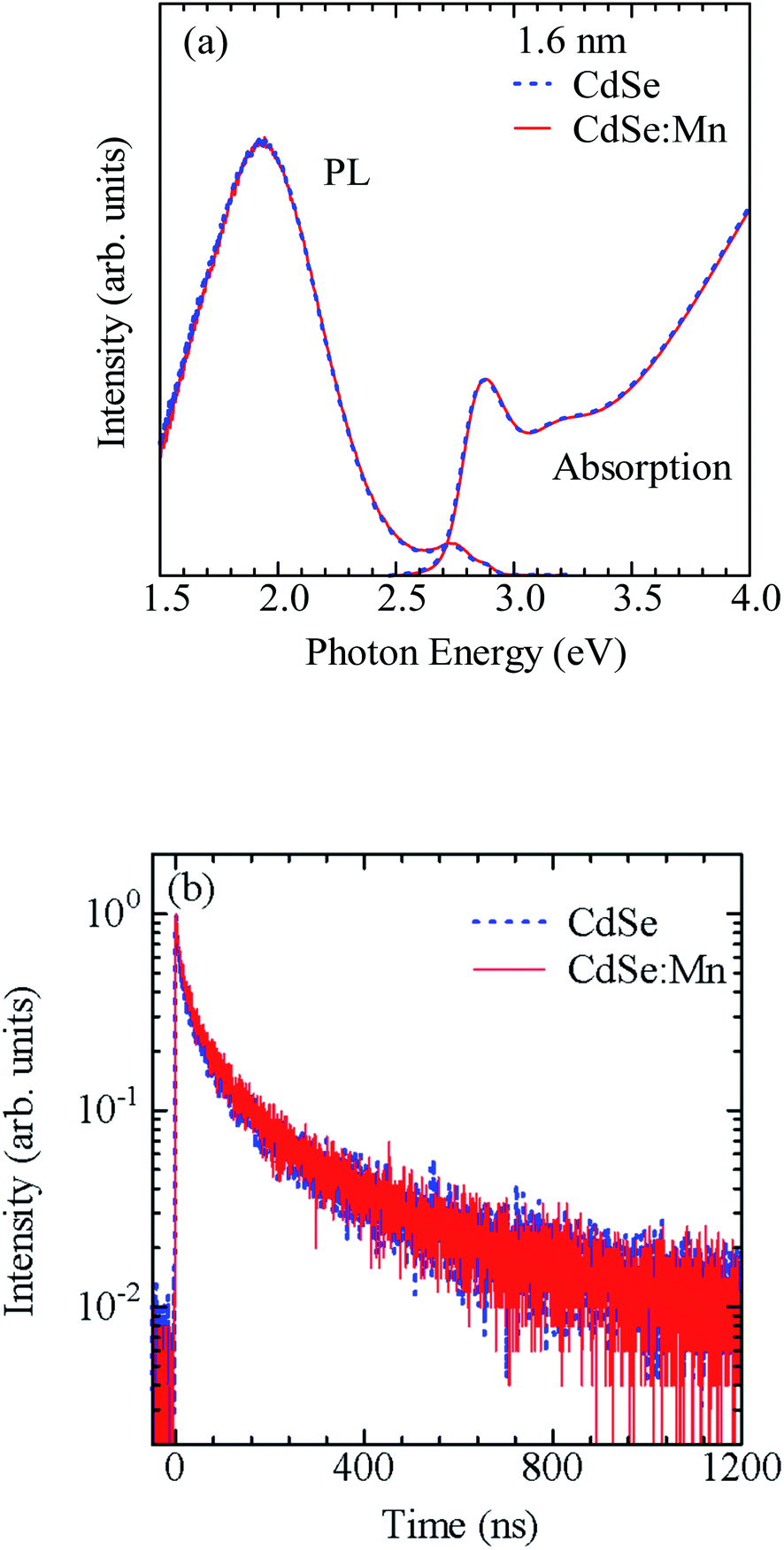
Based on the above consideration, we modified the preparation conditions as shown in Fig. S3.† (1) The addition and heating of the ZnS:Mn precursor solution were divided into two steps, and (2) the conditions for forming the ZnS:Mn shell layer were changed from 2 min at 120 °C to 15 min at 80 °C. Fig. 2a and b show the PL spectra of CdSe/ZnS:Mn/ZnS QDs prepared by changing the amount of ZnS:Mn shell precursor solution while fixing the CdSe core size at d = 1.6 nm. The molar ratio of Zn2+ to Mn2+ in the ZnS:Mn shell precursor solution was fixed at [Mn]/[Zn] = 0.15. The molar ratio of S2− ions in the ZnS:Mn shell precursor solution to Se2− ions in the CdSe core QD solution is defined as x = [S from ZnS:Mn]/[Se from CdSe core]. Hereafter, the value of x is referred to as the Mn preparation concentration.
 | ||
| Fig. 2 (a and b) PL spectra of CdSe/ZnS:Mn/ZnS QDs prepared using d = 1.6 nm CdSe core QDs and different amounts of ZnS:Mn shell precursor solution. Here, x = [S from ZnS:Mn shell precursor solution]/[Se from CdSe core precursor solution]. (c) Dependence of Mn PL intensity on x. | ||
Focusing on the PL spectra of CdSe/ZnS:Mn/ZnS QDs (x = 1) in Fig. 2a, a PL band with a peak near 2.2 eV, which is thought to be a defect-related PL, was observed because of the low Mn preparation concentration. For x = 2, the Mn PL intensity was stronger than that for x = 1 but still comparable to that of the band-edge PL. At higher x, the Mn PL intensity increased and became the main PL band for x > 4, as shown in Fig. 2b. Fig. 2c shows the plot for the x-dependent Mn PL intensity, which increases with x for x = 1–4 and decline at x > 5. Therefore, x = 4 is the optimal condition that maximises the Mn PL intensity for CdSe core QDs with d = 1.6 nm. The decrease in Mn PL intensity at x ≥ 5 is attributed to concentration quenching.65,66
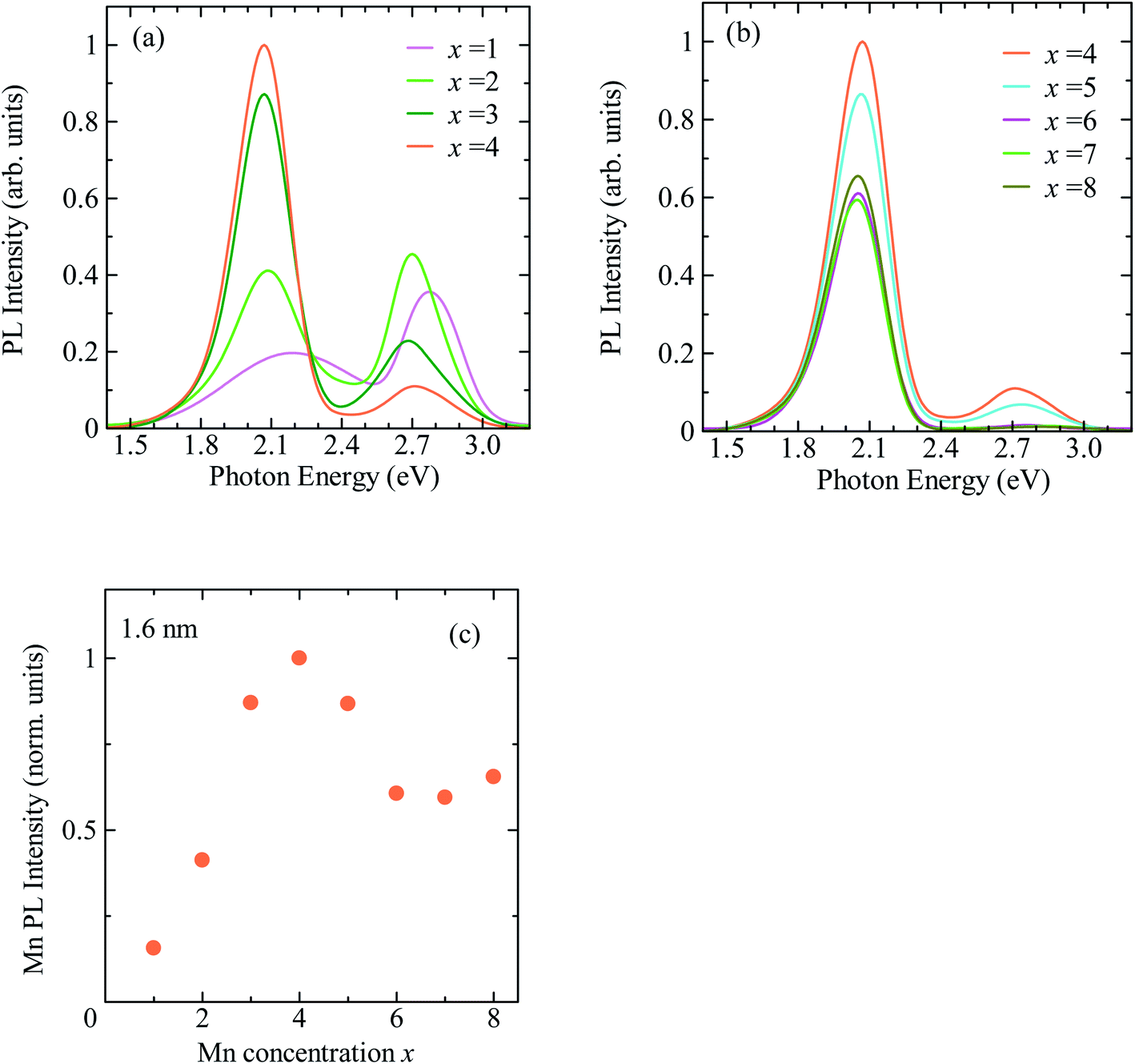
Fig. 3a shows the PL decay profiles detected at 2.0 eV for CdSe/ZnS:Mn/ZnS QDs synthesized with x = 1, 4, and 8. In all three cases, the PL profile shows a slow decay on the order of milliseconds, indicating that the PL band with a peak near 2 eV originates from the intra 3d-shell 4T1 → 6A1 transition of Mn2+.2,42,45 In the PL profile for x = 1, a fast decay component on the order of microseconds was observed in addition to the millisecond-order decay component. As shown in Fig. S4,† this fast decay component coincides with that of defect-related PL in the non-doped CdSe/ZnS QDs. Thus, the fast-decay component is considered to originate from the defect-related PL.
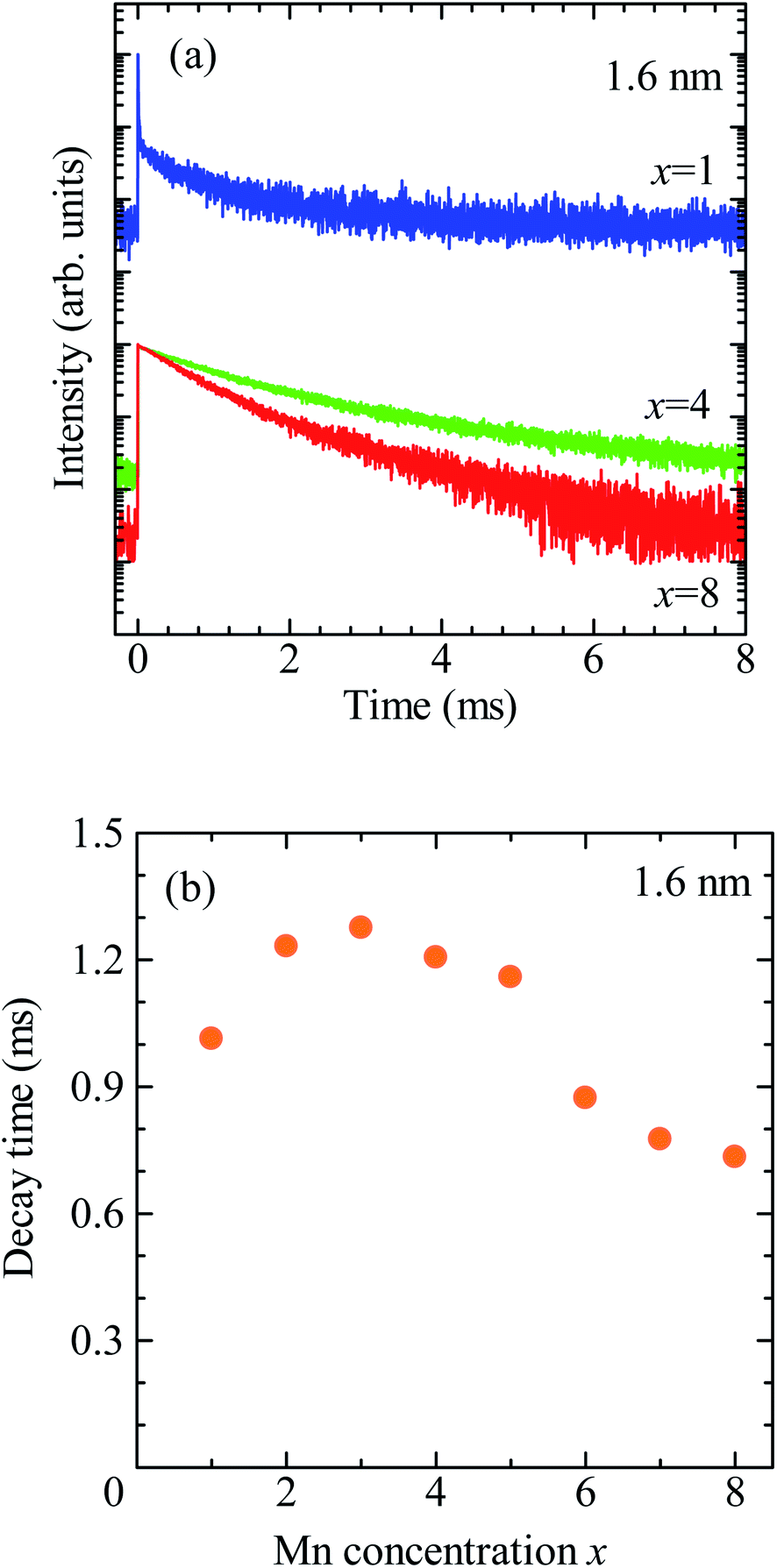 | ||
| Fig. 3 (a) PL decay profiles detected at 2.0 eV for CdSe/ZnS:Mn/ZnS QDs synthesized with x = 1, 4, and 8. (b) Dependence of average decay time of Mn PL on x. | ||
Next, we discuss the PL decay profile of CdSe/ZnS:Mn/ZnS QDs with x = 4, where the Mn PL intensity is at its maximum. The fast decay component on the order of microseconds was hardly observed, and a decay component on the order of milliseconds originating from Mn PL was observed. The PL decay profile was fitted using the sum of two single exponential functions I = A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2). The average lifetime was calculated using, 〈τ〉 = (A1τ12 + A2τ22)/(A1τ1 + A2τ2), where A1 and A2 represent the weight of each component. The obtained average decay time for x = 4 was 〈τx=4〉 = 2.0 ms. At the highest Mn doping level (x = 8), the PL decay profile shows faster decay than that for x = 4. Fig. 3b shows the plot for the average decay time of Mn PL against the x values. The intensity and decay time of Mn PL begin to decrease at the same x value, indicating that concentration quenching causes the decreased Mn emission intensity for x > 5. From these results, we conclude that the optimal condition for Mn2+ doping in CdSe QDs with a mean diameter of 1.6 nm is x = 4.
exp(−t/τ1) + A2exp(−t/τ2). The average lifetime was calculated using, 〈τ〉 = (A1τ12 + A2τ22)/(A1τ1 + A2τ2), where A1 and A2 represent the weight of each component. The obtained average decay time for x = 4 was 〈τx=4〉 = 2.0 ms. At the highest Mn doping level (x = 8), the PL decay profile shows faster decay than that for x = 4. Fig. 3b shows the plot for the average decay time of Mn PL against the x values. The intensity and decay time of Mn PL begin to decrease at the same x value, indicating that concentration quenching causes the decreased Mn emission intensity for x > 5. From these results, we conclude that the optimal condition for Mn2+ doping in CdSe QDs with a mean diameter of 1.6 nm is x = 4.
The PLQY of CdSe/ZnS:Mn/ZnS QDs with x = 4 was 51%. Typical TEM images of CdSe/ZnS:Mn/ZnS QDs are shown in Fig. 4a. A histogram of QD sizes is provided in Fig. 4b, which indicates a mean diameter of 3.0 nm. The absorption and PL spectra of Mn-doped CdSe/ZnS:Mn/ZnS QDs (x = 4) and non-doped CdSe/ZnS QDs are shown in Fig. 4c. Fig. 4d displays photographs of the two types of QDs under room light and UV excitation. The EDX measurements were used to estimate the Mn concentration in CdSe/ZnS:Mn/ZnS QDs to be 5 atom% of the total cation (Cd + Zn). The synthesized QDs were found to be stable even after one year, and no aggregation of QDs or decrease in the PL intensity were observed.
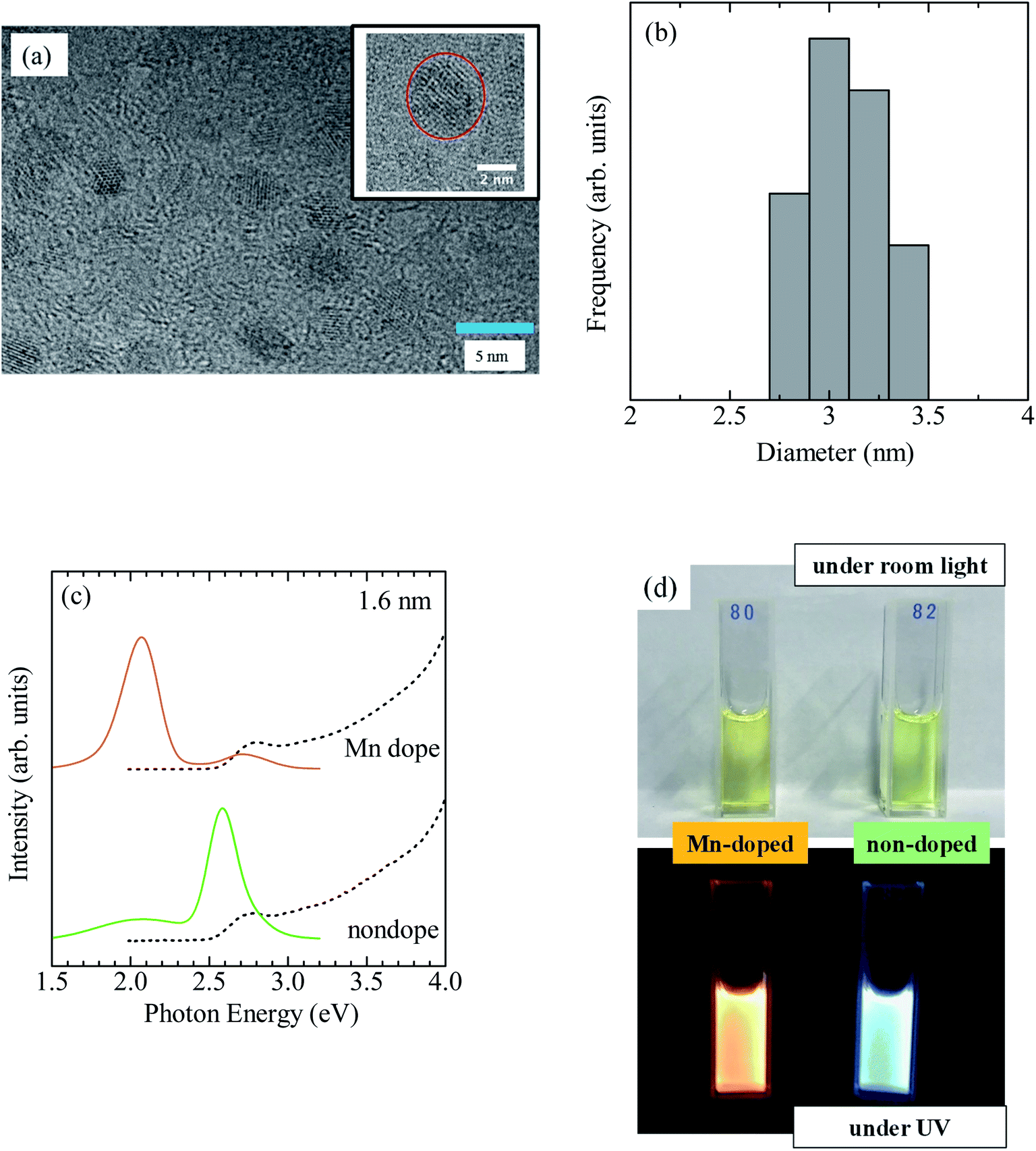 | ||
| Fig. 4 (a) TEM image of CdSe/ZnS:Mn/ZnS QDs (x = 4). The scale bar is 5 nm. The inset is a magnified image. (b) Histogram of the corresponding QD size. (c) Absorption (dashed curve) and PL (solid curve) of Mn-doped CdSe/ZnS:Mn/ZnS and non-doped CdSe/ZnS QDs. (d) Photographs of the QDs under room light and UV irradiation. | ||
Fig. 5a and b show Cd 3d and Se 3d XPS spectra, respectively, for CdSe core QDs with d = 1.6 nm. The XPS peaks observed at 404.5 and 411.2 eV correspond to the signals of Cd 3d5/2 and Cd 3d3/2, respectively.67 In addition, the XPS peak at 55.0 eV originates from Se 3d5/2. Fig. 5c shows Mn 2p XPS spectra of CdSe/ZnS:Mn core/shell QDs; the XPS signals corresponding to Mn 2p1/2 and 2p3/2 are clearly observed. The results of the XPS measurements for Cd 3d, Se 3d, and Zn 2p for CdSe/ZnSe:Mn/ZnS core/shell/shell QDs are shown in Fig. 5d, e, and f, respectively. The XPS signal intensities of Cd 3d and Se 3d for CdSe/ZnSe:Mn/ZnS QDs were lower than those for CdSe QDs. In contrast, the Zn 2p signal was clearly observed for CdSe/ZnSe:Mn/ZnS QDs. The composition ratios of Zn and Cd were estimated to be Zn: Cd = 0, 5.16, and 37.2 for CdSe, CdSe/ZnS:Mn, and CdSe/ZnS:Mn/ZnS QDs, respectively, by correcting Zn 2p and Cd 3d signal intensities with relative sensitivity factors. The Zn/Cd composition ratio was drastically increased due to the formation of the ZnS:Mn doping and ZnS capping shells. Therefore, the present XPS analysis results indicate the formation of multiple shells.
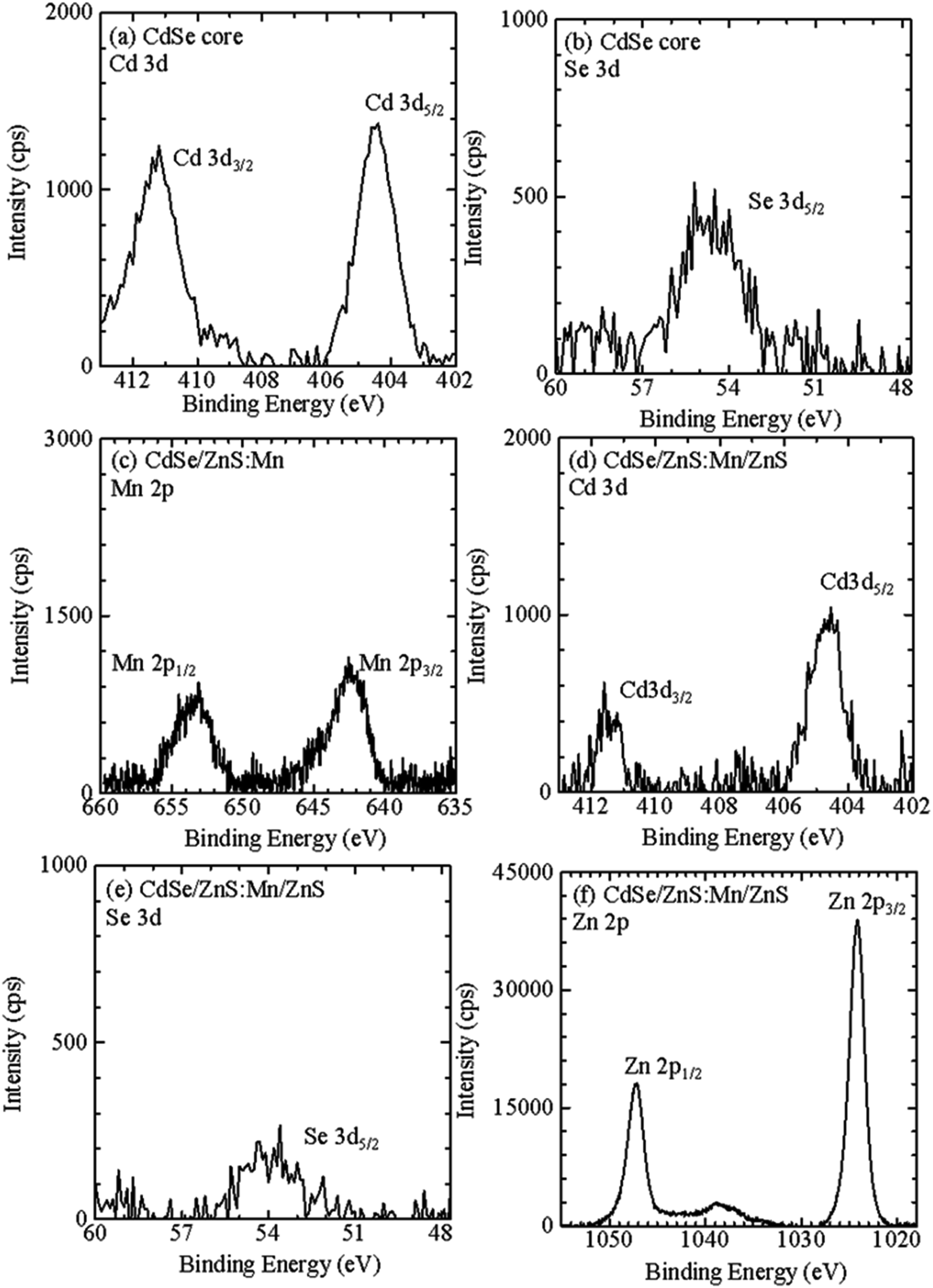 | ||
| Fig. 5 XPS spectra for (a) Cd 3d and (b) Se 3d of CdSe core QDs, for (c) Mn 2p of CdSe/ZnS:Mn core/shell QDs, and for (d) Cd 3d, (e) Se 3d, and (f) Zn 2p of CdSe/ZnS:Mn/ZnS core/shell/shell QDs. | ||
The CdSe/ZnS:Mn/ZnS QDs were synthesized using CdSe cores of different sizes (d = 1.7, 1.8, 1.9, 2.2, 2.4, and 2.7 nm) to investigate the optimum synthesis conditions for each case. Fig. S5 and S6† show the x-dependence of the PL spectra and Mn PL intensity, respectively. The optimum x values for d = 1.7, 1.8, 1.9, 2.2, 2.4, and 2.7 nm are 5, 4, 5, 4, 5, and 5, respectively. Fig. 6 shows the PL spectra of QDs synthesized under each optimum condition, and the corresponding decay profiles of Mn PL bands are shown in Fig. S7.† For d = 1.6–1.9 nm Mn PL is the main PL band, while for d = 2.2–2.7 nm the band-edge PL is stronger than Mn PL. According to Fig. S1,† a large core QD size means a low exciton energy and a smaller gap ΔE between it and the 4T1 state of Mn2+ dopant. Therefore, the thermal distribution from the 4T1 state of Mn2+ ions to the excitonic state increases, resulting in an increased fraction of the band-edge PL.42,45
 | ||
| Fig. 6 PL spectra of CdSe/ZnS:Mn/ZnS QDs synthesized with different core sizes (d = 1.6, 1.7, 1.8, 1.9, 2.2, 2.4, and 2.7 nm) and the corresponding optimum x values. | ||
As described above, the optimum x value did not change significantly even though the core size changed substantially between 1.6 and 2.7 nm. When the Mn2+ ions are directly doped into the host QDs, a longer heating time is needed to grow the QDs, which unfortunately induces a stronger self-purification effect that causes ejection of Mn2+ ions from the QDs and hinders Mn2+ doping. In contrast, as shown in Fig. 6, Mn doping can be achieved independently of the QD size when using the shell-doped method. In this method, undoped core QDs were synthesized at first, and then a shell layer containing the dopant was formed. This approach is versatile and may be applied to different host materials and dopant ions.
To confirm the broader applicability of the shell-doped method, Cu-doped ZnSe QDs (ZnSe/ZnS:Cu/ZnS) were synthesized. Fig. 7a shows the absorption and PL spectra of ZnSe/ZnS:Cu/ZnS QDs. For comparison, the spectra of the ZnSe/ZnS:Mn/ZnS and non-doped ZnSe/ZnS QDs are also shown. Although the three QDs exhibit similar absorption spectra, their PL spectra are significantly different. Cu-related PL band with a peak near 2.6 eV is observed for ZnSe/ZnS:Cu/ZnS QDs, whereas the band-edge PL with a peak near 3.3 eV and Mn PL with a peak near 2 eV are observed in non-doped QDs and Mn-doped QDs, respectively. Fig. 7b shows photographs of the water-soluble ZnSe/ZnS, ZnSe/ZnS:Cu/ZnS, and ZnSe/ZnS:Mn/ZnS QDs under the room light and UV excitation. The solution was highly transparent to visible light, and the emission colour of the QDs changed under UV irradiation, reflecting a shift in the origin of PL. Thus, we succeeded in controlling the PL properties of ZnSe QDs by doping Mn2+ or Cu2+ ions using the shell-doped method. Fig. S8a† shows a PL decay profile detected at 2.6 eV for the ZnSe/ZnS:Cu/ZnS QDs. The PL decay time of the ZnSe/ZnS:Cu/ZnS QDs is in the order of microseconds, which is consistent with the results reported so far for Cu-doped ZnSe QDs.33,36
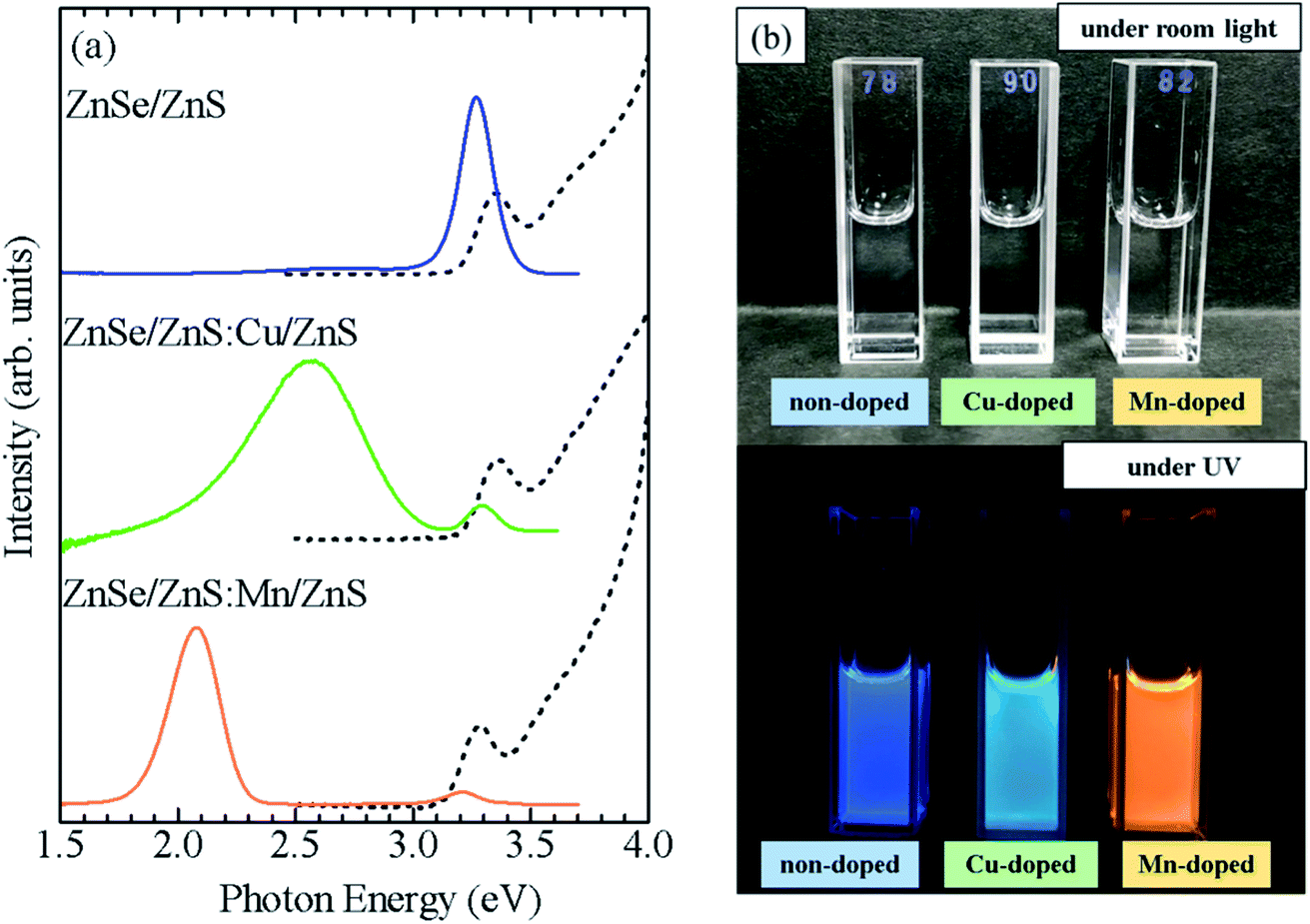 | ||
| Fig. 7 (a) Absorption (dashed curve) and PL (solid curve) spectra of Mn-doped ZnSe/ZnS:Mn/ZnS, Cu-doped ZnSe/ZnS:Cu/ZnS, and non-doped ZnSe/ZnS QDs. (b) Photograph of ZnSe/ZnS, ZnSe/ZnS:Cu/ZnS, and ZnSe/ZnS:Mn/ZnS QDs under room light and UV irradiation. | ||
Furthermore, CdSe/ZnS:Cu/ZnS QDs were synthesized using CdSe cores of two sizes (d = 1.6 and 2.7 nm). Fig. 8a shows the absorption and PL spectra, and photographs of the QD solutions under a room light and UV excitation are shown in Fig. 8b. The CdSe/ZnS:Cu/ZnS QDs with d = 1.6 and 2.7 nm showed PL bands with peaks near 1.9 and 1.5 eV, respectively. Because the Cu-related PL is due to the recombination of conduction band electron with the copper-localized hole, the energy of Cu-related PL depends on the diameter of the CdSe core as host.35 This differs from the Mn PL, in which the PL energy does not change with the QD size because the PL originates from electron transition in Mn ions. Fig. S8b† shows a PL decay profile for CdSe/ZnS:Cu/ZnS QDs. The PL decay time of ZnSe/ZnS:Cu/ZnS QDs is in the order of microseconds, which is consistent with the results reported so far for Cu-doped ZnSe QDs.33,36 These observations clearly demonstrate that the shell-doped method allows doping without any restriction in the material or size of the QDs.
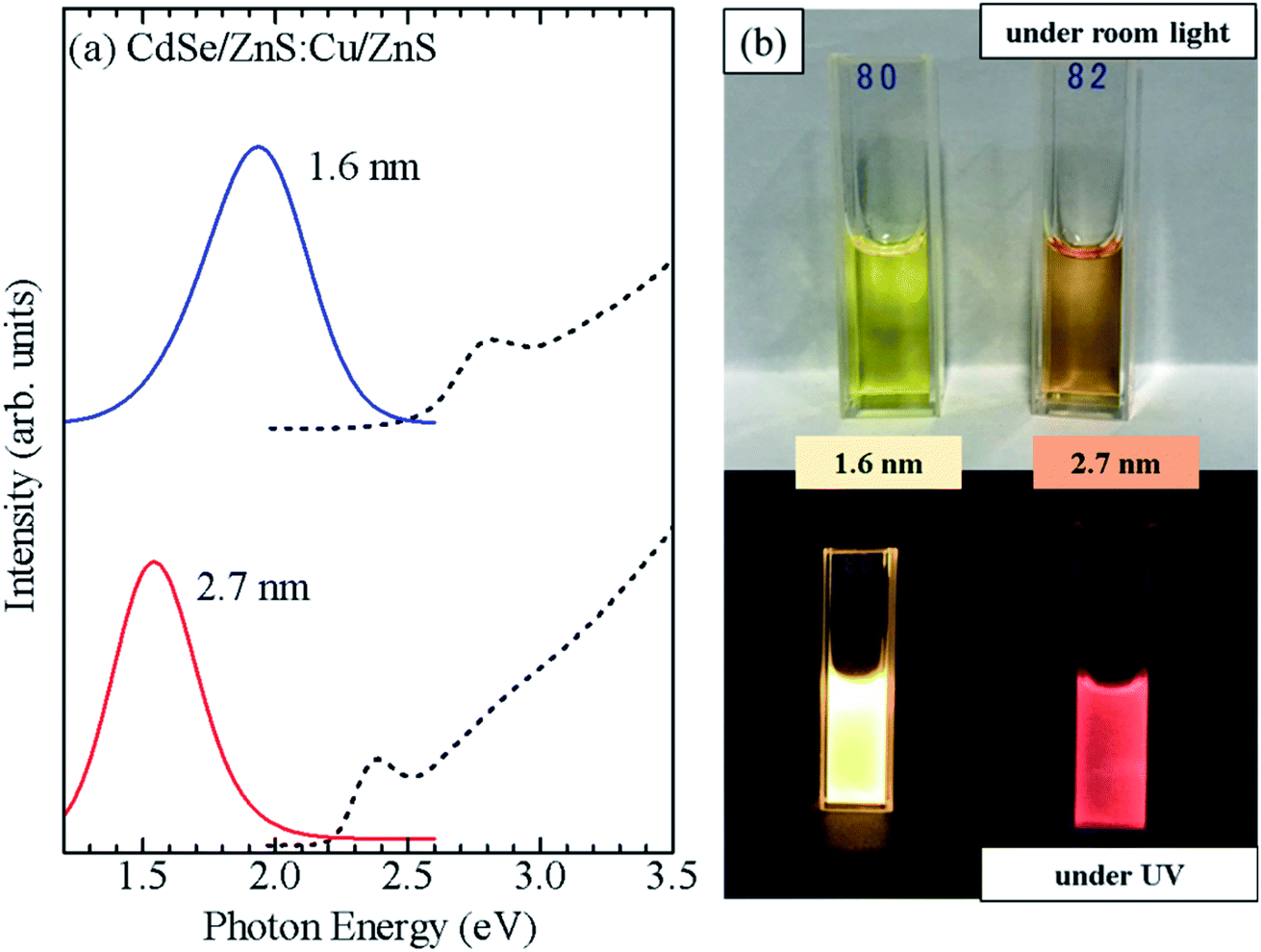 | ||
| Fig. 8 (a) Absorption (dashed curve) and PL (solid curve) spectra of CdSe/ZnS:Cu/ZnS QDs with core diameters of 1.6 and 2.7 nm. (b) Photograph of CdSe/ZnS:Cu/ZnS QDs synthesized using the CdSe core QDs with d = 1.6 and 2.7 nm and obtained under room light and UV irradiation. | ||
Conclusions
Herein, water-soluble CdSe/ZnS:Mn/ZnS core/shell/shell QDs were synthesized through a hydrothermal method to produce Mn2+-doped CdSe QDs with different sizes. The PL properties of doped QDs strongly depend on the energy gap between the exciton state (which can be controlled by the size of the CdSe core) and the 4T1 state of Mn2+ (which is independent of the QD size). For CdSe cores with d = 1.6–1.9 nm, energy transfer from excitons to Mn2+ is dominant due to the large ΔE, and Mn PL was observed as the main PL. In contrast, for CdSe cores with d = 2.2–2.7 nm, the band-edge PL was observed as the main PL band due to thermal activation from the 4T1 state of Mn2+ to the exciton state. Furthermore, CdSe/ZnS:Cu/ZnS and ZnSe/ZnS:Cu/ZnS QDs were synthesized using a similar approach with ZnS:Cu as the doping shell layer, and these QDs displayed Cu-related PL with different peak energies depending on the size and materials of the QDs. These results indicate that the core/doped shell/shell QD structure is applicable for synthesizing doped QDs regardless of the host material or the type of dopant.Author contributions
H. N. designed the experiment, prepared QDs, acquired and analysed optical spectra. K. E. acquired TEM images and XPS spectra. Y.-J. P. and D. K. provided conceptual advice. All authors contributed to the interpretation of the results and preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Grant-in-Aid for Scientific Research (B) (17H03538 and 20H02549) from the Japan Society for the Promotion of Science (KAKENHI). This work was also supported by the JST OPERA Program (JPMJOP1843).References
- U. Woggon, Optical Properties of Semiconductor Quantum Dots, Springer, New York, 1997 Search PubMed.
- R. Beaulac, S. T. Ochsenbein and D. R. Gamelin, in Nanocrystal Quantum Dots, ed. V. I. Klimov, CRC Press, New York, 2nd edn, 2010, ch.11, pp. 394–453 Search PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- V. I. Klimov, A. A. Mikhailovsky, S. Xu, A. Malko, J. A.Hollingsworth, C. A. Leatherdale, H.-J. Eisler and M. G. Bawendi, Science, 2000, 290, 314–317 CrossRef CAS PubMed.
- D. V. Talapin and J. Steckel, MRS Bull., 2013, 38, 685–691 CrossRef CAS.
- S. C. Erwin, L. Zu, M. I. Haftel, A. L. Efros, T. A. Kennedy and D. J. Norris, Nature, 2005, 436, 91–94 CrossRef CAS PubMed.
- D. J. Norris, A. L. Efros and S. C. Erwin, Science, 2008, 319, 1776–1779 CrossRef CAS PubMed.
- D. Bera, L. Qian, T. K. Tseng and P. H. Holloway, Materials, 2010, 3, 2260–2345 CrossRef CAS.
- N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 2574–2577 CrossRef CAS PubMed.
- H. Yang, S. Santra and P. H. Holloway, J. Nanosci. Nanotechnol., 2005, 5, 1364–1375 CrossRef CAS PubMed.
- N. Pradhan, D. M. Battaglia, Y. Liu and X. Peng, Nano Lett., 2007, 7, 312–317 CrossRef CAS PubMed.
- R. Beaulac, P. I. Archer, S. T. Ochsenbein and D. R. Gamelin, Adv. Funct. Mater., 2008, 18, 3873–3891 CrossRef CAS.
- L. W. Sun, H. Q. Shi, W. N. Li, H. M. Xiao, S. Y. Fu, X. Z. Cao and Z. X. Li, J. Mater. Chem., 2012, 22, 8221–8227 RSC.
- X. L. Yang, C. D. Pu, H. Y. Qin, S. J. Liu, Z. A. Xu and X. G. Peng, J. Am. Chem. Soc., 2019, 141, 2288–2298 CrossRef CAS PubMed.
- N. X. Ca, H. T. Van, P. V. Do, L. D. Thanh, P. M. Tan, N. X. Truong, V. T. K. Oanh, N. T. Binh and N. T. Hien, RSC Adv., 2020, 10, 25618–25628 RSC.
- P. K. Santra and P. V. Kamat, J. Am. Chem. Soc., 2012, 134, 2508–2511 CrossRef CAS PubMed.
- C. S. Erickson, L. R. Bradshaw, S. McDowall, J. D. Gilbertson, D. R. Gamelin and D. L. Patrick, ACS Nano, 2014, 8, 3461–3467 CrossRef CAS PubMed.
- L. R. Bradshaw, K. E. Knowles, S. McDowall and D. R. Gamelin, Nano Lett., 2015, 15, 1315–1323 CrossRef CAS PubMed.
- H. Nishimura, Y. X. Lin, M. Hizume, T. Taniguchi, N. Shigekawa, T. Takagi, S. Sobue, S. Kawai, E. Okuno and D. Kim, AIP Adv., 2019, 9, 025223 CrossRef.
- H. Nishimura, Y. X. Lin, M. Hizume, T. Taniguchi, N. Shigekawa, T. Takagi, S. Sobue, S. Kawai, E. Okuno and D. Kim, Chem. Lett., 2019, 48, 1081–1083 CrossRef CAS.
- H. Nishimura, T. Maekawa, K. Enomoto, N. Shigekawa, T. Takagi, S. Sobue, S. Kawai and D. Kim, J. Mater. Chem. C, 2021, 9, 693–701 RSC.
- J. Kossut and J. A. Gaj, Introduction to the Physics of Diluted Magnetic Semiconductors, Springer, Berlin, 2010 Search PubMed.
- C. Ronning, P. X. Gao, Y. Ding and Z. L. Wang, Appl. Phys. Lett., 2004, 84, 783–785 CrossRef CAS.
- A. Kudelski, A. Lemaître, A. Miard, P. Voisin, T. C. M. Graham, R. J. Warburton and O. Krebs, Phys. Rev. Lett., 2007, 99, 247209 CrossRef CAS PubMed.
- C. Le Gall, L. Besombes, H. Boukari, R. Kolodka, J. Cibert and H. Mariette, Phys. Rev. Lett., 2009, 102, 127402 CrossRef CAS PubMed.
- J. Kobak, T. Smoleński, M. Goryca, M. Papaj, K. Gietka, A. Bogucki, M. Koperski, J.-G. Rousset, J. Suffczyński, E. Janik, M. Nawrocki, A. Golnik, P. Kossacki and W. Pacuski, Nat. Commun., 2014, 5, 3191 CrossRef CAS PubMed.
- R. Fainblat, C. J. Barrow, E. Hopmann, S. Siebeneiche, V. A. Vlaskin, D. R. Gamelin and G. Bacher, Nano Lett., 2016, 16, 6371–6377 CrossRef CAS PubMed.
- J. K. Bindra, K. Shingh, J. van Tol, N. S. Dalal and G. F. Strouse, J. Phys. Chem. C, 2020, 124, 19348–19354 CrossRef CAS.
- N. Pradhan, D. Goorskey, J. Thessing and X. Peng, J. Am. Chem. Soc., 2005, 127, 17586–17587 CrossRef CAS PubMed.
- S. Jana, B. B. Srivastava, S. Acharya, P. K. Santra, N. R. Jana, D. D. Sarma and N. Pradhan, Chem. Commun., 2010, 46, 2853–2855 RSC.
- G. K. Grandhi and R. Viswanatha, J. Phys. Chem. Lett., 2013, 4, 409–415 CrossRef CAS PubMed.
- C. Wang, S. Xu, Y. Wang, Z. Wang and Y. Cui, J. Mater. Chem. C, 2014, 2, 660–666 RSC.
- R. Zeng, R. Shen, Y. Zhao, Z. Sun, X. Li, J. Zheng, S. Cao and B. Zou, CrystEngComm, 2014, 16, 3414–3423 RSC.
- H. Zhang, J. Liu, C. Wang, G. S. Selopal, D. Barba, Z. M. Wang, S. Sun, H. Zhao and F. Rosei, ACS Photo, 2019, 6, 2421–2431 CrossRef CAS.
- R. Viswanatha, S. Brovelli, A. Pandey, S. A. Crooker and V. I. Klimov, Nano Lett., 2011, 11, 4753–4758 CrossRef CAS PubMed.
- K. E. Knowles, H. D. Nelson, T. B. Kilburn and D. R. Gamelin, J. Am. Chem. Soc., 2015, 137, 13138–13147 CrossRef CAS PubMed.
- L. Jing, S. V. Kershaw, Y. Li, X. Huang, Y. Li, A. L. Rogach and M. Gao, Chem. Rev., 2016, 116, 10623–10730 CrossRef CAS PubMed.
- P. I. Archer, S. A. Santangelo and D. R. Gamelin, Nano Lett., 2007, 7, 1037–1043 CrossRef CAS PubMed.
- C. J. Barrows, V. A. Vlaskin and D. R. Gamelin, J. Phys. Chem. Lett., 2015, 15, 3076–3081 CrossRef PubMed.
- C. J. Barrows, R. Fainblat and D. R. Gamelin, J. Mater. Chem. C, 2017, 5, 5232–5238 RSC.
- J. Jiang, L.-A. T. Nguyen, T. D. Nguyen, D. H. Luong, D. Y. Kim, Y. Jin, P. Kim, D. L. Duong and Y. H. Lee, Phys. Rev. B, 2021, 103, 014441 CrossRef CAS.
- R. Beaulac, P. I. Archer, X. Liu, S. Lee, G. M. Salley, M. Dobrowolska, J. K. Furdyna and D. R. Gamelin, Nano Lett., 2008, 8, 1197–1201 CrossRef CAS PubMed.
- P. Wu and X.-P. Yan, Chem. Soc. Rev., 2013, 42, 5489–5521 RSC.
- B. Peng, J. W. May, D. R. Gamelin and X. Li, J. Phys. Chem. C, 2014, 118, 7630–7636 CrossRef CAS.
- R. Beaulac, P. I. Archer, J. van Rijssel, A. Meijerink and D. R. Gamelin, Nano Lett., 2008, 8, 2949–2953 CrossRef CAS PubMed.
- V. A. Vlaskin, N. Janssen, J. van Rijssel, R. Beaulac and D. R. Gamelin, Nano Lett., 2010, 10, 3670–3674 CrossRef CAS PubMed.
- G. M. Dalpian and J. R. Chelikowsky, Phys. Rev. Lett., 2006, 96, 226802 CrossRef PubMed.
- Al. L. Efros and M. Rosen, Annu. Rev. Mater. Sci., 2000, 30, 475–521 CrossRef CAS.
- M. Makkar and R. Viswanath, RSC Adv., 2018, 8, 22103–22112 RSC.
- R. N. Bhargava, D. Gallagher, X. Hong and A. Nurmikko, Phys. Rev. Lett., 1994, 72, 416–419 CrossRef CAS PubMed.
- D. J. Norris, N. Yao, F. T. Charnock and T. A. Kennedy, Nano Lett., 2001, 1, 3–7 CrossRef CAS.
- B. T. Luong, E. Hyeong, S. Ji and N. Kim, RSC Adv., 2012, 2, 12132–12135 RSC.
- N. Pradhan and X. Peng, J. Am. Chem. Soc., 2007, 11, 3339–3347 CrossRef PubMed.
- J. Yang, R. Fainblat, S. G. Kwon, F. Muckel, J. H. Yu, H. Terlinden, B. H. Kim, D. Iavarone, M. K. Choi, I. Y. Kim, I. Park, H. K. Hong, J. Lee, J. S. Son, Z. Lee, K. Kang, S. J. Hwang, G. Bacher and T. Hyeon, J. Am. Chem. Soc., 2015, 137, 12776–12779 CrossRef CAS PubMed.
- Y. Kanemitsu and H. Matsubara, Appl. Phys. Lett., 2002, 81, 535–537 CrossRef CAS.
- M. A. Malik, P. O'Brien and N. Revaprasadu, J. Mater. Chem., 2001, 11, 2382–2386 RSC.
- J. Eilers, E. Groeneveld, C. D. Donegá and A. Meijerink, J. Phys. Chem. Lett., 2012, 12, 1663–1667 CrossRef PubMed.
- N. O. Dantas, A. S. Silva, E. S. F. Freitas Neto and S. A. Lourenco, Phys. Chem. Chem. Phys., 2012, 14, 3520–3529 RSC.
- W. Liu, Q. Lin, H. Li, K. Wu, I. Robel, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2016, 138, 14954–14961 CrossRef CAS PubMed.
- M. C. De Siena, D. E. Sommer, S. E. Creutz, S. T. Dunham and D. R. Gamelin, Chem. Mater., 2019, 31, 7711–7722 CrossRef CAS.
- A. Wang, F. Muhammad, Y. Liu and Z. Deng, Chem. Commun., 2021, 57, 2677–2680 RSC.
- C. L. Gan, Y. P. Zhang, D. Battaglia, X. G. Peng and M. Xiao, Appl. Phys. Lett., 2008, 92, 241111 CrossRef.
- T. Lee, K. Noguchi, H. Nishimura and D. Kim, J. Phys.: Conf. Ser., 2019, 1220, 012028 CrossRef CAS.
- C. M. Donegá and R. Koole, J. Phys. Chem. C, 2009, 113, 6511–6520 CrossRef.
- C. Pu, J. Ma, H. Qin, T. Fu, Y. Niu, X. Yang, Y. Huang, F. Zhao and X. Peng, ACS Cent. Sci., 2016, 2, 32–39 CrossRef CAS PubMed.
- M. Greben, P. Khoroshyy, I. Sychugov and J. Balenta, Appl. Spectro. Rev., 2019, 54, 1–44 CrossRef.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, ed. J. Chastain, Physical Electronics, Minnesota, 1995 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08491g |
| This journal is © The Royal Society of Chemistry 2022 |