
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-free synthesis of 3,5-isoxazoles via 1,3-dipolar cycloaddition of terminal alkynes and hydroxyimidoyl chlorides over Cu/Al2O3 surface under ball-milling conditions†
Rafael A. Hernandez R. a,
Kelly Burchell-Reyesa,
Arthur P. C. A. Bragaa,
Jennifer Keough Lopeza and
Pat Forgione*ab
a,
Kelly Burchell-Reyesa,
Arthur P. C. A. Bragaa,
Jennifer Keough Lopeza and
Pat Forgione*ab
aDepartment of Chemistry and Biochemistry, Concordia University, 7141 rue Sherbrooke O., Montréal, QC, Canada H4B 1R6. E-mail: pat.forgione@concordia.ca
bCentre for Green Chemistry and Catalysis, McGill University, 801 rue Sherbrooke O., Montréal, QC, Canada H3A 0B8
First published on 23rd February 2022
Abstract
Scalable, solvent-free synthesis of 3,5-isoxazoles under ball-milling conditions has been developed. The proposed methodology allows the synthesis of 3,5-isoxazoles in moderate to excellent yields from terminal alkynes and hydroxyimidoyl chlorides, using a recyclable Cu/Al2O3 nanocomposite catalyst. Furthermore, the proposed conditions are reproducible to a 1.0-gram scale without further milling time variations.
The addition of oxygen or nitrogen-containing heterocycles in drug candidates has become a common feature of the recently approved drugs by the FDA.1,2 In particular, isoxazoles are common molecular scaffolds employed in medicinal chemistry due to the non-covalent interactions such as hydrogen bonding (through the N) and π–π stacking (by the unsaturated 5-membered ring).3–6 Within the isoxazole family, 3,5-isoxazoles (1) are regularly utilized as pharmacophores in medicinal chemistry.2,5,6 Selected examples including muscimol (GABAa agonist), isocarboxazib (antidepressant), isoxicam (anti-inflammatory), berzosertib (ATR kinase inhibitor), and sulfamethoxazole (antibiotic) are highlighted in Fig. 1.7–10
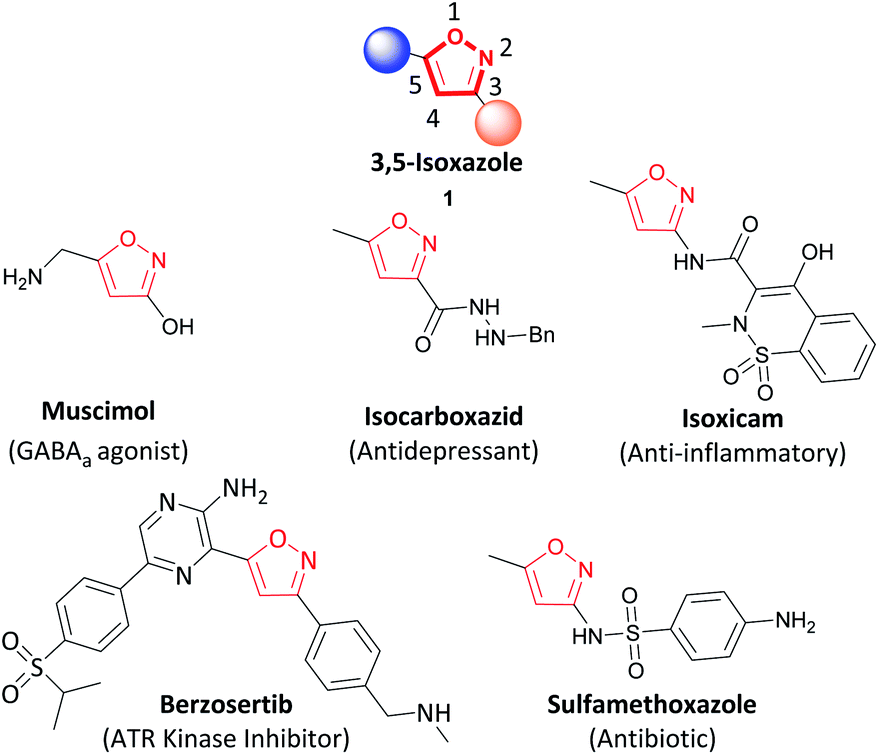 | ||
| Fig. 1 Examples of isoxazoles with pharmacological activity. | ||
Various methodologies to synthesize 3,5-isoxazoles have been developed over the years.7,9,11–13 Specifically, 1,3-dipolar cycloaddition between terminal alkynes (2) and nitrile oxides (4) formed in situ by deprotonation of hydroxyimidoyl chlorides (3) is a standard route to access 3,5-isoxazoles (1) (Fig. 2).7,9,14 Recent reports have sought to mitigate the environmental impact of this reaction by performing 1,3-dipolar cycloaddition under solvent-free conditions, using green solvents such as water or ionic liquids, under metal-free conditions, or using mild oxidants.14–28 However, these methodologies have a low atom economy, have a higher hazardous waste production, and are less energy efficient. Therefore, developing a greener methodology that enables rapid and efficient access to these scaffolds is highly desirable.
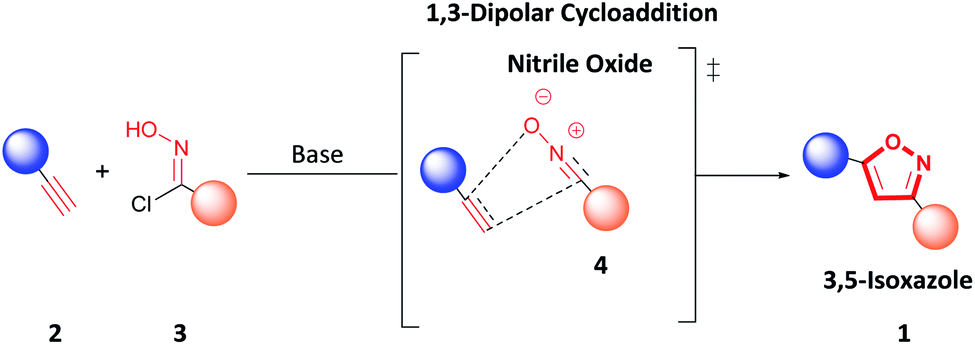 | ||
| Fig. 2 1,3-Dipolar cycloaddition of terminal alkynes and nitrile oxides. | ||
Mechanochemistry has been recognized as an environmentally friendly technique as reactions can be performed under solvent-free conditions. Additionally, in some instances, work-up and purification are simplified or absent from procedures, and the process consumes less energy than other solution-based techniques.29–33 The use of mechanochemical techniques to synthesize isoxazoles is limited. Sherin et al. reported a synthesis of 3,5-isoxazoles (7) by grinding in a mortar and pestle curcumin derivatives (5), hydroxylamine (6), and sub-stoichiometric amounts of acetic acid to form the 3,5-isoxazole (7) in short times and excellent yields (Fig. 3a).34 Likewise, Xu et al. studied the synthesis of trisubstituted isoxazoles (10) via 1,3-dipolar cycloaddition of N-hydroxybenzimidoyl chlorides (8) and N-substituted β-enamino carbonyl (9) compounds by ball-milling (Fig. 3b) in high yields, short reaction times, in the absence of catalyst and liquid additives.35 To our knowledge, mechanochemical synthesis of 3,5-isoxazoles (1) from terminal alkynes (2) and hydroxyimidoyl chloride (3) has not been reported (Fig. 3c). The proposed methodology employs a planetary ball-milling technique that provides a route to access in large scale, short reaction times, and high atom economy the corresponding 3,5-isoxazoles (1). Additionally, it utilizes synthetically accessible or commercially available motifs such as terminal alkynes (2) and hydroxyimidoyl chlorides (3) that are recurrent or easily installed in many substrates. Herein, we report a mechanochemical 1,3-dipolar cycloaddition using the planetary ball-mill to synthesize a wide range 3,5-isoxazoles from a broad library of alkynes and (E,Z)-N-hydroxy-4-nitrobenzimidoyl chloride (3a), ethyl (E,Z)-2-chloro-2-(hydroxyimino)acetate (3b), hydroxycarbonimidic dibromide (3c), or (E,Z)-N-hydroxy-4-methoxybenzimidoyl chloride (3d) in moderate to excellent yields, in short reaction time, and with less waste production than in solution based reactions (Fig. 3c).
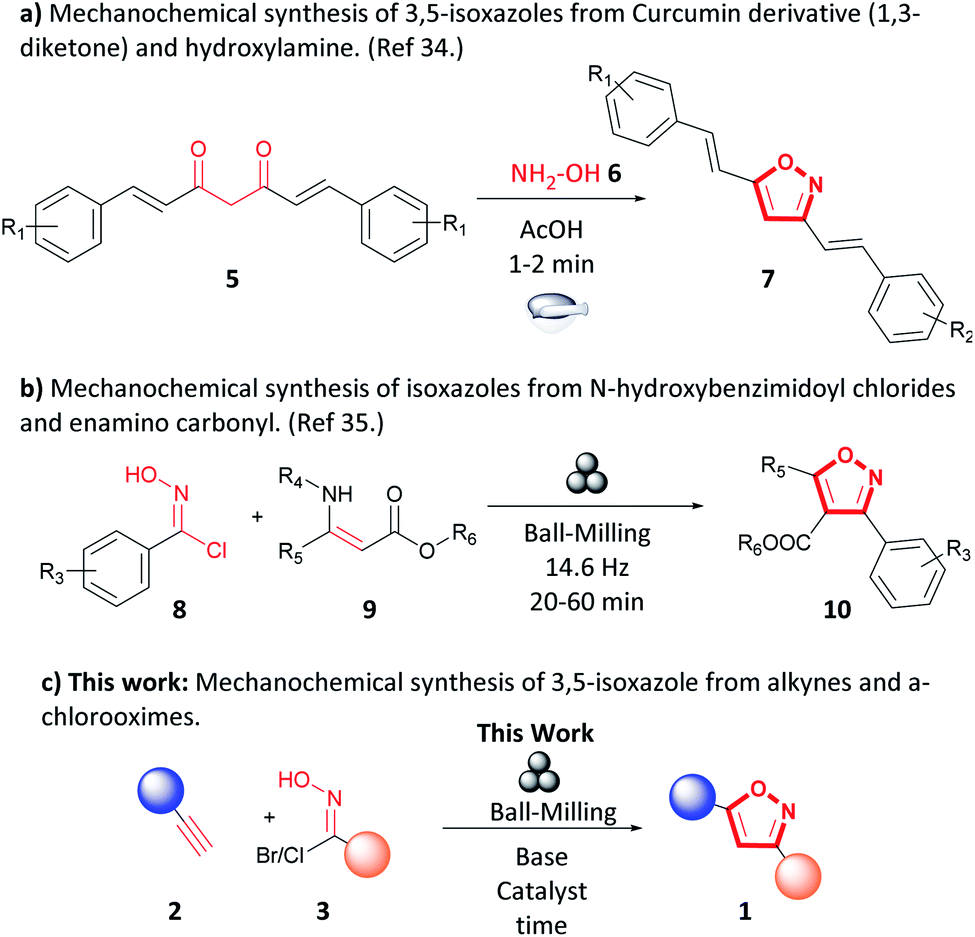 | ||
| Fig. 3 Previously reported synthesis of isoxazoles. | ||
We began our investigation by performing an optimisation of the 1,3-dipolar cycloaddition reaction between alkyne 2a and hydroxyimidoyl chlorides 3a by milling the selected substrates in a stainless-steel (SS) jar in the planetary ball-mill to obtain 3,5-isoxazole 1a (Table 1). During the optimization, the effect of milling time, amount of milling media, base, and equivalents of hydroxyimidoyl chlorides were studied to obtain the highest yield of 3,5-isoxazole 1a (Table 1). Optimization revealed the combination of 1.0 equivalent of alkyne 2a, 1.5 equivalents of hydroxyimidoyl chlorides 3a, and 2.0 equivalents of Na2CO3 while milling for 20 minutes with 8 SS balls provided the most effective conditions (Table 1, entry 1). Our first control experiments focused on optimizing the milling time (Table 1, entries 2–5). Milling the reagents for less than 20 minutes affords lower product yields (Table 1, entries 2 and 3). Conversely, milling the reagents longer than 20 minutes leads to a decrease in yield to about 60% (Table 1, entries 4 and 5). The strong abrasion of the SS milling media for extended periods could lead to a ring-opening by a reduction of the N–O to yield β-keto-enamine.36,37
|
||
|---|---|---|
| Entry | Changes from optimized conditions | Yieldb (%) of 1a |
| a Reaction Conditions: 0.166 mmol of 2a, 0.250 mmol of 3a, 0.332 mmol of Na2CO3, SS beaker (50 mL capacity), 8 × SS milling balls (10 mm diameter), 20 min milling, 60 Hz.b 1H-NMR yields were measured using 1,3,5-trimethoxybenzene as an internal standard. | ||
| 1 | None | 72 |
| 2 | Milling for 10 min, 7 SS balls | 59 |
| 3 | Milling for 15 min 7 SS balls | 64 |
| 4 | Milling for 30 min | 58 |
| 5 | Milling for 40 min | 60 |
| 6 | Using 1.0 equiv. of 3a | 65 |
| 7 | Using 2.0 equiv. of 3a | 57 |
| 8 | Using K2CO3 | 71 |
| 9 | Using Cs2CO3 | 71 |
| 10 | Using CaCO3 | 44 |
| 11 | Using Ag2CO3 | 18 |
| 12 | Using NEt3 | N.R. |
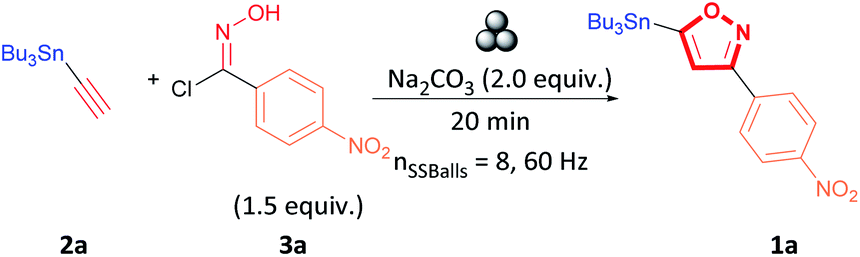
Having optimized the milling time, we next attempted to improve the yield by varying the equivalents of hydroxyimidoyl chlorides 3a since reaction stoichiometry has been shown to impact the product formed during mechanochemical reactions.38,39 3,5-isoxazole 1a was obtained in lower yields when using equimolar amounts alkyne 2a to hydroxyimidoyl chlorides 3a (entry 6, Table 1). Because nitrile oxides rapidly dimerize to form furoxans by a competing 1,3-dipolar cycloaddition.40–45 Likewise, increasing the equivalents of 18a from 1.0 to 2.0 equivalents lowered the yield of the reaction (entry 7, Table 1). We obtained the highest yield with 1.5 equivalents of the hydroxyimidoyl chlorides of 3a, and these conditions were used for further experiments (entry 1, Table 1). We next studied the effect of diverse carbonated bases in the reaction. We observed that changing the base did not improve the yield of the reaction, and Ag2CO3 was most detrimental to the reaction as it promoted furoxan formation (entries 8–11, Table 1).46,47 Using triethylamine (NEt3) proved impractical as the addition of NEt3 to hydroxyimidoyl chlorides was highly exothermic in the absence of solvent (entry 12, Table 1).
During the screening, we observed that milling time influences the reaction yield.35,48 Therefore, a milling time optimization for other alkyne and hydroxyimidoyl chloride combinations revealed that the most optimal milling time was determined to be between 10 and 30 minutes (see ESI† for milling time optimizations).
As shown in Fig. 4, stannanyl isoxazole 1a and 1b, silyl isoxazole 1c, and phenyl isoxazole 1d were synthesized with satisfactory yields under the proposed conditions. To explain these results, we suggest an electronic argument. The electron-withdrawing character of the metal substituents, stannyl or silyl of alkyne 2a and 2b, respectively, accelerates the reaction by deactivating the alkyne moiety.49–51 It is observed that alkyne 2a bearing the alkylstannane substituent has a more pronounced effect than the alkyne with the silyl substituent (2b). Therefore, alkyne 2a was the most reactive as it reacted with hydroxyimidoyl chlorides 3a and 3b to synthesize 3,5-isoxazole 1a and 1b respectively, in short times and excellent yields (Fig. 4). On the other hand, ethynyltrimethylsilane (2b) was less reactive as it could only react with a more labile hydroxyimidoyl chlorides 3b to form 3,5-isoxazole 1c (Fig. 4). Comparably, we suggest that the phenyl substituent of alkyne 1c increases the polarizability of the molecule, resulting in deactivating the alkyne moiety. As a result, phenylacetylene (1c) reacted in excellent yields with hydroxyimidoyl chlorides 1b.42 In addition, we observed that the electronic nature of the hydroxyimidoyl chloride substituent affects the reactivity of the nitrile oxide dipole. Hydroxyimidoyl chlorides 3a containing an aromatic substituent with strong electron-withdrawing groups decreased the reactivity of the nitrile oxide.52,53 Consequently, the nitrile oxide synthesized in situ from hydroxyimidoyl chlorides 3a could only react with tributyl(ethynyl)stannane (2a). On the other hand, hydroxyimidoyl chlorides 3b was the most reactive due to the bearing of a weaker electron-withdrawing group such as the ester functional group.43,52 Unfortunately, other alkynes containing substituents such as esters, pyridines, or substituted arenes were not tolerated under these conditions. Previous reports demonstrated the effect of copper catalyst or copper additives to accelerate the reaction and obtain the 3,5-isoxazoles in a regioselective manner.15,53–59 Therefore, we aimed to investigate the effect of copper additives or catalysts on this reaction.
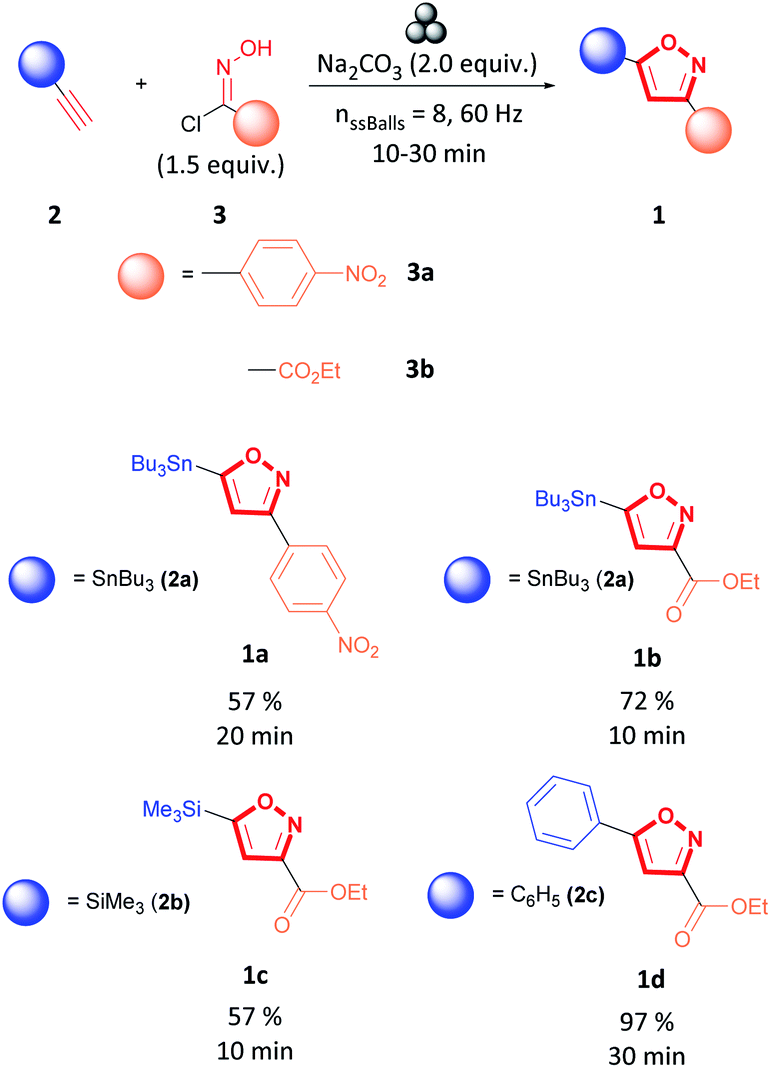 | ||
| Fig. 4 Catalyst-free mechanochemical synthesis of 3,5-isoxazoles. | ||
Although the mechanochemical synthesis of 3,5-isoxazoles using copper(II) catalyst is unprecedented, 1,2,3-triazoles have been synthesized in this way with copper(II) salts and copper(II) ions in alumina nanocomposites (Cu/Al2O3).60,61 We investigated the effect of Cu/Al2O3 (see ESI† for XPS spectrum) and copper salts using methyl propiolate (2d) and (E,Z)-2-chloro-2-(hydroxyimino)acetate (3b) as model substrates (Table 2).
|
||||
|---|---|---|---|---|
| Entry | Cu(II) | Equivalents | Time (min) | Yieldb (%) 19e |
| a Reaction conditions: 0.220 mmol of 2d, 0.330 mmol of 3b, 0.220 mmol of Na2CO3, 0.440 mmol (14 mol%) of Cu/Al2O3, SS beaker (50 mL capacity), 8 × SS milling balls (10 mm diameter), 60 Hz.b 1H NMR yields were measured using 1,3,5-trimethoxybenzene as an internal standard.c See ESI for solid-state characterization by FT-IR and MALDI-TOF-MS of reaction crude 1e. | ||||
| 1 | Cu/Al2O3 | 0.14 of Cu(II) | 10 | 73 |
| 20 | 76 | |||
| 30 | 79 | |||
| 40 | 64 | |||
| 50 | 56 | |||
| 2 | Cu(NO3)2·2.5H2O | 0.1 | 30 | 78 |
| 3 | Cu(NO3)2·2.5H2O | 1.0 | 30 | 84 |
| 4 | Cu(OAc)2·H2O | 1.0 | 30 | 88 |
| 5 | Cu(OTf)2 | 1.0 | 30 | 76 |
| 6 | CuCl2·H2O | 1.0 | 30 | 76 |
| 7 | Cu2CO3(OH)2 | 2.0 | 30 | 36 |
We observed a significant increase in yield and regioselective control when using sub-stoichiometric amounts of copper (0.14 equivalents or 14 mol%) of Cu/Al2O3 or 10 mol% of Cu(NO3)2·2.5H2O while milling the reagents for 30 minutes (entries 1 and 2, Table 2). Irreproducible yields were obtained by decreasing the catalytic loading to 7 mol% of Cu/Al2O3. In contrast, when the equivalents of Cu(NO3)2·2.5H2O were increased to 1.0 equivalent, we observed no significant increase in yield (entry 3, Table 2). Additionally, when investigating the effect of the counter anion on the copper(II), it was observed that Cu(OAc)2·H2O performs similarly to Cu(NO3)2·2.5H2O (entry 4, Table 2), while Cu(OTf)2 and CuCl2·H2O produced lower yields (entries 5 and 6, Table 2). Substituting Na2CO3 with Cu2CO3(OH)2 lowered the yield drastically (entry 7, Table 2). Interestingly, the addition of copper salts did not lead to homocoupling of the alkyne moiety by the Glaser reaction.62–64
We decided to continue our investigations using Cu/Al2O3 as the catalyst can be filtered and washed with solvent, thereby facilitating catalyst recovery and recycling (Fig. 5).60
 | ||
| Fig. 5 (a) Filtration of the Cu/Al2O3 catalyst after the first run. (b) Colour change of the Cu/Al2O3 catalyst after recycling. From left to right. (left) Fresh catalyst: blue. (middle) First recycle: green. (right) Second recycle: brown. | ||
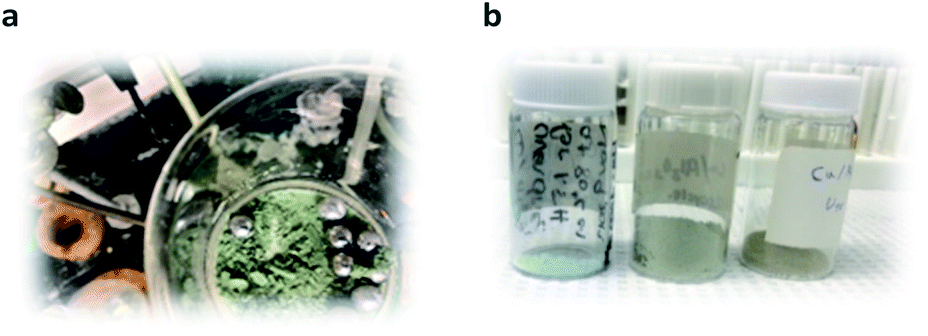
The Cu/Al2O3 catalyst effect was not exclusively beneficial for the cycloaddition with methyl propiolate (2d) (3,5-isoxazole 1e, Fig. 6). This system improves the reactivity of hydroxyimidoyl chlorides 3a, 3b, 3c, and 3d and other alkynes inaccessible under copper-free conditions, thus allowing access to a broader library of 3,5-isoxazoles (Fig. 6). Moreover, the presence of Cu/Al2O3 nanocomposite as part of the reaction conditions is not impaired by the presence of labile substituents such as silanes (1c, f–h), alkyl halides (1i–j), and boronic esters (1n) (Fig. 6). However, the presence of alkyl stannane substituents in the dipolarophile (2a) was not tolerated with Cu/Al2O3 catalyst, and no product was observed. Furthermore, Cu/Al2O3 enhances the reactivity of dipolarophiles bearing arenes with electron-donating substituents (EDG) (1o–q) and electron-withdrawing groups (EWG) (1n, 1r-2) when coupled with hydroxyimidoyl chlorides 3a and 3b. Additionally, pyridine substituents were more reactive towards the more reactive hydroxyimidoyl chlorides (3b) (3,5-isoxazole 1s, Fig. 6). Ethynyltrimethylsilane (1c) reacted efficiently with hydroxyimidoyl chlorides bearing EWG (3a, 3b, and 3c) to form the respective isoxazoles 1c,1f, and 1h, where silyl isoxazole 1c is obtained in higher yields compared to copper-free conditions (1c, Fig. 4). Hydroxyimidoil chloride bearing EDG (3d); resulted incompatible with terminal alkyne 2b and silyl isoxazole 1g was obtained in lower yields than with EWG in the hydroxyimidoyl chloride. However, terminal alkynes having an aliphatic substituent (2e and 2f) showed greater reactivity towards hydroxyimidoil chloride (3d) bearing EDG; consequently, aliphatic isoxazole 1j was obtained in higher yields than 1i. Then, we evaluated the impact of our conditions in the synthesis of 3,5-isoxazole 1f on a 1.0-gram scale (10.18 mmol). We were pleased to observe that the optimized Cu/Al2O3 conditions can be translated with excellent reproducibility from a 100 mg scale to a 1.0-gram scale without extending the milling time of the reagents (Fig. 6).
 | ||
| Fig. 6 Mechanochemical synthesis of 3,5-isoxazoles reaction scope. aAll shown yields are isolated yields. bReaction performed in 1.0-gram scale. | ||
The practicality of the proposed methodology allows the recovery of Cu/Al2O3 nanocomposite catalyst directly after the milling of the reagents. In addition, the catalyst recovery allowed investigating the reusability of the recovered catalyst. The Cu/Al2O3 was reused on four occasions, and it was observed that 3,5-isoxazole 1f was obtained successfully with only a minimal drop in yield with each subsequent use for the first two recycling cycles (Fig. 7). The decrease in yield is explained by the decrease in the concentration of active Cu species in the Cu/Al2O3 nanocomposite (see ESI†). ICP-MS analysis demonstrates that the Cu concentration of the first recycling represents a decrease of 1.24-fold (with respect to the fresh catalyst); thus, similar yields are obtained compared to the fresh catalyst (Fig. 7). However, the decrease in Cu concentration becomes more substantial for the second and third reuse with a decrease of 2.42 and 6.48-fold, respectively. Therefore, a considerable decrease in the yield of isoxazole 1f is observed. Furthermore, a change in the oxidation state and the bonding of the supported Cu(II) ions. X-ray Photoelectron Spectroscopy (XPS) analysis of the first and second recycled catalyst reveals that the characteristic satellite signals of Cu(II) found at about 942.8 eV are weak while the satellite signal at 963.2 eV is absent. Additionally, the 2p3/2 signal at about 933–934 eV is wider than in the fresh sample (see ESI for XPS spectra of the fresh, Fig. S3 for first recycling and Fig. S4† for second recycling). These observations suggest that the supported Cu(II) is reduced to Cu(0) and CuO is formed with each subsequent recycling.65–67
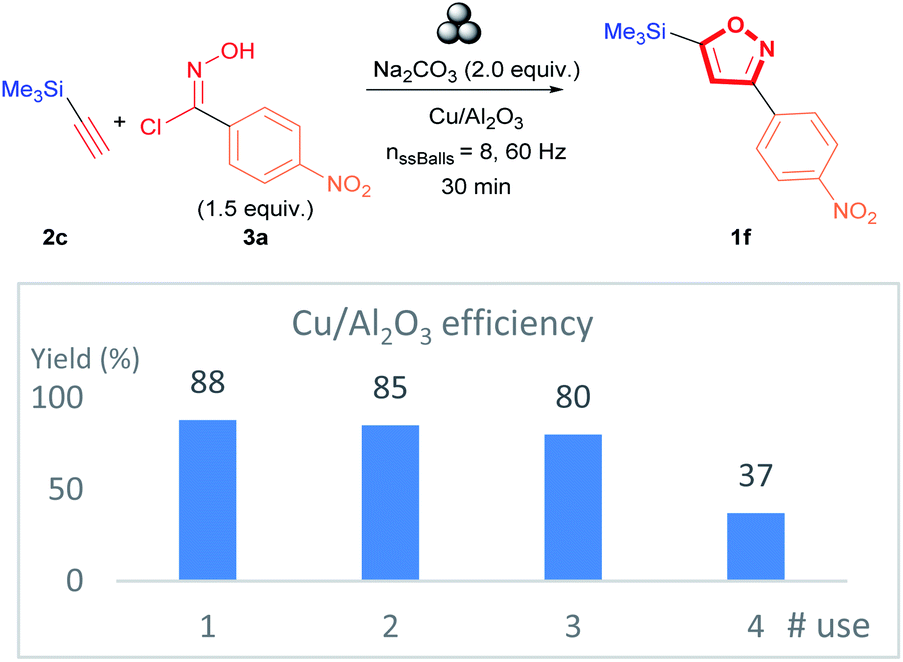 | ||
| Fig. 7 Cu/Al2O3 efficiency study in the synthesis of 3,5-isoxazole 1f. | ||
Lastly, we evaluated the sustainability of the proposed mechanochemical 1,3-dipolar cycloaddition conditions by comparing E-factor for the synthesis of 3,5 isoxazoles 1d and 1f to previously reported solution-based conditions (Fig. 8).54,68 Using E-factor, the values calculated for the planetary ball milling conditions (pathway a and c, Fig. 8) demonstrate the sustainability of this methodology compared to solution-based reactions (pathway b and d) (see ESI† for calculations). With our conditions, the absence of organic solvent is the most significant factor contributing to lowering the E-factor.69 Time differences were also another factor of comparison with previously reported solution-based conditions. Our mechanochemical conditions did not surpass 60 minutes, contrary to the reported solution-based conditions that require at least two hours to synthesize the desired 3,5-isoxazoles. Furthermore, our conditions did not show any sensitivity to oxygen or moisture present in the air as all reactions were performed in an open atmosphere.
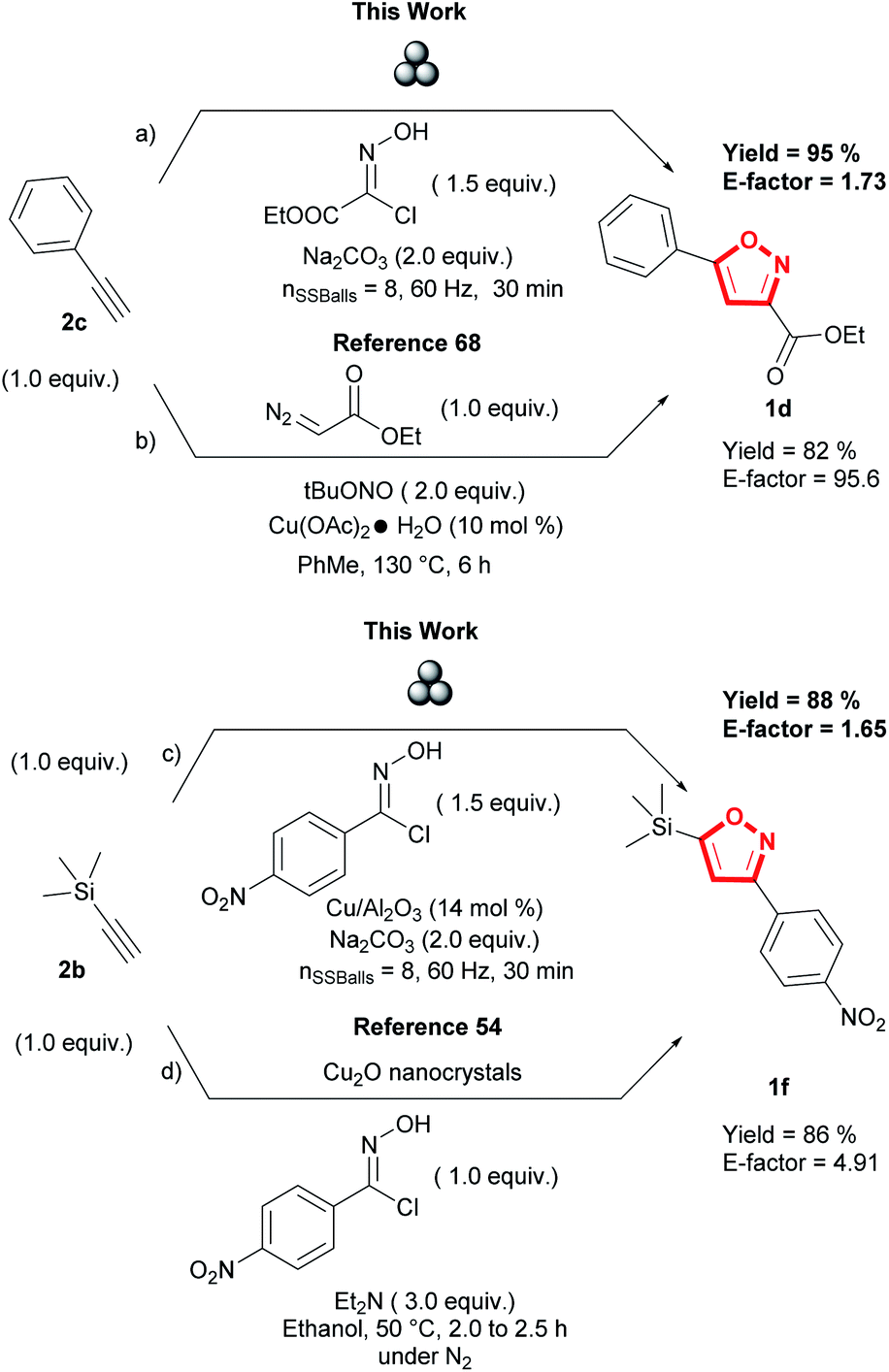 | ||
| Fig. 8 Comparative green metrics of the proposed methodology to previously reported solution-based methodologies. | ||
Conclusions
In conclusion, we have developed a scalable, solvent-free, and efficient mechanochemical synthesis of 3,5-isoxazoles via 1,3-dipolar cycloaddition from terminal alkynes and hydroxyimidoyl chlorides. We presented two methodologies; a catalyst-free methodology which scopes extended to dipolarophiles bearing alkyl stannane substituent. Under catalyst-free conditions, ethynyltrimethylsilane (2c) and phenylacetylene (2d) reacted satisfactorily with (E,Z)-2-chloro-2-(hydroxyimino)acetate (3b). Additionally, a Cu/Al2O3 mediated methodology allowed to react a broader range of dipolarophiles bearing electron-donating or electron-withdrawing substituents with any hydroxyimidoyl chloride. The reported methodology was scalable to a 1.0-gram scale without additional milling time variations. The Cu/Al2O3 catalyst was demonstrated to easily be recycled and reused three times with only a slight reduction in yield. The reported conditions require shorter reaction times, they had a lower E-factor, and no prevention was taken to air or moisture, making these methodologies less environmentally harmful and more practical than previously reported solution-based methodologies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Natural Sciences and Engineering Research Council (NSERC) and Le Fonds de Recherche du Québec, Nature et Technologies (FRQNT). Support was also kindly provided by the Centre for Green Chemistry and Catalysis (CGCC), and the Richard and Edith Strauss Foundation. Special thanks to Jiang Tian Liu and Fadil Taç for their valuable advice.Notes and references
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- M. D. Delost, D. T. Smith, B. J. Anderson and J. T. Njardarson, J. Med. Chem., 2018, 61, 10996–11020 CrossRef CAS PubMed.
- K. P. Rakesh, C. S. Shantharam, M. B. Sridhara, H. M. Manukumar and H. L. Qin, Med. Chem. Commun., 2017, 8, 2023–2039 RSC.
- P. P. Sharp, J. M. Garnier, D. C. S. Huang and C. J. Burns, Med. Chem. Commun., 2014, 5, 1834–1842 RSC.
- A. Sysak and B. Obmińska-Mrukowicz, Eur. J. Med. Chem., 2017, 137, 292–309 CrossRef CAS PubMed.
- J. Zhu, J. Mo, H. Lin, Y. Chen and H. Sun, Bioorg. Med. Chem., 2018, 26, 3065–3075 CrossRef CAS PubMed.
- M. Kim, Y. S. Hwang, W. Cho and S. B. Park, ACS Comb. Sci., 2017, 19, 407–413 CrossRef CAS PubMed.
- L. Gorecki, M. Andrs, M. Rezacova and J. Korabecny, Pharmacol. Ther., 2020, 210, 107518 CrossRef CAS PubMed.
- F. Hu and M. Szostak, Adv. Synth. Catal., 2015, 357, 2583–2614 CrossRef CAS.
- H. Zinnes, J. C. Sircar, N. Lindo, M. L. Schwartz, A. C. Fabian, J. Shavel, C. F. Kasulanis, J. D. Genzer, C. Lutomski and G. DiPasquale, J. Med. Chem., 1982, 25, 12–18 CrossRef CAS PubMed.
- S. Roscales and J. Plumet, Org. Biomol. Chem., 2018, 16, 8446–8461 RSC.
- L. Claisen and O. Lowman, Ber. Dtsch. Chem. Ges., 1888, 21, 1149–1157 CrossRef.
- J. Li, Z. Lin, W. Wu and H. Jiang, Org. Chem. Front., 2020, 7, 2325–2348 RSC.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- T. V. Hansen, P. Wu and V. V. Fokin, J. Org. Chem., 2005, 70, 7761–7764 CrossRef CAS PubMed.
- J. Li, J. Yu, W. Xiong, H. Tang, M. Hu, W. Wu and H. Jiang, Green Chem., 2020, 22, 465–470 RSC.
- D. Wang, F. Zhang, F. Xiao and G. J. Deng, Org. Biomol. Chem., 2019, 17, 9163–9168 RSC.
- D. R. Meena, B. Maiti and K. Chanda, Tetrahedron Lett., 2016, 57, 5514–5517 CrossRef CAS.
- S. B. Bharate, A. K. Padala, B. A. Dar, R. R. Yadav, B. Singh and R. A. Vishwakarma, Tetrahedron Lett., 2013, 54, 3558–3561 CrossRef CAS.
- J. M. Pérez and D. J. Ramón, ACS Sustainable Chem. Eng., 2015, 3, 2343–2349 CrossRef.
- A. M. Jawalekar, E. Reubsaet, F. Rutjes and F. van Delft, Chem. Commun., 2011, 47, 3198–3200 RSC.
- M. Vadivelu, S. Sampath, K. Muthu, K. Karthikeyan and C. Praveen, J. Org. Chem., 2019, 84, 13636–13645 CrossRef CAS PubMed.
- A. Yoshimura, K. R. Middleton, A. D. Todora, B. J. Kastern, S. R. Koski, A. V. Maskaev and V. V. Zhdankin, Org. Lett., 2013, 15, 4010–4013 CrossRef CAS PubMed.
- L. Lin, J. Zhang and R. Wang, Asian J. Org. Chem., 2012, 1, 222–225 CrossRef CAS.
- S. Mohammed, R. A. Vishwakarma and S. B. Bharate, RSC Adv., 2015, 5, 3470–3473 RSC.
- C. Kesornpun, T. Aree, C. Mahidol, S. Ruchirawat and P. Kittakoop, Angew. Chem., Int. Ed., 2016, 55, 3997–4001 CrossRef CAS PubMed.
- B. Touaux, F. Texier-Boullet and J. Hamelin, Heteroat. Chem., 1998, 9, 351–354 CrossRef CAS.
- M. Hu, Z. Lin, J. Li, W. Wu and H. Jiang, Green Chem., 2020, 22, 5584–5588 RSC.
- F. Schneider, T. Szuppa, A. Stolle, B. Ondruschka and H. Hopf, Green Chem., 2009, 11, 1894–1899 RSC.
- D. Tan, L. Loots and T. Friščić, Chem. Commun., 2016, 52, 7760–7781 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed, W. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- D. R. Sherin and K. N. Rajasekharan, Arch. Pharm. Chem. Life Sci., 2015, 908–914 CrossRef CAS PubMed.
- H. Xu, G. P. Fan, Z. Liu and G. W. Wang, Tetrahedron, 2018, 74, 6607–6611 CrossRef CAS.
- S. Auricchio, A. Bini, E. Pastormerlo and A. M. Truscello, Tetrahedron, 1997, 53, 10911–10920 CrossRef CAS.
- M. J. Rak, N. K. Saadé, T. Friščić and A. Moores, Green Chem., 2014, 16, 86–89 RSC.
- V. Štrukil, D. Margetic, M. D. Igrc, M. Eckert-Maksic and T. Frišcic, Chem. Commun., 2012, 48, 9705–9707 RSC.
- J. L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- J. W. Bode, Y. Hachisu, T. Matsuura and K. Suzuki, Tetrahedron Lett., 2003, 44, 3555–3558 CrossRef CAS.
- C. Grundmann and J. M. Dean, J. Org. Chem., 1965, 30, 2809–2812 CrossRef CAS.
- R. Huigens, Angew. Chem., Int. Ed., 1963, 2, 633–696 CrossRef.
- C. Grundmann and R. Richter, J. Org. Chem., 1967, 32, 2308–2312 CrossRef CAS PubMed.
- T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed.
- C. Grundmann and S. K. Datta, J. Org. Chem., 1968, 34, 2016–2018 CrossRef.
- P. F. Pagoria, M. X. Zhang, N. B. Zuckerman, A. J. DeHope and D. A. Parrish, Chem. Heterocycl. Compd., 2017, 53, 760–778 CrossRef CAS.
- M. X. Zhang, A. J. Dehope and P. F. Pagoria, Org. Process Res. Dev., 2019, 23, 2527–2531 CrossRef CAS.
- F. Schneider, A. Stolle, B. Ondruschka and H. Hopf, Org. Process Res. Dev., 2009, 13, 44–48 CrossRef CAS.
- T. Deb, J. Tu and R. M. Franzini, Chem. Rev., 2021, 121, 6850–6914 CrossRef CAS PubMed.
- E. Lukevics and P. Arsenyan, Chem. Heterocycl. Compd., 1998, 34, 1155–1169 Search PubMed.
- D. K. Heldmann and J. Sauer, Tetrahedron Lett., 1997, 38, 5791–5794 CrossRef CAS.
- M. S. Chiang and J. U. Lowe, J. Org. Chem., 1967, 32, 1577–1579 CrossRef.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- K. Chanda, S. Rej and M. H. Huang, Nanoscale, 2013, 5, 12494–12501 RSC.
- X. Di Wang, L. H. Zhu, P. Liu, X. Y. Wang, H. Y. Yuan and Y. L. Zhao, J. Org. Chem., 2019, 84, 16214–16221 CrossRef PubMed.
- V. V. S. T. M. Vishwanatha, J. Heterocycl. Chem., 2015, 52, 1823–1833 CrossRef.
- C. Chen and S. Cui, J. Org. Chem., 2019, 84, 12157–12164 CrossRef CAS PubMed.
- Y. Sun, A. Abdukader, H. Zhang, W. Yang and C. Liu, RSC Adv., 2017, 7, 55786–55789 RSC.
- Y. Li, M. Gao, B. Liu and B. Xu, Org. Chem. Front., 2017, 4, 445–449 RSC.
- N. Mukherjee, S. Ahammed, S. Bhadra and B. C. Ranu, Green Chem., 2013, 15, 389–397 RSC.
- R. Thorwirth, A. Stolle, B. Ondruschka, A. Wild and U. S. Schubert, Chem. Commun., 2011, 47, 4370–4372 RSC.
- K. S. Sindhu and G. Anilkumar, RSC Adv., 2014, 4, 27867–27887 RSC.
- R. Schmidt, R. Thorwirth, T. Szuppa and A. Stolle, Chem.–Eur. J., 2011, 17, 8129–8138 CrossRef CAS PubMed.
- L. Chen, B. E. Lemma, J. S. Rich and J. Mack, Green Chem., 2014, 16, 1101–1103 RSC.
- Y. Wen, W. Huang and B. Wang, Appl. Surf. Sci., 2012, 258, 2935–2938 CrossRef CAS.
- F. E. López-Suárez, A. Bueno-López, M. J. Illán-Gómez, A. Adamski, B. Ura and J. Trawczynski, Environ. Sci. Technol., 2008, 42, 7670–7675 CrossRef PubMed.
- J. B. Reitz and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 11467–11478 CrossRef CAS.
- X. Di Wang, L. H. Zhu, P. Liu, X. Y. Wang, H. Y. Yuan and Y. L. Zhao, J. Org. Chem., 2019, 84, 16214–16221 CrossRef PubMed.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08443g |
| This journal is © The Royal Society of Chemistry 2022 |