
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCycloaddition of Huisgen 1,4-dipoles: synthesis and rapid epimerization of functionalized spiropyrido[2,1-b][1,3]oxazine-pyrroles and related products†
Andrew R. Galeev‡
 ,
Anna A. Moroz‡,
Maksim V. Dmitriev* and
Andrey N. Maslivets*
,
Anna A. Moroz‡,
Maksim V. Dmitriev* and
Andrey N. Maslivets*
Department of Chemistry, Perm State University, ul. Bukireva 15, Perm, 614068, Russia. E-mail: dmax@psu.ru; koh2@psu.ru
First published on 22nd December 2021
Abstract
1,4-Dipolar cycloaddition has emerged as a powerful tool for the synthesis of various cyclic compounds. In the present work, 1H-pyrrole-2,3-diones are proposed as new dipolarophiles for 1,4-dipolar cycloaddition. Their [4 + 2] cycloaddition with dipoles generated from dimethyl acetylenedicarboxylate and pyridine was found to proceed regioselectively affording spiro[pyrido[2,1-b][1,3]oxazine-2,3′-pyrroles] as diastereomeric mixtures which exist in rapid equilibrium in solution. It was established that this phenomenon of rapid epimerization is a characteristic of other similar spiropyrido[2,1-b][1,3]oxazines and even related spiroquinolizines, which was demonstrated by the investigation of related products of previously reported, and reproduced in this work, 1,4-dipolar cycloaddition reactions.
Introduction
In the last two decades, researchers have shown an increased interest in 1,4-dipolar cycloaddition.1 Zwitterionic 1,4-dipolar species, that can be generated both by metal catalysis1a,b,2 and without catalysts,1d,e,3 have been successfully applied in the preparation of diverse carbo- and heterocycles. However, 1,4-dipole chemistry is still far less developed than 1,3-dipole chemistry.Within the field of 1,4-dipolar cycloaddition, the reactions of 1,4-dipoles, generated from dimethyl acetylenedicarboxylate (DMAD) or other activated acetylenes and azaheterocycles, the so-called Huisgen 1,4-dipoles, with carbonyl compounds (isatins,4 quinones,4b,5 aldehydes,4c,6 etc.7), alkenes,8 alkynes9 and imines4c,10 have been extensively studied (Scheme 1). Nevertheless, the search for new dipolarophiles, especially those containing several reaction sites, is still relevant.
 | ||
| Scheme 1 Cycloaddition of 1,4-dipoles, generated from acetylenes and heterocycles. | ||
We have a long-standing interest in the chemistry of 1H-pyrrole-2,3-diones,11 which contain at least three dipolarophilic sites providing their diverse reactivity. Recently, we have reported several 1,3-dipolar cycloaddition reactions involving an endo-cyclic double bond12 of 1H-pyrrole-2,3-diones as well as both C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C3O carbonyl groups (Scheme 2A),13 that differs from reactivity of related isatins14 (only the addition to C3O; Scheme 2B). To the best of our knowledge, the dipolar cycloaddition of 1H-pyrrole-2,3-diones with 1,4-dipoles has not been reported. Considering the above facts, we could expect the formation of at least three distinct types of skeletally diverse cycloadducts A–C in such reactions (Scheme 3). Herein, we report the first study on the 1,4-dipolar cycloaddition of 1H-pyrrole-2,3-diones with dipoles generated from DMAD and pyridine.
O and C3O carbonyl groups (Scheme 2A),13 that differs from reactivity of related isatins14 (only the addition to C3O; Scheme 2B). To the best of our knowledge, the dipolar cycloaddition of 1H-pyrrole-2,3-diones with 1,4-dipoles has not been reported. Considering the above facts, we could expect the formation of at least three distinct types of skeletally diverse cycloadducts A–C in such reactions (Scheme 3). Herein, we report the first study on the 1,4-dipolar cycloaddition of 1H-pyrrole-2,3-diones with dipoles generated from DMAD and pyridine.
 | ||
| Scheme 2 Cycloaddition of 1H-pyrrole-2,3-diones and isatins with 1,3-dipoles. | ||
Results and discussion
Initially, we carried out the reaction of 1,5-diphenyl-1H-pyrrole-2,3-dione 1a with DMAD 2a and pyridine in CHCl3 at room temperature (RT) and successfully isolated the spirocyclic [4 + 2] cycloadduct 3a as a diastereomeric mixture in a yield of 52% (Scheme 4). The structure of compound 3a was confirmed by the single crystal X-ray analysis (CCDC 2119396†). Noteworthy, only C3O cycloaddition product 3a (type B, Scheme 3) was detected in the reaction mixture by HPLC-MS, while regioisomeric products A and C (Scheme 3) were not observed, probably due to the fact that ketone C3O group is more electrophilic than lactam C2O group or enone C5C4 moiety. 1H NMR analysis (in CDCl3) of the reaction mixture, as well as isolated product 3a, showed a diastereomeric ratio (dr) of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. However, an additional set of signals belonging to unidentified minor impurities was observed in the 1H NMR spectrum of product 3a, which was pure by HPLC, and the intensity of these signals increased with time. We assumed that these impurities were the result of the compound decomposition15 in a CDCl3 solution which is known to be slightly acidic due to its degradation during storage. As expected, when the spectrum was recorded in DMSO-d6, the decomposition was not observed, but to our surprise, dr changed to 1.5:1. This dependence of dr from the solvents indicated the existence of a rapid equilibrium between the diastereomers in the solution. This fact was also confirmed by different dr values in the 1H NMR spectra of C6D6 and acetone-d6 solutions (Scheme 4).
:1. However, an additional set of signals belonging to unidentified minor impurities was observed in the 1H NMR spectrum of product 3a, which was pure by HPLC, and the intensity of these signals increased with time. We assumed that these impurities were the result of the compound decomposition15 in a CDCl3 solution which is known to be slightly acidic due to its degradation during storage. As expected, when the spectrum was recorded in DMSO-d6, the decomposition was not observed, but to our surprise, dr changed to 1.5:1. This dependence of dr from the solvents indicated the existence of a rapid equilibrium between the diastereomers in the solution. This fact was also confirmed by different dr values in the 1H NMR spectra of C6D6 and acetone-d6 solutions (Scheme 4).
 | ||
| Scheme 3 Possible pathways of cycloaddition of 1H-pyrrole-2,3-diones with Huisgen 1,4-dipoles. | ||
 | ||
| Scheme 4 Synthesis of cycloadduct 3a. | ||
It should be pointed out that the substituents with different electron-withdrawing properties can be readily introduced in 1H-pyrrole-2,3-diones, that may affect the regioselectivity of the dipolar cycloaddition. Therefore, despite the rapid epimerization of diastereomers in the solution, we performed the optimization of the reaction conditions and studied the substrate scope.
The model reaction of 1H-pyrrole-2,3-dione 1a with DMAD and pyridine proceeded smoothly in all tested solvents for 24 h at RT (Table 1). Moderate yields were observed in 1,2-dimethoxyethane and CH2Cl2 (Table 1, entries 2, 5). Other solvents showed similar yields, and ultimately, we chose EtOAc as a greener solvent among the tested ones.16 A 20% excess of DMAD and pyridine slightly improved the yield up to 85% (the optimal conditions, Table 1, entry 9).
| Entry | Solvent | Yieldb, % |
|---|---|---|
| a Reagents and conditions: 1a (0.1 mmol), 2a (0.1 mmol), pyridine (0.1 mmol), solvent (1 mL), in a capped vial.b Yields were determined by HPLC.c 1.1 equiv. of 2a and pyridine were used.d 1.2 equiv. of 2a and pyridine were used. | ||
| 1 | CHCl3 | 76 |
| 2 | 1,2-Dimethoxymethane | 56 |
| 3 | 1,4-Dioxane | 83 |
| 4 | Toluene | 83 |
| 5 | CH2Cl2 | 55 |
| 6 | Acetone | 81 |
| 7 | Ethyl acetate | 78 |
| 8c | Ethyl acetate | 82 |
| 9d | Ethyl acetate | 85 |
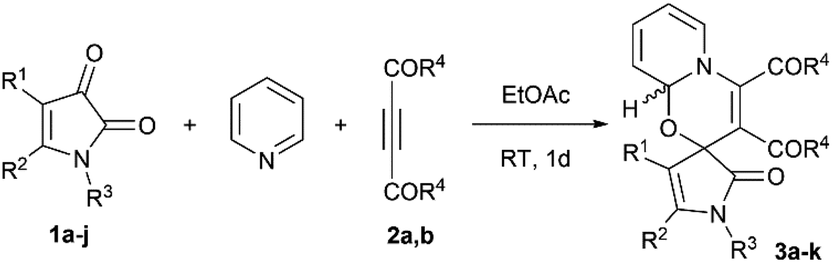
To explore the scope of the reaction, we investigated the cycloaddition of a variety of 1H-pyrrole-2,3-diones bearing electronically diverse substituents (Table 2). The reactions of 4-unsubstituted 5-aryl-1H-pyrrole-2,3-diones proceeded smoothly; the presence of electron-withdrawing or electron-donating groups in the 5-aryl substituent did not significantly affect the product yields (Table 2, entries 1–3, 3a–c). The introduction of an alkyl group into the 4-position of the 1H-pyrrole-2,3-diones also did not lead to a decrease in the yield (entry 4, 3d). The same regioselectivity was observed in the reactions of 5-aryl-1H-pyrrole-2,3-diones bearing EWGs in the 4-position which led to the products 3e–h in good yields (entries 5–8). The reaction was also tolerant to the substituents in N1 position. When two methoxycarbonyl substituents were introduced in the 4- and 5-positions of 1H-pyrrole-2,3-diones, the reaction was complicated by the formation of side-products, and the compounds 3i,j were isolated in lower yields (entries 9–10). Employing dibenzoylacetylene instead of DMAD, the product 3k was isolated in moderate yield (entry 11).
|
||||
|---|---|---|---|---|
| Entry | 1H-Pyrrole-2,3-dione (R1/R2/R3) | R4 | Product (yieldb) | dr |
| a Reagents and conditions: 1 (1 mmol), 2 (1.2 mmol), pyridine (1.2 mmol), ethyl acetate (10 mL), in a closed glass flask.b Isolated yields.c dr was determined by NMR analysis of isolated products in CDCl3.d dr was determined by NMR analysis of isolated products in DMSO-d6. | ||||
| 1 | 1a (H/Ph/Ph) | OMe | 3a (70%) | 5:1c |
| 2 | 1b (H/C6H4OMe-4/Ph) | OMe | 3b (82%) | 1.5:1d |
| 3 | 1c (H/C6H4Br-4/Ph) | OMe | 3c (79%) | 1.5:1d |
| 4 | 1d (Et/Ph/Ph) | OMe | 3d (84%) | 7.3:1d |
| 5 | 1e (COOEt/Ph/Ph) | OMe | 3e (80%) | 6.5:1c |
| 2.2:1d |
||||
| 6 | 1f (COOEt/Ph/C6H4Me-4) | OMe | 3f (76%) | 9:1c |
| 7 | 1g (COOEt/Ph/Me) | OMe | 3g (74%) | 2.3:1d |
| 8 | 1h (COPh/Ph/Bn) | OMe | 3h (77%) | 2.3:1c |
| 9 | 1i (COOMe/COOMe/Ph) | OMe | 3i (48%) | 1.5:1d |
| 10 | 1j (COOMe/COOMe/C6H4Me-4) | OMe | 3j (20%) | 5:1c |
| 11 | 1e (COOEt/Ph/Ph) | Ph | 3k (56%) | 5.3:1c |
Having established the substrate scope of the cycloaddition, we were interested in how well this diastereomeric equilibrium is described in the literature. The analysis of the literature revealed that the rapid equilibrium of [1,3]oxazines is known (Fig. 1). For example, during synthetic studies towards total synthesis of alkaloids of xestospongin/aguspongine family, Hoye et al. found that perhydropyrido[2,1-b][1,3]oxazines D exist in solution in rapid diastereomeric equilibrium through ring-chain tautomerism (Fig. 1).17 Similar epimerization in solution was observed on oxazolo[2,3-a]isoquinoline E in work by Seidel, Houk et al.18 Substituent effects on ring-chain tautomerism of naphth[1,2-e][1,3]oxazines F and G were studied in a series of works by Fülöp et al.19 Nevertheless, these cases deal with saturated or partially saturated pyrido[1,3]oxazine systems, while as a result of 1,4-dipolar cycloaddition to carbonyl group, unsaturated pyrido[1,3]oxazines are formed, for which the equilibrium between diastereomers and its consequences on observed dr have not been studied, and reported dr values could be incorrect. Considering this, we decided to investigate this possible epimerization on early reported cycloadducts obtained from Huisgen 1,4-dipoles (Fig. 1, structure H).
 | ||
| Fig. 1 Examples considering in this work and literature examples of [1,3]oxazines and oxazoles existing in the diastereomeric equilibrium. | ||
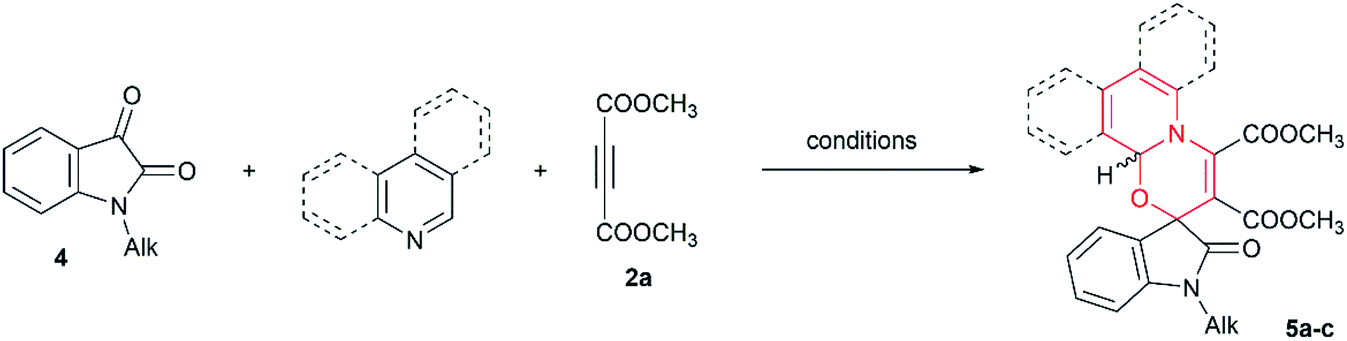
To this end, the model 1,4-dipolar cycloadditions of N-substituted isatins or 11H-indeno[1,2-b]quinoxalin-11-one with DMAD and azines (pyridine, isoquinoline and quinoline) were studied by HPLC and NMR methods (Tables 3 and 4).
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Compound, isolated yield | Heterocycle | Alk | Conditions, ref. | dr from literature by NMR (solvent) | dr observed in this work | ||||
| By HPLC for reaction mixture | By HPLC for isolated product | By NMR for isolated product in | ||||||||
| CDCl3 | K2CO3-treated CDCl3 | DMSO-d6 | ||||||||
| a The diastereomers were inseparable by HPLC.b The major diastereomer in the reaction mixture became the minor diastereomer after the isolation, probably due to the different solubility of the diastereomers. | ||||||||||
| 1 | 5a, 70% | Pyridine | Bn | DME, Ar, RT, 6 h (ref. 4c) | 5:1 (CDCl3–CCl4, 3:1) |
—a | —a | 4.8:1 |
4.8:1 |
1.2:1 |
| 2 | 5b, 86% | Isoquinoline | Bn | DCM, RT, 24 h (ref. 20b) | 2.4:1 (CDCl3) |
1:1 |
1.1:1 |
4.8:1 |
1:1 |
1:1 |
| 3 | 5c, 77% | Quinoline | All | Tol, 110 °C, 12 h (ref. 4b) | 4:1 (CDCl3) |
1.1:1 |
1:1.1b |
1:1 |
11 |
1:1 |
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Compound, isolated yield | Heterocycle | dr from literature by NMR (CDCl3) | dr observed in this work | ||||
| By HPLC for reaction mixture | By HPLC for isolated product | By NMR for isolated product in | ||||||
| CDCl3 | K2CO3-treated CDCl3 | DMSO-d6 | ||||||
| a The diastereomers were inseparable by HPLC.b The major diastereomer in the reaction mixture became the minor diastereomer after the isolation.c The change in dr was observed within 30 min after dissolution of compound 8b. | ||||||||
| 1 | 8a, 53% | Pyridine | Single diastereomer21 | —a | —a | 16:1 |
16:1 |
3.2:1 |
| 2 | 8b, 82% | Isoquinoline | Single diastereomer21 | 1.2:1 |
1:1.2b |
2.6:1→12:1c |
1.1:1 |
1.3:1 |
| 3 | 8c, 86% | Quinoline | 1.2:1 (ref. 21) |
1:1 |
1:1 |
1:1 |
1:1 |
1:1 |

We began our study with 1,4-dipolar cycloaddition of N-alkyl isatins 4. For the reaction with pyridine (Table 3, entry 1), HPLC analysis of the reaction mixture and the isolated product 5a showed a broad peak of inseparable diastereomers. The 1H NMR spectra of isolated product 5a (in CDCl3) showed a dr of 4.8:1 (with close agreement with the literature data,4c dr 5:1). The same dr value was observed in K2CO3-treated CDCl3, at the same time, a significantly different dr was observed in DMSO-d6 (1.2:1). These observations confirm the existence of a rapid epimerization as in the case of products 3a–k.
The reaction of N-benzylisatin with DMAD and isoquinoline has been reported two times,20 however, only one work20b provided information about dr. This reaction was successfully reproduced, and the HPLC analysis of the reaction mixture showed two well separated peaks of diastereomers in nearly 1:1 ratio (Table 3, entry 2). Isolation of diastereomeric mixture 5b by crystallization did not significantly change dr (HPLC data). The same dr was observed by NMR method in K2CO3-treated CDCl3 and DMSO-d6, and thus, this reaction showed no diastereoselectivity in contradistinction to the original work (dr 2.4:1).20b Nevertheless, significant change in dr (4.8:1) was observed in non-treated CDCl3, which clearly indicates the existence of acid-catalyzed isomerization of spiro[1,3]oxazino[2,3-a]isoquinoline 5b (entry 2). A related acid-catalyzed isomerization of spiropyrido[2,1-a]isoquinolines under the action of silica-gel or 37% HCl was recently reported by Cao et al.8d
Harsher conditions (110 °C) for three-component reaction of isatins, DMAD and quinoline have been reported,4b still HPLC analysis of the reaction mixture and isolated diastereomers 5c showed ca. 1:1 ratio, instead of reported 4:1 (Table 3, entry 3). The same dr was observed by NMR method in DMSO-d6 and K2CO3-treated CDCl3, even acidic CDCl3 did not change dr, and thus the products derived from quinoline are stable to acid-catalyzed isomerization. The discrepancy with reported dr probably arise from the fact that diastereomers 5c are separable, as judged by TLC, but were not separated in the original work,4b and incomplete collection of fractions during column chromatography could have happened.4b
Next, we investigated the reaction of readily accessible 11H-indeno[1,2-b]quinoxalin-11-one 6 with DMAD and azines (Table 4). The choice of this substrate was determined by the interesting fact that its reaction with DMAD and azines was known, however, the formation of spirolactones 7 rather than spirooxazines 8 was reported.21 Authors deduced structures of spirolactone products 7a–c based solely on IR, 1D NMR and mass spectra, without confirmation by single crystal X-ray analysis. By repeating the original work and performing X-ray analysis (CCDC 2119399 – 8b, CCDC 2119398 – 8c†), we were able to revise the reported structure 7 to be spirooxazines 8 (for full comparison with published data, see the ESI†).
For these reactions, HPLC analyses showed the results similar to N-alkyl isatins. In the case of pyridine, single broad peak was observed (Table 4, entry 1), and in the case of isoquinoline and quinoline, two separated peaks were observed in a nearly 1:1 ratio (Table 4, entries 2, 3). The 1H NMR spectra of pyridine product 8a showed good dr (about 16:1) in CDCl3 and 5-fold decreased dr in DMSO-d6 (Table 4, entry 1). Acid-catalyzed isomerization for isoquinoline product 8b was observed in untreated CDCl3: dr was 2.6:1 after dissolution, and it became 12:1 within 30 min at 303 K (entry 2). Quinoline product 8c showed stability to acid-catalyzed isomerization (entry 3).
Finally, we focused on 1,4-dipolar cycloaddition to CC bond with the expectation that the absence of aminal fragment will result in the increased stability of cycloadducts (Table 5). Reaction of isatylidene malononitrile 9 with DMAD and pyridine in refluxing THF was reported to result in the formation of spirocyclic quinolizine 10a with a dr of 10:1.8c HPLC analysis of the reaction mixture as well as isolated product showed single peak of inseparable diastereomers (Table 5, entry 1). The 1H NMR spectra of diastereomeric mixture in untreated CDCl3 or K2CO3-treated CDCl3 showed dr similar to the literature. At the same time, the 1H NMR spectrum in DMSO-d6 solution resulted in extensively broad peaks, among which two sets of signals could be seen in a ratio close to 1.5:1, that confirms the existence of a rapid equilibrium between diastereomers. Additional confirmation obtained with utilization of NOESY (EXSY) spectrum (see the ESI, S56†), which indicated the presence of the chemical exchange between diastereomers.22 Reaction of compound 9 with DMAD and quinoline was presented in the same work,8c and diastereomers were obtained in a ratio of 1.2:1 (similar to products 5c and 8c); therefore this reaction was not repeated in our study.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Compound, isolated yield | Heterocycle | dr from literature by NMR (CDCl3) | dr observed in this work | ||||
| By HPLC for reaction mixture | By HPLC for isolated product | By NMR for isolated product in | ||||||
| CDCl3 | K2CO3-treated CDCl3 | DMSO-d6 | ||||||
| a The diastereomers were inseparable by HPLC.b Compound 10b is unknown in the literature. | ||||||||
| 1 | 10a, 62% | Pyridine | 10.1:1 (ref. 8c) |
—a | —a | 9:1 |
9:1 |
1.5:1 |
| 2 | 10b, 76% | Isoquinoline | —b | 1.1:1 |
1.2:1 |
1.4:1 |
1.4:1 |
1.4:1 |

Reaction of isoquinoline, DMAD, and isatylidene malononitrile 9 was not reported. However, Cao et al. recently published a similar reaction of isoquinoline with CF3-propiolate and isatylidene malononitriles, which led to spirocyclic pyrido[2,1-a]isoquinolines with dr in ranges of 1:0.9 to 1:1.1.8a Adopting the conditions presented in the work,8c we were able to synthesize pyrido[2,1-a]isoquinoline 10b in 76% yield. HPLC analysis of the reaction mixture and isolated product showed dr of nearly 1:1 (Table 5, entry 2); finally, close dr (1.4:1) was observed in NMR spectra (CDCl3 and DMSO-d6). In contrast to [1,3]oxazine products 5b and 8b derived from isoquinoline, no acid-catalyzed isomerization of 10b took place in acidic CDCl3.
Based on 1,4-dipolar cycloaddition of pyrrole-2,3-diones and reproduced previously reported 1,4-dipolar cycloaddition reactions, several trends for this type of 1,4-cycloaddition can be derived:
(a) Addition of 1,4-dipoles derived from pyridine to CO or CC bonds results in an inseparable mixture of rapid interconverted diastereomers. These cycloaddition reactions are probably not diastereoselective, although apparent solvent-dependent dr values can be high.
(b) Isoquinoline-based 1,4-dipoles lead to almost equimolar diastereomeric mixtures in the case of CO dipolarophiles. Utilization of acidic CDCl3 (as a result of its decomposition) for recording NMR spectra can result in incorrect dr values. Addition of isoquinoline-based dipoles to CC bonds produces pyridoisoquinolines in a non-diastereoselective fashion, and no epimerization in acidic CDCl3 is observed. However, more acidic conditions may facilitate epimerization.8d
(c) In all cases of addition of quinoline dipoles to CO or CC bonds, equimolar mixture of diastereomers is observed, and their ratios are not affected by acidic CDCl3.23
Conclusions
In conclusion, we have shown that spiro[pyrido[2,1-b][1,3]oxazine-2,3′-pyrroles] can be synthesized as diastereomeric mixtures in 20–84% yields via the three-component reaction of 1H-pyrrole-2,3-diones, DMAD, and pyridine. Different substituents in 1H-pyrrole-2,3-diones did not affect the regioselectivity of 1,4-cycloaddition. Rapid equilibrium between synthesized diastereomers in solution was observed, that encouraged us to examine the existing literature by the repetition of known 1,4-cycloaddition reactions. From this analysis, several trends for this type of 1,4-cycloaddition were derived.The 1,4-dipolar cycloaddition reactions with other azaheterocycles or imines as well as other types of acetylenes and dipolarophiles were beyond the scope of this work, and their diastereoselectivity can be further revised.
As a general conclusion for the development of stereoselective methods, special emphasis should be paid to experimental data including analysis of reaction mixtures and the possible existence of isomeric equilibrium and factors influencing it.
Experimental section
The experimental procedure for the synthesis of compounds 3 and their characterisation data are given below. General information, characterisation data, NMR spectra, crystallographic data (for 3a, 3f, 8b and 8c) and experimental procedures for all other compounds are given in the ESI.†Deposition numbers CCDC 2119396 (for 3a), 2119397 (for 3f), 2119399 (for 8b), and 2119398 (for 8c) contain the supplementary crystallographic data for this paper.†
The general procedure for the synthesis of compounds 3
A round-bottom flask was charged with 1H-pyrrole-2,3-dione 1 (1 mmol), acetylene 2 (1.2 mmol), and anhydrous ethyl acetate (10 mL). Pyridine (97 μL, 1.2 mmol) was added to the resulted solution, and the reaction mixture was stirred at RT for 1 day in the closed flask. The solvent was evaporated under reduced pressure. The residue was purified by appropriate method as indicated below to afford 3.:1); mp 181–182 °C (decomp.); IR (cm−1): 1746, 1683, 1663, 1638, 1594, 1561; 1H NMR (400 MHz, CDCl3), mixture of inseparable diastereomers, d.r. = ∼5:1 (A:B): δ = 7.32–7.18 (6H, m, A + B), 7.18–7.13 (2H, m, A + B), 7.11–7.07 (2H, m, A + B), 6.59 (0.84H, dd, J = 3.4, 1.3 Hz, A), 6.41–6.23 (2H, m, A + B), 5.82 (0.16H, dd, J = 3.4, 1.3 Hz, B), 5.72 (0.84H, ddt, J = 10.0, 3.4, 1.1 Hz, A), 5.66–5.62 (0.32H, m, 2× B), 5.37–5.31 (0.84H, m, A), 5.30–5.26 (1H, m, A + B), 3.96 (2.52H, s, A), 3.91 (0.48H, s, B), 3.70 (2.52H, s, A), 3.68 (0.48H, s, B). 13C NMR (101 MHz, CDCl3): δ (A, major) = 175.2, 163.9, 163.6, 150.6, 144.9, 135.5, 130.9, 129.3, 128.8 (2C), 128.4 (2C), 127.8 (2C), 127.1, 126.9 (2C), 125.4, 124.8, 116.7, 107.5, 106.8, 101.7, 78.8, 78.6, 53.4, 52.1; 1H NMR (400 MHz, DMSO-d6), mixture of inseparable diastereomers, d.r. = 1.5:1 (A:B).: δ = 7.39–7.22 (6H, m, A + B), 7.19–7.12 (2H, m, A + B), 7.08–7.02 (2H, m, A + B), 6.57 (0.4H, dt, J = 7.5, 1.1 Hz, B), 6.53 (0.6H, dt, J = 7.6, 1.1 Hz, A), 6.41–6.26 (1.4H, m, A + B), 6.05 (0.6H, s, A), 6.00 (0.6H, dd, J = 3.4, 1.3 Hz, A), 5.75 (0.4H, ddt, J = 9.9, 3.4, 1.1 Hz, B), 5.68 (0.6H, ddt, J = 9.9, 3.5, 1.2 Hz, A), 5.45–5.40 (0.8H, m, 2× B), 5.39–5.33 (0.6H, m, A), 3.91 (1.2H, s, B), 3.88 (1.8H, s, A), 3.65 (1.2H, s, B), 3.62 (1.8H, s, A). 13C NMR (101 MHz, DMSO-d6): δ (A, major) = 175.1, 163.4, 162.5, 146.4, 143.6, 135.3, 130.1, 129.1, 128.6 (2C), 128.2 (2C), 127.3 (2C), 127.0, 126.8 (2C), 126.0, 125.1, 115.9, 108.4, 108.2, 101.0, 79.8, 79.2, 53.3, 52.0. 13C NMR (101 MHz, DMSO-d6): δ (B, minor) = 174.6, 163.0, 162.6, 149.5, 144.3, 135.0, 130.1, 129.1, 128.6 (2C), 128.2 (2C), 127.2 (2C), 127.0, 126.7 (2C), 125.2, 124.9, 116.1, 107.3, 105.7, 101.6, 77.9, 77.8, 53.4, 51.9; 1H NMR (400 MHz, acetone-d6), mixture of inseparable diastereomers, dr = ∼1.3:1 (A:B): δ = 7.37–7.20 (8H, m, A + B), 7.15–7.07 (2H, m, A + B), 6.55–6.52 (1.14H, m, 2× A), 6.48 (0.43H, dt, J = 7.6, 1.1 Hz, B), 6.39–6.28 (1H, m, A + B), 5.98 (0.43H, dd, J = 3.4, 1.3 Hz, B), 5.94 (0.43H, s, B), 5.71 (0.57H, ddt, J = 9.9, 3.3, 1.1 Hz, A), 5.67 (0.43H, ddt, J = 9.9, 3.4, 1.2 Hz, B), 5.40 (0.57H, ddd, J = 7.5, 6.2, 1.1 Hz, A), 5.33 (0.43H, ddd, J = 7.4, 6.0, 1.1 Hz, B), 5.30 (0.57H, s, A), 3.95 (1.7H, s, A), 3.91 (1.3H, s, B), 3.70 (1.7H, s, A), 3.68 (1.3H, s, B); 1H NMR (400 MHz, benzene-d6), mixture of inseparable diastereomers, d.r. = ∼3.2:1 (A:B): δ = 7.15–6.81 (10.76H, m, A + B), 6.38 (0.76H, dt, J = 7.5, 1.1 Hz, A), 6.24 (0.24H, dt, J = 7.5, 1.1 Hz, B), 5.84 (0.24H, ddt, J = 9.9, 6.1, 1.2 Hz, B), 5.78 (0.76H, ddt, J = 10.0, 6.0, 1.2 Hz, A), 5.54–5.46 (1H, m, A + B), 5.45–5.38 (0.24H, m, B), 5.34 (0.24H, s, B), 5.11 (0.76H, s, A), 4.91–4.86 (0.76H, m, A), 4.86–4.81 (0.24H, m, B), 3.57 (2.28H, s, A), 3.47 (0.72H, s, B), 3.29 (0.72H, s, B), 3.24 (2.28H, s, A); HRMS (ESI): m/z calcd for C27H22N2O6: 471.1551 [M + H]+; found: 471.1544.:1); mp 177–178 °C (decomp.); IR (cm−1): 1740, 1729, 1713, 1654, 1635, 1600, 1569; 1H NMR (400 MHz, DMSO-d6), mixture of inseparable diastereomers, d.r. = ∼1.5:1 (A:B): δ = 7.39–7.34 (2H, m, A + B), 7.30–7.25 (1H, m, A + B), 7.10–7.03 (4H, m, A + B), 6.87–6.80 (2H, m, A + B), 6.57 (0.4H, dt, J = 7.5, 1.1 Hz, B), 6.53 (0.6H, dt, J = 7.5, 1.1 Hz, A), 6.39 (0.4H, dd, J = 3.4, 1.3 Hz, B), 6.38–6.29 (1H, m, A + B), 5.98 (0.6H, dd, J = 3.4, 1.3 Hz, A), 5.95 (0.6H, s, A), 5.74 (0.4H, ddt, J = 9.9, 3.3, 1.1 Hz, B), 5.67 (0.6H, ddt, J = 9.9, 3.4, 1.1 Hz, A), 5.42 (0.4H, ddd, J = 7.3, 6.1, 1.1 Hz, B), 5.36 (0.6H, ddd, J = 7.3, 6.0, 1.1 Hz, A), 5.32 (0.4H, s, B), 3.91 (1.2H, s, B), 3.87 (1.8H, s, A), 3.72 (3H, s, A + B), 3.64 (1.2H, s, B), 3.62 (1.8H, s, A). 13C NMR (101 MHz, DMSO-d6): δ (A, major) = 175.2, 163.4, 162.5, 159.8, 146.1, 143.4, 135.5, 128.8 (2C), 128.6 (2C), 126.9, 126.9 (2C), 126.0, 125.1, 122.2, 115.9, 113.7 (2C), 108.6, 106.8, 100.9, 79.8, 79.3, 55.1, 53.2, 51.9. 13C NMR (101 MHz, DMSO-d6): δ (B, minor) = 174.7, 163.0 162.6, 159.7, 149.2, 144.2, 135.1, 128.7 (2C), 128.6 (2C), 126.9, 126.8 (2C), 125.2, 124.9, 122.4, 116.1, 113.7 (2C), 105.9, 105.6, 101.5, 77.9, 77.8, 55.1, 53.3, 51.9; HRMS (ESI): m/z calcd for C28H24N2O7: 501.1656 [M + H]+; found: 501.1657.:1); mp 173–174 °C (decomp.); IR (cm−1): 1739, 1709, 1684, 1652, 1633, 1595, 1567; 1H NMR (400 MHz, DMSO-d6), mixture of inseparable diastereomers, d.r. = ∼1.5:1 (A:B): δ = 7.52–7.43 (2H, m, A + B), 7.42–7.33 (2H, m, A + B), 7.33–7.24 (1H, m, A + B), 7.12–7.03 (4H, m, A + B), 6.58 (0.4H, d, J = 7.5 Hz, B), 6.54 (0.6H, d, J = 7.5 Hz, A), 6.39–6.29 (1.4H, m, A + B), 6.11 (0.6H, s, A), 5.99 (0.6H, d, J = 2.7 Hz, A), 5.75 (0.4H, B dd, J = 10.0, 3.2 Hz), 5.67 (0.6H, dd, J = 9.9, 3.3 Hz, A), 5.52 (0.4H, s, B), 5.46–5.40 (0.4H, m, B), 5.40–5.34 (0.6H, m, A), 3.91 (1.2H, s, B), 3.88 (1.8H, s, A), 3.64 (1.2H, s, B), 3.62 (1.8H, s, A). 13C NMR (101 MHz, DMSO-d6): δ (A, major) = 175.0, 163.3, 162.4, 145.3, 143.7, 135.1, 131.3 (2C), 129.3 (2C), 129.3, 128.7 (2C), 126.7 (2C), 126.6, 125.9, 125.2, 122.6, 115.9, 108.7, 108.2, 101.0, 79.9, 79.2, 53.3, 52.0. 13C NMR (101 MHz, DMSO-d6): δ (B, minor) = 174.5, 163.0, 162.5, 148.4, 144.4, 134.8, 131.3 (2C), 129.2 (2C), 128.7 (2C), 127.1 (2C), 125.2, 124.8, 122.5, 116.0, 108.1, 105.5, 101.7, 78.0, 77.7, 53.4, 51.9, two carbons were not separated/found; HRMS (ESI): m/z calcd for C27H21BrN2O6: 551.0635 [M + H]+; found: 551.0633.:1); mp 138–140 °C (decomp.); IR (cm−1): 1743, 1729, 1706, 1656, 1600, 1567; 1H NMR (400 MHz, DMSO-d6), mixture of inseparable diastereomers, d.r. = ∼7.3:1 (A:B): δ = 7.34–7.22 (5H, m, A + B), 7.21–7.13 (3H, m, A + B), 7.03–6.93 (2H, m, A + B), 6.68 (0.12H, d, J = 7.5 Hz, B), 6.62 (0.88H, d, J = 7.5 Hz, A), 6.43–6.35 (1H, m, A + B), 6.33 (0.88H, dd, J = 3.5, 1.2 Hz, A), 5.85–5.71 (1.12H, m, A + B), 5.49–5.38 (1H, m, A + B), 3.92 (2.64H, s, A), 3.87 (0.36H, s, B), 3.69 (3H, s, A + B), 2.28–2.19 (0.24H, m, B), 1.93 (1.76H, q, J = 7.5 Hz, A), 0.99 (0.36H, t, J = 7.5 Hz, B), 0.88 (2.64H, t, J = 7.5 Hz, A). 13C NMR (101 MHz, DMSO-d6): δ (A, major) = 173.7, 163.1, 162.7, 145.0, 143.0, 135.0, 129.7, 128.6 (3C), 128.4 (2C), 128.2 (2C), 126.7 (2C), 126.6, 125.2, 124.8, 119.1, 116.1, 106.0, 101.7, 79.3, 77.8, 53.4, 51.9, 17.0, 13.8; HRMS (ESI): m/z calcd for C29H26N2O6: 499.1864 [M + H]+; found: 499.1858.:1); mp 179–182 °C (decomp.); IR (cm−1): 1746, 1733, 1703, 1602, 1594, 1563; 1H NMR (400 MHz, CDCl3), mixture of inseparable diastereomers, d.r. = ∼6.5:1 (A:B): δ = 7.33–7.16 (8H, m, A + B), 7.08–6.89 (2H, m, A + B), 6.57–6.22 (3H, m, A + B), 5.75 (1H, dd [A], J = 10.0, 3.4 Hz, A + B), 5.50–5.21 (1H, m, A + B), 4.07–3.89 (5H, m, A + B), 3.77 (3H, s, A + B), 1.06 (2.6H, t, J = 7.1 Hz, A), 0.94 (0.4H, t, J = 7.1 Hz, B). 13C NMR (101 MHz, CDCl3): δ (A, major) = 174.3, 163.8, 163.6, 161.9, 159.8, 146.1, 134.1, 129.9, 129.7, 129.3 (2C), 128.9 (2C), 128.0 (2C), 127.97, 127.8 (2C), 125.7, 125.1, 116.3, 108.8, 107.2, 101.7, 79.0, 77.9, 60.0, 53.3, 52.2, 14.0; 1H NMR (400 MHz, DMSO-d6), mixture of inseparable diastereomers, d.r. = ∼2.2:1 (A:B): δ = 7.35–7.17 (8H, m, A + B), 7.11–7.04 (2H, m, A + B), 6.65 (0.31H, d, J = 7.6 Hz, B), 6.62 (0.69H, d, J = 7.5 Hz, A), 6.42–6.34 (1H, m, A + B), 6.32–6.25 (1H, m, A + B), 5.81–5.71 (1H, m, A + B), 5.49–5.37 (1H, m, A + B), 3.92 (2.07H, s, A), 3.89 (0.93H, s, B), 3.91–3.80 (2H, m, A + B), 3.73 (2.07H, s, A), 3.72 (0.93H, s, B), 0.94 (2.07H, t, J = 7.1 Hz, A), 0.79 (0.93H, t, J = 7.1 Hz, B). 13C NMR (101 MHz, DMSO-d6): δ (A, major) = 173.6, 163.1, 162.6, 160.8, 158.9, 145.7, 133.5, 129.4, 129.2, 128.8 (2C), 128.7 (2C), 128.1, 127.9 (2C), 127.6 (2C), 125.5, 125.3, 115.7, 108.1, 106.0, 101.4, 78.1, 77.0, 59.1, 53.3, 52.0, 13.5. 13C NMR (101 MHz, DMSO-d6): δ (B, minor) = 173.9, 163.8, 162.9, 162.3, 157.3, 143.0, 133.8, 129.3, 128.7 (2C), 128.6 (2C), 127.9 (2C), 127.5 (2C), 126.5, 125.3, 115.4, 111.1, 110.0, 101.0, 79.4, 77.9, 59.4, 53.3, 52.2, 13.2, two carbons were not separated/found; HRMS (ESI): m/z calcd for C30H26N2O8: 543.1762 [M + H]+; found: 543.1759.:1); mp 173–175 °C (decomp.); IR (cm−1): 1742, 1695, 1653, 1627, 1595, 1564; 1H NMR (400 MHz, CDCl3), mixture of inseparable diastereomers, d.r. = ∼9:1 (A:B): δ = 7.31–7.17 (5H, m, A + B), 7.04–6.95 (2H, m, A + B), 6.93–6.88 (2H, m, A + B), 6.49 (0.9H, dd, J = 3.5, 1.2 Hz, A), 6.42 (1H, dt [A], J = 7.5, 1.2 Hz, A + B), 6.34 (1.1H, ddt [A], J = 9.9, 6.0, 1.2 Hz, A + B), 5.74 (1H, ddt [A], J = 9.9, 3.6, 1.2 Hz, A + B), 5.36 (1H, ddd [A], J = 7.3, 6.0, 1.1 Hz, A + B), 4.05–3.94 (2H, m, A + B), 3.96 (2.7H, s, A), 3.91 (0.3H, s, B), 3.76 (2.7H, s, A), 3.75 (0.3H, s, B), 2.26 (2.7H, s, A), 2.23 (0.3H, s, B), 1.06 (2.7H, t, J = 7.1 Hz, A), 0.93 (0.3H, t, J = 7.1 Hz, B). 13C NMR (101 MHz, CDCl3): δ (A, major) = 174.4, 163.8, 163.6, 162.0, 160.1, 146.0, 137.9, 131.5, 130.0, 129.6 (3C), 129.3 (2C), 127.8 (2C), 127.7 (2C), 125.7, 125.1, 116.4, 108.6, 107.3, 101.6, 78.9, 77.9, 59.9, 53.3, 52.1, 21.2, 14.0; HRMS (ESI): m/z calcd for C31H28N2O8: 557.1918 [M + H]+; found: 557.1913.:1); mp 141–143 °C (decomp.); IR (cm−1): 1750, 1743, 1722, 1702, 1650, 1638, 1596, 1564; 1H NMR (400 MHz, DMSO-d6), mixture of inseparable diastereomers, d.r. = ∼2.3:1 (A:B): δ = 7.58–7.48 (3H, m, A + B), 7.41–7.31 (2H, m, A + B), 6.61 (0.3H, dt, J = 7.5, 1.1 Hz, B), 6.58 (0.7H, dt, J = 7.5, 1.1 Hz, A), 6.39–6.30 (1H, m, A + B), 6.27 (0.7H, dd, J = 3.4, 1.2 Hz, A), 6.25 (0.3H, dd, J = 3.5, 1.1 Hz, B), 5.74–5.63 (1H, m, A + B), 5.44–5.36 (1H, m, A + B), 3.90 (2.1H, s, A), 3.88 (0.9H, s, B), 3.84–3.73 (2H, m, A + B), 3.65 (2.1H, s, A), 3.63 (0.9H, s, B), 2.76 (2.1H, s, A), 2.75 (0.9H, s, B), 0.91 (2.1H, t, J = 7.1 Hz, A), 0.76 (0.9H, t, J = 7.1 Hz, B). 13C NMR (101 MHz, DMSO-d6): δ (A, major) = 174.31, 162.95, 162.66, 160.64, 159.72, 145.47, 129.75, 129.28, 128.20 (2C), 128.16 (2C), 125.40, 125.29, 115.61, 107.32, 106.17, 101.13, 77.89, 76.78, 58.82, 53.20, 51.83, 27.31, 13.44. 13C NMR (101 MHz, DMSO-d6): δ (B, minor) = 174.63, 163.52, 162.72, 162.35, 158.24, 142.96, 129.67, 129.48, 127.99 (2C), 126.43, 125.18, 115.37, 110.71, 109.26, 100.86, 79.24, 77.71, 59.14, 53.25, 51.99, 27.88, 13.17, two carbons were not separated/found; HRMS (ESI): m/z calcd for C25H24N2O8: 481.1605 [M + H]+; found: 481.1612.:1); mp 162–164 °C (decomp.); IR (cm−1): 1743, 1727, 1713, 1650, 1637, 1596, 1577, 1563; 1H NMR (400 MHz, CDCl3), mixture of inseparable diastereomers, d.r. = ∼2.3:1 (A:B): δ = 7.36–7.28 (2H, m, A + B), 7.23–7.17 (3H, m, A + B), 7.15–6.90 (10H, m, A + B), 6.52 (0.7H, dd, J = 3.5, 1.2 Hz, A), 6.44 (0.3H, d, J = 7.5 Hz, B), 6.37–6.26 (1.7H, m, A + B), 6.22 (0.3H, d, J = 3.5 Hz, B), 5.78–5.58 (1H, m, A + B), 5.40–5.23 (1H, m, A + B), 4.73–4.57 (2H, m, A + B), 3.93 (0.9H, s, B), 3.90 (2.1H, s, A), 3.60 (3H, s, A + B). 13C NMR (101 MHz, CDCl3): δ (A, major) = 190.6, 175.5, 164.0, 163.6, 158.2, 145.9, 139.6, 136.7, 130.8, 130.1, 129.6 (2C), 129.4, 128.8 (2C), 128.6 (2C), 128.1 (3C), 127.6 (2C), 127.4 (2C), 125.8, 125.5, 118.0, 116.1, 107.0, 101.6, 78.9, 78.9, 53.3, 52.0, 44.9. 13C NMR (101 MHz, CDCl3): δ (B, minor) = 163.2, 136.8, 131.3, 130.1, 129.8, 128.9 (2C), 128.6 (2C), 127.8 (2C), 127.7 (2C), 127.5 (2C), 126.3, 125.6, 116.0, 106.3, 101.5, 80.7, 53.3, 52.1, 45.6, eleven carbons were not separated/found; HRMS (ESI): m/z calcd for C35H28N2O7: 589.1969 [M + H]+; found: 589.1965.:1) and followed by crystallization from EtOH. Yellow solid (244 mg, 48% yield); Rf = 0.15 (toluene/EtOAc 8:1); mp 180–181 °C (decomp.); IR (cm−1): 1754, 1738, 1712, 1694, 1684, 1656, 1629, 1601, 1567; 1H NMR (400 MHz, DMSO-d6), mixture of inseparable diastereomers, d.r. = ∼1.5:1 (A:B): δ = 7.57–7.50 (2H, m, A + B), 7.49–7.43 (1H, m, A + B), 7.32–7.23 (2H, m, A + B), 6.67 (0.6H, d, J = 7.5 Hz, A), 6.64 (0.4H, d, J = 7.5 Hz, B), 6.45–6.34 (1H, m, A + B), 6.25 (0.4H, d, J = 3.3 Hz, B), 6.08 (0.6H, dd, J = 3.4, 1.2 Hz, A), 5.84–5.72 (1H, m, A + B), 5.53–5.48 (0.6H, m, A), 5.47–5.40 (0.4H, m, B), 3.93 (1.8H, s, A), 3.89 (1.2H, s, B), 3.703 (1.2H, s, A), 3.696 (1.8H, s, A), 3.69 (1.2H, s, B), 3.67 (3H, s, A + B), 3.60 (1.8H, s, A). 13C NMR (101 MHz, DMSO-d6): δ (A, major) = 172.3, 162.9, 162.3, 160.3, 160.1, 149.7, 146.4, 132.8, 129.5 (2C), 128.9, 126.0 (2C), 125.6, 125.1, 115.8, 108.9, 104.6, 102.2, 78.3, 76.2, 53.5, 53.2, 52.1, 51.5. 13C NMR (101 MHz, DMSO-d6): δ (B, minor) = 173.1, 163.3, 162.0, 161.8, 160.5, 148.4, 144.1, 132.8, 129.5 (2C), 129.1, 126.4 (2C), 126.0, 125.4, 115.5, 109.0, 108.1, 101.6, 79.7, 77.0, 53.5, 53.2, 52.3, 52.1; HRMS (ESI): m/z calcd for C25H22N2O10: 511.1347 [M + H]+; found: 511.1349.:1); mp 168–170 °C (decomp.); IR (cm−1): 1755, 1715, 1657, 1628, 1602, 1566; 1H NMR (400 MHz, CDCl3), mixture of inseparable diastereomers, d.r. = ∼5:1 (A:B): δ = 7.25–7.14 (4H, m, A + B), 6.46–6.21 (3H, m, A + B), 5.73 (0.83H, dd, J = 9.9, 3.5 Hz, A), 5.69–5.63 (0.17H, m, B), 5.43–5.36 (0.83H, m, A), 5.35–5.30 (0.17H, m, B), 3.95 (2.5H, s, A), 3.91 (0.5H, s, B), 3.76–3.70 (6.5H, m, A + B), 3.67 (2.5H, s, A), 2.37 (3H, s, A + B). 13C NMR (101 MHz, CDCl3): δ (A, major) = 173.3, 163.5, 163.4, 161.2, 161.2, 150.9, 146.7, 139.1, 130.9, 130.2 (2C), 126.4 (2C), 125.8, 124.8, 116.3, 109.4, 105.9, 102.3, 79.1, 77.1, 53.5, 53.2, 52.2, 51.7, 21.3; HRMS (ESI): m/z calcd for C26H24N2O10: 525.1504 [M + H]+; found: 525.1514.:1); mp 159–160 °C (decomp.); IR (cm−1): 1745, 1683, 1663, 1638, 1594, 1561; 1H NMR (400 MHz, CDCl3), mixture of inseparable diastereomers, d.r. = ∼5.3:1 (A:B): δ = 7.74–7.56 (1.84H, m, A + B), 7.51–7.44 (1H, m, A + B), 7.41–7.35 (1H, m, A + B), 7.34–7.01 (16.16H, m, A + B), 6.75 (0.16H, d, J = 3.4 Hz, B), 6.65 (0.84H, d, J = 3.8 Hz, A + B), 6.46 (0.16H, d, J = 7.6 Hz, A + B), 6.41 (1H, ddt [A], J = 9.8, 6.1, 1.0 Hz, A + B), 6.34 (0.84H, d, J = 7.7 Hz, A), 5.94–5.77 (1H, m, A + B), 5.33 (1H, ddd [A], J = 7.4, 6.0, 1.2 Hz, A + B), 4.18–3.95 (0.32H, m, B), 3.94–3.80 (1.68H, m, B), 1.02 (0.48H, t, J = 7.1 Hz, B), 0.82 (2.52H, t, J = 7.1 Hz, A). 13C NMR (101 MHz, CDCl3): δ (A, major) = 193.2, 190.4, 175.1, 164.0, 158.7, 147.9, 139.5, 138.0, 134.8, 134.4, 132.6, 130.3, 130.1 (4C), 129.29, 129.26 (2C), 128.9 (4C), 128.5 (2C), 128.1 (2C), 128.0, 127.6 (2C), 127.2, 126.1, 123.6, 115.8, 110.1, 101.7, 80.8, 79.8, 60.0, 13.8; HRMS (ESI): m/z calcd for C40H30N2O6: 635.2177 [M + H]+; found: 635.2178.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation, project # 19-13-00290.Notes and references
- (a) N. De and E. J. Yoo, ACS Catal., 2018, 8, 48 CrossRef CAS; (b) T.-R. Li, Y.-N. Wang, W.-J. Xiao and L.-Q. Lu, Tetrahedron Lett., 2018, 59, 1521 CrossRef CAS; (c) A. Mirzaei, G. Turczel, M. Nagyházi, V. Farkas, Á. Balla, H. Dang Vu and R. Tuba, Eur. J. Org. Chem., 2021, 2021, 326 CrossRef CAS; (d) V. Nair, A. Deepthi, D. Ashok, A. E. Raveendran and R. R. Paul, Tetrahedron, 2014, 70, 3085 CrossRef CAS; (e) B. A. Trofimov and K. V. Belyaeva, Tetrahedron Lett., 2020, 61, 151991 CrossRef CAS.
- (a) J. Du, Y.-D. Hua, Y.-J. Jiang, S. Huang, D. Chen, C.-H. Ding and X.-L. Hou, Org. Lett., 2020, 22, 5375 CrossRef CAS PubMed; (b) D. Garayalde and C. Nevado, ACS Catal., 2012, 2, 1462 CrossRef CAS; (c) B. M. Trost and Z. Jiao, J. Am. Chem. Soc., 2020, 142, 21645 CrossRef CAS PubMed.
- (a) A. Bhunia, D. Porwal, R. G. Gonnade and A. T. Biju, Org. Lett., 2013, 15, 4620 CrossRef CAS PubMed; (b) A. Bhunia, T. Roy, P. Pachfule, P. R. Rajamohanan and A. T. Biju, Angew. Chem., Int. Ed., 2013, 52, 10040 CrossRef CAS PubMed; (c) D. Liu, J. Sun, J. Xie, H. Shi and C.-G. Yan, J. Org. Chem., 2021, 86, 1827 CrossRef CAS PubMed; (d) M. S. Sammor, A. Q. Hussein, F. F. Awwadi and M. M. El-Abadelah, Tetrahedron, 2018, 74, 42 CrossRef CAS; (e) J. Zhang, H. Gao, J. Sun and C.-G. Yan, Eur. J. Org. Chem., 2014, 2014, 5598 CrossRef CAS; (f) V. E. Zhulanov, V. A. Vigovskaya, M. V. Dmitriev, P. S. Silaichev, A. N. Maslivets and M. Rubin, Org. Biomol. Chem., 2020, 18, 3382 RSC.
- (a) G. Liu, Y. Wu, J. Han, W. He, J. Chen, H. Deng, M. Shao, H. Zhang and W. Cao, Synthesis, 2018, 50, 4668 CrossRef CAS; (b) V. Nair, S. Devipriya and E. Suresh, Tetrahedron, 2008, 64, 3567 CrossRef CAS; (c) V. Nair, A. R. Sreekanth, N. Abhilash, A. T. Biju, B. Rema Devi, R. S. Menon, N. P. Rath and R. Srinivas, Synthesis, 2003, 2003, 1895 CrossRef; (d) I. R. Siddiqui, A. Srivastava, P. Rai, A. Srivastava and A. Srivastava, J. Heterocycl. Chem., 2015, 52, 1415 CrossRef CAS.
- V. Nair, A. R. Sreekanth, A. T. Biju and N. P. Rath, Tetrahedron Lett., 2003, 44, 729 CrossRef CAS.
- (a) H. Mehrabi and M. Hatami-Pour, Chin. Chem. Lett., 2014, 25, 1495 CrossRef CAS; (b) T. Sun, Q. Cai, M. Li, Z. Wang, J. Chen, H. Deng, M. Shao, H. Zhang and W. Cao, Tetrahedron, 2015, 71, 622 CrossRef CAS; (c) Z. Xu, T. Sun, Q. Cai, F. Ni, J. Han, J. Chen, H. Deng, M. Shao, H. Zhang and W. Cao, J. Fluorine Chem., 2016, 181, 45 CrossRef CAS.
- (a) K. V. Belyaeva, V. S. Gen, L. P. Nikitina, A. V. Afonin, D. V. Pavlov and B. A. Trofimov, Tetrahedron Lett., 2021, 153431 CrossRef CAS; (b) K. V. Belyaeva, L. P. Nikitina, V. S. Gen’, A. V. Afonin and B. A. Trofimov, Synthesis, 2021 DOI:10.1055/a-1644-2930; (c) A. A. Esmaeili, H. Vesalipoor, R. Hosseinabadi, A. F. Zavareh, M. A. Naseri and E. Ghiamati, Tetrahedron Lett., 2011, 52, 4865 CrossRef CAS; (d) M. Lei, W. Tian, R. Hu, W. Li and H. Zhang, Synthesis, 2012, 44, 2519 CrossRef CAS; (e) V. M. Muzalevskiy, Z. A. Sizova, K. V. Belyaeva, B. A. Trofimov and V. G. Nenajdenko, Molecules, 2019, 24, 3594 CrossRef PubMed.
- (a) Y. Hu, L. Ye, J. Chen, H. Zhang, H. Deng, J.-H. Lin and W. Cao, Eur. J. Org. Chem., 2021, 2021, 4405 CrossRef CAS; (b) V. Nair, B. Rema Devi and L. R. Varma, Tetrahedron Lett., 2005, 46, 5333 CrossRef CAS; (c) H.-B. Yang, X.-Y. Guan, Y. Wei and M. Shi, Eur. J. Org. Chem., 2012, 2012, 2792 CrossRef CAS; (d) M. Yu, Y. Wu, X. Peng, J. Han, J. Chen, Y. Kan, H. Deng, M. Shao, H. Zhang and W. Cao, J. Fluorine Chem., 2018, 216, 33 CrossRef CAS; (e) Y.-Y. Zhang, Y. Han, J. Sun and C.-G. Yan, ChemistrySelect, 2017, 2, 7382 CrossRef CAS.
- P. J. Abbott, R. M. Acheson, U. Eisner, D. J. Watkin and J. R. Carruthers, J. Chem. Soc., Chem. Commun., 1975, 155 RSC.
- (a) V. Nair, A. R. Sreekanth, N. Abhilash, M. M. Bhadbhade and R. C. Gonnade, Org. Lett., 2002, 4, 3575 CrossRef PubMed; (b) H.-W. Zhao, X.-Q. Chen, H.-L. Pang, T. Tian, B. Li, X.-Q. Song, W. Meng, Z. Yang, Y.-D. Zhao and Y.-Y. Liu, RSC Adv., 2016, 6, 61732 RSC.
- (a) E. A. Lystsova, E. E. Khramtsova and A. N. Maslivets, Symmetry, 2021, 13, 1509 CrossRef CAS; (b) V. V. Konovalova and A. N. Maslivets, Mini-Rev. Org. Chem., 2019, 16, 173 CrossRef CAS.
- (a) A. A. Moroz, V. E. Zhulanov, M. V. Dmitriev, D. N. Babentsev and A. N. Maslivets, Russ. J. Org. Chem., 2018, 54, 780 CrossRef CAS; (b) E. E. Stepanova, M. V. Dmitriev and A. N. Maslivets, Tetrahedron Lett., 2020, 61, 151595 CrossRef CAS; (c) E. E. Stepanova, M. V. Dmitriev and A. N. Maslivets, Russ. J. Org. Chem., 2021, 57, 32 CrossRef CAS.
- A. A. Moroz, V. E. Zhulanov, M. V. Dmitriev and A. N. Maslivets, Tetrahedron, 2020, 76, 130880 CrossRef CAS.
- (a) V. Nair, A. U. Vinod, N. Abhilash, R. S. Menon, V. Santhi, R. L. Varma, S. Viji, S. Mathew and R. Srinivas, Tetrahedron, 2003, 59, 10279 CrossRef CAS; (b) L. Tao, Z. Fan, X. Peng, J. Han, W. He, J. Chen, H. Deng, M. Shao, H. Zhang and W. Cao, Synthesis, 2018, 50, 4104 CrossRef CAS.
- The signals of impurities in the 1H NMR spectrum of 3a (a set of signals in the range of 8–9.5 ppm) are typical for enals formed by hydrolysis of the adduct of pyridine to acetylenes: B. A. Trofimov, L. V. Andriyankova, K. V. Belyaeva, L. P. Nikitina, A. V. Afonin and A. G. Mal'kina, Eur. J. Org. Chem., 2015, 2015, 7876 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- (a) T. R. Hoye and J. T. North, Tetrahedron Lett., 1990, 31, 4281 CrossRef CAS; (b) T. R. Hoye, J. T. North and L. J. Yao, J. Org. Chem., 1994, 59, 6904 CrossRef CAS.
- M. T. Richers, M. Breugst, A. Y. Platonova, A. Ullrich, A. Dieckmann, K. N. Houk and D. Seidel, J. Am. Chem. Soc., 2014, 136, 6123 CrossRef CAS PubMed.
- (a) L. Lázár and F. Fülöp, Eur. J. Org. Chem., 2003, 2003, 3025 CrossRef; (b) I. Szatmári, M. Heydenreich, A. Koch, F. Fülöp and E. Kleinpeter, Tetrahedron, 2013, 69, 7455 CrossRef; (c) I. Szatmári, T. A. Martinek, L. Lázár and F. Fülöp, Eur. J. Org. Chem., 2004, 2004, 2231 CrossRef; (d) I. Szatmári, T. A. Martinek, L. Lázár, A. Koch, E. Kleinpeter, K. Neuvonen and F. Fülöp, J. Org. Chem., 2004, 69, 3645 CrossRef PubMed.
- (a) V. Nair, A. R. Sreekanth, N. P. Abhilash, A. T. N. Biju, L. Varma, S. Viji and S. Mathew, ARKIVOC, 2005, 2005, 178 Search PubMed; (b) I. Yavari, Z. Hossaini, M. Sabbaghan and M. Ghazanfarpour-Darjani, Monatsh. Chem., 2007, 138, 677 CrossRef CAS.
- M. T. Maghsoodlou, S. M. Habibi-Khorassani, A. Moradi, N. Hazeri, A. Davodi and S. S. Sajadikhah, Tetrahedron, 2011, 67, 8492 CrossRef CAS.
- For utilization of NOESY to exchange studies, see: (a) D. X. Hu, P. Grice and S. V. Ley, J. Org. Chem., 2012, 77, 5198 CrossRef CAS PubMed; (b) S. R. Hussaini, A. Kuta, A. Pal, Z. Wang, M. A. Eastman and R. Duran, ACS Omega, 2020, 5, 24848 CrossRef CAS PubMed.
- See the ESI (S33 and S48†) for effects of HClconc and p-TSA on dr of products 5c and 8c..
Footnotes |
| † Electronic supplementary information (ESI) available: General information, characterisation data, copies of NMR spectra, and experimental procedures. CCDC 2119396 (3a), 2119397 (3f), 2119399 (8b), and 2119398 (8c). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra08384h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |