
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and biological evaluation of new derivatives of thieno-thiazole and dihydrothiazolo-thiazole scaffolds integrated with a pyrazoline nucleus as anticancer and multi-targeting kinase inhibitors†
Ismail M. M. Othmana,
Zahra M. Alamshanyb,
Nada Y. Tashkandib,
Mohamed A. M. Gad-Elkareema,
Somaia S. Abd El-Karim *c and
Eman S. Nossierd
*c and
Eman S. Nossierd
aDepartment of Chemistry, Faculty of Science, Al-Azhar University, Assiut 71524, Egypt
bDepartment of Chemistry, Faculty of Science, King Abdulaziz University, P.O. Box 42805, Jeddah 21551, Saudi Arabia
cDepartment of Therapeutic Chemistry, National Research Centre, Dokki, Cairo 12622, Egypt. E-mail: somaia_elkarim@hotmail.com
dDepartment of Pharmaceutical Medicinal Chemistry, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo 11754, Egypt. E-mail: dremannossier@azhar.edu.eg
First published on 22nd December 2021
Abstract
Deregulation of various protein kinases is considered as one of the important factors resulting in cancer development and metastasis, thus multi-targeting the kinase family is one of the most important strategies in current cancer therapy. This context represents the design and synthesis of two sets of derivatives bearing a pyrazoline-3-one ring conjugated either with a thieno[3,2-d]thiazole or with a dihydrothiazolo[4,5-d]thiazole scaffold via an NH linker, 3a–d and 5a–d respectively, using the pyrazolinone–thiazolinone derivative 1 as a key precursor. All the newly synthesized compounds were assessed in vitro for their anticancer activity against two cancer cell lines (MCF-7 and HepG-2). The safety profile of the most active cytotoxic candidates 1 and 3c was further examined against the normal cell line WI-38. The compounds 1 and 3c were further evaluated as multi-targeting kinase inhibitors against EGFR, VEGFR-2 and BRAFV600E, exhibiting promising suppression impact. Additionally, the latter compounds were investigated for their impact on cell cycle and apoptosis induction potential in the MCF-7 cell line. Moreover, the antimicrobial activity of all the new analogues was evaluated against a panel of Gram-positive and Gram-negative bacteria, yeast and fungi in comparison to streptomycin and amphotericin-B as reference drugs. Interestingly, both 1 and 3c showed the most promising microbial inhibitory effect. Molecular docking studies showed promising binding patterns of the compounds 1 and 3c with the prospective targets, EGFR, VEGFR-2 and BRAFV600E. Finally, additional toxicity studies were performed for the new derivatives which showed their good drug-like properties and low toxicity risks in humans.
1. Introduction
Despite the extensive research and rapid progress in drug science and chemotherapeutic agents for combating cancer, it is still one of the most leading causes of death worldwide.1 Statistics show that by 2040, cancer incidence will continue to rise to up to 29.5 million cases per year.2 Cancer treatment is still a major issue, owing to the toxicity, resistance, and lack of selectivity of the currently available anticancer medications.3 Dysregulated kinase function, which typically acts as an on/off switch for cellular proliferation and motility, is considered as one of the reasons for cancer. Mutation of kinases is responsible for cellular abnormalities leading to cancer initiation, progression or metastasis.4 Single and multiple kinase inhibitors are now considered as targeted therapeutic strategies for human malignancy treatment.5 Current drug discovery research shows that PIK3CA, BRAF, VEGR and epidermal growth factor receptor (EGFR) are key oncogenic kinase drug targets.5–8Epidermal growth factor receptor (EGFR) is a trans-membrane glycoprotein, belonging to a family which consists of four related receptor tyrosine kinases. It plays a key mediating role in cell signaling pathways including cell proliferation, apoptosis, angiogenesis, and metastatic spread.9 This critical role makes it a prominent target in cancer treatment. The overexpression of EGFR in a variety of human cancers, including head, neck, breast, lung, colorectal, prostate, renal, pancreas, ovary, and brain cancers results in poor treatment outcomes due to resistance to hormone therapy, cytotoxic drugs, and radiotherapy.10,11 As a result, international recommendations advocate anti-EGFR medicines as the first-line treatment in patients with advanced EGFR mutations, due to their higher efficacy and safety compared to standard chemotherapy.9,12,13
Furthermore, vascular endothelial growth factor receptor-2 (VEGFR-2), a transmembrane tyrosine kinase receptor, has been has been identified as the most important factor in inducing angiogenesis,14–16 which is considered as one of the defining features of tumor growth, invasion and metastasis. VEGFR-2 has long been recognized as the most important target in cancer anti-angiogenesis therapy.17,18 Several small molecule VEGFR-2 inhibitors were clinically approved or evaluated for cancer treatment.19–21
RAF is serine/threonine kinases which regulate ERK–MAPK pathway. There are three unique subtypes of RAF kinases in addition to homologues of BRAF.22–24 Many studies concluded that the BRAF serine/threonine kinase alterations have been detected in various types of human cancers that are linked to cell growth, survival and differentiation.25 BRAF gene (V600E) is the most abundant type of BRAF mutation in human cancers because of substitution of a valine for glutamic acid at position 600. Current researches showed that suppression of BRAF is a new era in human cancer therapeutic treatment.26
Drug discovery researches evidenced that heteroaromatic structures resemble various biologically active moieties in human bodies, like nucleic acids, hormones, and neurotransmitters. Accordingly, these scaffolds have been greatly utilized for designing different anticancer compounds which bind with different targets interrupting the biological pathways involved in cancer progress, making them as magnificent starting points for anticancer drug development.27
The drug design field ensures that thiazole is the most effective motif for the said target activity. Thiazoles have a superior anticancer activity because they have a better binding domain and less cytotoxicity in normal cells (physiological cells), but they also have site-specific mobility towards the malignant cells (pathological cell).28 Additionally, thiazole derivatives were reported to exert cytotoxic potency against several types of cancer disease via suppression of various kinases such as; JAK2 and EGFR, VEGFR and BRAF kinases.29–33 In addition, different researches confirmed the potent anticancer activities of the compounds containing thiophene and thiazole heterocycles via inhibition of BRAF kinase activity.34,35 Consequently, the combined substructures (thiophene and thiazole scaffolds) may produce synergistic effects to boost the anticancer activities without compromising their original effective qualities.27–29 Many clinically available thiazole-bearing antiproliferative drugs have marked their presence in the field of cancer chemotherapy depicting their anticancer activity profile through diverse mechanisms such as, tiazofurin,36 dasatinib,37 dabrafenib,38 patellamide A,39 ixabepilone and epothilone40,41 (Fig. 1).
 | ||
| Fig. 1 Some clinically approved thiazole-bearing anticancer drugs. | ||
In addition, pyrazole heterocycle is a privileged scaffold possessing diverse biological activities.42,43 It is emerged as a useful pharmacophore in the synthesis of potent anticancer drugs.44,45 Moreover, multiple studies identified various thiazolyl-pyrazole compounds as potential anticancer agents. As an example, Lv and coworkers have reported that the thiazolyl-pyrazoline derivative I displayed potent cytotoxic activity against breast cancer cell line (MCF-7) with IC50 of 0.07 μM via EGFR inhibition with IC50; 0.06 μM.46,47 On the other hand, Zhao et al. concluded that the 4,5-dihydropyrazole derivatives bearing thiazole and thiophene moieties IIa–c produced effective antiproliferative activity against WM266.4 and MCF-7 cell lines through BRAFV600E inhibitory activity.48 While Sadashiva and coworkers have investigated the dual anticancer activity against A549 and MCF-7 human cancer cell lines as well as antimicrobial activity of the derivatives bearing thiazole heterocycle linked with pyrazoline moiety via carbohydrazide linker as compounds IIIa–c.49 Furthermore, the thiazoline-pyrazolines IV–VI represented promising cytotoxic effects and different RTKs suppressing impact while, the thiazolopyrazolyl coumarin derivatives VIIa, b exhibited significant in vitro anticancer potentiality against different human cancer cell lines via a remarkable inhibition of VEGFR-2 with no noticeable toxicity towards the normal cells HFB4.50 In addition, Vaarla et al. have designed and synthesized the thiazolyl-3-arylpyrazole-4-carbaldehydes VIIIa, b as significant antimicrobial and cytotoxic agents against HeLa cell line and the docking simulation study validated different types of interactions with human microsomal cytochrome P450 (1z11.pdb) enzyme51 (Fig. 2).
 | ||
| Fig. 2 Examples of various thiazolyl-pyrazoline-based compounds as anticancer candidates of TK inhibition activity. | ||
Molecular hybridization of two or more bioactive pharmacophores in the same molecular architecture represents an optimistic strategy in discovery of novel anticancer drugs. This approach raises the possibility to synergize the anticancer efficacy via targeting two or more molecular proteins as a single entity, reduces the risk of drug–drug interactions as well as prevents the drug resistance obstacle.52 Prompted by the above considerations and in view of our continuous efforts in synthesis of heterocyclic derivatives of anticancer activity targeting different protein kinases,53–56 the ongoing study was focused on designing and synthesis of two sets of new analogues bearing pyrazoline-3-one ring conjugated either with the fused thieno[3,2-d]thiazole scaffold 3a–c or with dihydrothiazolo[4,5-d]thiazole scaffold 5a–c via NH linker as potential anticancer agents of multi-targeted tyrosine kinase inhibiting activity against EGFR, VEGFR-2 and BRAFV600E kinases. It was taken into account the impact of molecular orientation, ring size variation and the presence of heteroatoms that could participate in hydrogen bonding interactions with the binding pockets of the protein kinases under study (Fig. 3).
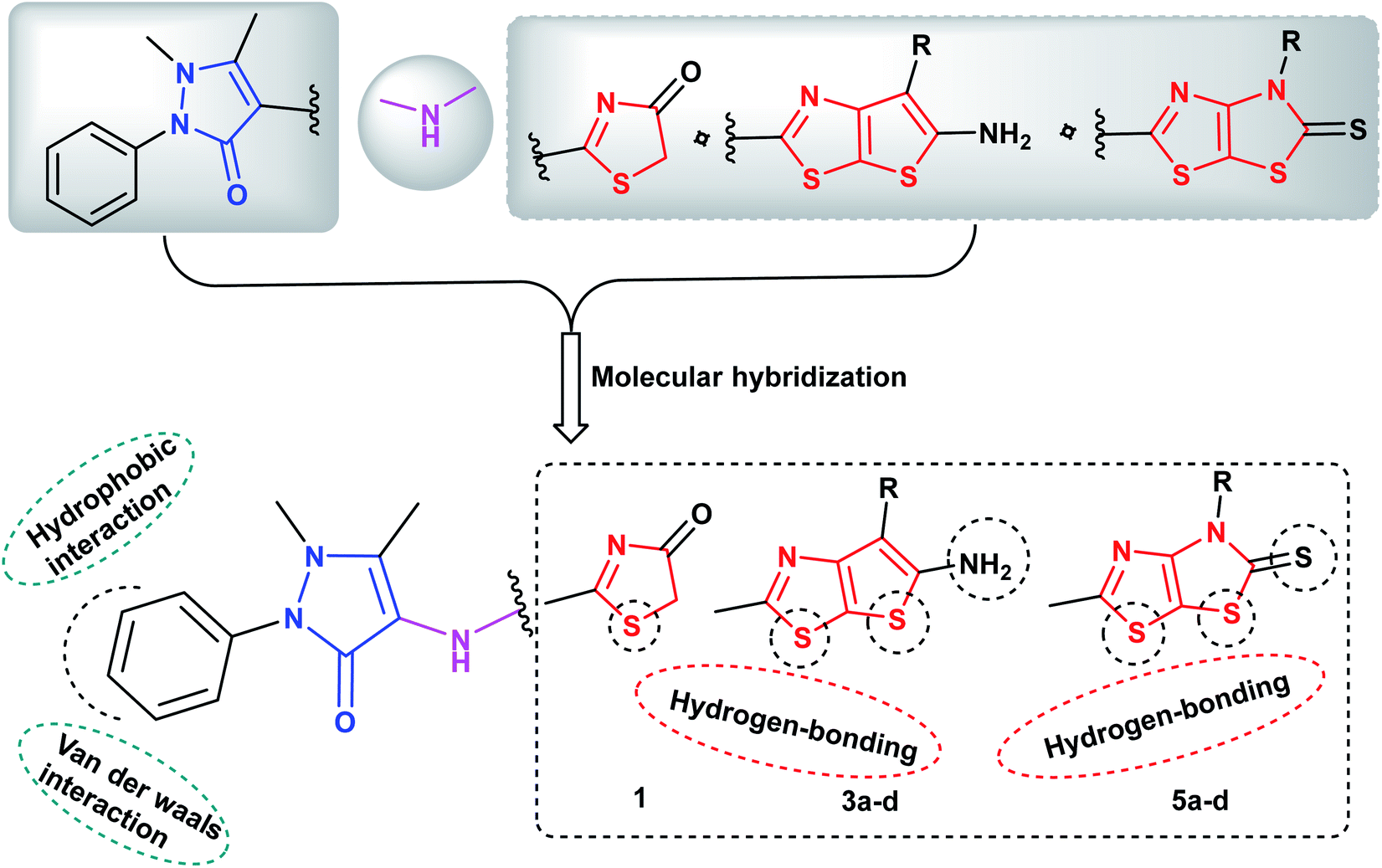 | ||
| Fig. 3 Proposed hypothetic model for pyrazolinyl-thieno[3,2-d]thiazole- and pyrazolinyl-thiazolo[4,5-d]thiazolidine-2-thione compounds 3a–d and 5a–d, respectively. | ||
All the newly synthesized compounds were assessed as anticancer candidates against human breast cancer cells (MCF-7) and human liver carcinoma cell line (HepG-2). The safety of the most promising anticancer candidates was also evaluated against the normal WI-38 cell line. Moreover, the most promising cytotoxic compounds were further evaluated as multitargeting protein kinases against EGFR, VEGFR-2 and f BRAFV600E and were investigated for their impact on cell cycle and apoptosis induction potential in MCF-7 cell line.
Immunosuppression which usually results due to anticancer drug regimen and destruction of the mucosal barrier due to utilizing invasive devices in cancer patients make them more vulnerable to different infectious diseases that need long-term prophylactic antibiotic regimens.57 Moreover, the efficiency of antibiotic regimen to combat infections in cancer patients is compromised due to the growth of drug resistant bacterial pathogens that dominate in neutropenic patients.58 Accordingly, novel anticancer agents that specifically suppress cancer cells having dual anticancer and antimicrobial activities are considered to act as prophylaxis against microbial infections alongside suppressing the growth of tumors in cancer patients.57,58 Accordingly, the new compounds were further subjected to antimicrobial investigation in comparison to amoxicillin trihydrate and clotrimazole as standard antibacterial and antifungal drugs, respectively, against a panel of Gram-positive, Gram-negative bacterial, yeast and fungal strains. In addition, minimum inhibitory concentrations were also assessed for the new candidates. Additional toxicity study was performed for the new derivatives which represented their good drug-likeness properties and low toxicity risks in humans.
2. Experimental
2.1. Chemistry
The instruments used for measuring the melting points, spectral data (IR, mass, 1H NMR and 13C NMR) and elemental analyses are provided in details in ESI.†The chemical names given for the prepared compounds are according to the IUPAC system. 2-Chloro-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)acetamide was prepared according to the reported method.59
Yield 86%, mp 242–244 °C; IR (νmax/cm−1): 3382 (NH), 3050 (CH-arom.), 2945 (CH-aliph.), 1685, 1655 (2CO); 1H NMR (DMSO-d6) δ: 2.15 (s, 3H, CH3), 3.21 (s, 3H, NCH3), 4.06 (s, 2H, CH2), 7.05–7.53 (m, 5H, Ar-H), 9.62 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): 12.11 (CH3), 32.04 (CH3), 35.16 (CH2), 116.10, 121.07, 122.69, 128.59, 131.84, 137.91, 158.00, 166.74 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 176.13 (CO); MS, m/z (%): 302 [M+] (14), 77 (100%); anal. calcd for C14H14N4O2S (302.35): C, 55.61; H, 4.67; N, 18.53; S, 10.61%. Found: C, 55.83; H, 4.89; N, 18.75; S, 10.84%.
O), 176.13 (CO); MS, m/z (%): 302 [M+] (14), 77 (100%); anal. calcd for C14H14N4O2S (302.35): C, 55.61; H, 4.67; N, 18.53; S, 10.61%. Found: C, 55.83; H, 4.89; N, 18.75; S, 10.84%.
2.1.2.1. 5-Amino-2-((1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)amino)thieno [3,2-d]thiazole-6-carbonitrile (3a). Yield: 72%; brown crystal; mp >300 °C; IR (νmax/cm−1): 3415, 3373 (NH2), 3226 (NH), 3067 (CH-arom.), 2985 (CH-aliph.), 2212 (CN), 1658 (CO); 1H NMR (DMSO-d6) δ: 2.15 (s, 3H, CH3), 3.07 (s, 3H, NCH3), 6.44 (s, 2H, NH2, D2O exchangeable), 7.35–7.58 (m, 5H, Ar-H), 9.25 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): 15.62 (CH3), 33.83 (CH3), 106.91, 109.67, 115.86, 118.20, 120.01, 122.69, 129.60, 130.74, 135.94, 139.17, 153.27, 156.13, 165.83 (C
O); MS, m/z (%): 382 [M+] (44), 77 (100); anal. calcd for C17H14N6OS2 (382.46): C, 53.39; H, 3.69; N, 21.97; S, 16.77%. Found: C, 53.60; H, 3.89; N, 21.74; S, 16.56%.
2.1.2.2. Ethyl 5-amino-2-((1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)amino) thieno[3,2-d]thiazole-6-carboxylate (3b). Yield: 75%; pale brown crystal; mp 285–287 °C; IR (νmax/cm−1): 3392, 3318 (NH2), 3237 (NH), 3057 (CH-arom.), 2942 (CH-aliph.), 1715, 1654 (2CO); 1H NMR (DMSO-d6) δ: 1.23 (t, J = 7.2 Hz, 3H, OCH2
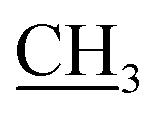 ), 2.18 (s, 3H, CH3), 3.11 (s, 3H, NCH3), 4.15 (q, J = 7.2 Hz, 2H, CH2, O
), 2.18 (s, 3H, CH3), 3.11 (s, 3H, NCH3), 4.15 (q, J = 7.2 Hz, 2H, CH2, O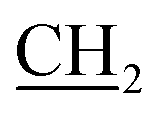 CH3), 6.70 (s, 2H, NH2, D2O exchangeable), 7.39–7.61 (m, 5H, Ar-H), 9.27 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): 12.35 (OCH2
CH3), 6.70 (s, 2H, NH2, D2O exchangeable), 7.39–7.61 (m, 5H, Ar-H), 9.27 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): 12.35 (OCH2 ), 16.24 (CH3), 31.70 (NCH3), 62.99 (O
), 16.24 (CH3), 31.70 (NCH3), 62.99 (O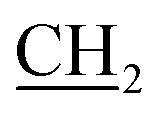 CH3), 109.10, 117.58, 120.61, 123.28, 125.01, 129.42, 131.03, 134.19, 139.81, 154.38, 158.18, 165.57 (CO), 168.73(CO, ester); MS, m/z (%): 429 [M+] (75), 228 (100); anal. calcd for C19H19N5O3S2 (429.52): C, 53.13; H, 4.46; N, 16.31; S, 14.93%. Found: C, 53.35; H, 4.67; N, 16.54; S, 14.72%.
CH3), 109.10, 117.58, 120.61, 123.28, 125.01, 129.42, 131.03, 134.19, 139.81, 154.38, 158.18, 165.57 (CO), 168.73(CO, ester); MS, m/z (%): 429 [M+] (75), 228 (100); anal. calcd for C19H19N5O3S2 (429.52): C, 53.13; H, 4.46; N, 16.31; S, 14.93%. Found: C, 53.35; H, 4.67; N, 16.54; S, 14.72%.
2.1.2.3. 5-Amino-2-((1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)amino) thieno[3,2-d]thiazole-6-carbohydrazide (3c). Yield: 70%; buff crystal; mp 293–295 °C; IR (νmax/cm−1): 3431, 3356, 3284, 3222 (2NH2), 3178 (2NH), 3069 (CH-arom.), 2937 (CH-aliph.), 1698, 1652 (2CO); 1H NMR (DMSO-d6) δ: 2.21 (s, 3H, CH3), 3.10 (s, 3H, NCH3), 5.91 (s, 2H, NH2, D2O exchangeable), 7.04–7.66 (m, 8H, Ar-H, NH, NH2, D2O exchangeable), 9.81 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): 13.75 (CH3), 33.92 (NCH3), 109.35, 114.51, 119.71, 123.61, 129.95, 131.08, 132.52, 137.40, 139.92, 151.73, 156.24, 166.78 (C
O), 169.89 (CO); MS, m/z (%): 415 [M+] (15), 214 (100); anal. calcd for C17H17N7O2S2 (415.49): C, 49.14; H, 4.12; N, 23.60; S, 15.43%. Found: C, 49.35; H, 4.33; N, 23.81; S, 15.64%.
2.1.2.4. 5-Amino-2-((1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)amino) thieno[3,2-d]thiazole-6-carbothioamide (3d). Yield: 73%; brown crystal; mp 297–299 °C; IR (νmax/cm−1): 3423, 3381, 3276, 3215 (2NH2), 3155 (NH), 3070 (CH-arom.), 2943 (CH-aliph.), 1661 (CO); 1H NMR (DMSO-d6) δ: 2.09 (s, 3H, CH3), 3.16 (s, 3H, NCH3), 6.82 (s, 2H, NH2, D2O exchangeable), 7.53–7.99 (m, 7H, Ar-H, NH2), 9.78 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): 12.42 (CH3), 32.37 (NCH3), 111.66, 117.31, 119.25, 121.95, 123.45, 129.61, 130.11, 132.93, 137.12, 151.32, 154.20, 165.52 (C
O), 183.72 (CS); MS, m/z (%): 417 [M+1] (29), 56 (100); anal. calcd for C17H16N6OS3 (416.54): C, 49.02; H, 3.87; N, 20.18; S, 23.09%. Found: C, 49.25; H, 3.65; N, 20.39; S, 23.31%.
2.1.3.1. 1,5-Dimethyl-2-phenyl-4-((4-phenyl-5-thioxo-4,5-dihydrothiazolo[4,5-d]thiazol-2-yl) amino)-1H-pyrazol-3(2H)-one (5a). Yield: 68%; dark brown crystal; mp 201–203 °C; IR (νmax/cm−1): 3335 (NH), 3078 (CH-arom.), 2936 (CH-aliph.), 1657 (CO), 1273 (C
S); 1H NMR (DMSO-d6) δ = 2.19 (s, 3H, CH3), 3.03 (s, 3H, NCH3), 7.05–7.62 (m, 6H, Ar-H), 7.85 (d, J = 7.2 Hz, 2H, Ar-H), 7.99 (d, J = 7.2 Hz, 2H, Ar-H), 10.13 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): 13.71 (CH3), 34.37 (NCH3), 116.48, 119.30, 120.55, 125.12, 127.26, 128.45, 129.76, 132.38, 136.23, 139.48, 143.16, 152.59, 157.38, 164.50 (CO), 185.12 (CS); MS, m/z (%): 451 [M+] (30), 452 [M+1] (25), 249 (100); anal. calcd for C21H17N5OS3 (451.59): C, 55.85; H, 3.79; N, 15.51; S, 21.30%. Found: C, 55.64; H, 3.57; N, 15.73; S, 21.51%.
2.1.3.2. 4-((4-Ethyl-5-thioxo-4,5-dihydrothiazolo[4,5-d]thiazol-2-yl)amino)-1,5-dimethyl-2-phenyl-1H-pyrazol-3(2H)-one (5b). Yield: 65%; brown crystal; mp 183–185 °C; IR (νmax/cm−1): 3352 (NH), 3085 (CH-arom.), 2978 (CH-aliph.), 1656 (CO), 1247 (C
S); 1H NMR (DMSO-d6) δ: 1.22 (t, J = 7.2 Hz, 3H, –CH2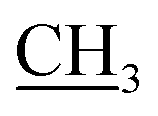 ), 2.65 (s, 3H, CH3), 3.01 (s, 3H, NCH3), 4.15 (q, J = 7.2 Hz, 2H,
), 2.65 (s, 3H, CH3), 3.01 (s, 3H, NCH3), 4.15 (q, J = 7.2 Hz, 2H, 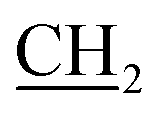 –CH3), 7.41–7.98 (m, 5H, Ar-H), 10.05 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): 13.92 (–CH2
–CH3), 7.41–7.98 (m, 5H, Ar-H), 10.05 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): 13.92 (–CH2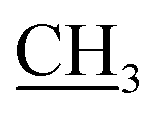 ), 18.35 (CH3), 32.66 (NCH3), 51.20 (
), 18.35 (CH3), 32.66 (NCH3), 51.20 (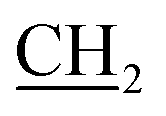 CH3), 118.19, 121.64, 124.03, 127.46, 130.63, 135.26, 146.10, 157.71, 157.96, 164.56 (CO), 184.27 (CS); MS, m/z (%): 404 [M+1] (28), 327 (100); anal. calcd for C17H17N5OS3 (403.54): C, 50.60; H, 4.25; N, 17.35; S, 23.84%. Found: C, 50.82; H, 4.46; N, 17.57; S, 23.63%.
CH3), 118.19, 121.64, 124.03, 127.46, 130.63, 135.26, 146.10, 157.71, 157.96, 164.56 (CO), 184.27 (CS); MS, m/z (%): 404 [M+1] (28), 327 (100); anal. calcd for C17H17N5OS3 (403.54): C, 50.60; H, 4.25; N, 17.35; S, 23.84%. Found: C, 50.82; H, 4.46; N, 17.57; S, 23.63%.
2.1.3.3. 1,5-Dimethyl-4-((4-methyl-5-thioxo-4,5-dihydrothiazolo[4,5-d]thiazol-2-yl)amino)-2-phenyl-1H-pyrazol-3(2H)-one (5c). Yield: 61%; reddish brown crystal; mp 189–191 °C; IR (νmax/cm−1): 3328 (NH), 3074 (CH-arom.), 2983 (CH-aliph.), 1652 (CO), 1261 (C
S); 1H NMR (DMSO-d6) δ: 2.27 (s, 3H, CH3), 3.05 (s, 3H, NCH3), 3.06 (s, 3H, NCH3), 7.33–7.68 (m, 5H, Ar-H), 10.14 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): 11.89 (CH3), 31.74 (NCH3), 34.38 (NCH3), 117.01, 121.82, 123.94, 128.43, 131.24, 138.42, 143.51, 150.29, 158.32, 165.06 (CO), 187.76 (CS); MS, m/z (%): 389 [M+] (55), 188 (100); anal. calcd for C16H15N5OS3 (389.52): C, 49.34; H, 3.88; N, 17.35; S, 24.70%. Found: C, 49.56; H, 3.67; N, 17.76; S, 24.91%.
2.1.3.4. 4-((4-(4-Chlorophenyl)-5-thioxo-4,5-dihydrothiazolo[4,5-d]thiazol-2-yl)amino)-1,5-dimethyl-2-phenyl-1H-pyrazol-3(2H)-one (5d). Yield: 69%; brown crystal; mp 211–213 °C; IR (νmax/cm−1): 3320 (NH), 3089 (CH-arom.), 2943 (CH-aliph.), 1662 (CO), 1287 (C
S); 1H NMR (DMSO-d6) δ: 2.12 (s, 3H, CH3), 3.02 (s, 3H, NCH3), 6.98 (d, J = 12.9 Hz, 2H, Ar-H), 7.29–7.34 (m, 1H, Ar-H), 7.52–7.78 (m, 6H, Ar-H), 9.43 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6): 12.18 (CH3), 32.86 (NCH3), 119.81, 122.42, 125.11, 128.02, 129.22, 130.16, 131.06, 133.82, 138.45, 141.73, 145.51, 158.18, 161.92, 165.87 (CO), 183.79 (CS); MS, m/z (%): 487, 485 [M+] (25, 70), 202 (100); anal. calcd for C21H16ClN5OS3 (486.03): C, 51.89; H, 3.32; Cl, 7.29; N, 14.41; S, 19.79%. Found: C, 51.68; H, 3.55; Cl, 7.51; N, 14.63; S, 19.58%.
2.2. Biological activity
2.2.5.1. Minimal inhibitory concentration (MIC) measurement. The bacteriostatic activity of the compounds was then evaluated using the two-fold serial dilution technique. Two-fold serial dilutions of the tested compounds solutions were prepared using the proper nutrient broth.67–69 More details in the ESI.†
2.3. Computational studies
3. Results and discussion
3.1. Chemistry
An efficient synthesis of the new thienothiazoles 3a–d and thiazolothiazoles 5a–d has been performed starting with (1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)carbamic chloride.59 Their synthetic routes were outlined in (Schemes 1 and 2). Refluxing 2-chloro-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)acetamide with ammonium thiocyanate in ethanol produced the corresponding thiazolidin-4-one derivative 1. The 1H-NMR spectrum of compound 1 appeared three singlet signals at δ 2.15, 3.21 and 4.06 ppm due to CH3, NCH3 and CH2 respectively and also, its 13C-NMR spectrum showed signals at δ 12.11, 32.04 and 35.16 ppm due to CH3, NCH3 and CH2 respectively.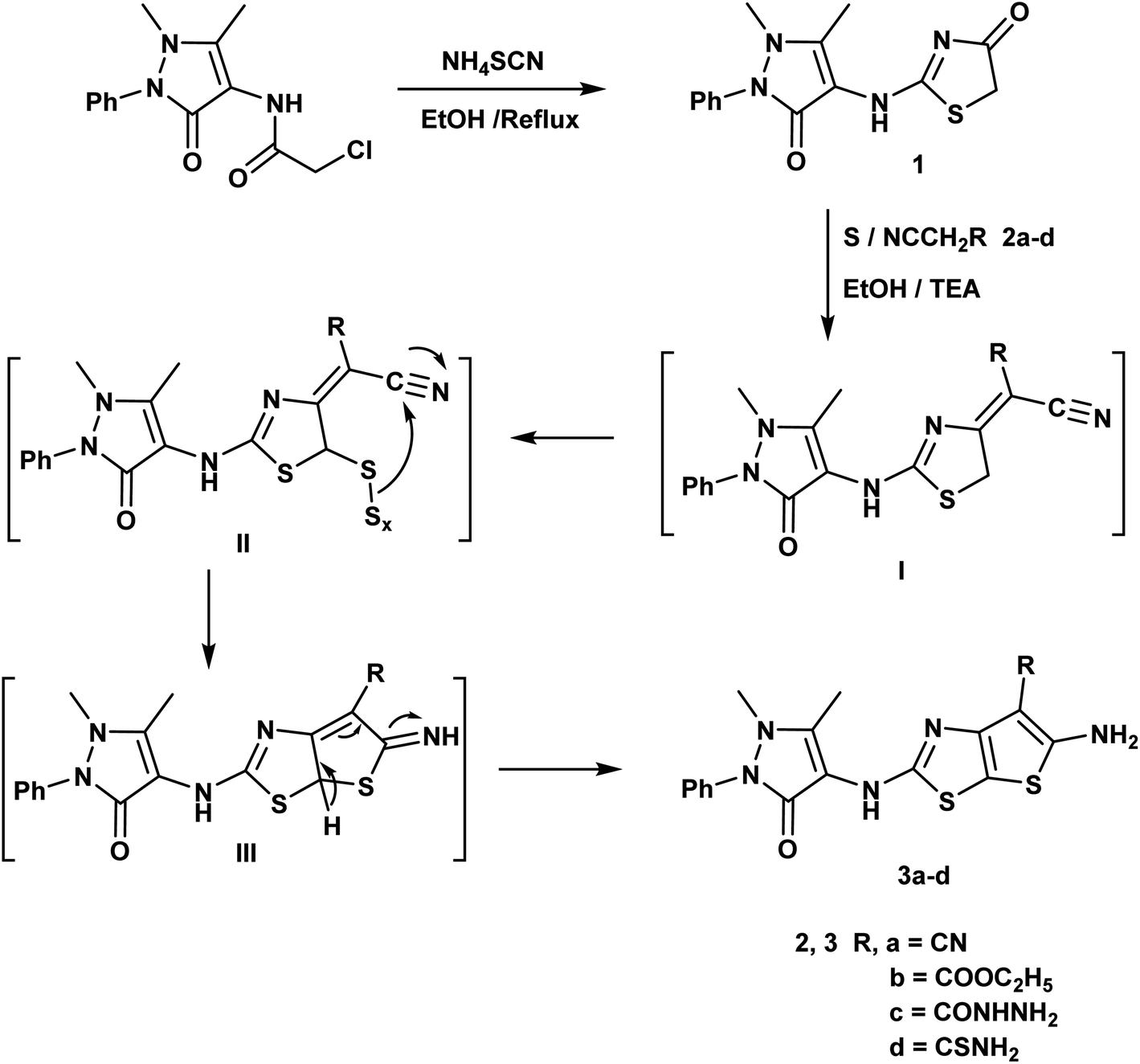 | ||
| Scheme 1 Synthesis of thienothiazole derivatives. | ||
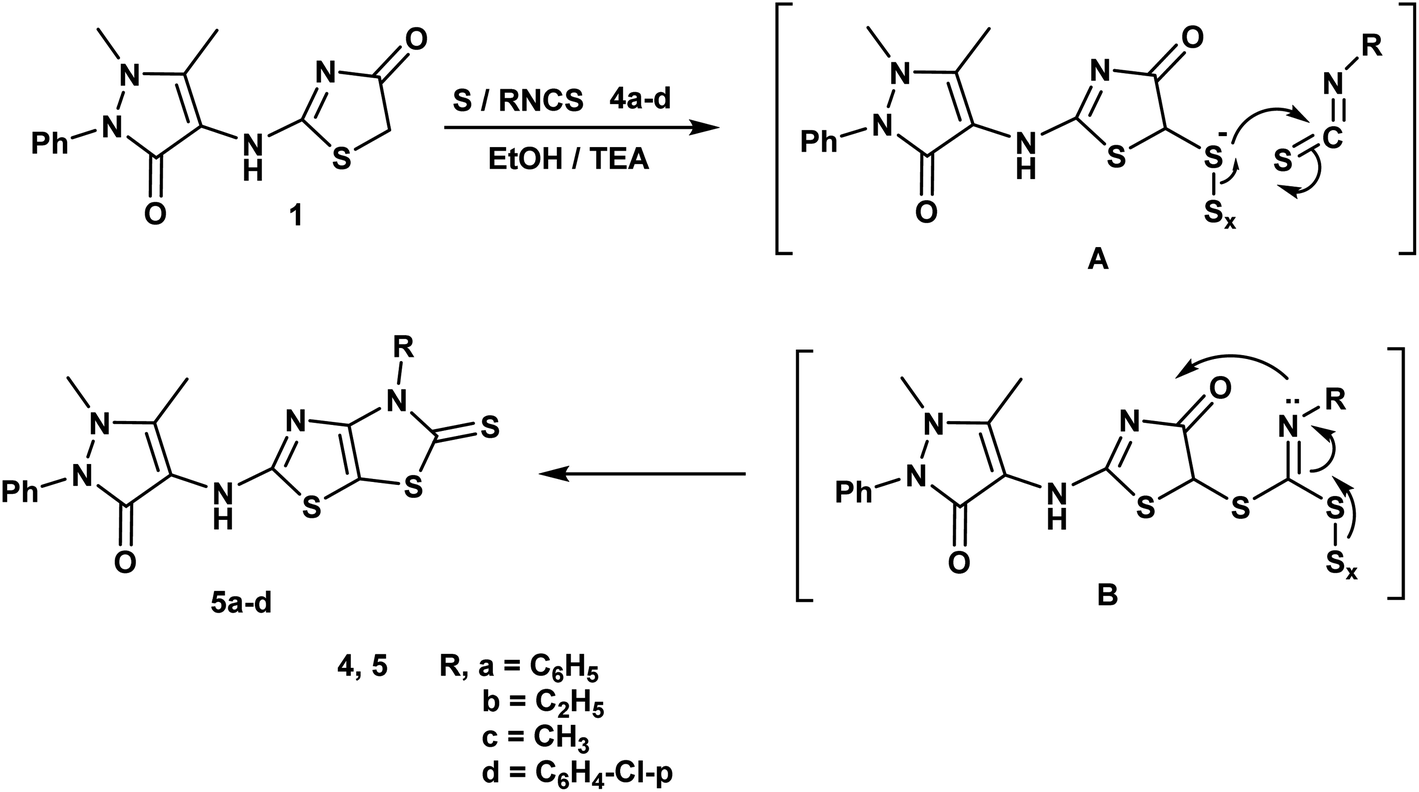 | ||
| Scheme 2 Synthesis of thiazolothiazole derivatives. | ||
The Gewald reaction is a condensation of ketone or aldehyde with acetonitriles substituted by a strong electron withdrawing group (such as ester, amide, and nitrile) in the presence of elemental sulfur and base to give a poly-substituted 2-amino-thiophene. So, the thieno[3,2-d]thiazole derivatives 3a–d were obtained via the treatment of key compound 1 according to Gewald reaction with a series of active methylene derivatives namely; malononitrile, ethyl cyanoacetate, cyanoacetohydrazide and/or cyanothioacetamide in the presence of sulfur and triethylamine. IR spectrum of the compound 3a showed bands at 3415, 3373 cm−1 referring to NH2 and strong absorption band at 2212 cm−1 due to CN. The 1H-NMR spectrum of compound 3b displayed triplet and quartet signals at δ 1.23 and 4.15 ppm according to the ester group (Scheme 1).
Furthermore, reaction of thiazolidin-4-one moiety 1 with different isothiocynate derivatives namely; phenyl isothiocyanate, ethyl isothiocyanate, methyl isothiocyanate and/or p-chloro phenyl isothiocyanate in the presence of sulfur and triethylamine afforded the corresponding dihydrothiazolo[4,5-d]thiazole derivatives 5a–d respectively. The 1H-NMR spectrum of compound 5b displayed triplet and quartet signals at δ 1.22 and 4.15 ppm according to the ethyl group and its 13C-NMR showed signals at δ 13.92, 51.20 and 184.27 ppm referring to ethyl and CS groups (Scheme 2).
3.2. Biological activity
| Compound no. | IC50a (μM) | IC50a (μM) | IC50a (μM) |
|---|---|---|---|
| MCF-7 | HepG-2 | WI-38 | |
| a IC50 values = mean ± SD of three independent determinations. | |||
| 1 | 4.02 ± 0.02 | 4.52 ± 0.04 | 60.44 |
| 3a | 27.58 ± 0.01 | 25.72 ± 0.03 | |
| 3b | 36.77 ± 0.02 | 39.02 ± 0.02 | |
| 3c | 8.35 ± 0.03 | 7.88 ± 0.01 | 54.72 |
| 3d | 18.19 ± 0.01 | 16.91 ± 0.01 | |
| 5a | 73.80 ± 0.04 | 65.19 ± 0.02 | |
| 5b | 43.94 ± 0.02 | 48.04 ± 0.03 | |
| 5c | 14.69 ± 0.03 | 11.85 ± 0.03 | |
| 5d | 88.56 ± 0.01 | 81.50 ± 0.03 | |
| Doxorubicin | 4.62 ± 0.01 | 5.66 ± 0.01 | |
The safety of the most active compounds 1, 3c towards the normal fibroblasts (WI-38) cell line was also examined. The tested compounds were found to be more selective to MCF-7 and HepG-2 cancer cell lines than to the normal cell line WI-38 as they revealed cytotoxicity with IC50 of 60.44, 54.72 μM. Therefore, they can be considered to be safe candidates against the normal cell line.
| Compound no. | IC50 (mean ± SEM) (μM) | ||
|---|---|---|---|
| EGFR | VEGFR-2 | BRAFV600E | |
| a IC50: compound concentration required to inhibit the enzyme activity by 50%, SEM: standard error mean; each value is the mean of three values. | |||
| Sorafenib | 0.025 ± 0.10 | 1.022 ± 0.15 | 0.040 ± 0.13 |
| 1 | 0.022 ± 1.00 | 2.470 ± 0.30 | 2.026 ± 0.50 |
| 3c | 0.017 ± 0.05 | 2.259 ± 0.45 | 0.088 ± 0.01 |
 | ||
| Fig. 4 Cell cycle analysis of and activation of apoptosis caused by the derivatives 1 and 3c against MCF-7 cells. | ||
 | ||
| Fig. 5 Apoptosis induction induced by the derivatives 1, 3c. | ||
| Mean diameter of inhibition zone (mean ± SEM) (mm) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd. no. | Gram +ve bacteria | Gram −ve bacteria | Fungi | |||||
| S. aureus 25923 | E. faecalis | S. pneumoniae 010010 | E. coli RCMB 010052 | S. typhi ATCC 14028 | P. aeruginosa | A. fumigatus RCMB 02568 | C. albicans ATCC 10231 | |
| a Streptomycin and Amphotericin B were used as standard drugs against the tested bacteria and fungi, respectively. | ||||||||
| 1 | 31 ± 0.17 | 28 ± 0.92 | 30 ± 0.87 | 26 ± 0.22 | 21 ± 0.54 | 27 ± 0.51 | 25 ± 0.88 | 22 ± 0.30 |
| 3a | 28 ± 0.15 | 13 ± 0.76 | 21 ± 0.69 | 25 ± 0.15 | 17 ± 0.95 | 22 ± 0.44 | 18 ± 0.25 | 12 ± 0.51 |
| 3b | 19 ± 0.68 | 20 ± 0.24 | 14 ± 0.84 | 15 ± 0.71 | 23 ± 0.79 | 17 ± 0.41 | 21 ± 0.22 | 11 ± 0.70 |
| 3c | 30 ± 0.47 | 27 ± 0.36 | 25 ± 0.17 | 28 ± 0.92 | 21 ± 0.16 | 23 ± 0.13 | 24 ± 0.15 | 18 ± 0.67 |
| 3d | 13 ± 0.95 | 22 ± 0.11 | 28 ± 0.45 | 22 ± 0.14 | 18 ± 0.13 | 20 ± 0.19 | 19 ± 0.45 | 16 ± 0.28 |
| 5a | 21.2 ± 0.25 | 13 ± 0.43 | NA | NA | 23 ± 0.05 | NA | 9 ± 0.53 | NA |
| 5b | 16 ± 0.88 | 27 ± 0.41 | 28 ± 0.12 | 10 ± 0.42 | NA | 18 ± 0.95 | NA | 12 ± 0.07 |
| 5c | 20 ± 0.08 | 24 ± 0.89 | 15 ± 0.86 | 25 ± 0.10 | 17 ± 0.78 | 22 ± 0.48 | 26 ± 0.17 | 19 ± 0.23 |
| 5d | 25 ± 0.13 | NA | NA | 15 ± 0.04 | NA | 13 ± 0.11 | NA | NA |
| aS | 26 ± 0.45 | 25 ± 0.38 | 22 ± 0.81 | 27 ± 0.56 | 23 ± 0.92 | 20 ± 0.53 | — | — |
| aA | — | — | — | — | — | — | 22 ± 0.72 | 20 ± 0.78 |
| Gram +ve bacteria | Gram −ve bacteria | Fungi | ||||||
|---|---|---|---|---|---|---|---|---|
| Cpd. no. | S. aureus 25923 | E. faecalis | S. pneumoniae 010010 | E. coli RCMB 010052 | S. typhi ATCC 14028 | P. aeruginosa | A. fumigatus RCMB 02568 | C. albicans ATCC 10231 |
| a Streptomycin and amphotericin B were used as standard drugs against the tested bacteria and fungi, respectively. | ||||||||
| 1 | 3.63 | 16.14 | 3.94 | 18.33 | 62.18 | 7.52 | 20.81 | 38.26 |
| 3a | 16.85 | 185.18 | 58.86 | 19.36 | 125.14 | 40.45 | 120.05 | 186.11 |
| 3b | 78.98 | 67.41 | 178.52 | 118.98 | 45.62 | 124.84 | 62.12 | 189.05 |
| 3c | 4.12 | 17.28 | 20.15 | 14.18 | 64.05 | 42.18 | 19.23 | 18.41 |
| 3d | 181.12 | 75.47 | 16.18 | 71.82 | 121.43 | 67.02 | 117.96 | 132.26 |
| 5a | 63.57 | 183.27 | — | — | 44.68 | — | 197.41 | — |
| 5b | 135.36 | 15.18 | 15.87 | 193.20 | — | 121.75 | — | 185.13 |
| 5c | 68.75 | 18.86 | 178.06 | 20.75 | 125.36 | 62.25 | 17.83 | 79.42 |
| 5d | 18.87 | — | — | 118.15 | — | 180.25 | — | — |
| aS | 16.75 | 19.48 | 65.33 | 9.18 | 47.12 | 68.25 | — | — |
| aA | — | — | — | — | — | — | 39.15 | 67.82 |
Based on the MIC records in Table 4, it has been detected that there is a wide variability in the antimicrobial potency of the newly tested members. Evidently, the pyrazolinyl-aminothiazol-one 1 and the thieno[3,2-d]thiazole-carbohydrazide derivative 3c produced the most potent broad spectrum antimicrobial activity against the test bacterial and fungal strains.
It has been detected that the compounds 1 and 3c exhibited selective antibacterial activity against the tested Gram-positive bacteria of 1.20–16 folds more potency than streptomycin of MIC range; 3.63–20.15 μM, MICstreptomycin; 16.75–65.33 μM. Conversely, the potency of the latter compounds decreased by 2 and 1.3 folds against the Gram-negative bacteria S. pneumoniae and E. coli of MIC range; 14.18–64.05 μM, MICstreptomycin; 9.18, 47.12 μM. On the other hand, P. aeruginosa strain represented greater selectivity towards 1, 3c than streptomycin of MIC values; 7.52, 42.18 μM, respectively, MICstreptomycin; 68.25 μM. In addition, the fungal and yeast strains under study exhibited 1.8–3.6 times more sensitivity towards the compounds 1 and 3c than the reference drug amphotericin B producing MIC range 18.41–38.26 μM, MICamphotericin B; 68.25 μM. Furthermore, the thieno [3,2-d]thiazole-6-carbonitrile analogue 3a produced equipotent antibacterial activity to streptomycin against S. aureus and 1.1, 1.6 folds more potency against S. pneumoniae and P. aeruginosa of MIC values; 16.85, 58.86 μM. On the other hand, the antifungal activity 3a was completely lost against the tested fungal and yeast strains. Overall, a majority of the target compounds showed moderate to weak activity against the tested kinds of susceptible strains.
The obtained results confirmed that the compounds 1 and 3c among the whole tested derivatives were the most promising antimicrobial agents against the examined Gram-positive bacteria and the fungal strains, but they produced a slightly weaker activity against the examined Gram-negative microbes.
3.3. Computational studies
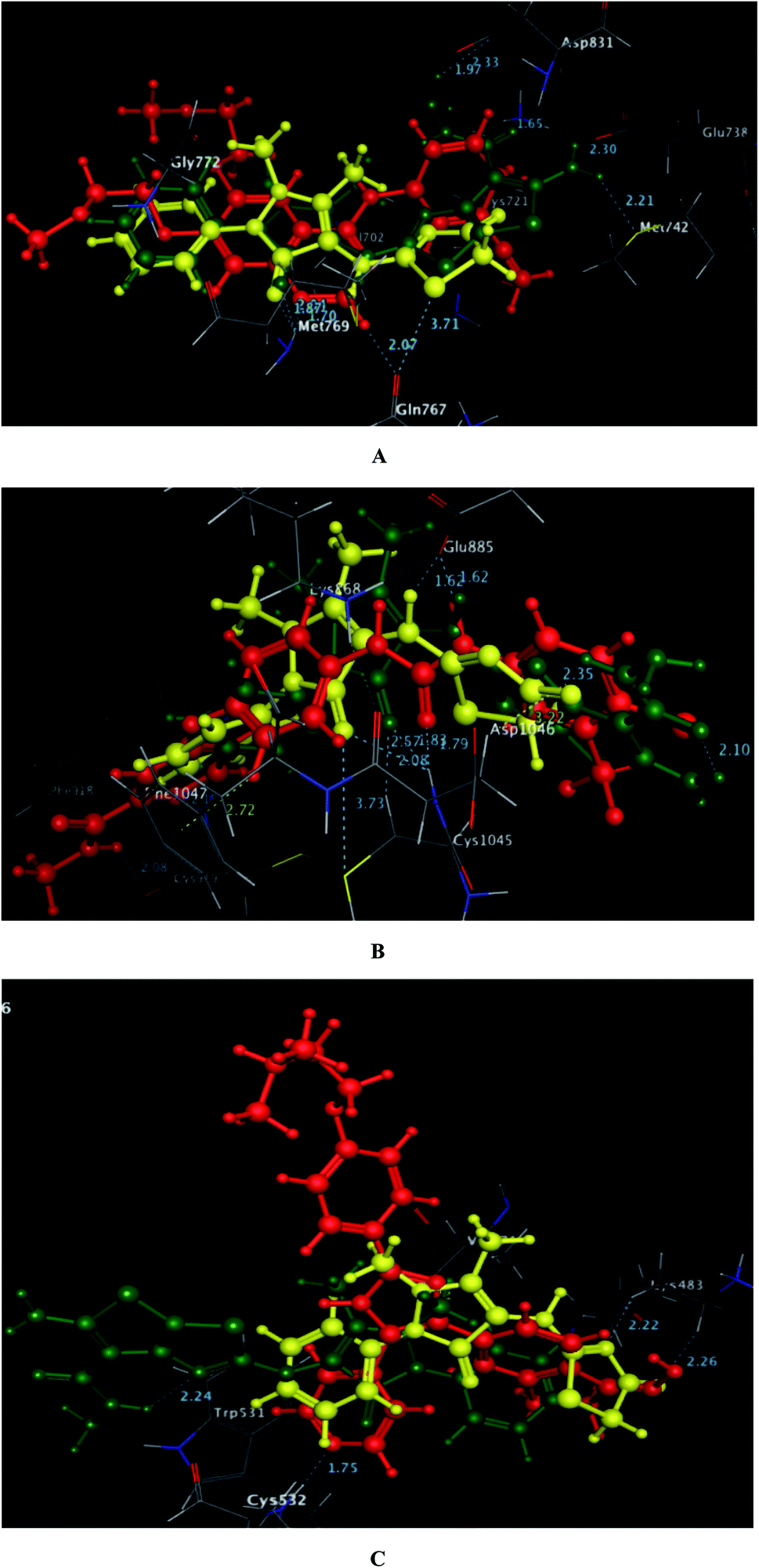
As depicted in Fig. 6, the targets 1 and 3c were bound to the active site of EGFR with energy scores of −11.67 and −11.83 kcal mol−1, respectively through formation of two H-bonds, one acceptor between the oxygen of the pyrazolone moiety and the backbone of the key amino acid Met769, and the other one was donor between the thiazolyl sulfur and the backbone of Gln767 (distance: 2.76 & 2.93 Å in compound 1 and 2.95 & 3.71 Å in compound 3c, respectively). Also, the pyrazolone centroid of compound 1 afforded arene-cation interaction with Gln772. On the other hand, the nitrogen of the amino group at p-2 of thienothiazole moiety in compound 3c gave two H-bond donors with the sidechains of Glu738 and Met742 (distance: 3.16 and 2.93 Å, respectively). Also, the carbohydrazide fragment improved fixation of compound 3c through formation of additional H-bonds. One was between the oxygen and the sidechain of Lys721 (distance: 2.50 Å, respectively), and the two others were between the amino nitrogen and the sidechain of Asp831 (distance: 2.80 and 3.14 Å, respectively).
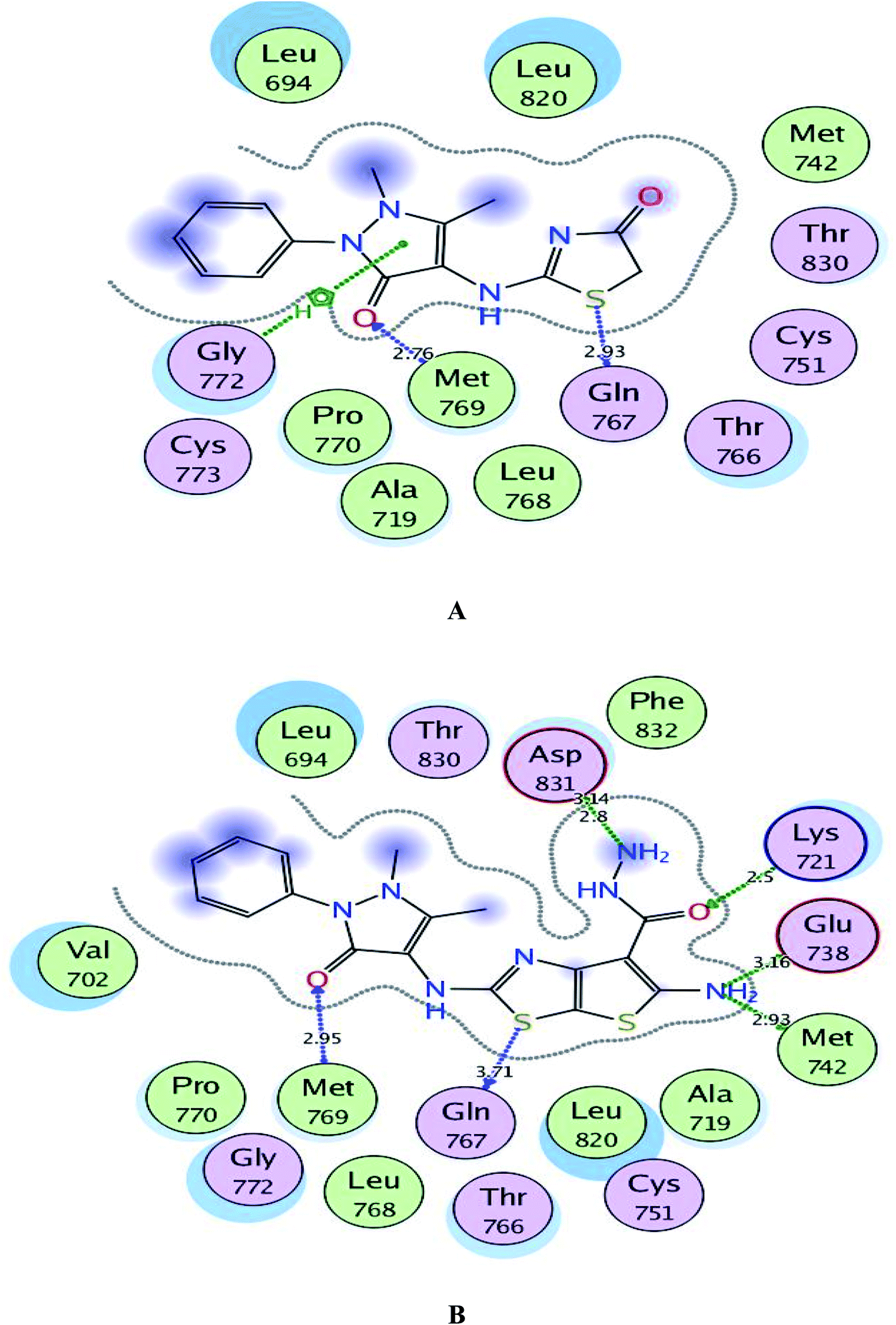 | ||
| Fig. 6 A and B maps illustrate the 2D binding features of the promising targets 1 and 3c within the active site of EGFR (PDB code: 1M17), respectively. | ||
Regarding to binding to VEGFR-2 active site in Fig. 7, it was observed that the pyrazolinone oxygen and NH linker in both compounds played an essential role in the fixation through formation of hydrogen bonding with the amino acids Cys1045, Asp1046 and Glu885. Moreover, the pyrazolinone scaffold in compound 3c displayed arene-cation interaction with Lys868. The targets 1 and 3c provided small energy scores of −10.63 and −11.22 kcal mol−1, respectively confirming their stability within the binding pocket.
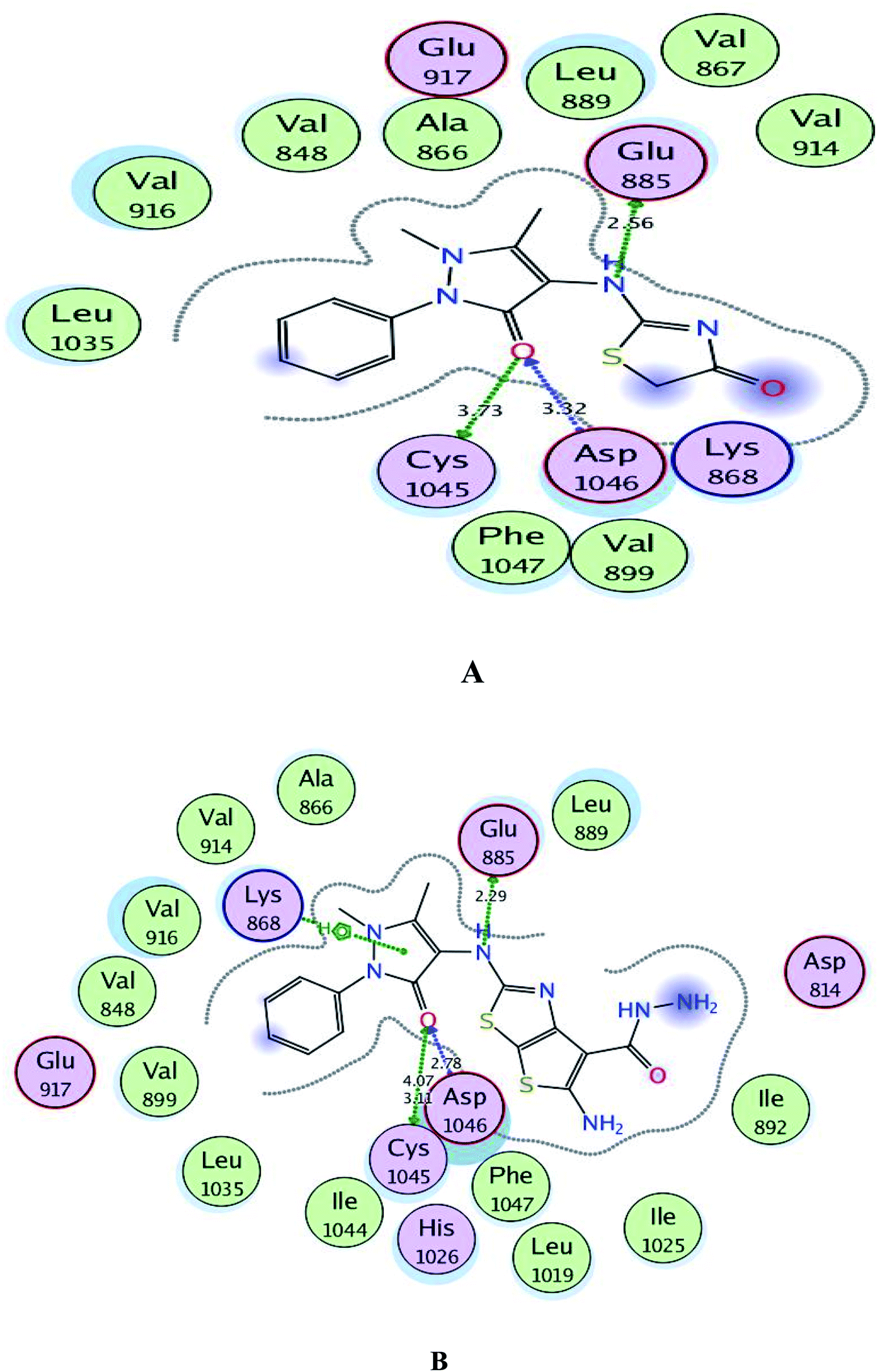 | ||
| Fig. 7 A and B maps illustrate the 2D binding features of the promising targets, 1 and 3c within the active site of VEGFR-2 (PDB code: 4ASD), respectively. | ||
By inspection of Fig. 8, the derivatives 1 and 3c were bound to the vicinity of BRAFV600E with energy scores of −10.25 and −10.92 kcal mol−1, respectively through arene-cation interaction between the pyrazolinone moiety and the amino acid Val471. Furthermore, the nitrogen and the carbonyl oxygen of thiazolinone scaffold in compound 1 exhibited two H-bond acceptors with the sidechain of Lys483 (distance: 3.20 and 3.19 Å, respectively). While the two nitrogens of the hydrazide fragment in the compound 3c revealed two H-bonds with the sidechain of Trp531 and the backbone of Cys532 (distance: 2.75 and 3.32 Å, respectively).
 | ||
| Fig. 8 A and B maps illustrate the 2D binding features of the promising targets, 1 and 3c within the active site of BRAFV600E (PDB code: 2FB8), respectively. | ||
Finally and regarding to the superimposition Fig. 9, it was noted that the presence of pyrazolinone scaffold linked to thiazolinone moiety in compound 1 or thieno[3,2-d]thiazole-6-carbohydrazide in compound 3c gave the chance for good fitting within the active sites of EGFR, VEGFR-2 and BRAFV600E with a relatively similar binding behavior as the original ligands erlotinib, sorafenib and SB-590885 through arene-cation and H-bond interactions.
| Comp. no. | Toxicity risks | Solubility | Drug-likeness | Drug score | |||
|---|---|---|---|---|---|---|---|
| Mutagenicity | Tumorigenicity | Irritancy | Reproductive effect | ||||
| 1 | Red | Red | Green | Green | −2.62 | 5.3 | 0.33 |
| 3a | Red | Red | Green | Green | −5.83 | 0.88 | 0.18 |
| 3b | Red | Red | Green | Green | −5.5 | 1.85 | 0.19 |
| 3c | Red | Red | Green | Green | −5.43 | 0.05 | 0.16 |
| 3d | Red | Red | Green | Green | −5.08 | 5.14 | 0.23 |
| 5a | Red | Red | Green | Green | −6.94 | 5.38 | 0.16 |
| 5b | Red | Red | Green | Green | −5.22 | 5.8 | 0.23 |
| 5c | Red | Red | Green | Green | −4.92 | 5.19 | 0.24 |
| 5d | Red | Red | Green | Green | −7.68 | 5.83 | 0.14 |
4. Conclusions
This context represents the design and synthesis of two sets of derivatives bearing pyrazoline-3-one ring integrated either with the thieno[3,2-d]thiazole or with thiazolo[4,5-d]thiazolidine-2-thione scaffolds via NH linker, 3a–d, 5a–d, respectively using the pyrazolinyl-thiazolinone derivative 1 as a key precursor. All the new derivatives were evaluated as cytotoxic candidates against two human cancer cell lines; breast MCF-7 and hepatocellular HepG-2, using doxorubicin as a reference drug. Compounds 1 and 3c revealed significant inhibitory activities against both cancer cell lines with IC50 s ranging from 4.02–8.35 μM, comparing to doxorubicin of IC50 s; 4.62, 5.66 μM. In addition, compounds 1 and 3c showed a promising safety profile against the normal WI38 cell line. Furthermore, the compounds were evaluated as multitargeting protein kinase inhibitors against EGFR, VEGFR-2 and BRAFV600E. Both compounds showed the most potent suppression effect against EGFR of IC50 s; 0.022, 0.017 μM, comparing to the reference drug sorafenib of IC50; 0.025 μM. The inhibitory activities of 1 and 3c decreased slightly against VEGFR-2 and BRAFV600E of IC50 range; 0.040–2.259 μM, IC50 sorafenib; 1.022, 0.040 μM. Moreover, compounds 1 and 3c were tested for their impact on cell cycle progression and induction of apoptosis in the MCF-7 cell line. It was investigated that they produce an apoptotic effect and cell cycle arrest. Additionally, all the new analogues were assessed as antibacterial and antifungal agents against a number of pathogenic Gram-positive, Gram-negative bacteria, yeast and fungi in comparison to streptomycin and amphotericin-B. Interestingly, both I and 3c appeared the most potent antimicrobial members against the examined microbial stains considering both compounds having dual anticancer and antimicrobial activities. Molecular docking study rationalized the promising suppression effect of 1 and 3c against EGFR, VEGFR-2 and BRAFV600E due to their good fitting and the binding energy scores in the active site of the tested kinases. Additional toxicity study was performed for all new derivatives which represented their good drug-likeness properties and medium toxicity risks in humans.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors are thankful to the Faculty of Pharmacy-Al-Azhar University/Egypt, for performing the spectral and biological data of the new target compounds.References
- J. Ferlay, M. Laversanne, M. Ervik, F. Lam, M. Colombet, L. Mery, M. Piñeros, A. Znaor, I. Soerjomataram and F. Bray, Global Cancer Observatory: Cancer Tomorrow, Lyon, France, International Agency for Research on Cancer, 2020, https://gco.iarc.fr/tomorrow Search PubMed.
- S. O. Zaraei, R. M. Sbenati, N. N. Alach, H. S. Anbar, R. El-Gamal, H. Tarazi, M. K. Shehata, M. S. Abdel-Maksoud, C.-H. Oh and M. I. El-Gamal, Eur. J. Med. Chem., 2021, 224, 113674 CrossRef CAS PubMed.
- R. Awasthi, A. Roseblade, P. M. Hansbro, M. J. Rathbone, K. Dua and M. Bebawy, Curr. Drug Targets, 2018, 19, 1696–1709 CrossRef CAS PubMed.
- Z. Du and C. M. Lovly, Mol. Cancer, 2018, 17, 58 CrossRef PubMed.
- N. Iqbal and N. Iqbal, Chemother. Res. Pract., 2014, 2014, 357027 Search PubMed.
- B. J. Druker, Trends Mol. Med., 2002, 8, S14–S18 CrossRef CAS PubMed.
- K. S. Bhullar, N. O. Lagarón, E. M. McGowan, I. Parmar, A. Jha, B. P. Hubbard and H. V. Rupasinghe, Mol. Cancer, 2018, 17, 1–20 CrossRef PubMed.
- H. Kittler and P. Tschandl, Br. J. Dermatol., 2018, 178, 26–27 CrossRef CAS.
- A. Ayati, S. Moghimi, S. Salarinejad, M. Safavi, B. Pouramiri and A. Foroumadi, Bioorg. Chem., 2020, 99, 103811 CrossRef CAS PubMed.
- R. R. Khattab, A. A. Hassan, D. A. A. Osman, F. M. Abdel-Megeid, H. M. Awad, E. S. Nossier and W. A. El-Sayed, Nucleosides, Nucleotides Nucleic Acids, 2021, 40, 1090–1113 CrossRef CAS PubMed.
- F. Ciardiello and G. Tortora, Clin. Cancer Res., 2001, 7, 2958–2970 CAS.
- R. R. Khattab, A. K. Alshamari, A. A. Hassan, H. H. Elganzory, W. A. El-Sayed, H. M. Awad, E. S. Nossier and N. A. Hassan, J. Enzyme Inhib. Med. Chem., 2021, 36, 504–516 CrossRef CAS PubMed.
- I. M. Othman, Z. M. Alamshany, N. Y. Tashkandi, M. A. Gad-Elkareem, M. M. Anwar and E. S. Nossier, Bioorg. Chem., 2021, 114, 105078 CrossRef CAS PubMed.
- P. Carmeliet, Oncology, 2005, 69, 4–10 CrossRef CAS PubMed.
- D. J. Hicklin and L. M. Ellis, J. Clin. Oncol., 2005, 23, 1011–1027 CrossRef CAS PubMed.
- Z. K. Otrock, J. A. Makarem and A. I. Shamseddine, Blood Cells, Mol., Dis., 2007, 38, 258–268 CrossRef CAS PubMed.
- A. S. Oguntade, F. Al-Amodi, A. Alrumayh, M. Alobaida and M. Bwalya, J. Egypt. Natl. Cancer Inst., 2021, 33, 1–11 CrossRef PubMed.
- D. H. Dawood, E. S. Nossier, M. M. Ali and A. E. Mahmoud, Bioorg. Chem., 2020, 101, 103916 CrossRef CAS PubMed.
- W. S. El-serwy, H. S. Mohamed, W. S. El-serwy, N. A. Mohamed, E. M. Kassem, K. Mahmoud and E. S. Nossier, ChemistrySelect, 2020, 5, 15243–15253 CrossRef CAS.
- L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu, T. Yuan, W. Yang, C. Tian, Z. Miao, T. Wang and S. Yang, Signal Transduction Targeted Ther., 2021, 6, 201 CrossRef PubMed.
- F. Wang, J. Molina, D. Satele, J. Yin, V. S. Lim and A. A. Adjei, Invest. New Drugs, 2019, 37, 658–665 CrossRef CAS PubMed.
- M. Y. Zhao, Y. Yin, X. W. Yu, C. B. Sangani, S. F. Wang, A. M. Lu, L. F. Yang, P. C. Lv, M. G. Jiang and H. L. Zhu, Bioorg. Med. Chem., 2015, 23, 46–54 CrossRef CAS PubMed.
- T. I. Bonner, S. B. Kerby, P. Sutrave, M. A. Gunnell, G. Mark and U. R. Rapp, Mol. Cell. Biol., 1985, 5, 1400–1407 CAS.
- T. Bonner, S. J. O'Brien, W. G. Nash, U. R. Rapp, C. C. Morton and P. Leder, Science, 1984, 223, 71–74 CrossRef CAS PubMed.
- P. C. Sharma, K. K. Bansal, A. Sharma, D. Sharma and A. Deep, Eur. J. Med. Chem., 2020, 188, 112016 CrossRef CAS PubMed.
- M. S. Abdel-Maksoud, M. I. El-Gamal, B. S. Lee, M. M. Gamal El-Din, H. R. Jeon, D. Kwon, U. M. Ammar, K. I. Mersal, E. M. Ali, K. T. Lee and K. H. Yoo, J. Med. Chem., 2021, 64, 6877–6901 CrossRef CAS PubMed.
- R. Bansal and A. Malhotra, Eur. J. Med. Chem., 2020, 12, 113016 Search PubMed.
- A. R. Sayed, S. M. Gomha, F. M. Abdelrazek, M. S. Farghaly, S. A. Hassan and P. Metz, BMC Chem., 2019, 13, 1–3 CrossRef CAS PubMed.
- X. Deng, X. Tan, T. An, Q. Ma, Z. Jin, C. Wang, Q. Meng and C. Hu, Molecules, 2019, 24, 682 CrossRef CAS PubMed.
- H. K. Mahmoud, T. A. Farghaly, H. G. Abdulwahab, N. T. Al-Qurashi and M. R. Shaaban, Eur. J. Med. Chem., 2020, 208, 112752 CrossRef CAS PubMed.
- C. M. Bandaru, N. Poojith, S. S. Jadav, M. V. B. Rao, K. S. Babu, R. Sreenivasulu and R. Alluri, Polycyclic Aromat. Compd., 2021 DOI:10.1080/10406638.2021.1939067.
- A. Ayati, S. Emami, A. Asadipour, A. Shafiee and A. Foroumadi, Eur. J. Chem., 2015, 97, 699–718 CrossRef CAS PubMed.
- P. C. Sharma, A. Jain, M. S. Yar, R. Pahwa, J. Singh and P. Chanalia, Arabian J. Chem., 2017, 10, S568–S575 CrossRef CAS.
- B. L. Flynn, G. P. Flynn, E. Hamel and M. K. Jung, Bioorg. Med. Chem. Lett., 2001, 11, 2341–2343 CrossRef CAS PubMed.
- M. S. Al-Said, M. S. Bashandy, S. I. Al-Qasoumi and M. M. Ghorab, Eur. J. Med. Chem., 2011, 46, 137–141 CrossRef CAS PubMed.
- P. Franchetti, L. Cappellacci, M. Grifantini, A. Barzi, G. Nocentini, H. Yang, A. O’Connor, H. N. Jayaram, C. Carrell and B. M. Goldstein, J. Med. Chem., 1995, 38, 3829–3837 CrossRef CAS PubMed.
- X. Li, Y. He, C. H. Ruiz, M. Koenig and M. D. Cameron, Drug Metab. Dispos., 2009, 37, 1242–1250 CrossRef CAS PubMed.
- S. Hu-Lieskovan, S. Mok, B. H. Moreno, J. Tsoi, L. Robert, L. Goedert, E. M. Pinheiro, R. C. Koya, T. G. Graeber, B. Comin-Anduix and A. Ribas, Sci. Transl. Med., 2015, 7, 279ra41 Search PubMed.
- M. A. Rashid, K. R. Gustafson, J. H. Cardellina, M. R. Boyd and F. Patellamide, J. Nat. Prod., 1995, 58, 594–597 CrossRef CAS PubMed.
- Y. Yao, S. Chen, X. I. Zhou, L. Xie and A. Chen, Oncol. Lett., 2014, 7, 541–547 CrossRef PubMed.
- K. H. Altmann, Mini-Rev. Med. Chem., 2003, 3, 149–158 CrossRef CAS PubMed.
- S. S. Abd El-Karim, H. S. Mohamed, M. F. Abdelhameed, A. E. Amr, A. A. Almehizia and E. S. Nossier, Bioorg. Chem., 2021, 111, 104827 CrossRef CAS PubMed.
- E. S. Nossier, S. M. El-Hallouty and E. R. Zaki, Int. J. Pharm. Sci., 2015, 7, 353–359 CAS.
- E. S. Nossier, S. S. Abd El-Karim, N. M. Khalifa, A. S. El-Sayed, E. S. Hassan and S. M. El-Hallouty, Molecules, 2018, 23, 3074 CrossRef PubMed.
- I. M. Othman, M. A. Gad-Elkareem, A. E. Amr, M. A. Al-Omar, E. S. Nossier and E. A. Elsayed, J. Enzyme Inhib. Med. Chem., 2020, 35, 1491–1502 CrossRef CAS PubMed.
- P. C. Lv, H. Q. Li, J. Sun, Y. Zhou and H. L. Zhu, Bioorg. Med. Chem., 2010, 18, 4606–4614 CrossRef CAS PubMed.
- P. C. Lv, D. D. Li, Q. S. Li, X. Lu, Z. P. Xiao and H. L. Zhu, Bioorg. Med. Chem. Lett., 2011, 21, 5374–5377 CrossRef CAS PubMed.
- M. Y. Zhao, Y. Yin, X. W. Yu, C. B. Sangani, S. F. Wang, A. M. Lu, L. F. Yang, P. C. Lv, M. G. Jiang and H. L. Zhu, Bioorg. Med. Chem., 2015, 23, 46–54 CrossRef CAS PubMed.
- R. Sadashiva, D. Naral, J. Kudva, S. M. Kumar, K. Byrappa, R. M. Shafeeulla and M. Kumsi, J. Mol. Struct., 2017, 1145, 18–31 CrossRef CAS.
- T. K. Mohamed, R. Z. Batran, S. A. Elseginy, M. M. Ali and A. E. Mahmoud, Bioorg. Chem., 2019, 85, 253–273 CrossRef CAS PubMed.
- K. Vaarla, R. K. Kesharwani, K. Santosh, R. R. Vedula, S. Kotamraju and M. K. Toopurani, Bioorg. Med. Chem. Lett., 2015, 25, 5797–5803 CrossRef CAS PubMed.
- M. O. Sarhan, S. S. Abd El-Karim, M. M. Anwar, R. H. Gouda, W. A. Zaghary and M. A. Khedr, Molecules, 2021, 26, 2273 CrossRef CAS PubMed.
- M. M. Anwar, S. S. Abd El-Karim, A. H. Mahmoud, A. E. Amr and M. A. Al-Omar, Molecules, 2019, 24, 2413 CrossRef CAS PubMed.
- S. S. Abd El-Karim, Y. M. Syam, A. M. El Kerdawy and T. M. Abdelghany, Bioorg. Chem., 2019, 86, 80–96 CrossRef CAS PubMed.
- A. M. Srour, N. S. Ahmed, S. S. Abd El-Karim, M. M. Anwar and S. M. El-Hallouty, Bioorg. Med. Chem., 2020, 28, 115657 CrossRef CAS PubMed.
- S. S. Abd El-Karim, M. M. Anwar, E. R. Zaki, S. A. Elseginy and Z. M. Nofal, Future Med. Chem., 2018, 10, 157–181 CrossRef CAS PubMed.
- P. Sharma, S. Kaur, B. S. Chadha, R. Kaur, M. Kaur and S. Kaur, BMC Microbiol., 2021, 21, 39 CrossRef CAS PubMed.
- G. Rodrigues, G. G. Silva, D. F. Buccini, H. M. Duque, S. C. Dias and O. L. Franco, Front. Microbiol., 2019, 10, 1690 CrossRef PubMed.
- V. Patil, K. Tilekar, S. Mehendale-Munj, R. Mohan and C. S. Ramaa, Eur. J. Med. Chem., 2010, 45, 4539–4544 CrossRef CAS PubMed.
- A. E. Amr, R. E. A. Mageid, M. El-Naggar, A. M. Naglah, E. S. Nossier and E. A. Elsayed, Molecules, 2020, 25, 1096 CrossRef PubMed.
- E. A. Abd El-Meguid, G. O. Moustafa, H. M. Awad, E. R. Zaki and E. S. Nossier, J. Mol. Struct., 2021, 1240, 130595 CrossRef.
- L. F. Brown, B. Berse, R. W. Jackman, K. Tognazzi, A. J. Guidi, H. F. Dvorak, D. R. Senger, J. L. Connolly and S. J. Schnitt, Hum. Pathol., 1995, 26, 86–91 CrossRef CAS PubMed.
- A. S. Hassan, G. O. Moustafa, H. M. Awad, E. S. Nossier and M. F. Mady, ACS Omega, 2021, 6, 12361–12374 CrossRef CAS PubMed.
- C. Perez, M. Pauli and P. Bazevque, Acta Biol. Med. Exp., 1990, 15, 113–115 Search PubMed.
- A. C. Scott, “Laboratory control of antimicrobial therapy”, in Mackie and MacCartney Practical Medical Microbiology, ed. J. G. Collee, J. P. Duguid, A. G. Fraser, and B. P. Marmion, Churchill Livingstone, Edinburgh, Scotland, 13th edn, 1989, vol. 2, pp. 161–181 Search PubMed.
- Y. M. Syam, M. M. Anwar, E. R. Kotb, S. A. Elseginy, H. M. Awad and G. E. Awad, Mini-Rev. Med. Chem., 2019, 19, 1255–1275 CrossRef CAS PubMed.
- H. E. Hashem, A. E. Amr, E. S. Nossier, E. A. Elsayed and E. M. Azmy, Molecules, 2020, 25, 2766 CrossRef CAS.
- E. M. Mohi El-Deen, E. A. Abd El-Meguid, E. A. Karam, E. S. Nossier and M. F. Ahmed, Antibiotics, 2020, 9, 695 CrossRef PubMed.
- E. M. Mohi El-Deen, E. A. Abd El-Meguid, S. Hasabelnaby, E. A. Karam and E. S. Nossier, Molecules, 2019, 24, 3650 CrossRef PubMed.
- A. A. El-Sayed, E. S. Nossier, A. A. Almehizia and A. E. Amr, J. Mol. Struct., 2022, 1247, 131285 CrossRef CAS.
- G. O. Moustafa, A. Shalaby, A. M. Naglah, M. M. Mounier, H. El-Sayed, M. M. Anwar and E. S. Nossier, Molecules, 2021, 26, 4573 CrossRef CAS PubMed.
- E. A. Abd El-Meguid, E. M. Mohi El-Deen, G. O. Moustafa, H. M. Awad and E. S. Nossier, Bioorg. Chem., 2021, 105504 CrossRef PubMed.
- M. A. Hawata, W. A. El-Sayed, E. S. Nossier and A. A. H. Abdel-Rahman, Biointerface Res. Appl. Chem., 2022, 12, 5217–5233 Search PubMed.
- A. E. G. E. Amr, E. A. Elsayed, M. A. Al-Omar, H. O. Badr Eldin, E. S. Nossier and M. M. Abdallah, Molecules, 2019, 24, 416 CrossRef PubMed.
- A. J. King, D. R. Patrick, R. S. Batorsky, M. L. Ho, H. T. Do, S. Y. Zhang, R. Kumar, D. W. Rusnak, A. K. Takle, D. M. Wilson and E. Hugger, Cancer Res., 2006, 66, 11100–11105 CrossRef CAS PubMed.
- The OSIRIS property explorer software, available from: http://www.organic-chem+istry.org/prog/peo/ Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08055e |
| This journal is © The Royal Society of Chemistry 2022 |