
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, crystal structures and spectroscopic properties of pure YSb2O4Br and YSb2O4Cl as well as Eu3+- and Tb3+-doped samples†
Ralf J. C. Lockea,
Felix C. Goerigka,
Martin J. Schäferb,
Henning A. Höppeb and
Thomas Schleid *a
*a
aInstitute of Inorganic Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
bInstitut für Physik, Augsburg University, Universitätsstraße 1, 86159 Augsburg, Germany
First published on 22nd December 2021
Abstract
The quaternary halide-containing yttrium(III) oxidoantimonates(III) YSb2O4Cl and YSb2O4Br were synthesised through solid-state reactions from the binary components (Y2O3, Sb2O3 and YX3, X = Cl and Br) at 750 °C in evacuated fused silica ampoules with eutectic mixtures of NaX and CsX (X = Cl and Br) as fluxing agents. YSb2O4Cl crystallizes tetragonally in the non-centrosymmetric space group P4212 with unit-cell parameters of a = 773.56(4) pm and c = 878.91(6) pm, whereas YSb2O4Br is monoclinic (space group: P21/c) with a = 896.54(6) pm, b = 780.23(5) pm, c = 779.61(5) pm and β = 91.398(3)°, both for Z = 4. The two new YSb2O4X compounds contain [YO8]13− polyhedra, which are connected via four common edges to form 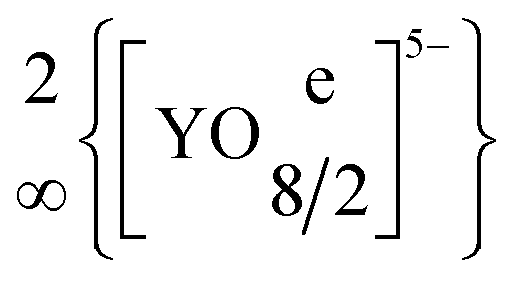 layers (d(Y3+–O2−) = 225–254 pm) without any Y3+⋯X− bonds (d(Y3+⋯X−) > 400 pm). Moreover, all oxygen atoms belong to ψ1-tetrahedral [SbO3]3− units, which are either connected to four-membered rings [Sb4O8]4− in the chloride (Y2[Sb4O8]Cl2 for Z = 2) or endless chains in the bromide (Y1/2(SbO2)Br1/2 for Z = 8) by common vertices. With distances of 307 pm in YSb2O4Cl and 326 pm in YSb2O4Br there are not even substantial bonding Sb3+⋯X− (X = Cl and Br) interactions at work. Luminescence spectroscopy on samples doped with trivalent europium and terbium showed an energy transfer from the oxidoantimonate(III) moieties as the sensitizer in the host structure onto the lanthanoid activators.
layers (d(Y3+–O2−) = 225–254 pm) without any Y3+⋯X− bonds (d(Y3+⋯X−) > 400 pm). Moreover, all oxygen atoms belong to ψ1-tetrahedral [SbO3]3− units, which are either connected to four-membered rings [Sb4O8]4− in the chloride (Y2[Sb4O8]Cl2 for Z = 2) or endless chains in the bromide (Y1/2(SbO2)Br1/2 for Z = 8) by common vertices. With distances of 307 pm in YSb2O4Cl and 326 pm in YSb2O4Br there are not even substantial bonding Sb3+⋯X− (X = Cl and Br) interactions at work. Luminescence spectroscopy on samples doped with trivalent europium and terbium showed an energy transfer from the oxidoantimonate(III) moieties as the sensitizer in the host structure onto the lanthanoid activators.
Introduction
In recent years, great attention has been paid to the structural diversity of rare-earth metal(III) oxidoarsenate(III) halides owing to the beneficial inorganic lone-pair antenna at the As3+ cations for luminescence applications.1 Several representatives are known with the formula RE5X3[AsO3]4 (RE = La–Lu, X = F–Br), but all their different crystal structures have the motif of isolated ψ1-tetrahedral [AsO3]3− anions in common. The fluoride derivatives (RE = Y, Ho, Tm–Lu)2,3 crystallize in the tetragonal space group P4/ncc with separated [AsO3]3− units that form a lone-pair channel along [001], while the chloride derivatives crystallize monoclinically (C2/c for La–Pr,4–6 P2/c for Nd5–7 and Sm2,6) with a layered structure, in which the [AsO3]3− units are linked to the chloride layers via weak secondary contacts. The three known bromide derivatives (RE = Pr, Sm, Eu)2 crystallize again in the monoclinic system with space group P2/c and similar coordination features as the chloride derivatives. The motif of ψ1-tetrahedra [AsO3]3− is also present in the oxide-halide representatives RE3OCl[AsO3]2 and RE3OBr[AsO3]2, which crystallize tetragonally in the space groups P42/mnm (RE = La)8,9 or P42nm (RE = Ce–Pr, Sm–Dy with X = Cl,2,4,10 RE = Ce, Nd, Sm, Gd, Tb with X = Br2). Like in the RE5F3[AsO3]4 cases (RE = Y, Ho, Tm–Lu), all RE3OX[AsO3]2 representatives have a lone-pair tunnel structure of [AsO3]3− units, but the rare-earth metal–oxygen linkage is different. Furthermore, there are RE5O4Cl[AsO3]2 members (RE = Nd, Pr),11,12 which crystallize monoclinically in C2/m. Not only compounds with isolated [AsO3]3− anions were synthesised, but also with additional oxidoarsenate(III) units of the pyroanionic species [As2O5]4− in the triclinic RE3X2[As2O5][AsO3] examples (RE = Sm–Gd with X = Cl,4,13 RE = Y, Ho–Yb with X = Br3,13). These also crystallize in a layered structure (space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), in which both the [AsO3]3− and the [As2O5]4− anions are bound to the halide layers via weak secondary contacts.
), in which both the [AsO3]3− and the [As2O5]4− anions are bound to the halide layers via weak secondary contacts.
The first rare-earth metal(III) oxidobismuthate(III) halide with the composition Nd0.5Bi2.5O4Cl14 was synthesised by Aurivillius. In this case, the rare-earth metal cation site is mixed with bismuth(III). Only ten years later, REBi2O4Cl phases (RE = Y, La, Nd)15 were the first synthesised representatives without mixed occupation of the layers. Oppermann et al. extended the spectrum of these representatives at first with ErBi2O4I16 considerably and found all except the cerium representatives with the composition REBi2O4X (RE = La, Pr–Nd, Sm–Lu for X = Cl–I).16,17 All these representatives crystallize in the tetragonal space group P4/mmm and form layered structures, in which the rare-earth metal cations are surrounded cube-shaped by eight oxygen atoms [REO8]13−. These cubes are linked to each other via common edges. The Bi3+ cations form square  [BiO4]5− (ax = axial) with four oxygen, which are linked to each other via their corners to form layers as well.
[BiO4]5− (ax = axial) with four oxygen, which are linked to each other via their corners to form layers as well.
The rare-earth metal(III) oxidoantimonate(III) halides have been neglected in previous research, except for La5F3[SbO3]4,18 which crystallizes analogously to the RE5F3 [AsO3]4 series (RE = Y, Ho, Tm–Lu), and SmSb2O4Cl19 postulated by Oppermann et al. to be the isotypic light homologue of SmBi2O4Cl17 with layers of corner-linked [SbO4]4− polyhedra 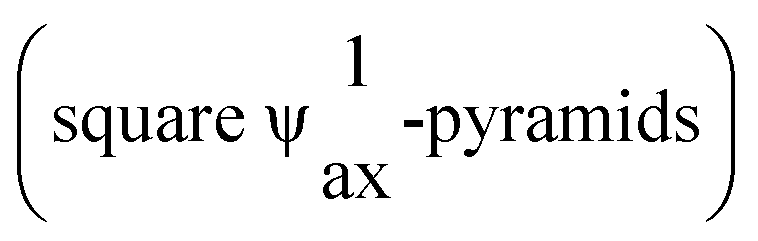 with axial lone pairs. It was not until 20 years later that the true composition could be elucidated as Sm1.3Sb1.7O4Cl,3,20 which crystallizes in principle analogously to the REBi2O4X family in the tetragonal space group P4/mmm. Furthermore, the analogous bromide derivative Sm1.5Sb1.5O4Br3,20 was discovered, in which there is also a mixed occupation of the antimony position of the Sb3+ with Sm3+ cations just like in Sm1.3Sb1.7O4Cl. In further studies, the derivatives of the other bromides with RE = Eu–Dy3,21–23 with the compositions RESb2O4Br were discovered. They crystallize in the monoclinic space group P21/c, but with a different linkage of the antimony–oxygen polyhedra. Here ψ1-tetrahedral [SbO3]3− units with only three oxygen atoms are present, which are linked to each other via two corners to form chains with the Niggli formula
with axial lone pairs. It was not until 20 years later that the true composition could be elucidated as Sm1.3Sb1.7O4Cl,3,20 which crystallizes in principle analogously to the REBi2O4X family in the tetragonal space group P4/mmm. Furthermore, the analogous bromide derivative Sm1.5Sb1.5O4Br3,20 was discovered, in which there is also a mixed occupation of the antimony position of the Sb3+ with Sm3+ cations just like in Sm1.3Sb1.7O4Cl. In further studies, the derivatives of the other bromides with RE = Eu–Dy3,21–23 with the compositions RESb2O4Br were discovered. They crystallize in the monoclinic space group P21/c, but with a different linkage of the antimony–oxygen polyhedra. Here ψ1-tetrahedral [SbO3]3− units with only three oxygen atoms are present, which are linked to each other via two corners to form chains with the Niggli formula  (v = vertex-sharing, t = terminal), not showing any mixed occupation with RE3+ cations. Moreover, the luminescence of trivalent europium and terbium will be investigated and discussed. The oxidoantimonate(III) host structure promises to provide an energy transfer to enhance the luminescence of the 4f–4f activators. Trivalent antimony cations themselves show an efficient 5s–5p excitation that can be used as an antenna for lanthanoid activators24–26 and antimony(III) compounds have proven to transfer energy previously.27,28
(v = vertex-sharing, t = terminal), not showing any mixed occupation with RE3+ cations. Moreover, the luminescence of trivalent europium and terbium will be investigated and discussed. The oxidoantimonate(III) host structure promises to provide an energy transfer to enhance the luminescence of the 4f–4f activators. Trivalent antimony cations themselves show an efficient 5s–5p excitation that can be used as an antenna for lanthanoid activators24–26 and antimony(III) compounds have proven to transfer energy previously.27,28
Results and discussion
The two rare-earth metal(III) oxidoantimonate(III) halides YSb2O4Cl and YSb2O4Br were formed from Y2O3 and Sb2O3 together with YX3 (X = Cl and Br) as colourless square platelets. YSb2O4Cl crystallizes in the tetragonal space group P4212, while YSb2O4Br adopts the monoclinic space group P21/c, just like the already known RESb2O4Br representatives with RE = Eu–Dy.3,21–23 The unit-cell parameters for YSb2O4Cl are a = 773.56(4) pm, c = 878.91(6) pm (c/a = 1.136), while a = 896.54(6) pm, b = 780.23(5) pm, c = 779.61(5) pm and β = 91.398(3)° apply for YSb2O4Br. The molar volumes of the bromides decrease from EuSb2O4Br (Vm = 84.36 cm3 mol−1) to DySb2O4Br (Vm = 82.57 cm3 mol−1) as consequence of the lanthanoid contraction.29 Despite being half as heavy the Y3+ cation can be classified by its ionic radius between Dy3+ and Ho3+,30 which also holds here, indicated with the molar volume of 82.08 cm3 mol−1 for YSb2O4Br. The molar volume of YSb2O4Cl is with 79.19 cm3 mol−1 considerably smaller as compared to the bromide derivative, due to the lighter and smaller halide anion.While the monoclinic YSb2O4Br shows two crystallographically distinct Y3+ positions, the tetragonal YSb2O4Cl only comprises one Y3+ position. In both cases, however, the Y3+ cations are surrounded by eight oxygen atoms that arrange themselves to square hemiprisms [YO8]13−. The [YO8]13− polyhedra are linked with four of their edges via the oxygen atoms to form two-dimensional infinite layers according to the Niggli formula  (e = edge-sharing, Fig. 1). These layers run parallel to the (001) plane in YSb2O4Cl and to the (100) plane in YSb2O4Br. The distances between yttrium and the oxygen atoms range between 227 and 253 pm in YSb2O4Cl or 225 and 253 pm in YSb2O4Br. These Y3+–O2− distances are in similar intervals as in yttrium sesquioxide Y2O3 (d(Y3+–O2−) = 225–234 pm) with bixbyite-type structure,31 where Y3+ resides in sixfold oxygen coordination.
(e = edge-sharing, Fig. 1). These layers run parallel to the (001) plane in YSb2O4Cl and to the (100) plane in YSb2O4Br. The distances between yttrium and the oxygen atoms range between 227 and 253 pm in YSb2O4Cl or 225 and 253 pm in YSb2O4Br. These Y3+–O2− distances are in similar intervals as in yttrium sesquioxide Y2O3 (d(Y3+–O2−) = 225–234 pm) with bixbyite-type structure,31 where Y3+ resides in sixfold oxygen coordination.
 | ||
Fig. 1 Two-dimensional infinite layers  of edge-linked square hemiprisms [YO8]13− in the tetragonal crystal structure of YSb2O4Cl (top) and in the monoclinic crystal structure of YSb2O4Br (bottom). of edge-linked square hemiprisms [YO8]13− in the tetragonal crystal structure of YSb2O4Cl (top) and in the monoclinic crystal structure of YSb2O4Br (bottom). | ||
The antimony(III) cations occupy one crystallographic position in YSb2O4Cl, while there are two different of them in YSb2O4Br. Common for both structures, the Sb3+ cations form ψ1-tetrahedral [SbO3]3− units with three oxygen atoms and the electron lone pair, but this is the only common feature, since in both structures they are linked differently to each other to achieve {[SbO2]−} motifs. In YSb2O4Br, their arrangement is already known from the representatives RESb2O4Br (RE = Eu–Dy),3,21–23 namely the linkage via two corners to form one-dimensional infinite chains according to the Niggli formula  (v = vertex-sharing, t = terminal; Fig. 2). The bridging oxygen atoms show distances of 203–213 pm to the antimony(III) cations and are thus significantly longer than the terminal antimony–oxygen distances of 193–195 pm. Moreover, the terminal O1 atoms of (Sb1)3+ exhibit distances to the next (Sb2)3+ cation within the chain of d(Sb2⋯O1) = 317 pm (Fig. 2, red), which is approximately the same as that of the terminal O2 atom of (Sb2)3+ to the next (Sb1)3+ cation, d(Sb1⋯O2) = 316 pm, between the chains (Fig. 2, yellow). These meandering chains propagate along [001] and lie parallel within the (100) plane.
(v = vertex-sharing, t = terminal; Fig. 2). The bridging oxygen atoms show distances of 203–213 pm to the antimony(III) cations and are thus significantly longer than the terminal antimony–oxygen distances of 193–195 pm. Moreover, the terminal O1 atoms of (Sb1)3+ exhibit distances to the next (Sb2)3+ cation within the chain of d(Sb2⋯O1) = 317 pm (Fig. 2, red), which is approximately the same as that of the terminal O2 atom of (Sb2)3+ to the next (Sb1)3+ cation, d(Sb1⋯O2) = 316 pm, between the chains (Fig. 2, yellow). These meandering chains propagate along [001] and lie parallel within the (100) plane.
 | ||
Fig. 2 One-dimensional infinite chains  of corner-linked ψ1-tetrahedra [SbO3]3− in the monoclinic crystal structure of YSb2O4Br, which run parallel to the [001] direction. The contacts of the terminal oxygen atoms to the next, not directly bonded Sb3+ cations are shown in red within the chains and in yellow between the chains. of corner-linked ψ1-tetrahedra [SbO3]3− in the monoclinic crystal structure of YSb2O4Br, which run parallel to the [001] direction. The contacts of the terminal oxygen atoms to the next, not directly bonded Sb3+ cations are shown in red within the chains and in yellow between the chains. | ||
The motif of chains occurs more frequently in crystal structures of ternary or quarternary antimony(III)–oxygen compounds. However, Sb3+ has than a coordination number of four and forms square ψ1-pyramids [SbO4]5− edge-linked according to 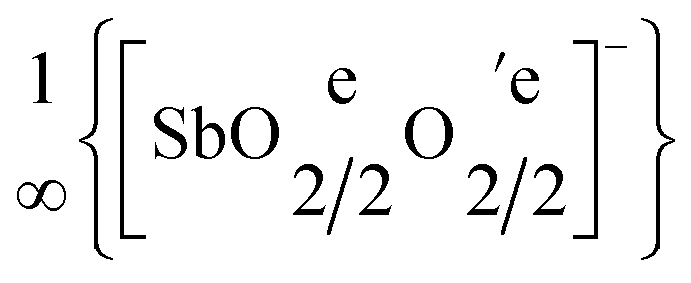 . Examples of representatives for this behaviour are ASbO2 (A = K–Cs),32,33 BaSb2O4Cl,34 PbSbO2Cl35 and ZnSbO2I.36 Oxygen antimony chains
. Examples of representatives for this behaviour are ASbO2 (A = K–Cs),32,33 BaSb2O4Cl,34 PbSbO2Cl35 and ZnSbO2I.36 Oxygen antimony chains  similar as in YSb2O4Br can be found in LiSbO2,37 but here they are twisted to spirals and not planar. In the YSb2O4Br structure there are four crystallographic different oxygen atoms, whereas in YSb2O4Cl we have only two different ones of them. Unlike the monoclinic compounds YSb2O4Br and RESb2O4Br (RE = Gd–Dy),3,21–23 in YSb2O4Cl four ψ1-tetrahedra [SbO3]3− form a closed ring according to {[Sb4O8]4−} by vertex-connections (Fig. 3). These rings lie within the (001) plane. The bridging oxygen atoms have distances of 204–210 pm to the Sb3+ cations. In contrast, the exo-standing terminal oxygen atoms show significantly shorter contacts of 194 pm just like it is the case for the monoclinic congeners. The terminal oxygen atoms O1 have a distance of d(Sb⋯O1) = 309 pm to the next non-covalently bonded Sb3+ cation, which is a shorter secondary contact than in the monoclinic YSb2O4Br representative. Discrete units of antimony and oxygen are relatively rare, but one example would be Na3[SbO3],38 where isolated ψ1-tetrahedral [SbO3]3− anions (d(Sb–O) = 189 pm, 3×) are present with their full C3v symmetry. The structural motif of separated
similar as in YSb2O4Br can be found in LiSbO2,37 but here they are twisted to spirals and not planar. In the YSb2O4Br structure there are four crystallographic different oxygen atoms, whereas in YSb2O4Cl we have only two different ones of them. Unlike the monoclinic compounds YSb2O4Br and RESb2O4Br (RE = Gd–Dy),3,21–23 in YSb2O4Cl four ψ1-tetrahedra [SbO3]3− form a closed ring according to {[Sb4O8]4−} by vertex-connections (Fig. 3). These rings lie within the (001) plane. The bridging oxygen atoms have distances of 204–210 pm to the Sb3+ cations. In contrast, the exo-standing terminal oxygen atoms show significantly shorter contacts of 194 pm just like it is the case for the monoclinic congeners. The terminal oxygen atoms O1 have a distance of d(Sb⋯O1) = 309 pm to the next non-covalently bonded Sb3+ cation, which is a shorter secondary contact than in the monoclinic YSb2O4Br representative. Discrete units of antimony and oxygen are relatively rare, but one example would be Na3[SbO3],38 where isolated ψ1-tetrahedral [SbO3]3− anions (d(Sb–O) = 189 pm, 3×) are present with their full C3v symmetry. The structural motif of separated  rings is also not novel, but found in valentinite (β-Sb2O3).39 Here they are further connected, not isolated, and show a twisted configuration. The Sb3+–O2− bond lengths in both compounds correspond well with typical antimony–oxygen distances in both crystalline forms of Sb2O3 (α: senarmontite: d(Sb–O) = 198 pm,40 β: valentinite: d(Sb–O) = 198–202 pm39).
rings is also not novel, but found in valentinite (β-Sb2O3).39 Here they are further connected, not isolated, and show a twisted configuration. The Sb3+–O2− bond lengths in both compounds correspond well with typical antimony–oxygen distances in both crystalline forms of Sb2O3 (α: senarmontite: d(Sb–O) = 198 pm,40 β: valentinite: d(Sb–O) = 198–202 pm39).
 | ||
| Fig. 3 Isolated rings [Sb4O8]4− of four cyclically vertex-linked ψ1-tetrahedra [SbO3]3− in the tetragonal crystal structure of YSb2O4Cl according to Y2[Sb4O8]Cl2. The contacts of the terminal oxygen atoms to the next, not directly bonded Sb3+ cations are shown in yellow. | ||
In YSb2O4Br there is only one crystallographic position for the halide anion, whereas in YSb2O4Cl two different ones of them are present. The halide anions show a minimum distance of d(Sb⋯Cl) = 307 pm to the nearest Sb3+ cation in YSb2O4Cl and of d(Sb⋯Br) = 326 pm in YSb2O4Br. Their distances to the nearest Y3+ cation amount to d(Y⋯Cl) = 420 pm for YSb2O4Cl and d(Y⋯Br) = 427 pm for YSb2O4Br. So at these distances, one can not speak of real coordination in either structure. Between each layer of Sb3+ cations there is a layer of halide anions, which in the case of YSb2O4Br spreads out parallel to the (100) plane, but parallel to the (001) plane in the case of YSb2O4Cl. This halide layer has no contact or connection to any other layer, neither via X−⋯Sb3+ nor via X−⋯Y3+ bonds. However, the layer of Sb3+ cations enjoys linkage to the layer of Y3+ cations via all oxygen atoms according to  in both yttrium(III) oxidoantimonate(III) halides YSb2O4X (X = Cl and Br).
in both yttrium(III) oxidoantimonate(III) halides YSb2O4X (X = Cl and Br).
Fig. 4 shows an extended unit cell of YSb2O4Br with depicted coordination spheres of the Y3+ and Sb3+ cations. The same applies to Fig. 5, which shows the extended unit cell of YSb2O4Cl.
 | ||
| Fig. 4 Extended tetragonal unit cell of YSb2O4Cl as viewed along [010]. | ||
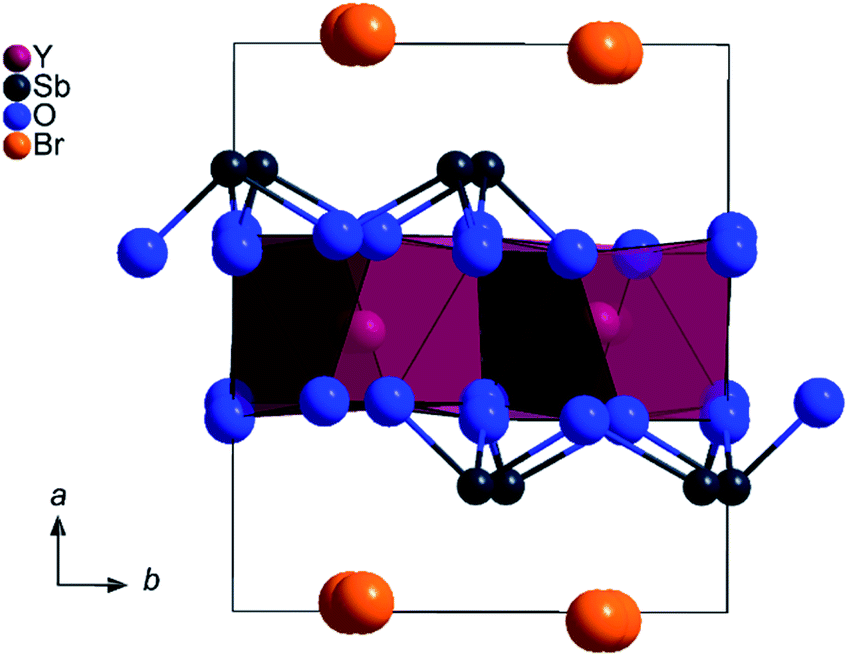 | ||
| Fig. 5 Extended monoclinic unit cell of YSb2O4Br as viewed along [001]. | ||
Since the yttrium cations are surrounded by oxidoantimonate layers, an energy transfer from these layers towards any cation doped on the yttrium site could be expected. This was verified via luminescence spectroscopy (Fig. 6 and 7), but apparently, the luminescence was quite different for all samples (Fig. 8).
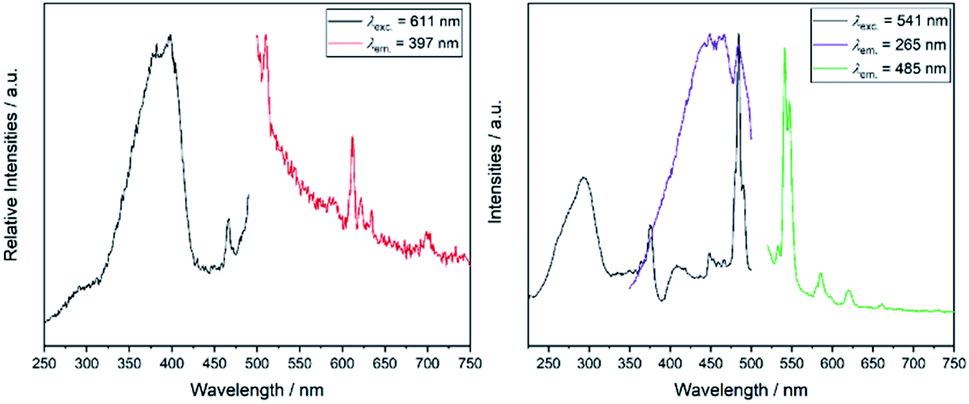 | ||
| Fig. 6 Fluorescence spectra of YSb2O4Cl doped with either Eu3+ (left) or Tb3+ (right). λexc. describes the wavelength used to record the excitation spectrum, whereas λem. represents the wavelength, at which the emission spectrum was recorded. | ||
 | ||
| Fig. 7 Luminescence spectra of YSb2O4Br doped with either Eu3+ (left) or Tb3+ (right). λexc. describes the wavelength used to record the excitation spectrum, whereas λem. represents the wavelength, at which the emission spectrum was recorded. | ||
 | ||
| Fig. 8 Comparison of the visible luminescence of the four samples YSb2O4Cl:Eu3+ (a), YSb2O4Cl:Tb3+ (b), YSb2O4Br:Eu3+ (c), and YSb2O4Br:Tb3+ (d). | ||
YSb2O4Cl:Eu3+ shows weak, orange-red luminescence. In the spectrum, the weak emission is represented by the low signal-to-noise-ratio. The excitation spectrum is dominated by the broad charge-transfer within the oxidoantimonate host structure peaking at 397 nm. Another band at 467 nm can be attributed to 4f–4f transitions of Eu3+. The emission spectrum features the main emission bands typical for trivalent europium. The band peaking at 612 nm, attributed to the emission 5D0→7F2, is much more intense than that for 5D0→7F1, normally located around 595 nm. This supports the experimentally obtained site symmetry of yttrium, as the comparably strong hypersensitive transition is a very good probe for the absence of a local inversion center.
The Tb3+-doped sample of YSb2O4Cl shows two different emissions (green and blue) that mix to give a turquoise colour impression. The broad emission in the blue regime with the maximum around 485 nm can be attributed to the emission of the host structure (3P1,2→1S0 transition of the Sb3+ lone-pair cation), which has been observed for LaOBr:Sb3+ (510 nm)3 and GdSb2O4Br (455 nm)21 as well and even matches with pure antimony(III) chlorides such as Cs2NaSbCl6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41 upon excitation between 255 to 280 nm. Three sharp bands assigned to the 4f–4f transitions 5D4→7FJ (J = 3, 4, 5) were also recorded. The excitation spectrum features two 4f–4f transitions with their respective maxima at 374 and 483 nm. The charge-transfer transition of the host structure is blue-shifted compared to the Eu3+-doped sample and peaks at 294 nm. In both spectra apparently an energy transfer between the host structure and the lanthanoid activator happens upon excitation to enhance the luminescence, but in the Tb3+-doped compound this transfer is obviously incomplete causing a characteristic turquoise emission colour.
41 upon excitation between 255 to 280 nm. Three sharp bands assigned to the 4f–4f transitions 5D4→7FJ (J = 3, 4, 5) were also recorded. The excitation spectrum features two 4f–4f transitions with their respective maxima at 374 and 483 nm. The charge-transfer transition of the host structure is blue-shifted compared to the Eu3+-doped sample and peaks at 294 nm. In both spectra apparently an energy transfer between the host structure and the lanthanoid activator happens upon excitation to enhance the luminescence, but in the Tb3+-doped compound this transfer is obviously incomplete causing a characteristic turquoise emission colour.
The oxidoantimonate bromides show a similar luminescence, when doped with trivalent europium or terbium, but significantly more intense (“heavy-atom effect”).1 YSb2O4Br:Eu3+ exhibits an excitation spectrum, in which the charge-transfer band of the oxidoantimonate host structure is even more dominating compared to any 4f–4f transition of Eu3+.
It is blue-shifted about 50 nm compared to the chloride. While the same bands were observed as in the oxidoantimonate chloride, their relative intensities are decidedly stronger.
The excitation band around 393 nm, normally the most prominent one, can be only seen as a slight shoulder. In the emission spectrum, the band attributed to the hypersensitive transition 5D0→7F2 is once again noticeably more intense as compared to the band of the 5D0→7F1 transition, since the yttrium cation occupies a site without inversion symmetry.
The emission spectrum of YSb2O4Br:Tb3+ consists of a very broad band, which we assign to the charge-transfer transition of the host structure. The sharp band of the 5D4→7F6 transition with a maximum around 540 nm is typically the most intense band in Tb3+ spectra, the other bands are not observed, due to restrictions on the recorded wavelength regime. Like in the case above, the excitation is blue-shifted and therefore not in the accessible region of the spectrum. Interestingly, the spectrum does not feature either the 4f–5d excitation or any of the typical 4f–4f transitions of Tb3+. This indicates that the Tb3+ cations are almost exclusively excited via the energy transfer from the oxidoantimonate(III) system (1S0→3P1,2 transition of the Sb3+ lone-pair cation and/or O2−→Sb3+ charge-transfer excitation).
Table 1 lists the most important crystallographic data for YSb2O4Br and YSb2O4Cl, while Table 2 gives the atomic coordinates, Wyckoff positions and equivalent isotropic displacement parameters. Table 3 contains selected bond lengths and interatomic distances for YSb2O4Cl and YSb2O4Br.
| YSb2O4Cl | YSb2O4Br | ||
|---|---|---|---|
| a This value also represents the Flack-x parameter for non-centrosymmetric crystal structures, from which it is transferred into BASF after the TWIN refinement41–43 as inversion twin. | |||
| Crystal system | Tetragonal | Monoclinic | |
| Space group | P4212 (no. 90) | P21/c (no. 14) | |
| Lattice constants | a/pm | 773.56(4) | 896.54(6) |
| b/pm | =a/pm | 780.23(5) | |
| c/pm | 878.91(6) | 779.61(5) | |
| β/° | 90 | 91.398(3) | |
| Formula units, Z | 4 | 4 | |
| X-ray density, Dx/g cm−3 | 5.454 | 5.803 | |
| Molar volume, Vm/cm3 mol−1 | 79.185 | 82.083 | |
| Diffractometer | κ-CCD (Bruker-Nonius) | ||
| Wavelength | λ = 71.07 pm (Mo-Kα) | ||
| F(000) | 760 | 832 | |
| Tmax/° | 27.48 | 27.39 | |
| hkl range (±hmax, ±kmax, ±lmax) | 10, 10, 11 | 11, 10, 10 | |
| Unique reflections | 606 | 1220 | |
| Absorption coefficient, μ/mm−1 | 21.56 | 27.64 | |
| Absorption correction | Program X-SHAPE 2.2145 |
||
| Rint/Rσ | 0.098/0.047 | 0.132/0.104 | |
| R1/R1 with |Fo| ≥ 4σ(Fo) | 0.049/0.040 | 0.118/0.060 | |
| wR2/goodness of fit (GooF) | 0.085/1.090 | 0.129/0.986 | |
| Structure determination and refinement | Program SHELX-9743,44 |
||
| Extinction coefficient, ε/10−6 pm−3 | — | 0.0008(2) | |
| ρmax/min/e− 10−6 pm−3 | 1.74/−1.71 | 2.16/−1.83 | |
| Batch scale factor (BASF)a | 0.45(6) | — | |
| CSD number | 2044973 | 2044975 | |
| Atom | Wyckoff site | x/a | y/b | z/c | Ueq/pm2 |
|---|---|---|---|---|---|
| Y1 | 2a | 0 | 0 | 0 | 120(6) |
| Y2 | 2c | 0 | 1/2 | 0.0153(4) | 118(6) |
| Sb | 8g | 0.24016(8) | 0.20182(8) | 0.28547(9) | 151(2) |
| O1 | 8g | 0.0629(9) | 0.2473(9) | 0.1351(11) | 165(17) |
| O2 | 8g | 0.4648(9) | 0.2483(9) | 0.1705(11) | 147(17) |
| Cl1 | 2b | 0 | 0 | 1/2 | 265(19) |
| Cl2 | 2c | 0 | 1/2 | 0.4929(9) | 250(18) |
| Y | 4e | 0.4922(2) | 0.2368(2) | 0.5018(2) | 232(5) |
| Sb1 | 4e | 0.77879(14) | 0.05212(15) | 0.75550(15) | 261(4) |
| Sb2 | 4e | 0.22116(14) | 0.00914(14) | 0.79184(15) | 240(4) |
| O1 | 4e | 0.6339(15) | 0.0071(13) | 0.5739(14) | 299(32) |
| O2 | 4e | 0.3698(14) | 0.1843(14) | 0.7440(14) | 285(29) |
| O3 | 4e | 0.6613(13) | 0.0069(13) | 0.9704(13) | 212(27) |
| O4 | 4e | 0.6624(14) | 0.2102(13) | 0.2595(15) | 281(30) |
| Br | 4e | 0.0153(2) | 0.2371(2) | 0.5044(2) | 329(5) |
| Distance | YSb2O4Cl | Distance | YSb2O4Br | ||
|---|---|---|---|---|---|
| Y1–O1 | (4×) | 230.3(10) | Y–O2 | (1×) | 224.5(12) |
| Y1–O2 | (4×) | 247.2(9) | Y–O1 | (1×) | 226.0(11) |
| Y2–O1 | (4×) | 227.3(9) | Y–O1′ | (1×) | 228.4(12) |
| Y2–O2 | (4×) | 253.6(9) | Y–O2 | (1×) | 234.9(11) |
| Y1⋯Sb | (4×) | 349.06(8) | Y–O4 | (1×) | 246.6(12) |
| Y2⋯Sb | (4×) | 366.9(2) | Y–O3 | (1×) | 252.5(11) |
| Sb–O1 | (1×) | 193.7(7) | Y–O4′ | (1×) | 252.7(12) |
| Sb–O2 | (1×) | 204.2(7) | Y–O3′ | (1×) | 252.9(10) |
| Sb–O2′ | (1×) | 209.6(7) | Y⋯Sb2 | (1×) | 351.0(2) |
| Cl1⋯Y1 | (2×) | 439.45(3) | Y⋯Sb1 | (1×) | 351.3(2) |
| Cl2⋯Y2 | (1×) | 419.8(9) | Y⋯Sb1′ | (1×) | 363.9(2) |
| Cl2⋯Y2′ | (1×) | 459.1(9) | Y⋯Sb2′ | (1×) | 367.5(2) |
| Cl1⋯Sb | (4×) | 307.31(7) | Sb1–O1 | (1×) | 193.0(12) |
| Cl2⋯Sb | (4×) | 320.5(5) | Sb1–O3 | (1×) | 203.1(10) |
| Cl2⋯Sb′ | (4×) | 347.8(4) | Sb1–O4 | (1×) | 212.9(11) |
| Sb2–O2 | (1×) | 195.1(12) | |||
| Sb2–O4 | (1×) | 204.9(11) | |||
| Sb2–O3 | (1×) | 211.3(11) | |||
| Br⋯Y | (1×) | 427.6(3) | |||
| Br⋯Y′ | (1×) | 468.9(3) | |||
| Br⋯Sb1 | (1×) | 325.8(2) | |||
| Br⋯Sb1′ | (1×) | 328.1(2) | |||
| Br⋯Sb1′′ | (1×) | 357.6(2) | |||
| Br⋯Sb1′′′ | (1×) | 357.6(2) | |||
| Br⋯Sb2 | (1×) | 319.7(2) | |||
| Br⋯Sb2′ | (1×) | 337.5(2) | |||
| Br⋯Sb2′′ | (1×) | 341.9(2) | |||
| Br⋯Sb2′′′ | (1×) | 364.4(2) |
Experimental section
Synthesis of YSb2O4Cl and YSb2O4Br
The yttrium(III) oxoantimonate(III) halides YSb2O4X (X = Cl and Br) were synthesised at elevated temperatures via solid-state reactions in evacuated silica ampoules (Quarz- und Glasbläserei Müller, Berlin-Adlershof; inner diameter: 10 mm, wall thickness: 1 mm, length: 40 mm). Yttrium oxide (Y2O3, ChemPur: 99.9%, 43.57 mg for the chloride, 39.51 mg for the bromide derivative), antimony sesquioxide (Sb2O3, ChemPur: 99.9%, 168.75 mg for the chloride, 153.00 mg for the bromide derivative) as well as yttrium chloride (YCl3, ChemPur: 99.9%, 37.68 mg) and yttrium bromide (YBr3, Aldrich: 99.9%, 57.49 mg) were used as reactants according to eqn (1). Eutectic mixtures of sodium chloride (NaCl, Merck: 99.9%, 126 mg) and cesium chloride (CsCl, Aldrich: 99.9%, 674 mg) were used for YSb2O4Cl and those of sodium bromide (NaBr, Merck: 99.9%, 203 mg) and cesium bromide (CsBr, ChemPur: 99.9%, 597 mg) for YSb2O4Br to improve the reaction speed and the crystal growth as fluxing agents. As doping material europium sesquioxide (Eu2O3, ChemPur: 99.9%, 1.7 mg) or terbium chloride (TbCl3, Aldrich: 99.9%, 1.0 mg) were used for YSb2O4Cl and europium sesquioxide (Eu2O3: ChemPur: 99.9%, 1.5 mg) or terbium bromide (TbBr3, Aldrich: 99.9%, 1.4 mg) for YSb2O4Br.
 | (1) |
The reactants were weighed into glassy silica ampoules under inert gas (argon) inside a glove box (Glovebox Systemtechnik, GS Mega E-line), sealed under dynamic vacuum and then subjected to a defined temperature program in a muffle furnace (Nabertherm, L 9/12). This was heated at a rate of 150 K h−1 to 750 °C, held there for two days, cooled with 5 K h−1 to 666 °C, held for another three days, cooled with 5 K h−1 to 530 °C, again held for two days, then cooled with 10 K h−1 to 480 °C and finally quenched to room temperature by cutting off the power to the closed furnace. The recovered product samples were washed with 500 ml demineralised water and then dried for 2 h in a drying oven at 120 °C. Under a stereomicroscope, colourless flat, square platelets were visible, clearly larger for YSb2O4Cl than for YSb2O4Br.
Single-crystal X-ray diffraction
Suitable crystals were selected from the samples for single-crystal X-ray diffraction experiments and fixed in glass capillaries (Hilgenberg, Malsfeld; outer diameter: 0.1 mm, wall thickness: 0.01 mm) with grease. The measurements were carried out with a κ-CCD four-circle diffractometer (Bruker-Nonius, Karlsruhe) at room temperature using Mo-Kα radiation. The program package Stoe X-Area 1.86 (2018) was used for data collection and integration. The crystal structures of YSb2O4Cl and YSb2O4Br were solved in the space groups P4212 and P21/c, respectively, by direct methods and refined with the SHELX-97 program package.42–45Luminescence spectroscopy
The luminescence spectra of all samples were measured using a Horiba Fluoromax-4 spectrometer scanning from 220 to 800 nm at room temperature. Therefore, finely ground powder samples were filled into the sample holder and subsequently placed in the sample chamber. These measurements were conducted and evaluated with the program FluorEssence.46 All excitation spectra were corrected to consider the xenon-lamp spectrum.Conclusions
With YSb2O4Br another representative of the known series RESb2O4Br (RE = Eu–Dy) could be synthesised and structurally characterised. Thus it also shows the structural motif of ψ1-tetrahedral [SbO3]3− groups linked to a meandering chain of the formula via common vertices. As first tetragonal representative of the rare-earth metal(III) oxoantimonate(III) halides YSb2O4Cl (space group: P4212) was obtained. Although its structure shows many similarities to the monoclinic YSb2O4Br (space group: P21/c) representative, it exhibits a different corner-linkage of the ψ1-tetrahedral [SbO3]3− units resulting in closed
via common vertices. As first tetragonal representative of the rare-earth metal(III) oxoantimonate(III) halides YSb2O4Cl (space group: P4212) was obtained. Although its structure shows many similarities to the monoclinic YSb2O4Br (space group: P21/c) representative, it exhibits a different corner-linkage of the ψ1-tetrahedral [SbO3]3− units resulting in closed  rings.
rings.
The luminescence spectra of samples doped with trivalent europium or terbium confirmed the lack of inversion symmetry around the yttrium cations in both structures, as well as an efficient energy transfer between the oxidoantimonate(III) layers and the lanthanoid(III)-activator cations.
Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Falk Lissner and Dr Ingo Hartenbach for the single-crystal X-ray diffraction measurements.Notes and references
- G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer-Verlag, Berlin, Heidelberg, 1994 Search PubMed.
- F. Ledderboge, Doctoral Dissertation, Univ. Stuttgart, 2016.
- F. C. Goerigk, Doctoral Dissertation, Univ. Stuttgart, 2021.
- D.-H. Kang, Doctoral Dissertation, Univ. Stuttgart, 2009.
- S. Schander, Doctoral Dissertation, Univ. Oldenburg, 2009.
- F. C. Goerigk, S. Schander, M. Ben Hamida, D.-H. Kang, F. Ledderboge, M. S. Wickleder and T. Schleid, Z. Naturforsch., B, 2019, 74, 497–506 CrossRef CAS.
- M. Ben Hamida and M. S. Wickleder, Z. Anorg. Allg. Chem., 2006, 632, 2195–2197 CrossRef CAS.
- D.-H. Kang, T. Schleid and Z. Anorg, Allg. Chem., 2007, 633, 1205–1210 CrossRef CAS.
- H. Ben Yahia, U. C. Rodewald and R. Pöttgen, Z. Naturforsch., B, 2010, 65, 1289–1292 CrossRef CAS.
- H. Ben Yahia, U. C. Rodewald and R. Pöttgen, Z. Naturforsch., B, 2009, 64, 896–900 CrossRef CAS.
- H. Ben Yahia, A. Villesuzanne, U. C. Rodewald, T. Schleid and R. Pöttgen, Z. Naturforsch., B, 2010, 65, 549–555 CrossRef CAS.
- D.-H. Kang, J. Wontcheu and T. Schleid, Solid State Sci., 2009, 11, 299–304 CrossRef CAS.
- F. C. Goerigk, S. Schander, M. S. Wickleder and T. Schleid, Z. Anorg. Allg. Chem., 2020, 646, 985–991 CrossRef CAS.
- B. Aurivillius, Chem. Scr., 1984, 24, 125–129 CAS.
- C. J. Milne, P. Lightfoot, J. D. Jorgensen and S. Short, J. Mater. Chem., 1995, 5, 1419–1421 RSC.
- M. Schmidt and H. Oppermann, Z. Anorg. Allg. Chem., 1999, 625, 544–546 CrossRef CAS.
- M. Schmidt, H. Oppermann, C. Henning, R. W. Henn, E. Gmelin and N. Söger, Z. Anorg. Allg. Chem., 2000, 626, 125–135 CrossRef CAS.
- A. G. Gukalova and M. N. Tseitlin, Kristallografiya, 1988, 33, 499–501 CAS.
- M. Schmidt, H. Oppermann, M. Zhang-Preße, E. Gmelin, W. Schnelle, N. Söger and M. Binnewies, Z. Anorg. Allg. Chem., 2001, 627, 2105–2111 CrossRef CAS.
- F. C. Goerigk and T. Schleid, Z. Anorg. Allg. Chem., 2010, 645, 1079–1084 CrossRef.
- F. C. Goerigk, V. Paterlini, K. V. Dorn, A.-V. Mudring and T. Schleid, Crystals, 2020, 10, 1089–1112 CrossRef CAS.
- R. J. C. Locke, Master Thesis, Univ. Stuttgart, 2021.
- R. J. C. Locke, F. C. Goerigk and T. Schleid, Z. Kristallogr., 2021, 41, 78–79 Search PubMed.
- G. Blasse and A. Bril, J. Chem. Phys., 1967, 47, 1920–1926 CrossRef CAS.
- E. K. Yukhno, L. A. Bashkirov, P. P. Pershukevich, I. N. Kandidatova, N. Mironova-Ulmane and A. Sarakovskis, J. Lumin., 2017, 182, 123–129 CrossRef CAS.
- S. Nigam, V. Sudarsan and R. K. Vatsa, Opt. Mater., 2011, 33(3), 558–562 CrossRef CAS.
- A. George, S. Gopi, E. Sreeja, T. Krishnappriya, A. C. Saritha, C. Joseph, N. V. Unnikrishnan and P. R. Biju, J. Mater. Sci.: Mater. Electron., 2020, 31, 423–434 CrossRef CAS.
- J. Wang, Y. Cheng, Y. Huang, P. Cai, S. I. Kim and H. J. Seo, J. Mater. Chem. C, 2014, 2, 5559–5569 RSC.
- A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, Walter-de-Gruyter-Verlag, Berlin, New York, 1985, pp. 91–100 Search PubMed.
- R. D. Shannon, Acta Crystallogr., 1975, 32, 751–767 CrossRef.
- M. G. Paton and E. N. Maslen, Acta Crystallogr., 1965, 19, 307–310 CrossRef CAS.
- C. Hirschle and C. Röhr, Z. Anorg. Allg. Chem., 2000, 626, 1305–1312 CrossRef CAS.
- C. Hirschle and C. Röhr, Acta Crystallogr., 1998, 54, 1219–1220 Search PubMed.
- F. Thuillier-Chevin, P. Maraine and G. Perez, Rev. Chim. Miner., 1980, 17, 102–109 CAS.
- L. G. Sillén and L. Melander, Z. Kristallogr., 1941, 103, 420–430 Search PubMed.
- N. Rück and A. Pfitzner, Z. Anorg. Allg. Chem., 2012, 638, 1586 CrossRef.
- B. P. de Laune, R. D. Bayliss and C. Greaves, Inorg. Chem., 2011, 50, 7880–7885 CrossRef CAS PubMed.
- H. D. Stöver and R. Hoppe, Z. Anorg. Allg. Chem., 1980, 468, 137–147 CrossRef.
- B. Antic, P. Oennerud, D. Rodic and R. Tellgren, Powder Diffr., 1993, 8, 216–220 CrossRef CAS.
- C. Svensson, Acta Crystallogr., 1975, 31, 2016–2018 CrossRef.
- E. W. J. L. Oomen, W. M. A. Smit and G. Blasse, Chem. Phys. Lett., 1987, 138, 23–28 CrossRef CAS.
- W. Herrendorf and H. Bärnighausen, HABITUS: Program for the Optimisation of the Crystal Shape for Numerical Absorption Correction in X-SHAPE, Version 1.06, Stoe, Darmstadt, 1999, Karlsruhe, 1993, Gießen, 1996 Search PubMed.
- G. M. Sheldrick, SHELXS-97 and SHELXL-97: Programs for the Solution and Refinement of Crystal Structures from X-Ray Diffraction Data, Göttingen, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. Weil, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 26–29 CrossRef CAS PubMed.
- FluorEssence, Steady State and Frequency Domain Software, Version 3.8.0.60, HORIBA Scientific, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08382a |
| This journal is © The Royal Society of Chemistry 2022 |