
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEstimations of Fe0/−1–N2 interaction energies of iron(0)-dicarbene and its reduced analogue by EDA-NOCV analyses: crucial steps in dinitrogen activation under mild conditions†
Sai Manoj N. V. T. Gorantla and
Kartik Chandra Mondal*
and
Kartik Chandra Mondal*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: csdkartik@iitm.ac.in
First published on 26th January 2022
Abstract
Metal complexes containing low valence iron atoms are often experimentally observed to bind with the dinitrogen (N2) molecule. This phenomenon has attracted the attention of industrialists, chemists and bio-chemists since these N2-bonded iron complexes can produce ammonia under suitable chemical or electrochemical conditions. The higher binding affinity of the Fe-atom towards N2 is a bit ‘mysterious’ compared to that of the other first row transition metal atoms. Fine powders of α-Fe0 are even part of industrial ammonia production (Haber–Bosch process) which operates at high temperature and high pressure. Herein, we report the EDA-NOCV analyses of the previously reported dinitrogen-bonded neutral molecular complex (cAACR)2Fe0–N2 (1) and mono-anionic complex (cAACR)2Fe−1–N2 (2) to give deeper insight of the Fe–N2 interacting orbitals and corresponding pairwise intrinsic interaction energies (cAACR = cyclic alkyl(amino) carbene; R = Dipp or Me). The Fe0 atom of 1 prefers to accept electron densities from N2 via σ-donation while the comparatively electron rich Fe−1 centre of 2 donates electron densities to N2 via π-backdonation. However, major stability due to the formation of an Fe–N2 bond arises due to Fe → N2 π-backdonation in both 1 and 2. The cAACR ligands act as a charge reservoir around the Fe centre. The electron densities drift away from cAAC ligands during the binding of N2 molecules mostly via π-backdonation. EDA-NOCV analysis suggests that N2 is a stronger π-acceptor rather than a σ-donor.
Introduction
Different forms of energy can be argued as the ingredients of life.1 Energy, life and information are linked to each other.2 Nitrogen is one of the most common and most essential elements for the sustainability of living organisms, plants and animals. The earth's atmosphere is mostly dominated (78%) by dinitrogen gas (N2) and yet most of the living organisms, plants and animals cannot directly utilize it according to their needs.3 This is due to the extreme inertness and non-polar nature of kinetically stable N2 molecules. The reduction of N2 to ammonia (NH3) and/or to other forms of N-containing compounds such as amino acids, nucleoside, nucleotides and most importantly peptides are keys to the existence of life on earth.4 Thermodynamically, reduction of areal N2 to relatively more stable NH3 is exothermic (−46 kJ mol−1 at 298 K). However, the binding of N2 and followed by activation of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond is challenging to chemists and bio-chemists. Interestingly, a few microorganisms in the nature have found a way to solve the problem5 providing around hundred twenty megatons of nitrogen source per year by nitrogen fixation. It is associated with some leguminous plants, like clover, beans, peas, alfalfa, lentils and lupins.6 The enzyme called nitrogenase possesses a bimetallic inorganic core V/Mo–Fe7S9C1− (co-factor) which is responsible for N2 binding in the reduced state.7,8 It catalytically produces ammonia via reductive protonation either by ‘distal’ or ‘alternative’ pathways with the loss of hydrogen gas during this process.7,8 The exact nature and mode of the dinitrogen binding and activation have not yet fully clear. N-containing nutrients are essential for the human race on the earth. Humans adopted domestic cultivation as early as 10
N bond is challenging to chemists and bio-chemists. Interestingly, a few microorganisms in the nature have found a way to solve the problem5 providing around hundred twenty megatons of nitrogen source per year by nitrogen fixation. It is associated with some leguminous plants, like clover, beans, peas, alfalfa, lentils and lupins.6 The enzyme called nitrogenase possesses a bimetallic inorganic core V/Mo–Fe7S9C1− (co-factor) which is responsible for N2 binding in the reduced state.7,8 It catalytically produces ammonia via reductive protonation either by ‘distal’ or ‘alternative’ pathways with the loss of hydrogen gas during this process.7,8 The exact nature and mode of the dinitrogen binding and activation have not yet fully clear. N-containing nutrients are essential for the human race on the earth. Humans adopted domestic cultivation as early as 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 BC.9 Fertilizers are now-a-days a must for effective cultivation. More than a century ago, Haber originally utilized an osmium catalyst under high temperature and pressure to produce 125 mL of NH3 per hour.10 In the following years Bosch and Mittasch developed efficient Fe3O4/FeO/α-Fe0 catalyst with high surface area for industrial production of twenty tonnes of NH3 per day in the next year at BASF company.10,11 The catalytic efficiency of iron is further promoted with K2O, CaO, SiO2, and Al2O3. At present global annual production rate of ammonia is 174 million tonnes.12 The actual Mittasch's catalyst (Fe3O4/FeO/α-Fe0) (Scheme 1) in Haber–Bosch catalytic cycle is the outer surface (α-Fe0) of very reactive Fe3O4/FeO/α-Fe0 particles having a body centre cubic (bcc) structure with two singly-occupied dz2 and dx2−y2 orbitals which do not participate in multicentre delocalized bonding with its neighbouring Fe0 atoms.13
000 BC.9 Fertilizers are now-a-days a must for effective cultivation. More than a century ago, Haber originally utilized an osmium catalyst under high temperature and pressure to produce 125 mL of NH3 per hour.10 In the following years Bosch and Mittasch developed efficient Fe3O4/FeO/α-Fe0 catalyst with high surface area for industrial production of twenty tonnes of NH3 per day in the next year at BASF company.10,11 The catalytic efficiency of iron is further promoted with K2O, CaO, SiO2, and Al2O3. At present global annual production rate of ammonia is 174 million tonnes.12 The actual Mittasch's catalyst (Fe3O4/FeO/α-Fe0) (Scheme 1) in Haber–Bosch catalytic cycle is the outer surface (α-Fe0) of very reactive Fe3O4/FeO/α-Fe0 particles having a body centre cubic (bcc) structure with two singly-occupied dz2 and dx2−y2 orbitals which do not participate in multicentre delocalized bonding with its neighbouring Fe0 atoms.13
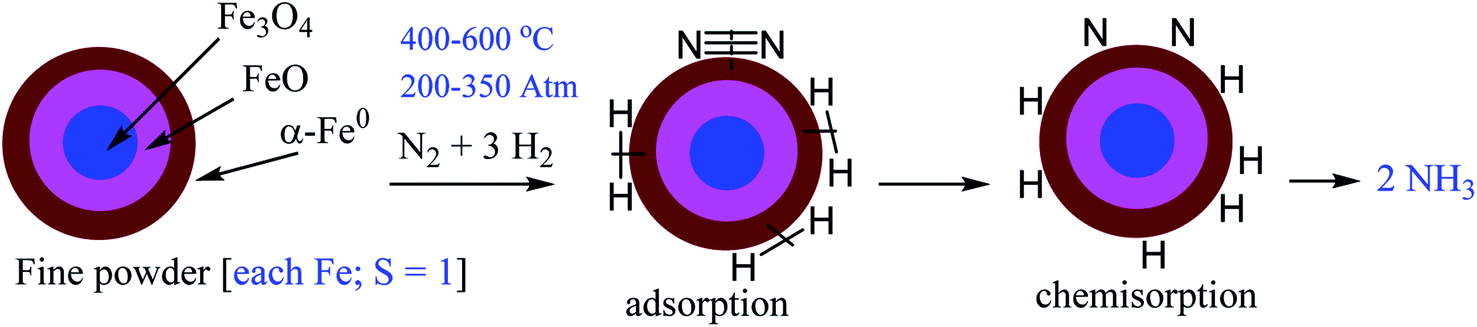 | ||
| Scheme 1 Illustration of H2 and N2 adsorption, dissociation and formation of NH3 in famous Haber–Bosch process showing α-Fe0 active catalytic surface. | ||
To obtain a favourable thermodynamic driving force for the reaction between N2 and H2 the industrial process is designed to occur at high pressure (Scheme 1). In 2007 Gerhard Ertl has been awarded the Nobel Prize in chemistry for his captivating decades long works on fundamental processes at the gas–solid interface involving Fe0–N2 adsorption on the α-Fe0 surface of Fe3O4/FeO/α-Fe0 catalyst. His work on ‘Interaction of nitrogen with iron surfaces’ clarified the long-standing confusion in Haber–Bosch process.14 It has been found that NN bond dissociates on pure Fe-surfaces forming Fe4N atomic-bilayer just above room temperature. He also explained the role of promoter K2O in Haber–Bosch ammonia synthesis process employing photoelectron spectroscopy and other experimental techniques. The pre-adsorbed potassium on the Fe-surface removes energy barrier of dissociative nitrogen chemisorption of N2 molecule.15 The overall yield of NH3 in Haber–Bosch industrial process is 97% when the unreacted gases are recycled again and again.10 However, at present this process consumes ∼2% of the total global energy supply. Additionally, it produces large amounts of greenhouse gases.16 Alternative synthetic methods which will produce NH3 in a much greener way are highly desired.17–41 Low valence and/or low valent metal complexes17–33,37–41 and low coordinate boron–carbene34–36 compounds are found to bind N2 which can be activated to obtain NH3 or N2H4 under milder conditions.17–41 The reductive protonation of bonded N2 of these complexes can lead to the formation of either NH3 or N2H4 or even a mixture of both. Formation of ammonia is more common due to its higher thermodynamic stability over hydrazine. It has been stated that π-backdonation from metal to N2 (M → N2) are very pivotal for the weakening of strong NN bond. Higher the electron densities on metal atom, higher is the π-backdonation from metal to N2 (Scheme 2). However, there is no report on the estimation of M–N2 interaction energies in a stable/isolable complex showing the extent of π-backdonation from M → N2 and σ-donation from N2 → M (M = transition metal; Scheme 2).42 The energy decomposition analysis-natural orbital for chemical valence (EDA-NOCV)43 analysis is a very powerful tool that can be employed for the estimation of intrinsic interaction energy (ΔEint) and pairwise orbital interaction energy (ΔEorb(n)). The triple bond of the free dinitrogen (N2) molecule is extremely strong. EDA-NOCV analysis of free N2 revealed that 30% of the total interaction energy between two N-atoms is only contributed by electrostatic energy (ΔEelstat). The remaining 70% is orbital interaction energy (ΔEorb) which is due to the covalent character of triple bond of N2 with zero dipole moment.43,44 The Pauli repulsion energy (ΔEPauli) between two interacting N-atoms is quite high due to repulsion between the electron clouds with similar spins (N–N = 1.102 Å). The Wiberg bond order has been computed to be 3.03. The orbital interaction energy (ΔEorb) is actually due to the covalent interactions in both σ (3σg+) + π (1πu) bonds. The former is 65.6% while the latter is nearly half (34.4%) of the former. They together (σ + π) give total orbital interaction energy (ΔEorb).43,44 Point to be noted that degenerate π*-orbitals (1πg) of N2, which are composed of px and py atomic orbitals, are high lying in energies (1πg; LUMO). The LUMO+1 is σ* orbital (3σu+) and HOMO is N–N σ orbital (3σg+) of N2.43,44
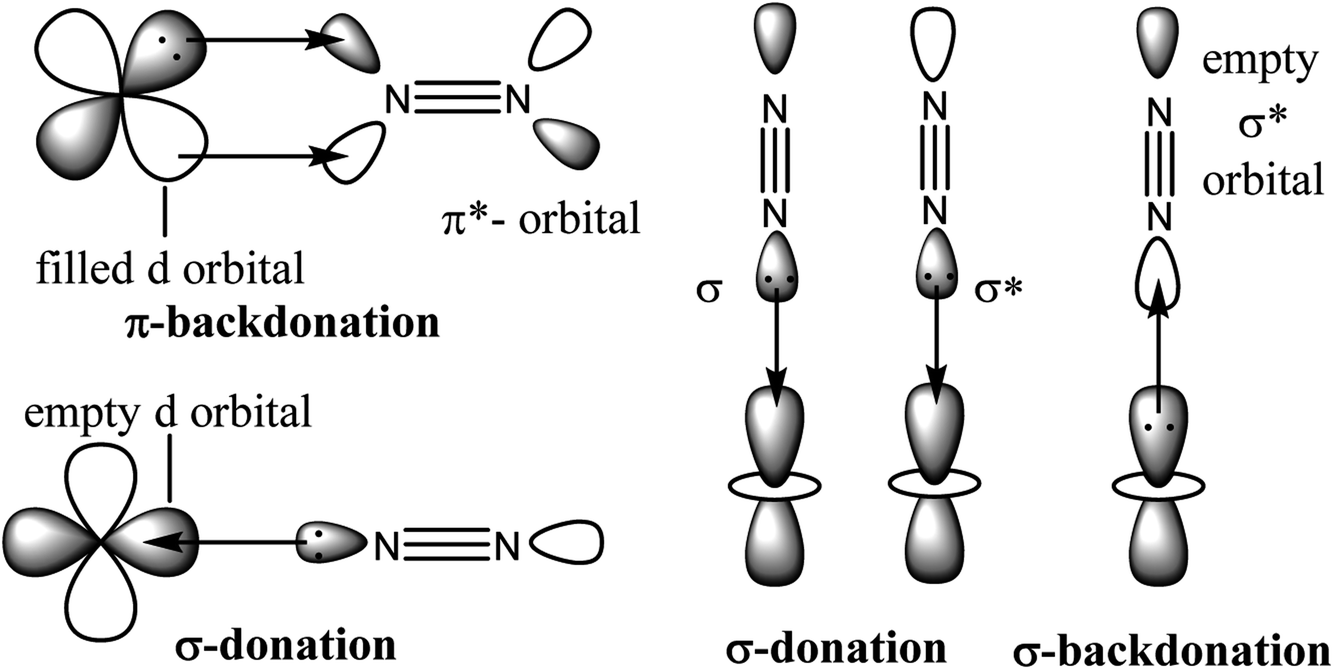 | ||
| Scheme 2 End-on interactions between the orbitals metal (M) and N2. | ||
Several metal complexes with low valence iron atoms containing coordinated N2 have been synthesized, isolated and characterized by X-ray single crystal structure determination.18–21,27–33,40,41 Additionally, they have been studied by different spectroscopic methods to shed light on their electronic structures. NBO calculations have been carried out to correlate the N–N bond lengths with those of experimentally obtained values to give emphasis on weakening of N–N bond due to M → N2 backdonation.18–21,27–33,40,41 However, there is till now no report42 on the exact nature of the M–N2 bonds and on their corresponding interaction energies (M = Fe and other transition metal) of stable/isolable dinitrogen bonded metal complexes. Herein, we report on the DFT, NBO, QTAIM calculations and EDA-NOCV analysis of previously reported dinitrogen-bonded (cAACR)2Fe0–N2 (1) and (cAACR)2Fe−1–N2 (2) complexes41 to give a deeper insight into the nature of M–N2 bonds and corresponding pairwise interaction energies (cAACR = cyclic alkyl(amino) carbene; R = Dipp41 or Me for our theoretical studies). The role of non-innocent cAAC ligands has also been discussed here.
Results and discussion
The spin of each Fe-atom of ferromagnetic α-iron metal is S = 1.10 The spin ground state of (cAACDipp)2Fe0 has been confirmed to be S = 1 by EPR and 57Fe Mössbauer spectroscopy by Peters et al.45 Additionally, they have experimentally shown41 (by UV/vis spectroscopy) that the N2 binds to (cAAC)2Fe0 in end-on fashion. This N2 binding at Fe-centre is highly temperature sensitive (<−80 °C).41 Experimentally the authors have isolated the elusive anionic (cAACDipp)2Fe−1–N2 species by reducing in situ formed precursor (cAACDipp)2Fe0–N2 (1′) with KC8 in the presence of 18-crown-6 ether below −95 °C with the chemical composition of [(cAACDipp)2Fe(N2)][K(18-crown-6)] (2′) (Scheme 3). The latter species (2′) has a ground state S = ½ confirmed by solution EPR measurements.41 This anionic complex has further been shown to catalytically produce NH3 below −95 °C. The reduction of dinitrogen to ammonia takes place upon treatment with N2, KC8 and HBArF4·2Et2O in ether medium. Only a little has been reported about their (1′–2′) aspects of chemical bonding.41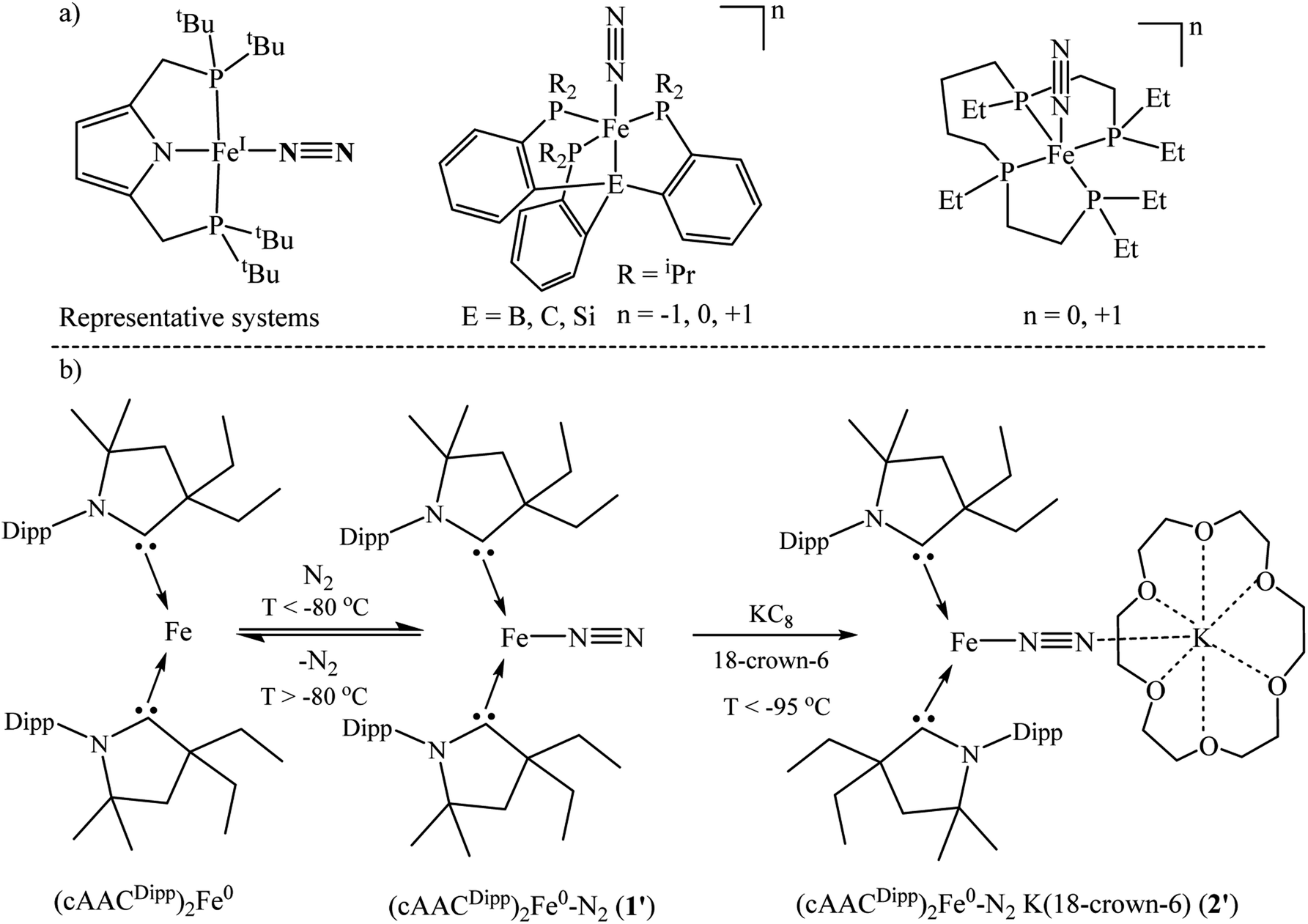 | ||
| Scheme 3 Representative Fe–N2 containing iron complexes.17,18,38,40,41,45 | ||
We have modelled and optimized (L)2Fe0–N2 as neutral (1; L = cAACMe) and anionic complexes (2; L = cAACMe) stabilized by cAACMe ligands to shed light on the bonding and stability of spectroscopically observed elusive neutral (cAACDipp)2Fe0–N2 (1′; cAACDipp) and crystallographically characterized [(cAACDipp)2Fe−1(N2)][K(18-crown-6)] complexes (2′) reported by Jonas Peters and co-workers. The Fe–N bond distance of [(cAACDipp)2Fe(N2)][K(18-crown-6)] is 1.777 Å while the N–N bond length of the bonded N2 is 1.035(4) Å which is slightly shorter than that of the free N2 (1.102 Å) molecule. X-ray crystallography of the complex 2′ revealed that Fe-centre adopted a distorted trigonal planar coordination geometry.41
The modelled neutral (cAACMe)2Fe–N2 complex (1) has been optimized in singlet (Fig. S1†), triplet (Fig. 1) and quintet electronic states (Fig. S1†). The calculations at BP86-D3(BJ)/Def2TZVPP level of theory in the gas phase suggest that triplet state is more stable by 11.53 and 20 kcal mol−1 over singlet and quintet states, respectively. The N–C–C–N torsion angle of 37.7° in complex 1 suggests that the two cAAC ligands are relatively perpendicular to each other, while the C–Fe–C bond angle of 149.3° shows that the geometry is lightly bent compared to that of (cAAC)2Fe0 containing a two coordinate Fe0 atom with CcAAC–Fe–CcAAC bond angle of 169.52(5)°.45 The Fe–N and N–N bond lengths of the simplified complex 1 are 1.801 Å and 1.140 Å respectively (with Me-group on N-atom of cAAC ligand). The two cAAC ligands are almost equidistant from the central Fe atom with a minor difference (Fig. 1). In contrast to the Fe–Mo cofactor (FeMoco) of nitrogenase enzyme,7,8 the two coordinate (cAAC)2Fe can bind to N2 even in resting condition below −80°41 or in other words without the external supply of electrons.
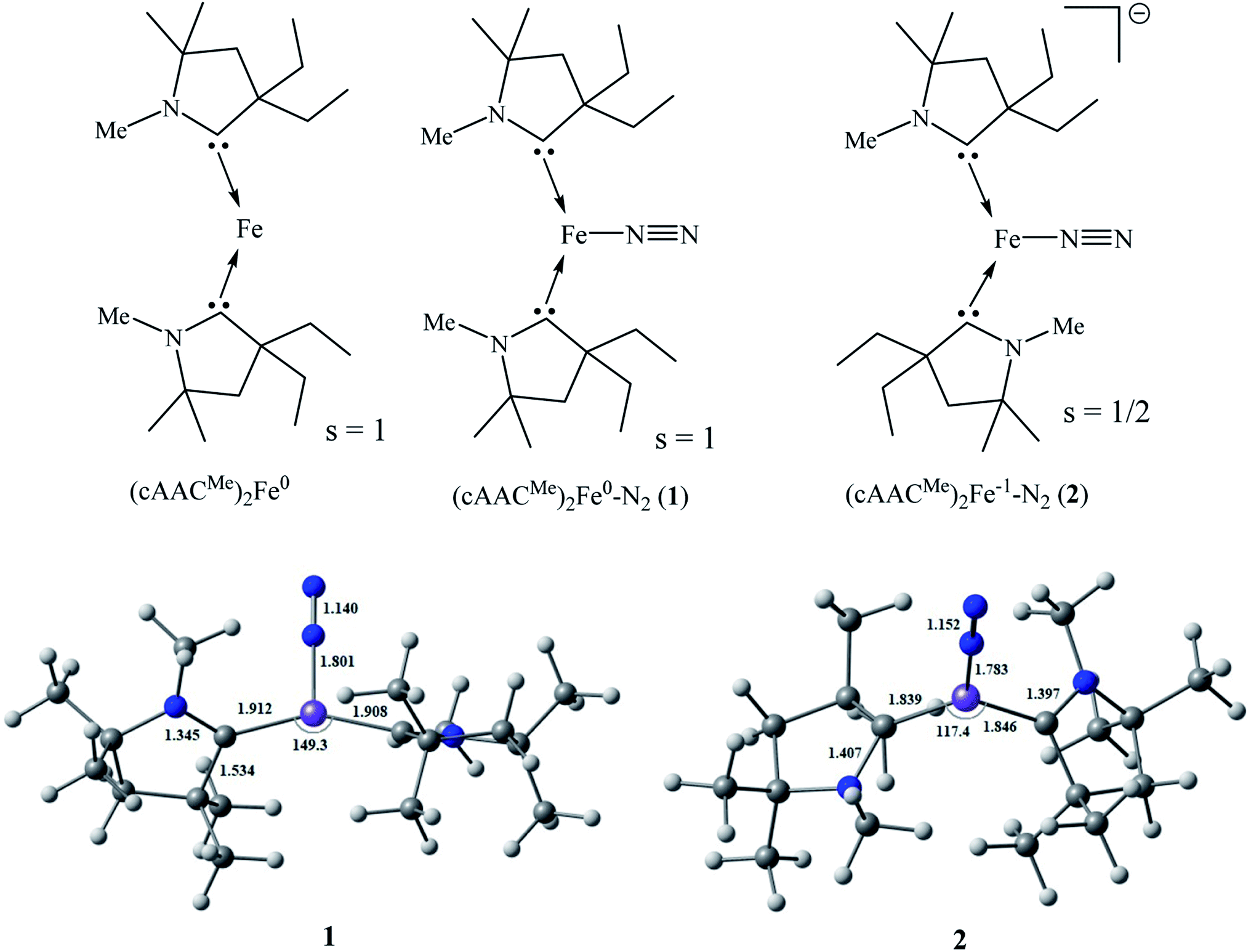 | ||
| Fig. 1 Optimized geometries of complex 1 in triplet state (s = 1) and mono-anionic complex 2 in doublet state (s = 1/2) at BP86-D3(BJ)/Def2-TZVPP level. | ||
Upon reduction, the geometry becomes more bent in complex 2 as indicated by the CcAAC–Fe–CcAAC bond angle of 117.4° (Fig. 1). This differs from the C–Fe–C bond angle (140.8°) of experimentally isolated [(cAACDipp)2Fe(N2)][K(18-crown-6)] (2′) due to to the steric effects of two cAACDipp ligands. We can reason the reduction in C–Fe–C bond angle in the modelled complex to the presence of less bulky substituents on cAAC ligand which reduces the steric repulsion. This reduction in C–Fe–C bond angle also slightly lower the C–Fe bond lengths in complex 2 compared to that of the reported structure. The Fe–N bond distance of modelled complex 2 (1.782 Å) correlated well with the reported value of 1.777 Å (2′). The geometrical parameters calculated at BP86-D3(BJ)/Def2TZVPP level agrees well with the experimental values with no major discrepancies. However, we have also performed geometry optimization of complexes 1 and 2 at the TPSS-D3(BJ)/Def2TZVPP and PW6B95-D3/Def2TZVPP level to compare and support the results. While the Fe–N bond lengths of complexes 1 and 2 calculated at TPSS-D3(BJ) level are 1.808 Å, 1.782 Å respectively (Fig. S1†), the calculated N–N bond lengths are 1.136 and 1.149 Å (Fig. S1†). The geometrical parameters calculated at TPSS-D3(BJ) match well with the results of the BP86-D3(BJ) level. However, the Fe–N bond lengths (1.899 and 1.864 Å) calculated at PW6B95-D3 level (Fig. S1†) differ significantly with those calculated at TPSS-D3(BJ), BP86-D3(BJ) and also experimental values. The calculated C–Fe–C bond angle of complex 2 at TPSS-D3(BJ) is also acute (120.7°), supporting the reason for the difference from the experimental bond angle as mentioned above. The N–C–C–N torsion angle of 145.1° indicates that the two carbene ligands are slightly more trans to each other in complex 2. The coordination geometries of Fe-centres reveal that both complexes 1 and 2 possess a distorted trigonal planar geometry as indicated by Σangle of 359.8° and 359.9° respectively and are in agreement with the reported structure (2′).41 The longer C–N bond lengths of 1.343–1.345 Å in complex 1 and 1.397–1.407 Å in complex 2 than that of 1.315(3) Å in free carbene, indicate spin delocalization onto ligands (CcAAC ← Fe).46 The dissociation of (cAACMe)2Fe–N2 bond [(cAACMe)2Fe–N2 → (cAACMe)2Fe + N2] in complex 1 and 2 is slightly endothermic (ΔG298 = 18.9–29.6 kcal mol−1) and the energy of dissociation is 30.1–40.26 kcal mol−1 (bond enthalpy) respectively. The electron affinity of 1 is 13.74 kcal mol−1.
We have employed charge and energy density methods like natural bond orbital (NBO), quantum theory of atoms in molecules (QTAIM) and energy decomposition analysis coupled with natural orbitals for chemical valence (EDA-NOCV) methods to study the nature of the Fe–N bond. The Wiberg bond index (WBI) of 0.82 (1), 0.92 (2) for Fe–N bond and 2.53 (1), 2.43 (2) for N–N bond of 1 and 2, respectively (Table 1). The Fe–N and N–N bond orders are consistent with the Fe–N and N–N bond lengths of both the complexes. The N–N bond orders (BO; 2.53 (1), 2.43 (2)) of N2 in complexes 1 and 2 are significantly smaller than those of free N2 molecule (BO = 3.03). This indicates the weakening of the N–N bond via π-backdonation after the binding of N2 with Fe-centres which is crucial for the activation of N2. Previous temperature dependent UV/vis studies showed that N2 binding at Fe0 centre could only happen below −80 °C.41 The computational results show that the spin density of the Fe–N bond with an electron occupancy of 0.99 is mostly concentrated on N-atom (80.6%) for complex 1. The calculation does not show bond occupancy for the Fe–N bond of complex 2. The CcAAC–Fe bonds of complex 1 and 2 show two occupancies representing σ- and π-interactions with spin density mostly concentrated on CcAAC (62.9–71.5%) for σ-interactions and Fe (68.4–75.3%) for π-interactions (Table 1). The natural charge distribution of (cAACMe)2Fe0 shows a negative charge on cAAC ligands and a positive charge on the Fe-atom suggesting π-backdonation (Fe → N2) is stronger than σ-donation (N2 → Fe). Upon binding to N2, the cAAC ligands develop a positive charge and the Fe-center of 1 shows an increase in positive charge as well, while the N2 ligand accumulates a negative charge, suggesting N2 is stronger π-acceptor than a σ-donor. This indicates that the charge transfer occurs in the direction cAAC → Fe → N2. It is well known that cAAC is a non-innocent ligand.46b,c It can control the charge flow and distribute the electron densities distributions based on the electronic situations and or requirement. Whereas the negative charge on cAAC, Fe and N2 of complex 2 suggests the delocalization of one electron-charge upon reduction of 1. The α-SOMO and α-SOMO−1 of complex 1 represent the two unpaired electrons residing in dxy and dx2−y2 of the triplet state. The α-SOMO−1 shows π-interaction of dx2y2 orbital of Fe and lone pair on cAAC ligand with πx-orbital of N2, while α-SOMO−2 indicates π-interaction of dyz orbital of Fe-atom with πy-orbital of N2 (Fig. S2†). Whereas α-SOMO of complex 2 represents an unpaired electron in dxz orbital of Fe showing small amount of interaction with πx orbital of N2. The α-SOMO−1, α-SOMO−2 and α-SOMO−3 indicate the interaction of dx2−y2, dz2 and dyz orbitals of Fe with πx and πy orbital of N2 (Fig. S3†). The QTAIM analysis shows a bond path (Fig. S4†) and considerable electron density ρ(r) along the Fe–N bond path in both complexes 1 and 2 (Table 2). Little increase in ρ(r) along Fe–N and CcAAC–Fe bond paths in complex 2 corroborates the delocalization of electron density and agrees with the charge distribution from NBO analysis. The ellipticity (εBCP = λ1/λ2 − 1) is a measure of bond order and in general, the εBCP of a single and triple bond is close to zero because of cylindrical contours of electron density ρ, while for double bond the value is greater than zero.47 This is due to the asymmetric distribution of electron density ρ perpendicular to the bond path for a double bond. The ellipticity (ε) values of 0.248 and 0.383 for the Fe–N bond of complexes 1 and 2 suggests the possible multiple bond character.
| Complex | Bond | ON | Polarization and hybridization (%) | WBI | qFe | qN2 | qcAAC | |
|---|---|---|---|---|---|---|---|---|
| 1 | Fe–N | 0.99 | Fe: 19.4 s(21.3),p(19.4),d(59.3) | N: 80.6 s(58.4), p(41.6) | 0.82 | 0.440 | −0.122 | 0.144 |
| C–Fe | 0.96 | Fe: 28.5 s(20.0), p(13.8), d(66.2) | C: 71.5 s(41.8), p(58.2) | 0.84 | ||||
| 0.91 | Fe: 75.3 s(0.0), p(4.7), d(95.3) | C: 24.7 s(0.8), p(99.2) | ||||||
| 2 | Fe–N | — | — | — | 0.92 | −0.223 | −0.275 | −0.510 |
| C–Fe | 0.97 | Fe: 37.1 s(20.6), p(8.3), d(71.1) | C: 62.9 s(45.3), p(54.7) | 1.09 | ||||
| 0.90 | Fe: 68.4 s(0.8), p(7.5), d(91.7) | C: 31.6 s(0.2), p(99.8) | ||||||
| (cAAC)2Fe | C–Fe | 0.91 | Fe: 14.6 s(42.5), p(50.0), d(7.5) | C: 85.4 s(39.5), p(60.5) | 0.88 | 0.245 | — | −0.244 |
| 0.90 | Fe: 98.9 s(0.4), p(0.2), d(99.4) | C: 1.1 s(5.6), p(94.4) | ||||||
| Bond | ρ(r) | ∇2ρ(r) | H(r) | V(r) | G(r) | εBCP |
|---|---|---|---|---|---|---|
| Fe–N(1) | 0.131 | 0.796 | −0.039 | −0.277 | 0.238 | 0.248 |
| C–Fe(1) | 0.124 | 0.342 | −0.056 | −0.196 | 0.140 | 0.318 |
| Fe–N(2) | 0.139 | 0.793 | −0.046 | −0.290 | 0.244 | 0.383 |
| C–Fe(2) | 0.142 | 0.376 | −0.072 | −0.237 | 0.165 | 0.107 |
The EDA-NOCV method48 is more appropriate in explaining the nature of the bond as one of the major strengths of the method is its ability to provide the best bonding model to represent the bonding situation in the equilibrium geometries.48 To give the best bonding description of Fe–N bond by EDA-NOCV method, we have considered neutral (cAACMe)2Fe fragment in electronic triplet state and neutral N2 fragment in electronic singlet state (1Σg+) for complex 1 and mono-anionic [(cAACMe)2Fe]− fragment in electronic doublet state and neutral N2 fragment in electronic singlet state (1Σg+) for complex 2 (Table 3). The instantaneous interaction (ΔEint) demonstrates the strength of the bond and ΔEint for Fe0-complex 1 (87.9 kcal mol−1) is significantly higher than that of Fe−1-complex 2 (63.8 kcal mol−1). This lowering of instantaneous interaction is favourable as the reduced Fe−1-complex 2 is the active species in the catalytic conversion of N2 → NH3 in the presence of strong proton donor. Note that the instantaneous interactions in 1 and 2 are moderately higher than their bond dissociation energies and the difference can be attributed to the preparative energy. The preparative energies emanate from the modifications in the geometry of the fragments from their equilibrium structure to the geometry in the compound and also from the electronic excitation to a reference state. The orbital (covalent) interactions marginally dominate the total attractive interactions in both complexes 1 (57.9%) and 2 (53.2%). The Fe–N bond of 1 is slightly more covalent in nature than that of 2. The electrostatic interactions contribute 40.2–45% and dispersion contribution provide 1.8–1.9% to the total attractive interactions (Table 3). We optimized complex 1 in diethyl ether solvent (see ref. 41) using the CPCM solvation model and performed an EDA-NOCV calculation to check the effect of the solvent on the bond strength (ΔEint). Interestingly, the results show that the effect of solvent is minimal and showed imperceptible differences in the bonding parameters like intrinsic interactions (87.5 kcal mol−1) and orbital interactions (−159.7 kcal mol−1) compared to that of the gas phase (87.9 and 161 kcal mol−1) (Table S1†).
| Energy | Interaction | (cAAC)2Fe (T) + [N2] (S); (1) | [(cAAC)2Fe]− (D) + [N2] (S); (2) |
|---|---|---|---|
| a The values in the parentheses show the contribution to the total attractive interaction ΔEelstat + ΔEorb + ΔEdisp.b The values in parentheses show the contribution to the total orbital interaction ΔEorb. | |||
| ΔEint | −87.9 | −63.8 | |
| ΔEPauli | 190.2 | 164.1 | |
| ΔEdispa | −5.2 (1.9%) | −4.1 (1.8%) | |
| ΔEelstata | −111.9 (40.2%) | −102.4 (45.0%) | |
| ΔEorba | −161.0 (57.9%) | −121.4 (53.2%) | |
| ΔEorb(1)b | (cAAC)2Fe ← N2 σ-donation | −61.4 (38.1%) | |
| (cAAC)2Fe → N2 π-backdonation | −39.4 (32.4%) | ||
| ΔEorb(2)b | (cAAC)2Fe → N2 π-backdonation | −53.6 (33.3%) | −34.0 (28.0%) |
| ΔEorb(3)b | (cAAC)2Fe → N2 π-backdonation | −33.2 (20.6%) | −25.2 (20.8%) |
| ΔEorb(4)b | (cAAC)2Fe ← N2 σ-donation | −7.5 (4.6%) | −14.7 (12.1%) |
| ΔEorb(rest)b | −5.3 (3.3%) | −8.1 (6.7%) | |
The total orbital interactions can further be broken down into pairwise contributions which can shed light on the type of interactions (Table 3). The deformation densities and the associated molecular orbitals provide insight into the direction of charge flow as shown in Fig. 2 and 3. The first orbital term (ΔEorb1) of complex 1 represents the σ-electron donation (Fe ← N2) from HOMO (3σg+) of N2 into vacant d-type anti-bonding orbital (LUMO; mixture of pz orbitals of CcAAC and d-orbital of Fe0 of 1) of (cAAC)2Fe0 along with the slight charge transfer from dxz orbital (HOMO−2) of Fe and contributes 38% of the total orbital interactions. The deformation densities of Fig. 2 (top, left) show charge flow from red region on N2 to blue region on (cAAC)2Fe0. The second and third orbital terms (ΔEorb2–3) designates π-backdonations (Fe → N2) from hybrid dz2–dx2 y2 orbital (HOMO) of Fe into vacant degenerate π* orbital LUMO (1πg) of N2 and partly into SOMO (dyz) of Fe center and from dxz orbital (HOMO−1) of Fe into vacant degenerate π* orbital LUMO′ 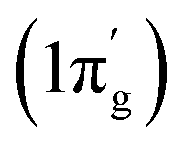 of N2 in second and third orbital terms respectively. The π-backdonations together contribute 53.9% to the total orbital interactions (Table 3). The weaker fourth orbital term (4.6%) is due to σ-electron donation (Fe ← N2) from HOMO−2 (2σu+) of N2 into vacant d-type orbital (LUMO) of Fe and the two Fe ← N2 σ electron donations (ΔEorb1, ΔEorb4) together contribute 42.6% (Fig. 2) of 1. Table 3 shows that the Fe0 → N2 π-backdonation (ΔEorb2–3) in 1 is nearly 10% higher than σ-donation (ΔEorb1,4). The fluctuations of electron clouds on cAAC ligands during the formation of σ- and π-bonds as expected since cAAC is known as both σ-donor and π-acceptor.46
of N2 in second and third orbital terms respectively. The π-backdonations together contribute 53.9% to the total orbital interactions (Table 3). The weaker fourth orbital term (4.6%) is due to σ-electron donation (Fe ← N2) from HOMO−2 (2σu+) of N2 into vacant d-type orbital (LUMO) of Fe and the two Fe ← N2 σ electron donations (ΔEorb1, ΔEorb4) together contribute 42.6% (Fig. 2) of 1. Table 3 shows that the Fe0 → N2 π-backdonation (ΔEorb2–3) in 1 is nearly 10% higher than σ-donation (ΔEorb1,4). The fluctuations of electron clouds on cAAC ligands during the formation of σ- and π-bonds as expected since cAAC is known as both σ-donor and π-acceptor.46
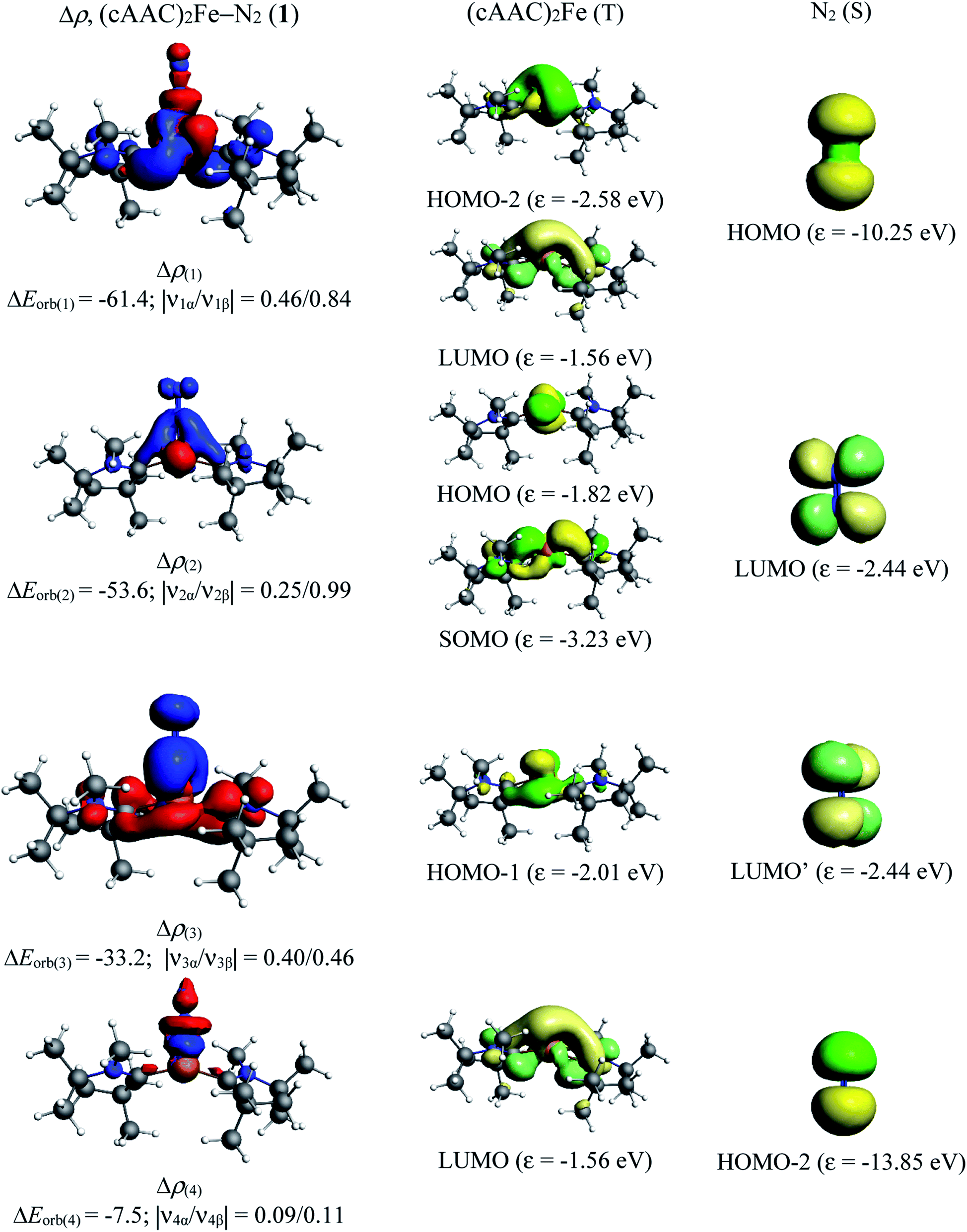 | ||
| Fig. 2 The shape of the deformation densities Δρ(1)–(4) that correspond to ΔEorb(1)–(4), and the associated MOs of (cAAC)2Fe–N2 (1) and the fragments orbitals of (cAAC)2Fe in triplet state and N2 in the singlet state at the BP86-D3(BJ)/TZ2P level. Isosurface values of 0.003 au for Δρ(1–4). The eigenvalues |νn| give the size of the charge migration in e. The direction of the charge flow of the deformation densities is red → blue. Energies are in kcal mol−1. | ||
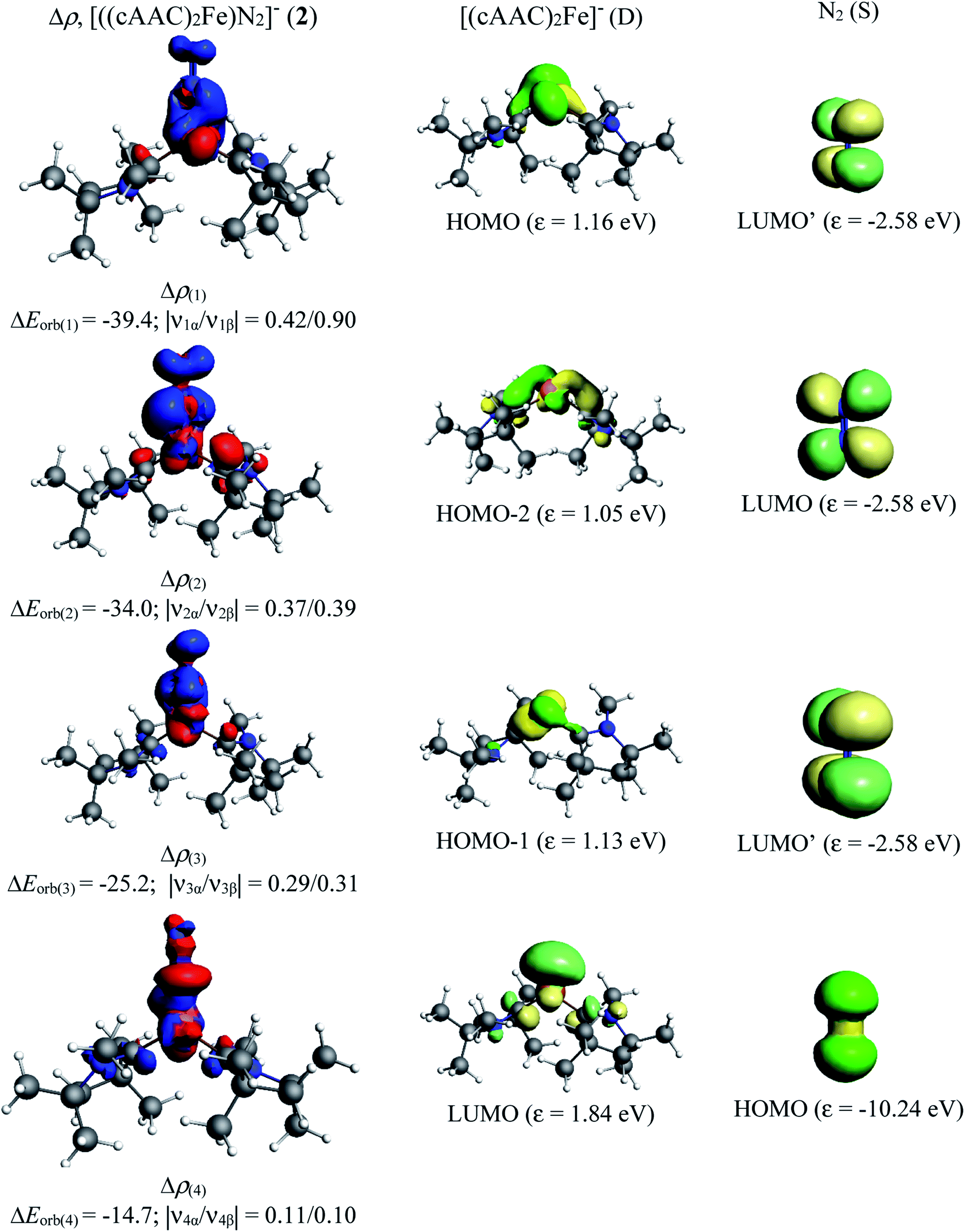 | ||
| Fig. 3 The shape of the deformation densities Δρ(1)–(4) that correspond to ΔEorb(1)–(4), and the associated MOs of [((cAAC)2Fe)N2]− (2) and the fragments orbitals of [(cAAC)2Fe]− in doublet state and N2 in the singlet state at the BP86-D3(BJ)/TZ2P level. Isosurface values of 0.003 au for Δρ(1–4). The eigenvalues |νn| give the size of the charge migration in e. The direction of the charge flow of the deformation densities is red → blue. Energies are in kcal mol−1. | ||
In contrast, the first three orbital terms (ΔEorb1–3) of complex 2 represent π-electron backdonations (Fe → N2). While the first orbital term (ΔEorb1) comes from the π-electron backdonation from dz2 orbital (HOMO) of Fe−1 into degenerate vacant π(py)* orbital (LUMO′) of N2, the second orbital term (ΔEorb2) is due to π-backdonation from dx2y2 orbital (HOMO-2) into vacant degenerate π* orbital (LUMO) (1πg) of N2. The third orbital term (ΔEorb3) is due to π-backdonation from dyz orbital (HOMO−1) of Fe−1 into vacant degenerate π* orbital LUMO′ 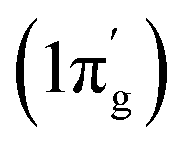 of N2. The three π-backdonations together contribute 81.2% of the total orbital contributions. The fourth orbital term (ΔEorb4) represents the σ electron donation (Fe ← N2) from HOMO (3σg+) of N2 into vacant d-type orbital (LUMO) of Fe and contributes 12.1% to the total orbital interactions (Fig. 3). It is to be observed that due to the mixing of orbitals of cAAC ligand, the shapes of d orbitals of Fe are slightly deformed and can be seen from the associated molecular orbitals of [(cAACMe)2Fe] fragment in Fig. 2 and 3. The mixing of the pz-orbital of cAAC ligands with d-orbitals of Fe-centre of 1 is much higher than that of 2.
of N2. The three π-backdonations together contribute 81.2% of the total orbital contributions. The fourth orbital term (ΔEorb4) represents the σ electron donation (Fe ← N2) from HOMO (3σg+) of N2 into vacant d-type orbital (LUMO) of Fe and contributes 12.1% to the total orbital interactions (Fig. 3). It is to be observed that due to the mixing of orbitals of cAAC ligand, the shapes of d orbitals of Fe are slightly deformed and can be seen from the associated molecular orbitals of [(cAACMe)2Fe] fragment in Fig. 2 and 3. The mixing of the pz-orbital of cAAC ligands with d-orbitals of Fe-centre of 1 is much higher than that of 2.
Overall, Fe → N2 π-backdonations are stronger than Fe ← N2 σ-donations in both complexes 1 and 2. The percentage of Fe → N2 π-backdonation in 2 is nearly one and half times higher than that of 1 and Fe ← N2 σ-donation in 2 is over nearly four times lower than that of 1. A close look at the deformation densities (Fig. 2 and 3) in 1–2 suggests that electronic effect of cAAC ligands is much lower in 2 than in 1 during the formations of σ- and π-bonds. The matrix isolated triplet M(N2)8 (M = Ca, Sr, Ba) species are also mainly stabilized by [M(dπ)] → (N2)8 π-backdonation.42 The EDA-NOCV results, in particular, the σ-donation and π-backdonations and the ellipticity values of QTAIM agrees well with each other and ascertain the speculation of the authors that “the ability of [(CAAC)2Fe]/[(CAAC)2Fe(N2)] to perform nitrogen fixation may arise from the relative flexibility of the system, which is capable of switching between two- and three-coordinate geometries, and allows the formation of highly covalent Fe–N2 multiple-bond interactions”.41
In conclusion, although a plethora of iron complexes are shown to bind N2 molecules in past. The nature of the bonding interactions between Fe-centre and N2 molecule has not been studied by EDA-NOCV. This study for the first time has provided a quantitative and detailed illustration orbital interactions to shed light on the engrossing Fe–N2 bond. The bonding interactions between (L)2Fen (n = 0, −1) and N2 fragments of two low coordinate and low valence Fe-complexes have been studied by DFT, NBO, QTAIM and EDA-NOCV analyses which revealed that Fe → N2 π-backdonations are major interactions for efficient N2 binding. However, the N2 → Fe σ-donation contributions is not negligible in both the complexes. Fe0 center of (L)2Fe0 of 1 is a better σ-acceptor than Fe−1 of 2 while Fe−1 center of (L)2Fe−1 of 2 is a much stronger π-backdonor due to its richness of electron densities in the latter. The Fe–N2 interaction energy of 1 is significantly higher than that of 2. These two Fe-complexes are an unprecedented set of complexes among the N2-bonded Fe-complexes18–21,27–33,40,41 which have been studied by EDA-NOCV calculations. The role of cAAC has been clearly shown by the deformation densities during N2 binding at Fe-centre (charge flow from red → blue). Our EDA-NOCV analysis will help the synthetic chemists to have much clearer view/understanding on the bonding interactions of captivating Fe–N2 bond and design a superior metal-complex for efficient N2 binding in their future studies.
Computational methods
Geometry optimizations and vibrational frequencies calculations of (cAAC)2Fe–N2 as neutral (1) and anionic complexes (2) in singlet, doublet, triplet and quintet electronic states has been carried out at the BP86-D3(BJ)/Def2TZVPP, for triplet and doublet states additionally at the TPSS-D3(BJ)/Def2TZVPP and PW6B95-D3/Def2TZVPP level49 in gas phase. The absence of imaginary frequencies assures the minima on potential energy surface. We have also optimized complex 1 in diethyl ether solvent using CPCM solvation model.49 All the calculations have been performed using Gaussian 16 program package.50 NBO51 calculations have been performed using NBO 6.0 (ref. 52) program to evaluate partial charges, Wiberg bond indices (WBI)53 and natural bond orbitals. The nature of Fe–N2 bonds in complexes 1 and 2 were analyzed by energy decomposition analysis (EDA)54 coupled with natural orbital for chemical valence (NOCV)55 using ADF 2018.105 program package.56 EDA-NOCV calculations were carried out at the BP86-D3(BJ)/TZ2P57 level using the geometries optimized at BP86-D3(BJ)/def2-TZVPP level. EDA-NOCV method involves the decomposition of the intrinsic interaction energy (ΔEint) between two fragments into four energy components as follows:| ΔEint = ΔEelstat + ΔEPauli + ΔEorb + ΔEdisp, | (1) |
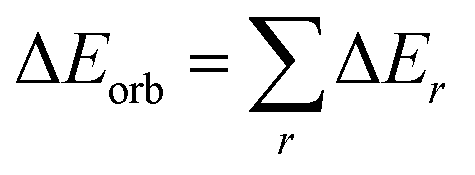 | (2) |
The combined EDA-NOCV method is able to partition the total orbital interactions into pairwise contributions of the orbital interactions which is important in providing a complete picture of the bonding. The charge deformation Δρk(r), which comes from the mixing of the orbital pairs ψk(r) and ψ−k(r) of the interacting fragments, gives the magnitude and the shape of the charge flow due to the orbital interactions (eqn (3)), and the associated orbital energy ΔEorb presents the amount of orbital energy coming from such interaction (eqn (4)).
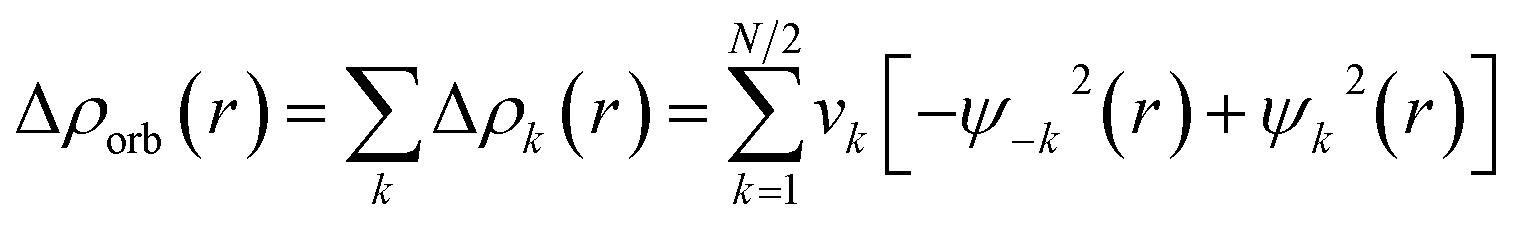 | (3) |
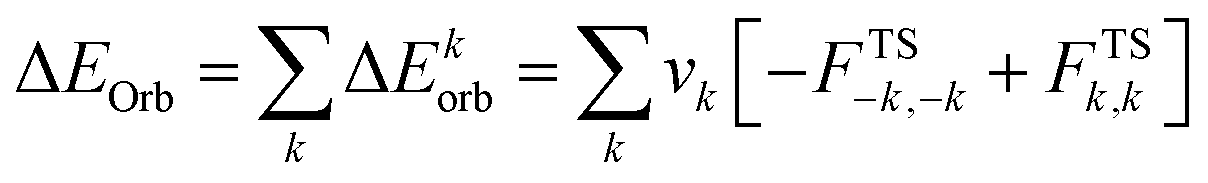 | (4) |
Readers are further referred to the recent reviews articles to know more about the EDA-NOCV method and its applications.48
Conflicts of interest
Authors do not have any conflict of interest.Acknowledgements
We thank Prof. Gernot Frenking and Prof. K. M. S for providing computational facilities. S. M. also thank Dr S. Pan. S. M. thanks CSIR for SRF. K. C. M thanks SERB for the ECR grant (ECR/2016/000890) and IIT madras for seed grant.References
- J. Wrigglesworth, Energy And Life, CRC Press, 1997, ISBN no. 9780748404339 Search PubMed.
- Exobiology: Matter, Energy, and Information in the Origin and Evolution of Life in the Universe, ed. J. Chela-Flores and F. Raulin, 1998, ISBN no. 978-94-011-5056-9 Search PubMed.
- P. Holland, Chem. Rev., 2020, 120, 4919 CrossRef CAS PubMed.
- N. Lehnert, B. W. Musselman and L. C. Seefeldt, Chem. Soc. Rev., 2021, 50, 3640 RSC.
- N. Lehnert, G. Coruzzi, E. Hegg, L. Seefeldt and L. Stein, NSF Workshop Report: Feeding the World in the 21st Century: Grand Challenges in the Nitrogen Cycle, National Science Foundation, Arlington, VA, Arlington, VA, 2016 Search PubMed.
- V. Smil, Enriching the Earth: Fritz Haber, Carl Bosch and the Transformation of World Food Production, MIT Press, Cambridge, 2001 Search PubMed.
- B. M. Hoffman, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2009, 42, 609 CrossRef CAS PubMed.
- J. L. Crossland and D. R. Tyler, Coord. Chem. Rev., 2010, 254, 1883 CrossRef CAS.
- M. A. Zeder, Curr. Anthropol., 2011, 52, S221–S235 CrossRef.
- M. Appl, "Ammonia", Ullmann's Encyclopedia of Industrial Chemistry, Weinheim, Wiley-VCH, 2006, DOI:10.1002/14356007.a02_143.pub2.
- A. Mittasch, Bemerkungen zur Katalyse, Ber. Dtsch. Chem. Ges. A and B Series, 1926, 59, 13–36 CrossRef.
- P. H. Pfromm, J. Renewable Sustainable Energy, 2017, 9, 034702 CrossRef.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, 2nd edn, 1997, pp. 1075–79, Butterworth-Heinemann, ISBN no. 9780750633659 Search PubMed.
- F. Bozso, G. Ertl, M. Grunze and M. Weiss, J. Catal., 1977, 49, 18 CrossRef CAS.
- G. Ertl, M. Weiss and S. B. Lee, Chem. Phys. Lett., 1979, 60, 391 CrossRef CAS.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331 RSC.
- S. D. Minteer, P. Christopher and S. Linic, ACS Energy Lett., 2019, 4, 163–166 CrossRef CAS.
- B. D. Matson and J. C. Peters, ACS Catal., 2018, 8, 1448 CrossRef CAS PubMed.
- N. B. Thompson, M. T. Green and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 15312 CrossRef CAS PubMed.
- Y. Lee, F. T. Sloane, G. Blondin, K. A. Abboud, R. García-Serres and L. J. Murray, Angew. Chem., Int. Ed., 2015, 54, 1499 CrossRef CAS PubMed.
- I. Čorić and P. L. Holland, J. Am. Chem. Soc., 2016, 138, 7200 CrossRef PubMed.
- L. A. Wickramasinghe, T. Ogawa, R. R. Schrock and P. Müller, J. Am. Chem. Soc., 2017, 139, 9132 CrossRef CAS PubMed.
- J. Fajardo Jr and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 16105 CrossRef PubMed.
- B. M. Lindley, R. S. van Alten, M. Finger, F. Schendzielorz, C. Würtele, A. J. M. Miller, I. Siewert and S. Schneider, J. Am. Chem. Soc., 2018, 140, 7922 CrossRef CAS PubMed.
- Y. Sekiguchi, K. Arashiba, H. Tanaka, A. Eizawa, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2018, 57, 9064 CrossRef CAS PubMed.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496 CrossRef CAS PubMed.
- I. Čorić, B. Q. Mercado, E. Bill, D. J. Vinyard and P. L. Holland, Nature, 2015, 526, 96 CrossRef PubMed.
- J. Rittle and J. C. Peters, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15898 CrossRef CAS PubMed.
- Q. J. Bruch, G. P. Connor, N. D. McMillion, A. S. Goldman, F. Hasanayn, P. L. Holland and A. J. M. Miller, ACS Catal., 2020, 10, 10826 CrossRef CAS.
- N. P. Mankad, M. T. Whited and J. C. Peters, Angew. Chem., Int. Ed., 2007, 46, 5768 CrossRef CAS PubMed.
- Y. Nishibayashi, Transition Metal-Dinitrogen Complexes: Preparation and Reactivity, Wiley-VCH Verlag GmbH & Co. KGaA, 2019, DOI: DOI:10.1002/9783527344260, First published: 25 January 2019, ISBN: 9783527344260.
- Y. Lee, N. P. Mankad and J. C. Peters, Proc. Natl. Acad. Sci. U. S. A., 2010, 2, 558 CAS.
- S. E. Creutz and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 1105 CrossRef CAS PubMed.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896 CrossRef PubMed.
- M. A. Legare, G. Belanger-Chabot, M. Rang, R. D. Dewhurst, I. Krummenacher, R. Bertermann and H. Braunschweig, Nat. Chem., 2020, 12, 1076 CrossRef CAS PubMed.
- D. L. J. Broere and P. L. Holland, Science, 2018, 359, 871 CrossRef CAS PubMed.
- T. A. Bazhenova and A. E. Shilov, Coord. Chem. Rev., 1995, 144, 69 CrossRef CAS.
- S. Kuriyama, K. Arashiba, K. Nakajima, Y. Matsuo, H. Tanaka, K. Ishii, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2016, 7, 12181 CrossRef CAS PubMed.
- M. Iwamoto, M. Akiyama, K. Aihara and T. Deguchi, ACS Catal., 2017, 7, 6924 CrossRef CAS.
- P. J. Hill, L. R. Doyle, A. D. Crawford, W. K. Myers and A. E. Ashley, J. Am. Chem. Soc., 2016, 138, 13521 CrossRef CAS PubMed.
- G. Ung and J. C. Peters, Angew. Chem., Int. Ed., 2015, 54, 532 CAS.
- Q. Wang, S. Pan, S. Lei, J. Jin, G. Deng, G. Wang, L. Zhao, M. Zhou and G. Frenking, Nat. Commun., 2019, 10, 3375 CrossRef PubMed.
- L. Zhao, S. Pan, N. Holzmann, P. Schwerdtfeger and G. Frenking, Chem. Rev., 2019, 119, 8781 CrossRef CAS PubMed.
- A. Krapp, F. M. Bickelhaupt and G. Frenking, Chem.–Eur. J., 2006, 12, 9196 CrossRef CAS PubMed.
- G. Ung, J. Rittle, M. Soleilhavoup, G. Bertrand and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53, 8427 CrossRef CAS PubMed.
- (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 CrossRef CAS PubMed; (b) S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357 CrossRef CAS PubMed; (c) K. C. Mondal, S. Roy and H. W. Roesky, Chem. Soc. Rev., 2016, 45, 1080 RSC.
- C. F. Matta and R. J. Boyd, The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, An Introduction to the Quantum Theory of Atoms in Molecules, Wiley, Hoboken, 2007, ch. 1 Search PubMed.
- (a) G. Frenking and F. M. Bickelhaupt, The Chemical Bond 1. Fundamental Aspects of Chemical Bonding, chap. The EDA Perspective of Chemical Bonding, Wiley-VCH, Weinheim, 2014, vol. 121 CrossRef; (b) L. M. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1345 Search PubMed; (c) L. Zhao, M. Hermann, W. H. E. Schwarz and G. Frenking, Nat. Rev. Chem., 2019, 3, 48 CrossRef CAS; (d) S. Pan and G. Frenking, Angew. Chem., Int. Ed., 2020, 59, 8756 CrossRef CAS PubMed; (e) J. Andrés, P. W. Ayers, R. A. Boto, R. Carbó-Dorca, H. Chermette, J. Cioslowski, J. Contreras-García, D. L. Cooper, G. Frenking, C. Gatti, F. Heidar-Zadeh, L. Joubert, Á. M. Pendás, E. Matito, I. Mayer, A. J. Misquitta, Y. Mo, J. Pilmé, P. L. A. Popelier, M. Rahm, E. Ramos-Cordoba, P. Salvador, W. H. E. Schwarz, S. Shahbazian, B. Silvi, M. Solà, K. Szalewicz, V. Tognetti, F. Weinhold and É. L. Zins, J. Comput. Chem., 2019, 40, 2248 CrossRef PubMed; (f) W. Yang, K. E. Krantz, L. A. Freeman, D. Dickie, A. Molino, G. Frenking, S. Pan, D. J. D. Wilson and R. J. Gilliard Jr, Angew. Chem., Int. Ed., 2020, 59, 3850 CrossRef CAS PubMed; (g) S. M. N. V. T. Gorantla, P. Parameswaran and K. C. Mondal, J. Comput. Chem., 2021, 42, 1159 CrossRef CAS PubMed; (h) S. M. N. V. T. Gorantla, S. Pan, K. C. Mondal and G. Frenking, Chem.–Eur. J., 2020, 26, 14211 CrossRef CAS PubMed; (i) L. Zhao, S. Pan, M. Zhou and G. Frenking, Science, 2019, 365, eaay5021 CrossRef CAS PubMed; (j) R. Saha, S. Pan, P. K. Chattaraj and G. Merino, Dalton Trans., 2020, 49, 1056 RSC.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef PubMed; (c) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed; (d) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (e) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (f) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC; (g) J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed; (h) Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2008, 4, 1849 CrossRef CAS PubMed; (i) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- (a) F. Weinhold and C. Landis, Valency and Bonding, A Natural Bond Orbital Donor – Acceptor Perspective, Cambridge University Press, Cambridge, 2005 Search PubMed; (b) C. R. Landis and F. Weinhold, The NBO View of Chemical Bonding, in The Chemical Bond: Fundamental Aspects of Chemical Bonding, ed. G. Frenking and S. Shaik, Wiley, 2014, pp. 91–120 Search PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429 CrossRef CAS PubMed.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1 CrossRef CAS.
- (a) M. Mitoraj and A. Michalak, Organometallics, 2007, 26, 6576 CrossRef CAS; (b) M. Mitoraj and A. Michalak, J. Mol. Model., 2008, 14, 681 CrossRef CAS PubMed.
- (a) ADF2017, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com Search PubMed; (b) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS.
- (a) E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142 CrossRef CAS PubMed; (b) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597 CrossRef CAS; (c) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables, figures, QTAIM, and optimized coordinates. See DOI: 10.1039/d1ra08348a |
| This journal is © The Royal Society of Chemistry 2022 |