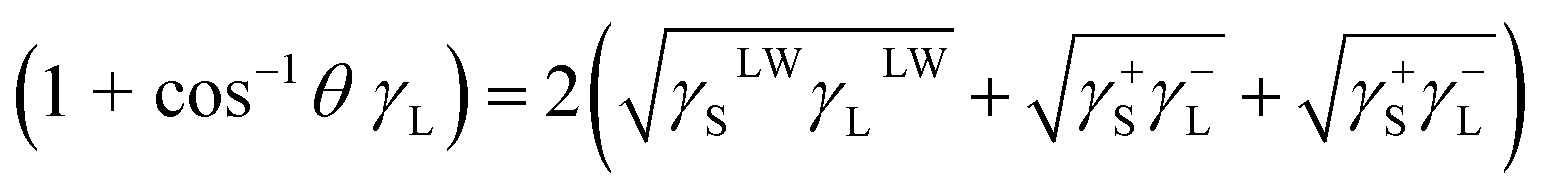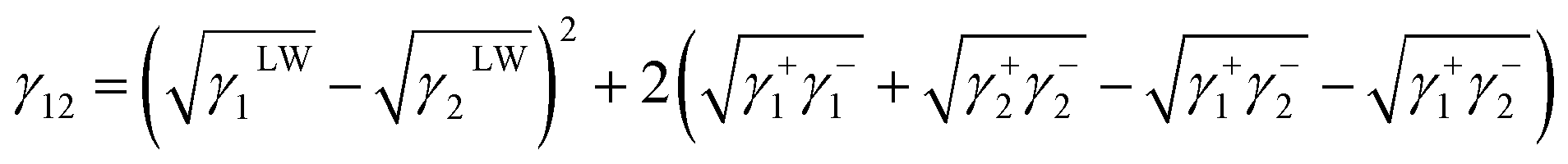DOI:
10.1039/D1RA07887A
(Paper)
RSC Adv., 2022,
12, 3454-3464
Tannic acid induces dentin biomineralization by crosslinking and surface modification†
Received
26th October 2021
, Accepted 16th January 2022
First published on 26th January 2022
Abstract
It is currently known that crosslinking agents can effectively improve the mechanical properties of dentin by crosslinking type I collagen. However, few scholars have focused on the influence of crosslinking agents on the collagen-mineral interface after crosslinking. Analysis of the Fourier transform infrared spectroscopy (FTIR) results showed that hydrogen bonding occurs between the tannic acid (TA) molecule and the collagen. The crosslinking degree of TA to collagen reached a maximum 41.28 ± 1.52. This study used TA crosslinked collagen fibers to successfully induce dentin biomineralization, and the complete remineralization was achieved within 4 days. The crosslinking effect of TA can improve the mechanical properties and anti-enzyme properties of dentin. The elastic modulus (mean and standard deviation) and hardness values of the remineralized dentin pretreated with TA reached 19.1 ± 1.12 GPa and 0.68 ± 0.06 GPa, respectively, which were close to those of healthy dentin measurements, but significantly higher than those of dentin without crosslinking (8.91 ± 1.82 GPa and 0.16 ± 0.01 GPa). The interface energy between the surface of collagen fibers and minerals decreased from 10.59 mJ m−2 to 4.19 mJ m−2 with the influence of TA. The current work reveals the importance of tannic acid crosslinking for dentin remineralization while providing profound insights into the interfacial control of biomolecules in collagen mineralization.
1. Introduction
As calcified hard tissues, dentin presents an intricate composition comprised of 70% (by weight) mineral, 20% organic matrix, and 10% water.1,2 Type I collagen fibers account for 90% of the organic matrix and are arranged into highly organized hierarchical structures. As a template for hydroxyapatite (HAP),3 type I collagen can have a number of structural roles, such as forming a strong and elastic mineralized scaffold to direct the nucleation and growth of crystals.4 The HAP mineral is embedded in the collagen and oriented along its longitudinal axis, which endows the hard tissue with excellent mechanical properties.5,6 Generally, due to the lack of self-repair ability, in the environment where cariogenic bacteria appear, hard tissues are easily attacked by bacteria-derived enzymes, and mineral loss leads to irreversible dentin destruction.7,8 Biomineralization is an approach that imitates natural biomineralization. Current research shows that through intrafibrillar mineralization, bare collagen fibers can be better protected, and excellent mechanical properties can be achieved.9,10 Novel remineralization methods are accepted for clinical use and show great promise for the future.11
Dentin biomineralization is a process completed under the regulation and induction of noncollagenous proteins (NCPs), known as nonclassical particle-mediated crystallization pathways.12,13 The pathways and mechanisms by which the amorphous phase mediates crystallization are the basis for dentin biomineralization.14 Amorphous calcium phosphate (ACP) was found to be the precursor phase that mediated crystallization pathways; precursor mineral phases exist in the liquid-like state and remain well hydrated.15,16 Charged polymers mimicking NCPs may make ACP precursors liquid-like and highly charged, which ultimately enables ACP precursors to enter collagen fibrils.17,18 In particular, recent studies have found that adjusting the collagen–mineral interface can accelerate the accumulation of minerals and reduce the duration of collagen mineralization. Shao and coworkers have reported that citrate molecules promote the binding of ACP to collagen by increasing wetting, thereby changing the properties of the mineralized interface.19 In addition, we found that as a surface modifier, polydopamine can make it easier for ACP to enter collagen fibrils and simultaneously reduce local heterogeneous nucleation barriers.20
However, so far, the process of remineralization remains too time-consuming for applications. It has been shown that host matrix metalloproteinases (MMPs) derived from dentin or saliva are readily capable of degrading the dentin matrix, which has been previously demineralized.21 Therefore, it is extremely important to provide a more efficient demineralized dentin collagen. The use of exogenous collagen crosslinkers induces further crosslinking of collagen and inhibits collagen degradation.22 Meanwhile, a more stable and dense collagen fiber network can effectively maintain the original minerals and promote collagen remineralization.23,24
Tannic acid (TA) is a plant-derived polyphenol composed of a complex mixture of polygalloyl glucose esters that has biological stability and high crosslinking potential for collagen.25 Bedran et al. found that the formation of hydrogen bonds between the amide NH group of collagen and hydroxyl group of TA enhanced the mechanical properties of dentin.26 In addition, as a novel desensitizer, the pyrogallol groups of TA combine with calcium ions to promote the mineral nucleation of dentin collagen.27
In this paper, we report that TA induces biomineralization of dentin by dual effects. The first objective of this study was to demonstrate that the TA modification of demineralized dentin collagen matrix is resistant to enzymatic degradation of collagenase and restores the biomechanical properties of the tissue. The second objective was to investigate the mechanism of promoting mineralization by inducing the intrafibrillar mineralization of dentin collagen crosslinked by TA.
2. Experimental methods
2.1 Material and reagents
Thymol, hexamethyldisilazane, phosphoric acid, CaCl2, Na2HPO4, poly-acrylic acid (PAA average Mw = 1800), NaCl, Tris, NaN3 and bacterial collagenase from Clostridium histolyticum (type I, ≥125 CDU per mg solid) were purchased form Sigma-Aldrich, USA. Fifty wt% of glutaraldehyde, TA, HCl, NaOH, ninhydrin were obtained from Aladdin, Shanghai, China. A hydroxyproline (HYP) detection kit was obtained from Jiancheng, Nanjing, China. Hank's Balanced Salt Solution (HBSS) was purchased from Solarbio, Beijing, China.
2.2 Specimen preparation
2.2.1 Preparation of collagen material and demineralized dentin. Assembling solutions (50 × 10−3 M glycine and 200 M KCl; the pH was preadjusted to 9.2 with 1.0 M NaOH) was prepared according to the method of Shao.19 Three microliters of the assembled collagen solution (0.2 g L−1) was used to prepare the collagen-loaded transmission electron microscopy (TEM) grids (n = 60). Collagen membranes were fabricated per the previous protocol.28 Two hundred milliliters (3 mg mL−1) of type I collagen stock was dripped on a hydrophobic membrane. Collagen membranes were neutralized with ammonia diffusion for 2 hours, and incubated at 37 °C for 20 hours. Then, the collagen membranes were rinsed with a large amount of deionized water until the pH of the supernatant was neutral. The 60 prepared collagen membranes were randomly divided into 3 groups for Fourier transform infrared spectroscopy (FTIR) detection (n = 20), mineralization testing (n = 20) and contact angle measurement experiments (n = 40).All experiments were performed in accordance with“international Ethical Guideline and Helsinki Declaration” and “national Ethics Censorship of Biomedical Research Involving Human Subject”, and were approved by the ethics committee of the First Affiliated Hospital of Zhejiang University School of Medicine. Informed consents were obtained from human participants of this study. We collected freshly extracted human non-caries third molars. An ultramicrotome (Boeckeler Instruments, Tucson, USA) was used to remove the crown enamel, and then the dentin was cut into discs approximately 1 mm thick below the enamel-dentinal Junction (EDJ). During the experiment, the prepared dentin was made into dentin slices with dimensions of 5 mm × 5 mm × 1 mm (n = 60) and dentin strips with dimensions of 10 mm × 0.8 mm × 0.8 mm (n = 168), and the dentin was polished using 600-, 1200- and 2000-grit silicon carbide papers, respectively. The dentin sample was immersed in 37% phosphoric acid for 15 s and rinsed with deionized water for 1 min to produce a 3–5 μm thick artificial demineralized dentin layer.
2.2.2 Specimen pre-treatment. Previous studies have shown that the optimal collagen performance when using 1 × 10−3 M TA.29 In this experiment, we used three distilled water samples to prepare a TA solution with a concentration of 1 × 10−3 M as a biomodifier for collagen. TEM grid-loaded collagen fibrils, collagen membranes and demineralized dentin were randomly divided into two groups: an experimental group and a control group. The experimental group and the control group were immersed in 1 × 10−3 M TA solution and deionized water for 2 hours at a constant temperature of 37 °C, and washed with deionized water three times.
2.2.3 Mineralization of collagen and dentin. The specific method of mineralizing liquid preparation was the same as in previous work.19 NaCl and Tris were used as buffers for preparing mineralized solutions. The calcium solution (20 mM CaCl2, 300 mM NaCl, and 100 mM Tris) was dripped into a Petri-dish, and then the designated amount of 10 g L−1 PAA stock solution was added into the calcium solution and mixed well. As a non-collagenous protein analog, PAA was added to this solution to stabilize ACP as nano-precursors. Twenty-five milliliters of phosphate-containing solution (0.12 M Na2HPO4) was slowly dropped into the same amount of calcium-containing solution, while using HCl solution and NaOH solution to adjust the pH to 7.4 ± 0.1. After mixing, the final concentrations of CaCl2, Na2HPO4, NaCl and PAA were 10.0 × 10−3 M, 6.0 × 10−3 M, 90 × 10−3 M, and 350 μg mL−1, respectively. NaN3 (0.02% (w/v)) was added to the mineralization solution to control the growth of microorganisms.The prepared solution was equally divided into two mineralized solutions for the experimental group and the control group. TEM-grid-loaded collagen fibrils were floated over the mineralization solution (partially demineralized dentin slices and collagen membranes were immersed in the mineralization solution) at 37 °C for a designated time. Remineralization was carried out at a constant temperature of 37 °C in an incubator. During the experiment of TEM-grid-loaded collagen fibrils and dentin, the remineralization solution was maintained, while the mineralization solution of the collagen membrane was changed after 5 days of mineralization. At designated time intervals (0 days, 2 days, and 4 days), 10 nickel meshes were removed from the mineralization solution of the control group and the TA group and sequentially washed with deionized water, 50% ethanol, and 100% ethanol. At the same time, 10 dentin slices were removed, washed with deionized water, dehydrated in 35%, 50%, 70%, 90% and 100% alcohol, and immersed in hexamethyldisilazane (HMDS) for 1 hour. Subsequently, each dentin slice was divided equally for TEM and scanning electron microscopy (SEM) observation. After 10 days of mineralization, the collagen membranes were taken out and washed three times with deionized water, and then freeze-dried for 24 hours.
2.3 Experimental part
2.3.1 Fourier transform infrared spectroscopy (FTIR). FTIR spectra were recorded for the collagen membranes without mineralization by an infrared spectrometer (Iraffinity-1, Shimadzu, Japan). The spectrum was obtained at 20 scans per film sample ranging from 4000 to 400 cm−1. Spectral data were analyzed with Origin 8.0 software.
2.3.2 Crosslinking degree. The crosslinking degree of the collagen membrane was measured by the ninhydrin method measuring free amino groups. During the experiment, the crosslinked collagen membrane was dried and weighed. Subsequently, the collagen membrane was heated with the ninhydrin solution for 15 minutes. The amount of free amino groups was then found from the optical absorbance of the solution at 570 nm using various known concentrations of glycine as a standard. Degree of crosslinking was calculated as follows:
| Crosslinking degree (%) = 100(Mo − Mt)/Mo |
where Mo is the amount of free amino groups in the non-crosslinked collagen membrane and Mt is the amount of amino groups remaining in the TA crosslinked collagen membrane.
2.3.3 Dentin collagen treated by collagenase. The HYP content in the solution was tested to evaluate the degree of dentin collagen degradation in each group. The demineralized dentin strips were randomly divided into four groups: A (demineralized dentin), B (dentin with TA cross-linking), C (dentin remineralization for 7 days), and D (dentin remineralization after TA crosslinking for 7 days). The experimental content and sample size are shown in Table 1. Each dentin strip was weighed before and after enzymatic degradation with an analytical balance (BS224S, Sartorius, China), and the percentage of mass loss of each dentin sample was calculated. Mass variation (wt%) was determined as the percentage of loss in mass for each specimen. The collagenase solution with a concentration of 0.1% (w/v) was composed of collagenase and Hank's Balanced Salt Solution (HBSS). The dentin strips in each group were immersed in collagenase solutions for 12 hours, 24 hours, 36 hours, 2 days, 3 days 4 days and 5 days of cumulative exposure time. The collagenase solution was changed every 12 hours for the first two days, and every 24 hours for the next 3 days. The HYP content in the supernatant was determined by the HYP assay kit (Table 1).
Table 1 Grouping and experimental content of enzymatic degradation
| Group (name, sample size) |
TA crosslinking |
Remineralization |
| A (demineralized dentin, n = 42) |
— |
— |
| B (dentin with TA crosslinking, n = 42) |
TA crosslinking for 2 hours |
— |
| C (dentin collagen without TA crosslinking and 7 days of remineralization, n = 42) |
— |
7 days of remineralization |
| D (dentin collagen with TA crosslinking and 7 days of remineralization, n = 42) |
TA crosslinking for 2 hours |
7 days of remineralization |
In addition, for monitoring the effect of TA on minerals, the remineralized dentin strips were demineralized with 1% ethylenediaminetetraacetic acid (EDTA) at pH 7.4 and room temperature.
2.3.4 Ultrastructure examination. Mineralized collagen fibrils were examined using TEM (HT-7700, Hitachi, Japan). Selected area electron diffraction (SAED) patterns and energy-dispersive spectroscope mapping were recorded using a high-resolution transmission electron microscope (HRTEM, JEM-2100F, JEOL, Japan). The ultrathin sections of calcified dentin disks were analyzed by TEM (JEM-1230, JEOL, Tokyo, Japan). The surface morphologies of the other half of the dentin disk and mineralized collagen membranes were examined using SEM (HITACHI, SU8010, Tokyo, Japan).
2.3.5 X-ray diffraction (XRD) characterization. X-ray diffraction was carried out on the remineralized dentin with and without TA crosslinking after 4 days of remineralization, and natural dentin and demineralized dentin were used as control groups. X-ray diffraction (XRD) analysis was carried out by a Rigaku Miniflex 600 diffractometer (Rigaku Corp, Tokyo, Japan) employing Cu K-α radiation at 40 kV and 30 mA. The data were collected in the 2θ range of 10–70° using 5° min−1 scan speed.
2.3.6 Mechanical properties. Whether to restore the hardness and modulus of dentin is the key to evaluating the success of dentin biomimetic mineralization. Therefore, we uesd nanoindentation was used by us to compare the mechanical properties of dentin by using a Nanoindenter G200 (Agilent, USA) and a Berko-vich diamond indenter (Agilent, USA). The nanoindentation tests were performed with the constant load method. Indentations were performed with a load of 10 mN and a time function of 10 s. Hardness and modulus of elasticity data were registered in GPa.
2.3.7 Thermo-gravimetric analysis (TGA). Thermal weight loss of mineralized collagen membranes and dentin powder were measured using a thermal gravimetric analyzer (SDT Q600, TA Instruments, USA) with a heating rate of 10 °C min−1 under a constant air flow rate of 100 mL min−1. Upon heating, mineralized collagen membrane specimens (3–5 mg) would proceed through two weight loss steps.
2.3.8 Contact angle measurement. The interface energy γ is measured using the static contact angle method developed by Owens. The contact angle is measured using the contact angle measuring instrument OCA15þ (Dataphysics, Germany). Contact angle (θ) measurements were performed with four different liquids (distilled water, ethylene glycol, n-octane and dimethyl sulfoxide) in the air-dried collagen membranes. Using the known γLLw, γL+ and γL− values, three times in the following equation (eqn (1)), γsLw, γs+ and γs− values for collagen and TA-collagen can be determined.| |
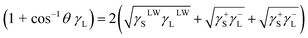 | (1) |
where L and S represent the liquid and collagen/TA-collagen, respectively. The interfacial energy between ACP and TA-collagen/collagen can be obtained as follows:| |
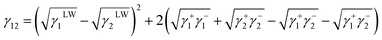 | (2) |
where 1 and 2 represent collagen and ACP, respectively.
3. Results and discussion
3.1 Analysis of TA crosslinking
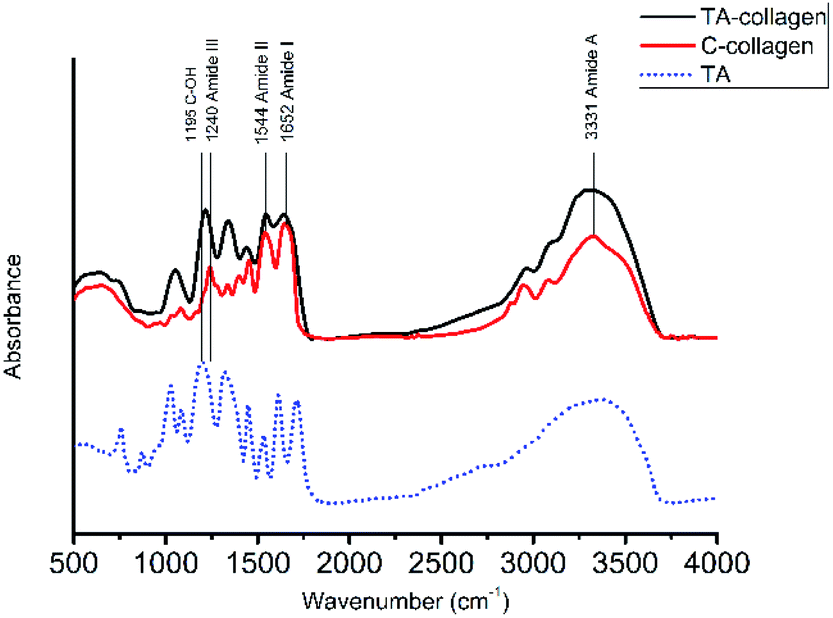
3.1.1 Fourier transform infrared spectroscopy. To investigate the strong interaction between collagen and TA, FTIR spectroscopy measurements of collagen, TA-collagen and TA were taken (Fig. 1), The characteristic absorption bands of 1655 cm−1, 1560 cm−1 and 1240 cm−1 are the FTIR spectra of amides I, II and III on collagen, respectively. These special peaks exist in the collagen after TA modification, indicating that the basic structure of collagen has not changed. The absorption peak of phenolic hydroxyl vibration due to TA binding to collagen was observed at 1217 cm−1.30 The main amide band characteristics of collagen peptide bonds are shown in Table 2. A slight redshift was shown in many characteristic absorption peaks assigned to TA-collagen, indicating that hydrogen bonding occurs between the TA molecule and the collagen. The frequency of stretching vibration may be weakened due to the formation of hydrogen bonds, reducing the electron cloud density. In addition, the observed shift of the amide III band stretching vibration to a lower wavenumber from 1240 to 1217 cm−1 and coincides with the peak position of the phenolic hydroxyl group of the TA. At the same time, the degree of crosslinking of the TA crosslinked collagen membrane has been calculated. According to the calculation formula, the crosslinking degree of the collagen membrane pretreated with TA was 41.28 ± 1.52, which proved that the type I collagen can be strongly crosslinked by TA.
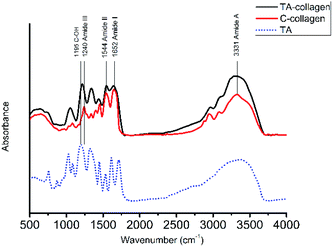 |
| | Fig. 1 FTIR for natural C-collagen (red line) and TA-collagen (black line) as well as for TA (blue dashed line). | |
Table 2 Band frequencies of C-collagen and TA-collagen
| Band |
Band frequency (cm−1) |
| C-collagen |
TA-collagen |
| Amide A |
3331 |
3317 |
| Amide I |
1652 |
1641 |
| Amide III |
1240 |
1217 |
| TA: C–OH |
— |
1195 |
3.1.2 Ultrastructural examination of collagen crosslinking. The ultrastructure of type I collagen fibrils exhibited the typical fibrillar structure on the TEM grids (Fig. 2a). The TEM images of type I collagen fibrils following treatment of TA showed that the collagen fibrils tended to aggregate, resulting in a reduction of interfibrillar spaces (Fig. 2c). From the SEM images, TA cross-linked self-assembly collagen membrane appeared to be more closely packed and had a dense and multiple network, when compared to the control group (Fig. 2d).
 |
| | Fig. 2 TEM images of the type I collagen fibrils without crosslinking (a) and with TA crosslinking (c). SEM images of the surface morphologies of self-assembly collagen without crosslinking (b) and with TA crosslinking (d). | |
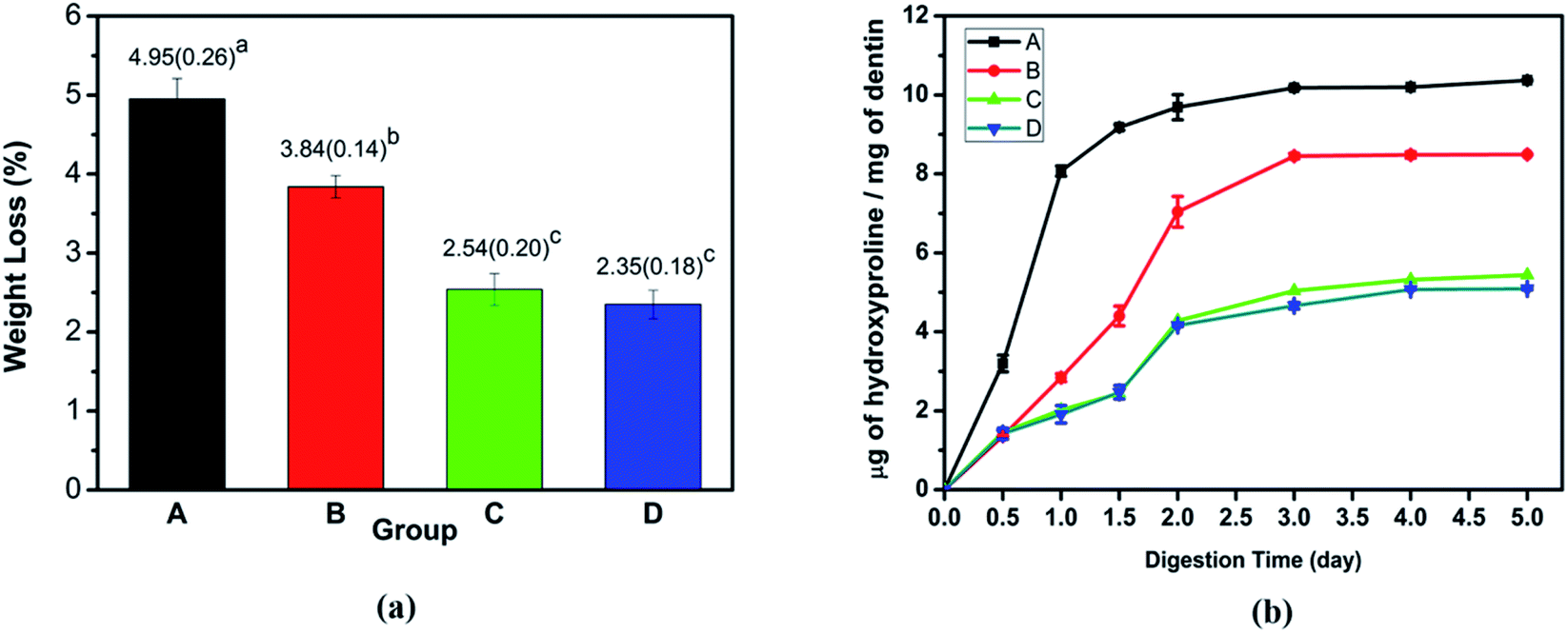
3.1.3 Dentin collagen treated by collagenase. After 5 days of collagenase, the collagen of demineralized dentin samples and TA crosslinked demineralized dentin revealed that the collagen fibers were sparsely arranged and the network structure collapsed (Fig. S1†). The demineralized dentin collagen matrix was almost completely destroyed, and the crosslinked dentin specimens showed that the well-formed collagen fibril structure could still be retained. However, for the TA-induced cross-linked collagen matrix, the collagen network exhibited a complete and dense collagen network structure (Fig. S1†). The results of the weight lost of the dentin dry weight after being placed in the collagenase solution for 5 days are shown in Fig. 3a. The mass loss of the TA crosslinked dentin group after demineralization was 3.84 ± 0.14%, which was significantly lower than that of the demineralized negative control group (4.95 ± 0.26%) (P < 0.05). After mineralization, the dentin weight loss of the two groups was much lower, and there was no significant difference in the weight lost of remineralized dentin in the presence or absence of TA crosslinking. The enzyme resistance of collagen was accurately identified by calculating the ratio of the HYP content in the digested supernatant to the mass loss of collagen matrix per mg dry weight. Compared with the demineralized dentin strips at each time point, TA crosslinking significantly reduced dentin HYP release (Fig. 3b). In addition, the HYP content in the mineralized dentin control group and the cross-linked group was significantly lower than that in the demineralized group, which was consistent with the dentin weight loss results of each group and consistent with the results of SEM observation results. In previous studies, it has been demonstrated that demineralization of calcium phosphate crystals and composites containing collagen can be performed by EDTA.31,32 Thermogravimetric analysis was performed on the mineral content of dentin after demineralization with 1% EDTA. There was no difference between the two groups after statistical analysis (p > 0.05) (Fig. S3†). Studies have shown that EDTA is a chelating agent that can demineralize dentin by combining with calcium ions of the dentine structure.32 Treatment with EDTA did not induce any effect on collagen conformation.33 Therefore, the mineralized dentin was demineralized by EDTA, and there was no significant difference in the remaining mineral weight between the dentin treated with TA and the control group. Due to the protective effect of TA on collagen, this result is different from collagenase treatment.
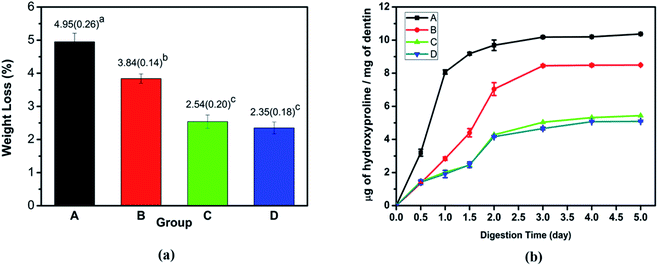 |
| | Fig. 3 Dentin collagen treated by collagenase: (a) the weight loss percentage and (b) the HYP content derived from the supernatant after 5 days of exposure. Group A: demineralized dentin, group B: dentin in the TA crosslinking, group C: dentin remineralization for 7 days, and group D: dentin remineralization after TA crosslinking for 7 days. | |
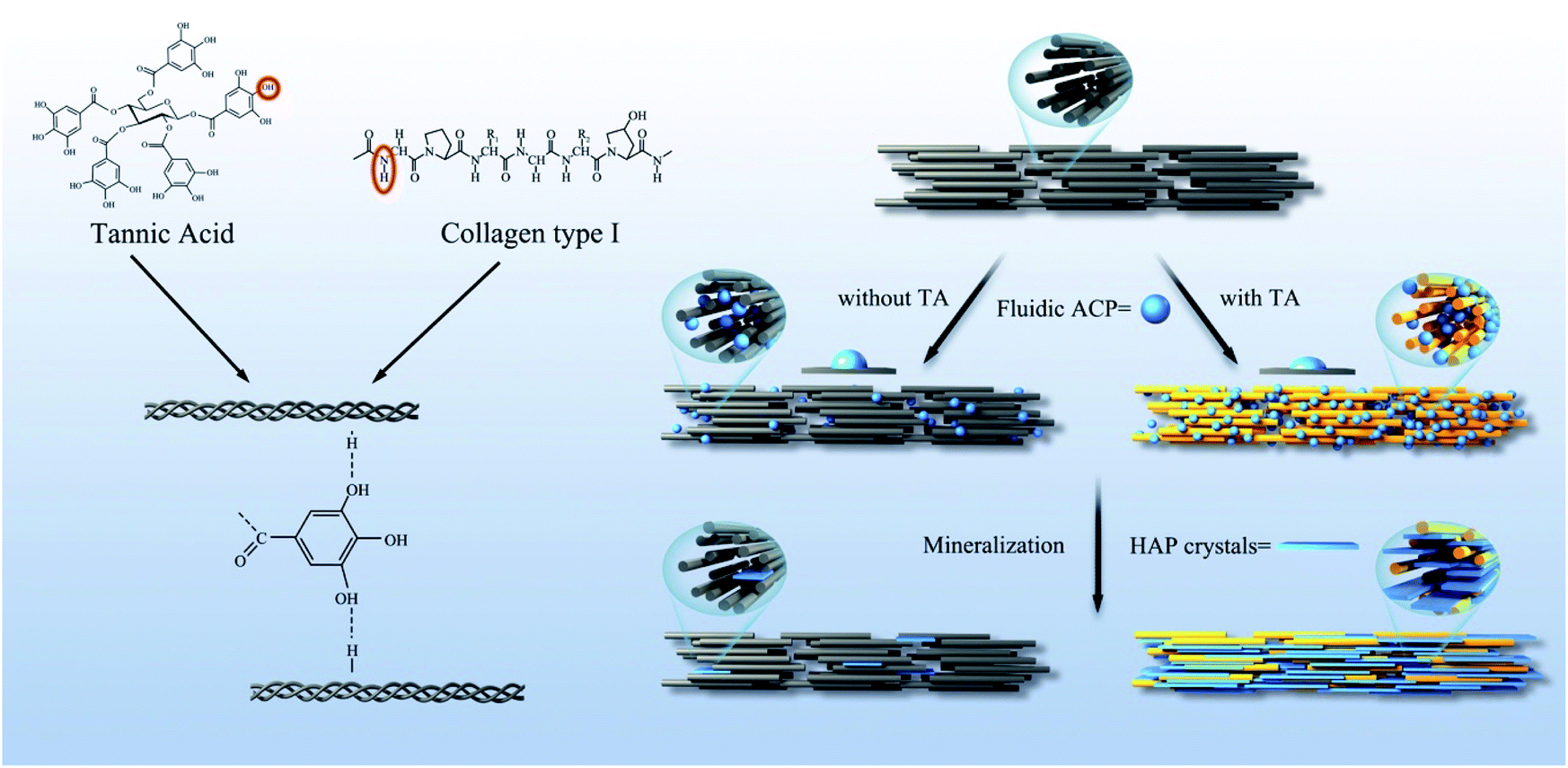
TA is a specific class of hydrolysable tannins, containing a penta galloyl glucose core, which has a five-arm polyphenol structure.34,35 The type I collagen which contains three repeating sequences of polypeptide α-chains, each consisting of more than 1000 amino acids. The interactions between proteins and polyphenols can involve hydrogen bonds, covalent bonds, ions, and hydrophobic bonds.36 A large number of phenolic hydroxyls in the polyphenol derivative crosslinker have a good modification effect on the collagen.37 TA crosslinked collagen mainly by noncovalent bonding under nonoxidized conditions.38 The H bond between amide NH and OH of the TA galloyl group was shown in Fig. 10.39
Analysis of the FTIR results confirmed that a slight redshift was shown in TA crosslinked collagen, indicating that hydrogen bond occurred between the TA molecule and the collagen. The absorption peak of phenolic hydroxyl vibration due to TA was observed. The crosslinking degree of the collagen membrane pretreated with TA reached a maximum 41.28 ± 1.52, which was also supported by the ultrastructure of type I collagen fibrils. The anti-enzymatic hydrolysis effect was tested on the crosslinked type I collagen of dentin. Therefore, TA can be used as a cross-linker in demineralized dentin collagen.
3.2 Influence of TA on mineralization
3.2.1 Mineralization of collagen materials. The mineralized C-collagen fibrils in the absence of TA pretreatment under TEM (Fig. 4a–d), which could identify the part of the high-density collagen-mineral composite structure and the low contrast region of unmineralized collagen. At the same time, according to the method of Smeets,40 the degree of mineralization is calculated to be 29.58 ± 3.82%. After TA-modified collagen fiber mineralization for 2 days, the mineralization degree of the TA group was 100%. The electron density of collagen fibers is significantly increased, consistent with the relatively uniform phosphate minerals in elemental mapping, in which the calcium and phosphorus elements overlap and are evenly distributed throughout the collagen fibers (Fig. 4e–h). The SAED date mode also confirmed that the mineral phase was HAP with specific diffusive rings corresponding to the (002), (211), and (004) diffractions (Fig. 4g). The SAED patterns in the (002), (211), and (004) diffraction planes indicated the presence of the HAP in the intrafibrillar region of collagen and were aligned orthogonally to the collagen c-axis.41
 |
| | Fig. 4 TEM images of the mouse tail type I collagen fibrils. (a) and (e) represent 2 days of mineralization of C-collagen and TA-collagen, respectively. (b) Enlarged image of C-collagen fibrils. (c) SAED pattern of b. (d) Element mapping of C-collagen fibrils. (f) Enlarged image of a TA-collagen fibril. (g) SAED patterns of panel f match those of HAP. (h) Element mapping analysis of TA-collagen fibrils. | |
As the in vivo mineralization of deposited crystals cannot be directly monitored, self-assembly collagen can be used as a model system to study the mineralization of dentin collagen fibers. The surface structure of the unmineralized collagen membrane was relatively smooth, showing a thin strip-like appearance. After mineralization, the collagen diameter was significantly thickened, and the rope-like structure was more pronounced after TA crosslinking. Energy dispersive X-ray spectrometer (EDS) analysis showed that obvious Ca and P signals appeared in the experimental group and control group after mineralization, and the Ca/P ratio was approximately 1.62, which proved that a large amount of HAP was compounded on the collagen after mineralization (Fig. S2†).
3.2.2 Remineralization of the dentin. Unlike self-assembled monolayer collagen fibers and reticulated collagen membranes, the dentin demineralization model is taken from the body, and the collagen environment can be further simulated. SEM results showed that the dentin collagen fibrils were completely exposed after 37% phosphoric acid treatment, and some collagen fiber networks collapsed due to a lack of mineral support (Fig. 5a and d). The TA-modified dentin was obviously changed into a rope-like structure and was strongly that is covered by the minerals after 2 days; coverage was more obvious after 4 days of mineralization (Fig. 5e and f). The acceleration of the mineralization process after TA treatment was strongly proven by the TEM characterization of the ultrathin section of dentin. After 2 days of mineralization, only partial remineralization of dentin without TA treatment was observed (Fig. 6b), while a relatively thick mineralization layer could be identified after four days of mineralization. The clear boundary was between the natural, partially mineralized and unrestored dentin layers (Fig. 6c). For the TA-modified dentin slices, most of the remineralization was achieved. After immersing the dentin slices in the remineralization solution for 2 days, a newly formed mineral layer of approximately 3–4 μm thick was observed (Fig. 6e), and the complete remineralization was achieved within 4 days (Fig. 6f). Therefore, TA treatment shortened the remineralization speed of dentin to 4 days, which greatly improves the possibility of clinical application of dentin restoration.
 |
| | Fig. 5 SEM images of the surface morphologies of remineralized dentin without (a)–(c) and with TA-pretreatment (d)–(f) for 0 day, 2 days, and 4 days. | |
 |
| | Fig. 6 TEM images of remineralized dentin without (a–c) and with TA-pretreatment (d–f) for 0 day, 2 days, and 4 days. N: natural dentin. R: remineralized dentin. | |
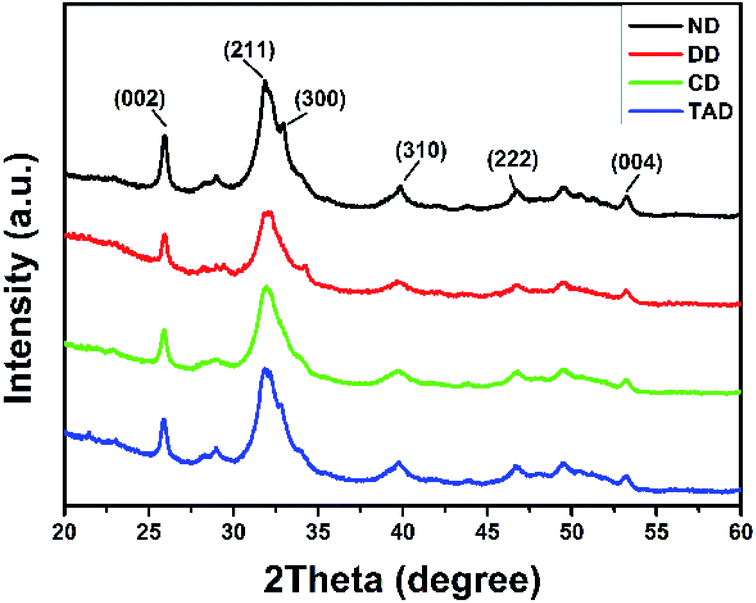
Fig. 7 showed the XRD results of the surface of the dentin disks. After demineralization, the characteristic HAP peaks decreased, and the low crystallinity comes from the incomplete demineralization of the dentin. After repair, significant characteristic HAP diffraction peaks were observed on surface of both remineralization dentin (C-dentin) and TA crosslinked remineralization dentin (TA-dentin) indicating that the precipitates were HAP. Especially, the XRD pattern of TA-dentin exhibited the characteristic HAP diffraction peaks (300) at 2θ = 33, which was not found in C-dentin.
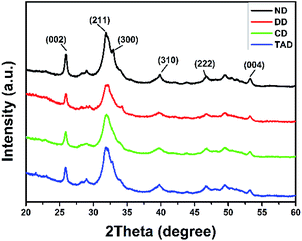 |
| | Fig. 7 XRD patterns of the dentin surface of natural dentin (black line), demineralized dentin (red line), C-dentin (green line), and the TA-dentin (blue line). | |
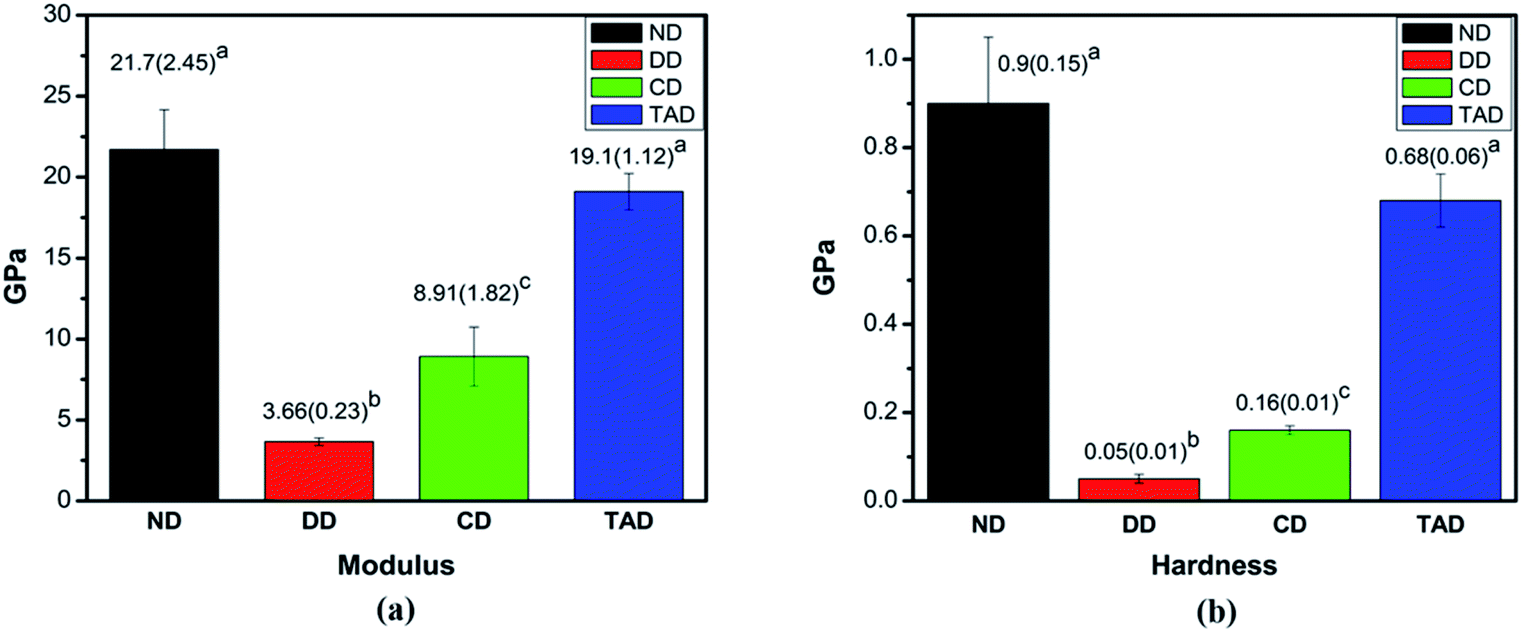
3.2.3 Mechanical properties. The typical elastic modulus and hardness of remineralized dentin for 4 days, natural dentin and control demineralized dentin are presented (Fig. 8). The attained elastic modulus (mean and standard deviation) and hardness (mean and standard deviation) for each group are displayed in Fig. 6a and b, respectively. The values of TA-dentin were 19.1 ± 1.12 GPa and 0.68 ± 0.06 GPa, respectively, which were close to those of healthy dentin measurements (21.7 ± 2.45 GPa and 0.9 ± 0.15 GPa) but significantly higher than those of C-dentin (8.91 ± 1.82 GPa and 0.16 ± 0.01 GPa). Statistical analysis depicted significant differences in nanomechanical properties between the 4 days of remineralization with TA dentin when compared among the other groups (P < 0.05), but no significant difference from healthy dentin. TA crosslinking of the dentin collagen offers similar mechanical properties as natural dentin, whereas the demineralization dentin and of C-dentin are clearly different. Clearly, the remineralization of the dentin crosslinked by TA essentially restores the elastic modulus and hardness of the demineralized dentin to normal.
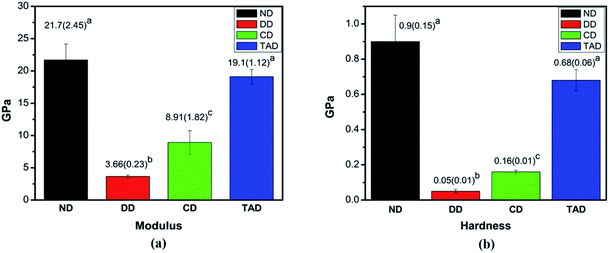 |
| | Fig. 8 Nanomechanical properties of dentin. (a) The elastic modulus of different types of dentin (GPa). (b) The hardness of different types of dentin (GPa). Abbreviations: ND: natural dentin, DD: demineralized dentin, CD: 4d remineralization of C-dentin, and TAD: 4 days remineralization of TA-dentin. | |
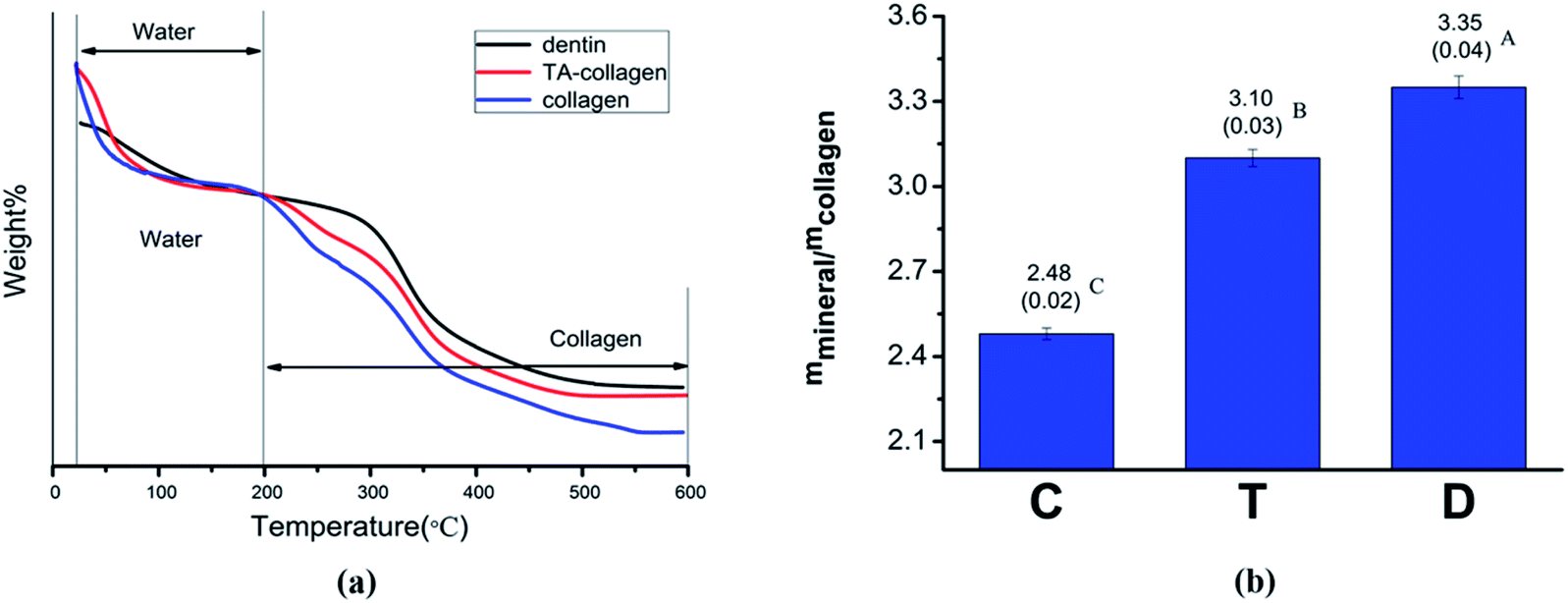
3.2.4 Thermogravimetric analysis. TGA curves of mineralized collagen membranes and dentin powder are shown in Fig. 9. During the temperature rise from 25 °C to 600 °C, the change in mineral weight was mainly divided into two steps (Fig. 9a). When the temperature rose from 0 to 200 °C, a rapid weight loss occurred due to water loss. In the second stage, 200–600 °C, weight loss was attributed to collagen. We believe that after the temperature reached 600 °C, the remaining substances are inorganic minerals. The TGA curve plotted using a temperature of 200 °C as a reference shows that the collagen membrane without TA treatment loses the most weight compared to the normal dentin and TA-crosslinked membrane (Fig. 9a). By calculating the relative mineral content of the collagen fibers (mineral/collagen weight ratio), the value of the TA-treated collagen group was significantly higher than that of the control group (Fig. 9b).2
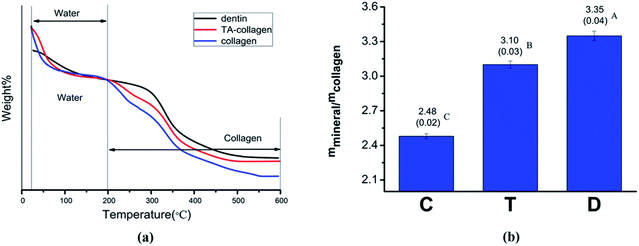 |
| | Fig. 9 (a) Thermogravimetric analysis of dentin, mineralized collagen and mineralized TA-collagen. (b) The mineral contents of collagen fibrils. C: control group mineralized collagen fibrils, T: mineralized collagen fibrils pretreated with TA, and D: dentin. IBM SPSS 24.0 software was used to perform the statistical analyses. There was a significant difference between C and T (P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) <0.01), and between T and D (P<0.01). <0.01), and between T and D (P<0.01). | |
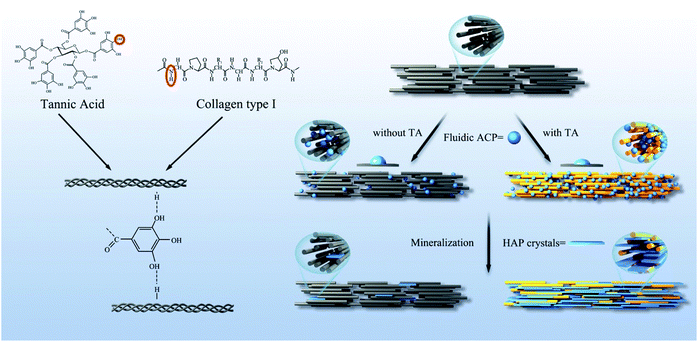 |
| | Fig. 10 Scheme of the crosslinking reaction mechanism between collagen and TA, and the process of collagen mineralization was promoted by TA. | |
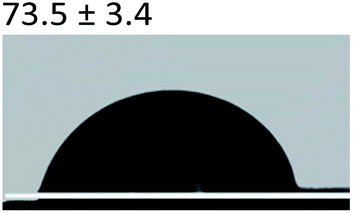
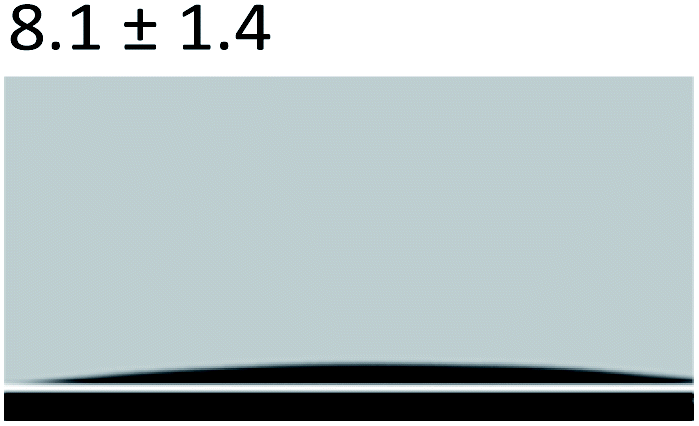
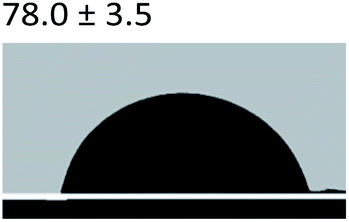
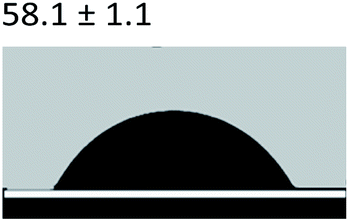
3.2.5 Contact angle measurement. A large amount of data indicates that biomineralization frequently occurs via amorphous precursor phases, which crystallize into the final stable biominerals.14,42 The heterogeneous nucleation process of biomineralization involves a minimum energy barrier through the intermediate mineralization pathway, which is significantly lower than direct nucleation. Studies have shown that additive molecules such as citrate or silicate can alter the ACP surface to promote phase transition.19,43 Therefore, reducing the nucleation energy barrier may be an effective means to promote mineralization. It was calculated that, as shown in Tables 3 and 4, the interfacial energy of the mineralization system after TA crosslinking decreased from 10.59 mJ m−2 to 4.19 mJ m−2. This means that when the interfacial energy between the ACP and the collagen is reduced, the contact angle between the two components is reduced and the wettability is increased, resulting in a lower heterogeneous nucleation barrier and accelerating phase transition.
Table 3 Content angle with the probing liquid
| Content angle of probing liquid (°) |
Distilled water |
Ethylene glycol |
N-Octane |
| C-collagen |
 |
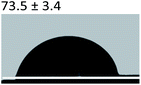 |
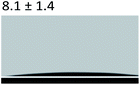 |
| TA-collagen |
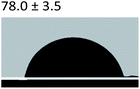 |
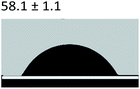 |
 |
Table 4 Surface tension components (mJ m−2) used in the calculations of interfacial energy between collagen and ACPγsLw, γs+ and γs−
| |
γ |
γLw |
γ+ |
γ− |
γij |
| C-collagen |
22.06 |
21.40 |
0.6 |
0.18 |
10.59 |
| TA-collagen |
28.53 |
21.62 |
1.04 |
11.49 |
4.19 |
According to the results, the mechanism of TA promoting remineralization in this study can be described as follows. The experiments demonstrated that the crosslinking of the collagen fibrils during the TA pretreatment, and the triple helix conformation of collagen was not destroyed. Collagen was considered as a template for the growth of mineral crystals. TA binds to the endogenous protease cleavage site and prevents enzyme substrate interactions, protecting the exposed collagen from solubilization by proteases. In addition, we confirmed that the interface energy between ACP and collagen was decreased after TA treatment, which facilitated the heterogeneous formation of ACP precursors on collagen. The lower interface promoted more ACP to penetrate into collagen fibers and subsequent intrafibrillar mineralization by a wetting effect. TA provided additional nucleation sites for calcium phosphate crystals. TA facilitated the mineralization of dentin by binding to calcium ions and accumulating them at the interface due to the presence of abundant phenolic hydroxyls, thereby accelerating the formation of HAP (Fig. 10).44
4. Conclusions
The biomimetic mineralization of dentin is rarely transformed into clinical application based on the slow restoration of the polymer-induced liquid-precursor (PILP) phase. The demineralized dentin collagen fiber network degrades when attacked by MMPs.45 This research demonstrates that the mechanical properties and enzymatic resistance of collagen improved after the dentin was treated with TA. In addition, the interfacial energy can be reduced, which makes it easier for the liquid nanoprecursor to enter the collagen fiber and promotes the mineralization process. Therefore, TA adjusts the mineralization interface through the wetting effect, this finding may provide unique insight for the crosslinking agent in promoting mineralization.
Author contributions
Weijing Kong and Qiaolin Du: methodology, investigation, writing – original draft,writing – review & editing. Yinan Qu: investigation, formal analysis. Changyu Shao: validation. Chaoqun Chen: software. Jian Sun: project administration. Caiyun Mao: funding acquisition. Ruikang Tang: resources. Xinhua Gu: conceptualization, supervision, funding acquisition, methodology, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge project funding provided by the Public Welfare Technology Application Research Project of Zhejiang Province (LGF18H140001), Natural Science Foundation of Zhejiang Province (LY20H140002), Medical and Health Science and Technology Project of Zhejiang Province (2018KY073, 2019KY383).
References
- A. Linde, Anat. Rec., 1989, 224, 154–166 CrossRef CAS PubMed.
- J. Ma, J. Wang, X. Ai and S. Zhang, Biotechnol. Adv., 2014, 32, 744–760 CrossRef CAS PubMed.
- K. Gelse, E. Poschl and T. Aigner, Adv. Drug Delivery Rev., 2003, 55, 1531–1546 CrossRef CAS PubMed.
- F. Nudelman, A. J. Lausch, N. A. J. M. Sommerdijk and E. D. Sone, J. Struct. Biol., 2013, 183, 258–269 CrossRef CAS PubMed.
- T. A. Ulrich, A. Jain, K. Tanner, J. L. MacKay and S. Kumar, Biomaterials, 2010, 31, 1875–1884 CrossRef CAS PubMed.
- J. B. Forien, C. Fleck, P. Cloetens, G. Duda, P. Fratzl, E. Zolotoyabko and P. Zaslansky, Nano Lett., 2015, 15, 3729–3734 CrossRef CAS PubMed.
- V. Hass, I. V. Luque-Martinez, M. F. Gutierrez, C. G. Moreira, V. B. Gotti, V. P. Feitosa, G. Koller, M. F. Otuki, A. D. Loguercio and A. Reis, Dent. Mater., 2016, 32, 732–741 CrossRef CAS PubMed.
- L. B. He, Y. Hao, L. Zhen, H. L. Liu, M. Y. Shao, X. Xu, K. N. Liang, Y. Gao, H. Yuan, J. S. Li, J. Y. Li, L. Cheng and C. van Loveren, J. Struct. Biol., 2019, 207, 115–122 CrossRef CAS PubMed.
- Y. Liu, L. Tjaderhane, L. Breschi, A. Mazzoni, N. Li, J. Mao, D. H. Pashley and F. R. Tay, J. Dent. Res., 2011, 90, 953–968 CrossRef CAS PubMed.
- L. F. Barbosa-Martins, J. P. Sousa, L. A. Alves, R. P. W. Davies and R. M. Puppin-Rontanti, Materials, 2018, 11 Search PubMed.
- J. D. Featherstone, M. Fontana and M. Wolff, J. Dent. Res., 2018, 97, 125–127 CrossRef CAS PubMed.
- A. George and A. Veis, Chem. Rev., 2008, 108, 4670–4693 CrossRef CAS PubMed.
- A. Rao, J. K. Berg, M. Kellermeier and D. Gebauer, Eur. J. Mineral., 2014, 26, 537–552 CrossRef CAS.
- W. J. Jin, S. Q. Jiang, H. H. Pan and R. K. Tang, Crystals, 2018, 8, 48 CrossRef.
- M. J. Olszta, D. J. Odom, E. P. Douglas and L. B. Gower, Connect. Tissue Res., 2003, 44(1), 326–334 CrossRef CAS PubMed.
- M. A. S. Melo, S. F. F. Guedes, H. H. K. Xu and L. K. A. Rodrigues, Trends Biotechnol., 2013, 31, 459–467 CrossRef CAS PubMed.
- M. J. Olszta, X. G. Cheng, S. S. Jee, R. Kumar, Y. Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., 2007, 58, 77–116 CrossRef.
- F. Nudelman, K. Pieterse, A. George, P. H. H. Bomans, H. Friedrich, L. J. Brylka, P. A. J. Hilbers, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS PubMed.
- C. Y. Shao, R. B. Zhao, S. Q. Jiang, S. S. Yao, Z. F. Wu, B. Jin, Y. L. Yang, H. H. Pan and R. K. Tang, Adv. Mater., 2018, 30 Search PubMed.
- Y. N. Qu, T. Y. Gu, Q. L. Du, C. Y. Shao, J. Wang, B. Jin, W. J. Kong, J. Sun, C. Q. Chen, H. H. Pan, R. K. Tang and X. H. Gu, ACS Biomater. Sci. Eng., 2020, 6, 3327–3334 CrossRef CAS PubMed.
- C. Chaussain-Miller, F. Fioretti, M. Goldberg and S. Menashi, J. Dent. Res., 2006, 85, 22–32 CrossRef CAS PubMed.
- T. R. Aguiar, C. M. P. Vidal, R. S. Phansalkar, I. Todorova, J. G. Napolitano, J. B. McAlpine, S. N. Chen, G. F. Pauli and A. K. Bedran-Russo, J. Dent. Res., 2014, 93, 417–422 CrossRef CAS PubMed.
- T. M. Du, X. F. Niu, Z. W. Li, P. Li, Q. L. Feng and Y. B. Fan, Int. J. Biol. Macromol., 2018, 113, 450–457 CrossRef CAS PubMed.
- C. M. P. Vidal, T. R. Aguiar, R. Phansalkar, J. B. McAlpine, J. G. Napolitano, S. N. Chen, L. S. N. Araujo, G. F. Pauli and A. Bedran-Russo, Acta Biomater., 2014, 10, 3288–3294 CrossRef CAS PubMed.
- A. C. Machado, E. Dezan Junior, J. E. Gomes-Filho, L. T. Cintra, D. B. Ruviere, R. Zoccal, C. A. Damante and E. G. Jardim Junior, J. Appl. Oral Sci., 2012, 20, 414–418 CrossRef PubMed.
- A. K. B. Bedran-Russo, K. J. Yoo, K. C. Ema and D. H. Pashley, J. Dent. Res., 2009, 88, 807–811 CrossRef CAS PubMed.
- D. X. Oh, E. Prajatelistia, S. W. Ju, H. J. Kim, S. J. Baek, H. J. Cha, S. H. Jun, J. S. Ahn and D. S. Hwang, Sci. Rep., 2015, 5, 10884 CrossRef CAS PubMed.
- J. Wang, Y. N. Qu, C. Q. Chen, J. Sun, H. H. Pan, C. Y. Shao, R. K. Tang and X. H. Gu, Mater. Sci. Eng., C, 2019, 104, 109959 CrossRef CAS PubMed.
- P. Velmurugan, E. R. A. Singam, J. R. Rao and V. Subramanian, Biopolymers, 2014, 101, 471–483 CrossRef CAS PubMed.
- M. Jastrzebska, J. Zalewska-Rejdak, R. Wrzalik, A. Kocot, I. Mroz, B. Barwinski, A. Turek and B. Cwalina, J. Biomed. Mater. Res., Part A, 2006, 78, 148–156 CrossRef CAS PubMed.
- O. I. Ulusoy and G. Gorgul, Aust Endod J., 2013, 39, 66–72 CrossRef PubMed.
- M. P. Gashti, M. Bourquin, M. Stir and J. Hulliger, J. Mater. Chem. B, 2013, 1, 1501–1508 RSC.
- M. G. Gandolfi, P. Taddei, A. Pondrelli, F. Zamparini, C. Prati and G. Spagnuolo, Materials, 2018, 12, 25 CrossRef PubMed.
- X. Zhang, M. D. Do, P. Casey, A. Sulistio, G. G. Qiao, L. Lundin, P. Lillford and S. Kosaraju, J. Agric. Food Chem., 2010, 58, 6809–6815 CrossRef CAS PubMed.
- J. Guo, W. Sun, J. P. Kim, X. Lu, Q. Li, M. Lin, O. Mrowczynski, E. B. Rizk, J. Cheng, G. Qian and J. Yang, Acta Biomater., 2018, 72, 35–44 CrossRef CAS PubMed.
- C. Simon, K. Barathieu, M. Laguerre, J. M. Schmitter, E. Fouquet, I. Pianet and E. J. Dufourc, Biochemistry, 2003, 42, 10385–10395 CrossRef CAS PubMed.
- S. Varma, J. P. Orgel and J. D. Schieber, Biophys. J., 2016, 111, 50–56 CrossRef CAS PubMed.
- L. Wu, H. Shao, Z. Fang, Y. Zhao, C. Y. Cao and Q. Li, ACS Biomater. Sci. Eng., 2019, 5, 4272–4284 CrossRef CAS PubMed.
- F. H. Heijmen, J. S. du Pont, E. Middelkoop, R. W. Kreis and M. J. Hoekstra, Biomaterials, 1997, 18, 749–754 CrossRef CAS PubMed.
- P. J. M. Smeets, A. R. Finney, W. Habraken, F. Nudelman, H. Friedrich, J. Laven, J. J. De Yoreo, P. M. Rodger and N. Sommerdijk, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7882–E7890 CrossRef CAS PubMed.
- W. Traub, T. Arad and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9822–9826 CrossRef CAS PubMed.
- S. S. Yao, B. A. Jin, Z. M. Liu, C. Y. Shao, R. B. Zhao, X. Y. Wang and R. K. Tang, Adv. Mater., 2017, 29 Search PubMed.
- Y. N. Wang, S. Q. Jiang, H. H. Pan and R. K. Tang, CrystEngComm, 2016, 18, 379–383 RSC.
- J. Guo, Y. Ping, H. Ejima, K. Alt, M. Meissner, J. J. Richardson, Y. Yan, K. Peter, D. von Elverfeldt, C. E. Hagemeyer and F. Caruso, Angew. Chem., Int. Ed., 2014, 53, 5546–5551 CrossRef CAS PubMed.
- J. Sun, C. Q. Chen, H. H. Pan, Y. Chen, C. Y. Mao, W. Wang, R. K. Tang and X. H. Gu, J. Mater. Chem. B, 2014, 2, 4544–4553 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07887a |
| ‡ Weijing Kong and Qiaolin Du contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d and
Xinhua Gu
d and
Xinhua Gu