
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, solid state self-assembly driven by antiparallel π⋯π stacking and {⋯H–C–C–F}2 dimer synthons, and in vitro acetyl cholinesterase inhibition activity of phenoxy pendant isatins†
Saba Mehreena,
Aman Ullah b,
Humaira Nadeemc,
Necmi Deged and
Muhammad Moazzam Naseer*a
b,
Humaira Nadeemc,
Necmi Deged and
Muhammad Moazzam Naseer*a
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad, 45320, Pakistan. E-mail: moazzam@qau.edu.pk
bDepartment of Agricultural, Food and Nutritional Science, 4-10 Agriculture/Forestry Centre, University of Alberta, Edmonton, AB T6G 2P5, Canada
cDepartment of Pharmaceutical Chemistry, Riphah Institute of Pharmaceutical Sciences, Riphah International University, G-7/4, Islamabad, Pakistan
dOndokuz Mayıs University, Faculty of Arts and Sciences, Department of Physics, Kurupelit, 55139, Samsun, Turkey
First published on 12th January 2022
Abstract
A series of novel phenoxy pendant isatins PI1–12 have been synthesized in excellent yields by a simple nucleophilic substitution reaction involving isatins and 1-(2-bromoethoxy)-4-substituted benzenes, and characterized by their FT-IR, 1H NMR, 13C NMR and GC-MS data, and in the case of PI4 by its single crystal X-ray analysis. The solid-state structure of PI4 showed an intriguing and unique 1D-supramolecular chain-based self-assembled structure, the driving force of which is mainly the strong antiparallel π⋯π stacking and {⋯H–C–C–F}2 dimer synthons. This compound not only highlights the potential of the isatin moiety in forming strong antiparallel π⋯π stacking interactions but also provides a platform to have considerable insight into the nature, strength and directionality of much debated π–π and C–H⋯F–C interactions. The in vitro biological studies revealed that three phenoxy pendant isatins PI1, PI2 and PI4 are highly potent inhibitors of acetylcholinesterase enzyme with IC50 values of 0.52 ± 0.073 μg ml−1, 0.72 ± 0.012 μg ml−1 and 0.68 ± 0.011 μg ml−1, respectively, showing comparable activity to the standard drug, donepezil (IC50 = 0.73 ± 0.015 μg ml−1). A simple and efficient synthesis of phenoxy pendant isatins PI1–12 from inexpensive and commercially available starting materials, and their high potential of acetyl cholinesterase inhibition provide an attractive opportunity to find more effective medication for Alzheimer's disease (AD).
1 Introduction
Despite tremendous progress, the π⋯π interaction remains one of the most debated non-covalent interactions in supramolecular chemistry.1–5 These interactions which are mainly present in aromatic systems attract attention because of their potential applications in different areas of materials and biological sciences in addition to the fundamental understanding of their complicated nature.1–5 For instance, they have been found to play an important role in the outstanding photoconductivity in organic crystals, designing molecular shuttles, efficient charge transport channels and sensing explosives based on nitroaromatics.6–12 They are also vital non-covalent interactions in structure and properties of biomolecules.13–20 Similarly, the organic fluorine C–H⋯F interactions represent another most debated interaction in contemporary research.21–28 The nature of this interaction is still a controversial subject.21–28 According to Pauling's definition of the hydrogen bond, F atom should be strong H-bond acceptor compared to O and N atoms owing to its high electronegativity.29 But practically, it is not the case and organic fluorine is still considered as a poor H-bond acceptor in supramolecular chemistry,21–28 although many efforts have been made to highlight the importance of this interaction in crystal packing.30–38 Therefore, new model systems with unique structural features are highly desired to have considerable insight into the nature, strength and directionality of these debated π⋯π and C–H⋯F–C interactions.Isatin is undoubtedly considered as a magic nucleus in medicinal chemistry owing to diverse applications of its derivatives that include anti-bacterial, anti-fungal, anti-cancer, anti-tubercular, anti-diabetic, neuroprotective, anti-convulsant, anti-HIV, analgesic, anti-oxidant, anti-inflammatory, anti-glycation, anti-malarial and anti-anxiety.39–41 Most of these derivatives have been obtained either by taking the advantage of nucleophilic nature of its NH or by utilizing the reactivity of its 3-carbonyl group.39–41 Recently, it has been found in a theoretical analysis that isatin nucleus has an interesting duality having electron rich six membered ring and electron deficient five-membered ring.42 This feature impart it a unique tendency to form strong antiparallel π–π stacking interactions, making it more attractive from crystal engineering view point. However, these antiparallel π⋯π stacking interactions are not observed in most of its 3-carbonyl derivatives due to the presence of nearby bulky groups and competing interactions.
Alzheimer's disease (AD) is a global problem that has been estimated to effect 115 million people by 2050.43–45 This is a progressive neurodegenerative disorder of the brain that leads to multiple cognitive impairments such as loss of memory, judgment and learning capability.43–45 According to the cholinergic hypothesis, the decreased cognitive and mental function is linked to the loss of cortical cholinergic neurotransmission which can be improved by the inhibition of acetylcholinesterase (an enzyme responsible for the degradation of the neurotransmitter acetylcholine).46 Consequently, most of the available drugs to treat this disease are acetylcholinesterase inhibitors.47 However, these drugs have various undesirable side effects urging researchers to design and explore novel acetylcholinesterase inhibitors.48
In this context and as continuation of our research interests in the synthesis of bioactive isatin derivatives,49–51 and non-covalent interactions52–57 herein, we report the synthesis of a series of new phenoxy pendant isatins PI1–PI12, the solid-state structure of PI4 and in vitro acetyl cholinesterase inhibition activity of synthesized phenoxy pendant isatins. The solid-state self-assembly reported here that is mainly driven by antiparallel π⋯π stacking and {⋯H–C–C–F}2 dimer synthons may serve as the model for detailed understanding of nature, strength, and directionality of much debated π⋯π stacking and C–H⋯F–C interactions.
2 Results and discussion
2.1 Chemistry
The synthesis of phenoxy pendant isatins PI1–12 was initiated with the preparation of 1-(2-bromoethoxy)-4-substituted benzenes B1–4 (Scheme 1). The treatment of substituted phenols A1–4 with large excess of 1,2-dibromoethane at 50 °C in CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF (9:1) mixture provided desired intermediates B1–458–60 in 62–70% isolated yields. The intermediates B1–4 were finally reacted with isatin, 5-chloroisatin and 5-bromoisatin in DMF solvent in the presence of K2CO3 as base at 60 °C to furnish phenoxy pendant isatins PI1–12 in excellent (83–85%) yields (Scheme 1). All the synthesized phenoxy pendant isatins were characterized by their FTIR, 1H & 13C NMR and GC-MS data and in case of PI4 unambiguously by its single crystal X-ray technique.
:DMF (9:1) mixture provided desired intermediates B1–458–60 in 62–70% isolated yields. The intermediates B1–4 were finally reacted with isatin, 5-chloroisatin and 5-bromoisatin in DMF solvent in the presence of K2CO3 as base at 60 °C to furnish phenoxy pendant isatins PI1–12 in excellent (83–85%) yields (Scheme 1). All the synthesized phenoxy pendant isatins were characterized by their FTIR, 1H & 13C NMR and GC-MS data and in case of PI4 unambiguously by its single crystal X-ray technique.
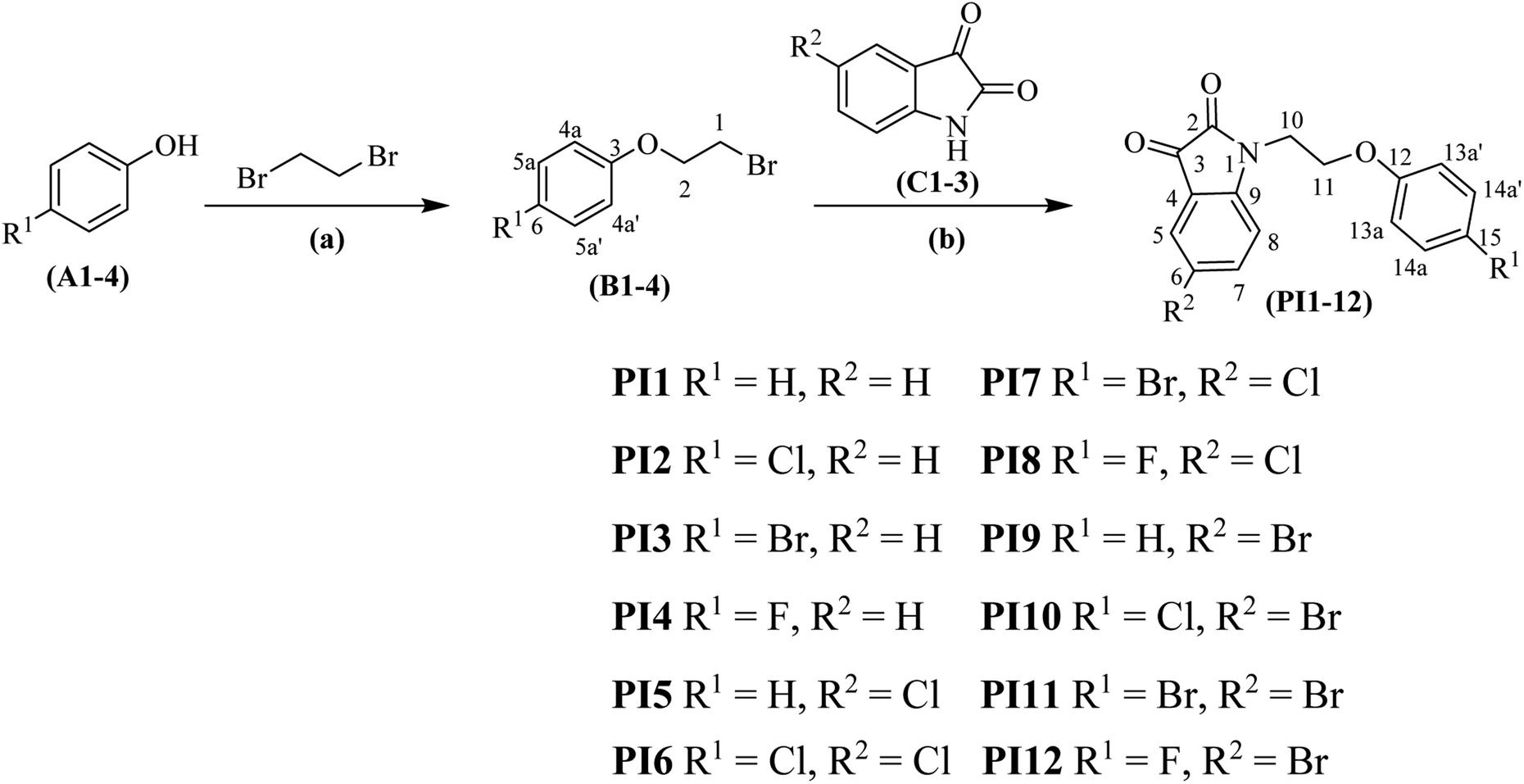 | ||
| Scheme 1 Synthesis of phenoxy pendant isatins PI1–12. Reaction conditions: (a) K2CO3/CH3CN:DMF(9:1)/50–90 °C, (b) K2CO3/DMF/60 °C; numbering of atoms in the structures are just for 1H & 13C NMR signal assignment purpose (see Experimental section for details). | ||
In the IR spectra, the formation of products PI1–12 was indicated by the appearance of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (amide) stretchings in the range from 1682 to 1692 cm−1. Similarly, appearance of ketonic carbonyl (CO) stretchings around 1728–1743 cm−1 indicates the products PI1–12 formation. Along with it, disappearance of N–H stretchings of secondary amide of isatins C1–3 in the range from 3310 to 3350 cm−1 further confirms the formation of products PI1–12. Appearance of Csp3–H stretchings from 2875 to 2878 cm−1 and 2926 to 2951 cm−1 corresponds to the presence of two methylenes in the target molecules. Further confirmation of structures was provided by the 1HNMR data of compounds PI1–12. The absence of N–H protons (usually observed around 11.23 ppm in isatins) indicate the presence of phenoxy pendants. Furthermore, two methylene protons adjacent to the nitrogen atom of isatin of this pendant were observed as triplets around 4.02–4.08 ppm whereas another two other methylene protons adjacent to oxygen of phenoxy group appeared slightly deshielded as triplets around 4.17 to 4.21 ppm. Finally, all aromatic protons were observed as expected in the range from 6.83 to 7.76 ppm as multiplet. Similarly, the appearance of CO (ketone) carbon was observed in the range from 182.38 to 183.81 ppm in 13C NMR spectra. The CO (amide) carbons resonated around 158.37–159.89 ppm. The peaks around 65.38–66.88 ppm were referred to methylenic carbon which are somewhat deshielded due to their direct attachment with oxygen atom. The other methylenic carbons resonate in the range from 39.68 to 39.98 ppm. Aromatic carbons of isatin and phenolic ring were appeared in the range from 111.81–158.75 ppm. These 13C NMR signals along with 1H NMR and FTIR data confirmed the formation of phenoxy pendant isatins PI1–12. Final confirmation of their structure was obtained by observing the molecular ion peak of all the compounds in their GC-MS spectra (see Fig. S1† and experimental section for details).
O (amide) stretchings in the range from 1682 to 1692 cm−1. Similarly, appearance of ketonic carbonyl (CO) stretchings around 1728–1743 cm−1 indicates the products PI1–12 formation. Along with it, disappearance of N–H stretchings of secondary amide of isatins C1–3 in the range from 3310 to 3350 cm−1 further confirms the formation of products PI1–12. Appearance of Csp3–H stretchings from 2875 to 2878 cm−1 and 2926 to 2951 cm−1 corresponds to the presence of two methylenes in the target molecules. Further confirmation of structures was provided by the 1HNMR data of compounds PI1–12. The absence of N–H protons (usually observed around 11.23 ppm in isatins) indicate the presence of phenoxy pendants. Furthermore, two methylene protons adjacent to the nitrogen atom of isatin of this pendant were observed as triplets around 4.02–4.08 ppm whereas another two other methylene protons adjacent to oxygen of phenoxy group appeared slightly deshielded as triplets around 4.17 to 4.21 ppm. Finally, all aromatic protons were observed as expected in the range from 6.83 to 7.76 ppm as multiplet. Similarly, the appearance of CO (ketone) carbon was observed in the range from 182.38 to 183.81 ppm in 13C NMR spectra. The CO (amide) carbons resonated around 158.37–159.89 ppm. The peaks around 65.38–66.88 ppm were referred to methylenic carbon which are somewhat deshielded due to their direct attachment with oxygen atom. The other methylenic carbons resonate in the range from 39.68 to 39.98 ppm. Aromatic carbons of isatin and phenolic ring were appeared in the range from 111.81–158.75 ppm. These 13C NMR signals along with 1H NMR and FTIR data confirmed the formation of phenoxy pendant isatins PI1–12. Final confirmation of their structure was obtained by observing the molecular ion peak of all the compounds in their GC-MS spectra (see Fig. S1† and experimental section for details).
2.2 X-ray crystallographic studies and solid-state self-assembly
Good quality single crystals of phenoxy pendant isatins PI4 were obtained from its concentrated DMSO solution within a week at room temperature. The details of X-ray crystallographic data are provided in Table S1.† The phenoxy pendant isatin PI4 was crystallized in the triclinic crystal lattice with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The molecular structure (ORTEP diagram) of PI4 including the crystallographic numbering is presented as Fig. 1.
space group. The molecular structure (ORTEP diagram) of PI4 including the crystallographic numbering is presented as Fig. 1.
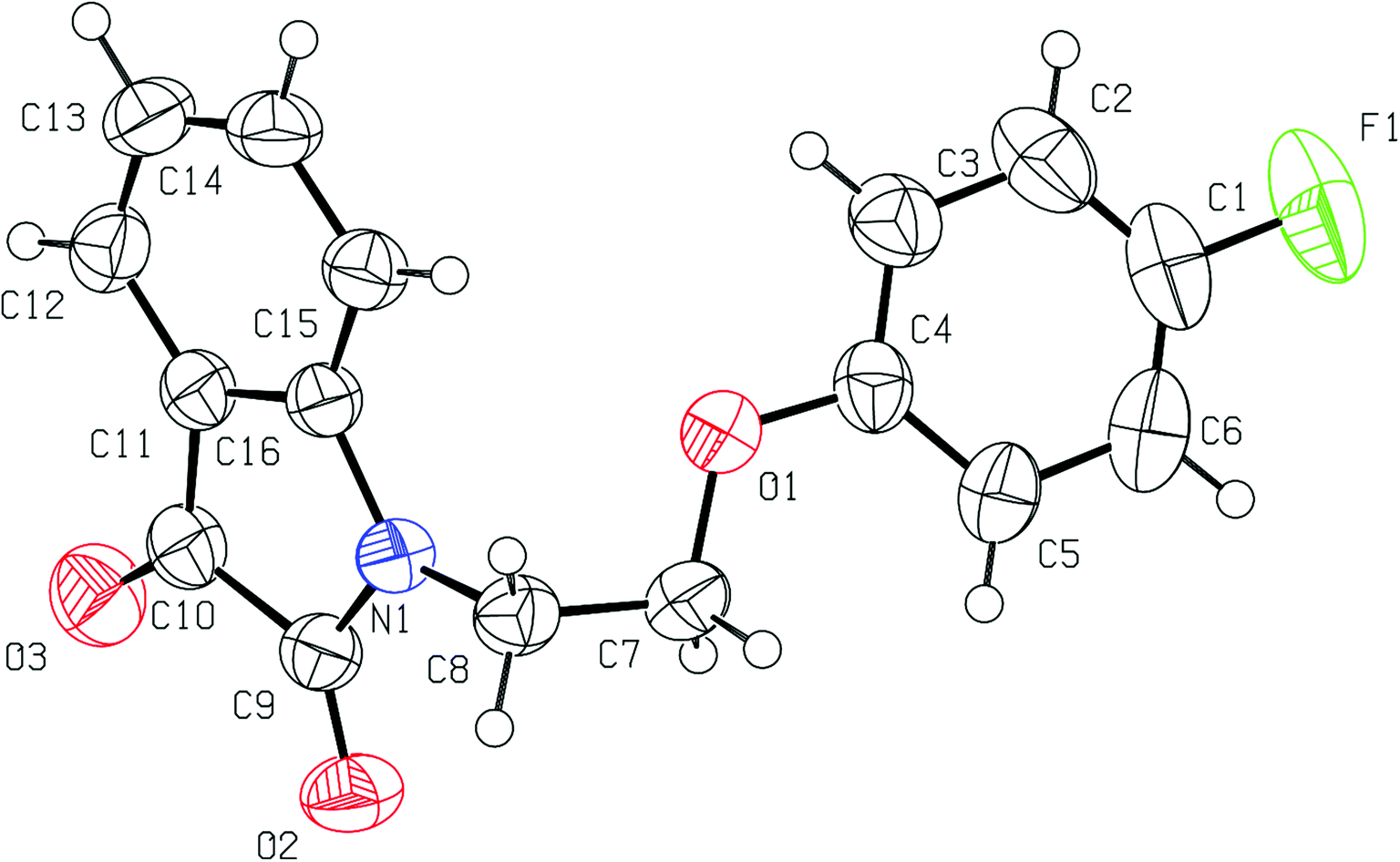 | ||
| Fig. 1 The molecular structure (ORTEP diagram) of phenoxy pendant isatin PI4. Displacement ellipsoids are drawn at the 50% probability level. | ||
In this compound, isatin and phenoxy rings are bridged by an ethylene moiety lie almost perpendicular to each other. The central ethylene moiety is present in staggered gauche conformation with the two aryl rings having a dihedral angle of O(1)–C(7)–C(8)–N(1) 64.46(14)°. The lone pair of nitrogen of isatin may be resonating marginally towards the phenyl ring and to the 3-carbonyl group [N(1)–C(16) 1.4214(16) Å, C(16)–C(11) 1.4000(19) Å, C(11)–C(10) 1.4663(19) Å, C(10)–O(3) 1.2088(17) Å]. However, it is delocalizing slightly more towards the nearby 2-carbonyl group which is quite evident from the relevant N–C [N(1)–C(9) 1.3684(17) Å] and CO [C(9)–O(2) 21.2177(16) Å] bond distances. This delocalization and the presence of two highly electronegative oxygen atoms makes the five membered ring of isatin an electron deficient ring and offers an interesting duality due to the presence of fused electron rich six-membered ring. Owing to this structural feature, isatin moiety is capable of strong antiparallel π–π stacking interactions. This structural feature of isatin, which is considered as privileged nucleus in medicinal chemistry because of diverse applications of its derivatives, remains largely unexplored although this can be very interesting with respect to crystal engineering applications.
Another important and interesting feature of compound PI4 is the presence of 4-fluoro-substituent on the phenoxy pendant that provides an opportunity to analyze the H-bond acceptor ability of organic fluorine in the presence of carbonyl oxygen acceptors. Crucially, the inductive nature of highly electronegative fluorine atom makes the nearby (Ar)CH a good H-bond donor. An expected consequence of this is the facile formation of a centrosymmetric R22(8) {⋯H–C–C–F}2 dimer synthon.
Owing to the special structural features of PI4 (vide supra), its solid-state self-assembly is dominated mainly by two types of non-covalent interactions i.e., antiparallel π⋯π stacking (3.548 Å distance between the centre of stacked five- and six-membered rings) and {⋯H–C–C–F}2 dimer [C(2)–H(2)⋯F(1) 2.669 Å] synthons. Owing to these two interactions, the 1D-supramolecular chains are formed (Fig. 2A). These 1D-supramolecular chains by means of antiparallel π⋯π stacking and a CH-π [C(8)–H(8B)⋯C(1) 2.897 Å] interaction extend themselves to a 2D-sheet like structure (Fig. 2B). These 2D-sheets then connects to the neighboring sheets by means of three different types of CH–O [C(13)–H(13)⋯O(2) 2.523 Å, C(8)–H(8A)⋯O(2) 2.801 Å and C(7)–H(7B)⋯O(2) 2.599 Å] interactions providing an overall a 3D-network structure (Fig. 2C).
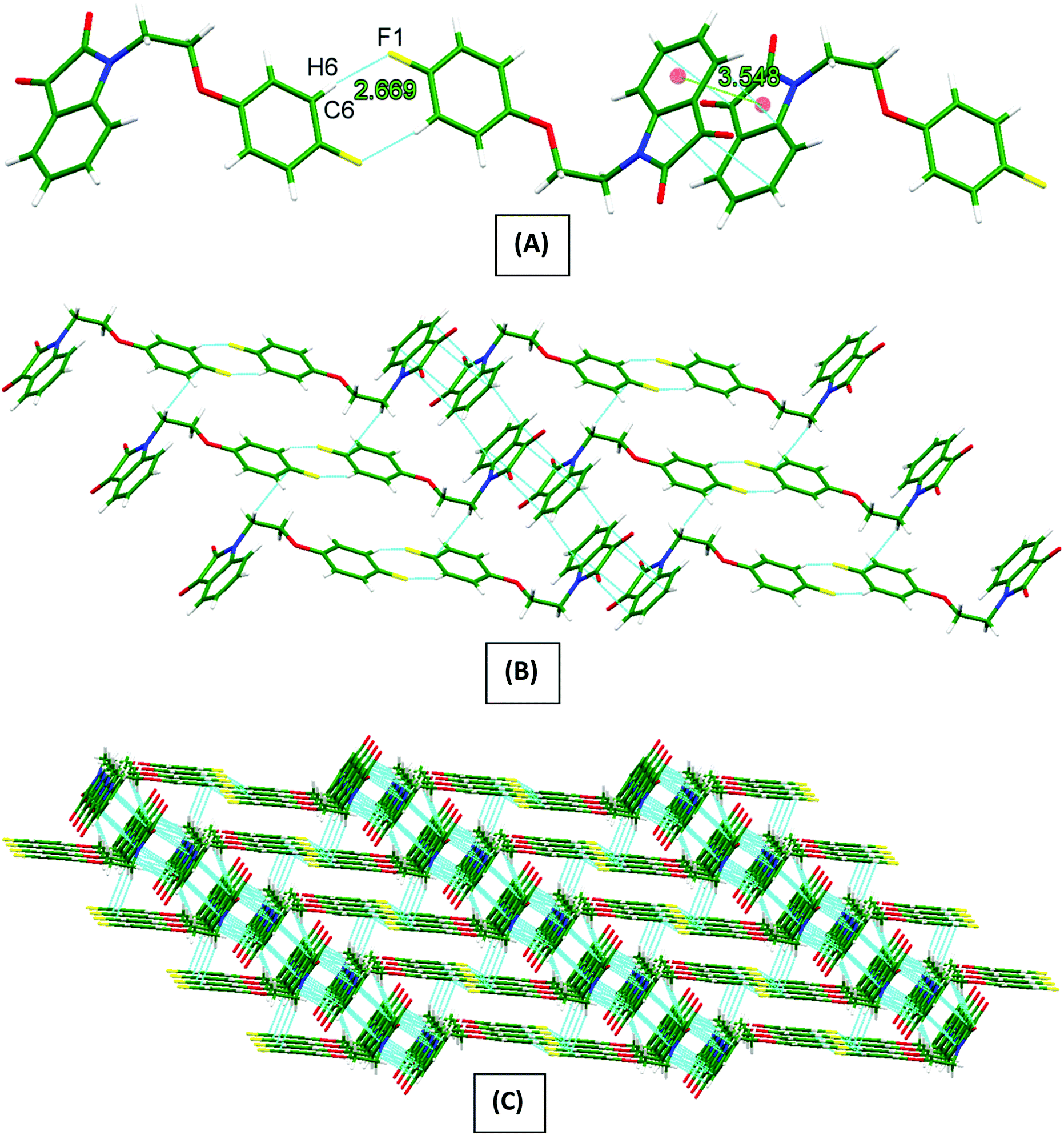 | ||
| Fig. 2 Molecular packing of phenoxy pendant isatin PI4 in the solid state, (A) showing 1D-supramolecular chain formed through antiparallel π⋯π stacking and {⋯H–C–C–F}2 dimer synthons; (B) showing 2D-sheet like structure; (C) showing 3D-network structure. | ||
In general, it is believed that the linear hydrogen bonds (150° < ϴ < 180°) are structurally more significant because of the dipole-monopole and dipole–dipole contribution to the electrostatic energy (maximum at ϴ = 180° and zero at ϴ = 90°).61 Careful analysis of the bond angles ϴ in Table 1 reveals that the hydrogen bonds involved in the formation of {⋯H–C–C–F}2 dimer synthon are relatively linear and therefore more significant when compared to the (Ar)C–H⋯O and (sp3)C–H⋯O interactions. The angle ϕ is used to analyze the lone-pair directionality of the acceptor atom. The angle ϕ for carbonyls acceptors have been observed in the range of 120–140°, indicating the sidewise approach of the H-bond donors.62 Likewise, it is well established that a halogen substituent exhibits an anisotropic distribution of the electron density, bearing a positive σ-hole at the extension of the C–halogen bond and a negative charge density perpendicular to the σ-bond.63 As shown in Table 2, angle ϕ in case of (sp3)C–H⋯O interactions is slightly less than the range discussed above for the carbonyl acceptors, indicating the nature of these interactions as ‘structure guided’. Keeping in view the linearity of (Ar)C–H⋯F and the formation of self-complimentary {⋯H–C–C–F}2 dimer synthon in the solid-state structure of PI4, it can be anticipated that this dimer synthon is robust and strong enough to play the main structure guiding role, consequently it is mainly driving this solid state self-assembly along with antiparallel π⋯π stacking interactions.

|
|||
|---|---|---|---|
| Compounds | R1 | R2 | IC50 (μg ml−1) ± SEM |
| a ND = not determined, SEM = standard error of mean. | |||
| PI1 | H | H | 0.52 ± 0.073 |
| PI2 | Cl | H | 0.72 ± 0.012 |
| PI3 | Br | H | ND |
| PI4 | F | H | 0.68 ± 0.011 |
| PI5 | H | Cl | ND |
| PI6 | Cl | Cl | 0.91 ± 0.015 |
| PI7 | Br | Cl | ND |
| PI8 | F | Cl | ND |
| PI9 | H | Br | 0.91 ± 0.083 |
| PI10 | Cl | Br | 0.98 ± 0.055 |
| PI11 | Br | Br | 1.14 ± 0.013 |
| PI12 | F | Br | 1.31 ± 0.083 |
| Donepezil | — | — | 0.73 ± 0.015 |
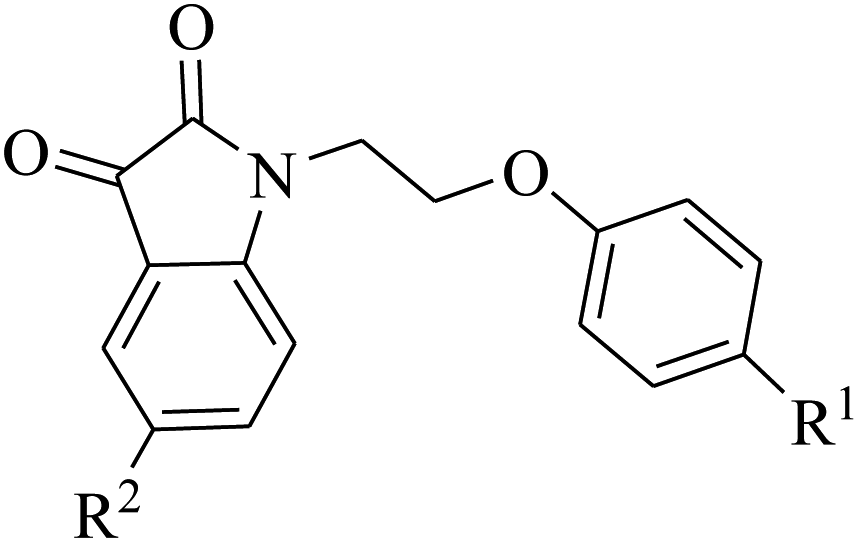
2.3 Acetyl cholinesterase inhibition of phenoxy pendant isatin PI1–12
The phenoxy pendant isatins PI1–12 were screened for their acetyl cholinesterase inhibitory activity following the literature protocol with slight modification64 using donepezil as the standard reference drug. The IC50 values were only calculated for those phenoxy pendant isatins PI1–12 which showed greater than 50% inhibition in the assay (Table S2 and Fig. S1 in ESI†). The IC50 values of all the active compounds are quoted in Table 2. Out of twelve compounds tested, eight i.e., PI1, PI2, PI4, PI6, PI9, PI10, PI11 and PI12 were proved to be active against acetyl cholinesterase. Three compounds of the series i.e., PI1, PI2 and PI4 showed comparable activity (IC50 = 0.52 ± 0.073 μg ml−1, 0.72 ± 0.012 μg ml−1 and 0.68 ± 0.011 μg ml−1, respectively) to the reference inhibitor, donepezil (IC50 = 0.73 ± 0.015 μg ml−1). All other active compounds of the series PI6, PI9, PI10, PI11 and PI12 appeared to have moderate to good acetyl cholinesterase inhibitory activity with IC50 values of 0.91 ± 0.015 μg ml−1, 0.91 ± 0.083 μg ml−1, 0.98 ± 0.055 μg ml−1, 1.14 ± 0.013 μg ml−1 and 1.31 ± 0.083 μg ml−1, respectively when compared to standard reference inhibitor, donepezil (IC50 = 0.73 ± 0.015 μg ml−1) (Table 2). The structure–activity relationship (SAR) studies in the phenoxy pendant isatins PI1–12 revealed that the presence of halogen substituents at position 5 of isatin has detrimental effect on acetyl cholinesterase inhibitory activity and hence most active compounds of the series (PI1, PI2 and PI4) are those compounds derived from simple isatin. Same is the case with the halogen substitution on phenoxy pendant. The most active compound of the series i.e. PI1 [(IC50 = 0.52 ± 0.073 μg ml−1) compared to donepezil (IC50 = 0.73 ± 0.015 μg ml−1)] has no halogen substitution on both isatin and phenoxy pendant. With the introduction of chlorine substituent on phenoxy pendant, the activity of compound PI2 was slightly decreased (IC50 = 0.72 ± 0.012 μg ml−1). Similarly, the replacement of chlorine with bromine on phenoxy pendant resulted in significant loss in activity (IC50 was not determined because of less than 50% inhibition). The least unfavorable effect on activity was observed in case of fluorine substitution on the phenoxy pendant where the IC50 of 0.68 ± 0.011 μg ml−1 was observed for compound PI4. This may be due to comparable sizes of hydrogen and fluorine atoms, although fluorine due to its high electronegativity, result in considerable changes in the electronic properties of the molecule.383 Conclusions
In summary, we have synthesized a series of novel phenoxy pendant isatins (PI1–12) in excellent yields from simple and commercially available starting materials and characterized by their FTIR, 1H NMR, 13C NMR and GC-MS data. The single crystal X-ray analysis of PI4 provided an intriguing and unique solid-state structure driven mainly by the strong antiparallel π⋯π stacking and {⋯H–C–C–F}2 dimer synthons. Because molecular self-assemblies stabilized mainly by these two interactions and in the absence of other strong non-covalent interactions are rare, this structure may serve as model to understand the nature, strength and directionality of recently much debated π–π and C–H⋯F–C interactions. Furthermore, this solid-state structure highlights the potential of isatin moiety (capable of forming strong antiparallel π⋯π stacking interactions) for crystal engineering applications. The in vitro biological evaluation of these phenoxy pendant isatins (PI1–12) provided three (PI1, PI2 and PI4) highly potent inhibitors of acetylcholinesterase enzyme that are comparable in activity to the standard drug, donepezil. As highlighted by Sussman, the most remarkable feature of the structure of Torpedo californica acetylcholinesterase (TcAChE), and of other AChEs, is about 20 Å long gorge called ‘active-site gorge’ consisting of 14 aromatic residues that makes ∼40% of the surface of the gorge.65 Therefore, the molecules capable of interacting with this active site can prevent acetylcholinesterase induced Aβ aggregation and hence the compounds containing planar and aromatics scaffolds are potential targets for the development of anti-AD agents.46–48 In this context, the high acetylcholinesterase inhibitory activity of the phenoxy pendant isatins may be attributed to the strong ability of isatin moiety for π–π interactions. The structure–activity relationship (SAR) studies however indicate that the presence of halogen substituents at position 5 of isatin or position 4 of phenoxy pendant are not beneficial for this activity.4 Experimental
4.1 General procedure for the synthesis of 1-(2-bromoethoxy)-4-substituted benzenes (B1–4)
A reported modified procedure was utilized for the synthesis of 1-(2-bromoethoxy)-4-substituted benzenes (B1–4). To a stirred solution of substituted phenol (10.0 mmol) in CH3CN:DMF (9:1), anhydrous potassium hydroxide (12.0 mmol) was added, the mixture after stirring for 30 minutes was then added to 1,2-bromoethane (50.0 mmol). The stirring of the resulting mixture was then continued at 50 °C for 24 hours. TLC was used to monitor the progress of reaction. The cold distilled water (25 ml) was added to the reaction mixture upon completion and crude product was extracted with EtOAc (3 × 30 ml), washed with brine and distilled water (20 ml) and dried over anhydrous MgSO4. The combined organic layers were concentrated under vacuum and column chromatography (n-hexane: ethyl acetate, 85:15) was employed to get the pure 1-(2-bromoethoxy)-4-substituted benzenes (B1–4) which solidified at low temperature.
4.1.1.1 Transparent liquid. Yield: 62%; m.p.: 31–32 °C (lit. 31–34 °C);58 1H NMR (300 MHz, CDCl3): δ (ppm) 7.28–7.37 (2H, m, aromatic), 6.94–7.05 (3H, m, aromatic), 4.32 (2H, t, 3J = 6.3 Hz, H-2), 3.68 (2H, t, 3J = 6.3 Hz, H-1). 13C NMR (75 MHz, CDCl3) δ (ppm): 29.31 (C-1), 67.75 (C-2), 114.74 (C-4a, 4a′), 121.47 (C-3), 129.57 (C5a–5a′), 158.07 (C-6).
4.1.2.1 Brown liquid. Yield: 64%; m.p.: 39–40 °C (lit. 40–42 °C);59 1H NMR (300 MHz, CDCl3): δ (ppm) 7.24–7.28 (2H, m, aromatic), 6.80–6.83–(2H, m, aromatic), 4.21 (2H, t, 3J = 6.0 Hz, H-2), 3.60 (2H, t, 3J = 6.0 Hz, H-1). 13C NMR (75 MHz, CDCl3) δ (ppm): 29.25 (C-1), 68.15 (C-2), 116.10 (C-3), 116.88 (C-4a, 4a′), 129.32 (C-5a, 5a′), 156.72 (C-6).
4.1.3.1 Off-white liquid. Yield: 62%; m.p.: 57–58 °C (lit. 56–58 °C);58 1H NMR (300 MHz, CDCl3): δ (ppm) 7.38–7.43 (2H, m, aromatic), 6.79–6.84 (2H, m, aromatic), 4.26 (2H, t, 3J = 6.3 Hz, H-2), 3.64 (2H, t, 3J = 6.0 Hz, H-1). 13C NMR (75 MHz, CDCl3) δ (ppm): 28.95 (C-1), 68.06 (C-2), 113.66 (C-3) 116.56 (C-4a, 4a′), 132.43 (C-5a, 5a′), 157.21 (C-6).
4.1.4.1 Transparent liquid. Yield: 70%; m.p.: 58–59 °C (lit. 58–60 °C);60 1H NMR (300 MHz, CDCl3): δ (ppm) 6.96–7.03 (2H, m, aromatic), 6.85–6.91 (2H, m, aromatic), 4.27 (2H, t, 3J = 6.3 Hz, H-2), 3.64 (2H, t, 3J = 6.3 Hz, H-1). 13C NMR (75 MHz, CDCl3) δ (ppm): 29.14 (C-1), 68.60 (C-2), 115.96 (d, 3J = 8.25 Hz, C-4a, 4a′), 115.99 (d, 2J = 23.25 Hz, C-5a, 5a′), 154.21 (d, 4J = 2.25 Hz, C-3), 157.63 (d, 1J = 237.75 Hz, C-6).
4.2 General procedure for the synthesis of phenoxy pendant isatins (PI1–12)
To a solution of isatin (5 mmol) in DMF (10 ml) was added anhydrous potassium carbonate (1 mmol). To this mixture after stirring for 30 minutes at room temperature was slowly added the B1 (5.5 mmol) and the stirring was further continued for 6 hours at 60 °C. TLC was used to monitor the progress of the reaction. Ice cold distilled water (25 ml) was added to the reaction mixture upon completion of the reaction with the precipitation of crude product, which was filtered, washed, dried and recrystallized from ethanol to yield the pure phenoxy pendant isatins (PI1–12).4.2.1.1 Orange red. Yield: 85%; m.p.: 113–114 °C; FTIR ῡ (cm−1); 2929, 2878 (Csp3–H), 1730 (ketone C
O), 1687 (amide CO), 1608, 1484 (aromatic CC), 1351 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.65–7.71 (1H, m, H-7), 7.52–7.55 (1H, m, H-5), 7.33 (1H, d, 3J = 8.1 Hz, H-8), 7.22–7.28 (2H, m, H-14a, 14a′), 7.13 (1H, t, 3J = 7.5 Hz, H-6), 6.86–6.93 (3H, m, H-15, H-13a, 13a′), 4.21 (2H, t, 3J = 5.1 Hz, H-10), 4.07 (2H, t, 3J = 5.4 Hz, H-11). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.81 (C-10), 65.38 (C-11), 111.86 (C-8), 114.84 (C-13a, 13a′), 117.83 (C-15), 117.89 (C-4), 123.66 (C-6), 124.81 (C-5), 129.97 (C-14a, 14a′), 138.59 (C-7), 151.44 (C-9), 158.46 (C-12), 158.75 (C-2), 183.66 (C-3). EI-MS (m/z): 267 (267.28), 174, 146, 132, 120, 77 (100%), 65, 51. Anal. calcd. for C16H13NO3: C, 71.90; H, 4.90; N, 5.24. Found: C, 71.85; H, 5.08; N, 4.99.
4.2.2.1 Mustard yellow. Yield: 84%; m.p.: 152–153 °C; FT-IR ῡ (cm−1); 2926, 2875 (Csp3–H), 1728 (ketone C
O), 1682 (amide CO), 1612, 1488 (aromatic CC), 1351 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.66–7.72 (1H, m, H-7), 7.54 (1H, m, H-5), 7.39–7.45 (2H, m, H-14a, 14a′), 7.32 (1H, d, 3J = 7.8 Hz, H-8), 7.13 (1H, t, 3J = 7.5 Hz, H-6), 6.83–6.89 (2H, m, H-13a, 13a′), 4.21 (2H, t, 3J = 5.1 Hz, H-10), 4.07 (2H, t, 3J = 5.4 Hz, H-11). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.97 (C-10), 65.88 (C-11), 111.81 (C-8), 117.19 (C-13a, 13a′), 117.27 (C-15), 117.89 (C-4), 123.66 (C-6), 124.81 (C-5), 132.60 (C-14a, 14a′), 138.58 (C-7), 151.39 (C-9), 158.04 (C-12), 158.74 (C-2), 183.62 (C-3). EI-MS (m/z): 301 (301.72), 174, 146, 154, 132 (100%), 127, 105, 77, 51. Anal. calcd. for C16H12ClNO3: C, 63.69; H, 4.01; N, 4.64. Found: C, 63.77; H, 4.24; N, 4.56.
4.2.3.1 Yellow. Yield: 84%; m.p.: 147–150 °C; FT-IR ῡ (cm−1); 2928, 2880 (Csp3–H), 1729 (ketone C
O), 1682 (amide CO), 1612, 1489 (aromatic CC), 1352 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.65–7.71 (1H, m, H-7), 7.55 (1H, d, 3J = 7.5 Hz, H-5), 7.39–7.44 (2H, m, H-14a, 14a′), 7.32 (1H, d, 3J = 7.8 Hz, H-8), 7.13 (1H, t, 3J = 7.5 Hz, H-6), 6.83–6.88 (2H, m, H-13a, 13a′), 4.21 (2H, t, 3J = 5.1 Hz, H-10), 4.07 (2H, t, 3J = 5.4 Hz, H-11). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.96 (C-10), 65.88 (C-11), 111.82 (C-8), 117.20 (C-13a, 13a′), 117.28 (C-15), 117.90 (C-4), 123.66 (C-6), 124.81 (C-5), 132.60 (C-14a, 14a′), 138.59 (C-7), 151.39 (C-9), 158.04 (C-12), 158.75 (C-2), 183.62 (C-3). EI-MS (m/z): 345 (345.18), 199, 174, 157, 146, 132 (100%), 105, 77, 65, 51. Anal. calcd. for C16H12BrNO3: C, 55.51; H, 3.49; N, 4.05. Found: C, 55.73; H, 3.16; N, 3.85.
4.2.4.1 Orange yellow. Yield: 83%; m.p.: 127–130 °C; FT-IR ῡ (cm−1); 2928, 2877 (Csp3–H), 1729 (ketone C
O), 1685 (amide CO), 1612, 1486 (aromatic CC), 1352 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.65–7.71 (1H, m, H-7), 7.54 (1H, d, 3J = 7.5 Hz, H-5), 7.32 (1H, d, 3J = 7.8 Hz, H-8), 7.13 (1H, t, 3J = 7.8 Hz, H-6), 7.05–7.10 (2H, m, H-14a, 14a′), 6.87–6.91 (2H, m, H-13a, 13a′), 4.20 (2H, t, 3J = 5.1 Hz, H-10), 4.06 (2H, t, 3J = 5.4 Hz, H-11). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.96 (C-10), 66.11 (C-11), 111.84 (C-8), 116.19 (d, 3J = 6 Hz, C-14a, 14a′), 116.29 (d, 2J = 24.75 Hz, C-13a, 13a′), 117.89 (C-4), 123.65 (C-6), 124.81 (C-5), 138.59 (C-7), 151.41 (C-9), 154.81 (d, 4J = 2.25 Hz, C-12), 157.07 (d, 1J = 234.75 Hz, C-15), 158.74 (C-2), 183.64 (C-3). EI-MS (m/z): 285 (285.27), 174, 146, 138, 132 (100%), 111, 105, 95, 77, 65, 57, 51. Anal. calcd. for C16H12FNO3: C, 67.37; H, 4.24; N, 4.91. Found: C, 67.57; H, 4.45; N, 4.83.
4.2.5.1 Orange red. Yield: 85%; m.p.: 137–139 °C; FT-IR ῡ (cm−1); 2926, 2878 (Csp3–H), 1743 (ketone C
O), 1688 (amide CO), 1609, 1487 (aromatic CC), 1351 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.74 (1H, dd, 3J = 8.4 Hz, 4J = 2.1 Hz, H-7), 7.62 (1H, d, 3J = 2.1 Hz, H-5), 7.55–7.59 (3H, m, H-14a, 14a′, H-15), 7.38 (1H, d, 3J = 8.4 Hz, H-8), 6.86–6.93 (2H, m, H-13a, 13a′), 4.19 (2H, t, 3J = 5.1 Hz, H-10), 4.08 (2H, t, 3J = 5.1 Hz, H-11). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.67 (C-10), 65.61 (C-11), 114.28 (C-15), 114.83 (C-8), 119.63 (C-13a, 13a′), 121.33 (C-6), 124.61 (C-4), 127.24 (C-14a, 14a′), 127.78 (C-5), 129.96 (C-7), 137.70 (C-9), 149.66 (C-12), 159.64 (C-2), 183.81 (C-3). EI-MS (m/z): 301 (301.72), 208, 180, 166, 132, 120, 111, 105, 93, 77 (100%), 65, 51. Anal. calcd. for C16H12ClNO3: C, 63.69; H, 4.01; N, 4.64. Found: C, 63.56; H, 4.37; N, 4.46.
4.2.6.1 Mustard yellow. Yield: 84%; m.p.: 181–183 °C; FT-IR ῡ (cm−1); 2929, 2878 (Csp3–H), 1743 (ketone C
O), 1692 (amide CO), 1610, 1492 (aromatic CC), 1353 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.73 (1H, dd, 3J = 8.4 Hz, 4J = 2.4 Hz, H-7), 7.58 (1H, d, 3J = 2.1 Hz, H-5), 7.36 (1H, d, 3J = 8.4 Hz, H-8), 7.28–7.32 (2H, m, H-14a, 14a′), 6.89–6.92 (2H, m, H-13a, 13a′), 4.19 (2H, t, 3J = 4.8 Hz, H-10), 4.07 (2H, t, 3J = 4.8 Hz, H-11). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.98 (C-10), 66.17 (C-11), 113.68 (C-4), 116.67 (C-13a, 13a′), 119.29 (C-8), 124.22 (C-5), 125.02 (C-15), 127.79 (C-7), 129.70 (C-14a, 14a′), 137.38 (C-6), 150.01 (C-9), 157.33 (C-12), 158.53 (C-2), 182.53 (C-3). EI-MS (m/z): 335 (336.17), 208, 180, 166, 154, 127, 111, 99, 75 (100%), 63, 51. Anal. calcd. for C16H11Cl2NO3: C, 57.17; H, 3.30; N, 4.17. Found: C, 57.37; H, 3.04; N, 3.99.
4.2.7.1 Yellow. Yield: 84%; m.p.: 191–193 °C; FT-IR ῡ (cm−1); 2951, 2878 (Csp3–H), 1742 (ketone C
O), 1689 (amide CO), 1608, 1488 (aromatic CC), 1352 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.73 (1H, dd, 3J = 8.4 Hz, 4J = 2.4 Hz, H-7), 7.60 (1H, d, 3J = 2.1 Hz, H-5), 7.35–7.43 (3H, m, H-8, H-14a, 14a′), 6.84–6.87 (2H, m, H-13a, 13a′), 4.19 (2H, t, 3J = 4.8 Hz, H-10), 4.07 (2H, t, 3J = 4.8 Hz, H-11).13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.95 (C-10), 66.12 (C-11), 113.67 (C-4), 117.20 (C-13a, 13a′), 117.38 (C-15), 119.29 (C-8), 124.23 (C-5), 127.78 (C-7), 132.60 (C-14a, 14a′), 137.38 (C-6), 150.00 (C-9), 157.77 (C-12), 158.53 (C-2), 182.52 (C-3). EI-MS (m/z): 381 (380.62), 208, 180, 172, 166 (100%), 139, 111, 91, 75, 61. Anal. calcd. for C16H11BrClNO3: C, 50.49; H, 2.91; N, 3.68. Found: C, 50.69; H, 3.12; N, 3.35.
4.2.8.1 Orange yellow. Yield: 83%; m.p.: 154–155 °C; FT-IR ῡ (cm−1); 2926, 2878 (Csp3–H), 1736 (ketone C
O), 1688 (amide CO), 1606, 1489 (aromatic CC), 1353 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.73 (1H, dd, 3J = 8.4 Hz, 4J = 2.4 Hz, H-7), 7.59 (1H, d, 3J = 2.1 Hz, H-5), 7.37 (1H, d, 3J = 8.4 Hz, H-8), 7.06–7.11 (2H, m, H-14a, 14a′), 6.86–6.91 (2H, m, H-13a, 13a′), 4.17 (2H, t, 3J = 4.8 Hz, H-10), 4.06 (2H, t, 3J = 4.8 Hz, H-11). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.68 (C-10), 66.35 (C-11), 113.69 (C-4), 116.18 (d, 3J = 6.75 Hz, C-13a, 13a′), 116.28 (d, 2J = 24.75 Hz, C-14a, 14a′), 119.27 (C-8), 124.22 (C-5), 127.79 (C-7), 137.39 (C-6), 150.03 (C-9), 154.80 (d, 4J = 2.25 Hz, C-12), 157.02 (d, 1J = 225 Hz, C-15), 158.65 (C-2), 182.54 (C-3). EI-MS (m/z): 319 (319.71), 208, 180, 138, 111, 95 (100%), 77, 65. Anal. calcd. for C16H11ClFNO3: C, 60.11; H, 3.47; N, 4.38. Found: C, 60.38; H, 3.57; N, 3.99.
4.2.9.1 Orange red. Yield: 85%; m.p.: 183–184 °C; FT-IR ῡ (cm−1); 2951, 2877 (Csp3–H), 1736 (ketone C
O), 1689 (amide CO), 1597, 1498 (aromatic CC), 1353 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.73 (1H, dd, 3J = 8.4 Hz, 4J = 2.1 Hz, H-7), 7.59 (1H, s, H-5), 7.37 (1H, d, 3J = 8.4 Hz, H-8), 7.06–7.11 (3H, m, H-14a, 14a′, 15), 6.87–6.91 (2H, m, H-13a, 13a′), 4.18 (2H, t, 3J = 5.1 Hz, H-10), 4.06 (2H, t, 3J = 5.1 Hz, H-11). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.68 (C-10), 66.35 (C-11), 113.69 (C-15), 116.23 (C-8), 116.45 (C-13a, 13a′), 119.27 (C-6), 124.22 (C-4), 127.80 (C-14a, 14a′), 137.39 (C-5), 150.03 (C-7), 154.79 (C-9), 158.52 (C-12), 158.65 (C-2), 182.54 (C-3). EI-MS (m/z): 346 (346.18), 252, 225, 120, 93, 77 (100%), 65. Anal. calcd. for C16H12BrNO3: C, 55.51; H, 3.49; N, 4.05. Found: C, 55.77; H, 3.59; N, 3.77.
4.2.10.1 Mustard yellow. Yield: 84%; m.p.: 170–171 °C; FT-IR ῡ (cm−1); 2927, 2878 (Csp3–H), 1736 (ketone C
O), 1685 (amide CO), 1607, 1490 (aromatic CC), 1353 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.86 (1H, dd, 3J = 8.4 Hz, 4J = 2.1 Hz, H-7), 7.70 (1H, d, 3J = 1.8 Hz, H-5), 7.28–7.33 (3H, m, H-14a, 14a′, H-8), 6.88–6.92 (2H, m, H-13a, 13a′), 4.19 (2H, t, 3J = 5.1 Hz, H-10), 4.06 (2H, t, 3J = 4.8 Hz, H-11). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.95 (C-10), 66.18 (C-11), 114.11 (C-4), 116.74 (C-13a, 13a′), 119.68 (C-8), 124.93 (C-5), 125.01 (C-15),126.96 (C-7), 129.71 (C-14a, 14a′), 140.18 (C-6), 150.38 (C-9), 157.33 (C-12), 158.37 (C-2), 182.38 (C-3). EI-MS (m/z): 379 (380.62), 252, 225, 155, 127, 99 (100%), 77, 65. Anal. calcd. for C16H11BrClNO3: C, 50.49; H, 2.91; N, 3.68. Found: C, 50.64; H, 3.14; N, 3.58.
4.2.11.1 Yellow. Yield: 84%; m.p.: 191–193 °C; FT-IR ῡ (cm−1); 2929, 2878 (Csp3–H), 1737 (ketone C
O), 1685 (amide CO), 1606, 1487 (aromatic CC), 1353 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.86 (1H, dd, 3J = 8.4 Hz, 4J = 2.1 Hz, H-7), 7.69 (1H, d, 3J = 2.1 Hz, H-5), 7.40–7.43 (2H, m, H-14a, 14a′), 7.32 (1H, d, 3J = 8.4 Hz, H-8), 6.84–6.87 (2H, m, H-13a, 13a′), 4.19 (2H, t, 3J = 5 Hz, H-10), 4.06 (2H, t, 3J = 4.8 Hz, H-11). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.81 (C-10), 66.12 (C-11), 114.11 (C-4), 115.31 (C-5), 117.20 (C-13a, 13a′), 117.28 (C-15), 119.68 (C-8), 126.96 (C-7), 132.60 (C-14a, 14a′), 140.18 (C-6), 150.38 (C-9), 157.77 (C-12), 158.37 (C-2), 182.38 (C-3). EI-MS (m/z): 424 (425.07), 252, 225, 198, 171, 77, 65, 63 (100%). Anal. calcd. for C16H11Br2NO3: C, 45.21; H, 2.61; N, 3.30. Found: C, 45.10; H, 2.75; N, 3.06.
4.2.12.1 Orange yellow. Yield: 83%; m.p.: 139–141 °C; FT-IR ῡ (cm−1); 2924, 2876 (Csp3–H), 1737 (ketone C
O), 1687 (amide CO), 1607, 1490 (aromatic CC), 1353 (–CH2– bending). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.86 (1H, dd, 3J = 7.5 Hz, H-7), 7.70 (1H, s, H-5), 7.32 (1H, d, 3J = 8.4 Hz, H-8), 7.06–7.11 (2H, m, H-14a, 14a′), 6.87–6.91 (2H, m, H-13a, 13a′), 4.17 (2H, t, 3J = 4.5 Hz, H-10), 4.06 (2H, t, 3J = 4.8 Hz, H-11). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 39.68 (C-10), 66.34 (C-11), 114.13 (C-13a, 13a′), 115.31 (C-14a, 14a′), 116.14 (C-6), 116.45 (C-8), 119.67 (C-4), 126.96 (C-5), 140.19 (C-7), 150.41 (C-9), 154.82 (C-15), 158.36 (C-12), 159.89 (C-2), 182.21 (C-3). EI-MS (m/z): not appeared (346.17), 252, 225, 138, 83 (100%), 77, 65. Anal. calcd. for C16H11BrFNO3: C, 52.77; H, 3.04; N, 3.85. Found: C, 52.93; H, 3.35; N, 3.48.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SM is thankful to HEC for funding under IRSIP program and MMN is grateful to The World Academy of Sciences (TWAS) for financial support through Project No. 13-419 RG/PHA/AS_CUNESCO FR: 3240279216. SM is also grateful to Hafiz Aamir Ali Kharl for his guidance to AChE inhibition studies.References
- G. R. Desiraju and A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 473–482 CrossRef.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430–3434 CrossRef CAS PubMed.
- J. W. Bloom and S. E. Wheeler, Angew. Chem., Int. Ed., 2011, 50, 7847–7849 CrossRef CAS PubMed.
- A. Banerjee, A. Saha and B. K. Saha, Cryst. Growth Des., 2019, 19, 2245–2252 CrossRef CAS.
- H. Li, X. Zhang and W. Zu, J. Appl. Phys., 2014, 115, 054510 CrossRef.
- W. Yu, X.-Y. Wang, J. Li, Z.-T. Li, Y.-K. Yan, W. Wang and J. Pei, Chem. Commun., 2013, 49, 54–56 RSC.
- M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833–4856 CrossRef CAS PubMed.
- R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef CAS.
- L. Liu, J. Hao, Y. Shi, J. Qiu and C. Hao, RSC Adv., 2015, 5, 3045–3053 RSC.
- D. Yan, A. Delori, G. O. Lloyd, T. Friščić, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 12483–12486 CrossRef CAS PubMed.
- T. Chen, M. Li and J. Liu, Cryst. Growth Des., 2018, 18, 2765–2783 CrossRef CAS.
- S. Burley and G. Petsko, Adv. Protein Chem., 1988, 39, 125–189 CrossRef CAS PubMed.
- G. B. McGaughey, M. Gagné and A. K. Rappé, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS PubMed.
- R. Bhattacharyya, U. Samanta and P. Chakrabarti, Protein Eng., 2002, 15, 91–100 CrossRef CAS PubMed.
- N. Kannan and S. Vishveshwara, Protein Eng., 2000, 13, 753–761 CrossRef CAS PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- M. Nishio, Y. Umezawa, J. Fantini, M. S. Weiss and P. Chakrabarti, Phys. Chem. Chem. Phys., 2014, 16, 12648–12683 RSC.
- S. Burley and G. A. Petsko, Science, 1985, 229, 23–28 CrossRef CAS PubMed.
- W. Saenger, in Principles of Nucleic Acid Structure, Springer, 1984, pp. 1–8 Search PubMed.
- J. A. Howard, V. J. Hoy, D. O'Hagan and G. T. Smith, Tetrahedron, 1996, 52, 12613–12622 CrossRef CAS.
- V. R. Thalladi, H.-C. Weiss, D. Bläser, R. Boese, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1998, 120, 8702–8710 CrossRef CAS.
- J. D. Dunitz and R. Taylor, Chem.–Eur. J., 1997, 3, 89–98 CrossRef CAS.
- A. R. Choudhury and T. N. Guru Row, Cryst. Growth Des., 2004, 4, 47–52 CrossRef CAS.
- D. Chopra and T. G. Row, CrystEngComm, 2008, 10, 54–67 RSC.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC.
- D. Chopra, Cryst. Growth Des., 2012, 12, 541–546 CrossRef CAS.
- A. Vulpetti and C. Dalvit, Chem.–Eur. J., 2021, 27, 8764–8773 CrossRef CAS PubMed.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, NY, 1960 Search PubMed.
- V. Vasylyeva and K. Merz, Cryst. Growth Des., 2010, 10, 4250–4255 CrossRef CAS.
- A. Schwarzer and E. Weber, Cryst. Growth Des., 2008, 8, 2862–2874 CrossRef CAS.
- A. Schwarzer, W. Seichter, E. Weber, H. Stoeckli-Evans, M. Losada and J. Hulliger, CrystEngComm, 2004, 6, 567–572 RSC.
- P. Panini, T. Mohan, U. Gangwar, R. Sankolli and D. Chopra, CrystEngComm, 2013, 15, 4549–4564 RSC.
- M. T. Kirchner, D. Bläser, R. Boese and G. R. Desiraju, CrystEngComm, 2009, 11, 229–231 RSC.
- A. R. Choudhury and T. N. G. Row, CrystEngComm, 2006, 8, 265–274 RSC.
- G. Kaur, P. Panini, D. Chopra and A. Roy Choudhury, Cryst. Growth Des., 2012, 12, 5096–5110 CrossRef CAS.
- M. Karanam and A. R. Choudhury, Cryst. Growth Des., 2013, 13, 4803–4814 CrossRef CAS.
- G. Kaur and A. R. Choudhury, CrystEngComm, 2015, 17, 2949–2963 RSC.
- R. Kakkar, MedChemComm, 2019, 10, 351–368 RSC.
- R. Nath, S. Pathania, G. Grover and M. J. Akhtar, J. Mol. Struct., 2020, 128900 CrossRef CAS.
- G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed.
- M. N. Ahmed, M. Arif, F. Jabeen, H. A. Khan, K. A. Yasin, M. N. Tahir, A. Franconetti and A. Frontera, New J. Chem., 2019, 43, 8122–8131 RSC.
- Alzheimer's Disease International, The global voice on dementia, The Global Impact of Dementia 2013–2050, p. 1 Search PubMed.
- N. Khunnawutmanotham, N. Chimnoi, P. Saparpakorn and S. Techasakul, Bioorg. Chem., 2016, 65, 137–145 CrossRef CAS PubMed.
- Q. Zhang, C. Hao, Y. Miao, Y. Yun, X. Sun, Y. Pan, J. Sun and X. Wang, New J. Chem., 2021, 45, 17287–17300 RSC.
- A. B. Rodgers, Alzheimer's disease: unraveling the mystery, National Institutes of Health, 2002 Search PubMed.
- C. Dan, P. Ya-fei, L. Chuan-jun, X. Yun-feng and J. Yu-ren, Virtual screening of acetylcholinesterase inhibitors, IntechOpen, 2012 Search PubMed.
- (a) S. Behrens, G. B. Rattinger, S. Schwartz, J. Matyi, C. Sanders, M. S. DeBerard, C. G. Lyketsos and J. T. Tschanz, Int. Psychogeriatr., 2018, 30, 1499–1507 CrossRef PubMed; (b) Alzheimer's Association, FDA-Approved treatments for Alzheimer's, July, 2007, Retrieved September 21, 2015, from http://www.alz.org/texascapital/documents/topicsheet_treatments.pdf Search PubMed.
- I. Abbasi, H. Nadeem, A. Saeed, H. A. A. Kharl, M. N. Tahir and M. M. Naseer, Bioorg. Chem., 2021, 105385 CrossRef CAS PubMed.
- S. Khatoon, A. Aroosh, A. Islam, S. Kalsoom, F. Ahmad, S. Hameed, S. W. Abbasi, M. Yasinzai and M. M. Naseer, Bioorg. Chem., 2021, 110, 104816 CrossRef CAS PubMed.
- H. Pervez, N. Khan, J. Iqbal, S. Zaib, M. Yaqub, M. N. Tahir and M. M. Naseer, Heterocycl. Commun., 2018, 24, 51–58 CAS.
- M. Zia, S. Hameed, A. Frontera, E. Irran and M. M. Naseer, CrystEngComm, 2021, 23, 3144–3151 RSC.
- M. M. Naseer, M. Hussain, A. Bauzá, K. M. Lo and A. Frontera, ChemPlusChem, 2018, 83, 881–885 CrossRef CAS PubMed.
- M. M. Naseer, A. Bauzá, H. Alnasr, K. Jurkschat and A. Frontera, CrystEngComm, 2018, 20, 3251–3257 RSC.
- M. Hussain, A. Bauzá, A. Frontera, K. M. Lo and M. M. Naseer, CrystEngComm, 2018, 20, 150–154 RSC.
- H. Pervez, M. Ahmad, T. B. Hadda, L. Toupet and M. M. Naseer, J. Mol. Struct., 2015, 1098, 124–129 CrossRef CAS.
- R. Jawaria, M. Hussain, Z. Shafiq, H. B. Ahmad, M. N. Tahir, H. A. Shad and M. M. Naseer, CrystEngComm, 2015, 17, 2553–2561 RSC.
- Y. Pájaro, Á. Sathicq, E. Puello-Polo, A. Pérez, G. Romanelli and J. Trilleras, J. Chem., 2017, 2017, 6175315 CrossRef.
- Z. Zhang, C. Qiu, Y. Xu, Q. Han, J. Tang, K. P. Loh and C. Su, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- M. Vettorazzi, L. Vila, S. Lima, L. Acosta, F. Yépes, A. Palma, J. Cobo, J. Tengler, I. Malik and S. Alvarez, Arch. Pharm., 2019, 352, 1800298 CrossRef PubMed.
- G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441–449 CrossRef CAS PubMed.
- R. Custelcean, N. L. Engle and P. V. Bonnesen, CrystEngComm, 2007, 9, 452–455 RSC.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- I. Silman and J. L. Sussman, Curr. Opin. Pharmacol., 2005, 5, 293–302 CrossRef CAS PubMed.
- H. Dvir, I. Silman, M. Harel, T. L. Rosenberry and J. L. Sussman, Chem.-Biol. Interact., 2010, 187, 10–22 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2090877. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra08286h |
| This journal is © The Royal Society of Chemistry 2022 |