
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of novel bone targeting peptide–drug conjugate of 13-aminomethyl-15-thiomatrine for osteoporosis therapy†
Jia Su‡
a,
Chao Liu‡ b,
Haohao Baib,
Wei Congb,
Hua Tangb,
Honggang Hub,
Li Su*b,
Shipeng He*b and
Yong Wang*a
b,
Haohao Baib,
Wei Congb,
Hua Tangb,
Honggang Hub,
Li Su*b,
Shipeng He*b and
Yong Wang*a
aDepartment of Orthopaedics, Wenzhou Hospital of Integrated Traditional Chinese and Western Medicine, Zhejiang, China. E-mail: wyhpi008@163.com
bInstitute of Translational Medicine, Shanghai University, Shanghai, China. E-mail: suli1020@shu.edu.cn; heshipeng@shu.edu.cn
First published on 20th December 2021
Abstract
13-Aminomethyl-15-thiomatrine (M19) previously developed by our research group was a promising candidate for novel anti-osteoporosis drug development. However, the application of M19 was limited by its unsatisfactory druggability including poor chemical stability, excessively broad pharmacological activity and some degree of cytotoxicity. To solve these problems, M19-based bone targeting and cathepsin K sensitive peptide–drug conjugates (BTM19-1, BTM19-2 and BTM19-3) were developed to realize precise drug release in the bone tissue. Subsequent studies showed a rapid drug release process via cathepsin K digestion but sufficient stability over several hours in chymotrypsin. Besides, greatly improved chemical stability and strong hydroxyapatite binding affinity were also demonstrated. In biological evaluation studies, these PDCs showed less cytotoxicity and similar osteoclast inhibitory activity compared with the prototype drug. The optimal BTM19-2 could serve as a suitable candidate for further osteoporosis therapy research.
Introduction
Osteoporosis, a bone disease characterized by systematic loss of bone mass and deterioration of bone microstructure, affects 200 million people worldwide and causes 1.6 million hip fractures and 7.4 million other fractures every year, and is regarded as a public health problem.1–3 Nowadays, many drugs have been developed and used clinically for osteoporosis therapy, such as bisphosphonates, selective estrogen receptor modulators, teriparatide and so on, which have already achieved certain efficacy in clinical application. However, there were still some limitations and side effects in long-term use and dose maintenance.4–6Matrine was a quinolizidine alkaloid isolated from the roots of Sophora flavescens, Sophora tonkinensis and Sophora alopecuroides which showed a variety of pharmacological activities including antibacterial, anti-arrhythmia, anti-tumor, anti-fibrosis and anti-inflammatory, etc.7–11. In our previous study, M19 was developed and represented the new generation of highly active matrine derivative because of its great inhibitory effect on pro-inflammatory cytokine TNF-α and NF-κB transcriptional activity, making it a promising anti-inflammatory drug candidate.9 Recently, we showed that M19 could block NF-κB, AKt, MAPK and other signalling pathways by stabilizing ribosomal protein S5 (RPS5), thereby inhibiting RANKL-induced osteoclast differentiation and alleviating bone loss in ovariectomized mice.12 However, direct development of M19 into anti-osteoporosis drug was generally limited. First of all, the excessively broad pharmacological activity of M19 may bring the risk of off-target effects.13–15 Besides, the biological particularity of bone tissue, such as high tissue density and poor permeability, brought great difficulties to drug delivery.16 More importantly, the druggability of M19 was not satisfactory owing to its poor chemical stability and strong alkalinity. Therefore, novel drug design strategies need to be applied to realize its role as anti-osteoporosis drug.
Peptide–drug conjugates (PDCs), novel prodrug modification strategy, have been widely employed in the development of anti-malignant tumor drugs.17–21 By covalently coupling functional peptides to drugs with specific linker, PDCs could selectively deliver drugs to target cells/tissues/organs, reducing systemic toxicity and improving pharmacokinetic and pharmacodynamic parameters.22,23 Inspired by their promising success in targeted cancer therapy, we envisioned that conjugation with bone targeting peptide would make M19 to have bone-targeting characteristics as well as improve its anti-osteoporosis potency. Herein, M19-based bone targeting PDCs were rationally developed by coupling M19 with bone targeting peptide and cathepsin K sensitive smart linker through suitable spacers. These PDCs showed excellent specificity for hydroxyapatite (HA), the composition of bone tissue and tooth, and inhibitory activity on osteoclast differentiation, providing a valuable example for overcoming the shortcomings of natural product source compounds and improve their druggability.
Results and discussion
Rational design of M19-based bone targeting PDCs
To rational design bone-targeting PDCs, bone-targeting selective peptides and enzyme sensitive linkers need to be considered. First of all, as a special connective tissue, bone tissue was composed of a variety of cells and intercellular bone matrix with high inorganic salt content which was mainly composed of HA. In previous studies, tetracycline,24 bisphosphonates,25 hydroxymalonic acids26 and small heterocyclic compounds27 were often used in design of bone targeting drug molecules or drug carriers. In recent years, more and more peptide sequences with excellent bone targeting ability have been found, such as polyaspartic acid (hydroxyapatite targeting),28 Ser–Asp–Ser–Ser–Asp (osteoblasts specific factor-2 targeting),29 Ser–Thr–Phe–Thr–Lys–Ser–Pro (hematopoietic stem cell targeting)30 and (Asp–Ser–Ser)6 (hydroxyapatite targeting),31 etc. Compared with bone-targeting small molecules, these peptides have clear mechanism, high targeting affinity and low side effects.32 Since M19 has been found excellent inhibitory effect on osteoclasts, rather than promotion effect on osteoblasts, moderate length polyaspartic acids (Asp–Asp–Asp–Asp–Asp–Asp), whose HA binding affinity was comparable to tetracycline and calcein,28 was chosen as the target peptide sequence for this work.Secondly, it was reported that osteoclasts effectively degraded extracellular matrix proteins through secretion of cathepsin K, which was a crucial protease for the degradation of bone organic matrix collagen type I and type II and specifically distributed in Howship's lacunae formed by osteoclasts and osteocytes.33,34 Therefore, peptide sequences sensitive to cathepsin K could be served as cleavable linker of PDCs for drug releasing in bone tissue. At present, some peptide substrates such as GGGMGPSGPWGGK35 and GHPGGPQGKC36 have been employed in researches of bone relative material development. However, amino acids would remain on the material after specific degradation which was not suitable for PDCs use. Notably, Bossard and co-workers37 found that a series of short peptide (1–3 amino acids) were specific substrates of cathepsin K. And among them, the substrate Cbz–Leu–Arg–AMC (AMC, 7-amino-4-methylcoumarin) could quickly and specifically release AMC under the treatment of cathepsin K with high apparent secondary rate constant, making dipeptide Leu–Arg an ideal cleavable linker in this work.
In addition, in order to increase the overall hydrophilicity of PDCs and avoid steric hindrance, a polyethylene glycol spacer and 1,6-self-immolative p-aminobenzyloxycarbonyl (PABC) structure were introduced between the polyaspartic acids, sensitive dipeptide and drug molecule (Fig. 1).
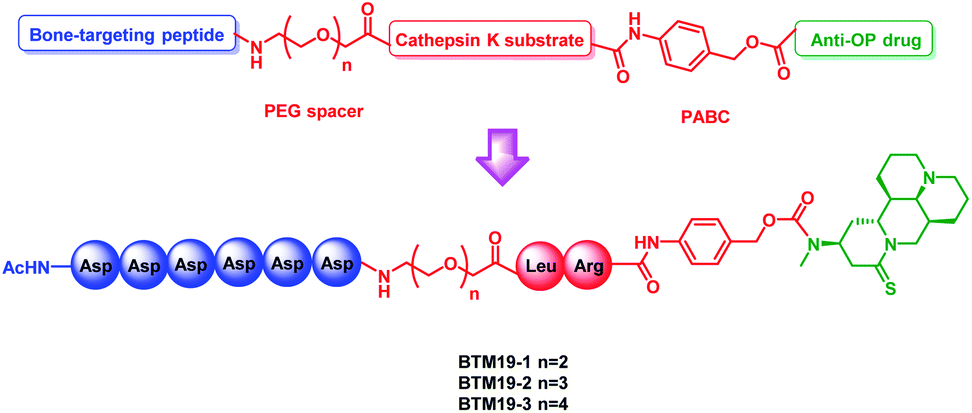 | ||
| Fig. 1 General design of the novel M19-based bone targeting PDCs. PEG = polyethylene glycol; PABC = p-aminobenzyloxycar-bonyl. | ||
Synthesis of M19-based bone targeting PDCs
To begin with, thiosophocarpine (2) was obtained by reacting commercially available sophocarpine (1) with Lawesson's reagent in toluene solution. In this step, viscous insoluble by-products generated in the reaction system need to be removed continuously to ensure the smooth progress of the reaction. Subsequent Michael addition reaction between 2 and methylamine provided M19 in a yield of 65% (Scheme 1A).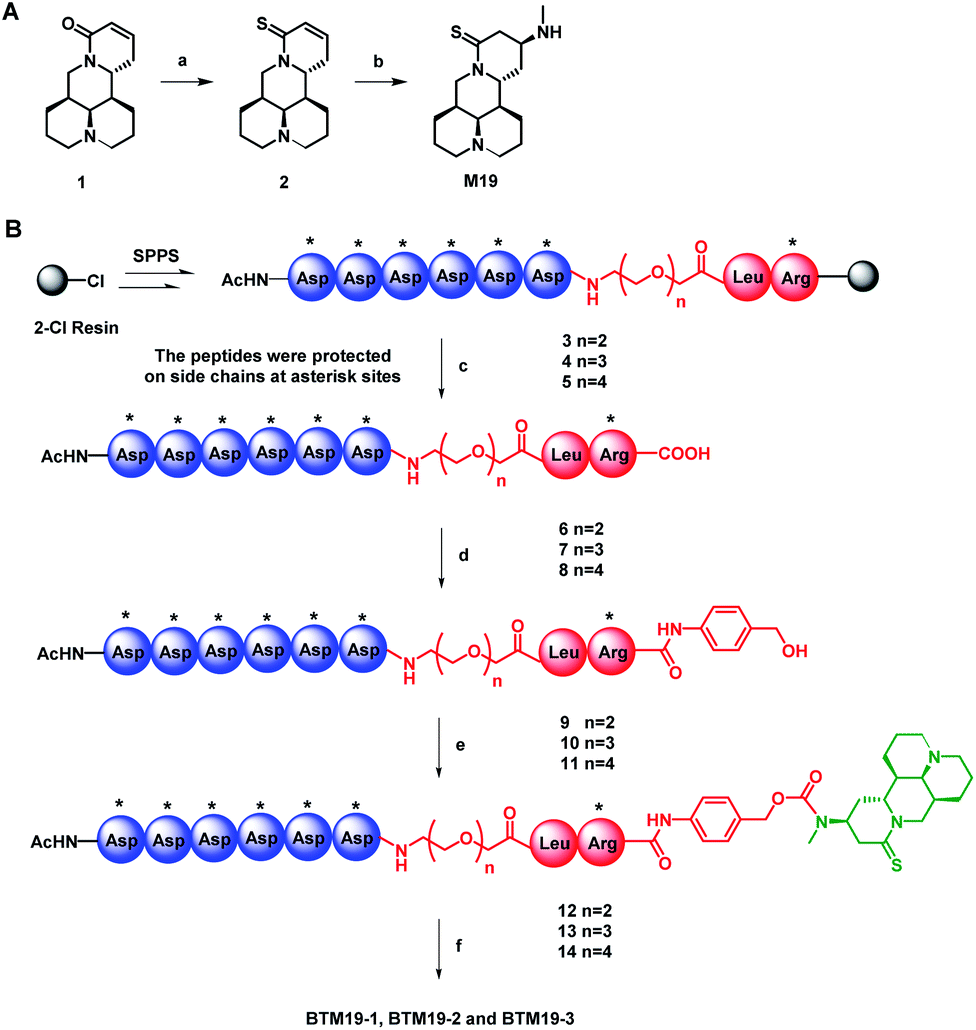 | ||
Scheme 1 The synthesis route of M19 (A) and PDCs (B). Reagents and conditions: (a) Lawesson's reagent, toluene, reflux, 2 h, 37%; (b) methylamine ethanol solution, rt, 12 h, 65%; (c) TFE/DCM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v), rt, 4 h, 80–83%; (d) 4-aminophenyl methanol, HOBt, DIC, DMF, rt, 2 h, 74–79%; (e) (i) triphosgene, activated carbon, THF, rt, 12 h; (ii) M19, Et3N, DMF, rt, 12 h, 78–86% in two steps; (f) TFA/water/EDT/TIPs (95:2:2:1, v/v/v/v), rt, 2 h, 60–63%. The resin-bound peptides were protected on side chains at asterisk sites: tert-butyl (tBu; for Asp) and (2,2,4,6,7-pentamethyl-2,3-dihydro-1-benzofuran-5-yl)sulfonyl (Pbf; for Arg). :3, v/v), rt, 4 h, 80–83%; (d) 4-aminophenyl methanol, HOBt, DIC, DMF, rt, 2 h, 74–79%; (e) (i) triphosgene, activated carbon, THF, rt, 12 h; (ii) M19, Et3N, DMF, rt, 12 h, 78–86% in two steps; (f) TFA/water/EDT/TIPs (95:2:2:1, v/v/v/v), rt, 2 h, 60–63%. The resin-bound peptides were protected on side chains at asterisk sites: tert-butyl (tBu; for Asp) and (2,2,4,6,7-pentamethyl-2,3-dihydro-1-benzofuran-5-yl)sulfonyl (Pbf; for Arg). | ||
With the prototype drug in hand, the next step was the synthesis of on-resin peptide precursors Asp(OtBu)6–PEGn–Leu–Arg(Pbf)–Resin (3–5) concluding poly-aspartic acid polypeptide, polyethylene glycol of different lengths and leucine–arginine sequence through standard SPPS process using [(6-chloro-1H-benzotriazol-1-yl)oxy](dimethylamino)-N,N-dimethylmethani-minium hexafluorophosphate (HCTU) and N,N-diisopropylethyl-amine (DIPEA) as coupling reagents. After cleavage from the resin with trifluoroethanol, full protected intermediates Asp(OtBu)6–PEGn–Leu–Arg(Pbf) (6–8) were coupled with 4-aminobenzyl alcohol by 1H-1,2,3-benzotriazole (HOBt) and N,N′-diisopropylcarbodiimide to construct 1,6-self-immolative p-aminobenzyloxycarbonyl (PABC) linker containing intermediates Asp(OtBu)6–PEGn–Leu–Arg(Pbf)–PABC (9–11). The following reaction between 9–11 with triphosgene and M19 in the presence of DIPEA given protected PDCs Asp(OtBu)6–PEGn–Leu–Arg(Pbf)–PABC–M19 (12–14). Finally, global deprotection of compounds 12–14 with K reagent obtained the target PDCs (BTM19-1, BTM19-2 and BTM19-3). Pure products were obtained as a freeze-dried powder with a good chemical yield of 59–63% after purification by preparative reverse phase chromatography and subsequent lyophilization (Scheme 1B).
Chemical stability studies of M19-based bone targeting PDCs
In our previous research, we found that the chemical stability of M19 was unsatisfactory. It spontaneously converted to thiosophocarpine through retro-Michael reaction in the solution at room temperature. Therefore, we firstly conducted chemical stability investigation of the synthesized PDCs (Fig. 2A and ESI Fig. S1†). M19 and BTM19-1, BTM19-2 and BTM19-3 were dissolved in water/MeCN solutions and incubated for 14 days at room temperature respectively. As shown in Fig. 2A, M19 decomposed into thiosophocarpine as anticipation with a half-life of 1.7 days. While PDCs were relatively stable within 14 days' incubation without any significant degradation. It suggested that, benefitting from PDCs construction, the chemical stability of prototype drug M19 was greatly improved. One possible explanation was that the newly generated amide bond structure stabilized the lone pair of the 13-position aminomethyl group via electron withdrawing conjugate.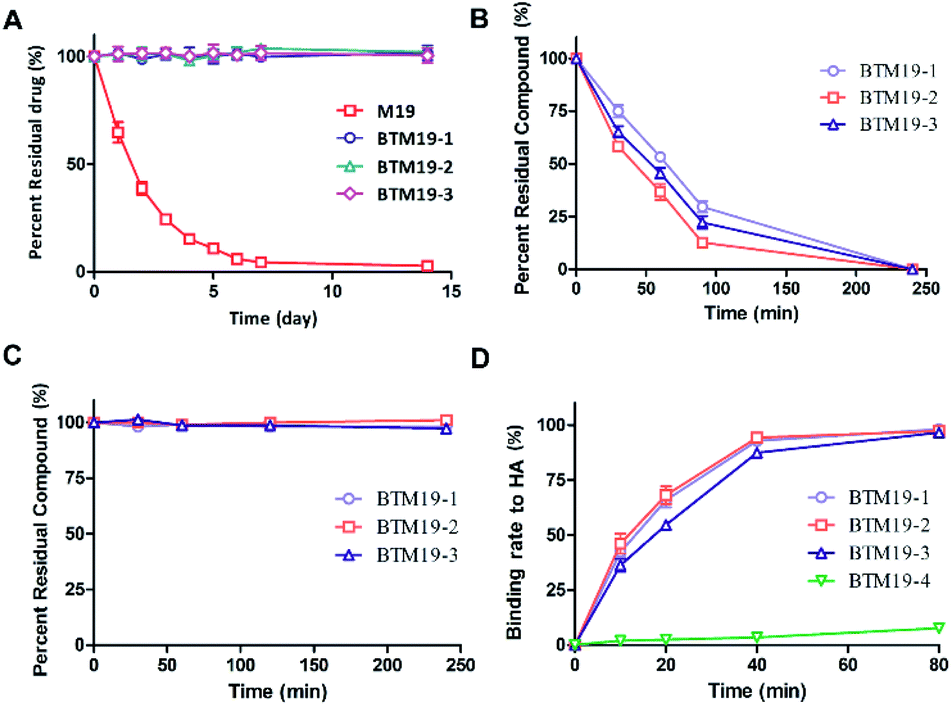 | ||
| Fig. 2 (A) Quantitative data of chemical stability study of M19 vs. PDCs in water/MeCN solution at rt; (B) quantitative data of drug release study of PDCs at pH 5.5 and 37 °C with cathepsin K; (C) quantitative data of proteolytic stability study of PDCs under a-chymotrypsin treatment C; (D) quantitative data of binding study of PDCs with hydroxyapatite at pH = 5.5 and 37 °C; data points were displayed as the mean value SEM of duplicate independent experiments. | ||
Drug release properties of M19-based bone targeting PDCs with cathepsin K
According to our design, the bone targeting PDCs should release M19 under conditions prevailing around osteoclasts by the cathepsin K digestion and following degradation of 1,6-self-immolative PABC linker. Besides, the pH optimum of cathepsin K was documented to be pH 5.5 depending on the nature of the substrate.38 Thus, we investigated the drug release properties in an acidic system to simulate the Howship's lacunae in bones (Fig. 2B and ESI Fig. S2†). As shown in Fig. 2B, under the specific conditions (cathepsin K 5 nM, 100 mM Na acetate at pH 5.5 containing 20 mM cysteine and 5 mM EDTA), both prodrugs were cleavaged and released prototype M19. The half-life was 54 minutes for BTM19-1, 48 minutes for BTM19-2 and 51 minutes for BTM19-3 and all the PDCs could be completely cleavaged after 240 minutes' treatment.Enzymatic stability studies of M19-based bone targeting PDCs
The protease stability was an important criterion for biological activity of synthesized PDCs, which ensured the transportation of active cargo to the target cell, tissue and/or organ.39 To determine their hydrolytic enzymes tolerance, BTM19-1, BTM19-2 and BTM19-3 were incubated with chymotrypsin at 37 °C and analysed by HPLC (Fig. 2C and ESI Fig. S3†). It was found that the cathepsin K cleavable PDCs were stable under these conditions with no M19 or other degradation products observed after 240 minutes' exposure.Hydroxyapatite binding affinity of M19-based bone targeting PDCs
Subsequently, we assessed the affinity between the novel bone targeting PDCs and the bone mineral HA. Fifteen equivalents of HA were incubated with BTM19-1, BTM19-2 and BTM19-3 at 37 °C and pH 5.5 to simulate the acidic circumstance in the Howship's lacunae. As shown in Fig. 2D and S4 (ESI†), approximately 50% of both PDCs were bound to hydroxyapatite after 20 minutes' treatment and it reached over 95% after 80 minutes. In order to have a more comprehensive understanding, the negative control BTM19-4 with a scrambled peptide sequence was successfully realized (ESI Scheme S1†). When BTM19-4 was incubated with HA powder under the same condition, only very weak binding could be observed after 80 minutes' incubation. These results suggested that the high affinity between M19 based PDCs with HA was generated from the polyaspartic acid moiety rather than M19 itself.Cytotoxicity of M19-based bone targeting PDCs
In previous studies, M19 performed a certain degree of cytotoxicity, especially to liver cells. Thus, CCK-8 analysis was performed on RAW264.7 cells to test their potential toxicity (ESI Fig. S5†). The results showed that the prototype drug M19 exhibited about 20% cell viability inhibition at concentration of 10 μM, while little significant cytotoxicity at a concentration range of 1–10 μM was observed for all PDCs, indicated that the introduction of the bone-targeting moiety reduced the toxicity of the prototype drug, enlightening us that the cytotoxicity of M19 may originated from 13-position aminomethyl group, especially related to its alkalinity.In vitro osteoclast inhibition activity of M19-based bone targeting PDCs
The next move was to evaluate their biological activity against RANKL-induced osteoclast differentiation on RAW264.7 cells via tartrate-resistant acid phosphatase (TRAP) staining (Fig. 3 and ESI Fig. S6†). Different concentrations of PDCs and M19 were co-cultured with RANKL and colony stimulating factor 1 (CSF-1) in RAW264.7 cells. After 3 days' treatment, both compounds exhibited dose-dependently inhibitory effects on the TRAP activity and the IC50 was 2.41 μM (M19), 4.38 μM (BTM19-1), 2.62 μM (BTM19-2) and 2.84 μM (BTM19-3). These results indicated that, with the exception of BTM19-1, the osteoclast suppression effect produced by the M19-based PDCs was equivalent to that of the free M19, confirming the bone-targeted PDC modification strategy did not significantly affect the activity of the prototype drug. However, BTM19-1 exhibited a heavily decreased activity. A reasonable speculation might be that the steric hindrance caused by its shorter PEG linker prevented cathepsin K from substrates cleaving, which delayed the release of the prototype drug.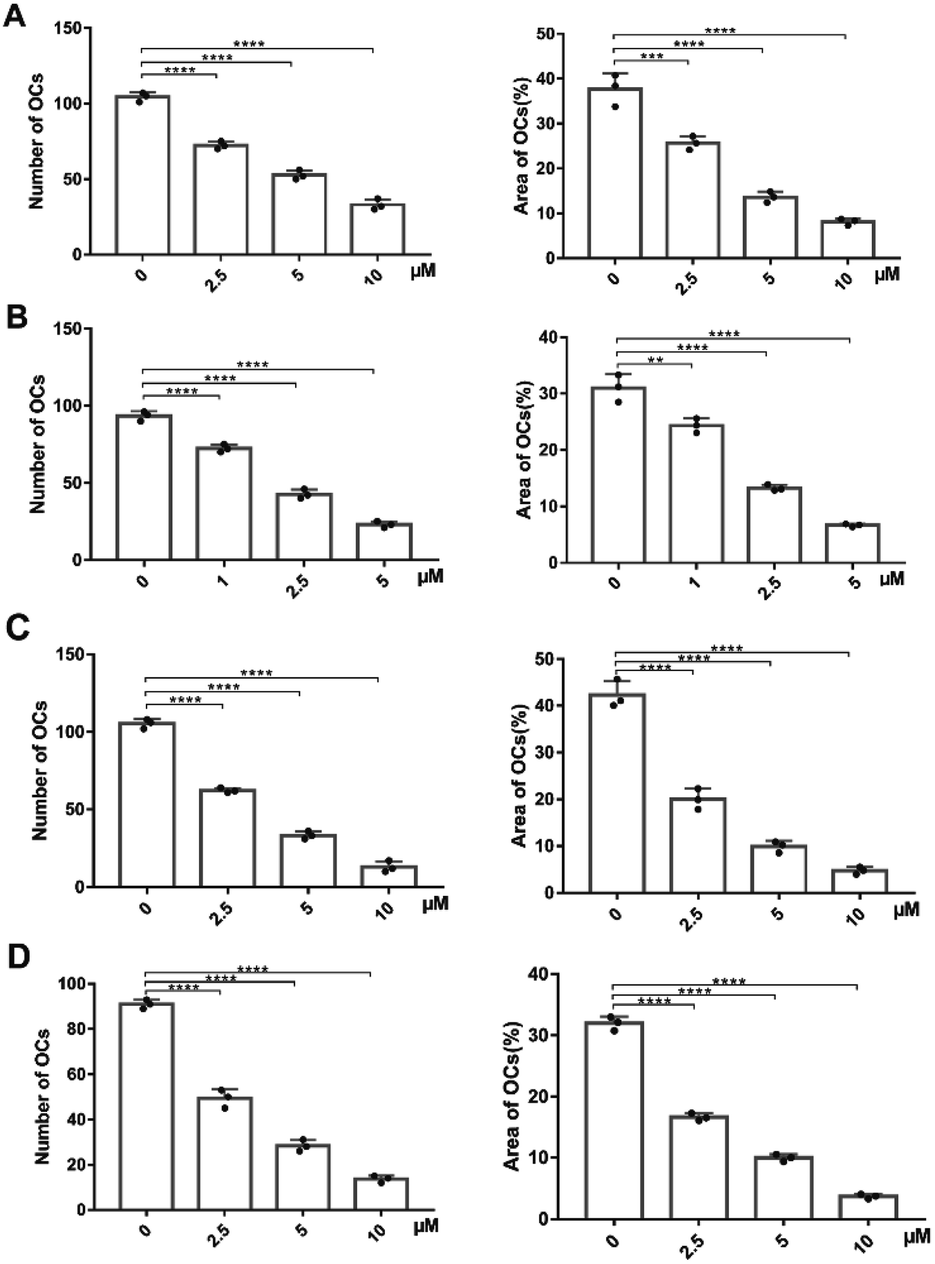 | ||
| Fig. 3 Quantitative data of TRAP-positive cells formation from RAW264.7 cells with the treatment of M19 (A), BTM19-1 (B), BTM19-2 (C) and BTM19-3 (D) respectively based on numbers (left) and area (right) (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). | ||
Experimental
General methods
:1–10:1, DCM/MeOH) to afford 2 as a yellow powder (3.9 g, 37%). 1H-NMR (600 MHz, CDCl3): δ 6.47–6.46 (m, 1H), 6.08–6.05 (m, 1H), 5.09 (dd, J = 6 Hz, J = 18 Hz, 1H), 4.33 (s, 1H), 3.03 (s, 1H), 2.92 (s, 2H), 2.68–2.64 (m, 1H), 2.36–2.31 (m, 2H), 2.05 (s, 3H), 1.85–1.80 (m, 3H), 1.72–1.69 (m, 1H), 1.63–1.55 (m, 1H), 1.50–1.42 (m, 3H); 13C-NMR (600 MHz, CDCl3): δ 189.76, 131.95, 127.59, 63.77, 57.01, 54.34, 50.79, 39.96, 34.92, 27.47, 26.55, 24.96, 20.89, 20.35; ESI-MS m/z calculated for C15H22N2S 262.15; found [M + H]+ = 263.2.:1–10:1, DCM/MeOH) to afford 3 as a yellow powder (2.9 g, 65%). 1H-NMR (600 MHz, CDCl3): δ 5.43 (dd, J = 6 Hz, J = 12 Hz, 1H), 4.26–4.24 (m, 1H), 3.52 (t, J = 12 Hz, 1H), 3.24–3.21 (m, 1H), 2.97–2.92 (m, 1H), 2.88–2.80 (m, 3H), 2.24 (s, 3H), 2.17 (s, 1H), 2.04–1.98 (m, 2H), 1.94–1.88 (m, 4H), 1.74–1.71 (m, 2H), 1.64–1.61 (m, 2H), 1.59–1.55 (m, 2H), 1.48–1.44 (m, 3H); 13C-NMR (600 MHz, CDCl3): δ 195.46, 63.55, 56.98, 56.96, 55.58, 50.90, 49.39, 47.91, 42.69, 35.63, 33.64, 30.38, 27.63, 26.56, 21.12, 20.59; ESI-MS m/z calculated for C16H27N3S 293.19; found [M + H]+ = 294.2.:3, v/v) was added to the resin at room temperature. After stirring for 2 hours, the cleavage cocktail was collected and concentrated. The chilled diethyl ether was added to the concentrated residue to precipitate the crude peptides. The peptide suspensions were centrifuged for 3 minutes at 3000 rpm and then the clear solution was decanted to afford 6–8 and 15.6 as a white powder (1.43 g, 82%). HR-MS m/z calculated for C81H132N12O28S 1752.8995; found [M + 2H]2+ = 877.4609.
7 as a white powder (1.5 g, 83%). HR-MS m/z calculated for C83H136N12O29S 1796.9257; found [M + 2H]2+ = 899.4961.
8 as a white powder (1.47 g, 80%). HR-MS m/z calculated for C85H140N12O30S 1840.9519; found [M + 2H]2+ = 921.4898.
16 as a white powder (1.6 g, 87%). HR-MS m/z calculated for C90H150N14O24S 1843.0668; found [M + 2H]2+ = 922.5041.
9 as a white powder (1.12 g, 79%). HR-MS m/z calculated for C88H139N13O28S 1857.9573; found [M + 2H]2+ = 929.9841.
10 as a white powder (1.18 g, 75%). HR-MS m/z calculated for C90H143N13O29S 1901.9835; found [M + 2H]2+ = 951.9905.
11 as a white powder (1.15 g, 74%). HR-MS m/z calculated for C92H147N13O30S 1946.0098; found [M + 2H]2+ = 974.0215.
16 as a white powder (125 g, 74%). HR-MS m/z calculated for C97H157N15O24S 1948.1247; found [M + 2H]2+ = 975.0709.
12 as a white powder (1.1 g, 78%). HR-MS m/z calculated for C105H164N16O29S2 2177.1292; found [M + 2H]2+ = 1089.5528.
13 as a white powder (1.07 g, 85%). HR-MS m/z calculated for C107H168N16O30S2 2221.1554; found [M + 2H]2+ = 1111.5709.
14 as a white powder (1.09 g, 86%). HR-MS m/z calculated for C109H172N16O31S2 2265.1816; found [M + 2H]2+ = 1133.5828.
17 as a white powder (1.05 g, 76%). HR-MS m/z calculated for C114H182N18O25S2 2267.2965; found [M + 3H]3+ = 756.7441.
:2:2:1, v/v/v/v) was added to the Asp(OtBu)6–PEGn–Leu–Arg(Pbf)–PABC–M19 at room temperature. After stirring for 2 hours, the solvent was concentrated. The chilled diethyl ether was added to the concentrated residue to precipitate the crude peptides. The peptide suspensions were centrifuged for 3 minutes at 3000 rpm and then the clear solution was decanted. The resulting white residues were dissolved in MeCN/H2O, analysed by HPLC and HR-MS and purified by RP-HPLC to afford BTM19-1, BTM19-2, BTM19-3 and BTM19-4.BTM19-1 as a white powder (492 mg, 62%). HR-MS m/z calculated for C68H100N16O26S 1588.6715; found [M + H]+ = 1589.6329.
BTM19-2 as a white powder (493 mg, 63%). HR-MS m/z calculated for C70H104N16O27S 1632.9678; found [M + 2H]2+ = 817.4979.
BTM19-3 as a white powder (443 mg, 60%). HR-MS m/z calculated for C72H108N16O28S 1676.7240; found [M + 2H]2+ = 839.3631.
BTM19-4 as a white powder (458 mg, 59%). HR-MS m/z calculated for C80H126N18O20S 1690.9116; found [M + 2H]2+ = 846.4346.
Chemical stability studies
To investigate the degradation rates, BTM19-1, BTM19-2, BTM19-3 and M19 were dissolved in water/MeCN solution (9:1, v/v, 0.1 mg mL−1) and stirred at room temperature respectively. The percentage of residue was monitored by HPLC at 0, 1, 2, 3, 4, 5, 6, 7 and 14 days.
Drug release studies
820 mg sodium acetate, 242 mg cysteine and 146 mg EDTA were dissolved in 100 mL PBS buffer solution and adjusted pH = 5.5 to afford buffer solution. 10 μL of 0.5 mg mL−1 cathepsin K active human solution was diluted 2000 times for later use. 1 μM BTM19-1, BTM19-2 and BTM19-3 were respectively dissolved with 1 mL buffer solution and then diluted 10 times for later use. 100 μL of cathepsin K solution and PDCs solution was mixed and incubated in 37 °C water bath. Aliquots (20 μL) were taken after 0, 0.5, 1, 1.5 and 4 hours and analyzed by HPLC.Hydroxyapatite binding assay
65.2 mg of sodium phosphate and 870 mg of sodium chloride was dissolved in 100 mL PBS buffer solution and adjusted pH = 5.5 to afford buffer solution. 1 μM BTM19-1, BTM19-2 and BTM19-3 were respectively dissolved with 1 mL buffer solution and diluted 3 times for later use. 15 mg of HA was added to 1 mL of PDCs solution and incubated in 37 °C water bath. Aliquots (20 μL) were taken after 0, 10, 20, 40, and 80 minutes and analyzed by HPLC.Enzymatic stability studies
222 mg of calcium chloride was dissolved in 1000 mL PBS buffer solution and adjusted pH = 7.4 to afford buffer solution. 5 mg of chymotrypsin was dissolved in 1 mL the above buffer solution and diluted 1000 times for later use. 1 μM BTM19-1, BTM19-2 and BTM19-3 were respectively dissolved with 1 mL buffer solution to prepare the stock solution. 1950 μL of the chymotrypsin solution was mixed with 50 μL of PDCs solution and incubated in 37 °C water bath. Aliquots (20 μL) were taken after 0, 0.5, 1, 2 and 4 hours and analysed by HPLC.Cytotoxicity test
RAW264.7 cells were cultured in 96 well plates (1 × 105 mL−1, 200 μL per well) for 48 hours co-cultured with designed PDCs and M19 at the indicated concentration (0, 1.25, 2.5, 5 and 10 μM). Then, 10 μL of CCK-8 reagent was added and incubated for 4 hours in a 37 °C protected from light. The absorbance (OD) at 450 nm was recorded on the microplate reader. Cell survival rate = OD value of the experimental group/OD value of the blank control group; cytotoxic effect = 1 − cell survival rate.In vitro osteoclast inhibition test
Murine osteoclast precursors from 8 week-old C57BL/6 male mice were cultured in 96 well plates (1 × 105 mL−1, 200 μL per well) for 3 days co-cultured with 20 ng mL−1 murine CSF-1 and 300 ng mL−1 sRANKL with or without treatment of designed PDCs and M19 at the indicated concentration (0, 1.25, 2.5, 5 and 10 μM). Then, the fixed cells were stained using tartrate-resistant acid phosphatase kit (Sigma). TRAP-positive multinucleated cells were observed by a microscope (OLYMPUS-BX53).Conclusions
In summary, current efforts of PDCs were mainly focused on the targeted therapy of various malignant tumor, however, the research works of anti-osteoporotic PDCs have not been reported. These tailor-made thiomatrine derivative based bone-targeted PDCs for osteoporosis therapy developed in this work showed high cathepsin K sensitivity and affinity for hydroxyapatite in vitro. Besides, they not only performed satisfactory chemical and enzyme stability, but also lower cytotoxicity and similar osteoclasts inhibitory activity compared to the prototype drug. The optimal PDC BTM19-2 was a suitable candidate for further evaluation in osteoporosis therapy research.Author contributions
Shipeng He and Yong Wang designed the experiments; Jia Su and Chao Liu conducted the experiments; Honggang Hu revised the manuscript; Haohao Bai, Wei Cong, Fei Gao and Hua Tang analyzed the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 91849129 and 82003567) and the sponsored by Shanghai Sailing Program (No. 20YF1414100).References
- O. Johnell and J. A. Kanis, Osteoporosis Int., 2006, 17, 1726–1733 CrossRef CAS PubMed.
- M. Lorentzon, J. Intern. Med., 2019, 285, 381–394 CrossRef PubMed.
- F. Salamanna, M. Maglio, V. Borsari, M. P. Landini and M. Fini, Trends Endocrinol. Metab., 2021, 32, 672–679 CrossRef CAS PubMed.
- T. D. Rachner, S. Khosla and L. C. Hofbauer, Lancet, 2011, 377, 1276–1287 CrossRef CAS.
- T. Matsumoto and I. Endo, J. Bone Miner. Metab., 2021, 39, 91–105 CrossRef CAS PubMed.
- D. Bonn, Lancet, 2004, 363, 786–787 CrossRef.
- Y. J. Zhou, Y. J. Guo, X. L. Yang and Z. L. Ou, J. Cancer, 2018, 9, 1357–1364 CrossRef PubMed.
- B. Zhang, Z. Y. Liu, Y. Y. Li, Y. Luo, M. L. Liu, H. Y. Dong, Y. X. Wang, Y. Liu, P. T. Zhao, F. G. Jin and Z. C. Li, Eur. J. Pharm. Sci., 2011, 44, 573–579 CrossRef CAS PubMed.
- H. Hu, S. Wang, C. Zhang, L. Wang, L. Ding, J. Zhang and Q. Wu, Bioorg. Med. Chem. Lett., 2010, 20, 7537–7539 CrossRef CAS PubMed.
- L. M. Gao, Y. X. Han, Y. P. Wang, Y. H. Li, Y. Q. Shan, X. Li, Z. G. Peng, C. W. Bi, T. Zhang, N. N. Du, J. D. Jiang and D. Q. Song, J. Med. Chem., 2011, 54, 869–876 CrossRef CAS PubMed.
- H. Y. Gao, G. Y. Li, M. M. Lou, X. Y. Li, X. Y. Wei and J. H. Wang, J. Inflammation, 2012, 9, 16 CrossRef CAS PubMed.
- X. Chen, X. Zhi, L. Cao, W. Weng, P. Pan, H. Hu, C. Liu, Q. Zhao, Q. Zhou, J. Cui and J. Su, Cell Death Dis., 2017, 8, e3037 CrossRef PubMed.
- Y. Zou, M. Sarem, S. Xiang, H. Hu, W. Xu and V. P. Shastri, BMC Cancer, 2019, 19, 949 CrossRef PubMed.
- J. Li, J. Xu, Y. Lu, L. Qiu, W. Xu, B. Lu, Z. Hu, Z. Chu, Y. Chai and J. Zhang, Molecules, 2016, 21, 649 CrossRef PubMed.
- J. Xu, Y. Qi, W. H. Xu, Y. Liu, L. Qiu, K. Q. Wang, H. G. Hu, Z. G. He and J. P. Zhang, Int. Immunopharmacol., 2016, 36, 59–66 CrossRef CAS PubMed.
- H. Hirabayashi and J. Fujisaki, Clin. Pharmacokinet., 2003, 42, 1319–1330 CrossRef CAS PubMed.
- S. D. Chipman, F. B. Oldham, G. Pezzoni and J. W. Singer, Int. J. Nanomed., 2006, 1, 375–383 CrossRef CAS PubMed.
- E. S. Kim, D. Kim, S. Nyberg, A. Poma, D. Cecchin, S. A. Jain, K. A. Kim, Y. J. Shin, E. H. Kim, M. Kim, S. H. Baek, J. K. Kim, T. R. Doeppner, A. Ali, J. Redgrave, G. Battaglia, A. Majid and O. N. Bae, Sci. Rep., 2020, 10, 699 CrossRef CAS PubMed.
- D. Mahalingam, G. Wilding, S. Denmeade, J. Sarantopoulas, D. Cosgrove, J. Cetnar, N. Azad, J. Bruce, M. Kurman, V. E. Allgood and M. Carducci, Br. J. Cancer, 2016, 114, 986–994 CrossRef CAS PubMed.
- T. B. Andersen, C. Q. López, T. Manczak, K. Martinez and H. T. Simonsen, Molecules, 2015, 20, 6113–6127 CrossRef CAS PubMed.
- C. P. Leamon, M. A. Parker, I. R. Vlahov, L. C. Xu, J. A. Reddy, M. Vetzel and N. Douglas, Bioconjugate Chem., 2002, 13, 1200–1210 CrossRef CAS PubMed.
- H. Su, J. M. Koo and H. Cui, J. Controlled Release, 2015, 219, 383–395 CrossRef CAS PubMed.
- W. Ma, A. G. Cheetham and H. Cui, Nano Today, 2016, 11, 13–30 CrossRef CAS PubMed.
- D. D. Perrin, Nature, 1965, 208, 787–788 CrossRef CAS PubMed.
- P. C. Bulman Page, J. P. G. Moore, I. Mansfield, M. J. McKenzie, W. B. Bowler and J. A. Gallagher, Tetrahedron, 2001, 57, 1837–1847 CrossRef.
- G. Caselli, M. Mantovanini, C. A. Gandolfi, M. Allegretti, S. Fiorentino, L. Pellegrini, G. Melillo, R. Bertini, W. Sabbatini, R. Anacardio, G. Clavenna, G. Sciortino and A. Teti, J. Bone Miner. Res., 1997, 12, 972–981 CrossRef CAS PubMed.
- T. M. Willson, B. R. Henke, T. M. Momtahen, P. L. Myers, E. E. Sugg, R. J. Unwalla, D. K. Croom, R. W. Dougherty, M. K. Grizzle, M. F. Johnson, K. L. Queen, T. J. Rimele, J. D. Yingling and M. K. James, J. Med. Chem., 1996, 39, 3030–3034 CrossRef CAS PubMed.
- S. Kasugai, R. Fujisawa, Y. Waki, K. Miyamoto and K. Ohya, J. Bone Miner. Res., 2000, 15, 936–943 CrossRef CAS PubMed.
- Y. Sun, X. Ye, M. Cai, X. Liu, J. Xiao, C. Zhang, Y. Wang, L. Yang, J. Liu, S. Li, C. Kang, B. Zhang, Q. Zhang, Z. Wang, A. Hong and X. Wang, ACS Nano, 2016, 10, 5759–5768 CrossRef CAS PubMed.
- G. S. Nowakowski, M. S. Dooner, H. M. Valinski, A. M. Mihaliak, P. J. Quesenberry and P. S. Becker, Stem Cells, 2004, 22, 1030–1038 CrossRef CAS PubMed.
- H. Wu, G. Zhang, B. S. Guo, T. Tao, G. Li, K. M. Lee, L. K. Hung and L. Qin, Bone, 2010, 47, S413–S414 CrossRef.
- T. A. Stone and C. M. Deber, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 577–585 CrossRef CAS PubMed.
- V. Everts, W. Korper, K. A. Hoeben, I. D. Jansen, D. Bromme, K. B. Cleutjens, S. Heeneman, C. Peters, T. Reinheckel, P. Saftig and W. Beertsen, J. Bone Miner. Res., 2006, 21, 1399–1408 CrossRef CAS PubMed.
- V. Everts, J. M. Delaissé, W. Korper, D. C. Jansen, W. Tigchelaar-Gutter, P. Saftig and W. Beertsen, J. Bone Miner. Res., 2002, 17, 77–90 CrossRef CAS PubMed.
- C. W. Hsu, R. M. Olabisi, E. A. Olmsted-Davis, A. R. Davis and J. L. West, J. Biomed. Mater. Res., Part A, 2011, 98, 53–62 CrossRef PubMed.
- K. Chang and F. Jaffer, J. Nucl. Cardiol., 2008, 15, 417–428 CrossRef PubMed.
- M. J. Bossard, T. A. Tomaszek, S. K. Thompson, B. Y. Amegadzie, C. R. Hanning, C. Jones, J. T. Kurdyla, D. E. McNulty, F. H. Drake, M. Gowen and M. A. Levy, J. Biol. Chem., 1996, 271, 12517–12524 CrossRef CAS PubMed.
- K. Date, H. Sakagami and K. Yura, Sci. Rep., 2021, 11, 12023 CrossRef CAS PubMed.
- K. Hochdörffer, K. Abu Ajaj, C. Schäfer-Obodozie and F. Kratz, J. Med. Chem., 2012, 55, 7502–7515 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08136e |
| ‡ Jia Su and Chao Liu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |