
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of all-atom force fields in viral capsid simulations and properties†
Ruijie D. Teo * and
D. Peter Tieleman*
* and
D. Peter Tieleman*
Centre for Molecular Simulation and Department of Biological Sciences, University of Calgary, Calgary, AB T2N 1N4, Canada. E-mail: ruijiedarius.teo@ucalgary.ca; tieleman@ucalgary.ca
First published on 21st December 2021
Abstract
As the past century has been characterized by waves of viral pandemics, there is an ever-growing role for molecular simulation-based research. In this study, we utilize all-atom molecular dynamics to simulate an enterovirus-D68 capsid and examine the dependency of viral capsid dynamics and properties on AMBER and CHARMM force fields. Out of the six force fields studied, we note that CHARMM36m and CHARMM36 generate secondary structures that are most consistent with protein structural data and sample the largest conformational space. The ion distribution and radius of gyration of the capsid are similar across all force fields investigated.
1. Introduction
Since the turn of the decade, Enterovirus D68 (EV-D68), a non-enveloped virus belonging to the Picornaviridae family,1 has been associated with a rise in cases worldwide,2 including an unprecedented 2014 outbreak occurring in the United States. The EV-D68 virion consists of an exterior pseudo icosahedral (T = 3) protein capsid encapsulating a positive-sense single-stranded RNA; the capsid itself consists of 60 copies of viral protein (VP) 1–4.3 In addition to causing acute flaccid myelitis, a paralytic condition similar to poliomyelitis, EV-D68 causes respiratory illnesses (and in some cases, death).Molecular dynamics (MD) simulations have improved our understanding of viruses, ranging from influenza A H1N1 to HIV-1.4 Investigating these processes using MD simulations, however, can be computationally challenging due to the mesoscale nature of a viral capsid such as EV-D68 which comprises of 240 protein monomers. An appropriate force field suitable for these simulations must be selected, along with the availability of adequate high-performance computing resources (in our case, the Cedar 3.6-petaflop supercomputer).
The myriad of additive protein force fields for MD simulations, such as AMBER, CHARMM and GROMOS, share the same functional form Etotal = Ebonded + Enon-bonded + Eother,5 where Ebonded represents bonded interactions (bond stretching, angle bending, dihedral rotation), Enon-bonded represents non-bonded interactions (electrostatics, dispersion, Pauli repulsion), and Eother represents other terms such as polarization effects and the coupling between bond lengths and angles. Compared to AMBER's energy function, the CHARMM dihedral energy function has a quadratic dependence on the improper dihedral angle and the angle bending energy function includes an additional Urey–Bradly term.5 While CHARMM does not scale 1,4-non-bonded interactions, AMBER scales it by 0.5.5 These differences arise due to the way these parameters are optimized and fitted to experimental and quantum mechanical (QM) data. For instance, partial charges in the AMBER and CHARMM force field are fitted to reproduce the electrostatic potential (gas-phase) and dimerization energies respectively. Likewise, AMBER φ/ψ dihedral parameters are fitted to structured peptide6 data while CHARMM φ/ψ parameters are fitted to MD trajectories of myoglobin7 and X-ray crystallographic data.8
The recent cryogenic electron microscopy (cryo-EM) structure3 of the EV-D68 capsid (Fig. 1) provides a good platform to assess the performance of six commonly used force fields on a mesoscale structure and their influence on structural dynamics and properties. To the best of our knowledge, there is only one other study9 that examined the dependence of viral capsid structure on the choice of force fields, albeit the GROMOS53a6 and ff03 (AMBER) force field. Thus, this study investigates the dependence of the EV-D68 capsid structure and properties on six atomistic AMBER and CHARMM force fields (ff99SB*-ILDNP,10–13 ff14SB,14 ff03*,12,15 C22*,7,16 C36,17 and C36m18), and the suitability of force fields in simulating these mesoscale systems is discussed below. Simulations were performed using 256 processors, with an average of 4.31 ns per day and occupying 4.04 terabytes of disk space in total (see Section 2 for details).
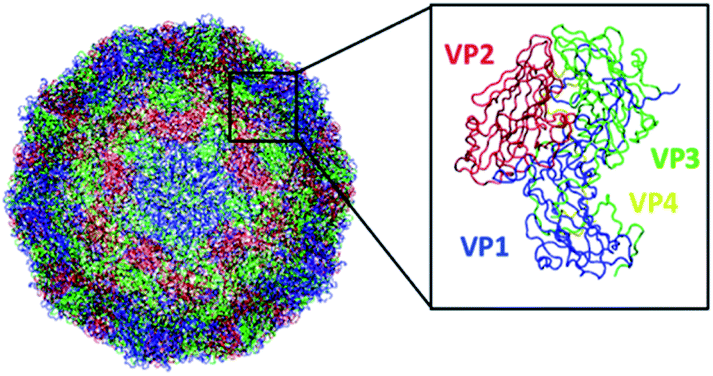 | ||
| Fig. 1 Structure of the EV-D68 capsid (PDB 6CSG) consisting of 60 copies of viral protein 1 (VP1), VP2, VP3, and VP4. This figure was generated using VMD. | ||
2. Computational methods
The structure belonging to the capsid of the human EV-D68 full native virion (without RNA) was taken from PDB ID 6CSG with a resolution of 2.17 Å. Missing residues were added using MODELLER.19,20 followed by six molecular dynamics (MD) simulations corresponding to the usage of different AMBER and CHARMM force fields: ff99SB*-ILDNP,10–13 ff14SB,14 ff03*,12,15 C22*,7,16 C36,17 and C36m.18 For each system, the final number of atoms is 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 169301. Frames were saved every 5000 steps or 10 ps, occupying 4.04 terabytes of disk space in total. Atomistic MD simulations were performed with the respective force field and TIP3P water21 using the GROMACS package, version 2020.2.22 As recommended for CHARMM force fields, C36(m) was used with the standard TIP3P water model while C22* was used with the CHARMM-modified water model.23 The system was neutralized and the NaCl concentration was set to 0.15 M, mimicking physiological concentration. The viral capsid starting structure was minimized for 50000 steps to remove any unnatural clashes, and two NVT (100 ps) and NPT (1 ns) equilibration runs were carried out. This was followed by a production run of 400 ns, where energies and compressed coordinates were saved every 5000 steps. The V-rescale24 temperature coupling algorithm was used for all runs with a time constant of 0.1 ps, while the Berendsen25 and the Parrinello–Rahman26,27 pressure coupling algorithm were used for the NPT and production run respectively (with a time constant of 2.0 ps). The smooth Particle-Mesh Ewald (PME)28 method with a 1.2 nm Coulomb cut-off was used to treat long-range electrostatic interactions. The force-switching method29 was used for van der Waals interactions, with a decay between 1.0 and 1.2 nm. Bonds with hydrogen atoms were constrained using the SHAKE algorithm.30 The pressure and temperature were set at 1.0 bar and 310 K respectively for all runs, with a time step of 2 fs. MD snapshots from the final 50 ns of each production run were extracted every 10 ps for analysis (unless indicated). The electrostatic potential surface was obtained using eF-surf.31,32 Secondary structure analysis was performed using the DSSP module.33,34 The single linkage algorithm,35 where a structure was added to a cluster when its distance to any member of the cluster is smaller than 0.2 nm, was used for cluster analysis.
169301. Frames were saved every 5000 steps or 10 ps, occupying 4.04 terabytes of disk space in total. Atomistic MD simulations were performed with the respective force field and TIP3P water21 using the GROMACS package, version 2020.2.22 As recommended for CHARMM force fields, C36(m) was used with the standard TIP3P water model while C22* was used with the CHARMM-modified water model.23 The system was neutralized and the NaCl concentration was set to 0.15 M, mimicking physiological concentration. The viral capsid starting structure was minimized for 50000 steps to remove any unnatural clashes, and two NVT (100 ps) and NPT (1 ns) equilibration runs were carried out. This was followed by a production run of 400 ns, where energies and compressed coordinates were saved every 5000 steps. The V-rescale24 temperature coupling algorithm was used for all runs with a time constant of 0.1 ps, while the Berendsen25 and the Parrinello–Rahman26,27 pressure coupling algorithm were used for the NPT and production run respectively (with a time constant of 2.0 ps). The smooth Particle-Mesh Ewald (PME)28 method with a 1.2 nm Coulomb cut-off was used to treat long-range electrostatic interactions. The force-switching method29 was used for van der Waals interactions, with a decay between 1.0 and 1.2 nm. Bonds with hydrogen atoms were constrained using the SHAKE algorithm.30 The pressure and temperature were set at 1.0 bar and 310 K respectively for all runs, with a time step of 2 fs. MD snapshots from the final 50 ns of each production run were extracted every 10 ps for analysis (unless indicated). The electrostatic potential surface was obtained using eF-surf.31,32 Secondary structure analysis was performed using the DSSP module.33,34 The single linkage algorithm,35 where a structure was added to a cluster when its distance to any member of the cluster is smaller than 0.2 nm, was used for cluster analysis.
3. Results and discussion
3.1 Capsid structure stabilizes and deviates the least for ff14SB (AMBER) and C22* (CHARMM)
Compared to the initial structure (after minimization), we find that the difference in the min–max RMSD values (in units of nm) is smallest for ff14SB (0.18, 0.25), followed by ff99SB*-ILDNP (0.20, 0.29), and ff03* (0.15, 0.30), indicating that the structure from the ff14SB run stabilizes the quickest (Fig. 2a). For the CHARMM family, the difference is smallest for C22* (0.21, 0.32), followed by C36m (0.19, 0.44), and C36 (0.19, 0.45) (Fig. 2b). The difference is noticeably larger for C36m and C36. The average RMSD (in nm), which shows how much the initial structure is preserved across the production run, is ordered (from smallest to largest) as follows: ff14SB (0.233 ± 0.01), ff99SB*-ILDNP (0.261 ± 0.02), ff03* (0.264 ± 0.02), C22* (0.281 ± 0.02), C36m (0.368 ± 0.06), C36 (0.374 ± 0.06). Consistent with the min–max RMSD values, ff14SB provides the smallest standard deviation (0.01) of the RMSD while C36m and C36 provide the largest values (0.06). Overall, the MD trajectories stabilize more quickly and show a smaller structural deviation (less fluctuation) for the AMBER force fields than CHARMM.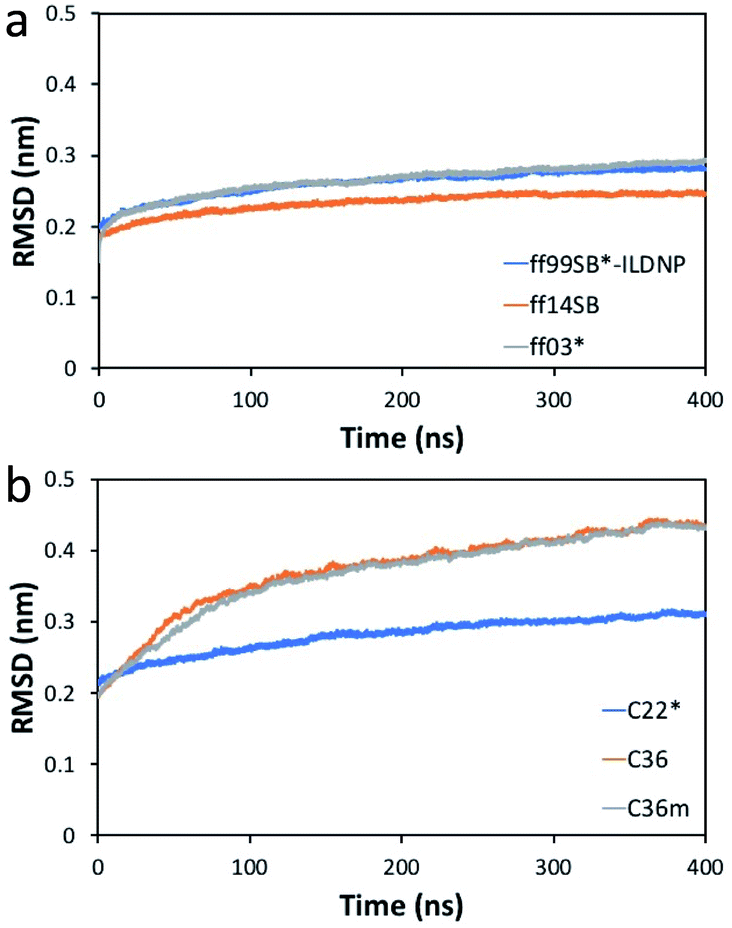 | ||
| Fig. 2 RMSD (nm) of the EV-D68 capsid relative to the initial structure versus time (ns) for the (a) AMBER and (b) CHARMM force fields. | ||
3.2 Radius of gyration varies slightly across all force fields
The radius of gyration (Rg) indicates the average position of the amino acid atoms relative to the capsid's center. Hence, it is equivalent to the average of the inner and outer radius of the capsid. We find that Rg stabilizes very quickly, with small standard deviations across the AMBER and CHARMM force fields (Fig. S1†). The average Rg value and its standard deviation across the production run is ordered (from smallest to largest, in units of nm) as follows: C36 (13.200 ± 0.006), C36m (13.200 ± 0.006), ff14SB (13.232 ± 0.003), ff03* (13.238 ± 0.004), ff99SB*-ILDNP (13.248 ± 0.004), C22* (13.282 ± 0.004). The difference in the average Rg value between C36 and C22* is only 0.082 nm, or 0.82 Å, while the standard deviations range between 0.03–0.06 Å, indicating that the change in capsid radius is small during the production run. It is noted that although the difference of 0.82 Å is small, it is not statistically insignificant as the difference is more than an order of magnitude larger than the standard deviations.Nonetheless, we expect Rg to remain fairly constant throughout the run, consistent with the small RMSDs of <0.5 nm (see Fig. 2). Thus, the choice of force field does not seem to significantly impact Rg.
3.3 C36m and C36 sample the largest conformational space
Consistent with Fig. 2, we find that the number of clusters formed from the production run using C36 provided the highest number of 19, followed by C36m with 16 clusters (Table S1†). ff14SB provided the least number of clusters (2). While RMSD can indicate the degree of conformational sampling based on how closely the MD trajectories align with the initial structure, principal component analysis (PCA) provides a more direct measure. With PCA, the two dominant modes (eigenvectors or principal components) associated with the largest concerted motions of the capsid are identified and the MD snapshots are projected onto the two modes (Fig. 3). Across the AMBER force fields, we find that the conformer distribution along the subspace is similar. For CHARMM, C22* has a relatively compact distribution, while both C36 and C36m sample more regions, indicating an increase in conformational mobility and regions of the phase space with energy minima that are more accessible. From Fig. S6,† we also find that the first twenty eigenvectors capture 71–74% of the protein motion for the AMBER family (71% for ff14SB, 72% for ff03*, 74% for ff99SB*-ILDNP), and 79–87% for the CHARMM family (79% for C22, 84% for C36m, 87% for C36). The first two eigenvectors account for 37–45% of the collective motions for AMBER (37% for ff03*, 42% for ff14SB, 45% for ff99SB*-ILDNP), and 45–53% for CHARMM (45% for C36m, 47% for C22, 53% for C36). This indicates that capsid motion modeled by CHARMM is represented by fewer dominant modes than AMBER.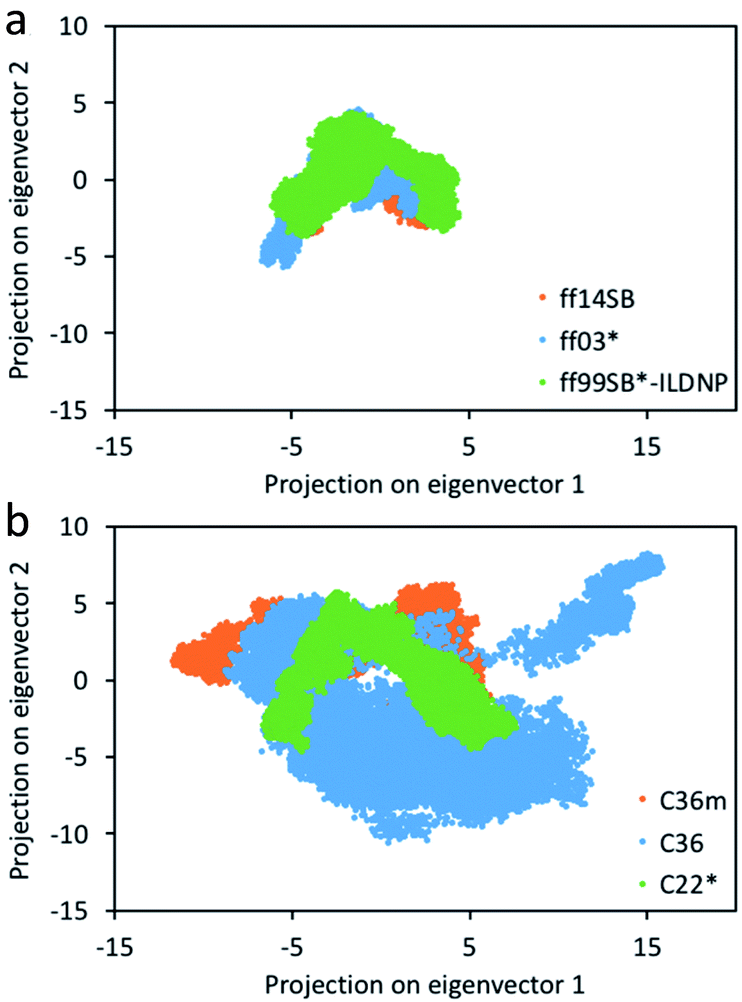 | ||
| Fig. 3 Projection of the coordinates from the 400 ns MD simulation on the top two eigenvectors (principal components) for the (a) AMBER and (b) CHARMM force fields. See Fig. S2–S5† for unobscured projections. | ||
3.4 AMBER and CHARMM force fields result in varying secondary structures
From our previous analysis above, ff14SB and C36m result in different degrees of conformational sampling and energy landscapes. Any variation in the secondary structure content (and preference) between the energy minima of ff14SB and C36m could influence the conformational stability and number of conformations accessible outside of the minima. To investigate this, we first carried out secondary structure analysis on the capsid structure corresponding to the energy minima. We find that ff14SB has a higher percentage of β (β-sheet and β-bridge) and helical (α-helix and 3-helix) content that C36m (Table S2†), while C36m has a slightly higher enrichment in bend and random coil structures. Extending this analysis to all force fields over the course of the production run, we note that the AMBER force fields are enriched in β and helical content, while C22* has the most helical content within the CHARMM family (Table S3†). ff03* and ff14SB has the highest bend and turn content respectively.Ramachandran plots (Fig. 4) were constructed for the six force fields with several notable differences – (1) the α− region is oversampled in the AMBER force fields, (2) a portion of the β region where ϕ ∈ [−180°, −130°] and ψ ∈ [100°, 150°] is least favored in C36m and C36, (3) the αL region is more populated for the AMBER force fields and least populated for C36m and C36, and 4) the α–β transition region is more populated for the AMBER force fields, with the least population noted in C36m and C36, followed by C22*. ff03* and ff99SB*-ILDNP contains modified backbone potential from ff03 (ref. 12) and ff99SB respectively;16 ff99SB*-ILDNP additionally contains modified side-chain torsion potential for the ILDNP amino acids. In a similar vein, C22* contains modified backbone torsion potentials from C22.16 ff14SB adjusts the φ and ψ backbone parameters and refits all twenty amino acid side chain dihedral parameters from ff94.14 The exploration of conformational space by the AMBER force fields and C22* is correlated with the extent of sampling in the β and helical regions. High α and β content would stabilize local conformations (minima), creating metastable states that increases the sampling time of the conformational landscape. We note that the αR and β regions of C22* (Fig. 4f) are less populated than the AMBER family, thus correlating with a less compact distribution in conformational space (Fig. 3b). This supports a previous study36 that attributed the increase in helicity for ff14SB to its higher torsional barriers. This indicates that C36m and C36 has the lowest torsional barriers of the six force fields studied, since they show the lowest population of helices. More importantly, the α− region is also nonphysical, as it is not present from high-resolution protein crystallographic data.36,37 In this regard, using the C36m or C36 force fields would help suppress non-native conformations arising from this region. This appears consistent as C36 corrects the overpopulation α-helices found in C22/CMAP (correction map).17 There seems to be little difference in the αL distribution and secondary structure between C36m and C36, as C36m was developed for intrinsically disordered proteins.18 In addition, both C36m and C36 show the least sampling in the transition region, followed by C22*, and little noticeable difference among the AMBER force fields. Compared to representative Ramachandran plots (see Fig. 3 of ref. 37), both C36m and C36 seem to match most closely.
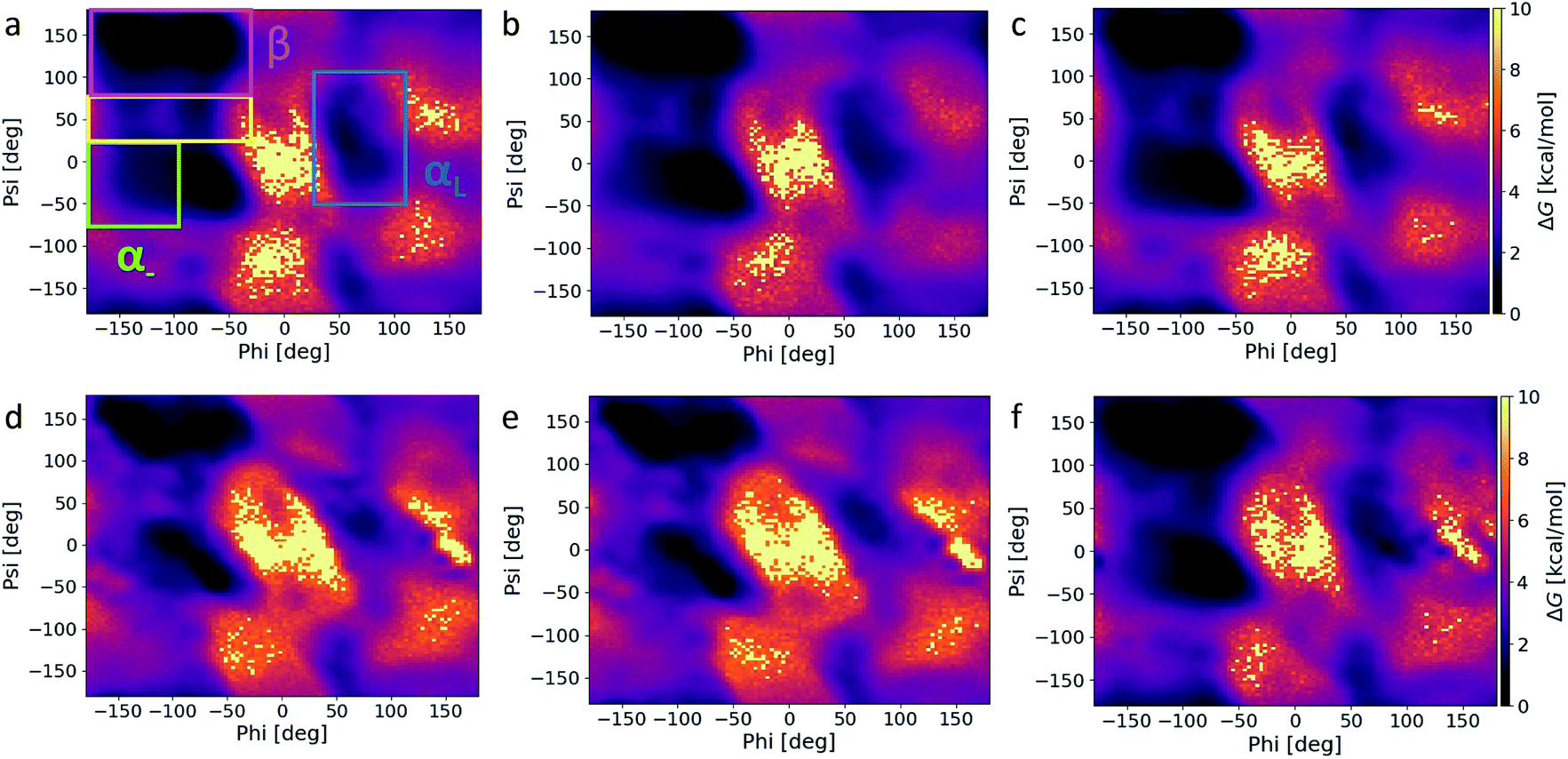 | ||
| Fig. 4 Ramachandran plots corresponding to (a) ff14SB, (b) ff03*, (c) ff99SB*-ILDNP, (d) C36m, (e) C36, and (f) C22* MD snapshots over the course of the 400 ns simulation. The α− region (green) consists of ϕ ∈ [−180°, −100°] and ψ ∈ [−67°, 25°], the β region (pink) consists of ϕ ∈ [−180°, −45°] and ψ ∈ [75°, 180°], the transition region (yellow) consists of ϕ ∈ [−180°, −45°] and ψ ∈ [25°, 75°], and the αL region (blue) consists of ϕ ∈ [30°, 110°] and ψ ∈ [−50°, 110°]. | ||
3.5 Regions of ion accumulation are consistent across all force fields
Shown in Fig. S7† are the electrostatic potential maps for the outward- and inner-facing orientation of a capsomer. The outer-surface of the capsid has a mixture of positive and negative regions (Fig. S7a†), while most of the inner-surface of the capsid has a negative electrostatic potential (Fig. S7b†). The largest peak at 11 nm (with respect to the capsid's center of mass) corresponds to the screening cloud formed by the Na+ ions in the presence of the negatively-charged inner radius (Fig. S7c and d†). The Na+ and Cl− distribution dips to a minimum at around 12–13 nm, before rising at 14–16 nm, corresponding to the interaction with the outer radius. The slightly higher accumulation of Na+ ions than Cl− ions indicates that the outer radius likely has an overall negative potential. The ion distribution plateaus beyond 16 nm, further away from the charged capsid. These behaviors are replicated using other force fields (see Fig. S8†) and the location of the peaks do not change significantly, indicating that the capsid's radii are conserved across the AMBER and CHARMM families.4. Conclusions
For capsid-related studies that seek to explore specific binding properties, such as capsid-host factor interactions, or physical properties such as normal modes, the CHARMM family, especially C36m and C36, is recommended due to more accurate structure modelling (as noted from the Ramachandran plots). The caveat in using C36m or C36 to model large mesoscale structures is that this requires sufficient computational power and storage resources, as we showed that the MD trajectory requires a longer time to stabilize. In this study, we do not find any noticeable difference between C36m and C36 in terms of secondary structure. For other global properties like the radius of gyration, the choice of force field appears to be less sensitive. Other properties like water permeation rates could be heavily influenced by capsid structure, which we aim to explore in future studies. These studies also include comparing simulation data with experimental data of the EV-D68 capsid when available.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. D. T. acknowledges postdoctoral support from the Canadian Institutes of Health Research. The D. P. T.'s group is supported by the Natural Sciences and Engineering Research Council (Canada) and the Canada Research Chairs Program. Support from Compute Canada, the Canada Foundation for Innovation and partners is also gratefully acknowledged. In addition, R. D. T. would like to thank Justin Lemkul, Vladimir Farafonov, and Dmitry Nerukh for helpful discussions.References
- H. Cassidy, R. Poelman, M. Knoester, C. C. Van Leer-Buter and H. G. M. Niesters, Front. Microbiol., 2018, 9, 2677 CrossRef PubMed.
- A. Fall, N. Ndiaye, M. M. Jallow, M. A. Barry, C. S. B. Touré, O. Kebe, D. E. Kiori, S. Sy, M. Dia, D. Goudiaby, K. Ndiaye, M. N. Niang and N. Dia, Sci. Rep., 2019, 9, 13881 CrossRef PubMed.
- Y. Liu, J. Sheng, A. L. W. van Vliet, G. Buda, F. J. M. van Kuppeveld and M. G. Rossmann, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E12209–E12217 CrossRef CAS PubMed.
- J. D. Durrant, S. E. Kochanek, L. Casalino, P. U. Ieong, A. C. Dommer and R. E. Amaro, ACS Cent. Sci., 2020, 6, 189–196 CrossRef CAS PubMed.
- O. Guvench and A. D. MacKerell, in Molecular Modeling of Proteins, ed. A. Kukol, Humana Press, Totowa, NJ, 2008, pp. 63–88, DOI:10.1007/978-1-59745-177-2_4.
- A. Okur, B. Strockbine, V. Hornak and C. Simmerling, J. Comput. Chem., 2003, 24, 21–31 CrossRef CAS PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- A. D. Mackerell Jr, M. Feig and C. L. Brooks III, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef PubMed.
- E. Tarasova, V. Farafonov, R. Khayat, N. Okimoto, T. S. Komatsu, M. Taiji and D. Nerukh, J. Phys. Chem. Lett., 2017, 8, 779–784 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CrossRef CAS PubMed.
- A. E. Aliev, M. Kulke, H. S. Khaneja, V. Chudasama, T. D. Sheppard and R. M. Lanigan, Proteins, 2014, 82, 195–215 CrossRef CAS PubMed.
- R. B. Best and G. Hummer, J. Phys. Chem. B, 2009, 113, 9004–9015 CrossRef CAS PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang and P. Kollman, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS PubMed.
- S. Piana, K. Lindorff-Larsen and D. E. Shaw, Biophys. J., 2011, 100, L47–L49 CrossRef CAS PubMed.
- J. Huang and A. D. MacKerell Jr, J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- B. Webb and A. Sali, Curr. Protoc. Bioinf., 2016, 54, 5. 6.1–5.6.37 Search PubMed.
- A. Šali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- E. Lindahl, M. J. Abraham, B. Hess and D. Van Der Spoel, Zenodo, 2020 DOI:10.5281/zenodo.3773801.
- S. Boonstra, P. R. Onck and E. van der Giessen, J. Phys. Chem. B, 2016, 120, 3692–3698 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. v. Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- S. Nosé and M. L. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- P. J. Steinbach and B. R. Brooks, J. Comput. Chem., 1994, 15, 667–683 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- K. Kinoshita, J. i. Furui and H. Nakamura, J. Struct. Funct. Genomics, 2002, 2, 9–22 CrossRef CAS PubMed.
- K. Kinoshita and H. Nakamura, Bioinformatics, 2004, 20, 1329–1330 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- W. G. Touw, C. Baakman, J. Black, T. A. H. te Beek, E. Krieger, R. P. Joosten and G. Vriend, Nucleic Acids Res., 2014, 43, D364–D368 CrossRef PubMed.
- J. C. Gower and G. J. S. Ross, J. R. Stat. Soc. Ser. C Appl. Stat., 1969, 18, 54–64 Search PubMed.
- D. Wang and P. E. Marszalek, J. Chem. Theory Comput., 2020, 16, 3240–3252 CrossRef CAS PubMed.
- S. A. Hollingsworth and P. A. Karplus, Biomol. Concepts, 2010, 1, 271–283 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S3 and Fig. S1–S8. See DOI: 10.1039/d1ra08431c |
| This journal is © The Royal Society of Chemistry 2022 |