
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic strategy and structure–activity relationship (SAR) studies of 3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole (YC-1, Lificiguat): a review
Ko-Hua Yu and
Hsin-Yi Hung *
*
School of Pharmacy College of Medicine, National Cheng Kung University, Tainan 701, Taiwan. E-mail: z10308005@email.ncku.edu.tw
First published on 20th December 2021
Abstract
Since 1994, YC-1 (Lificiguat, 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole) has been synthesized, and many targets for special bioactivities have been explored, such as stimulation of platelet-soluble guanylate cyclase, indirect elevation of platelet cGMP levels, and inhibition of hypoxia-inducible factor-1 (HIF-1) and NF-κB. Recently, Riociguat®, the first soluble guanylate cyclase (sGC) stimulator drug used to treat pulmonary hypertension and pulmonary arterial hypertension, was derived from the YC-1 structure. In this review, we aim to highlight the synthesis and structure–activity relationships in the development of YC-1 analogs and their possible indications.
1. Introduction
YC-1 (Lificiguat, 3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole) is an indazole derivative whose structure contains furyl and benzyl substituents at positions 1 and 3 (Fig. 1). In 1994, YC-1 was first synthesized and reported by Kuo's group and exhibited antiplatelet activity with a 70% inhibition of platelet aggregation induced at a concentration of 30 μmol L−1.1,2 YC-1 is a potential antiplatelet agent for prevention and treatment of vascular embolisms. The mechanisms of action of the antiplatelet activity of YC-1 include direct stimulation of platelet-soluble guanylate cyclase (sGC) and indirect elevation of platelet cyclic guanosine monophosphate (cGMP) levels.1 Since the discovery of YC-1 in 1994, many other molecular targets of YC-1 have been identified. YC-1 is a hypoxia-inducible factor-1 (HIF-1) inhibitor3,4 that exhibits anticancer activities,5–7 reverse chemoresistance,8,9 retinal neovascularization inhibition,10,11 neural protection12,13 and decreased allergic inflammatory gene expression.14 In addition, Carroll et al.15 found that YC-1 displays inhibitory activity against vascular endothelial growth factor (VEGF) for treating inflammation.16 Additionally, YC-1 was found to inhibit the activity of the nuclear factor kappa light chain enhancer of activated B cells (NF-κB),17 which is a critical signaling pathway for learning and memory,18 as well as bone formation.19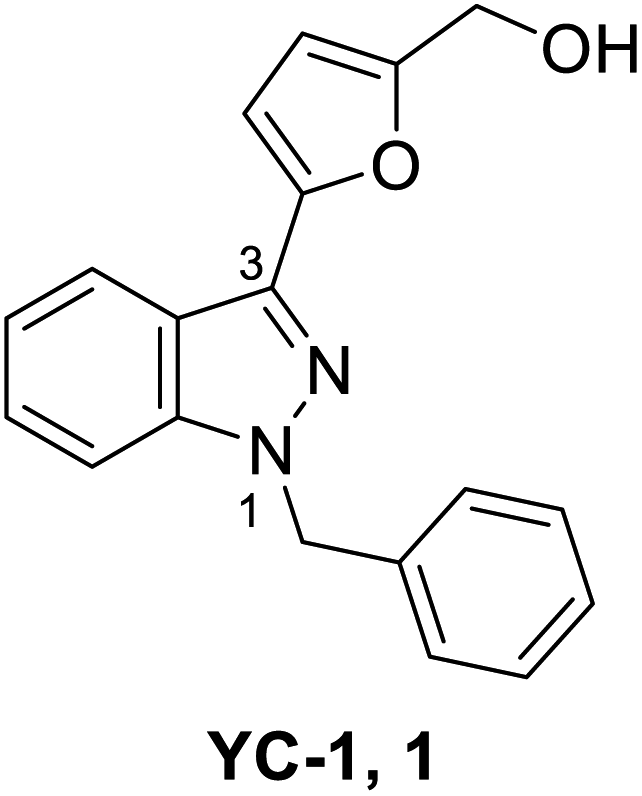 | ||
| Fig. 1 Structure of YC-1 (1). | ||
The structure–activity relationship (SAR) of YC-1 and its derivatives have been studied in the past decade. However, in the past reviews20,21 of YC-1, only the relevant pharmacological activities were discussed, leaving open the synthesis strategy and SAR. Some valuable YC-1 derivatives have even been developed into clinical drugs, such as Riociguat. Riociguat, whose structure is based on YC-1, is a clinical drug indicated for pulmonary artery hypertension (PAH) via stimulating soluble guanylate cyclase (sGC). Thus, we mainly focus on the chemical synthesis of YC-1, the synthesis of its derivatives, and discussion of the SAR in this review.
Based on an overview of all the chemical syntheses for YC-1 and its derivatives, the starting materials are easily available and, inexpensive raw materials.2,22–24 The derivatives are modified mainly on the functional groups in aromatic and furyl rings and the number of nitrogen atoms. The chemical modification reactions comprise reduction, oxidation, esterification, substitution reactions, Suzuki coupling, and direct arylation. In the following section of this review, we comprehensively describe the synthetic methods, biological activities, and the SAR of YC-1 and summarize the most effective structure for various biological activities (HIF-1, sGC, VEGF and NF-κB), which will be beneficial for future research and development of YC-1 derivatives with optimized bioactivity.
2. Synthetic strategies of 3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole (YC-1) and YC-1 derivatives
The synthesis of YC-1 derivatives has attracted considerable attention in organic and medicinal chemistry due to their excellent bioactivities. Different synthetic strategies have been developed depending on the desired chemical structure. Here, we summarize several synthetic strategies used in the synthesis of YC-1 (Scheme 1).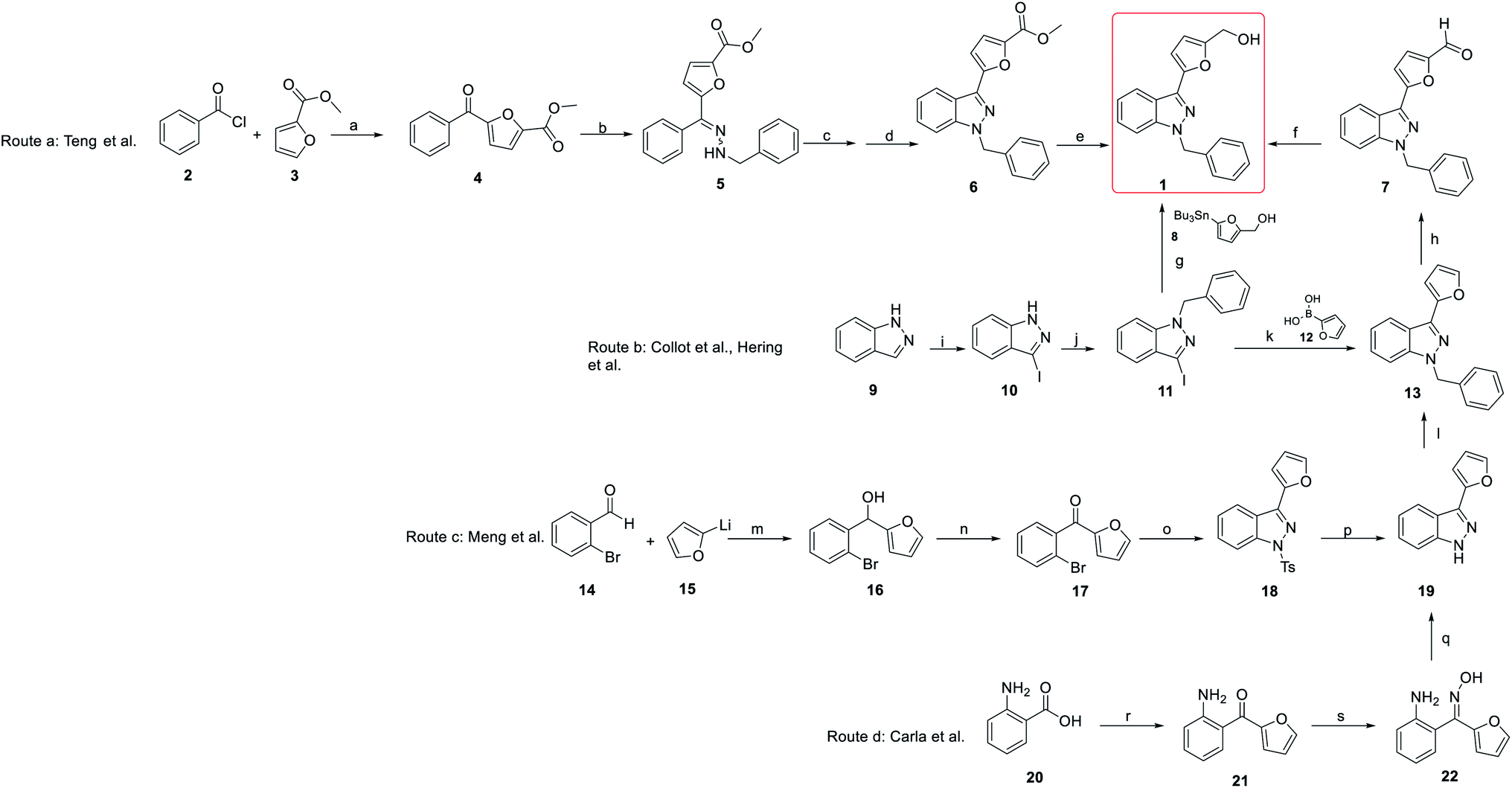 | ||
| Scheme 1 Summary of synthesis of YC-1 (1). Route a: synthesis by Teng et al. (2002); route b: synthesis by Collot et al. and Hering et al. (1999, 2006); route c: synthesis by Meng et al. (2017); route d: synthesis by Carla et al. (2008). Reagents and conditions: (a) FeCl3; (b) PhCH2NHNH2, HOAc; (c) Pb(OAc)4; (d) BF3Et2O; (e) Ca(BH4)2; (f) NaBH4, MeOH, rt; (g) Pd2(dba)3, AsPh3, DMF; (h) POCl3, DMF, 0 °C to 80 °C; (i) I2, KOH, DMF; (j) BnBr, t-BuOK, Bu4NI, THF; (k) Pd(PPh3)4, NaHCO3, DME, reflux; (l) ArCH2Br, t-BuOK, THF, 0 °C to rt; (m) THF, −80 °C; (n) MnO2, CH2Cl2; (o) (i) TsNHNH2, HCl, MeOH; (ii) Cu2O, i-AmOH, reflux; (p) Mg, MeOH; (q) MsCl, Et3N, CH2Cl2; (r) furan, n-BuLi, THF, −78 °C to 0 °C; (s) HO–NH2 HCl, pyridine, MeOH, reflux. | ||
The synthesis of YC-1 was first described by Teng et al. (2002).25,26 Their synthetic plan started from benzoyl chloride and ethyl furan-2-carboxylate catalyzed by ferric chloride to obtain carbonyl coupling compound 4 (route a). Then, methyl 5-benzoylfuran-2-carboxylate (4) was converted to hydrazine compound 5 using benzyl hydrazine and acetic acid. Compound 5 underwent a cyclization reaction catalyzed by lead(IV) acetate [Pb(OAc)4] and then boron trifluoride (BF3) to obtain compound 6. Finally, the ester group was reduced to a hydroxyl group via calcium borohydride [Ca(BH4)2] to yield the final compound 1 (YC-1), and the total yield was 4.3%.
In the original synthesis route of YC-1, the corresponding indazole from cyclization of the hydrazine compound had several drawbacks, including too many steps, use of organic metal reagents, and low yield. Collot et al.,27 Hering et al.28 and Takeuchi et al.22 modified the synthetic process of YC-1 starting from indazole (9) (route b).29 First, indazole mixed with iodine (I2), and potassium hydroxide (KOH) in dimethylformamide (DMF) was used to install the iodo group at position 3. Next, benzyl bromide (BnBr), potassium tert-butoxide (t-BuOK), and tetrabutylammonium iodide (Bu4NI) in tetrahydrofuran (THF) were reacted with compound 10 to obtain benzyl substituent 11. Hering28 and Takeuchi22 prepared YC-1 via stannane reaction using palladium catalysis from 1-benzyl-3-iodoindazole, and the yield increased to 74%. However, organostannanes are generally considered inappropriate for drug synthesis due to their toxicity. Collot avoided organostannane reagents. Instead, Suzuki coupling reaction of 11 with boronic acid substituted with aromatic compound (12) was carried out to obtain 13 by Vilsmeier–Haack reaction.27 Reduction of the formyl group with sodium borohydride was applied to obtain YC-1, and the yield improved to 79%.
Even though the yield improved to above 70%, there were still doubts about the use of heavy metals in the total synthesis. Hence, in 2008, Carla et al. developed a synthesis method that did not involve the use of heavy metals.24,30 In this report, all of the reagents used were less toxic and less expensive than those used in previous studies (route d). The starting materials were 2-aminobenzoic acid (20) and furan, which reacted with n-butyllithium in THF to obtain carbonyl compound 21. Compound 21 reacted with hydroxylamine hydrochloride to give oxime compound 22, which underwent a cyclization reaction using methanesulfonyl chloride (MsCl) to give compound 19. The final product, YC-1, was prepared following formylation and reduction of compound 19.27 This synthetic route not only reduced heavy metal usage but also retained the total yield.
In addition, in 2004 and 2007, Inamoto et al.31 and Sakamoto et al.32 reported the first method for preparing indazole through intramolecular cyclization of Pd-catalyzed ortho-bromine tosylhydrazone under the action of a base and ligand. However, the reaction must be carried out under alkaline conditions, and compound 24 was formed by Bamford–Stevens reaction, where the E-form was the main product. The E-form isomer cannot obtain the corresponding product (25); in contrast, the Z-form can give better reaction results (Scheme 2). In 2017, Meng et al. pointed out for the first time that indazole (25) can be obtained by the reaction of an E-form or Z-form tosylhydrazone isomer with cuprous oxide (Cu2O) by heating (Scheme 2), which is suitable for a variety of functional groups and has an extremely high yield.23
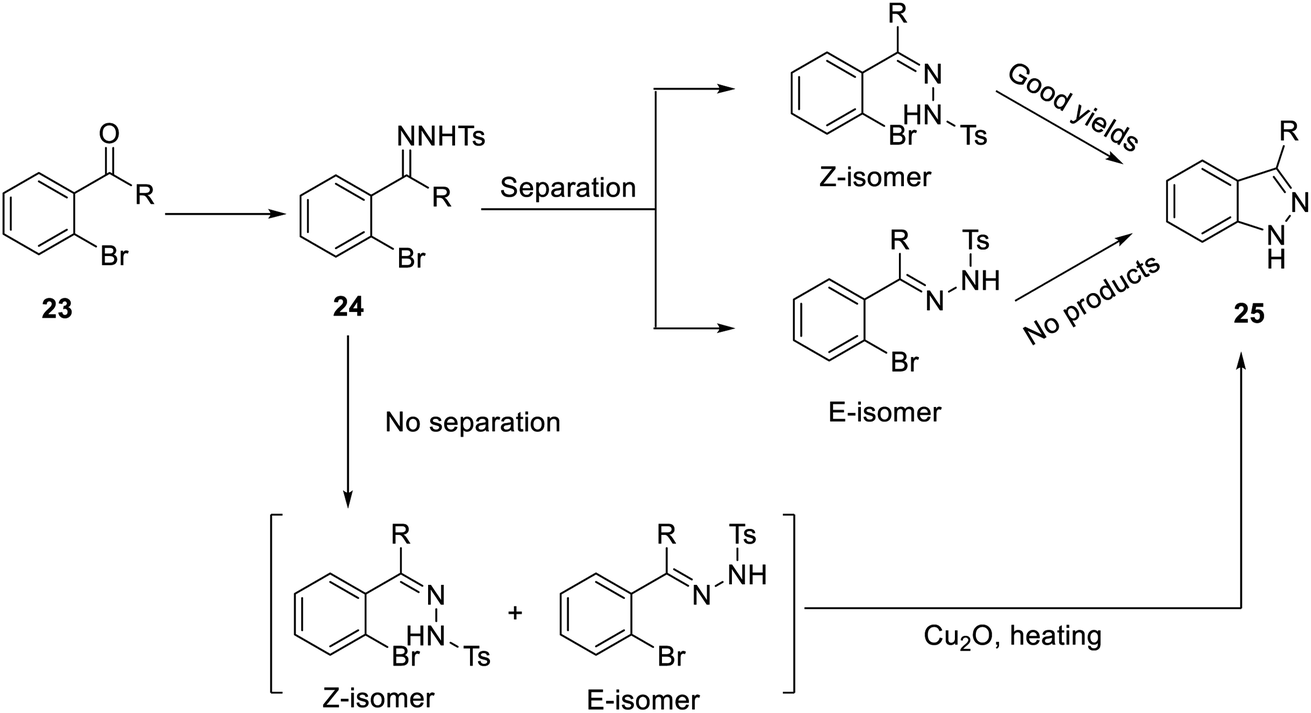 | ||
| Scheme 2 Synthesis of indazole (25), an intermediate of YC-1 synthesis. | ||
Meng's group also reported another synthetic strategy (Scheme 1, route c) based on 2-bromobenzaldehyde (14). (2-Bromophenyl)(furan-2-yl)methanone (17) was prepared through affinity addition and oxidation, after which it was cyclized by tosylhydrazone to form indazole compound 18. Removing tosyl and adding benzyl groups was obtained compound 19. Subsequently, 19 was subjected to Vilsmeier–Haack reaction and reduced to obtain YC-1 (1).
3. Structure–activity relationship (SAR) result of YC-1
3.1. Soluble guanylate cyclase (sGC)
YC-1 was first discovered as a novel sGC stimulator that is influenced by NO or NO donors.33 Upon sGC stimulation, several physiological events occur, including inhibition of the contraction of vascular smooth muscles and inhibition of platelet adhesion and aggregation.2,34 YC-1 may also potentiate the action of endogenous and exogenous carbon monoxide (CO) on sGC and stimulate sGC with NO in a synergistic manner. In addition, administration of YC-1 may affect age-related learning and memory dysfunction via the NO/cGMP signaling pathway, which sGC crucially contributes to.18,35,36 For example, in Morris water maze (MWM) avoidance response tests, YC-1-treated group (1 mg kg−1) improved rodent learning behavior and performed a higher avoidance response rate in which the mean avoidance response was 18.7% and 33.3% for the control and YC-1-treated rats, respectively, on day 11.37Moreover, YC-1 has been found to have an important regulatory effect on sGC and cGMP signaling in the cardiovascular system, where YC-1 directly stimulates sGC and then activates NO production that subsequently relaxes vascular smooth muscles.38,39 Furthermore, YC-1 inhibits focal adhesion kinase and proliferative factor transforming growth factor β1 (TCF-β1) to reduce the growth of vascular smooth muscle.40 In addition, YC-1 prevents oxidized LDL-mediated apoptosis and induces Hsp70 expression.39
Due to many stimulate activities related to sGC, the SAR discussion in the next section is divided into two parts: relaxation of vascular smooth muscles and antiplatelet activities.
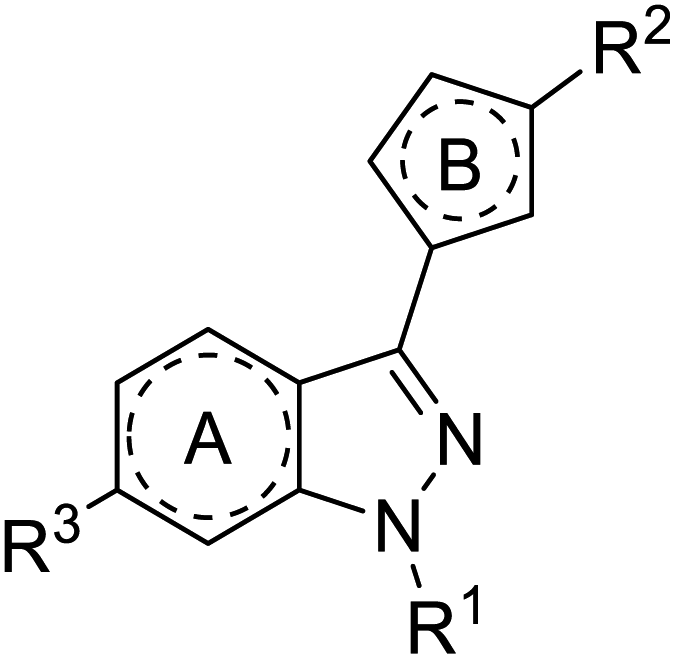 | ||
| Fig. 2 Generic structure to represent SAR for YC-1. | ||
According to the structure of YC-1, Straub et al. discussed the SAR based on 1-N-substituent whose synthesis followed route b in Scheme 1 and replaced the benzyl bromide functional group to obtain compound 26 (Fig. 3)26,41. Among all the derivatives, only fluoro or cyano substitution at the ortho position of the benzene ring led to better inhibitory activity, evidenced from 26a and 26e (IC50 = 4.9 and 8.7 μM, respectively). If the substitution was at the meta or para position, this inhibitory activity was reduced, such as in compounds 26b, 26c and 26f (IC50 = 10, 19 and 26 μM, respectively). The dual substitution of fluoro also reduced inhibition activity, for which the IC50 value of 26d was 15 μM. However, only 5-pyrimidinyl (26i) had good inhibitory activity among all of the heterocyclic substitutions (26g–26i), where the IC50 value was 3.2 μM. According to the results shown in Fig. 3, the substituted position of fluoro or cyano plays an important role in the relaxation of preconstricted aortic rings.
 | ||
| Fig. 3 Effect of 1-N-substituted derivatives (26) on relaxation of preconstricted aortic rings. | ||
In the core structure of YC-1 (heterocycle A), Straub et al. studied the impact of different cores on relaxation effects on preconstricted aortic rings (Fig. 4).26,41 The synthesis of compound 27 followed route b in Scheme 1 and replaced the indazole starting material. The results showed that more nitrogen atoms in the heterocycles contributed to little increases in inhibitory activity (27f, IC50 = 9.4 μM). The most effective compound was pyrazolopyridinyl pyrimidine (27b), with an IC50 value of 1.7 μM. Unfortunately, other pyrazolopyridine derivatives exhibited inferior activities, such as 27c, 27d and 27e (IC50 values of 6.9, 9.8, and 20.4 μM, respectively). Therefore, the position of nitrogen has a close relationship with inhibitory activity. A compound with 1H-pyrazolo[4,3-c]pyridine (27b) was found to be more potent than one with indazole (27a) in the core structure.
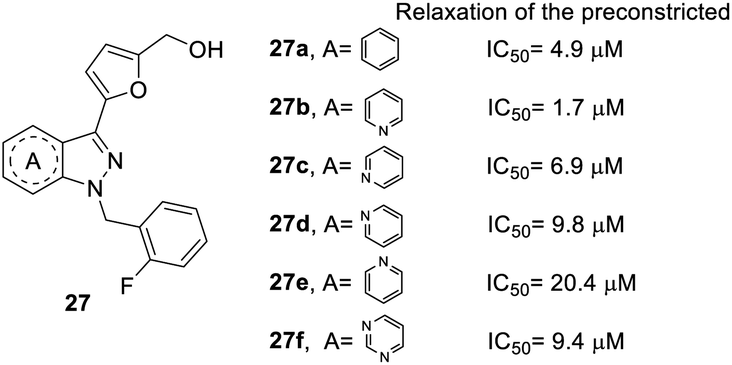 | ||
| Fig. 4 Effect of YC-1 derivatives (27) on relaxation of preconstricted aortic rings. | ||
Next, when R2 was substituted as compound 28, whose synthesis followed Scheme 1, route b, only the hydroxymethyl side chain retained inhibitory activity (Fig. 5). For example, compound 27b with a hydroxymethyl side chain resulted in good inhibitory activity (IC50 = 1.7 μM), but the inhibitory activity was reduced (IC50 = 9.3 and 8.0 μM, respectively) when the hydroxymethyl group was replaced with hydrogen (28a) or sulfonamide (28b).
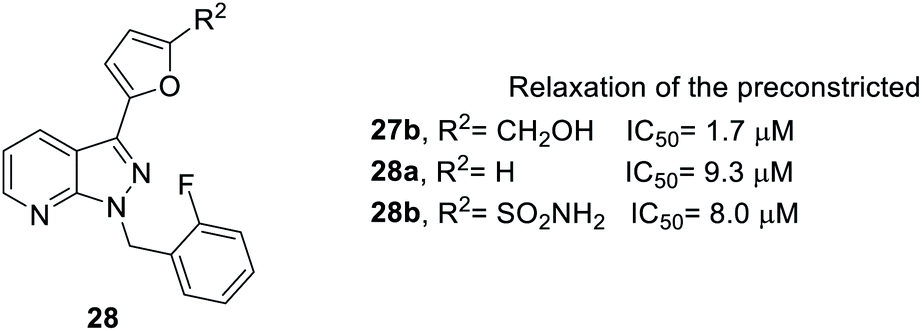 | ||
| Fig. 5 Effect of YC-1 derivatives (28) on relaxation of preconstricted aortic rings. | ||
As for heterocycle B, the synthesis of the derivatives followed route b in Scheme 1 and replaced compound 12 to other heterocycles. When the furan ring of compound 28 was replaced with isoxazole (29a–c), 1H-pyrazole (29d–e) or other heterocycles (29f–k), better inhibitory effects were observed (Fig. 6). For instance, the isoxazole of compound 29a–c (IC50 = 0.9, 1.2 and 1.5 μM, respectively) and 1H-pyrazole of compound 29e (IC50 = 0.6 μM) manifested better activity than the furan-substituted functional group (28b, IC50 = 1.7 μM). Furthermore, dinitrogen-containing heterocycles B [imidazole (29e–f) and pyrimidine (29i–k)] increased inhibitory performance (IC50 = 0.6, 1.8, 1.25, 1.3 and 0.27 μM, respectively). However, tetrazole-substituted 29g significantly reduced this activity (IC50 = 8.6 μM).
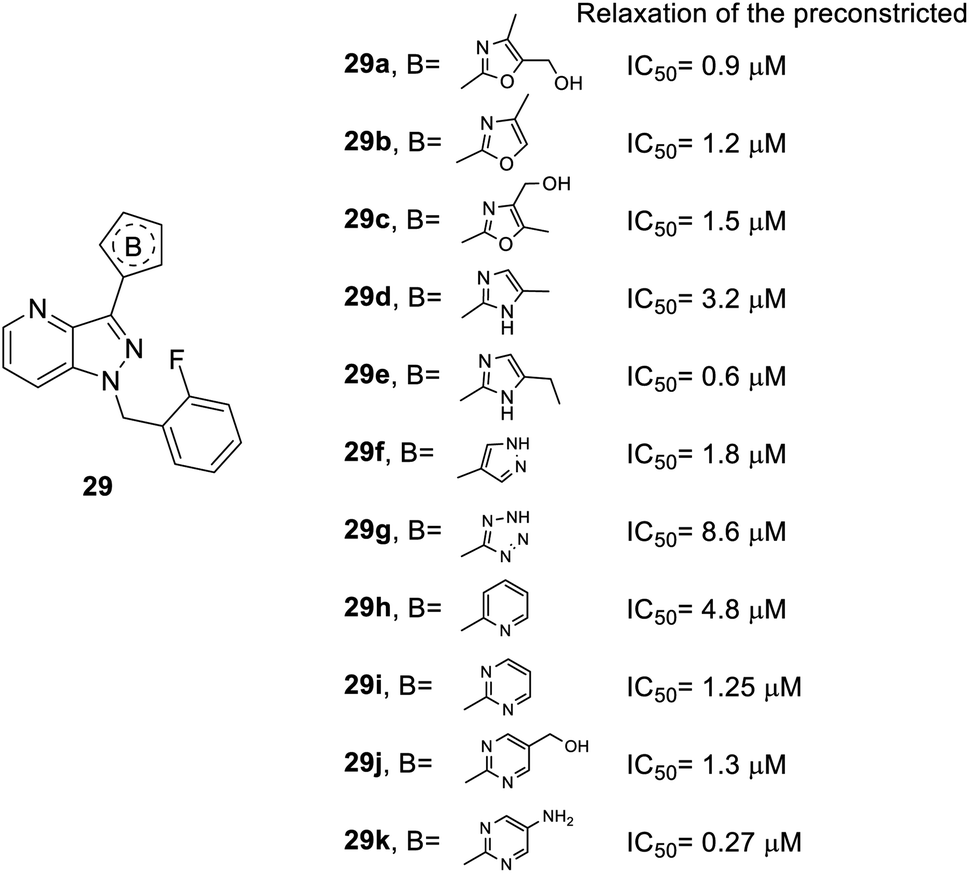 | ||
| Fig. 6 Effect of heterocycle B-substituted YC-1 derivatives (29) on relaxation of preconstricted aortic rings. | ||
In summary (Fig. 7), in terms of the core structure, the best structure is pyrazolopyridinyl pyrimidine, which further develops into Riociguat. At R1, the best substituent is the ortho fluoro benzyl group. The best substituent for the R2 position is a hydroxymethyl or ether group, and the worst is sulfonamide. The presence of pyrimidine ring as A ring as well as R1 substitution gave considerable IC50 values in compounds, 27b, 26i, 29e, and 29k. More nitrogen-substituted rings have shown enhancement in activity for relaxation effects on preconstricted aortic rings. In the ring B position, heterocycles are generally effective except for tetrazole, and 6-membered rings are better than 5-membered rings (Fig. 7).
 | ||
| Fig. 7 SAR of YC-1 analogs on relaxation of preconstricted aortic rings. | ||
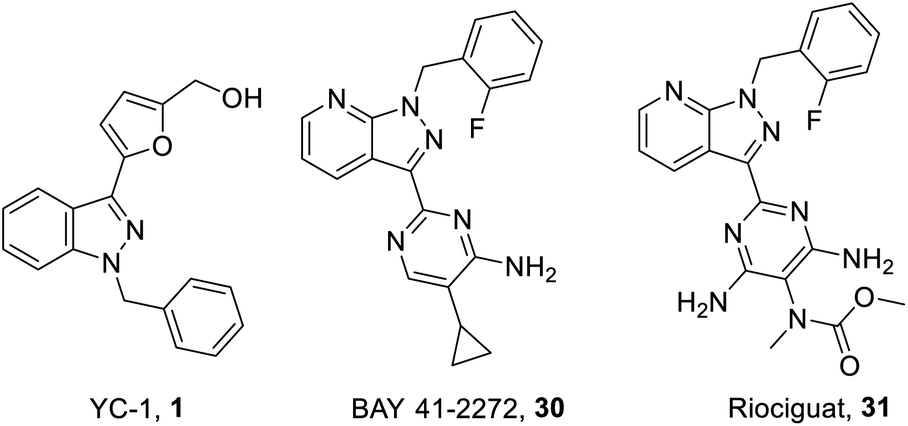 | ||
| Fig. 8 Structure of YC-1 (1), BAY 41-2272 (30) and Riociguat (31). | ||
On the other hand, one of the YC-1 derivatives (Bay 41-2272, 30,) Fig. 8 has been developed into a clinically used drug by Bayer Pharmaceuticals as a new pyrazolopyridinyl pyrimidine derivative (Riociguat, 31). This drug acts on the NO-sGC-cGMP pathway to effectively dilate blood vessels and inhibit smooth muscle hyperplasia. The synthesis of Riociguat-like, pyrazolopyridinyl pyrimidine derivatives was published in a report in 2009 by Mittendorf et al. (Scheme 3).43
 | ||
| Scheme 3 Synthesis of YC-1 analogs (42–44). Reagents and conditions: (a) TFA, 1,4-dioxane, reflux, 8 h; (b) TFA, 1,4-dioxane, reflux, 3 days; (c) NH3/MeOH, rt, 2 days; (d) TFAA/pyridine, pyridine, rt, 8 h; (e) MeONa/MeOH, rt, 2 h; (f) and (i) NH4Cl/HOAc, MeOH, reflux, 8 h; (ii) Na2CO3/H2O, rt; (g) HOAc, 100 °C, 2 h; (h) toluene, reflux, overnight; (j) t-BuOK, [Pd2dba3], (rac)-BINAP, toluene, 70 °C, overnight; (i) neat, 105 °C, slight vacuum, 3–12 h. | ||
Mittendorf et al. started from ethyl cyanopyruvate (33) and 2-fluorobenzylhydrazine (32) to generate aminopyrazole (34). The subsequent 3-dimethylaminoacrolein (35) underwent cyclo-condensation to give pyrazolopyridine 36 with a 50% overall yield. Then, the ester group of 39 was transformed into an amide using ammonia in methanol and reacted with trifluoroacetic anhydride (TFAA) to give cyano compound 38. Compound 38 underwent a pinner reaction with sodium methoxide, followed by substitution with ammonium chloride to give 40. Condensation of amidine compound 40 and 2-bromomalondialdehyde in acetic acid resulted in 4,6-unsubstituted 41; then, Buchwald–Hartwig reaction was applied to obtain morpholine substituted compound 42. In addition, amidine compound 40 reacted with alkyl-substituted malonic dinitrile under neat conditions to obtain diaminopyrimidines (43) with a yield of 40–70%. The nitrile compounds were transformed into the corresponding enol ether. Condensation of the enol ether and amidine 40 yielded 44. The structure–activity relationship of the pyrazolopyridinyl pyrimidine derivatives (42–44) are discussed later in this work.
The development of Bay 41-2272 and Riociguat were based on the SAR results mentioned above, and sGC stimulator 29k with pyrazolopyridinyl pyrimidine structure was identified and found to be suitable for further research. The results for additional pyrazolopyridinyl pyrimidine derivatives on the relaxation of preconstricted aortic rings are shown in Fig. 9.43 Overall, pyrazolopyridinyl pyrimidine derivatives (42–44) are more active than YC-1. In most cases, the diamino analogs (44) are slightly more potent than their monoamino (43) counterparts. For example, the inhibitory effect of 44a (IC50 = 0.11 μM), a diamino analog, is better than that of monoamino compound 43a (IC50 = 0.27 μM). The same pattern was observed for compounds 43b/44b and 43f/44c. At the R1 position of 42–44, a smaller steric group led to better inhibitory activity. n-Propyl substituted 43c (IC50 = 0.25 μM) exhibited better inhibitory activity than n-hexyl substituted 43d (IC50 = 1.04 μM). The bulky t-butyl substituted 43e reduced inhibitory activity (IC50 = 0.71 μM). In addition, cyclic substitution, such as cyclopropyl 43f (IC50 = 0.30 μM) and cyclopentyl 43g (IC50 = 0.90 μM) also decreased potency. However, conversion of an alkyl group to cyano substitution (43h) slightly reduced the potency (IC50 = 0.39 μM). When installing a linker into 43h as compound 43i with a 6-cyanoheptyl substituent, the inhibitory activity was greatly reduced (IC50 = 1.29 μM). Furthermore, the electron-donating groups exhibited better activity than the electron-withdrawing substitution. For instance, methoxy substituted 43j (IC50 = 0.34 μM) was superior to the fluoro substituted 43k (IC50 = 0.48 μM).
 | ||
| Fig. 9 Effects of pyrazolopyridinyl pyrimidine derivatives on relaxation of preconstricted aortic rings. | ||
In summary, the SAR of pyrazolopyridinyl pyrimidine derivatives is shown in Fig. 10. Generally, more amino substituents result in better inhibitory activity. Furthermore, all substituents are sensitive to steric effects. Thus, a smaller cycloalkane or a shorter alkyl led to better activity. At the R1 position, the best substituents are N-morpholino and ethyl, and the worst is cyano substitution.
 | ||
| Fig. 10 SAR of pyrazolopyridinyl pyrimidine derivatives on relaxation of preconstricted aortic rings. | ||
In addition to the inhibitory activity on preconstricted aortic rings, Lien et al. also studied the effects of the derivatives on SNP-induced apoptosis of rat aortic smooth muscle cells (Fig. 11).42 YC-1 almost completely reversed SNP-induced apoptosis of VSMCs (cell survival: 94.7%) at 30 μM.42 Conversion of the hydroxymethyl (YC-1, 1) into ether (45a) at the R2 position retained the activity. Conversion of the hydroxymethyl to amino (45b) and diethylamino (45c) substitution significantly reduced the activity, while conversion into a methyl group (45d) produced a cytotoxic effect in the VSMCs, resulting in a significant reduction in the anti-apoptotic effects. Finally, replacement of the hydroxymethyl by an ester (45e) only slightly reduced the activity.26,41,42
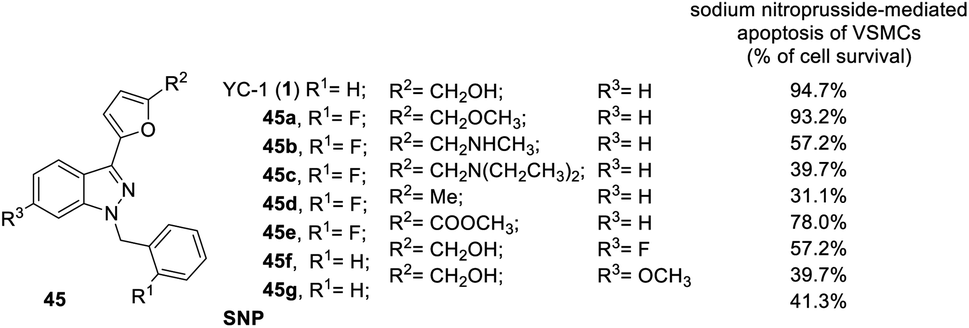 | ||
| Fig. 11 Anti-apoptotic effect of YC-1 derivatives (45) on the sodium nitroprusside-mediated apoptosis of VSMCs. | ||
Insertion of any atom into the YC-1 indazole structure will reduce the cell survival rate.41 For example, conversion of the R3 positions with a bridging dioxomethylene group led to even lower activity (cell survival: 39.7%). Introduction of fluoro (45f) or methoxy (45g) groups into the R3 position slightly reduced the potency of the inhibitory VEGF.
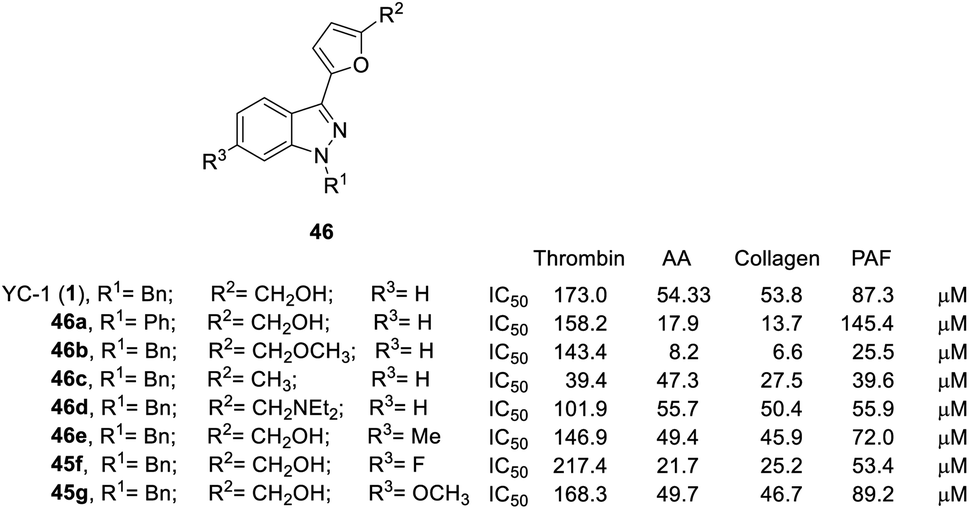 | ||
| Fig. 12 Effects of YC-1 derivatives (46a–e, 45f–g) on platelet aggregation induced by thrombin, arachidonic acid (AA), collagen, and platelet-activating factor (PAF). | ||
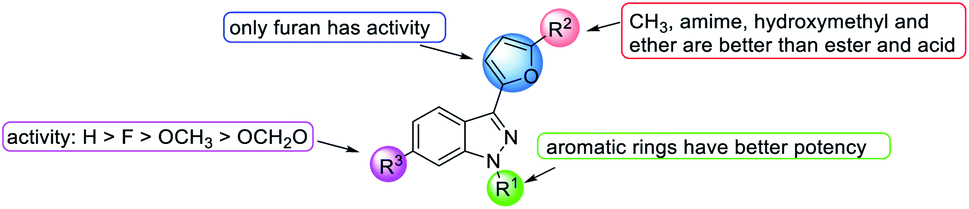 | ||
| Fig. 13 Effects of the SAR of YC-1 analogs on antiplatelet activities. | ||
Conversion of the 1-benzyl group of YC-1 to hydrogen significantly reduced the antiplatelet activity. Thus, aromatic rings are needed at the R1 position in a broad spectrum of antiplatelet activities.
Conversion of the alcohol group into an ether (46b) at the R2 position resulted in considerably increased activity. In addition, amine-substituted 46d enhanced the inhibitory activity toward thrombin- and PAF-induced aggregation but was inactive against collagen- and AA-induced aggregation. Introduction of an ester resulted in weak inhibitory antiplatelet activities as high as 300 μM. Finally, the acid derivatives were also less potent than their hydroxymethyl counterparts (YC-1, 1). When the furan was converted into a benzene ring, the activity was significantly reduced.
However, introduction of an F atom at the R3 position (45f) dramatically enhanced the inhibition of collagen-, AA- and PAF-induced aggregation but reduced the inhibitory effect on thrombin-induced aggregation. Methyl (46e) and methoxy (45g) substituted derivatives did not alter the platelet aggregation inhibition activity. The derivatives with bridging dioxomethylene groups significantly diminished the activity.
3.2. Hypoxia-inducible factor (HIF)
Hypoxia-inducible factor (HIF) is a transcription factor that induces the expression of various genes to resist tissue hypoxia.4 Cellular adaptability to hypoxia is an important strategy for tumor survival.44 In many studies, hypoxic conditions have been associated with malignancy in various cancers. Chemicals and drugs targeting HIF-1α have been discovered, and YC-1 is one of them. Moreover, under hypoxia conditions, YC-1 can inhibit HIF-1α activity and achieve anticancer effects.3 As an inhibitor of HIF-1, YC-1 has manifested excellent effects in many different cancer cells,45 including lung cancer,4,5 bladder cancer,46 breast cancer,6 canine lymphoma,47 chemoresistance,8,9 colorectal cancer,7 gastric carcinoma,48,49 hepatocellular carcinoma cancer (HCC),50–53 preeclampsia,54 ovarian cancer,55 and non-small cell lung cancer (NSCLC).50,56–60In addition, HIF-1α is upregulated in lung parenchyma and alveolar macrophages in asthma and is also viewed as a major regulator of inflammation. YC-1 can inhibit the increase in HIF-1α expression controlling allergen-induced airway diseases in (AIAD) mice.14 YC-1 can also decrease blood eosinophilia, the aryl hydrocarbon receptor (AHR), and allergic inflammatory gene expression: IL-13, IL-5, myeloperoxidase, and nitric oxide synthase (iNOS).14
However, YC-1 downregulated HIF-1α expression, a key component in osteogenesis, results in significant inhibition of regenerative bone formation.61–63 In addition, YC-1 partially eliminates the protection ability of FG-4592, which stabilizes HIF-1α expression in spinal cord and PC-12 cells.64
Furthermore, under hypoxic/ischemic conditions in the brain, YC-1 can induce the regulatable subunit of HIF-1α, a potent therapeutic target in cerebral ischemia.65 In 2011, Yan et al. found that inhibition of HIF-1 expression improves the ischemia-induced blood–brain barrier (BBB) disruption but does not influence brain edema.66 Moreover, YC-1 suppressed the release of neutrophil, inflammatory factor and the activation of the HMGB1/TLR4/NF-NF-κB signaling pathway to protect BBB integrity.67 Therefore, YC-1 has a protective effect on the cerebral vascular system.66,67
HIF-1α also leads to an imbalance in the expression of the angiogenic protein that is considered to be the primary factor in hypoxic conditions prevalent in preeclampsia.54,68 In 2015, Brownfoot et al. discovered that YC-1 significantly decreases soluble fms-like tyrosine kinase-1 (sFlt1), soluble endoglin (sENG) secretion and endothelial dysfunction in primary human tissues, which is a novel drug target for treating preeclampsia.69
The first structure–activity relationship study of YC-1 on HIF-1α inhibition in 2011 discussed the aromatic ring and the R1 and R2 functional groups of YC-1,70 and in 2012, Takeuchi et al. further researched SAR studies of the N-terminal aromatic ring.22
In order to test the effects of YC-1 derivatives (47–65) on the inhibition of HIF-1 transcriptional activity, a cell-based HRE reporter gene assay was applied in HRE (X5)-Luc transfected HeLa cells. The main chemical modifications of the derivatives are located on the different substituents of heterocycles A, B, R1, and R2 (Fig. 2), including different heterocycles, electron donor or withdrawal groups, halide substitution, and other structures, which follow synthetic route a in Scheme 1 to give YC-1 derivatives (47–48). The SAR discussions are summarized in Fig. 14–16 and Schemes 5–8.
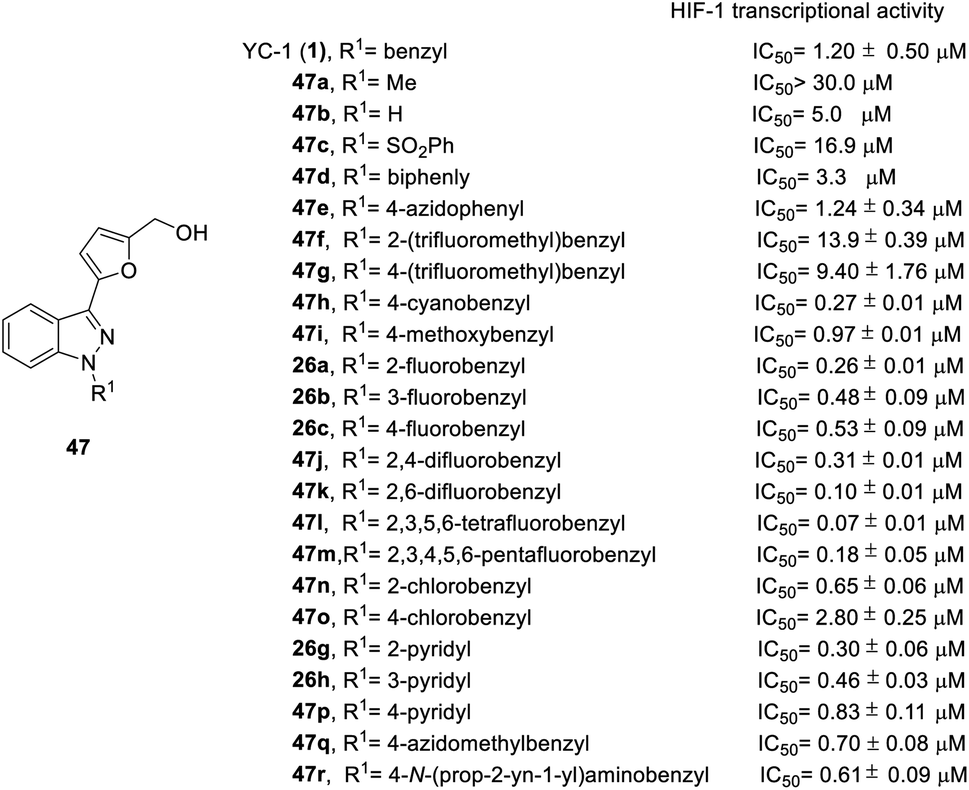 | ||
| Fig. 14 The effect of YC-1 derivatives (47) on HIF-1 transcriptional activity. | ||
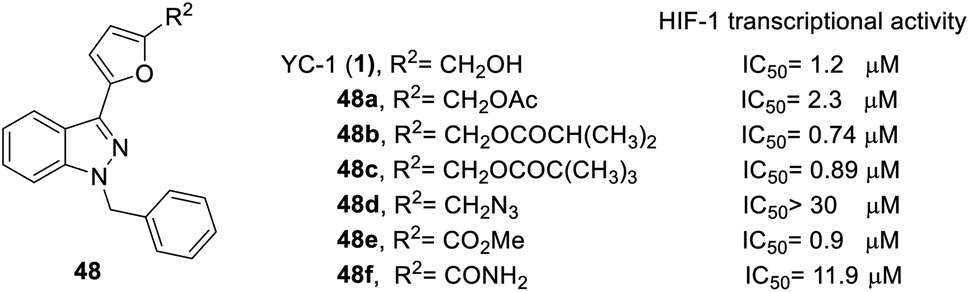 | ||
| Fig. 15 The effect of YC-1 derivatives (48) on HIF-1 transcriptional activity. | ||
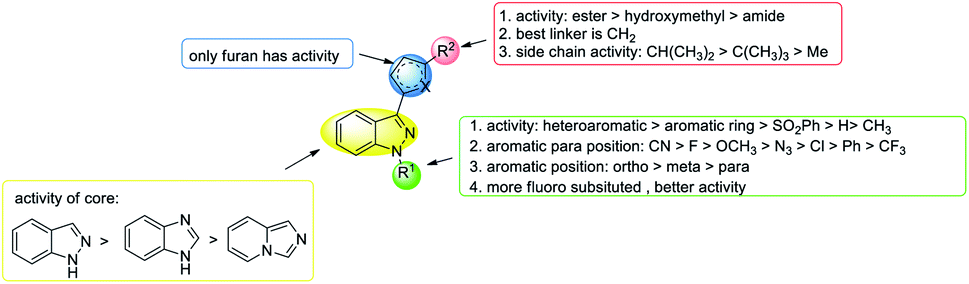 | ||
| Fig. 16 SAR of YC-1 analogs for HIF-1 transcriptional activity. | ||
Firstly, the activity of pyridyl substitution was better than that of YC-1 (benzyl, 1). Introduction of trifluoromethyl substitution (47f–g) at both the para and ortho positions resulted in reduced activity. In addition, fluorination was also an effective modification for inhibiting HIF-1 transcriptional activity. Among the monofluoro-substituted compounds (26a–c), ortho derivative 26a showed better inhibitory activity than meta (26b) and para (26c) derivatives. In addition, ortho-difluoro-substituted derivative 47k exhibited significant inhibition (IC50 = 0.10 μM) compared with para, ortho-difluoro-substituted derivative 47j (IC50 = 0.31 μM). However, pentafluoro-substituted derivative 47m also showed marked inhibition but was less potent than 47l. These results indicated that fluoro substitution at the ortho position is crucial for inhibiting the transcriptional activity of HIF-1.
At the heterocycle B position, only furan substitution led to inhibitory activity. Other heterocycles, such as isoxazole, oxazole, thiazole, etc., lost their inhibitory activity.
Almost all the R2 altered substitutions displayed reduced activity, except for the esters. The ester derivatives (48b, 48c and 48f) had better inhibitory ability (IC50: 0.74, 0.89 and 0.9 μM, respectively).
In addition, from 2014 to 2015, Chen et al.71 and Masoud et al.72 was based on YC-1 structure molecular modeling and synthesized two series of N-substituted indole derivatives 53 and 58 by using either a mixture of 1H-benzo[d]imidazol-2-amine (49) or 2-(azidomethyl)-1H-benzo[d]imidazole (55) and various benzyl bromide (50) in ethanol at room temperature to obtain intermediates 51 and 56. Subsequently, the intermediates were reacted with substituted acetyl halide (52) or substituted with ethynylbenzene (57) to produce 53 and 58 derivatives, respectively (Schemes 4 and 5).
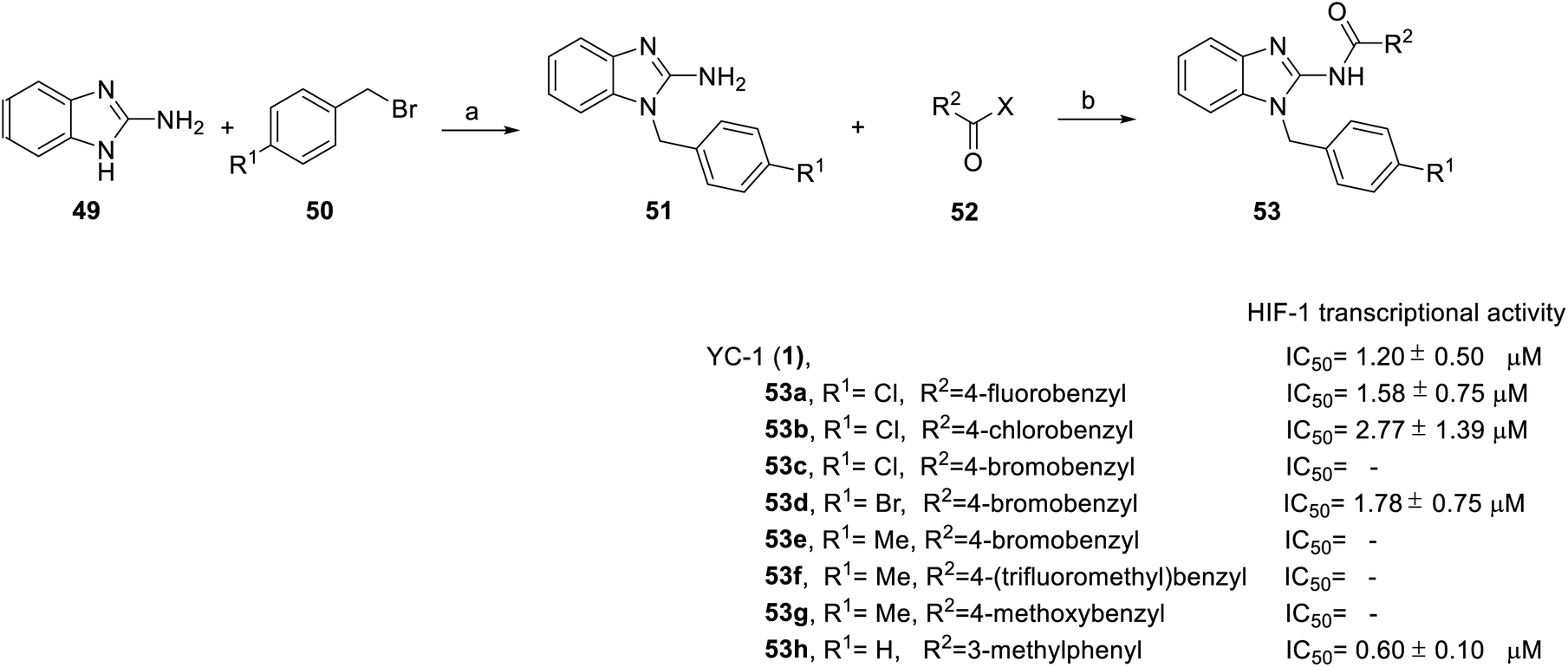 | ||
| Scheme 4 Synthesis and HIF-1 transcriptional activity effect of YC-1 analogs (53). Reagents and conditions: (a) KOH, EtOH, rt, 18 h; (b) EDCA, HOBt, DMF, rt, 24 h. | ||
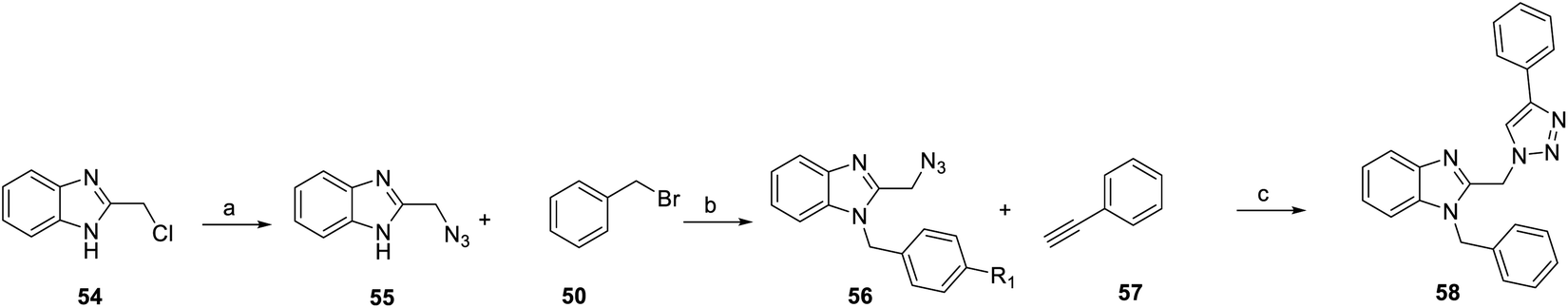 | ||
| Scheme 5 Synthesis of YC-1 analogs (58). Reagents and conditions: (a) NaN3, DMSO, rt, 5 h; (b) KOH, EtOH, rt, 18 h; (c) CuSO4·5H2O, sodium ascorbate, DCM/water/t-BuOH, rt, 24 h. | ||
Among the compounds (53) in this series, the most potent was 53h, with an IC50 value of 0.6 μM (Scheme 4). For R1 substituents, halogen substitution was more potent than alkyl substitution. For example, bromo substituted 53d (IC50 = 1.78 μM) exhibited better inhibitory activity than methyl substituted derivatives (53e–g). In the R2 position, fluoro-substituted 53a exhibited better inhibitory activity (IC50 = 1.58 μM) than bromo- or chloro-substituted derivatives (53b and 53c). Bioisosteric replacement of an amide linker to triazole ring (58) tended to extend conformational flexibility. However, none of the triazole series compounds (58) showed more than a 50% inhibition of HIF-1α expression.72
In 2016, Fuse et al. synthesized another series of imidazopyridine derivatives, 63 and 64.73 In this series, 1-iodoimidazo[1,5-a] pyridine (59) was used as the starting material, followed by Suzuki reaction to obtain compound 60. Direct acylation catalyzed by Pd(OAc)2 was applied for the formation of compounds 61 and 62. Finally, sodium borohydride reduction was used to achieve final products 63 and 64 (Scheme 6).
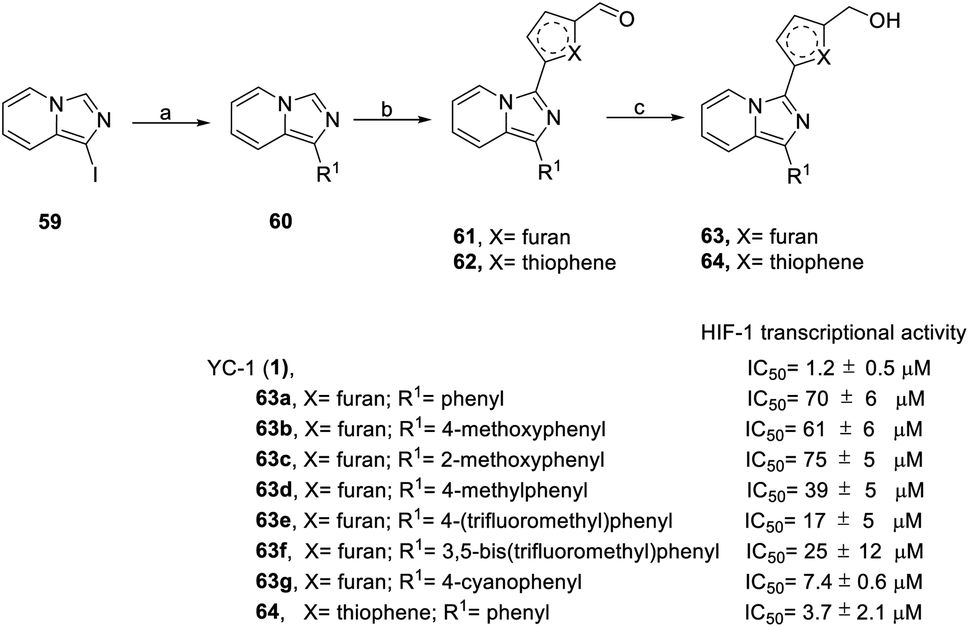 | ||
| Scheme 6 Synthesis and HIF-1 transcriptional activity effect of YC-1 analogs (63–64). Reagents and conditions: (a) 5 mol% Pd(PPh3)4, arylboronic acids, K2CO3, 1,4-dioxane/H2O, 110 °C, microwave, 1 h; (b) 5 mol% Pd(OAc)2, 5-iodofuran-2-carbaldehyde (for 61) or 5-bromothiophene-2-carbaldehyde (for 62), CsOAc, 100 °C, microwave, 1.5 h; (c) NaBH4, MeOH, rt, 5 min. | ||
In the SAR study (Scheme 6), derivatives 63b–63d (IC50 = 39–75 μM) at the R1 position containing electron-donating groups, such as methoxyphenyl and methylphenyl, had similar levels of inhibitory activity to 63a (IC50 = 70 μM). Derivatives 63e–63g containing electron-withdrawing compounds, such as (trifluoromethyl)phenyl and cyanophenyl, exerted higher levels of inhibitory activity. Despite the similar steric bulkiness of the substituents at the R1 position, the inhibitory activity was 63d (4-methoxyphenyl) < 63e (4-(trifluoromethyl)phenyl) < 63g (4-cyanophenyl) in increasing order. In the case of heterocycle X substitution, thiophene substituted 64 (IC50 = 3.7 μM) exerted a better effect than furan substituted 63a (IC50 = 70 μM). However, these derivatives were all less potent than YC-1.
In conclusion, in terms of the core derivatives, indazole has better inhibitory activity than benzoimidazole and imidazopyridine. For heterocycle B substitution, only furan has HIF-1 inhibitory activity. Among the R1 functional groups, the most effective is heterocyclic substitution. Furthermore, the best substitution position is the ortho position of the aromatic ring with fluoro substitution. Also, at the R2 position, a hydroxymethyl group is a more effective substituent than the hydroxymethyl group. Finally, the most suitable side chain structure is isobutene (Fig. 16).
Recently, Fuse et al. synthesized indeno[2,1-c]pyrazolone compounds to test their HIF-1 transcriptional inhibitory activity via the use of a dual luciferase assay under hypoxic condition, as well as to test their antiproliferative activity using an MTT assay ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)) in HeLa cells under normoxic conditions.74
Pyrazole (65) reacted with 66 to give 1-N-substituted pyrazole 67. Compound 69 was obtained using nucleophilic addition from 67 and 68, followed by cyclization to give compound 70 (Scheme 7).
 | ||
| Scheme 7 Synthesis, cytotoxicity of HeLa cells and HIF-1 transcriptional activity of indeno[2,1-c]pyrazolones (70). Reagents and conditions: (a) Cu2O, CsCO3, DMF, 100 °C, 22 h; (b) n-BuLi, THF; (c) (i) K2CO3, ICI, CHCl3, rt, 16 h; (ii) Pd(PPh3)2Cl2, NaOAc, DMA, 130 °C, 20 h. | ||
Introduction of a nitrogen atom to the X position deteriorated HIF-1 inhibitory activity, for example, as in 70b. In the R1 position, the ring activating groups, such as methyl (70c), methoxy (70d) and hydroxy (70e), at the para position improved HIF-1 inhibition activity. These indeno[2,1-c]pyrazolones derivatives (70) didn't show strong toxicity, with the exception of compound 70e (cytotoxicity IC50 = 2.2 μM). Compound 70e showed impressive HIF-1 inhibitory activity and cytotoxicity under normoxic conditions, suggesting that other biological processes also contribute to antiproliferative effects.
3.3. Vascular endothelial growth factor (VEGF)
Vascular endothelial growth factor (VEGF) is a signaling protein that stimulates blood vessel formation.75 Without the nourishment of blood vessels, the size of a tumor mass cannot exceed 2 mm3; therefore, angiogenesis is essential for tumor progression.76,77 Additionally, the exudation process requires vascularization during metastasis.Since VEGF is overexpressed in various human cancers and is released by a variety of tumor cells, it has been identified as the most important antiangiogenic factor contributing to tumor progression. When a drug can inhibit VEGF production or block its receptor signaling, it also leads to a significant inhibition of tumor growth, such as is the case with YC-1.
YC-1 blocks the induction of erythropoietin (EPO) and VEGF mRNA by inhibiting the hypoxia accumulation of HIF-1α under hypoxic conditions. According to studies by Yan et al., in experimental central retinal venous occlusion (CRVO) in rhesus monkeys, the macular edema thickness was significantly decreased after YC-1 injection (90 μl, 200 μM) at 1 week. Meanwhile, the concentration of IL-6, IL-8, VEGF, the inflammation markers, were significantly decreased in YC-1 injection group. YC-1 showed anti-inflammatory and anti-angiogenesis-related expression and significantly alleviated macular edema.16
Temirak et al., Abdullaziz et al. synthesized a series of YC-1 analogs 73–78 (Scheme 8) and discussed the SAR with cytotoxicity and VEGF levels.78–80
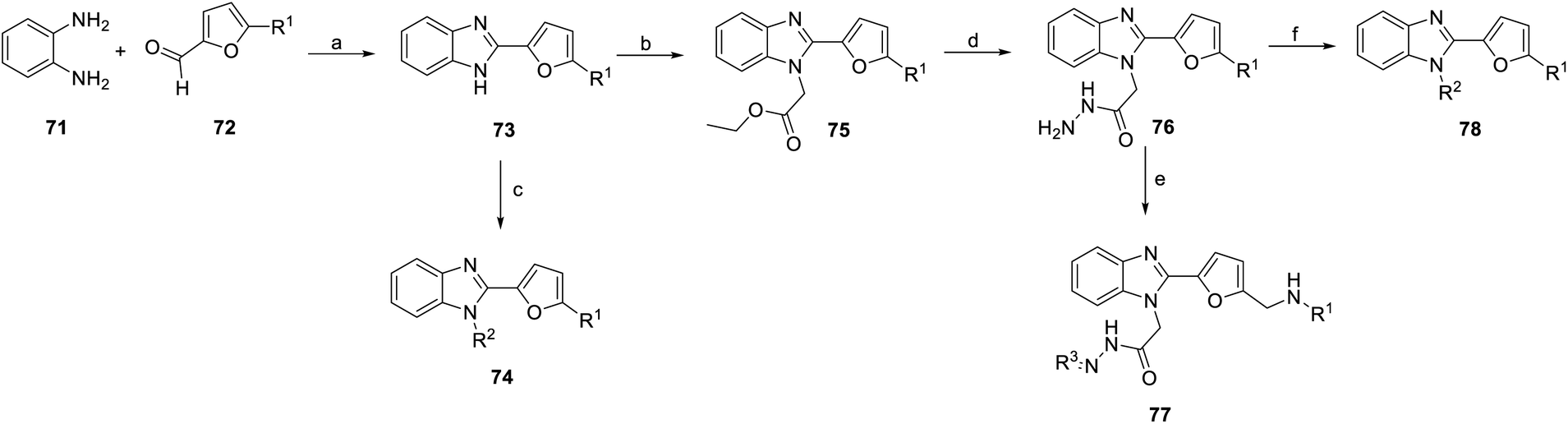 | ||
| Scheme 8 Synthesis of benzimidazole derivatives (73–78). Reagents and conditions: (a) p-TsOH, DMF, 100 °C; (b) ethyl bromoacetate, K2CO3, acetone, rt; (c) chloro compounds, K2CO3, acetone, rt; (d) NH2NH2, ethanol, 90 °C; (e) EtOAc, KOH, ethanol, reflux; (f) CS2, KOH, ice followed by reflux. | ||
1,2-Phenylenediamine (71) was condensed with furfural compound 72 in p-toluenesulfonic acid and DMF to obtain 5-methyl derivatives 73. Then, compound 73 was reacted with various chloro compounds with K2CO3 in acetone at room temperature to give N-substituted product 74. Under the same reaction conditions, treatment of 1H-benzimidazoles 73 with ethyl bromoacetate can provide N-alkylated products 75. Next, compound 76 was obtained from compound 75 with hydrazine hydrate in ethanol. Compound 76 was reacted with potassium hydroxide (KOH) and ethanol to obtain compound 77. Hydrazide compound 76 was cyclized with carbon disulfide in potassium hydroxide and ethanol to obtain oxadiazole ring compound 78 (Scheme 8).
Benzimidazole derivatives (73–78) were tested for cytotoxicity in MCF-7 human breast cancer cells. An ELISA kit was used to measure the level of human VEGF in MCF-7 cells, and the inhibition percentage was calculated based on the level of untreated cells.81,82 The SAR was discussed based on the R1, R2, R3 positions and aromatic substituents, as shown in Fig. 17–19.78–80
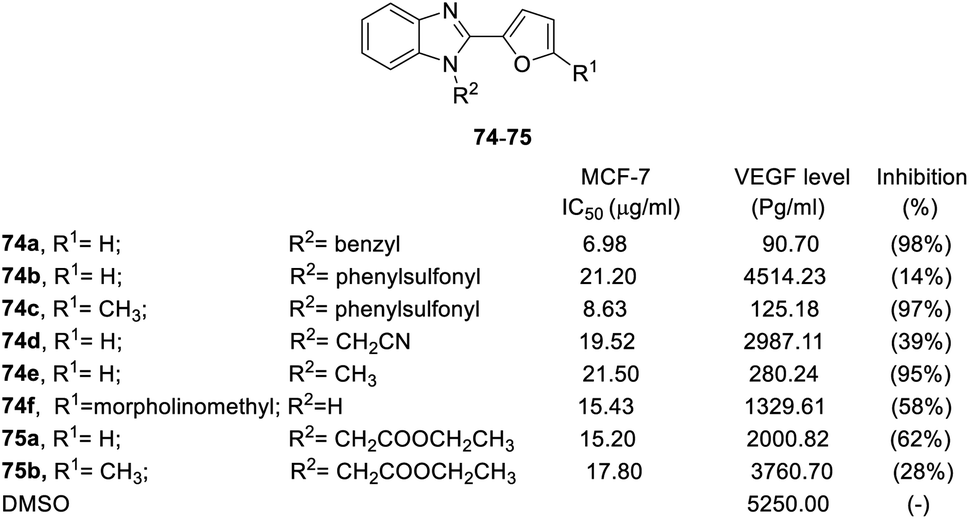 | ||
| Fig. 17 Effects of the compounds 74–75 on the MCF-7 breast cancer cell line and the VEGF level (pg mL−1) in the MCF-7 breast cancer cell line. | ||
The results showed that steric effect has an extreme impact on the R1 position. Conversion to methyl at the R1 position significantly enhanced VEGF inhibition activity. For example, R1 methyl compounds 74c significantly enhanced VEGF inhibition (VEGF inhibition was 97%), and VEGF inhibition of hydrogen compounds 74b and 76b ranged from 14–23%. Morpholinomethyl substitution 74f led to a moderate inhibition effect. At the R2 position, the conversion of hydrogen to benzyl significantly increased the activity (74a, VEGF inhibition = 98%). The cyanomethyl compound (74d) slightly decreased VEGF inhibition activity. Conversion into ethoxy methanoneyl (75) diminished the activity, where VEGF inhibition ranged from 28–62% (Fig. 17).
When the N-terminal functional group was converted to hydrazineyl, benzoylhydrazineyl, benzylhydrazineyl and methylcarbamothioylhydrazineyl substitutions, the derivatives had weak anticancer activity, such as compounds 76a–d. On the other hand, phenyl at the R3 position (77a, VEGF inhibition = 5%) had no VEGF inhibition activity, but phenyls with substitutes were potent functional groups for VEGF inhibition (77b–d, VEGF inhibition was above 96%). Conversion to a heterocycle at the R3 position decreased activity, including pyridine (77e, VEGF inhibition 58%) and furan (77f, VEGF inhibition 30%). Side chains at the R3 position played important roles in inhibitory activity. For instance, methyl side chains of compounds 77g and 77h led to worse VEGF inhibition (VEGF inhibition = 31% and 19%, respectively) (Fig. 18).
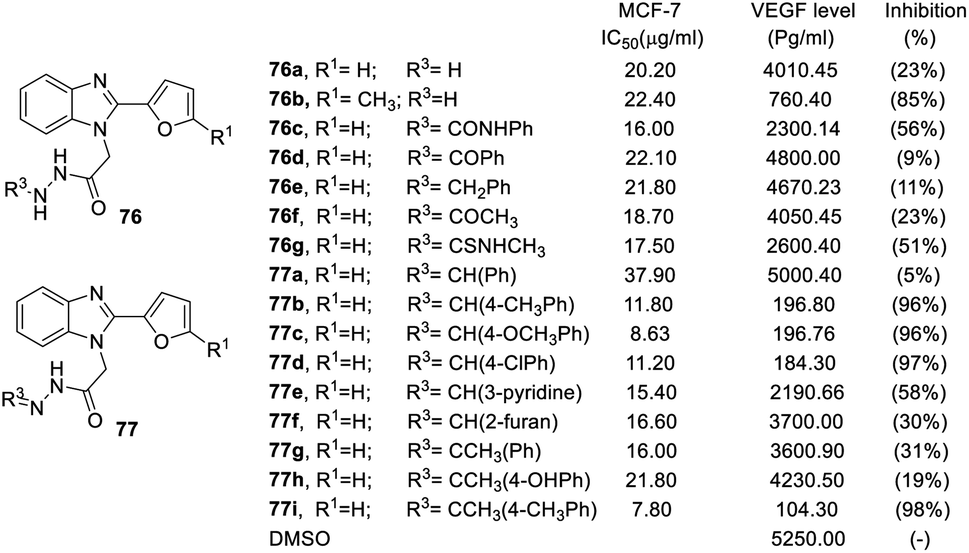 | ||
| Fig. 18 Effect of the compounds 76–77 on the breast cancer cell line MCF-7 and the VEGF level (pg mL−1) in the breast cancer cell line MCF-7. | ||
 | ||
| Fig. 19 Effects of the compounds 78 on the MCF-7 breast cancer cell line and the VEGF level (pg mL−1) in the breast cancer MCF-7 cell line. | ||
Finally, replacement of the R2 position with a heterocycle dramatically reduced the inhibitory potency of VEGF, where VEGF inhibition was less than 41%, including oxadiazole (78b, 78c, 78d), thiadiazole (78e) and imidazole (78f). However, some of heterocycles in the R2 position retained inhibitory effects, such as a 91% and 86% inhibition on triazole (78a) and thiazolidine (78g), respectively. In addition, heterocycles with a side chain can improve activity. For example, 2-ethoxy-2-oxoethyl (78h) and 2-hydrazineyl-2-oxoethyl (78i) displayed excellent inhibitory activity on VEGF levels (92% and 96%, respectively).
The SAR of benzimidazole derivatives on VEGF inhibition is summarized in Fig. 20. At the R1 position, methyl was best for VEGF inhibitory activity. However, the R2 position needs to maintain an aromatic ring or heterocycle with a side chain to have good activity.
 | ||
| Fig. 20 Effects of the SAR of benzimidazole derivatives on VEGF inhibition. | ||
3.4. Nuclear factor kappa light chain enhancer of activated B cells (NF-κB)
In 2014, Kuo et al. synthesized a series of YC-1 analogs 79 and discussed the SAR with osteoclast inhibition.83 To examine the effects of the inhibitory activity of each derivative on osteoclast formation, each derivative was added to bone marrow-derived osteoclasts (from male rats) for testing. In the presence of RANKL (50 ng mL−1) and M-CSF (20 ng mL−1), 10 μM of different test compounds were added for 5 days. After 5 days, the formation of osteoclasts was confirmed using tartrate-resistant acid phosphatase staining (TRAP staining). Among these derivatives, only 79f, with a furan ring and 2-(dimethylamino)ethyl, inhibited osteoclastogenesis by over 90% (Fig. 21).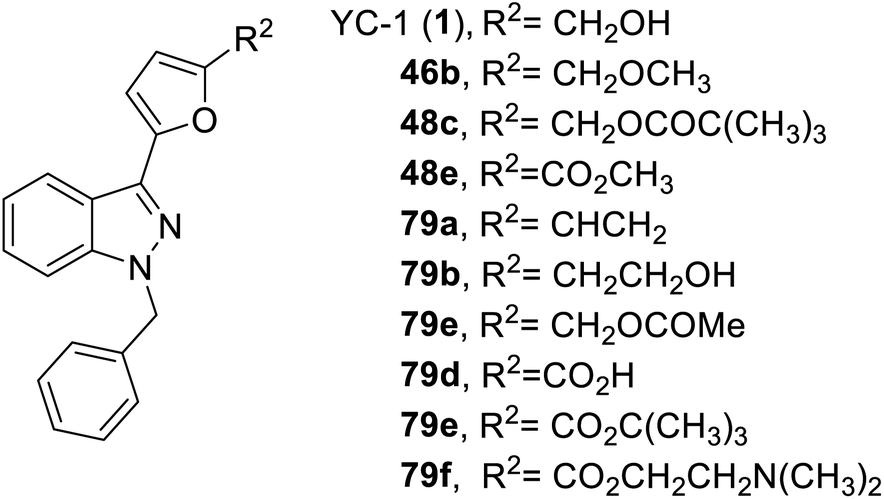 | ||
| Fig. 21 Structure of YC-1 analogs (48, and 79). | ||
4. Conclusions
Since YC-1 was discovered in 1994, there have been discussions on many related physiological activities as well as SAR. Riociguat was even developed based on YC-1 activity. The 83 articles, including 4 patent literatures, selected and discussed in this article confirm its wide use.In this review, we summarized the synthetic strategies of YC-1 derivatives based on various substituents of YC-1, systematically reviewed their biological activities, including sGC, HIF-1, VEGF, and NF-κB, and described the SAR in detail. Many chemists have modified the compound structures associated with these pharmacological activities to obtain more derivatives with greater biological activity in areas including anticancer, reverse chemoresistance, neovascularization, neural protection, asthma, learning and memory, and bone formation. However, most of the studies were on HIF-1 and sGC. The latest research indicated that YC-1 and NF-κB inhibition are significantly related, but there are few relevant studies. We believe that the inhibition of NF-κB by YC-1 derivatives is worth focusing on.
From a future perspective, to promote the innovation of YC-1 analog drugs, green, combinatorial, and diversity-oriented synthetic methods are essential for developing large-scale analog libraries for high-throughput drug screening and SAR research. In addition, although YC-1 has a wide range of applications, no studies have focused on molecular docking methods. Relevant studies in the future will help to explain the related multipharmacology and will also lead to the discovery of relevant multitarget drugs for clinical use.
Author contributions
Data curation, K.-H. Y., investigation, K.-H. Y., H.-Y. H. Project administration, H.-Y. H.; resources, H.-Y. H.; supervision, H.-Y. H.; writing – original draft, K.-H. Y.; writing – review and editing, H.-Y. H. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Notes and references
- C. C. Wu, F. N. Ko, S. C. Kuo, F. U. Lee and C. M. Teng, Blood, 1994, 84, 4226–4233 CrossRef.
- J. C. Lien, F. Y. Lee, L. J. Huang, T. M. Huang, S. C. Tsai, C. M. Teng, C. C. Wu, F. C. Cheng and S. C. Kuo, J. Med. Chem., 2001, 44, 3746–3749 CrossRef PubMed.
- E. J. Yeo, Y. S. Chun, Y. S. Cho, J. Kim, J. C. Lee, M. S. Kim and J. W. Park, J. Natl. Cancer Inst., 2003, 95, 516–525 CrossRef CAS PubMed.
- T. Bollinger, S. Gies, J. Naujoks, L. Feldhoff, A. Bollinger, W. Solbach and J. Rupp, J. Leukocyte Biol., 2014, 96, 305–312 CrossRef PubMed.
- T. C. Hsia, W. H. Liu, W. W. Qiu, J. Luo and M. C. Yin, Molecules, 2014, 19, 19892–19906 CrossRef PubMed.
- L. Bergandi, S. Canosa, G. Pittatore, F. Silvagno, S. Doublier, G. Gennarelli and C. Benedetto, Biol. Reprod., 2019, 100, 1521–1535 CrossRef CAS PubMed.
- X. Li, J. Xing, H. Wang and E. Yu, Biosci. Rep., 2019, 39(5), BSR20180268 CrossRef CAS PubMed.
- W. L. Chen, C. C. Wang, Y. J. Lin, C. P. Wu and C. H. Hsieh, J. Transl. Med., 2015, 13, 389–402 CrossRef PubMed.
- S. Kaowinn, I. R. Cho, J. Moon, J. Soh, H. Y. Kang, C. R. Jung, S. Oh, H. Song, S. S. Koh and Y. H. Chung, Int. J. Oncol., 2015, 46, 2076–2082 CrossRef PubMed.
- J. Wu, X. Ke, N. Ma, W. Wang, W. Fu, H. Zhang, M. Zhao, X. Gao, X. Hao and Z. Zhang, Drug Des., Dev. Ther., 2016, 10, 3071–3081 CrossRef CAS PubMed.
- X. Ke, J. Wu, W. Wang, H. Zhang, N. Ma, W. Fu, M. Zhao, X. Gao, X. Hao and Z. Zhang, Int. J. Biol. Sci., 2016, 12, 1363–1371 CrossRef PubMed.
- X. Li, N. Lu, R. Tan, J. An, Z. Cai, X. Hu, F. Wang, H. Wang, C. Lu and H. Lu, J. Mol. Neurosci., 2018, 66, 238–250 CrossRef PubMed.
- W. T. Lee, S. H. Tai, A. C. Lee, Y. W. Lin, H. Y. Hung, S. Y. Huang, Y. S. Wu and E. J. Lee, Mol. Med. Rep., 2018, 17, 6490–6496 Search PubMed.
- E. McEachern, C. Dewitz, S. Shin, K. Akong, D. G. Nagle, D. H. Broide, P. Akuthota and L. E. Crotty Alexander, J. Clin. Immunol., 2017, 176, 94–99 CrossRef PubMed.
- Y. Liang, C. E. Carroll, I. Benakanakere, C. Besch-Williford and S. M. Hyder, Int. J. Oncol., 2013, 4, 179–187 Search PubMed.
- J. An, Z. Yan, Q. Shang, N. Zhou and J. Ma, Curr. Eye Res., 2018, 43, 526–533 CrossRef PubMed.
- W. S. Lee, M. S. Chang, B. C. Chen, J. R. Sheu and C. H. Lin, Mol. Pharmacol., 2004, 66, 561–571 Search PubMed.
- K. C. Liang, W. L. Chien, C. M. Teng, S. C. Kuo, F. Y. Lee and W. M. Fu, Mol. Pharmacol., 2003, 63, 1322–1328 CrossRef PubMed.
- C. B. Yeh, J. W. Wang, S. J. Chou, K. C. Lu, T. H. Chu, W. Y. Chen, J. L. Chien, M. H. Yen, T. H. Chen and J. F. Shyu, J. Bone Miner. Metab., 2018, 36, 508–518 CrossRef PubMed.
- Y. S. Chun, E. J. Yeo and J. W. Park, Cancer Lett., 2004, 207, 1–7 CrossRef CAS PubMed.
- C. H. Wu, C. H. Pan and M. J. Sheu, Vasc. Biol., 2019, 5 DOI:10.5772/intechopen.84572.
- M. Hori, A. Takeuchi, S. Sato, H. S. Ban, T. Kuchimaru, S. Kizaka-Kondoh, T. Yamori and H. Nakamura, Med. Chem. Commun., 2012, 3, 1455–1461 RSC.
- T. Meng, K. Yuanfang and C. Bingje, China Patent, 106316958A, 2015 Search PubMed.
- C. M. Counceller, C. C. Eichman, B. C. Wray, C. Brenda and J. P. Stambuli, Org. Lett., 2008, 10, 1021–1023 CrossRef CAS PubMed.
- J. C. Lien, F. Y. Lee, L. J. Huang, T. M. Huang, S. C. Tsai, C. M. Teng, C. C. Wu, F. C. Cheng and S. C. Kuo, J. Med. Chem., 2001, 44, 3746–3749 CrossRef PubMed.
- A. Straub, C. Furstner, U. Niewohner, T. Jaetsch, A. Feurer, R. Kast, J. P. Stasch, E. Perzborn, J. Hutter and K. Dembowsky, European Patent, EP0932403A1, 1996 Search PubMed.
- V. Collot, P. Dallemagne, B. Patrick, R. Philippe and S. Rault, Tetrahedron, 1999, 55, 6917–6922 CrossRef CAS.
- K. W. Hering, J. D. Artz, W. H. Pearson and M. A. Marletta, Bioorg. Med. Chem. Lett., 2006, 16, 618–621 CrossRef CAS PubMed.
- X. Yang, J. Yu, C. Wu and W. Su, J. Org. Chem., 2020, 85, 1009–1021 CrossRef PubMed.
- C. M. Teng, F. Y. Lee and S. C. Kuo, US Pat., US 7345079B2, 2002 Search PubMed.
- M. Katsuno, K. Inamoto, T. Yoshino, L. Suzuki, K. Hiroya and T. Sakamoto, Chem. Lett., 2004, 33(8), 1026–1027 CrossRef.
- K. Inamoto, M. Katsuno, T. Yoshino, Y. Arai, K. Hiroya and T. Sakamoto, Tetrahedron, 2007, 63, 2695–2711 CrossRef CAS.
- O. V. Evgenov and J. P. Stasch, Handb. Exp. Pharmacol., 2013, 218, 279–313 Search PubMed.
- S. C. Kuo, F. Y. Lee and C. M. Teng, US Pat, US5574168A, 1996 Search PubMed.
- T. Utkan, I. Komsuoglu Celikyurt, C. Ozer, N. Gacar and F. Aricioglu, Med. Sci. Monit. Basic Res., 2014, 20, 130–137 CrossRef PubMed.
- K. C. Liang, W. L. Chien, C. M. Teng, S. C. Kuo, F. Y. Lee and W. M. Fu, Eur. J. Neurosci., 2005, 21, 1679–1688 CrossRef PubMed.
- K. C. Liang, W. L. Chien and W. M. Fu, Eur. J. Pharmacol., 2008, 590, 233–240 CrossRef PubMed.
- D. A. Tulis, Curr. Med. Chem., 2004, 2, 343–359 CAS.
- K. J. Peyton, X. M. Liu, N. N. Mendelev, H. Wang, D. A. Tulis and W. Durante, Mol. Pharmacol., 2009, 75, 208–217 CrossRef PubMed.
- W. C. Chang, C. H. Wu, G. Y. Chang, S. C. Kuo and C. M. Teng, J. Pharmacol. Sci., 2004, 94, 252–260 CrossRef PubMed.
- A. Straub, J. P. Stasch, A. A. Cristina, B. B. Jordi, B. Ducke, A. Feurer and C. Fürstner, Bioorg. Med. Chem. Lett., 2001, 11, 781–784 CrossRef CAS PubMed.
- F. Y. Lee, J. C. Lien, L. J. Huang, S. L. Pan, J. H. Guh, C. M. Teng and C. C. Kuo, J. Med. Chem., 2002, 45, 4947–4949 CrossRef PubMed.
- S. Weigand, J. Mittendorf, C. Alonso-Alija, E. Bischoff, A. Feurer, M. Gerisch, A. Kern, D. Knorr, D. Lang, K. Muenter, M. Radtke, H. Schirok, K. H. Schlemmer, E. Stahl, A. Straub, F. Wunder and J. P. Stasch, ChemMedChem, 2009, 4, 853–865 CrossRef PubMed.
- P. Vaupel and M. Höckel, J. Natl. Cancer Inst., 2001, 93, 266–276 CrossRef PubMed.
- T. Harada, K. Hirose, Y. Wada, M. Sato, K. Ichise, M. Aoki, T. Kato, K. Takeda and Y. Takai, J. Radiat. Res., 2020, 61, 524–534 CrossRef CAS PubMed.
- C. L. Shui, C. Liu, Q. Wang, H. Luo and C. G. Gu, Braz. J. Med. Biol. Res., 2017, 51, e6768–e6775 Search PubMed.
- M. Igase, S. Kambayashi, K. Kobayashi, A. Kimura, T. Shimokawa Miyama, K. Baba, S. Noguchi, T. Mizuno and M. Okuda, J. Vet. Med. Sci., 2015, 77, 1405–1412 CrossRef PubMed.
- R. X. Wang, X. W. Ou, M. E. Kang and J. Q. Shi, Eur. Rev. Med. Pharmacol. Sci., 2018, 22, 8237–8247 Search PubMed.
- Y. Kitajima, K. Wakiyama, T. Tanaka, M. Kaneki, K. Yanagihara, S. Aishima, J. Nakamura and H. Noshiro, Sci. Rep., 2017, 7, 12653–12665 CrossRef PubMed.
- J. Wu, P. Li, L. Zhao and X. W. Feng, Sleep Breath., 2016, 20, 167–173 CrossRef PubMed.
- J. Kong, X. Ding, W. Xu, S. Dong, Y. Du, C. Yao, J. Gao, S. Ke, S. Wang and W. Sun, Oncol. Lett., 2018, 16, 5230–5236 Search PubMed.
- Y. Liu, X. Wan, Y. Zhao, X. Sun, D. Fan and L. Guo, PLoS One, 2017, 12, e0184213 CrossRef PubMed.
- X. Li, W. Zhao and Z. Li, Pharmazie, 2017, 71, 524–529 Search PubMed.
- Y. Zhang, H. J. Zhao, X. R. Xia, F. Y. Diao, X. Ma, J. Wang, L. Gao, J. Liu, C. Gao, Y. G. Cui and J. Y. Liu, Clin. Chim. Acta, 2019, 489, 203–211 CrossRef CAS PubMed.
- P. Lin, L. Sun, Z. Qin, Y. Liu, L. L. Deng and C. Lu, Exp. Biol. Med., 2015, 240, 1434–1445 CrossRef PubMed.
- H. Hu, X. K. Miao, J. Y. Li, X. W. Zhang, J. J. Xu, J. Y. Zhang, T. X. Zhou, M. N. Hu, W. L. Yang and L. Y. Mou, Eur. J. Pharmacol., 2020, 874, 172961–172977 CrossRef CAS PubMed.
- J. Sakakibara-Konishi, Y. Ikezawa, H. Mizugaki, S. Oizumi and M. Nishimura, Int. J. Clin. Oncol., 2017, 22, 59–69 CrossRef PubMed.
- F. Takahashi, F. Nurwidya, I. Kobayashi, A. Murakami, M. Kato, K. Minakata, T. Nara, M. Hashimoto, S. Yagishita, H. Baskoro, M. Hidayat, N. Shimada and K. Takahashi, Biochem. Biophys. Res. Commun., 2014, 455, 332–338 CrossRef PubMed.
- J. Zhou, Q. Jin, X. Xu, F. Huang and W. Xu, Oncol. Lett., 2019, 1, 4034–4043 Search PubMed.
- W. Wu, J. Wan, Y. Huang, W. Ge and S. Liu, Oncol. Rep., 2016, 36, 659–668 CrossRef PubMed.
- L. Ye, C. Zhou, C. Jiang, J. Bai, Y. Chi and H. Zhang, Tumor Biol., 2015, 36, 9179–9188 CrossRef PubMed.
- H. Furusho, H. Sasaki, D. B. Rider, J. M. Dobeck, W. P. Kuo, A. Fujimura, S. Yoganathan, K. Hirai, S. Xu, K. Sasaki and P. Stashenko, J. Endod., 2019, 45, 181–188 CrossRef PubMed.
- J. Shao, C. Wan, S. R. Gilbert, R. C. Riddle, F. Long, R. S. Johnson, E. Schipani and T. L. Clemens, Ann. N. Y. Acad. Sci., 2010, 1192, 322–326 CrossRef PubMed.
- K. Zhou, K. Wu, Y. Wang, Y. Zhou, N. Tian, Y. Wu, D. Chen, D. Zhang, X. Wang, H. Xu and X. Zhang, Brain Res., 2016, 163, 19–26 Search PubMed.
- Y. Tang, M. Bernaudin, M. Reilly, E. Petit and F. R. Sharp, J. Biol. Chem., 2002, 277, 39728–39738 CrossRef PubMed.
- J. Yan, B. Zhou, S. Taheri and H. Shi, PLoS One, 2011, 6, e27798–e27808 CrossRef CAS PubMed.
- L. Kong, Y. Ma, Z. Wang, N. Liu, G. Ma, C. Liu, R. Shi and G. Du, Int. Immunopharmacol., 2021, 94, 107507–107519 CrossRef CAS PubMed.
- H. M. Brandon, A. Rajakumar, A. Daftary, R. Ness and K. P. Conrad, Placenta, 2004, 25, 763–769 CrossRef PubMed.
- S. Tong, F. C. Brownfoot, N. J. Hannan, R. Hastie, P. Cannon, L. Tuohey and T. J. Kaitu'u-Lino, Mol. Cell. Endocrinol., 2015, 41, 202–208 Search PubMed.
- N. J. Kim, H. An, J. W. Jung, H. Jang, J. W. Park and Y. G. Suh, Bioorg. Med. Chem. Lett., 2011, 21, 6297–6300 CrossRef PubMed.
- J. Wang, J. Chen, P. Schwab, K. T. Park, T. N. Seagroves, L. K. Jennings, D. D. Miller and W. Li, Anticancer Res., 2014, 34, 3891–3904 Search PubMed.
- J. Wang, G. N. Masoud, J. Chen, D. Miller and W. Li, Anticancer Res., 2015, 35, 3849–3859 Search PubMed.
- S. Fuse, T. Ohuchi, Y. Asawa, S. Sato and H. Nakamura, Bioorg. Med. Chem. Lett., 2016, 26, 5887–5890 CrossRef CAS PubMed.
- S. Fuse, K. Suzuki, T. Kuchimaru, T. Kadonosono, H. Ueda, S. Sato, S. Kizaka-Kondoh and H. Nakamura, Bioorg. Med. Chem., 2020, 28, 115207–115213 CrossRef CAS PubMed.
- S. J. Galli, D. R. Senger, A. M. Dvorak, C. A. Perruzzi, V. S. Harvey and H. F. Dvorak, Science, 1983, 219, 983–985 CrossRef PubMed.
- M. O'Reilly, S. Parangi, G. Christofori, L. Holmgren, J. Grosfeld, J. Folkman and D. Hanahan, Proc. Natl. Acad. Sci. U.S.A., 1996, 93, 2002–2007 CrossRef PubMed.
- L. Holmgren, M. S. O'Reilly and J. Folkman, Nat. Med., 1995, 1, 149–153 CrossRef CAS PubMed.
- A. Temirak, Y. M. Shaker, F. A. F. Ragab, M. M. Ali, S. M. Soliman, J. Mortier, G. Wolber, H. I. Ali and H. I. El Diwani, Arch. Pharm., 2014, 347, 291–304 CrossRef CAS PubMed.
- Y. M. Shaker, A. Temirak, F. A. Ragab, M. M. Ali, H. I. Ali and H. I. El Diwani, Eur. J. Med. Chem., 2014, 87, 868–880 CrossRef PubMed.
- M. A. Abdullaziz, H. T. Abdel-Mohsen, A. M. El Kerdawy, F. A. F. Ragab, M. M. Ali, S. M. Abu-bakr, A. S. Girgis and H. I. El Diwani, Eur. J. Med. Chem., 2017, 136, 315–329 CrossRef CAS PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J. Langley, P. Cronise, A. Vaigro-Wolff, M. Gray-Goodrich, H. Campbell, J. Mayo and M. Boyd, J. Natl. Cancer Inst., 1991, 83, 757–766 CrossRef CAS PubMed.
- T. H. Lin, T. H. Kuo, R. S. Yang, S. C. Kuo, W. M. Fu and H. Y. Hung, J. Med. Chem., 2015, 58, 4954–4963 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |