
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT study on the electronic structure and optical properties of an Au-deposited α-Fe2O3 (001) surface
Yong Shia,
Cuihua Zhao *ab,
Xing Chena,
Cuiting Chena,
Xi Zhoua and
Jianhua Chenab
*ab,
Xing Chena,
Cuiting Chena,
Xi Zhoua and
Jianhua Chenab
aSchool of Resources, Environment and Materials, Guangxi University, Nanning 530004, China
bGuangxi Key Laboratory of Processing for Non-ferrous Metal and Featured Materials, Guangxi University, Nanning 530004, China. E-mail: xiaocui2000110@163.com
First published on 16th February 2022
Abstract
The electronic structure and optical properties of gold clusters deposited on an α-Fe2O3 surface were studied by using density functional theory (DFT), with a special emphasis on the influence of Au cluster sizes. There is a strong interaction between Au clusters and the α-Fe2O3 surface, and the binding energy increases with an increase of Au cluster size. The Au atoms of the gold cluster are bonded to the iron atoms of the α-Fe2O3 surface for the Au/α-Fe2O3 system, and the electrons transfer from the Au cluster to the α-Fe2O3 surface with the largest number of electrons transferred for 4Au/α-Fe2O3. The peaks of the refractive index, extinction coefficient and dielectric function induced by Au clusters appear in the visible range, which results in the enhanced optical absorption for the Au/α-Fe2O3 system. The optical absorption intensifies with increasing Au cluster size in the visible range, showing a maximum value for 4Au/α-Fe2O3. Further increasing the Au cluster size above 4Au results in a decrease in absorption intensity. The results are in good agreement with those of the refractive index, extinction coefficient and dielectric function.
1 Introduction
Long-term exposure to high concentrations of volatile organic compounds (VOCs) could cause serious damage to the lungs and induce various cancers.1–3 In recent years, several studies have been reported on the removal of VOCs.4,5 After years of development, some removal methods have been developed, such as incineration,6 condensation,7 adsorption and absorption8 and so on. However, these methods have their own shortcomings, which limits their application in practice. For example, incineration and condensation have good cost performance only for medium and high VOC concentrations.9 Adsorption and absorption only transfer VOCs to another medium, and do not destroy them. Furthermore, the adsorption part of the adsorbent could be clogged and filled by moist airflow. Herein, photo-catalytic oxidation has attracted increasing attention for its effective efficiency and low budget for low VOC concentrations.Various photo-catalysts (TiO2, ZnS, CdS, SnO2 and WO3) have been developed by the continuous efforts. However, most oxides of these semiconductors can only absorb UV light, which severely limits their practical applications. Although sulfides have a lower band gap and better stability than oxides, these materials are very unstable in liquid media.10 Compared with other photocatalytic materials, α-Fe2O3 possesses a lower band gap (2.2 eV) with a strong capacity of light absorption in the visible region. In addition, α-Fe2O3 is stable in most aqueous solutions (pH > 3) and is one of the cheapest semiconductor materials available.11,12 Therefore, it has been investigated as a potential photocatalytic material in recent years. For example, Dong et al. discovered grain-like α-Fe2O3 nanostructures can efficiently degrade the congo red dyes.13 Nevertheless, the photocatalytic activity of α-Fe2O3 is limited by high position of valence, weak diffusion capacity of photo-generated holes and low conductivity.14,15 In order to overcome these defects, a lot of research has been carried out, including surface modification,16,17 photosensitization,18,19 and doping with different materials,20,21 etc.
Noble metal deposition22 is an effective strategy to overcome the limitation of high recombination rate and poor electrical conductivity of electron–hole for α-Fe2O3. The deposition of noble metals on the catalyst surface by local surface plasmon resonance (LSPR) makes the metal nanoparticles have a strong absorption effect on the photon energy.23 Generally, the photocatalytic reaction is determined by three main energy transfer mechanisms, including the orbital leap of charge, the enhancement of the nearby electric field and the scattering mechanism. Liu et al.24 synthesized porous α-Fe2O3 decorated by Au nanoparticles, which exhibits a much higher response in comparison to pure α-Fe2O3. Li et al. found25 that the Schottky barrier formed between precious metals and semiconductors effectively promotes the separation of light-induced electrons and holes, and the photocurrent is significantly enhanced on the visible-light catalytic oxidation of refractory organic compounds of Au/α-Fe2O3. Nevertheless, the degradation rate of organic compounds decreases with the increase of the loading amounts. The overloaded Au particles turns into the recombination center of photogenerated electrons and holes, and the generation of photocurrent is inhibited with the decrease of quantum yield. Lin et al.26 successfully loaded Au and Pd on the surface of α-Fe2O3 by the reduction method of polyvinyl alcohol, and the photocatalytic degrading VOCs efficiency of modified α-Fe2O3 could be significantly improved because of the large optical response range. Combined with photo-assisted growth, gold nanoparticles were deposited successfully on α-Fe2O3 nanocrystals through a cysteine-linked seed planting approach by Cao et al.,27 and the size of the gold particles on α-Fe2O3 nanocrystals was regulated from 2 to 20 nm through a photoinduced seed mediation scheme. The Au deposited α-Fe2O3 nanocrystals not only exhibit efficient catalysis properties for 4-NP reduction but also have great efficiency for visible-light-driven photocatalytic dye degradation and water oxidation.
Metal clusters consist of several to hundreds of atoms have been popularized very much in recent years.28,29 It is usually that metal nanoclusters possess a very small size that is comparable to the Fermi wavelength (the de Broglie wavelength of the conduction electrons present near the Fermi energy level) compared with nanoparticles. The size of metal clusters (Ag and Au) is usually smaller than 2 nm.28 The metal nanoclusters are more like photonic supramolecules than the plasmonic nanoparticle analogue. For quantum confinement, the metal nanoclusters exhibit discrete molecular-like energy levels and a size-dependent LUMO–HOMO gap, meanwhile, they lose the related properties of the bulk ones, such as the SPR effect, which means the surface electrons in metal nanoclusters no longer oscillate together. Hence, the chemical and physical properties of the metal nanoclusters change to make the energy transfer more different and complicated from their larger counterparts.30 For bulk Ag metal nanoparticles in the aqueous phase, the Ag+/Ag redox potential is reported to be +0.79 V (vs. NHE) but is −1.80 V (vs. NHE) for Ag nanoclusters.31 Carolina et al.32 investigated the structures and the growth processes of small Cu, Ag and Au clusters on the Fe-terminated (0001) surface of α-Fe2O3 using DFT method. Hon et al.33 studied Au nanoclusters supported on the α-Fe2O3 (0001) surface by the PBE+U calculations, and found that the periphery atoms of Au10 particles become oxidized through the dissociation of O2 at the metal-oxide interface. Nguyen et al.34 studied the different properties of Au monomers and dimers on the defect-free and defective terminations of α-Fe2O3 (0001) surface and their interaction with CO through DFT calculation. The results showed that the range of charge states of Au atoms is somewhere in between 0 and 1, besides, the most active site for CO oxidation is probable Au replacing a Fe in the Fe2O3 surface. Pabisiak et al.35 investigated the adsorption of small Aun nanocluster on the iron terminated Fe2O3(0001) surface and CO on the Aun cluster supported on the Fe2O3(0001) surface. They got the conclusion that the interaction of CO molecule with Aun cluster on the hematite surface is stronger than that with pure Fe2O3(0001) surface. Overall, the photocatalytic activity of noble metal cluster deposition on α-Fe2O3 surface could be enhanced via facilitating the electron movement on the heteroboundary surface and hindering the recombination of photogenerated electron holes, and the amount and size of loaded metals cluster are of significance to the catalytic activity of α-Fe2O3.
Although Au cluster deposition on α-Fe2O3 (0001) surface have been studied, the microscopic mechanism of the influence of Au cluster size on α-Fe2O3 photocatalytic performance haven't been described in detail. Here, we focus on the electronic structures and optical properties of Au clusters deposited α-Fe2O3 surface by using first principle method based on density functional theory (DFT), with a special emphasis on the influences of Au cluster size. This study will provide an important theoretical basis for the application of metal-enhanced photocatalytic materials in industry.
2 Computational methods and models
2.1 Computational method
All calculations were carried out by using the CASTEP (Cambridge Sequential Total Energy Package) module in the MS (material studio) software package by Payne et al.36 The exchange-correlation functional of the generalized gradient approximation (GGA), developed by Perdew and Wang (PW91),37 A Monkhorst–Pack k-point sampling density of 3 × 3 × 1 and a plane wave cutoff energy of 340 eV were used for all calculations based the test results. Under these parameters, the lattice constant and band gap of iron oxide are close to the experimental values, and the system energy is relatively low, which was described in detail elsewhere.38 The interactions between valence electrons and ionic core were represented with ultrasoft pseudopotentials.39 The dipole correction with self-consistent was used in calculation. In order to decrease the error of calculated band gap, the calculations used the DFT+U method with U value of 3.5 eV based the test. Valence electrons configuration considered in this study included O 2s2 2p4, Fe 3d6 4s2 and Au 5d10 6s1 states. The convergence tolerances for geometry optimization calculations were set to the maximum displacement of 0.002 Å, the maximum force of 0.05 eV Å−1, the maximum energy change of 2.0 × 10−5 eV per atom and the maximum stress of 0.1 GPa, and the self-consistent field (SCF) convergence tolerance was set to 2.0 × 10−6 eV per atom.2.2 Computational model
Hematite (α-Fe2O3) possesses a typical rhombohedral structure with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c. Each Fe atom coordinates with adjacent six O atoms, and each O atom coordinates with four Fe atoms, forming an corundum crystal structure, which has been described in detail elsewhere.40 The lattice constants of the α-Fe2O3 bulk are a = b = 5.088 Å, and c = 14.138 Å, which are close to the experimental results (a = b = 5.0355 Å, and c = 13.747 Å). In addition, the bandgap of the Fe2O3 bulk is 2.265 eV, which is in accord with the experimental value (1.9–2.2 eV).41 These results were described in detail by our group.42 α-Fe2O3 (001) surface was cut from the bulk with the optimum unit cell volume. The adjacent mirror images of the slab can be separated under a vacuum environment, the thickness of vacuum layer in the vertical direction is set as 15 Å, and the slab thickness was selected as 18 layers by testing the surface energies of α-Fe2O3 (001). The α-Fe2O3 surface was modeled using 2 × 2 × 1 supercell geometries for better research of Au particle deposited α-Fe2O3 surface. The surface energies of α-Fe2O3 (001) with varying slab thicknesses were tested to determine the slab size and finally established the most stable slab model, as shown in Fig. 1.
c. Each Fe atom coordinates with adjacent six O atoms, and each O atom coordinates with four Fe atoms, forming an corundum crystal structure, which has been described in detail elsewhere.40 The lattice constants of the α-Fe2O3 bulk are a = b = 5.088 Å, and c = 14.138 Å, which are close to the experimental results (a = b = 5.0355 Å, and c = 13.747 Å). In addition, the bandgap of the Fe2O3 bulk is 2.265 eV, which is in accord with the experimental value (1.9–2.2 eV).41 These results were described in detail by our group.42 α-Fe2O3 (001) surface was cut from the bulk with the optimum unit cell volume. The adjacent mirror images of the slab can be separated under a vacuum environment, the thickness of vacuum layer in the vertical direction is set as 15 Å, and the slab thickness was selected as 18 layers by testing the surface energies of α-Fe2O3 (001). The α-Fe2O3 surface was modeled using 2 × 2 × 1 supercell geometries for better research of Au particle deposited α-Fe2O3 surface. The surface energies of α-Fe2O3 (001) with varying slab thicknesses were tested to determine the slab size and finally established the most stable slab model, as shown in Fig. 1.
 | ||
| Fig. 1 Supercell model of α-Fe2O3 (001) surface. (a) Main view; (b) top view. Numbers indicate the distance in Å. | ||
3 Results and discussion
3.1 Configurations of Au/α-Fe2O3 (001) surface
In order to determine the stable configuration of Au deposited on α-Fe2O3 surface, we studied the deposition of one Au atom on α-Fe2O3 surface with different sites, including the top deposition of Au atom on iron of α-Fe2O3 surface, top deposition on oxygen, and vacancy deposition of α-Fe2O3 surface, and calculated their binding energies. The results show that the top deposition of Au atom on iron of α-Fe2O3 surface is the most stable. As a result, we built the deposition models of multiple Au atoms by putting Au atoms at the top of iron atom of α-Fe2O3 surface. Fig. 2 shows the configurations of Au/α-Fe2O3 surface with different Au atoms. For single Au atom deposited on α-Fe2O3 surface, Au1 atom is bonded to Fe1 atoms of the α-Fe2O3 surface, and the bond length of Au1–Fe1 is 2.515 Å (Fig. 2(a)). For 2Au/α-Fe2O3, on the one hand, Au1 atom is bonded to Au2 atom with a bond length of 2.686 Å; on the other hand, Au1 and Au2 atoms are bonded to Fe1 and Fe2 atoms of the α-Fe2O3 surface, respectively, and the bond lengths of Au1–Fe1 and Au2–Fe2 are 2.816 Å and 2.784 Å, respectively (Fig. 2(b)). Nguyen et al.34 studied the deposition of Au monomers and dimers on the Fe2O3 (0001) surface. They found that Au atom is bonded to the iron atom of α-Fe2O3 surface for monomers, and the bond length of Au–Fe is 2.49 Å. As for Au dimer, Au1 atom is bonded to Au2 atom with a bond length of 2.56 Å. In addition, Au1 and Au2 atoms are bonded to Fe and O atoms of the α-Fe2O3 surface, respectively, and the bond length of Au1–Fe is 2.66 Å. Pabisiak et al.35 also investigated small Au clusters structures supported on iron termination of α-Fe2O3(0001) surface. They found that Au atom for Au monomer is bonded to an iron of surface with a bond length of 2.52 Å. The length of Au1–Au2 bond for Au dimer is 2.59 Å, and Au1 and Au2 atoms are bonded to iron and oxygen atoms of the α-Fe2O3 surface, respectively with the Au1–Fe bond of 2.67 Å and Au2–O bond of 2.17 Å. For the deposition of a gold atom on α-Fe2O3 surface, our result is similar with those of Nguyen and Pabisiak et al. Au is bonded to an iron of the surface with a bond length of about 2.5 Å. However, there are some differences for dimers. Two Au atoms are bonded to two Fe atoms of the surface in our study, while in Nguyen and Pabisiak's study, two Au atoms are bonded to Fe and O atoms of the surface, respectively. The reason may be different modeling methods. Two Au atoms are simultaneously placed on the surface of α-Fe2O3 in our modeling, while in Nguyen and Pabisiak's modeling, the Au dimer was constructed with the second Au atom positioned on the neighboring site of the first Au atom which was already adsorbed on α-Fe2O3 surface. For the difference, we will further study in the future. For 3Au/α-Fe2O3, three Au atoms are bonded to each other, forming a triangular cluster structure, meanwhile, they are also bonded to three iron atoms of α-Fe2O3 surface (Fe1, Fe2 and Fe3), respectively. The lengths of newly formed Au1–Au2, Au1–Au3, Au2–Au3, Au1–Fe1, Au2–Fe2 and Au3–Fe3 bonds are 2.726 Å, 2.743 Å, 2.729 Å, 2.776 Å, 2.772 Å and 2.767 Å, respectively (Fig. 2(c)). For 4Au/α-Fe2O3, the Au1 atom is bonded to Au3 and Au4 atoms, and Au3 atom is bonded to Au2 atom besides Au1 atom. In addition, Au3 and Au2 are also bonded to Fe3 and Fe2 of α-Fe2O3 surface, respectively. The lengths of newly formed Au4–Au1, Au1–Au3, Au3–Au2, Au2–Fe2 and Au3–Fe3 bonds are 2.717 Å, 2.740 Å, 2.779 Å, 2.814 Å and 2.793 Å, respectively (Fig. 2(d)). As for 5Au/α-Fe2O3, Au1, Au3 and Au5 atoms form a triangular structure. Au4 atom is bonded to Au1 and Au2 atoms, and Au4 and Au2 atoms are bonded to Fe1 and Fe2, respectively. All bond lengths are shown in Fig. 2(e).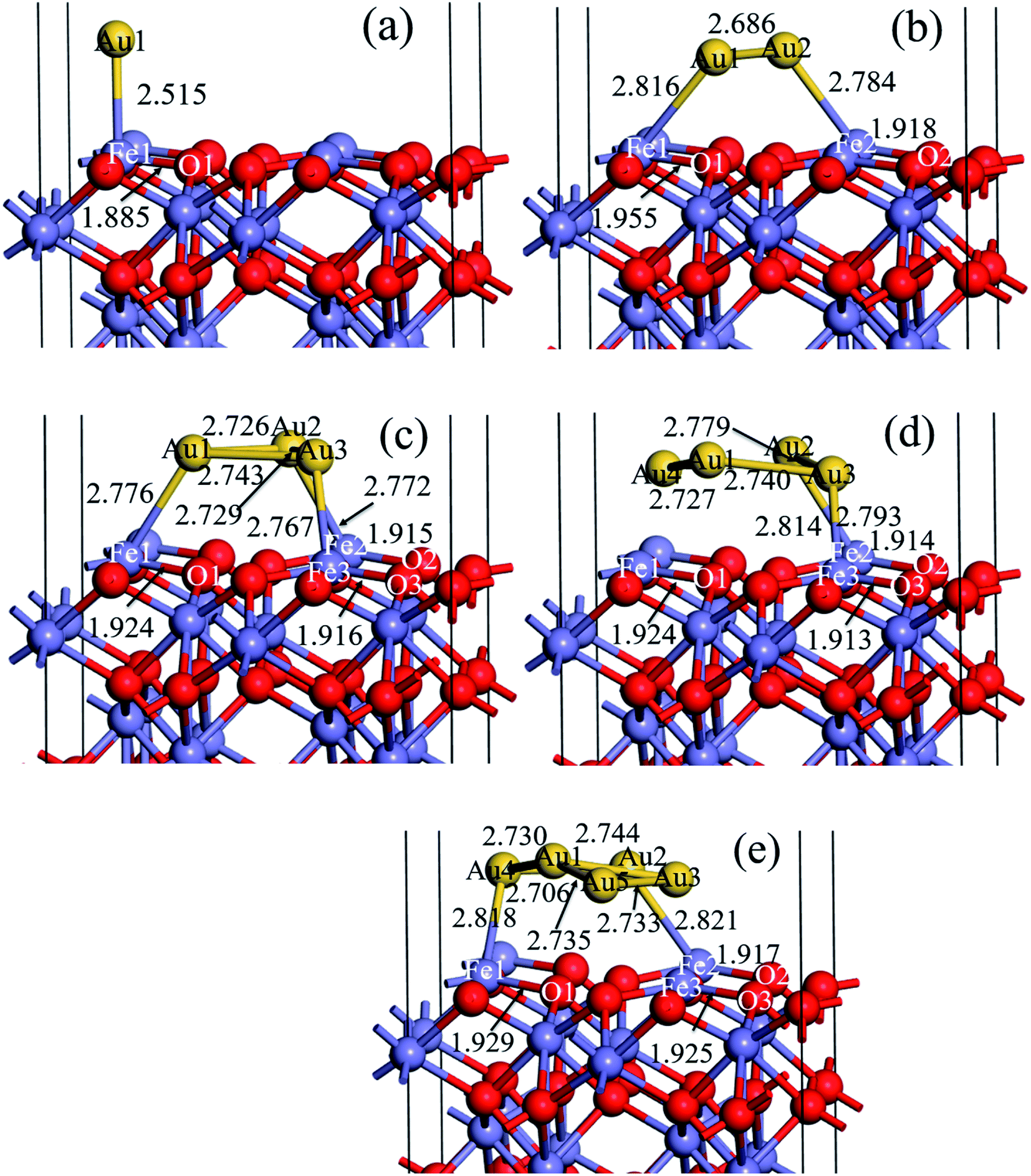 | ||
| Fig. 2 Equilibrium configuration of (a) 1Au/α-Fe2O3 (001) surface, (b) 2Au/α-Fe2O3 (001) surface (c) 3Au/α-Fe2O3 (001) surface surface, (d) 4Au/α-Fe2O3 (001) surface, (e) 5Au/α-Fe2O3 (001) surface. Numbers indicate the distance in Å. | ||
It is found from Fig. 2 that Au atoms all interact with iron atoms of α-Fe2O3 surface. Kiejna et al.43 investigated the structure and electronic properties of clean and Au or Pd covered α-Fe2O3 (0001) surface with spin-polarized density functional theory (DFT) and the DFT+U method. The results showed that both Au and Pd atoms bind strongly to hematite surfaces and induce large changes in their geometry. For the iron terminated surface, the binding energy per adatom increases with Au and Pd coverage, whereas the opposite trend is observed for the oxygen terminated one, which is consistent with our Au/α-Fe2O3 configurations. In addition, it is also observed that the bond lengths of Au–Fe for Fig. 2(a) and (c) are slightly smaller than that for Fig. 2(b), (d) and (e), indicating that the interactions between Au and Fe of α-Fe2O3 surface of Fig. 2(a) and (c) may be stronger than those of other models. However, the bonding conditions are different in Au/α-Fe2O3 systems with different number of gold atoms, resulting in their different stability. In addition to the interaction between gold and gold, there is an Au–Fe bond for 1Au/α-Fe2O3, two Au–Fe bonds for 2Au/α-Fe2O3, and three Au–Fe bonds for 3Au/α-Fe2O3, that is, the number of Au–Fe bonds increases with increasing the number of gold atoms. However, the number of Au–Fe bonds not only does not increase, but decreases when the number of gold atoms increases to four and five. There are two Au–Fe bonds for 4Au/α-Fe2O3 and 5Au/α-Fe2O3.
To assure the strength of interaction between Au and α-Fe2O3 surface in different systems, the binding energies of Au/α-Fe2O3 with different number of gold atoms are calculated (Table 1). The binding energies of Au particle deposited on α-Fe2O3 surfaces were calculated as:
| Ebin = EAu/surface − nEAu − Esurface | (1) |
The Mulliken bond population can reflect the strength of covalent bonds, the larger the bond population is, the stronger the covalent bond is. Tables 2–4 show the Mulliken bond populations of Fe–Au and Au–Au bonds for 3Au/α-Fe2O3, 4Au/α-Fe2O3 and 5Au/α-Fe2O3, respectively. It is observed that the populations of three Fe–Au bonds of 3Au/α-Fe2O3, 4Au/α-Fe2O3 and 5Au/α-Fe2O3 are similar, however, populations of three Fe–Au bonds (Fe1–Au1, Fe2–Au2) of 3Au/α-Fe2O3 are largest (0.14), and followed by two bonds (Fe1–Au4 and Fe2–Au2) of 5Au/α-Fe2O3 (0.13) and two bonds (Fe2–Au2 and Fe3–Au3) of 4Au/α-Fe2O3 (0.12) in order, showing the strength of Fe–Au bond for 3Au/α-Fe2O3 is slightly larger compared to those of 4Au/α-Fe2O3 and 5Au/α-Fe2O3. In addition, it is also seen that Mulliken bond populations of Au–Au in all models are much larger than those of Fe–Au, suggesting that the interaction between Au and Au is much stronger than that between Fe and Au. Therefore, the binding energy is in increasing order of 1Au/α-Fe2O3, 2Au/α-Fe2O3, 3Au/α-Fe2O3, 4Au/α-Fe2O3 and 5Au/α-Fe2O3 due to the increase of Au–Au bond (Table 1).
| Bond types | Population | Bond length (Å) |
|---|---|---|
| Fe1–Au1 | 0.14 | 2.776 |
| Fe2–Au2 | 0.14 | 2.772 |
| Fe3–Au3 | 0.14 | 2.767 |
| Au1–Au2 | 0.35 | 2.726 |
| Au1–Au3 | 0.34 | 2.743 |
| Au2–Au3 | 0.34 | 2.729 |
| Bond types | Population | Bond length (Å) |
|---|---|---|
| Fe2–Au2 | 0.12 | 2.814 |
| Fe3–Au3 | 0.12 | 2.793 |
| Au1–Au4 | 0.35 | 2.727 |
| Au1–Au3 | 0.28 | 2.740 |
| Au2–Au3 | 0.27 | 2.779 |
| Au1–Au2 | 0.26 | 2.804 |
| Au2–Au4 | 0.26 | 2.804 |
| Bond types | Population | Bond length (Å) |
|---|---|---|
| Fe1–Au4 | 0.13 | 2.818 |
| Fe2–Au2 | 0.13 | 2.821 |
| Au2–Au4 | 0.37 | 2.730 |
| Au1–Au4 | 0.34 | 2.706 |
| Au1–Au3 | 0.25 | 2.744 |
| Au1–Au5 | 0.30 | 2.735 |
| Au3–Au5 | 0.41 | 2.733 |
3.2 Electronic structures
The energy band and corresponding PDOS of up-spin and down-spin of pure α-Fe2O3 surface was studied previously.38 The band gap of pure α-Fe2O3 surface depends on the top valence band of the spin-up band and the bottom of the conduction band of the spin-down band, and the value is 2.0 eV. The spin-up band is divided into three groups. The group between −9 eV and −5 eV is mainly from Fe 3d and O 2p, only with few contributions from Fe 4s, Fe 4p and O 2s. The group between −5 eV and 0 eV is from O 2p, Fe 3d with a few contributions from Fe 4s and Fe 4p. The group in the conduction band is mainly from Fe 4p, Fe 4s, O 2p and O 2s. For the spin-down band, there is one group in the valence band, which is mainly from O 2p and Fe 3d with a few contributions from Fe 4p and Fe 4s. The band between 2 eV and 5 eV in the conduction band is mainly Fe 3d and O 2p with few contributions from Fe 4p, O 2s and Fe 4s, while that in the range of 5 eV to 8 eV is mainly from Fe 4p and Fe 4s with few contributions from O 2p and O 2s.Fig. 3 shows the energy band and PDOS of spin-up (a) and spin-down (b) for 1Au/α-Fe2O3, spin-up (c) and spin-down (d) for 2Au/α-Fe2O3 surface, spin-up (e) and spin-down (f) for 3Au/α-Fe2O3 surface, spin-up (g) and spin-down (h) for 4Au/α-Fe2O3 surface, spin-up (i) and spin-down (j) for 5Au/α-Fe2O3 surface. For up-spin band and DOS, it is observed that the energy structure and corresponding DOS of the valence band with small Au cluster (1Au/α-Fe2O3) are similar with that of pure α-Fe2O3.38 There are two groups for the valence band of 1Au/α-Fe2O3. The group between −6 and 0 eV is mainly composes of O 2p and Fe 3d with few contribution from Fe 4s and Fe 4p, while that between −9 and −5 eV is mainly from Fe 3d and O 2p. The two groups are connected to each other with increasing the size of Au cluster (from 2Au to 5Au), and the larger the Au cluster, the more the two groups overlap (Fig. 3(a), (c), (e), (g) and (i)). Compared to pure α-Fe2O3,38 Au/α-Fe2O3 systems present the different energy structure for the conduction band. A new energy level at 0.30 eV for 1Au/α-Fe2O3 appears near the Fermi level, which is mainly from O 2p, Fe 3d, Au 6s and Au 6p. The number of new energy levels increases with increasing the size of Au cluster. There are two new energy levels for 2Au/α-Fe2O3 and 3Au/α-Fe2O3, and they move away from the Fermi level relative to 1Au/α-Fe2O3 (Fig. 3(c) and (e)). The energy level at 1.72 eV is mainly from O 2p and Fe 3d, while that at 3.01 eV is mainly from O 2p and Fe 4p. For 4Au/α-Fe2O3, there are four new energy levels about 1.71, 1.90, 2.35 and 3.27 eV respectively. It is also found that the DOS peaks about 3.27 eV and 2.35 eV are weak, and the DOS curves corresponding to energy levels at about 1.71 eV and 1.90 eV are connected to each other to form a DOS peak, which is from O 2p, Fe 3d, Au 6s and Au 6p (Fig. 3(g)). As for 5Au/α-Fe2O3, there are also four new energy levels, however, these energy levels, including all energy levels of the conduction band move toward to Fermi level relative to those of 4Au/α-Fe2O3. The DOS curve corresponding to the energy level closest to the Fermi level overlaps with DOS curve of the valence band. The other three energy levels are located at 0.71, 1.63, 1.86 eV, respectively, and their corresponding DOSs are very weak. Changeable rule of DOS curve of Au cluster with different sizes is similar with that of α-Fe2O3 matrix. It is found that the spin-up DOS of Au cluster for the valence band is divided into two groups for 1Au/α-Fe2O3. The group between −1 and 0 eV is mainly from Au 5d with few contribution of Au 6s, and that between −8 and −1 eV is attributed to Au 6s. DOS curve of the conduction band between 3 to 6 eV for Au cluster is mainly from Au 6p with few contribution of Au 6s, and that with large localization near Fermi level is attributed to Au 6s, Au 6p and Au 5d (illustration in Fig. 3(a)). The two groups of the valence band are connected to each other with an increase of Au cluster size from 2Au to 5Au. There are two DOS peaks of Au cluster with large localization for 2Au/α-Fe2O3 and 3Au/α-Fe2O3 (illustration in Fig. 3(c) and (e)), three DOS peaks for 4Au/α-Fe2O3 and 5Au/α-Fe2O3 (illustration in Fig. 3(g) and (i)), which are from Au 6s, Au 6p and Au 5d.
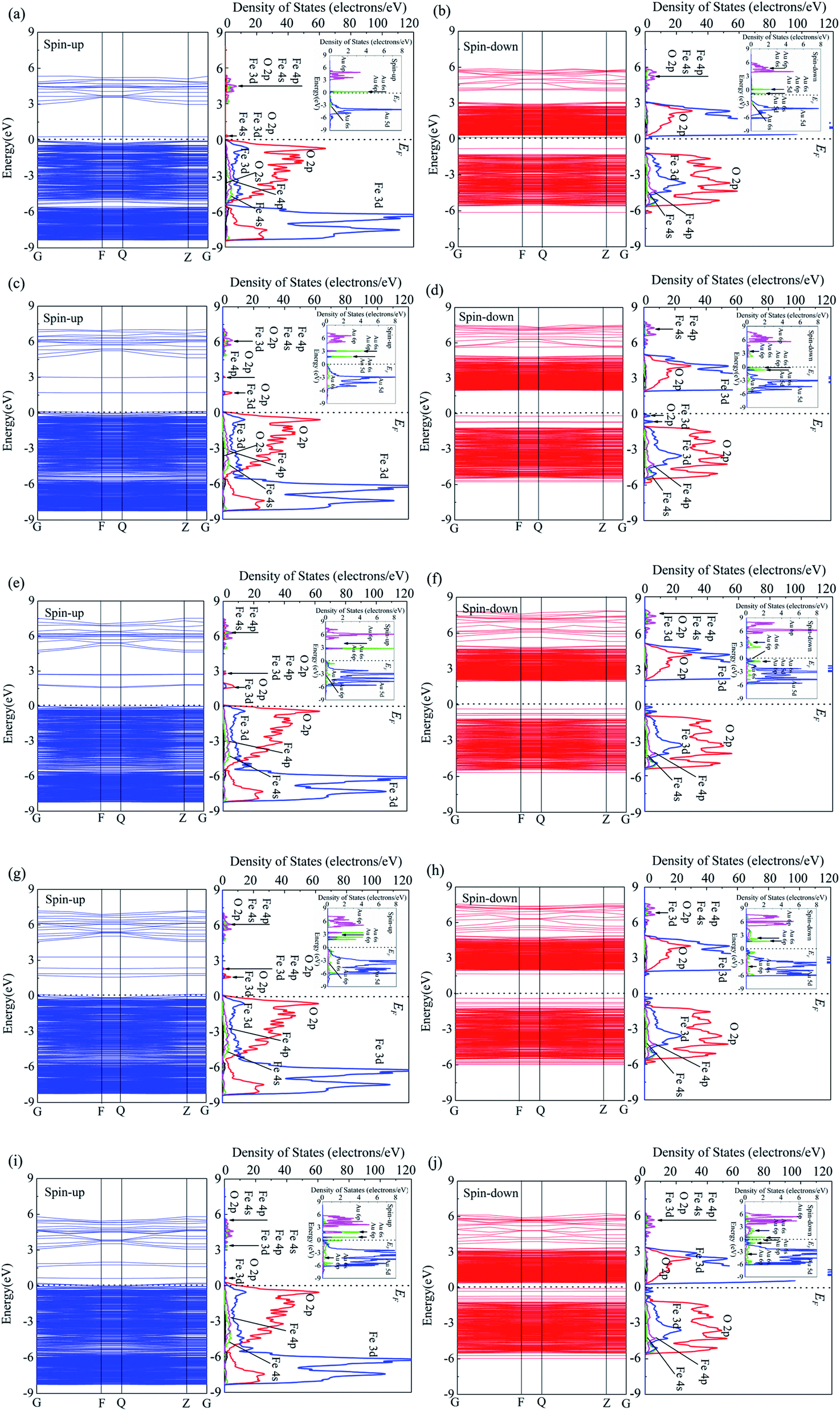 | ||
| Fig. 3 Energy band and PDOS of spin-up (a) and spin-down (b) for 1Au/α-Fe2O3 (001) surface, spin-up (c) and spin-down (d) for 2Au/α-Fe2O3 (001) surface, spin-up (e) and spin-down (f) for 3Au/α-Fe2O3 (001) surface, spin-up (g) and spin-down (h) for 4Au/α-Fe2O3 (001) surface and spin-up (i) and spin-down (j) for 5Au/α-Fe2O3 (001) surface. | ||
For down-spin band, it is seen that two groups of the conduction band for 1Au/α-Fe2O3 move toward Fermi level relative to that of pure α-Fe2O3.38 However, they move away from the Fermi level with increasing the size of Au cluster from 2Au to 4Au, and the interval between two groups decreases. When 5Au are deposited in α-Fe2O3 surface, the energy levels of the conduction band are very close to Fermi level. It is also observed that DOS curves change little with increasing the size of Au cluster from 1Au to 5Au, although there is a large shift for the energy levels of the conduction band. Unlike up-spin energy structure, new energy levels appears at the top of the valence band of down-spin band for Au/α-Fe2O3 systems. There is a new energy level about −0.82 eV for 1Au/α-Fe2O3, which is from Fe 3d and Fe 4p. The number of new energy level increases with increasing the size of Au cluster from 2Au to 5Au. There are two new energy levels for 2Au/α-Fe2O3 at the top of the valence band, three for 3Au/α-Fe2O3, four for 4Au/α-Fe2O3 and five for 5Au/α-Fe2O3. Two new energy level for 2Au/α-Fe2O3 corresponds to two DOS peaks, which are from Fe 3d, O 2p, Fe 4s and Fe 4p. Three new energy levels for 3Au/α-Fe2O3 only exhibit two DOS peaks because the two energy levels far from the Fermi level are very close, so they overlap each other to form a peak corresponding to Fe 3d and O 2p. The peak closest to Fermi level also is from Fe 3d and O 2p. Similarly, due to the overlap of DOS curves, four energy levels for 4Au/α-Fe2O3 and five energy levels for 5Au/α-Fe2O3 exhibit two DOS peaks and three DOS peaks, respectively. Two DOS peaks for 4Au/α-Fe2O3 are from Fe 3d and O 2p, and three DOS peaks for 5Au/α-Fe2O3 are from Fe 3d and Fe 4s.
The down-spin DOS of Au cluster for the conduction band is divided into two groups for 1Au/α-Fe2O3. The group between 0 and 3 eV is very weak, and the group between 3 and 6 eV is mainly from Au 6p with few contribution of Au 6s. DOS curve between 3 and 6 eV for 1Au/α-Fe2O3 shifts to high energy with increasing Au cluster size from 2Au to 3Au, however, it shifts to low energy when the Au cluster size continues to increase from 4Au to 5Au. The DOS curve between 0 and 3 eV corresponds to Au 6s, Au 6p and Au 5d, which becomes stronger with an increase of Au cluster size from 2Au to 5Au. The down-spin DOS curve of Au cluster for the valence band can also be divided into two parts for 1Au/α-Fe2O3. The part between −7 and −2 eV is mainly from Au 5d with few contribution of Au 6s. There are two sharp peaks in the range of −2 to 0 eV. One near Fermi level corresponds Au 6s and Au 6p, and the other is from Au 6s and Au 5d. The two peaks are close to each other with an increase of Au cluster size from 2Au to 4Au, and become weak. However, the peaks become stronger when five Au atoms are deposited on α-Fe2O3 surface.
Fig. 4 shows the atomic Mulliken charge population of Au/α-Fe2O3 systems. It is seen that from Fig. 4(a) the charge of Au is 0.18e when 1Au is deposited on α-Fe2O3 surface, suggesting that the electrons transfer from Au to α-Fe2O3 surface. After 2Au depositing, the charges of Au1 and Au2 are 0.05e and 0.09e, respectively, and the total charge is 0.14e (Fig. 4(b)). After 3Au depositing, the charges of Au1, Au2 and Au3 are 0.04e, 0.04e and 0.05e, respectively, and the total charge is 0.13e (Fig. 4(c)). As shown in Fig. 4(d), the charges of 4Au are 0.06e, 0.03e, 0.09e, and 0.13e, respectively, and the total charge is 0.31e. As for 5Au/α-Fe2O3, it is observed that the charges of 5Au are 0.05e, 0.03e, 0.1e, −0.01e, and 0.01e respectively with the total charge of 0.18e (Fig. 4(e)). According to above analysis, Au atoms lose electrons after Au clusters depositing on α-Fe2O3 surface, suggesting the electrons transfer from Au clusters to the α-Fe2O3 surface for Au/α-Fe2O3 system. And the total number of the charges for Au cluster decreases with increasing the Au cluster size from 1Au to 3Au (from 0.18e to 0.13e), and increases again when the Au cluster increases to 4Au (0.31e). The total charges of Au cluster with 5Au is 0.18e, which is the same with that of 1Au. Therefore, for Au/α-Fe2O3 systems, the number of charge transfer between Au cluster and α-Fe2O3 surface is the largest for 4Au/α-Fe2O3 (0.31e), which suggests the interaction between Au clusters and Fe2O3 surface for 4Au/α-Fe2O3 is the strongest.
 | ||
| Fig. 4 Mulliken atomic charge population of (a) 1Au/α-Fe2O3 (001) surface, (b) 2Au/α-Fe2O3 (001) surface (c) 3Au/α-Fe2O3 (001) surface surface, (d) 4Au/α-Fe2O3 (001) surface, (e) 5Au/α-Fe2O3 (001) surface. | ||
3.3 Optical properties
Fig. 5 shows real parts (a) and imaginary parts (b) of complex refractive index for pure and Au-deposited α-Fe2O3 (001) surface. The complex refractive index can be expressed as follows:| N = n + ik | (2) |
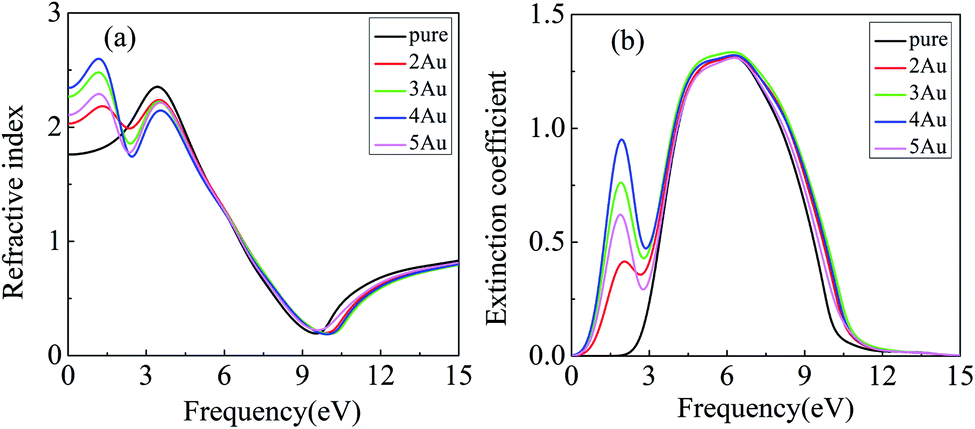 | ||
| Fig. 5 Real parts (a) and imaginary parts (b) of complex refractive index for pure and Au-deposited α-Fe2O3 (001) surface. | ||
The real (n) and imaginary (k) parts of complex refractive index are the refractive index and extinction coefficient, respectively. It is seen from Fig. 5 that there is a sharp peak of the refractive index with a value of 2.35 in the 3.37 eV and a gentle peak of extinction coefficient with a value of 1.31 in the 6.13 eV for pure α-Fe2O3 surface. When Au clusters are deposited on the α-Fe2O3 surface, the curves of refractive index and extinction coefficient change a lot. Notably, there are two peaks for the refractive index and extinction coefficient in the range of 0 to 10 eV. It is obvious that the first peaks for the refractive index at about 1.15 eV and extinction coefficient at about 6.08 eV are induced by Au cluster. The peak value of the refractive index for 2Au/α-Fe2O3, 3Au/α-Fe2O3, 4Au/α-Fe2O3 and 5Au/α-Fe2O3 are 2.18, 2.47, 2.60 and 2.29, respectively. The result shows that the intensity of the first peak increases with increasing Au cluster size from 2Au to 4Au, and decreases with further increasing Au cluster size above 4Au, exhibiting a maximum value with 4Au. The changeable rule of the extinction coefficient with the increase of Au cluster size is similar with that of the refractive index. The first peak of the extinction coefficient intensifies with an increase of Au cluster size up to 4Au, but weakens with increasing further Au cluster above 4Au.
Fig. 6 shows real parts (a) and imaginary parts (b) of complex dielectric function for pure and Au-deposited α-Fe2O3 surface. The complex dielectric function was using the following equation:
| ε = ε1 + iε2 | (3) |
| ε = N2 | (4) |
 | ||
| Fig. 6 Real parts (a) and imaginary parts (b) of complex dielectric function for pure and Au-deposited α-Fe2O3 (001) surface. | ||
Therefore, the relation between the real part (ε1) and imaginary part(ε2) of complex dielectric function (ε) and refractive index (n) and extinction coefficient (k) is:
| ε1 = n2 − k2 | (5) |
| ε2 = 2nk | (6) |
We can infer from formulas (4) and (5) that the real part of dielectric function increases with an increase of refractive index and a decrease of extinction coefficient, and the imaginary part increases with increasing the refractive index and extinction coefficient. It is observed from Fig. 6 that the real part and imaginary part of the complex dielectric function are similar to the change of refractive index and extinction coefficient for pure and Au-deposited α-Fe2O3, respectively. The real part of the dielectric function at the frequency of 0 eV corresponds to the static dielectric constant. It is found that the static dielectric constants of 2Au-deposited, 3Au-deposited, 4Au-deposited and 5Au-deposited α-Fe2O3 surface are 4.14, 5.14, 5.49 and 4.43, respectively, which is far larger than that of pure α-Fe2O3 (3.11), suggesting that the deposition of Au clusters lead to an increase of static dielectric constant of the α-Fe2O3 surface. Furthermore, it is also observed that the Au cluster size has a great influence on the static dielectric constant of α-Fe2O3. The static dielectric constant increases with increasing Au cluster size up to 4Au, but decreases with increasing further the Au cluster size above 4Au. There is a shark peak of the real part at about 3.26 eV and a sharp peak of the imaginary part at about 5.2 eV for pure α-Fe2O3. After the deposition of Au clusters, there are two peaks for the real part and imaginary part of the complex dielectric function. Obviously, the peak at about 3.26 eV for the real part corresponds to the peak of pure α-Fe2O3, while the peak at 1.12 eV is induced by Au clusters. It is also found from Fig. 6(a) that the peak from α-Fe2O3 matrix is weakened due to the influence of Au clusters, and that of 4Au/α-Fe2O3 weakens the most, while the peak induced by Au clusters intensities with increasing Au cluster size up to 4Au, but weaken with increasing further Au cluster size beyond 4Au, showing a maximum value for 4Au (peak value: 2Au–Fe2O3, 4.72; 3Au–Fe2O3, 6.04; 4Au–Fe2O3, 6.62; 5Au–Fe2O3, 5.21). The peak at about 4.58 eV for the imaginary part corresponds to that of pure α-Fe2O3, while the peak at 1.85 eV is induced by Au clusters (Fig. 6(b)). It is found that the peak from α-Fe2O3 matrix has not much change with different Au cluster size, while the peak induced by Au cluster intensities first, and then weakens with an increase of Au cluster size, showing a maximum for 4Au (peak value: 2Au–Fe2O3, 1.69; 3Au–Fe2O3, 3.29; 4Au–Fe2O3, 4.16; 5Au–Fe2O3, 2.53), which is consistent with those of the real part and imaginary part of complex refractive index.
The optical absorption of pure and Au-deposited α-Fe2O3 surface are shown in Fig. 7. It is found that the pure α-Fe2O3 surface shows a weak absorption in the range of 380 to 800 nm, however, the optical absorption is obviously enhanced after Au clusters deposition. The intensity of optical absorption increases with increasing the Au cluster size up to 4Au, but weakens with increasing further Au cluster size above 4Au, showing a maximum value for 4Au/α-Fe2O3. The optical absorption coefficient (η) is related to the extinction coefficient (k) by:
| η = 2kω/c | (7) |
 | ||
| Fig. 7 Optical absorption of pure and Au-deposited α-Fe2O3 (001) surface. | ||
4 Conclusion
Based on the density functional theory (DFT), we studied the electronic structure and optical property of pure and Au-deposited α-Fe2O3 surface, and focused on the influence of Au cluster size. Au atoms are prone to interact with iron atoms of α-Fe2O3 surface, which results in a great change of the structure of α-Fe2O3 surface. The length of Fe–O bond of Au/α-Fe2O3 surface is obviously larger compared with those of pure α-Fe2O3 surface. The bandgap and semiconducting properties change after Au cluster depositing. The energy level in the conduction band with the corresponding PDOS curve shifts to Fermi level, and the part near to Fermi level is split, and. The series of new energy level for Au/α-Fe2O3 appears near the Fermi level, which is mainly from O 2p, Fe 3d, Au 6s and Au 6p. In addition, Au atoms lose electrons after Au clusters are deposited on α-Fe2O3 surface, suggesting that the electrons transfer from Au clusters to the α-Fe2O3 surface for Au/α-Fe2O3 system, and the number of charge transfer between Au clusters and α-Fe2O3 for 4Au/α-Fe2O3 is the largest (0.31e). The new peaks of complex refractive index, complex dielectric function and optical absorption due to Au clusters are observed in the visible range for Au-deposited α-Fe2O3 system, and the changing trend of their curves is similar. In addition, the intensities of complex refractive index, complex dielectric function and optical absorption are enhanced with increasing Au cluster size, showing a maximum value with 4Au. Increasing Au cluster size above 4Au results in a decrease in their intensities.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 51964004) and the high-performance computing platform of Guangxi University.References
- J. C. Little, A. T. Hodgson and A. J. Gadgil, Atmos. Environ., 1993, 28, 227–234 CrossRef.
- W. Lindinger, A. Hansel and A. Jordan, Int. J. Mass Spectrom. Ion Processes, 1998, 173, 191–241 CrossRef CAS.
- Y. Song, M. Shao, Y. Liu, S. Lu, W. Kuster, P. Goldan and S. Xie, Environ. Sci. Technol., 2007, 41, 4348–4353 CrossRef CAS PubMed.
- R. Atkinson and J. Arey, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS PubMed.
- T. Singh, N. Srivastava, P. K. Mishra, A. K. Bhatiya and N. L. Singh, Mater. Sci. Forum, 2016, 855, 20–32 Search PubMed.
- K. Kante, M. Florent, A. Temirgaliyeva, B. Lesbayev and T. J. Bandosz, Carbon, 2019, 146, 568–571 CrossRef CAS.
- D. P. Das and K. M. Parida, J. Mol. Catal. A: Chem., 2007, 276, 17–23 CrossRef CAS.
- F. G. Edwards and N. Nirmalakhandan, Water Sci. Technol., 1996, 34, 565–571 CrossRef CAS.
- J. J. Pei and J. J. Zhang, HVACR Res., 2011, 17, 476–503 CAS.
- M. Mishra, H. Park and D. M. Chun, Adv. Powder Technol., 2016, 27, 130–138 CrossRef CAS.
- G. Y. Zhang, Y. Feng, Y. Y. Xu, D. Z. Gao and Y. Q. Sun, Mater. Res. Bull., 2012, 47, 625–630 CrossRef CAS.
- M. Mishra and D. M. Chun, Appl. Catal., A, 2015, 498, 126–141 CrossRef CAS.
- Y. Dong, L. Xing, F. Hu, A. Umar and X. Wu, Vacuum, 2018, 150, 35–40 CrossRef CAS.
- F. J. Morin, Phys. Rev., 1951, 83, 1005–1010 CrossRef CAS.
- J. Kennedy and K. Frese, J. Electrochem. Soc., 1978, 125, 709–714 CrossRef CAS.
- H. B. Qi, D. W. Liu and H. Li, Mater. Sci. Forum, 2016, 852, 14–21 Search PubMed.
- A. Quintanilla, N. Menéndez, J. Tornero, J. Casas and J. J. Rodríguez, Appl. Catal., B, 2008, 81, 105–114 CrossRef CAS.
- H. Shan, C. Liu, L. Li, J. B. Zhang, H. Y. Li, Z. Liu, X. B. Zhang, X. Q. Bo and X. Chi, ACS Appl. Mater. Interfaces, 2013, 5, 6376–6380 CrossRef CAS PubMed.
- B. Palanisamy, C. M. Babu, B. Sundaravel, S. Anandan and V. Murugesan, J. Hazard. Mater., 2013, 252–253, 233–242 CrossRef CAS PubMed.
- F. B. Li, X. Z. Li, C. S. Liu and T. X. Liu, J. Hazard. Mater., 2007, 149, 199–207 CrossRef CAS PubMed.
- G. K. Pradhan, S. Martha and K. M. Parida, ACS Appl. Mater. Interfaces, 2012, 4, 707–713 CrossRef CAS PubMed.
- D. L. Allara, A. F. Hebard, F. J. Padden, R. G. Nuzzo and D. R. Falcone, J. Vac. Sci. Technol., A, 1983, 1, 376–382 CrossRef CAS.
- J. Song, R. F. Guan, X. R. Liu, C. F. Jiang and G. H. Hou, J. Phys. Chem. Solids, 2018, 3697, 31619–31620 Search PubMed.
- X. H. Liu, J. Zhang, X. Z. Guo, S. H. Wu and S. R. Wang, Nanotechnology, 2010, 21, 095501 CrossRef PubMed.
- D. D. Li, X. L. Yan, M. M. Yang, C. X. Luo, P. W. Li, J. Hu, G. Li, H. B. Jiang and W. D. Zhang, J. Alloys Compd., 2018, 775, 150–157 CrossRef.
- H. X. Lin, Y. X. Liu, J. G. Deng, K. F. Zhang, X. Zhang, S. H. Xie, X. T. Zhao, J. Yang, Z. Han and H. X. Dai, J. Environ. Sci., 2018, 70, 74–86 CrossRef CAS PubMed.
- S. W. Cao, J. Fang, M. M. Shahjamali, Z. Wang and C. Xue, CrystEngComm, 2012, 14, 7229–7235 RSC.
- W. T. Chen, Y. J. Hsu and P. V. Kamat, J. Phys. Chem. Lett., 2012, 3, 2493–2499 CrossRef CAS PubMed.
- K. Jug, B. Zimmermann, P. Calaminici and A. M. Köster, J. Chem. Phys., 2002, 116, 4479 CrossRef.
- M. Fang, X. L. Tan, Z. X. Liu, B. W. Hu and X. K. Wang, Research, 2021, 10, 34133 Search PubMed.
- C. M. Aikens, S. Z. Li and G. C. Schatz, J. Phys. Chem. C, 2008, 112, 11272–11279 CrossRef CAS.
- C. E. Zubieta, L. G. Aquino-Linarez, S. A. Fuente, P. G. Belelli and R. M. Ferullo, Comput. Mater. Sci., 2020, 173, 109392 CrossRef CAS.
- S. W. Hon, L. Thomas, G. Jones and D. J. Willock, Res. Chem. Intermed., 2015, 41, 9587–9601 CrossRef.
- M. T. Nguyen, M. F. Camellone and R. Gebauer, J. Chem. Phys., 2015, 143, 034704 CrossRef PubMed.
- T. Pabisiak, M. J. Winiarski and A. Kiejna, J. Chem. Phys., 2016, 144, 044704 CrossRef PubMed.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045–1097 CrossRef CAS.
- J. P. Perdew and W. Yue, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- C. T. Chen, C. H. Zhao, X. Zhou, J. H. Chen, L. G. Chen and F. T. C. Li, Vacuum, 2021, 188, 110164 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed.
- M. C. Toroker, D. K. Kanan, N. Alidoust, L. Y. Isseroff, P. Liao and E. A. Carter, Phys. Chem., 2011, 13, 16644–16654 CAS.
- M. Cao, T. Liu, S. Gao, G. Sun, X. Wu, C. Hu and Z. L. Wang, Angew. Chem., Int. Ed., 2010, 36, 4197–4201 Search PubMed.
- C. H. Zhao, B. S. Li, X. Zhou, J. H. Chen and H. Q. Tang, Metals, 2021, 11, 424 CrossRef CAS.
- A. Kiejna and T. Pabisiak, J. Phys.: Condens. Matter, 2012, 24, 095003 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |