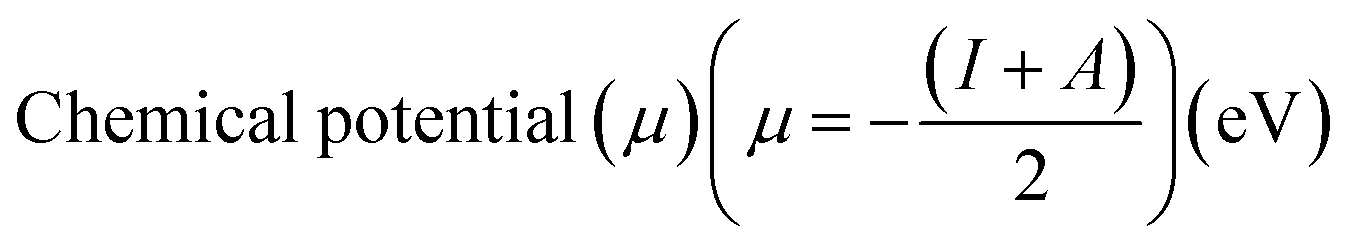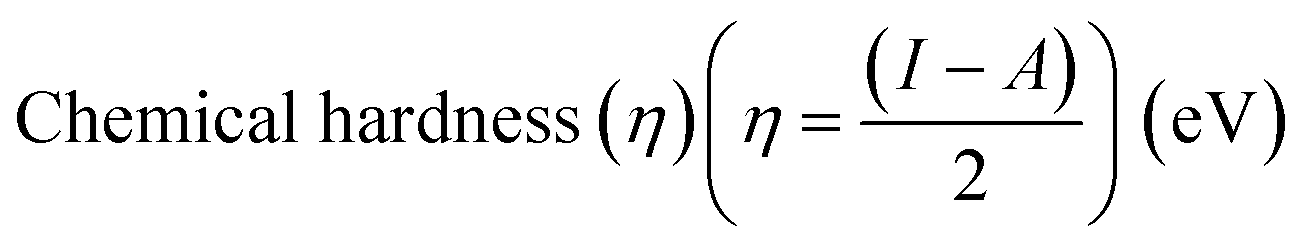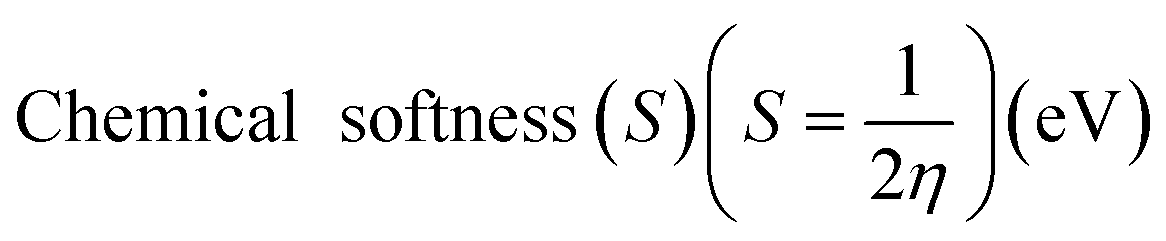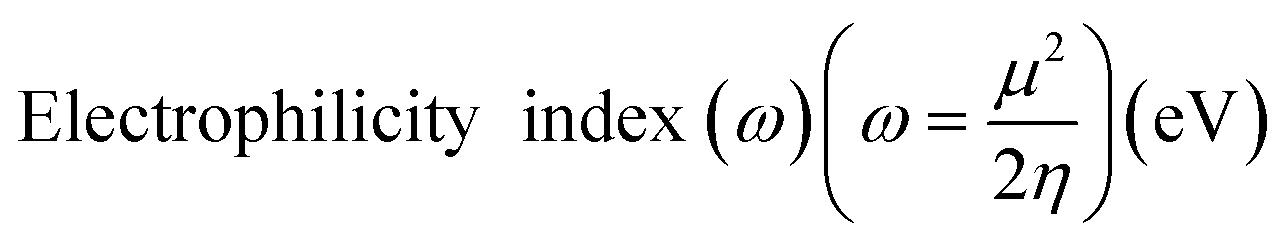DOI:
10.1039/D1RA07364H
(Paper)
RSC Adv., 2022,
12, 5357-5368
Exploration of plant-derived natural polyphenols toward COVID-19 main protease inhibitors: DFT, molecular docking approach, and molecular dynamics simulations†
Received
3rd October 2021
, Accepted 19th January 2022
First published on 14th February 2022
Abstract
Recent outbreaks of coronavirus have brought serious challenges to public health around the world, and it is essential to find effective treatments. In this study, the 3C-like proteinase (3CLpro) of SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) has been considered as an important drug target because of its role in viral replication. We initially optimized 251 compounds at the PM7 level of theory for docking with 3CLpro, and then we selected the top 12 compounds for further optimization with the B3LYP-D3/6-311G** method and obtained the top four compounds by further molecular docking. Quantum chemistry calculations were performed to predict molecular properties, such as the electrostatic potential and some CDFT descriptors. We also performed molecular dynamics simulations and free energy calculations to determine the relative stability of the selected four potential compounds. We have identified key residues controlling the 3CLpro/ligand binding from per-residue based decomposition of the binding free energy. Convincingly, the comprehensive results support the conclusion that the compounds have the potential to become a candidate for anti-coronavirus treatment.
1 Introduction
The COVID-19 acute respiratory tract disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported in December 2019 in Wuhan, China.1–3 The disease was declared a pandemic by the World Health Organization (WHO) on March 11, 2020.4 In the absence of potential and specific therapeutics, COVID-19 cases were rapidly increasing, and over 232 million cases with more than 4.75 million deaths were reported up to September 2021.5 To date, though the development of such drugs is underway,6,7 there are still no commonly used effective drugs for treating such CoVs.
SARS-CoV-2 is a member of the Coronaviridae family, the same family for other beta coronaviruses, such as Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) and Middle East Respiratory Syndrome Coronavirus (MERS-CoV).8–10 Recent studies have shown that SARS-CoV-2 shares ∼89% sequence similarity with other SARS-CoVs.8 SARS-CoV-2 contains two kinds of proteins, i.e., 4 structural proteins (spike, envelope, membrane, and nucleocapsid) and 16 nonstructural proteins. In the autoproteolytic cleavage catalyzed by PLpro enzymes (papain-like cysteine protease) and 3CLpro enzymes (3C-like proteinase), SARS-CoV-2 genetic material enters the host cell and borrows ribosomes to translate into 16 nonstructural proteins. Because of its mechanistic significance, 3CLpro, also known as Mpro, is a central target for the effective inhibitors (antiviral drugs) against SARS-CoV-2 and other known CoVs.11 The crystal structure of main protease 3CLpro of SARS-CoV-2 in the complex with an inhibitor N3 (PDB code 7BQY, shown in Fig. 1 with PyMOL12) has been revealed, with the resolution of 1.7 Å.13 This protease has been considered to be a promising therapeutic target for COVID-19 since 3CLpro plays an important role in the viral replication process while no close homolog exists in humans.10,14
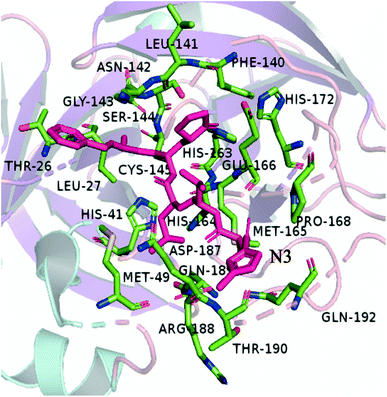 |
| | Fig. 1 The 3D model shows the main residues that are part of the active site of the Mpro. | |
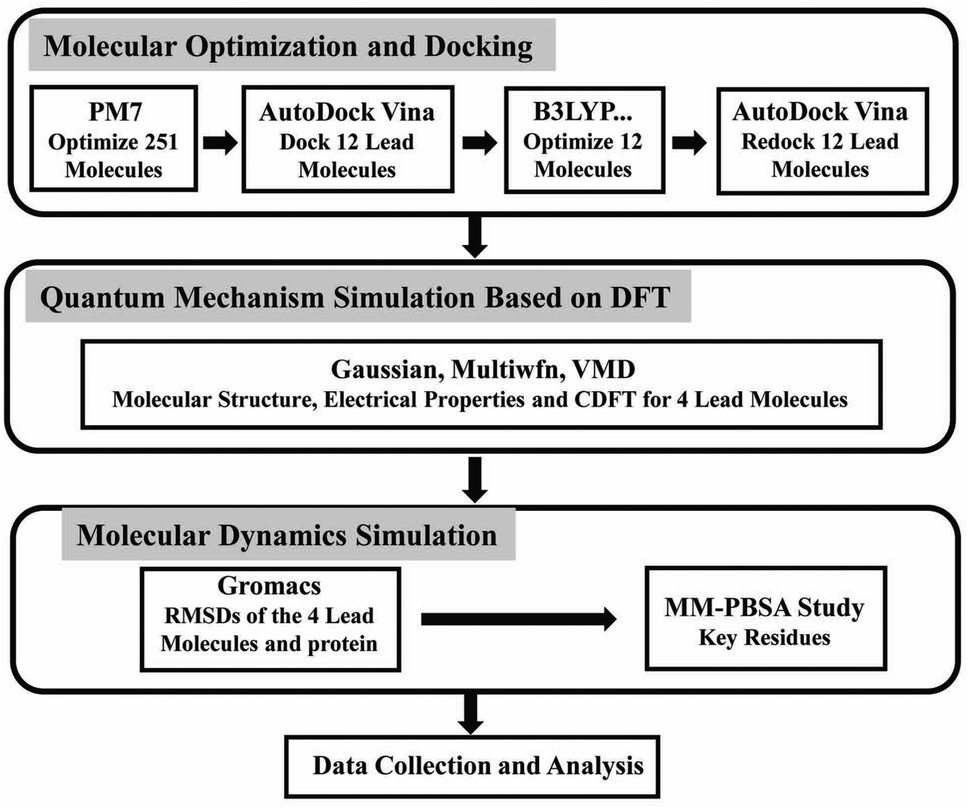
Natural compounds have been used for medical treatments since ancient times because of their low cost, minimum side-effects, and abundant therapeutic resources. In some previous studies, the compounds based on natural products were discovered as inhibitors against viruses, such as polio-virus type 1, the RNA-dependent RNA polymerase (RdRp), respiratory synchronization (sin-SISH-Uhl), and most notably as purine-based inhibitors of viral replication complex component.15,16 The flavonoids are phenolic compounds that are among the most plentiful and prevalent natural compounds. There are ten major sub-groups of flavonoids, i.e., aurones, biflavonoids, catechins, flavanones, flavanols, chalcones, flavanonols, flavans, flavones and isoflavones.17 Baicalin (a known flavonoid) has an EC50 value of 12.5–25 μg ml−1 at 48 h without causing significant cytotoxicity.18 Flavonoids luteolin and quercetin have the ability to prevent SARS-CoV from infecting host cells.19 Gallocatechin gallate (GCG) display good inhibition toward 3CLpro with IC50 values of 47 μM.20 Because of these properties, flavonoids could be potential candidates to interfere with the life cycle of coronaviruses.21,22 According to in vtiro studies, epigallocatechin gallate (EGCG) inhibited 3CLpro by 85% at a concentration of 200 μM, and had an IC50 measure of 73 ± 2 μM.20 The anthocyanin of red-fleshed potato inactivated both influenza viruses A (IVA) and B (IVB).23 Hence, flavonoids, as other natural products, continue to be a promising source of anti-coronavirus therapeutics. Traditional Chinese medicine has a long history and has accumulated rich experience in the use of natural medicines.24 After the careful literature search on the topics related to flavonoids and viral diseases (specifically the coronavirus diseases), we established our screening library with a total of 251 kinds of polyphenols included. In light of the long and expensive process of developing compounds into approved drugs and the rapidly evolving nature of the epidemic, a combination of computational methodologies is a promising approach for identifying potential drug candidates from compound libraries.25,26 In the present paper (as shown in Fig. 2), firstly, we optimized the molecules in our screening library. In order to further study the composition characteristics of the molecules, the compounds based on docking scores were studied by the quantum mechanics method. We also used molecular dynamics simulation to identify the stability of promising compounds with Mpro. Then, we performed binding energy calculations using the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method to evaluate the binding affinity of the compounds and key residues. In silico studies were performed for identifying bioactive compounds that can inhibit COVID-19 Mpro effectively, as well as providing useful information for future studies.
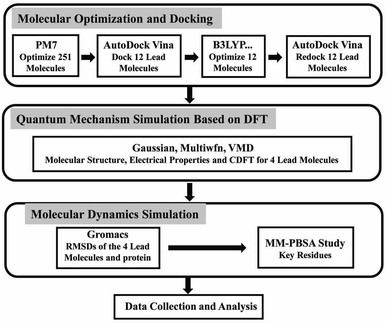 |
| | Fig. 2 The flow chart of screening anti-coronavirus compounds from the small molecule library and the methods of studying the properties of the screened molecules. | |
2 Methods
2.1 Molecular docking studies
The X-ray crystal structure of 3CLpro in complex with the inhibitor N3 (PDB ID 7BQY) with the resolution of 1.7 Å was used for our study, and the crystal structure was prepared by removing the N3 ligand and adding hydrogen atoms, as well as computing the Gasteiger charge using the AutoDock v4.2 program.27
Our research focuses on flavonoids and viral diseases (specifically Coronavirus diseases), and in order to broaden the scope of our search we collected compounds related to flavonoids from Chinese herbs. The SDF structures of the selected 251 ligands (shown in Table S1†) were retrieved from the PubChem database (https://pub-chem.ncbi.nlm.nih.gov/). We adopted the PM7 method implemented in Gaussian 16 (ref. 28) to optimize these 251 compounds, and the optimized geometries were used to do docking study with the Autodock Vina program.29 Based on the scores of docking, we screened 12 compounds out of the 251 candidates for the further geometry optimization with the B3LYP-D3/6-311G** method, which is more reliable than PM7. Finally, a second docking for the 12 compounds was performed with Autodock Vina, and 4 top-score compounds were selected. The ligands were saved and converted to PDBQT format using Raccoon.30 The top 4 of 12 highest binding energy and best-docked conformation were considered for the further quantum chemistry calculations. A grid box with dimensions 60 × 60 × 60 centered at N3 in protein (X = 9.269, Y = −1.127, and Z = 22.118) was used as the search area.
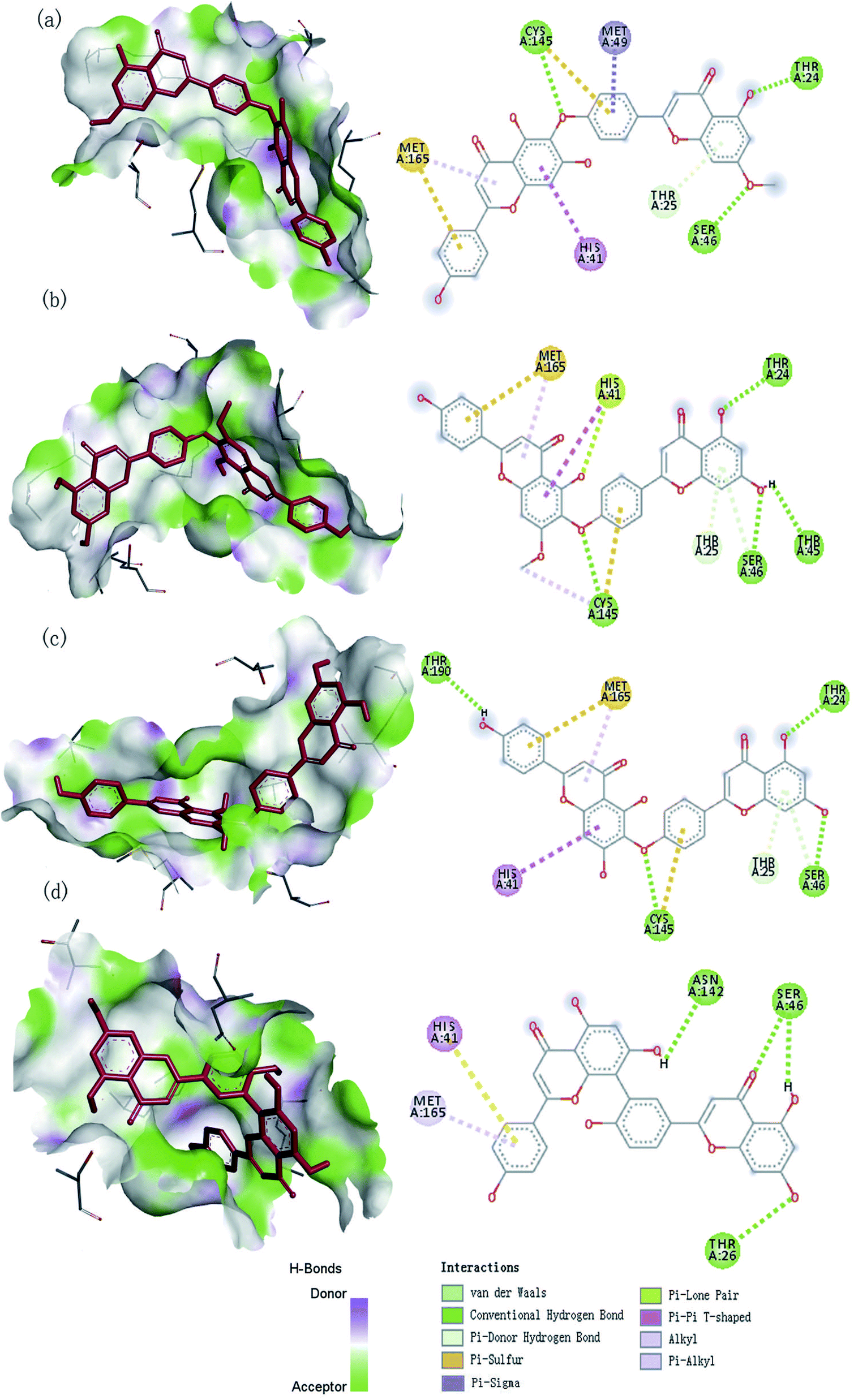
We used the software Discovery Studio Visualizer31 to illustrate the interactions between the best four ligands and proteins, including hydrogen bonds, atom accessibility, and hydrophobic interactions (Fig. 3).
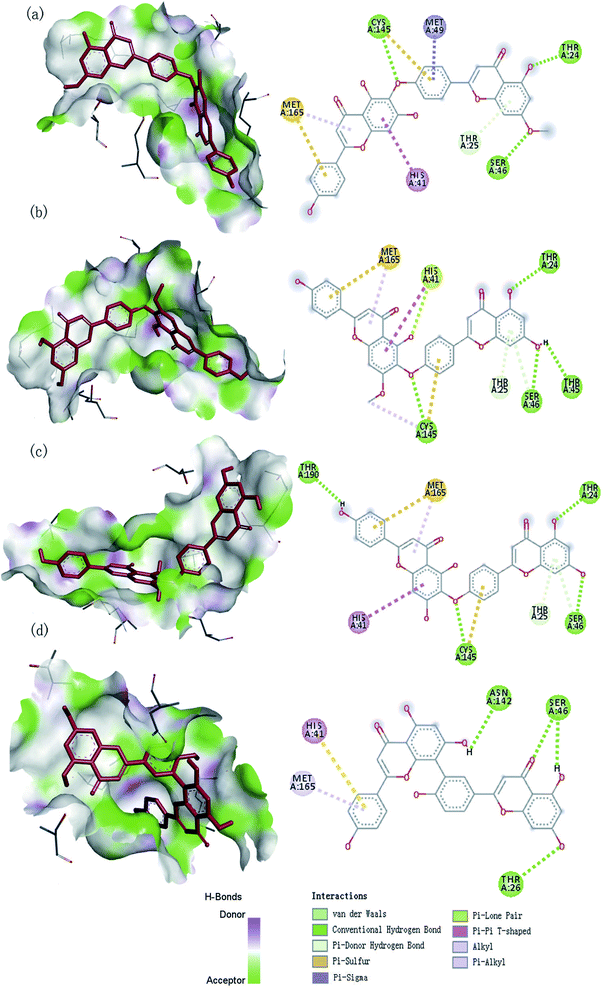 |
| | Fig. 3 The 3D and 2D interaction diagram of SARS-CoV-2 Mpro with four compounds: (a) neocryptomerin, (b) isocryptomerin, (c) hinokiflavone, (d) amentoflavone. | |
2.2 Quantum chemical calculations
In order to predict molecular properties, we have carried out the related quantitative calculation.32 Among various computational methodologies, DFT has become the preferred method for investigating molecular properties, which has been extensively used to compute the electronic structure properties. Because of comparatively better results and less computational cost,33 we choose the DFT/B3LYP methods34 for the optimization of the screened compounds against SARS-CoV-2 Mpro using Gaussian 16 software.
Electrostatic Potential (ESP)35 were generated for the selected molecules with Multiwfn,36 and the ESP surfaces are shown by VMD37 to analyze the electrostatic induced weak interaction. According to the theory of the Conceptual Density Functional Theory (CDFT), we calculated the chemical parameters to show more molecular properties within the framework of the HSAB theory, including EHOMO, ELUMO, ionization potential (IP), electron affinity (EA), energy gap (Eg), electronegativity (χ), chemical potential (μ), chemical hardness (η), chemical softness (S), electrophilicity (ω) and nucleophilicity (N). The ionization energy and electron affinity are related to the HOMO and LUMO energies, respectively, based on Koopmans' theorem.38 The related formulas are as follows:
| | |
Ionization potential (I) (I = −EHOMO) (eV)
| (1) |
| | |
Electron affinity (A) (A = −ELUMO) (eV)
| (2) |
| | |
Energy gap (ΔE) (ΔE = ELUMO − EHOMO) (eV)
| (3) |
| |
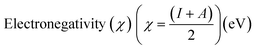 | (4) |
| |
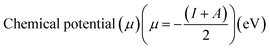 | (5) |
| |
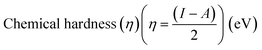 | (6) |
| |
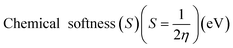 | (7) |
| |
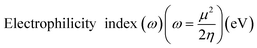 | (8) |
2.3 Molecular dynamics simulation
The four ligands with the best docking score were selected for the MD simulation using Gromacs39 to evaluate their intermolecular interactions with 3CLpro and the stability of the complexes. The ligands were parameterized with the general AMBER force field (GAFF)40 using the Acpype program.41 The restrained electrostatic potential (RESP) approach at the B3LYP/6-311G** level was applied to calculate the atomic partial charges of the analogs with the assistance of Gaussian16 software.42 Protein topology and coordinate files were generated using the Amber99SB force field43 provided in Gromacs.39 The protein–ligand complex was contained in a dodecahedron and solvated with an explicit TIP3P44 water model within a periodic boundary box of distance 1.0 nm fixed in between the protein and dodecahedron box, and the Na+ ions were added to neutralize the solvated system that followed by quick energy minimization with the steepest descent minimization algorithm. This was followed by a restrained constant number of particles, volume, and temperature (NVT) ensemble equilibration for 500 ps and then simulated for 1 ns in the NPT ensemble at 310 K (ref. 45) and 1.0 bar.46 The particle mesh Ewald method was used to calculate the long-range electrostatics.47 Modified Berendsen thermostat and Parrinello–Rahman barostat were used for temperature and pressure coupling, respectively. Finally, unrestrained 100 ns production simulations were carried out for the systems at 310 K and 1 bar atmospheric pressure. For MD simulation analysis, the results of RMSD and bonding interactions were performed using Gromacs tools.48
2.4 Binding free energies
Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) has been regarded as a competitive method in the field of binding free energy calculation,49 and is applied to estimate the relative binding free energies for the four final compounds and to evaluate their relative stability of the biomolecular structures against SARS-CoV-2 in our studying.50,51 We use g_mmpbsa that was developed to enable the use of the MM-PBSA method in conjunction with the Gromacs package to do the calculation.52 In general terms, the binding free energy of the protein with ligand in solvent can be expressed as:| | |
ΔGbinding = Gcomplex− (Gprotein + Gligand)
| (9) |
where, Gcomplex is the total free energy of the protein–ligand complex, and Gprotein and Gligand are total free energies of the isolated protein and ligand in solvent, respectively.
g_mmpbsa can also be used to estimate the energy contribution per residue to the binding energy. To decompose the binding energy, at first ΔEMM, ΔGpolar and ΔGnon-polar were separately calculated for each residue and were then summed up to obtain the contribution of each residue to the binding energy.
3 Results and discussion
3.1 Molecular docking analysis
Molecular docking is an effective tool in drug design,53 which speeds up this process. We use molecular docking technology to screen compounds based on their docking scores and identify the key interactions of ligands within the active site of a target protein.53 The docking scores of the first 12 compounds were shown in Table 1. Based on docking energy values of −9.7, −9.7, −9.5, and −9.5 kcal mol−1, respectively, the four compounds (isocryptomerin, hinokiflavone, amentoflavone, and neocryptomerin) with favorable binding affinities in the active pocket of SARS-CoV-2 were selected for the further studying.
Table 1 Binding residues of top 12 compounds at 4 Å area in the active pocket of SARS-CoV-2 Mpro
| S. no. |
Compounds |
Binding energy (kcal mol−1) |
Interacting residues |
| 1 |
Isocryptomerin |
−9.7 |
Ala191, Glu166, Gln192, Pro168Thr190, Gln189, Leu167, Arg188, Mer165, Asp187, His41, Met49, His164, Cys145, Leu27, Thr25, Ser46, Thr45, Thr24, Thr26 |
| 2 |
Hinokiflavone |
−9.7 |
Pro168, Gln192, Thr190, Ala191Gln189, Arg188, Met165, Asp187, His164, Met49, Thr26, Thr24, Thr45, Ser46, Thr25, Leu27, Gly143, Cys145, Asn142, His41, Glu166, Leu167 |
| 3 |
Amentoflavone |
−9.5 |
Asn142, Ser46, Thr26, His41, Met165, Arg188, Asp187, Tyr54, His164, Cys145, Gly143, Thr24, Thr25, Met49, Pro168, Glu166, Gln189 |
| 4 |
Neocryptomerin |
−9.5 |
Cys145, Ser46, Thr24, Met165, Met49, His41, Thr25, Ala191, Arg188, Pro168, Gln189, Thr190, Gln192, Leu167, Glu166, Asn142, Thr45, Thr26, Leu27, His164, Asp187 |
| 5 |
Robustaflavone |
−9.3 |
Leu141, Gly143, Ser144, Cys145, Asn142, Thr25, Thr26, Leu27, His163, Phe140, His172, His164, His41, Leu167, Met165, Glu166, Gln189, Pro168, Thr190, Gln192, Ala191 |
| 6 |
Podocarpusflavone A |
−9.3 |
Met49, His41, Cys145, His164, Gln189, Met165, Arg188, Leu167, Thr190, Ala191, Gln192, Pro168, His163, Glu166, His172, Asn142, Leu141, Phe140, Ser144 |
| 7 |
Cupressiflavone |
−9.3 |
Asp197, Arg131, Lys137, Thr198, Asn238, Thr199, Leu289, Asp289, Leu287, Tyr239, Leu272, Tyr237 |
| 8 |
Isoginkgetin |
−9.2 |
Asn142, Phe140, Ser144, Leu141, His163, Cys143, Glu166, Gln189, His164, His41, Met165, Arg188, Met49, Leu167, Gln192, Asp187, Thr190, Pro168, Ala191 |
| 9 |
Podocarpusflavone B |
−9.1 |
Thr25, Ser46, Met49, Leu141, Leu27, Gly143, Asn142, Ser144, Cys145, Phe140, Gln189, Glu166, His163, His172, His164, His41, Met165, Asp187, Tyr54, Arg188 |
| 10 |
Procyanidin A2 |
−9.0 |
Leu167, Glu166, Pro168, Met165, Gln189, Agr188, Asp187, His41, His163, His172, Phe140, Asn142, Cys145, Met49, Ser144, Thr25, Gly143, Thr26, Leu27, His164 |
| 11 |
Robustaflavone |
−8.9 |
Leu141, Gly143, Asn142, Thr25, Thr26, Cys145, His163, Leu27, His164, His41, Phe140, His172, Glu166, Met165, Leu167, Gln189, GPro168, Thr190, Gln192, Ala191 |
| 12 |
Sesguoiafiavone |
−8.6 |
Thr26, Thr25, Met49, Gly143, Ser46, Asn142, Cys145, His41, His164, Glu166, Met165, Asp187, Glu189, Arg188, Tyr54 |
The observed hydrogen bond interactions and hydrophobic interactions were shown in Fig. 3. Neocryptomerin fits well the active cavity of 3CLpro and is stabilized by three hydrogen bonds (at the sites of Cys145, Ser46, and Thr24). Besides, some residues like Met165, (Pi–alkyl interaction), Met49 (Pi–sigma interaction), Cys145 (Pi–sulfur interactions), His41 (Pi–Pi T-shaped interactions), and Thr25 (Pi–donor hydrogen bond interactions) are found to undergo hydrophobic interactions. Amentoflavone forms Hydrogen bonds with Asn142, Ser46, and Thr24, while forming multiple interactions with His41 and Met165. Four hydrogen bonds of 3CLpro/hinokiflavone are formed within the active site, while His41 (Pi–Pi T-shaped interactions), Cys145 (Pi–sulfur interactions), and Met165 (Pi–alkyl interactions) also interact with the compound. The main residues of isocryptomerin forming hydrogen bonds with proteins include Cys145, Ser46, Thr45, and Thr24, while other interactions are also spotted, such as Pi–sulfur, alkyl, Pi–Pi T-shaped, Pi–donor hydrogen bond, and Pi–lone pair interactions between the isocryptomerin and Mpro. After the docking screening, four compounds were found to have substantial binding energy toward the 3CLpro, and are expected to inhibit the 3CLpro activity, thus being the candidates for further study.
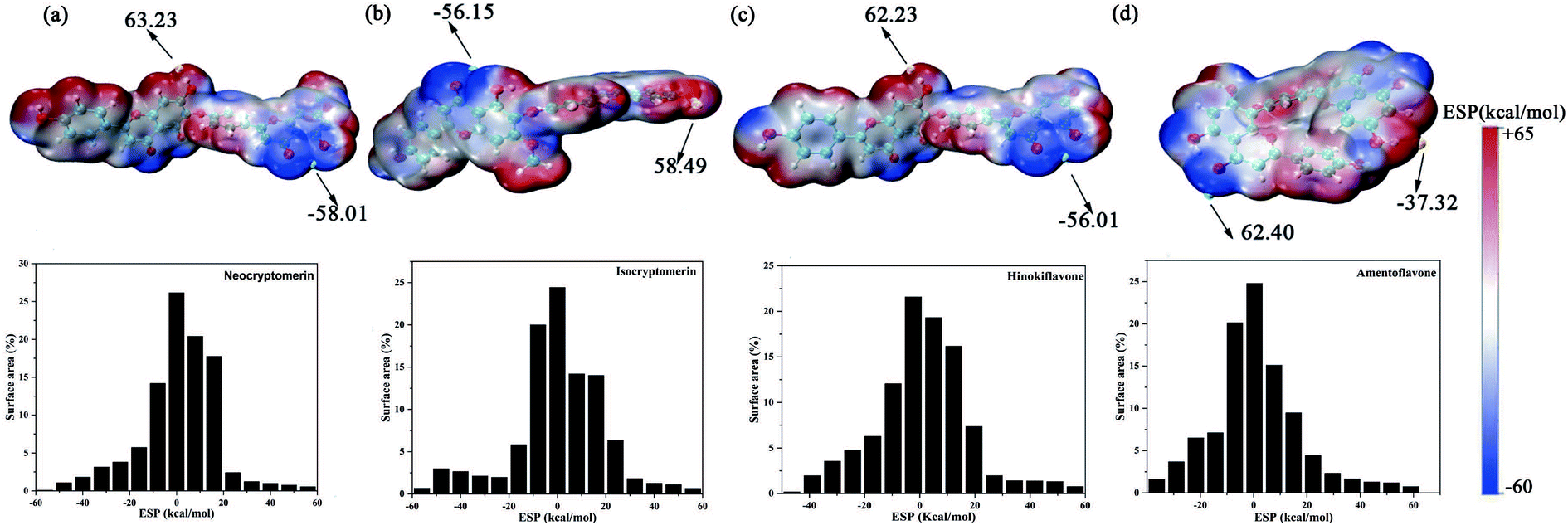
3.2 Electrostatic potential (ESP)
Studying the ESP of various compounds is useful for finding new chemical or biochemical compounds that can be used in the discovery and design of new therapeutics and for developing them into useful medicines. The interactions between biomolecules and compounds mainly include hydrogen bonds and halogen bonds dominated by electrostatic interactions, and ESP is the real space function that is most closely related to the electrostatic effect, and it is very suitable to analyze the electrostatic-induced weak interaction. Therefore, the analysis of electrostatic potential is helpful to understand the charge-dependent properties in the molecular structure of the complex.35,54 Fig. 4 shows the ESP distribution of the molecule as well as the active sites and comparative reactivity of the atoms. The negative region is presented in blue, and the positive region in red. The histogram in Fig. 4 shows that the large portion vdW surface of them have ESP value within −10 to +10 kcal mol−1. Only a tiny part of the vdW surface has ESP value larger than +60 kcal mol−1 or less than −65 kcal mol−1. Even small areas with minima and maxima of ESP can be very important to establish hydrogen bonds or electrostatic interactions. The most negative region corresponds to the oxygen in 4-Oxo-4h-1-benzopyran, and the most positive region to the hydrogen in hydroxyl. Surface local minima and maxima of ESP for neocryptomerin, isocryptomerin, hinkiflavone and amentoflavone are labeled by green and orange spheres, respectively, with their values shown in Fig. 4. These extremes are the nucleophilic and electrophilic sites, which are most likely to be active with residues. As expected, the ESP results are consistent with the hydrogen bond and polar interactions depicted in Fig. 3.
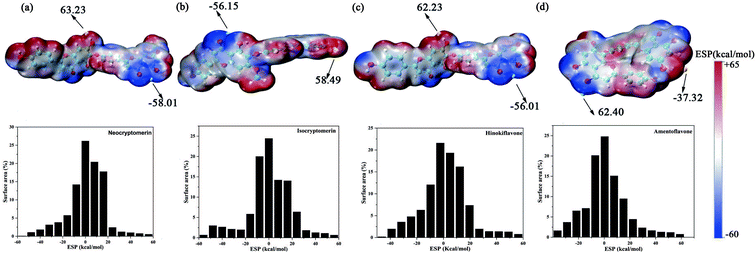 |
| | Fig. 4 ESP mapped molecular vdW surface of the selected compounds and the area percentage in each ESP range: (a) neocryptomerin, (b) isocryptomerin, (c) hinokiflavone and (d) amentoflavone. | |
3.3 HOMO–LUMO gap
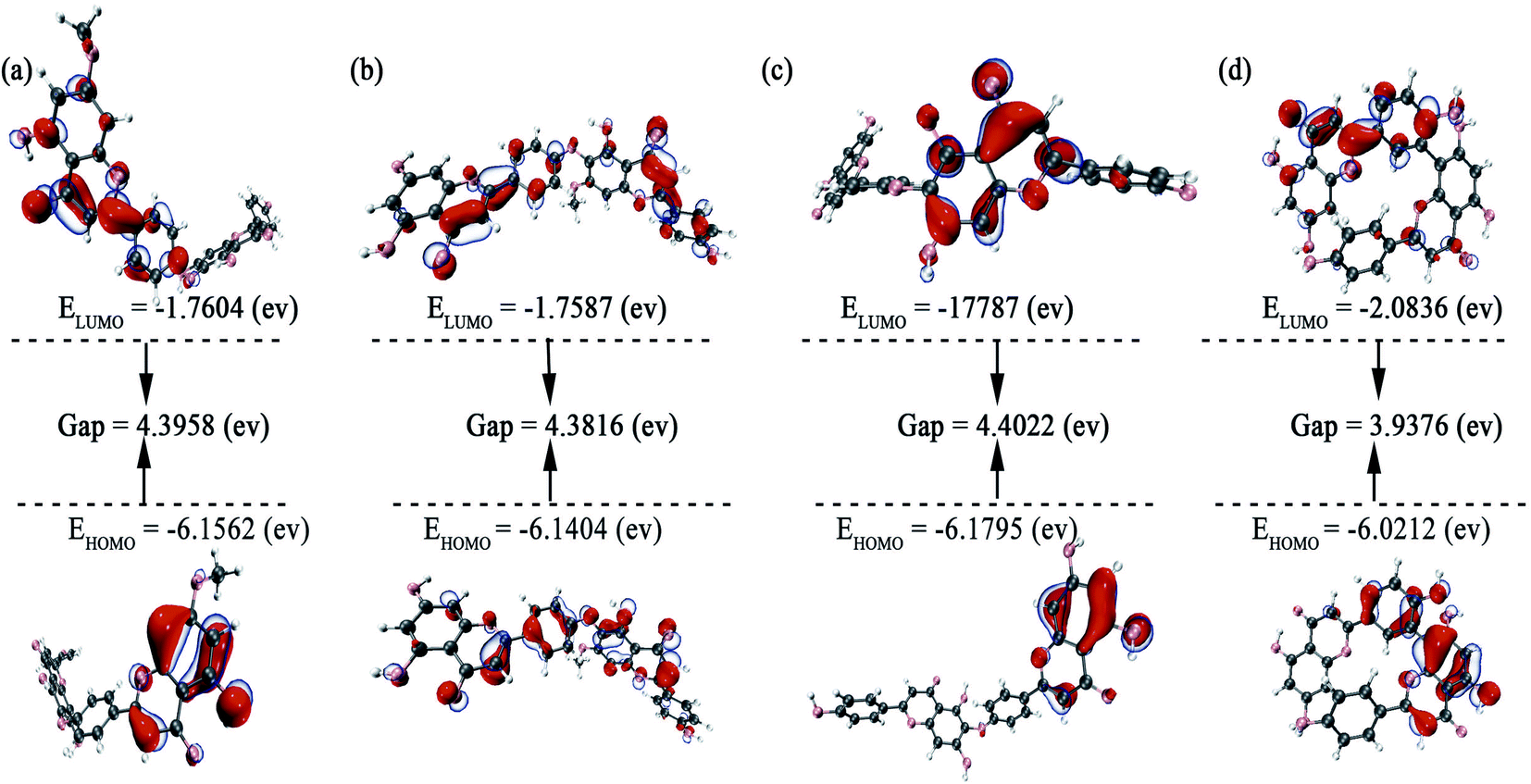
According to the frontier molecular orbital theory, the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) are the keys to determining the chemical reaction of the system.55 Therefore, the study of frontier orbitals of the drug molecules can provide important information for the exploration of the mechanism of action and the identification of active sites.56 HOMO–LUMO energy gaps of similar systems indicate the stability of molecules, with a smaller energy gap representing a more unstable molecule.57 The HOMO and LUMO orbitals for the screened compounds and their HOMO–LUMO energy gaps are shown in Table 2 and Fig. 5, and LUMO–HOMO energy gaps decreased in the following order: amentoflavone < isocryptomerin < hinokifavone < neocryptomerin. The smaller the gap is, the easier it interacts with the protein.58,59
Table 2 The Conceptual Density Functional Theory (CDFT) using DFT calculated at B3LYP-D3/6-311G**
| CDFT descriptors |
Neocryptomerin |
Hinokiflavone |
Isocryptomerin |
Amentoflavone |
| E_HOMO (eV) |
−6.16 |
−6.14 |
−6.18 |
−6.02 |
| E_LUMO (eV) |
−1.76 |
−1.76 |
−1.78 |
−2.08 |
| Ionization potential (eV) |
6.16 |
6.14 |
6.18 |
6.02 |
| Electron affinity (eV) |
1.76 |
1.76 |
1.78 |
2.08 |
| Energy gap (eV) |
4.40 |
4.38 |
4.40 |
3.94 |
| Mulliken electronegativity (eV) |
3.96 |
3.95 |
3.98 |
4.05 |
| Chemical potential (eV) |
−3.96 |
−3.95 |
−3.98 |
−4.05 |
| Hardness (eV) |
6.47 |
6.46 |
6.48 |
6.43 |
| Softness (eV − 1) = 1/hardness |
0.15 |
0.15 |
0.15 |
0.16 |
| Electrophilicity index (eV) |
1.21 |
1.21 |
1.22 |
1.28 |
| Nucleophilicity index (eV) |
2.97 |
2.98 |
2.94 |
3.10 |
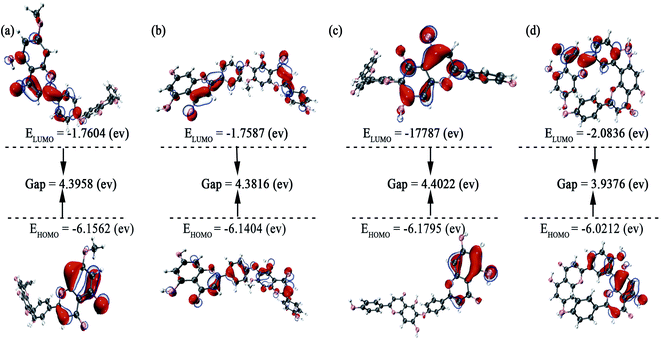 |
| | Fig. 5 Frontier molecular orbitals for (a) neocryptomerin, (b) isocryptomerin, (c) hinokiflavone, and (d) amentoflavone inhibitor. | |
3.4 CDFT
As we all know, the quantum chemical parameters of the compound are very important for their practical applications. Therefore, for the selected four potential inhibitors, we calculate some physical and chemical properties defined by the Conceptual Density Functional Theory (CDFT),59 such as EHOMO, ELUMO, ionization potential (IP), electron affinity (EA), electronegativity (χ), chemical potential (μ), chemical hardness (η), chemical softness (S), electrophilicity (ω) and nucleophilicity (N). Compounds can be studied using CDFT to predict reactive sites and the reactivity, as well as their properties.38,60 By comparing the parameters we calculated, we can further screen the promising compounds. Another functional (M06-2X) has also been included for comparison (shown in Table S2 in ESI†), and the CDFT results are comparable with those from the B3LYP.
Amentoflavone is more reactive than others because of its low chemical hardness (h) and naturally high softness (S) based on Pearson's HSAB principle.61 From Table 2, since the hardness values η of amentoflavone (6.42 eV) and hinokiflavone (6.46 eV) are smaller than those of neocryptomerin (6.47 eV) and isocryptomerin (6.48 eV), amentoflavone and hinokiflavone are less stable or more reactive than two others. Electronegativity (χ) and chemical potential (μ) measure the ability of an atom or a functional group to attract electrons.62 These two values in Table 2 indicate that amentoflavone has the most electron attracting ability, while hinokiflavone possesses the least electronegativity. The electrophilicity index defines the tendency of an electrophile to acquire a given amount of electron density and the resistance for a molecule to exchange electron density with the environment.63 Thus, among all compounds, amentoflavone shows higher electrophilicity at 1.27 eV than other three compounds, and hinokiflavone shows only less value at 1.21 eV. Thus, amentoflavone and hinokiflavone are more active than neocryptomerin and isocryptomerin in terms of polar reactions activity.
The screened compounds are characterized using quantitative chemical parameters in this part. Calculations were done to establish relevant data so that we can gain a better understanding of the properties of the compound.
3.5 Molecular dynamics (MD) simulation
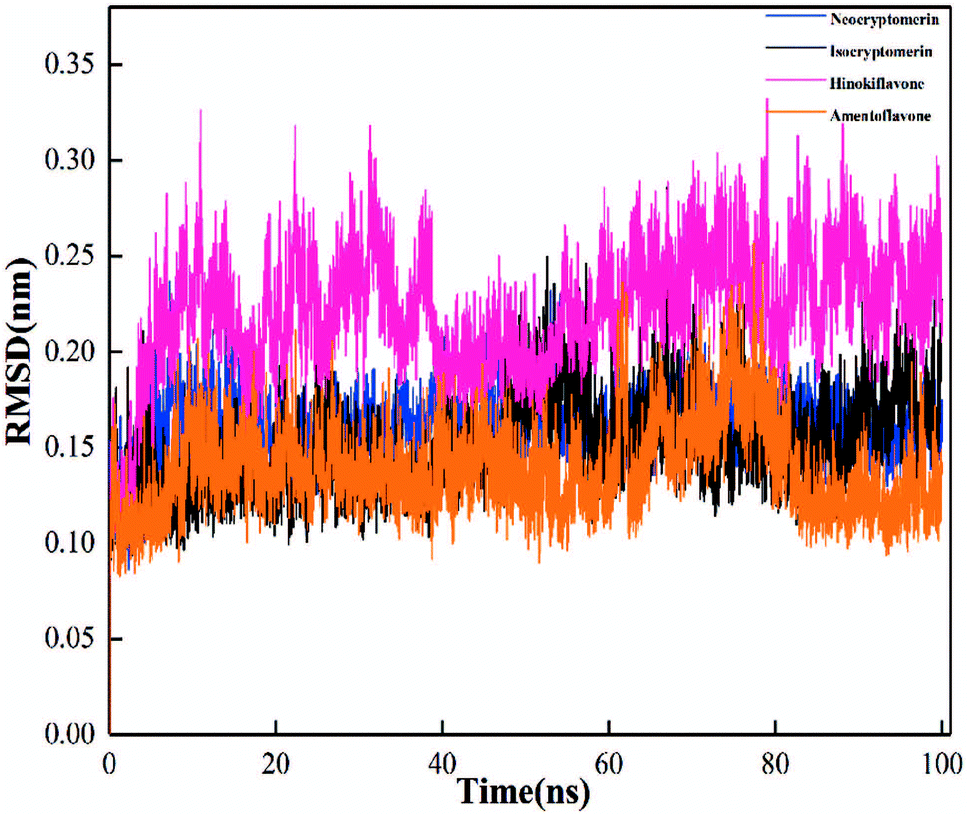
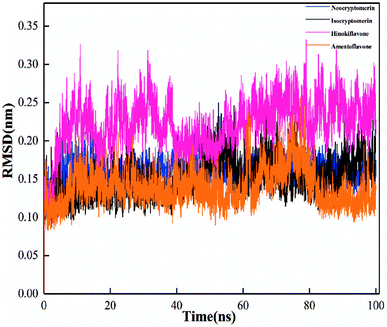
In order to evaluate the stability of the initial structure of the protein and ligand complex, we proceed with the molecular dynamics simulation. In the MD simulation, the root-mean-square deviations (RMSDs) of backbone atoms relative to their respective initial positions are observed (Fig. 6), and the average RMSD values for neocryptomerin, isocryptomerin, hinokiflavone, and amentoflavone are 0.16 nm, 0.15 nm, 0.23 nm, and 0.14 nm, respectively.
 |
| | Fig. 6 Root-mean-square deviation (RMSD) of the backbone atoms from the initial structure for four compounds (a–d) towards the SARS-CoV-2 main protease (Mpro) over 100 ns MD simulations. | |
The highest RMSD deviation (0.33 nm) is obtained for the 3CLpro/hinokiflavone system, while the least RMSD (0.09 nm) is obtained for 3CLpro/amentoflavone. As can be seen in Fig. 6, all compounds tend to the equilibrium state after 80 ns. Neocryptomerin, isocryptomerin, and amentoflavone display an average RMSD of <0.2 nm. However, hinokiflavone shows higher fluctuations as compared to others, and an average RMSD of <0.3 nm was noted from Fig. 6. 3CLpro/neocryptomerin has a slight acceptable fluctuation at 50 ns, and this system is stable throughout the simulation. For 3CLpro/isocryptomerin, a slight disturbance between 50 and 55 ns is shown, but it remains stable during the simulation. 3CLpro/hinokiflavone, which fluctuates slightly before 60 ns, gains stability and enters the production stage after 60 ns. In the case of 3CLpro/amentoflavone, it fluctuates slightly at about 75 ns, but it is stable at other time periods. The data in Fig. 6 indicate that all these screened compounds are stable in the entire simulation process except for some negligible small fluctuations, proving that these compounds are able to bind steadily to the proteins without affecting the overall topology of SARS-CoV-2 Mpro.
3.6 Binding free energy analysis
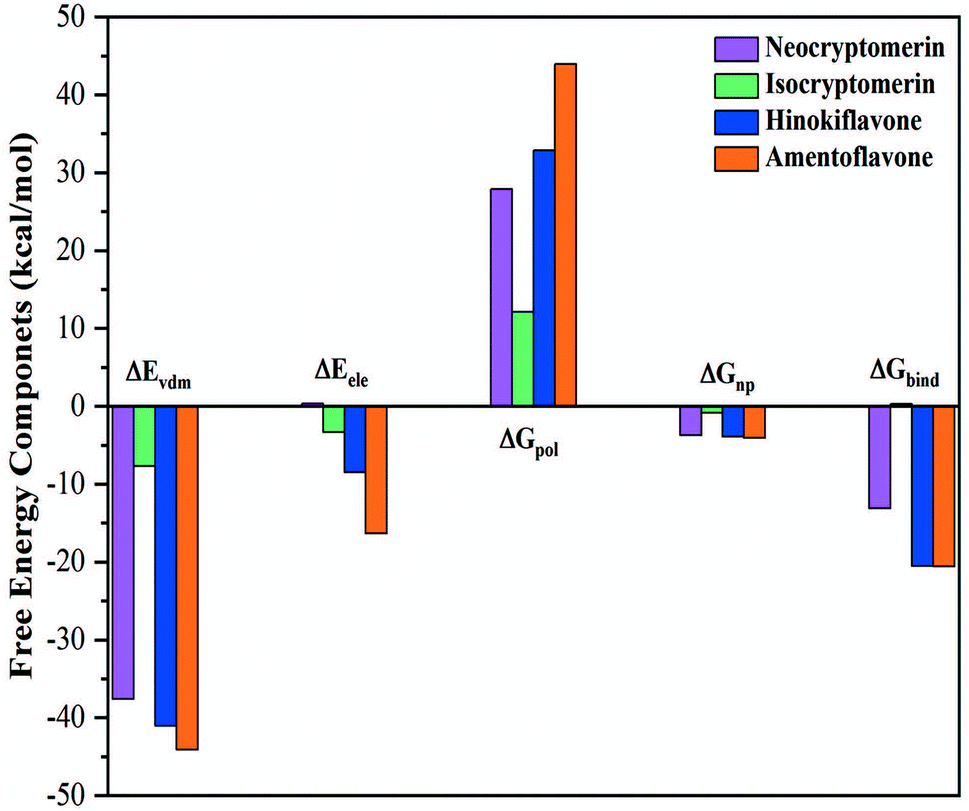
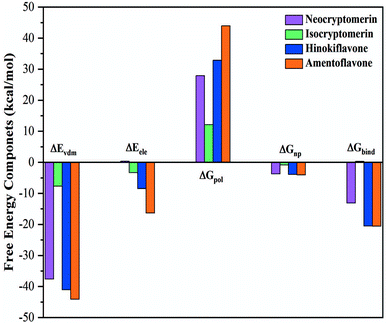
The binding free energies and energy decomposition analyses for the four screened compounds are evaluated with the help of Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) approach.50 Through the production process, the coordinates and energy values are gathered every 100 ps. It provides individual components, such as van der Waals energy, electrostatic energy, polar solvation energy, non-polar solvation energy, and total binding free energy of all the systems. In Fig. 7, the components of the binding free energy for the inhibitors in the presence of 3CLpro are plotted, and the corresponding data are presented in Table 3.
 |
| | Fig. 7 Energy components (kcal mol−1) for the binding of four compounds to 3CLpro receptor, including van der Waals interaction (ΔEvdW), electrostatic interaction (ΔEele), polar solvation energy (ΔGpol), non-polar solvation energy (ΔGnp), and estimated binding affinity (ΔGbind). | |
Table 3 Energetic components of the binding free energy (kcal mol−1) for SARS-CoV-2-inhibitor complexes calculated using MM-PBSA
| Components |
Neocryptomerin |
Isocryptomerin |
Hinokiflavone |
Amentoflavone |
| ΔEvdw |
−37.62 |
−7.66 |
−41.06 |
−44.08 |
| ΔEele |
0.35 |
−3.33 |
−8.45 |
−16.36 |
| ΔGpol |
27.91 |
12.12 |
32.90 |
43.96 |
| ΔGnp |
−3.71 |
−0.82 |
−3.90 |
−4.079 |
| ΔGbind |
−13.10 |
0.30 |
−20.50 |
−20.56 |
This study indicates that the van der Waals attraction, electrostatic interactions (except for isocryptomerin), and non-polar solvation energy are the main contributions to the binding, whereas the polar solvation free energy weakens the complexation. Table 3 shows that the estimated binding free energies for the 3CLpro/amentoflavone, 3CLpro/hinokiflavone and 3CLpro/neocryptomerin complexes are −20.56, −20.50, and −13.10 kcal mol−1, respectively, while that of 3CLpro/isocryptomerin complex is positive (+0.30 kcal mol−1). In short, the binding affinity to 3CLpro decreases in the following order: amentoflavone > hinokiflavone > neocryptomerin > isocryptomerin.
It is further revealed in Table 3 that the van der Waals attraction and non-polar solvation energy of the 3CLpro/neocryptomerin complex are almost the same as those of the 3CLpro/amentoflavone complex and the 3CLpro/hinokiflavone complex, but its electrostatic energy (+0.35 kcal mol−1) is positive, different from other complexes. Thus, the total binding energy is less. For the 3CLpro/isocryptomerin complex, the van der Waals energy (−7.66 kcal mol−1), which plays an important role in stability, allows its binding energy to be positive. Amentoflavone has the highest binding energy and is relatively stable, while hinokiflavone has its binding energy very close to amentoflavone. From Table 3, it can be seen that the stability-promoting components for the two compounds (amentoflavone and hinokiflavone) are van der Waals energy, electrostatic interaction, and non-polar solvation energy, while the stability-inhibiting component is polar solvation energy. The binding energies for other complexes are also calculated and shown in Table S3 in ESI.† Overall, amentoflavone, hinokiflavone, and neocryptomerin can be considered as the leading compounds of rational compounds against COVID-19.
3.7 Sum-per-residues binding free energy decomposition analysis
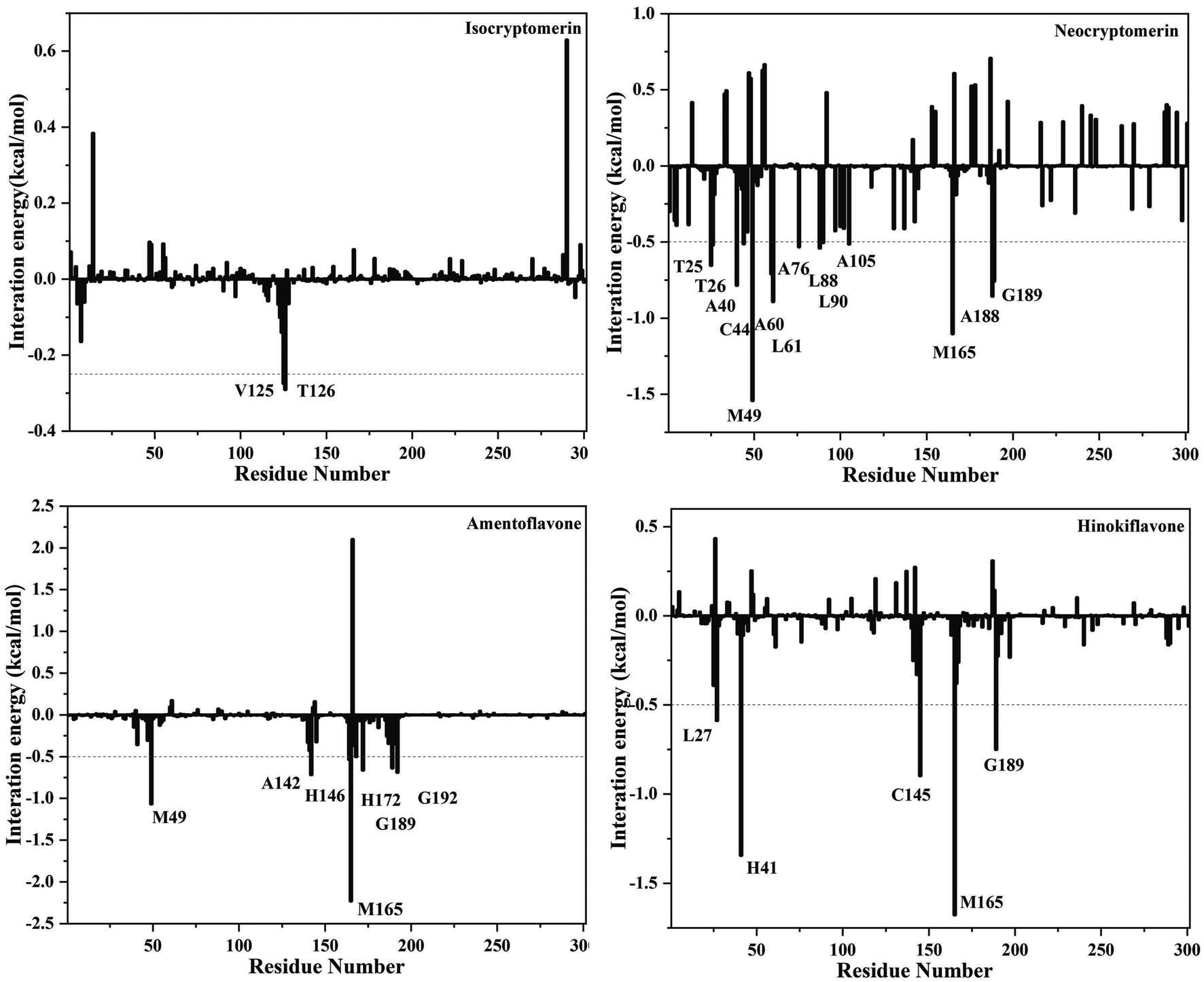
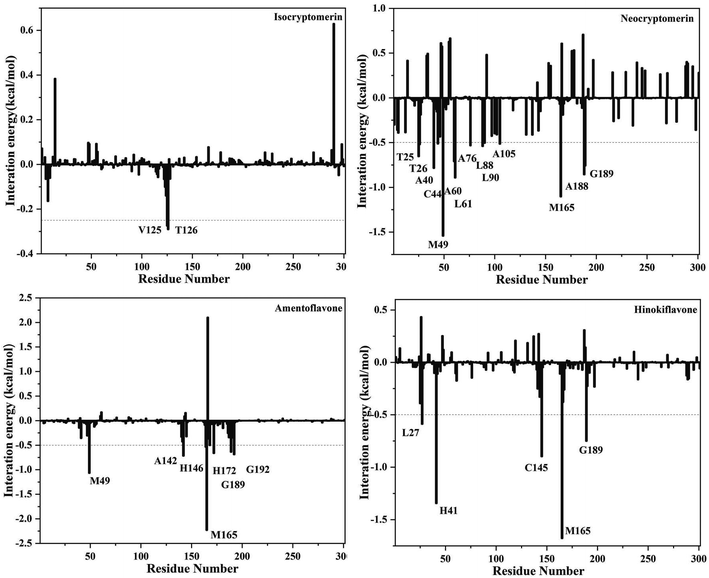
In order to identify the crucial residues responsible for stabilizing the interactions between the inhibitors and 3CLpro, binding free energy contributions from individual residues were decomposed with the MM-PBSA method. Decomposing the binding free energy on a per-residue basis into contributions from internal energy, polar solvation energy, and non-polar solvation energy for residues, as shown in Fig. 7. Key residues with a cut-off value of −0.5 kcal mol−1 were identified from the free energy binding spectra.
It is evident from Fig. 8 that several hotspot residues (MET-49, ASN-142, HIS-164 MET-165, PRO-168, HIS-172, GLN-189, GLN-192) contribute favorably to the binding of amentoflavone. It can further be observed from Fig. 8 that the catalytic dyad, MET-49 (−1.06 kcal mol−1) and MET-165 (−2.23 kcal mol−1), contribute significantly to the binding of amentoflavone. For the 3CLpro/neocryptomerin complex, THR-25, THR-26, ARG-40, CYS-44, MET-49, ARG-60, LYS-61, ARG-76, LYS-88, LYS-90, ARG-105, MET-165, ARG-188, GLN-189 have major contributions for the interaction, and MET-49 (−1.54 kcal mol−1) and MET-165 (−1.10 kcal mol−1) also play a major role for the stability of compounds. For the 3CLpro/hinokiflavone complex, LEU-27, HIS-41, CYS-145, MET-165, and GLN-189 residues are the primary driving force of binding energy, and MET-165 (−1.68 kcal mol−1) and HIS-41 (−1.34 kcal mol−1) play a key role in stabilizing compounds. In the case of the 3CLpro/isocryptomerin, there are two residues VAL-125 (−0.28 kcal mol−1) and TYR-126 (−0.29 kcal mol−1) with binding energy values higher than −0.25 kcal mol−1. This might be the reason for the poor affinity of isocryptomerin against 3CLpro.
 |
| | Fig. 8 Decomposition of the binding free energy into contributions from individual residues for 3CLpro complexed with neocryptomerin, isocryptomerin, hinokiflavone and amentoflavone. | |
The results showed that MET-49 and MET-165 are the key residues, which provided a clue for further research. Overall, the identification of hotspot residues from our analysis can facilitate the discovery of new selective inhibitor against COVID-19.
4 Conclusions
Computational medicinal chemistry has become an important element of modern drug research. The sudden outbreak of the new coronavirus has caused panic all over the world and seriously threatened the health of human beings. However, there is no effective drug now, and the discovery of an effective drug is imminent. Computer-aided drug discovery (CADD) methodologies have emerged as powerful tools in the drug discovery process and could reduce the cost of research and, most importantly, speed up drug discovery. In order to have a more comprehensive understanding of the properties of the drug candidates, we not only did molecular dynamics research but also calculated their molecular properties through quantum chemistry calculations. First of all, we screened 12 candidates from 251 compounds through molecular docking, and the first 4 compounds were selected after further structural optimization. These selected four molecules were calculated by the DFT/B3LYP methods to predict LUMO/HOMO gap, ESP, and other properties related to molecular interacting abilities (EHOMO, ELUMO, ionization potential, electron affinity, electronegativity, chemical potential, chemical hardness, chemical softness, electrophilicity, and nucleophilicity). After that, molecular dynamics simulation was carried out to evaluate the stability of the selected compounds, and we have also investigated the hotspot residues controlling the receptor-ligand binding. Based on the results of our study, hinokiflavone, and amentoflavone are considered as promising compounds against SARS-CoV-2. This is coherent with the previous findings that these two compounds have antiviral effects. We found that the compound hinokiflavone and its derivatives have antagonistic effects against influenza64 and dengue virus,65,66 while amentoflavone and its derivatives have inhibitory effects against CVB3,67 HBA,68 dengue virus66 and SARS-CoV.69 Our findings suggest that these two compounds might serve as a good starting point for further investigations of potential drugs against SARS-CoV-2. We hope that our findings in the present paper will be helpful for the experimental study to develop effective drugs against SARS-CoV-2.
Author contributions
Conceptualization, Y. M. and M. Z.; methodology, Y. M.; software, Y. M.; validation, Y. M., and Y. T.; formal analysis, Y. M.; investigation, Y. M.; resources, Y. M.; data curation, Y. M.; writing—original draft preparation, Y. M.; writing—review and editing, Y. M.; visualization, Y. M. and H. Q.; supervision, M. Z., X. G., F. Y., and C. W.; project administration, M. Z., and F. Y.; funding acquisition, M. Z. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by the National Natural Science Foundation of China (grants No. 10904111, 11604238), the Tianjin Natural Science Foundation (11JCYBJC14500), and the Science and Technology Development Fund of Tianjin Education Commission for Higher Education (2019KJ175). We thank all colleagues in our laboratory for helpful discussions and technical assistance.
Notes and references
- J. T. Wu, K. Leung and G. M. Leung, Lancet, 2020, 395, 689–697 CrossRef CAS.
- W. Ji, W. Wang, X. Zhao, J. Zai and X. Li, J. Med. Virol., 2020, 92, 433–440 CrossRef CAS PubMed.
- P. Zhou, X. L. Yang, X. G. Wang, B. Hu, L. Zhang, W. Zhang, H. R. Si, Y. Zhu, B. Li, C. L. Huang, H. D. Chen, J. Chen, Y. Luo, H. Guo, R. D. Jiang, M. Q. Liu, Y. Chen, X. R. Shen, X. Wang, X. S. Zheng, K. Zhao, Q. J. Chen, F. Deng, L. L. Liu, B. Yan, F. X. Zhan, Y. Y. Wang, G. F. Xiao and Z. L. Shi, Nature, 2020, 579, 270–273 CrossRef CAS PubMed.
- D. Cucinotta and M. Vanelli, Acta Biomed., 2020, 91, 157–160 Search PubMed.
- D. Worldometer, COVID-19 coronavirus pandemic, https://www.worldometers.info/coronavirus/, 2020, 2 October 2021, date last accessed Search PubMed.
- F. Kabinger, C. Stiller, J. Schmitzová, C. Dienemann, G. Kokic, H. S. Hillen, C. Höbartner and P. Cramer, Nat. Struct. Mol. Biol., 2021, 28, 740–746 CrossRef CAS PubMed.
- E. Mahase, BMJ, 2021, 374, n2083 CrossRef PubMed.
- A. Grifoni, J. Sidney, Y. Zhang, R. H. Scheuermann, B. Peters and A. Sette, Cell Host Microbe, 2020, 27, 671–680 CrossRef CAS PubMed.
- A. Vincent, L. Awada, I. Brown, H. Chen, F. Claes, G. Dauphin, R. Donis, M. Culhane, K. Hamilton, N. Lewis, E. Mumford, T. Nguyen, S. Parchariyanon, J. Pasick, G. Pavade, A. Pereda, M. Peiris, T. Saito, S. Swenson, K. Van Reeth, R. Webby, F. Wong and J. Ciacci-Zanella, Zoonoses Public Health, 2014, 61, 4–17 CrossRef CAS PubMed.
- J. Chen, D. Liu, L. Liu, P. Liu, Q. Xu, L. Xia, Y. Ling, D. Huang, S. Song, D. Zhang, Z. Qian, T. Li, Y. Shen and H. Lu, J. Zhejiang Univ., 2020, 49, 215–219 Search PubMed.
- K. Anand, G. J. Palm, J. R. Mesters, S. G. Siddell, J. Ziebuhr and R. Hilgenfeld, EMBO J., 2002, 21, 3213–3224 CrossRef CAS PubMed.
-
(a) L. DeLano, The PyMOL molecular graphics system, DeLano Scientific, San Carlos, CA, 2002, http://www.pymol.org Search PubMed;
(b) R. A. Laskowski and M. B. Swindells, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S. H. Jung, J. Med. Chem., 2016, 59, 6595–6628 CrossRef CAS PubMed.
- L. T. Lin, W. C. Hsu and C. C. Lin, J. Tradit., Complementary Altern. Med., 2014, 4, 24–35 CrossRef PubMed.
- J. Shawon, Z. Akter, M. M. Hossen, Y. Akter, A. Sayeed, M. Junaid, S. S. Afrose and M. A. Khan, Curr. Pharm. Des., 2020, 26, 5241–5260 CrossRef CAS PubMed.
- J. B. Harborne and J. M. Tom, The flavonoids: advances in research, Springer, 2013 Search PubMed.
- F. Chen, K. H. Chan, Y. Jiang, R. Y. Kao, H. T. Lu, K. W. Fan, V. C. Cheng, W. H. Tsui, I. F. Hung, T. S. Lee, Y. Guan, J. S. Peiris and K. Y. Yuen, J. Clin. Virol., 2004, 31, 69–75 CrossRef CAS PubMed.
- L. Yi, Z. Li, K. Yuan, X. Qu, J. Chen, G. Wang, H. Zhang, H. Luo, L. Zhu, P. Jiang, L. Chen, Y. Shen, M. Luo, G. Zuo, J. Hu, D. Duan, Y. Nie, X. Shi, W. Wang, Y. Han, T. Li, Y. Liu, M. Ding, H. Deng and X. Xu, J. Virol., 2004, 78, 11334–11339 CrossRef CAS PubMed.
- T. T. Nguyen, H. J. Woo, H. K. Kang, V. D. Nguyen, Y. M. Kim, D. W. Kim, S. A. Ahn, Y. Xia and D. Kim, Biotechnol. Lett., 2012, 34, 831–838 CrossRef CAS PubMed.
- G. L. Russo, I. Tedesco, C. Spagnuolo and M. Russo, Semin. Cancer Biol., 2017, 46, 1–13 CrossRef CAS PubMed.
- C. Spagnuolo, S. Moccia and G. L. Russo, Eur. J. Med. Chem., 2018, 153, 105–115 CrossRef CAS PubMed.
- K. Hayashi, m. Mori, y. M. Knox, t. Suzutan, m. Ogasawara, i. Yoshida, k. Hosokawa, a. Tsukui and m. AzumA, Food Sci. Technol. Res., 2003, 9, 242–244 CrossRef CAS.
- E. Britannica, Britannica Book of the Year 2009, Encyclopaedia Britannica, Inc., 2009 Search PubMed.
- A. C. Anderson, Chem. Biol., 2003, 10, 787–797 CrossRef CAS PubMed.
- S. J. Macalino, V. Gosu, S. Hong and S. Choi, Arch. Pharmacal Res., 2015, 38, 1686–1701 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian (Revision A.03) 16, Gaussian Inc., Pittsburgh, PA, 2016 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11, 905–919 CrossRef CAS PubMed.
- BIOVIA, Dassault Systèmes, Discovery Studio Visualizer (Revision 20.1.0.19295), Dassault Systèmes, San Diego, 2019 Search PubMed.
- S. Medjahed, S. Belaidi, S. Djekhaba, N. Tchouar and A. Kerassa, J. Bionanosci., 2016, 10, 118–126 CrossRef CAS.
- R. Abbasoglu, J. Mol. Model., 2006, 12, 991–995 CrossRef CAS PubMed.
- C.-Y. Yam, X. Zheng and G. Chen, J. Comput. Theor. Nanosci., 2006, 3, 857–863 CrossRef CAS.
- P. C. Rathi, R. F. Ludlow and M. L. Verdonk, J. Med. Chem., 2020, 63, 8778–8790 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1874 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- A. W. S. Da Silva and W. F. Vranken, BMC Res. Notes, 2012, 5, 1–8 CrossRef PubMed.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- S. A. Showalter and R. Brüschweiler, J. Chem. Theory Comput., 2007, 3, 961–975 CrossRef CAS PubMed.
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- S. Melchionna, G. Ciccotti and B. Lee Holian, Mol. Phys., 2006, 78, 533–544 CrossRef.
- H. J. Berendsen, J. v. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- E. Wang, H. Sun, J. Wang, Z. Wang, H. Liu, J. Z. Zhang and T. Hou, Chem. Rev., 2019, 119, 9478–9508 CrossRef CAS PubMed.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan and W. Wang, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- L. Li, C. Li, S. Sarkar, J. Zhang, S. Witham, Z. Zhang, L. Wang, N. Smith, M. Petukh and E. Alexov, BMC Biophys., 2012, 5, 1–11 CrossRef PubMed.
- R. Kumari, R. Kumar, C. Open Source Drug Discovery and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- N. S. Pagadala, K. Syed and J. Tuszynski, Biophys. Rev., 2017, 9, 91–102 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- I. Fleming, Frontier orbitals and organic chemical reactions, Wiley, 1977 Search PubMed.
- B. Babu, J. Chandrasekaran, B. Mohanbabu, Y. Matsushita and M. Saravanakumar, RSC Adv., 2016, 6, 110884–110897 RSC.
- K. S. Thanthiriwatte and K. N. De Silva, J. Mol. Struct., 2002, 617, 169–175 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1988, 110, 2092–2097 CrossRef CAS.
- M. Salihović, S. Huseinović, S. Špirtović-Halilović, A. Osmanović, A. Dedić, Z. Ašimović and D. Završnik, Bull. Chem. Technol. Soc. Bosnia Herzegovina, 2014, 42, 31–36 Search PubMed.
- P. Geerlings, E. Chamorro, P. K. Chattaraj, F. De Proft, J. L. Gázquez, S. Liu, C. Morell, A. Toro-Labbé, A. Vela and P. Ayers, Theor. Chem. Acc., 2020, 139, 1–18 Search PubMed.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- J. Padmanabhan, R. Parthasarathi, V. Subramanian and P. Chattaraj, J. Phys. Chem. A, 2007, 111, 1358–1361 CrossRef CAS PubMed.
- R. G. Parr, L. v. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- K. Miki, T. Nagai, T. Nakamura, M. Tuji, K. Koyama, K. Kinoshita, K. Furuhata, H. Yamada and K. Takahashi, Heterocycles, 2008, 75, 879–886 CrossRef CAS.
- P. Coulerie, C. Eydoux, E. Hnawia, L. Stuhl, A. Maciuk, N. Lebouvier, B. Canard, B. Figadère, J.-C. Guillemot and M. Nour, Planta Med., 2012, 78, 672–677 CrossRef CAS PubMed.
- P. Coulerie, M. Nour, A. Maciuk, C. Eydoux, J.-C. Guillemot, N. Lebouvier, E. Hnawia, K. Leblanc, G. Lewin and B. Canard, Planta Med., 2013, 79, 1313–1318 CrossRef CAS PubMed.
- S. Wilsky, K. Sobotta, N. Wiesener, J. Pilas, N. Althof, T. Munder, P. Wutzler and A. Henke, Arch. Virol., 2012, 157, 259–269 CrossRef CAS PubMed.
- F. Li, X. Song, G. Su, Y. Wang, Z. Wang, J. Jia, S. Qing, L. Huang, Y. Wang and K. Zheng, Viruses, 2019, 11, 466 CrossRef CAS PubMed.
- Y. B. Ryu, H. J. Jeong, J. H. Kim, Y. M. Kim, J.-Y. Park, D. Kim, T. T. H. Naguyen, S.-J. Park, J. S. Chang and K. H. Park, Bioorg. Med. Chem., 2010, 18, 7940–7947 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07364h |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yulian Taoa,
Hanyang Qua,
Cuihong Wangb,
Fei Yan*a,
Xiujun Gaoa and
Meiling Zhang
a,
Yulian Taoa,
Hanyang Qua,
Cuihong Wangb,
Fei Yan*a,
Xiujun Gaoa and
Meiling Zhang