
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid quantitative 1H–13C two-dimensional NMR with high precision†
Yu-Shan Wua,
Bai-Xiang Lib and
Ying-Yun Long *b
*b
aJilin Business and Technology College, Changchun 130507, China
bState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China. E-mail: yylong@ciac.ac.cn
First published on 14th February 2022
Abstract
Two dimensional (2D) 1H–13C heteronuclear single-quantum correlation (HSQC) spectroscopy has recently been proposed for quantitative determination of typical linear low density polyethylene (LLDPE) with high accuracy. It requires highly precise measurement to achieve further reliable quantification. In this context, this paper aims at determining conditions that allow the achievement of high precision. On the basis of the optimized parameters, two time-saving strategies, nonuniform sampling (NUS) and band-selective HSQC are evaluated on model polyolefins in terms of repeatability. Precision better than 0.3% and 5% for ethylene content (E mol%) and 1-hexene content (H mol%) of the model poly(ethylene-co-1-hexene)s are obtained with 50% NUS or band-selective HSQC. Moreover, dramatic precision enhancements can be achieved with the combination of band-selective HSQC and 50% NUS, in which repeatabilities better than 0.15% and 2.5% for E mol% and H mol% are observed. The experiment times are reduced to about 0.5 h. These methods open important perspectives for rapid, precise and accurate quantitative analysis of complex polymers.
Introduction
One-dimensional (1D) 1H nuclear magnetic resonance (NMR) is a powerful tool for quantitative analysis in a wide range of domains, from drug analysis1,2 to natural products3–5 or metabolomics analysis.6–8 Nevertheless, precise quantitative analysis of complex mixtures is often difficult due to the presence of large overlap between peaks.9 In this case, quantitative 13C NMR is useful and is widely described,10–12 since the 13C NMR spectrum has a wider chemical shift range. But 13C NMR typically requires long experiment times and large amounts of sample to give spectra with sufficient signal-to-noise ratios (SNRs) for quantification, owing to the weak sensitivity of the 13C nuclei and very long relaxation delays needed.13,14 Interestingly, two dimensional (2D) spectroscopy offers an alternative, as it offers resonances with much better discrimination than 1D NMR that is essential for quantitative studies of complex spectra. There has been a continuous interest among chemists in applying 2D NMR experiments for the quantitative analysis of complicated mixtures in recent years.15–30In spite of their high potentialities, quantitative 2D NMR is restricted by limitations. The cross peak intensity in 2D NMR is not directly proportional to the concentration, which is affected by relaxation times, evolution time, 1JC–H couplings, JH–H couplings, T1 and T2 relaxations, off-resonance effects, etc.14,15 Various studies have been reported to address this issue. They are mostly based on theoretical calculations,24,25 external calibration or adding an internal standard compound.21,31 These methods lead to high accuracy and high precision for small molecules in pharmaceutics, metabolomics or natural products,14 but most of them are not suitable for quantifying polymer samples, owning to the more complicated factors arising from the molecular weight and the molecular weight distribution.19 If a low molecular weight compound is added to a polymer as internal standard, the resonances from an internal standard references compound of a low molecular weight can have rather different 1JC–H couplings, different 13C chemical shift and different proton T2 profiles from the polymer. To achieve accurate signal quantification in such a system, errors of all types need to be corrected. As a consequence, there has been a very limited example describing the use of 2D NMR for quantitative analysis of polymers up to date. Heikkinen reported the successful use of quantitative HSQC (Q-HSQC) for the elucidation of wood lignin,32,33 a complex polymer. Later, Crestini and coworkers used the improved quick quantitative HSQC (QQ-HSQC) for quantitative evaluation of milled softwood and hardwood.34 However; these methods require specific acquisition and processing programs that are not commercially available.14
Recently, Keresztes and Coates reported the use of band-selective 2D HSQC for rapid characterization of the stereo-regularity of polyacrylonitrile (PAN) and polypropylene (PP).35 We described the first use of 2D 1H–13C HSQC for quantitative analysis of poly(ethylene-co-1-hexene), reducing the experiment time to less than an hour with a high accuracy.36 As for a quantitative NMR method, both a high accuracy and a high precision are indispensable to achieve reliable quantification. However, the precision has not been mentioned yet among the examples related with the use of 2D NMR for quantitative analysis of polymers. Herein we are interested in evaluating the precision of 2D HSQC for quantitative analysis of poly(ethylene-co-1-hexene).
According to the previous reports, the limitation of precision mainly comes from the experimental duration.23 They generate additional noise in the indirect dimension, namely “t1 noise”, because of the long time interval separating the physical acquisition of two successive FIDs.37 As a consequence of the “t1 noise”, SNR is always low in F1 dimension, which then strongly affects the precision of quantitative 2D experiments. Consequently, many faster 2D acquisition strategies have been proposed, and most of them are applied in biological extracts and natural products.22,38–43 All the time-saving strategies are to shorten the duration of multidimensional NMR experiments.21,22,39 With these strategies, a precision of a few percent is reached in a number of applications.44–46 Giraudeau reported that a precision of a few per mil could be reached with a 10 ppm HSQC experiment for a small molecule ibuprofen.17 This paper aims at analyzing more complicated molecules, poly(ethylene-co-1-hexene) with high molecular weight and inhomogeneous composition. The typical linear low density polyethylene (LLDPE), poly(ethylene-co-1-hexene) with 1-hexene incorporation ≤10 mol%, are used as model samples. We want to determinate conditions, allowing the acquisition of 2D HSQC for quantitative analysis of these polymers with a high precision in terms of repeatability. It considers several approaches that have been proposed to reduce the duration of heteronuclear 2D experiments, namely band-selective 2D HSQC, 2D HSQC with non-uniform sampling (NUS) and the combination of band-selective HSQC with NUS. For comparison, the precision of traditional 2D HSQC for quantitative analysis of poly(ethylene-co-1-hexene) is also evaluated. Notably, band-selective 2D HSQC with 50% non-uniform sampling (NUS) enables the reduction of experiment time from more than 23 hours to less than 0.5 h with precision better than 2.5%.
Result and discussion
Traditional 2D HSQC
In our previous work, we have optimized the acquisition and process parameters of traditional 2D 1H–13C HSQC, making it to be a method for the content determination of poly(ethylene-co-1-hexene) with accuracy up to 99.5%.36 Herein, we evaluate the precision of this method based on the optimized parameters. Two typical linear low density polyethylene (LLDPE) samples are chosen as models, with 1-hexene incorporation of 4.08 mol% (EH-1) and 8.60 mol% (EH-2), respectively. The quantitative 13C NMR and 1H NMR spectra are shown in Fig. S1–S4,† while the composition and monomer sequence distributions are listed in Table S1.† The chemical shift assignments and their regions, together with the equations for the determination of triads content, were listed in ESI.†As described above, short experiment duration is the key to obtain a high precision.47,48 The experiment time is always affected by the number of scans (NS), the number of F1 increments (TD) and the recovery delay (D1). The most basic approach to reduce experiment time is to reduce these three parameters to their minimum acceptable values. The optimal acquisition conditions obtained previously indicated that NS could be chosen to 8 and D1 to 2 seconds to preserve a sufficiently high SNR and high accuracy. TD was reduced to 512 while preserving a good resolution along F1. With these conditions leading to a 158 min experiment time, an acceptable precision can be reached for the polymer composition as presented in Fig. 1. The values of CV are between 0.23% and 7.14% for the composition of EH-1 and between 0.23% and 3.48% for EH-2, respectively. The better repeatability of EH-2 might stem from its higher signal volumes of the typical regions. The values of CV for ethylene content (E mol%) are lower than those for 1-hexene content (H mol%), probably due to the higher SNR of the signal in region D (F2: δ 1.30–1.45 ppm, F1: δ 29.0–31.0 ppm) related with the E mol% calculation. The average repeatability is close to 4% and 2% for the triads of EH-1 and EH-2, respectively, with a maximum value of 7.14% (Table 1), which is still reasonable for a number of semi-quantitative applications. However, this does not fully meet the high-precision criteria required for the quantitative analysis. It can be seen from Fig. 1 that the TD can be reduced to 128 without significantly affecting the precision, thus resulting in a shorter experiment time. However, such a small TD value leads to an obvious resolution decrease in F1 (Fig. S5†) and thus relatively low accuracy. As a consequence, it appears more reasonable to consider alternative sampling strategies to reduce the experiment duration while preserving a high accuracy. Due to this purpose, we choose to evaluate the impact of two approaches to reduce the duration of 2D HSQC experiments. The approaches described below are well-described in the literature and easily implemented in commercial software. All of them rely on a partial sampling of the indirect dimension.
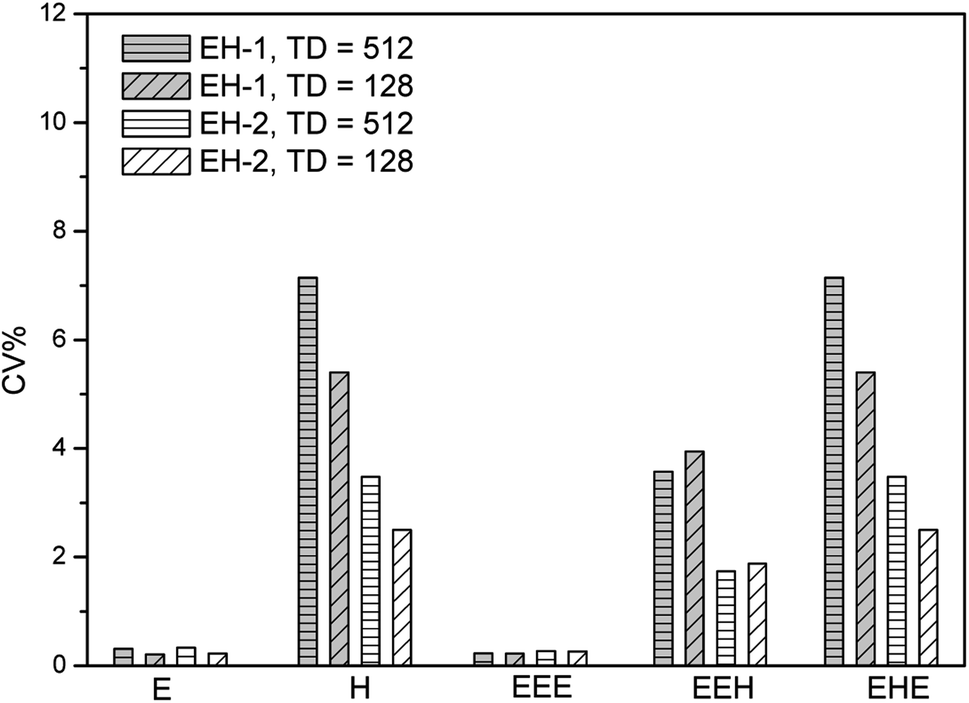 | ||
| Fig. 1 Precision of 2D 1H–13C HSQC experiments evaluated from their repeatability, measured on the sample EH-1 and EH-2, on a 400 MHz Bruker spectrometer equipped with a 5 mm BBFO SMART probe. | ||
| Sample | EH-1 | EH-2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | Triads | Content | Triads | Content | |||||||
| EEE | EEH | EHE | E | H | EEE | EEH | EHE | E | H | ||
| HSQC (TD = 512), 158 min | Mean | 0.874 | 0.084 | 0.042 | 0.958 | 0.042 | 0.742 | 0.172 | 0.086 | 0.914 | 0.086 |
| SD | 0.002 | 0.003 | 0.002 | 0.003 | 0.003 | 0.002 | 0.003 | 0.003 | 0.003 | 0.003 | |
| CV% | 0.23 | 3.57 | 7.14 | 0.31 | 7.14 | 0.27 | 1.74 | 3.48 | 0.33 | 3.48 | |
| HSQC (TD = 128), 40 min | Mean | 0.894 | 0.076 | 0.037 | 0.963 | 0.037 | 0.763 | 0.158 | 0.079 | 0.921 | 0.079 |
| SD | 0.002 | 0.003 | 0.002 | 0.002 | 0.002 | 0.002 | 0.003 | 0.002 | 0.002 | 0.002 | |
| CV% | 0.22 | 3.94 | 5.40 | 0.21 | 5.40 | 0.26 | 1.90 | 2.53 | 0.22 | 2.53 | |
| HSQC (TD = 512)/NUS 50%, 80 min | Mean | 0.877 | 0.082 | 0.041 | 0.959 | 0.041 | 0.745 | 0.170 | 0.085 | 0.915 | 0.085 |
| SD | 0.002 | 0.002 | 0.002 | 0.002 | 0.003 | 0.001 | 0.002 | 0.002 | 0.002 | 0.002 | |
| CV% | 0.22 | 2.42 | 4.86 | 0.20 | 4.86 | 0.14 | 1.18 | 2.36 | 0.22 | 2.36 | |
| bs-HSQC (TD = 256 ), 58 min | Mean | 0.871 | 0.086 | 0.043 | 0.957 | 0.043 | 0.736 | 0.176 | 0.088 | 0.912 | 0.088 |
| SD | 0.001 | 0.002 | 0.002 | 0.002 | 0.002 | 0.002 | 0.002 | 0.002 | 0.001 | 0.002 | |
| CV% | 0.13 | 2.32 | 4.65 | 0.21 | 4.65 | 0.27 | 1.13 | 2.22 | 0.12 | 2.22 | |
Non-uniform sampling
One of the most famous technique is the non-uniform sampling (NUS),49–54 which permits high-resolution spectra to be obtained from short data records, drastically reducing experiment time to several minutes by acquiring data points in the time domain in non-consecutive time increments.55 In order to yield clean artifact-free spectra, NUS must be associated with an appropriate processing procedure such as Compressed Sensing (CS) which are well-described in the literature and easily implemented in commercial software. It has enabled advanced applications of multidimensional NMR spectroscopy to study metabolomics,53 biological macromolecules56,57 and natural products.58,59 Moreover, 2D NMR method with NUS is reported to be successfully applied in a few examples of quantitative analysis. For example, Cooper used 2D HSQC with 25% NUS for the successful quantitative analysis of active lignans in Sambucus williamsii.20 We have used NUS/CS for quantitative analysis of poly(ethylene-co-1-hexene), successfully reducing the experiment time to less than a hour. Here we evaluate the precision of this method.Several percentages of NUS are tested. The representative data are presented in Fig. 2 as a plot of repeatability CV against the monomer and triads content. The NUS rate of 50% is found to obtain the smallest CV values. The average repeatability is about 2.5% and 1.3% for the composition (E mol% and H mol%) of EH-1 and EH-2, respectively, which is increased by more than half. The values of CV are between 0.20% and 4.86% for the composition of EH-1 and between 0.14% and 2.36% for EH-2, respectively. In addition, the content of triads EEH and EHE also show much higher precision than those from traditional HSQC. Similarly, the precision of the content of E and EEE is very satisfactory (better than 0.25%, see Fig. 2). Notably, it takes only one half of the traditional 2D HSQC experiment time to obtain a spectrum with a similar high F1 resolution to the one that is acquired with regular sampling (Fig. 3a vs. 3b). Therefore, while traditional 2D 1H–13C HSQC is an interesting tool for quantitative analysis of polyolefins due to its high accuracy and acceptable repeatability, the introduction of NUS can speed up the analysis of these polymers with its improved precision.
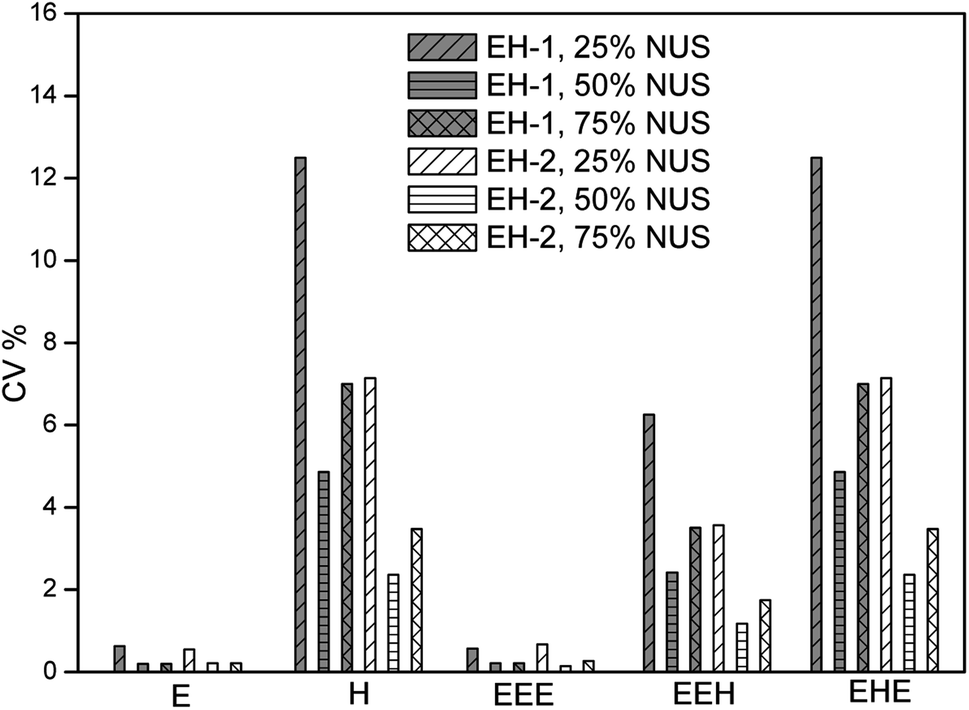 | ||
| Fig. 2 Precision of 2D 1H–13C HSQC with different NUS level evaluated from their repeatability, measured on the sample EH-1 and EH-2, on a 400 MHz Bruker spectrometer equipped with a 5 mm BBFO SMART probe. | ||
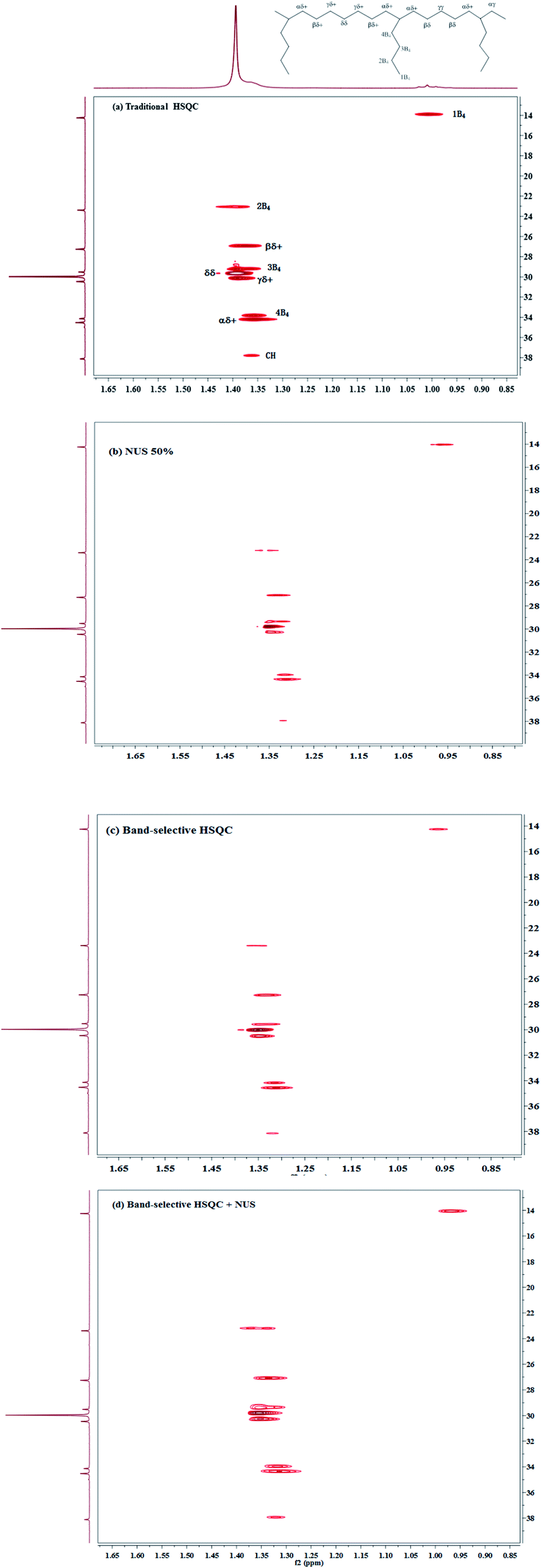 | ||
| Fig. 3 1H–13C HSQC spectra of poly(ethylene-co-1-hexene) EH-2 in 1,1,2,2-tetrachloroethane-d4 at 383 K, recorded with NS = 8, D1 = 2 s, on a 400 MHz Bruker spectrometer equipped with a BBFO probe, illustrating the time-saving strategies whose precision is evaluated in this study. (a) Traditional 2D HSQC spectrum recorded with a 75 ppm spectrum width and 512 increments in F1 dimension. (b) Spectrum recorded with a NUS rate of 50% and 512 increments in F1 dimension. (c) Band-selective 2D HSQC spectrum recorded with a 28 ppm spectral width and 256 increments in F1 dimension. (d) Band-selective 2D HSQC spectrum recorded with a 28 ppm spectral width, 256 increments in F1 dimension, and a NUS rate of 50%. | ||
Band-selective HSQC
The other way to reduce measurement time is the use of band-selective 2D HSQC, which reduces the spectra width in the indirect dimension and only includes a narrow band centered on the chemical shift range of interest. This method combines the improved sensitivity of 1H detection with the capability to approach natural linewidth resolution in the 13C NMR dimension. This benefit is mainly used for structure elucidation of complex structure. We applied this procedure for the quantitative content determination of poly(ethylene-co-1-hexene), by choosing a 28 ppm spectral width (SW) along the 13C dimension where all the peaks except the solvent were in the range. As a consequence of this smaller SW, a smaller number of indirect increments (TD = 256) can be used while preserving good resolution in F1. The corresponding spectrum is shown in Fig. 3c. Compared with the traditional 2D HSQC with 512 increments in F1 dimension, it shows excellent agreement regarding the composition and triad monomer sequence distributions (Table 1), by saving about 60% experiment time, thus demonstrating that band-selective HSQC is compatible with the accuracy.The average precision of the monomer content (E% and H%) are about 2.4% and 1.2% for EH-1 and EH-2, respectively. The CV values obtained from triads EEE show much higher precision than those from triads EEH and EHE (CV: 0.1% vs 1.1–2.3%), similar to the results obtained by traditional HSQC. Significantly, the precision of the content of E is extremely high (about 0.1%), which can match the high-precision criteria required for the quantitative analysis. We therefore decided to keep this reduced 28 ppm spectra width for the rest of the study.
Band-selective HSQC with 50% NUS
The third way to reduce the experiment time while preserving good resolution in F1 is to combine the two approaches described above. Combining NUS with a reduced 28 ppm spectral width, we adjusted the acquisition parameters, such as TD (F1), NS and the amount of NUS level to reduce the experiment time to less than half an hour. The typical spectra are shown in Fig. 3d and 4. Obviously, there are more noises observable in the HSQC spectra with NUS rate of 25% than those with NUS rate of 50% (Fig. 4A vs. 4B and C). Fortunately, the noises are not overlapped with signals that used for content calculation. The corresponding results are summarized in Table 2. It is indicated that all the precisions of sample EH-2 are better than EH-1. The precision decreases with the monomer content, probably stem from the relatively low signal-to-noise of their corresponding signals. The precisions of H, EEH and EHE are calculated from signals assigned to βδ+, while those of E and EEE are calculated from signals attributable to δδ, γδ+, βδ+ and 2B4. The values of CV are between 0.11% and 2.42% for the composition of EH-1 and between 0.10% and 1.17% for EH-2, respectively, when the HSQC spectra are obtained with TD of 512 in the indirect dimension and the NS is decreased to 4. It indicates that the concentration of the polymer is high enough for the achievement of good 1H NMR spectrum and the resolution of the 2D spectra plays a more important role for the precision and accuracy. However, further increasing the TD to 1024 in the indirect dimension does not work much more for the precision (Table 2). In addition, the TD number can be reduced to a value as small as 128 without significantly affecting the relative volume of each region, while preserving a resolution identical to the traditional one. The corresponding spectrum is shown in Fig. 3d. On the whole, both excellent precision and satisfactory accuracy can be obtained under the conditions that TD and NS are not less than 256 and 4 with 50% NUS. As a consequence of this smaller SW and smaller number of indirect increments or smaller number of scans, the experiment time reduces to less than 0.5 h. As can be seen in Fig. 5, the average precision of the composition content are 1.3% and 0.7% for EH-1 and EH-2, respectively, which is very excellent. The precision of the content of E and EEE is still extremely high (about 0.1%), comparable to the one measured with band-selective HSQC. Especially, the results highlight dramatic precision enhancements in the content of H and triads EHE, EEH, which probably benefit from the less experiment time and thus less t1 noise. In addition, the combination of band-selective HSQC with NUS is compatible with the high accuracy for the content determination of EH-1 and EH-2. It appears that the combination of band-selective HSQC with NUS is more convenient and more efficient for similar quantitative analysis of polyolefins.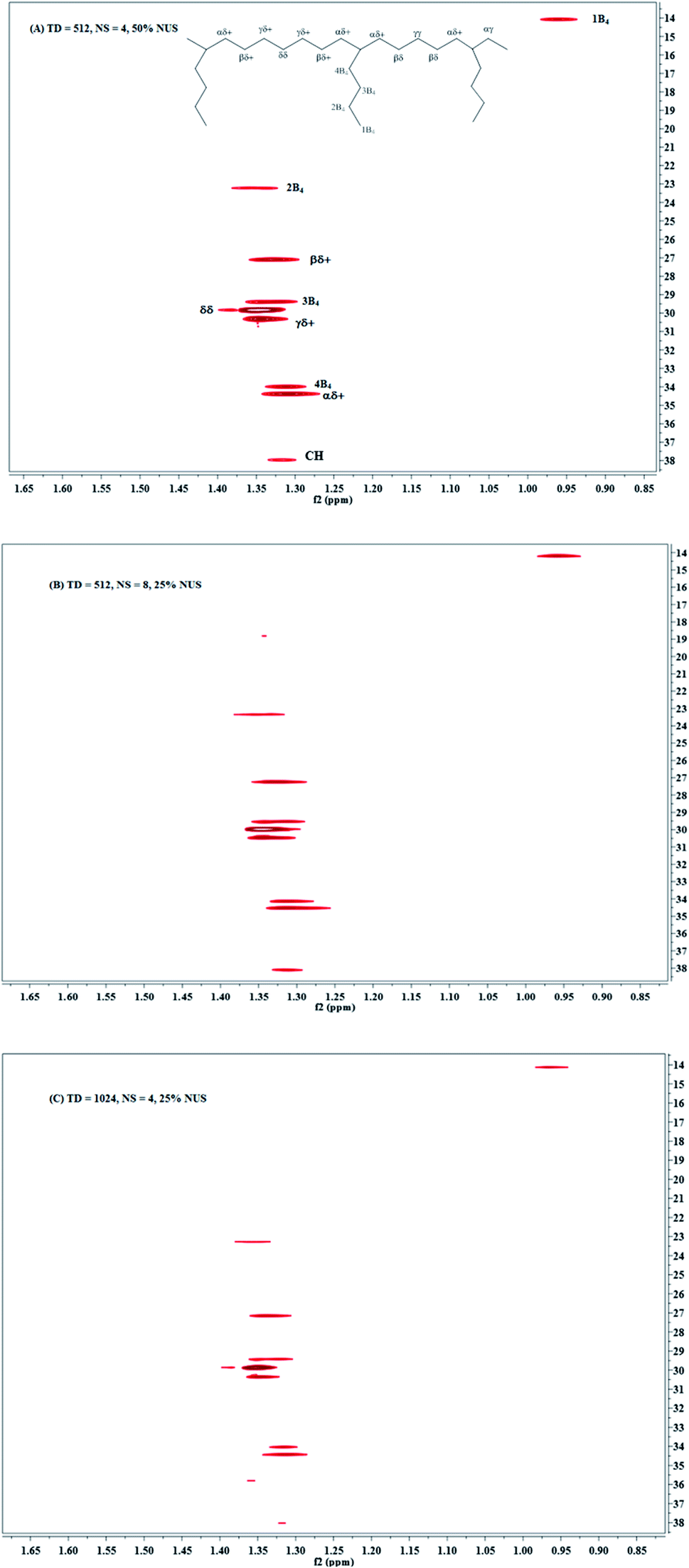 | ||
| Fig. 4 Band-selective HSQC spectra of poly(ethylene-co-1-hexene) in 1,1,2,2-tetrachloroethane-d4 at 383 K, with a 28 ppm spectral width in 13C dimension, on a 400 MHz Bruker spectrometer equipped with a BBFO probe during 29 min (A) TD = 512, NS = 4, 50% NUS; (B) TD = 512, NS = 8, 25% NUS; (C). TD = 1024, NS = 4, 25% NUS. | ||
| Sample | EH-1 | EH-2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | Triads | Content | Triads | Content | |||||||
| EEE | EEH | EHE | E | H | EEE | EEH | EHE | E | H | ||
| bs-HSQC/50% NUS, TD = 128, NS = 16 | Mean | 0.891 | 0.066 | 0.033 | 0.967 | 0.033 | 0.772 | 0.152 | 0.076 | 0.924 | 0.076 |
| SD | 0.003 | 0.002 | 0.002 | 0.003 | 0.002 | 0.003 | 0.002 | 0.002 | 0.003 | 0.002 | |
| CV% | 0.33 | 3.03 | 6.06 | 0.33 | 6.06 | 0.26 | 1.32 | 2.64 | 0.33 | 2.64 | |
| bs-HSQC/NUS 50%, TD = 256, NS = 8 | Mean | 0.877 | 0.082 | 0.041 | 0.959 | 0.041 | 0.739 | 0.174 | 0.087 | 0.913 | 0.087 |
| SD | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | |
| CV% | 0.14 | 1.25 | 2.50 | 0.12 | 2.50 | 0.15 | 0.60 | 1.15 | 0.12 | 1.15 | |
| bs-HSQC/25% NUS, TD = 512, NS = 8 | Mean | 0.886 | 0.076 | 0.038 | 0.962 | 0.038 | 0.754 | 0.164 | 0.082 | 0.918 | 0.082 |
| SD | 0.003 | 0.002 | 0.002 | 0.003 | 0.002 | 0.003 | 0.002 | 0.002 | 0.003 | 0.002 | |
| CV% | 0.34 | 2.63 | 5.26 | 0.33 | 5.26 | 0.40 | 1.22 | 2.44 | 0.33 | 2.44 | |
| bs-HSQC/50% NUS, TD = 512, NS = 4 | Mean | 0.877 | 0.082 | 0.041 | 0.959 | 0.041 | 0.745 | 0.170 | 0.085 | 0.915 | 0.085 |
| SD | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | |
| CV% | 0.11 | 1.21 | 2.42 | 0.11 | 2.42 | 0.10 | 0.58 | 1.17 | 0.11 | 1.17 | |
| bs-HSQC/25% NUS, TD = 1024, NS = 4 | Mean | 0.876 | 0.084 | 0.042 | 0.958 | 0.042 | 0.742 | 0.172 | 0.086 | 0.914 | 0.086 |
| SD | 0.002 | 0.001 | 0.001 | 0.002 | 0.001 | 0.002 | 0.001 | 0.001 | 0.002 | 0.001 | |
| CV% | 0.22 | 1.25 | 2.50 | 0.22 | 2.50 | 0.26 | 0.60 | 1.20 | 0.26 | 1.20 | |
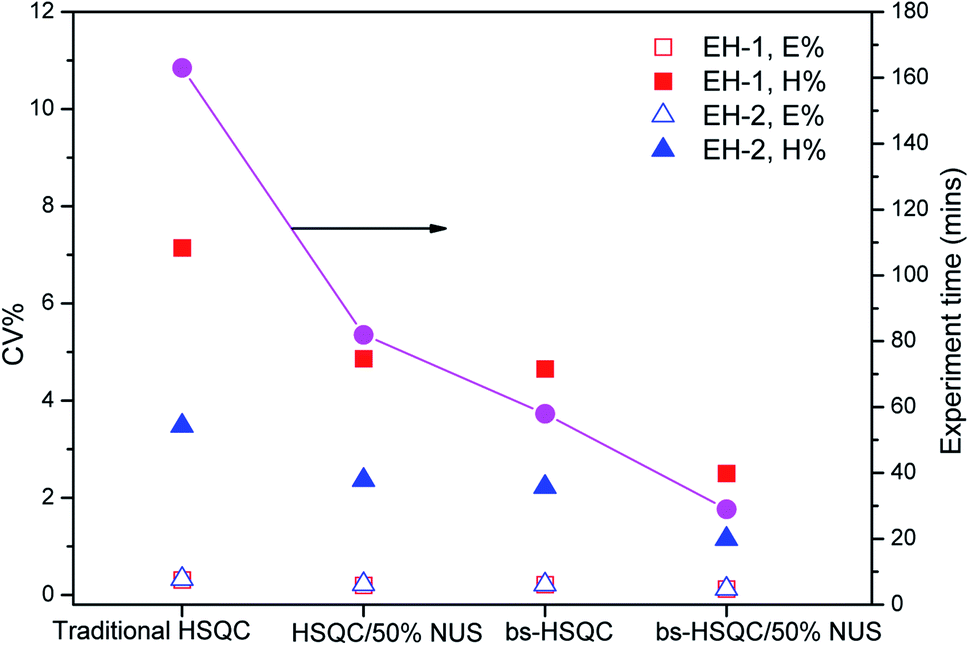 | ||
| Fig. 5 Plots of precision CV and experiment time versus different methods used for the determination of monomer contents of sample EH-1 and EH-2. The hollow and solid squares plot the precision of E% and H% of sample EH-1, while the hollow and solid triangles represent the precision of E% and H% of sample EH-2, respectively. The solid circles indicate the experiment time of different methods. | ||
Experimental
Sample preparation
Poly(ethylene-co-1-hexene) samples were obtained from Jilin Petro-Chemical Industry Corporation. The number molecular weights of sample EH-1 and EH-2 are about 38 kg mol−1 and 32 kg mol−1, while their polydispersities of the average molecular weight are 2.8 and 2.7, respectively, determined by Gel Permeation Chromatography (GPC).A poly(ethylene-co-1-hexene) sample of 45 mg were placed in the NMR tube and dissolved with 0.6 mL C2D2Cl4 solvent in order to obtain a concentration of 75 mg mL−1. This concentration corresponds to the conditions typically used in polyolefin analysis. The sample was sealed and horizontally positioned in a hot air oven at 383 K for about 10 hours until a homogenous solution was observed.
NMR experiments
All the NMR acquisitions were performed at 383 K on a Bruker Avance III 400.13 MHz spectrometer equipped with a 5 mm BBFO SMART probe, utilizing Topspin 3.6 with the standard Bruker library pulse programs. Before acquisition, all NMR samples were thermally equilibrated at 383 K for at least 15 min inside the spectrometer, the probe was automatically tuned and matched, and the 90° hard pulses for both 1H and 13C were carefully calibrated. All series of experiments were performed with automatic shimming to preserve the B0 field homogeneity.The traditional 2D HSQC spectra
The HSQC experiment (Bruker pulse program: hsqcetgpsp.3) was performed at 383 K, by using 16 dummy scans; 8 scans; 2.0 s relaxation delay; 126 JCH1 value; 203 receiver gain; 6.5 ppm (F2) and 75 ppm (F1) spectral width; transmitter offset was set at 3.2 ppm (F2) and 40 ppm (F1); chemical shift region is from 0 ppm to 6.5 ppm in F2 and from 5 ppm to 80 ppm in F1. The spectra were acquired with 256, 512 or 1024 t1 increments. The FIDs were recorded with 1024 or 2048 data points. A 1H 90° hard pulse was automatically calibrated before acquisition.Zero filling was applied to 2048 in F2 and 1024 in F1; a 90° shifted squared sine bell function was applied in both dimensions. The integration of 2D peak volumes was performed using the integration routine in the Bruker software. All the integration results are the average of five experiments. We chose to rely on the direct integration of 2D signals. The integration box widths were carefully adjusted for all experiments so that to obtain the same level of truncation for all peaks. Peak volumes were exported to Microsoft Excel 2010 for statistical analysis.
2D HSQC spectra with nonuniform sampling
The acquisition parameters of 2D HSQC with NUS were the same with those of traditional HSQC except that the NUS mode was used. HSQC with 25%, 35%, 50% and 75% level of NUS were performed with these parameters. An exponential weighting was applied to this NUS scheme. Zero filling was applied to 2048 in F2 and 1024 in F1; a 90° shifted squared sine bell function was applied in both dimensions. The integration of 2D peak volumes was performed using the integration routine in the Bruker software. All the integration results are the average of five experiments. We chose to rely on the direct integration of 2D signals. The integration box widths were carefully adjusted for all experiments so that to obtain the same level of truncation for all peaks. Peak volumes were exported to Microsoft Excel 2010 for statistical analysis.Band-selective 2D HSQC spectra
Band-selective HSQC experiments were performed using the shsqcetgpsisp2.2 pulse program of the manufacturer's pulse program library with band-selective shaped 13C refocusing pulse. The shape form Q3.1000 was chosen. To achieve selective excitation over a frequency range of 2817 Hz (28 ppm), the length of the pulse was determined to 1750.4 us with a power of 0.690 W for the used probe head. The experiments were performed at 383 K, by using 16 dummy scans; 8 scans; 2.0 s relaxation delay; 126 1JCH value; 203 receiver gain; 6.5 ppm (F2) and 28 ppm (F1) spectral width; transmitter offset was set at 3.2 ppm (F2) and 27.0 ppm (F1); chemical shift region is from 0 ppm to 6.5 ppm in F2 and from 13.0 ppm to 41.0 ppm in F1; 512, 256 or 128 increments in F1. A 1H 90° hard pulse was automatically calibrated before acquisition. Zero filling was applied to 2048 in F2 and 1024 in F1; a 90° shifted squared sine bell function was applied in both dimensions. The integration of 2D peak volumes was performed using the integration routine in the Bruker software. All the integration results are the average of five experiments. We chose to rely on the direct integration of 2D signals. The integration box widths were carefully adjusted for all experiments so that to obtain the same level of truncation for all peaks. Peak volumes were exported to Microsoft Excel 2010 for statistical analysis.Band-selective 2D HSQC spectra with NUS acquisition mode
The acquisition parameters of band-selective HSQC with NUS were the same with those of band-selective HSQC except that the NUS mode was used. HSQC with 25%, 35%, 50% and 75% level of NUS were performed with these parameters. An exponential weighting was applied to this NUS scheme. Zero filling was applied to 2048 in F2 and 1024 in F1; a 90° shifted squared sine bell function was applied in both dimensions. The integration of 2D peak volumes was performed using the integration routine in the Bruker software. All the integration results are the average of five experiments. We chose to rely on the direct integration of 2D signals. The integration box widths were carefully adjusted for all experiments so that to obtain the same level of truncation for all peaks. Peak volumes were exported to Microsoft Excel 2010 for statistical analysis.The repeatability and precision
To evaluate the variability of the data, the same 2D NMR experiment were performed in the same laboratory and obtained with the same 400 MHz spectrometer. Five replicates were performed on five independent samples. Replicates were obtained at the same concentration and temperature, and in the same solvent, with the same relaxation times and coupling constants. Each spectrum was recorded after independent field homogenization and 1H pulse calibration; under these conditions each run was independent. Mean values, standard deviation (SD) and coefficient of variation (CVs) were calculated on each composition and triads separately. CVs were calculated by dividing standard deviation by the general mean, while
Conclusions
We have described the use of 2D 1H–13C HSQC as a tool for the rapid determination of a typical LLDPE at triads level. It demonstrates that a very high precision can be achieved with much less experiment time, because of the introduction of time-saving strategies. The combination of 2D band-selective HSQC with NUS not only preserves excellent agreement with the results from quantitative 13C NMR spectrum regarding the composition, but also greatly increases the precision in monomer and triads content. The average CVs of composition is lower than 2.0%, with the highest precision up to 0.1%, thus satisfying the conditions that needed for the fast quantitative determination of polyolefins. This simple and reliable method not only supports the structure elucidation of polymers, but also simultaneously provides the content of composition and monomer sequence. Especially, this method is well suited for polyolefins because of their narrow range of JC–H coupling constants, T2, 13C NMR spectra and 1H NMR spectra windows covered.We believe the application of band-selective HSQC with NUS is useful for speeding up the quantitative microstructure determination of polymers, and then speeding up the batch-to-batch quality control in polymer production in industry. While these approaches display high accuracy and high precision for quantitative analysis, information on quaternary carbons is not accessible via this method. In this case, one could consider using alternative 2D acquisition strategies such as heteronuclear-multiple bond correlation (HMBC). Future work will focused on the application of 2D NMR for analyzing microstructures of a wide variety of more complex polymers and quantitative comparison of results obtained from quantitative 1D 13C NMR.
Author contributions
Ying-Yun Long: conceptualization, methodology, supervision, funding acquisition, projection administration. Yu-Shan Wu: writing original draft, formula analysis, investigation, data analysis. Bai-Xiang Li: resources, investigation, visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for subsidy provided by the Science and Technology Department of Jilin Province (No. 20200703022ZP).References
- U. Holzgrabe, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 229–240 CrossRef CAS PubMed.
- J. K. Kwakye, Talanta, 1985, 32, 1069–1071 CrossRef CAS PubMed.
- E. Tenailleau, P. Lancelin, R. J. Robins and S. Akoka, J. Agric. Food Chem., 2004, 52, 7782–7787 CrossRef CAS PubMed.
- F. Le Grand, G. George and S. Akoka, J. Agric. Food Chem., 2005, 53, 5125–5129 CrossRef CAS PubMed.
- G. F. Pauli, Phytochem. Anal., 2001, 12, 28–42 CrossRef CAS PubMed.
- J. C. Lindon, J. K. Nicholson, E. Holmes and J. R. Everett, Concepts Magn. Reson., 2000, 12, 289–320 CrossRef CAS.
- D. S. Wishart, TrAC, Trends Anal. Chem., 2008, 27, 228–237 CrossRef CAS.
- S. Zhang, G. A. Nagana Gowda, V. Asiago, N. Shanaiah, C. Barbas and D. Raftery, Anal. Biochem., 2008, 383, 76–84 CrossRef CAS PubMed.
- P. Giraudeau, Magn. Reson. Chem., 2014, 52, 259–272 CrossRef CAS PubMed.
- T. H. Mareci and K. N. Scott, Anal. Chem., 1977, 49, 2130–2136 CrossRef CAS.
- J. N. Shoolery, Prog. Nucl. Magn. Reson. Spectrosc., 1977, 11, 79–93 CrossRef.
- D. J. Cookson and B. E. Smith, J. Magn. Reson., 1984, 57, 355–368 CAS.
- H. N. Cheng, in Modern Methods of Polymer Characterization, ed. H. G. Barth and J. W. Mays, John Wiley & Sons, New York, 1991, pp. 409–494 Search PubMed.
- H. Koskela, Quantitative 2D NMR Studies, Annual Reports on NMR Spectroscopy, vol. 66, pp. 1–31, ISSN 0066-4103, DOI:10.1016/S0066-4103(08)00401-8.
- P. Giraudeau, Magn. Reson. Chem., 2017, 55, 61–69 CrossRef CAS PubMed.
- H. Koskela, I. Kilpeläinen and S. Heikkinen, J. Magn. Reson., 2005, 174, 237–244 CrossRef CAS PubMed.
- E. Msrtineau, S. Akoka, R. Boisseau, B. Delanoue and P. Giraudeau, Anal. Chem., 2013, 85, 4777–4783 CrossRef PubMed.
- S. Massou, C. Nicolas, F. Letisse and J.-C. Portais, Phytochemistry, 2007, 68, 2330–2340 CrossRef CAS PubMed.
- L. Zhang and G. Gellerstedt, Magn. Reson. Chem., 2007, 45, 37–45 CrossRef CAS PubMed.
- H. H. Xiao, J. Lv, D. Mok, X. S. Yao, M. S. Wong and R. Cooper, J. Nat. Prod., 2019, 82, 1733–1740 CrossRef CAS PubMed.
- I. A. Lewis, S. C. Schommer, B. Hodis, K. A. Robb, M. Tonelli, W. Westler, M. Sussman and J. L. Markley, Anal. Chem., 2007, 79, 9385–9390 CrossRef CAS PubMed.
- W. Gronwald, M. S. Klein, H. Kaspar, S. R. Fagerer, N. NÜrnberger, K. Dettmer, T. Bertsch and P. J. Oefner, Anal. Chem., 2008, 80, 9288–9297 CrossRef CAS PubMed.
- K. Hu, J. J. Ellinger, R. A. Chylla and J. L. Markley, Anal. Chem., 2011, 83, 9252–9360 Search PubMed.
- R. K. Rai, P. Tripathi and N. Sinha, Anal. Chem., 2009, 81, 10232–10238 CrossRef CAS PubMed.
- K. Bingol, F. Zhang, L. Brüschweiler-Li and R. Brüschweiler, Anal. Chem., 2013, 85, 6413–6420 CrossRef PubMed.
- S. Yang, J. L. Wen, T. Q. Yuan and R. C. Sun, RSC Adv., 2014, 4, 57996–58004 RSC.
- X. J. Chen, B. Wang, P. L. Huang, J. L. Wen and R. C. Sun, RSC Adv., 2016, 6, 45315–45325 RSC.
- T. Y. Chen, B. Wang, X. J. Shen, H. Y. Li, Y. Y. Wu, J. L. Wen, Q. Y. Liu and R. C. Sun, RSC Adv., 2017, 7, 3376–3387 RSC.
- X. Wang, Y. Z. Guo, J. H. Zhou and G. W. Sun, RSC Adv., 2017, 7, 8314–8322 RSC.
- R. Sharma, N. Gogna, H. Singh and K. Dorai, RSC Adv., 2017, 7, 29860–29870 RSC.
- E. Martineau, I. Tea, S. Akoka and P. Giraudeau, NMR Biomed., 2012, 25, 985–992 CrossRef CAS PubMed.
- S. Heikkinen, M. M. Toikka, P. T. Karhunen and I. Kilpeläinen, J. Am. Chem. Soc., 2003, 125, 4362–4367 CrossRef CAS PubMed.
- H. Koskela, I. Kilpeläinen and S. Heikkinen, J. Magn. Reson., 2005, 174, 237–244 CrossRef CAS PubMed.
- M. Sette, R. Wechselberger and C. Crestini, Chem.–Eur. J., 2011, 17, 9529–9535 CrossRef CAS PubMed.
- B. J. Tiegs, S. Sarkar, A. M. Condo, J. I. Keresztes and G. W. Coates, ACS Macro Lett., 2016, 5, 181–184 CrossRef CAS.
- Y. Y. Long, J. Lv, B. X. Li and Y. G. Liu, Polymer, 2021, 229, 123993 CrossRef CAS.
- G. A. Morris, J. Magn. Reson., 1992, 100, 316–328 CAS.
- K. Hu, T. P. Wyche, T. S. Bugni and J. L. Markley, J. Nat. Prod., 2011, 74, 2295–2298 CrossRef CAS PubMed.
- G. A. N. Gowda, F. Tayyari, T. Ye, Y. Suryani, S. Wei, N. Shanaiah and D. Raftery, Anal. Chem., 2010, 82, 8983–8990 CrossRef CAS PubMed.
- D. Jeannerat, Magn. Reson. Chem., 2003, 41, 3–17 CrossRef CAS.
- A. S. Stern, K.-B. Li and J. C. Hoch, J. Am. Chem. Soc., 2002, 124, 1982–1993 CrossRef CAS PubMed.
- K. Bingol and R. Brüschweiler, Anal. Chem., 2014, 86, 47–57 CrossRef CAS PubMed.
- I. A. Lewis, R. H. Karsten, M. E. Norton, M. Tonelli, W. M. Westler and J. L. Markley, Anal. Chem., 2010, 82, 4558–4563 CrossRef CAS PubMed.
- K. Hu, W. M. Westler and J. L. Markley, J. Am. Chem. Soc., 2011, 133, 1662–1665 CrossRef CAS PubMed.
- H. Koskela, O. Heikkilä, I. Kilpeläinen and S. Heikkinen, J. Magn. Reson., 2010, 202, 24–33 CrossRef CAS PubMed.
- P. Giraudeau, G. S. Remaud and S. Akoka, Anal. Chem., 2009, 81, 479–484 CrossRef CAS PubMed.
- G. A. Morris, J. Magn. Reson., 1992, 100, 316–328 CAS.
- A. F. Mehlkopf, D. Korbee, T. A. Tiggelman and R. Freeman, J. Magn. Reson., 1984, 58, 315–323 CAS.
- J. C. J. Barna, E. D. Laue, M. R. Mayger, J. Skilling and S. J. P. Worrall, J. Magn. Reson., 1987, 73, 69–77 CAS.
- D. J. Holland, M. J. Bostock, L. F. Gladden and D. Nietlispach, Angew. Chem., Int. Ed., 2011, 50, 6548–6551 CrossRef CAS PubMed.
- C. M. Thiele and W. Bermel, J. Magn. Reson., 2012, 216, 134–143 CrossRef CAS PubMed.
- M. R. Palmer, C. L. Suiter, G. E. Henry, J. Rovnyak, J. C. Hoch, T. Polenova and D. Rovnyak, J. Phys. Chem. B, 2015, 119, 6502–6515 CrossRef CAS PubMed.
- A. L. Guennec, P. Giraudeau and S. Caldarelli, Anal. Chem., 2014, 86, 5946–5954 CrossRef CAS PubMed.
- Y. Y. Long, Y. X. Wang, B. X. Li, Y. G. Li and Y. S. Li, Polymer, 2019, 169, 185–194 CrossRef CAS.
- J. C. Hoch, M. W. Maciejewski, M. Mobli, A. D. Schuyler and A. S. Stern, Acc. Chem. Res., 2014, 47, 708–717 CrossRef CAS PubMed.
- S. Hyberts, S. Robson and G. Wagner, J. Biomol. NMR, 2013, 55, 167–178 CrossRef CAS PubMed.
- M. Palmer, B. Wenrich, P. Stahlfeld and D. Rovnyak, J. Biomol. NMR, 2014, 58, 303–314 CrossRef CAS PubMed.
- A. Le Guennec, I. Tea, I. Antheaume, E. Martineau, B. Charrier, M. Pathan, S. Akoka and P. Giraudeau, Anal. Chem., 2012, 84, 10831–10836 CrossRef CAS PubMed.
- R. Dass, W. Kozmiński and K. Kazimierczuk, Anal. Chem., 2015, 87, 1337–1343 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08423b |
| This journal is © The Royal Society of Chemistry 2022 |