
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDonor moieties with D–π–a framing modulated electronic and nonlinear optical properties for non-fullerene-based chromophores†
Muhammad Nadeem Arshadab,
Muhammad Khalid *c,
Ghulam shabbirc,
Mohammad Asadab,
Abdullah M. Asiriab,
Maha M. Alotaibia,
Ataualpa A. C. Bragad and
Anish Khanab
*c,
Ghulam shabbirc,
Mohammad Asadab,
Abdullah M. Asiriab,
Maha M. Alotaibia,
Ataualpa A. C. Bragad and
Anish Khanab
aChemistry Department, Faculty of Science, King Abdulaziz University, Jeddah 21589, P.O. Box 80203, Saudi Arabia
bCenter of Excellence for Advanced Material Research (CEAMR), King Abdulaziz University, Jeddah 21589, P.O. Box 80203, Saudi Arabia
cDepartment of Chemistry, Khwaja Fareed University of Engineering & Information Technology, Rahim Yar Khan, 64200, Pakistan. E-mail: khalid@iq.usp.br; muhammad.khalid@kfueit.edu.pk; khalidhej@hotmail.com
dDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, São Paulo, 05508-000, Brazil
First published on 2nd February 2022
Abstract
Herein, a series of non-fullerene-based substantial chromophores (FHD1–FHD6) with a D–π–A framework was designed from a synthesized non-fullerene compound (FH) via structural tailoring with various donor moieties. The FH and its designed derivatives were optimized with frequency analysis at the M06/6-311G (d,p) level to confirm their true minima on potential energy surfaces. These optimized geometries were utilized to perform further analyses, such as absorption, natural bonding orbital (NBO), frontier molecular orbital (FMO), and nonlinear orbital (NLO) analyses at the aforesaid level. Quantum chemical study revealed that all the designed chromophores exhibited a lower band gap than that of the parent molecule with the exception of FHD3. Furthermore, density of states (DOS) analysis supported the findings from the FMO study, and this agreement revealed that the efficient charge was transferred from the HOMO to the LUMO. The NBO investigations disclosed that all the compounds comprised donor moieties with positive charges and acceptors having negative charges. Interestingly, π-conjugated linkers were also found with positive charges, showing an effective donating property. These NBO findings explicated that FHD1–FHD6 exhibited an efficient push–pull mechanism. The λmax values of the designed chromophores were observed to be greater than the reference compound. The average polarizability 〈α〉, first hyperpolarizability (βtot), and second hyperpolarizability 〈γ〉 values of FHD2 were found to be 2.170 × 10−22, 3.150 × 10−27, and 4.275 × 10−32 esu, respectively, while all the other derivatives had been reported in the relevant range. Efficient NLO data revealed that FH-based derivatives may contribute significantly toward NLO technology.
Introduction
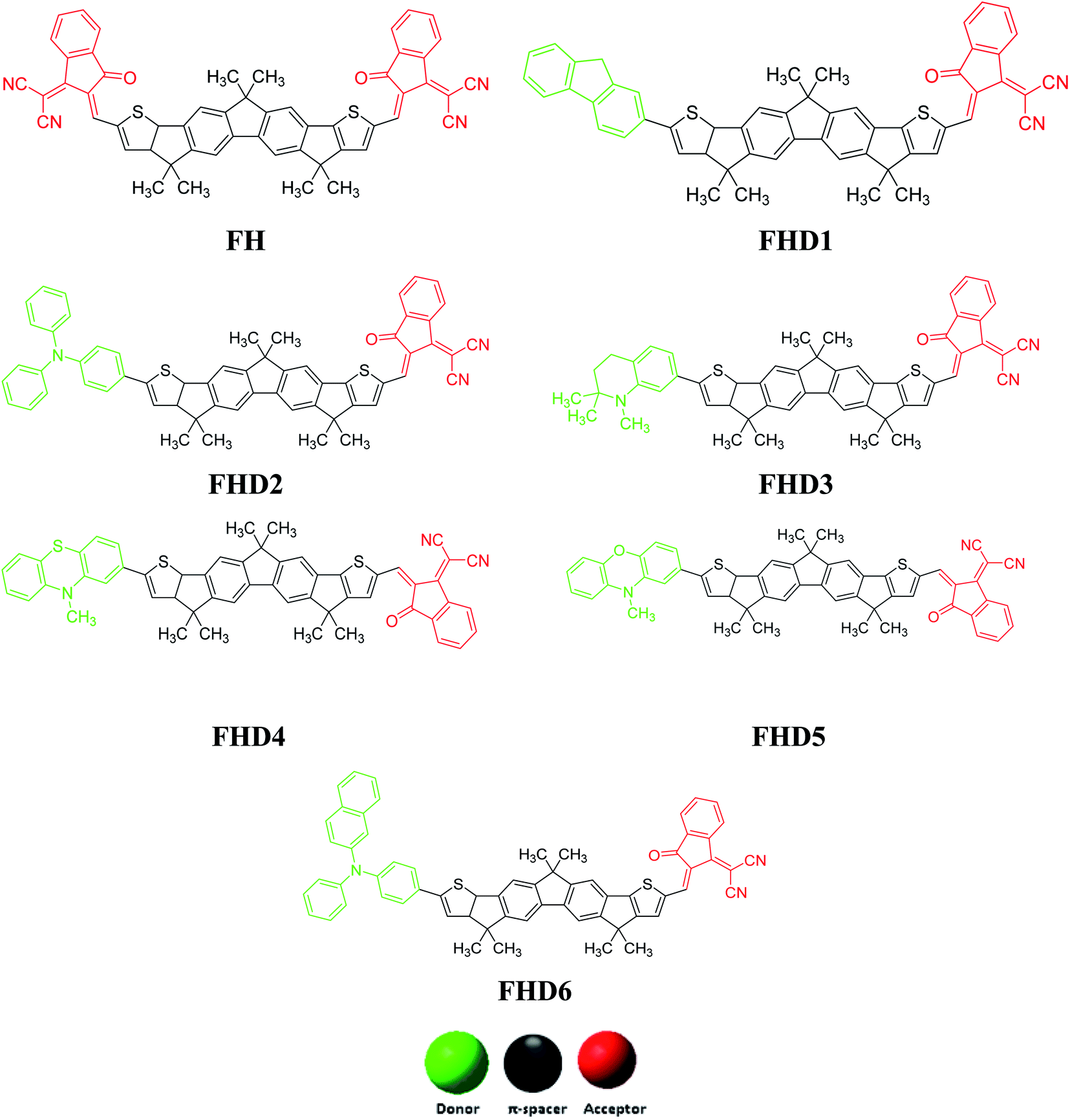
The promising optoelectronic properties of low-cost, facile-synthesized, fused-structured polycyclic aromatic hydrocarbons (PAHs) have been well explored by many researchers in recent years. These molecules (PAHs) are reported to have significant thermal stability and solubility with a facile synthetic procedure.1 Although, research in PAHs has primarily been restricted to their synthesis and photoluminescence processes, it is currently being widened to organic electronics and frequently to their use as building block materials for opto-electronics.2 Moreover, PAHs are also considered as promising scaffolds based on their various properties, including polarization, molecular wires, liquids crystals, organic electronics, and optical applications.3 A literature review revealed that different classes of polycyclic aromatic hydrocarbons have been computed and they have shown promising nonlinear optical (NLO) and electronic properties.1–8 In fact, the above-mentioned materials have attracted attention for their strong two-photon absorption properties in recent years. Their absorption properties may be affected by their molecular weight, symmetry, and solubility parameters. Nowadays, an enormous amount of work is devoted to improving the absorption characteristics via employing the sophisticated approach of molecular designing.9 Organic compounds with extended π-conjugation displaying excellent NLO, fluorescence emission, and one-photon absorption are being extensively employed in two-photon photodynamic treatment,10 light-emitting diodes,11 and 3D optical data storage.12 The designed compounds with proper arrangements of π-conjugation play an important role in improving the NLO response, based on their expanded π-electronic delocalization and enhanced donor to acceptor intramolecular charge transfer via π-spacers.6,13 The induction of conjugated electron acceptor and donor end groups unsymmetrically may extend their electron-accepting or -donating strength, leading to increased absorption. Their facile synthesis along with structural variation have established a decent core element in organic π-conjugation compounds utilized in NLO devices. These compounds with appropriate geometric structures, one/two-photon physical characteristics, energy levels, and thermodynamic properties have been investigated and have attained promising results, indicating that these compounds have potential as NLO optical materials.14Organic materials with established NLO characteristics can enable the modeling of these molecules.15 Suitable donor, π-spacer, and acceptor parts with enhanced conjugation are needed for attaining the appealing architecture (D–π–A) of NLO materials.16 Compounds with good NLO properties have been designed based on theoretical/experimental investigations with the following points in focus: planar D–π–A system, bond length alternation approach, heterocyclic compounds auxiliary acceptors/donors model, and twisted π-electronic arrangements.17,18 In view of the above methodology, NLO responses are simulated upon D or A strength optimization with conjugated bridge enhancement via π-spacer modification. These donor/acceptor electronic associations are sufficient to amplify the oscillator strength of transition matrix substance toward CT transition. Novel D–π–A based non-fullerene compounds were designed that showed a red-shifted absorption spectrum of the type used extensively in organic solar cell (OSC) devices. These acceptors (non-fullerene type) possess a D–π–A structure,19–21 containing several intrinsic benefits, comprising broader absorption spectra as well as charge transportability.19–21 The central part (π) and “D” and “A” moieties may play a key role in D–π–A kinds of molecules,22 and provides appropriate access to tune HOMO/LUMO computations and absorption by altering the central “D” or end group “A” moiety, either separately or all together in which the central “D” moiety performs a crucial role regarding the appropriate molecular packing and its morphology. There are numerous efficient approaches that can be employed to alter the donor D moiety to accomplish enhanced absorption, like conjugation length expansion along with π bridge blocks modifications.23–27
Keeping in view the significant properties of non-fullerene compounds, the compound 2,2′-((2Z,2′Z)-((4,4,7,7,12,12-hexamethyl-7,12-dihydro-4H-thieno[2′′,3′′:1′,2′]indeno [5′,6′:5,6]-s-indaceno[1,2-b]thien-2,9-diyl)bis(methaneylylidene))bis(3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile (FH) was selected as a reference compound for our NLO study, whose synthetic procedure was reported by Nailiang et al. in 2020.2 Such an NLO study for FH and its designed derivatives has not been performed so far. Therefore, a series of compounds named FHD1–FHD6 were designed using FH as the reference molecule. Subsequently, density functional theory (DFT)-based NLO calculations were performed for FH and FHD1–FHD6 for the first time. For the comprehensive understanding of the NLO properties of the aforementioned compounds, theoretical calculations, including the optimization, FMO, TDMs, binding energy (Eb), NBO, density of states (DOS), UV-Vis, and MEP, were performed at the M06 level with the 6-311G(d,p) basis set. The promising DFT-based findings were used to explore the novel designed compounds for their efficient NLO properties. It is expected that the current research work could pay a pivotal role for aiding theoretical as well as experimental researchers in the field of NLO-based technology.
Computational procedure
In this work, different types of electronic analyses, absorption spectra, and NLO parameters of non-fullerene chromophore (FH) and its derivatives were determined by utilizing DFT computations. The quantum chemical studies of the chromophores were performed at the M06/6-311G(d,p) level using the Gaussian 09 program package.28 The NBO 3.1 program package was used for the natural bond orbital (NBO) analysis by applying the above-mentioned level of DFT and the same basis set.29 Time-dependent density functional theory (TDDFT) computations were accomplished at the M06/6-311G (d, p) level for the FMO, MEP, TDM, and UV-Vis spectral investigations. Gauss view 6.0 was used for organizing the input files.30 Moreover, for interpretation of the further results, Avogadro31 and Chemcraft32 were utilized. The dipole moment was determined by the following eqn (1).33| μ = (μx2+ μy2+ μz2)1/2 | (1) |
The average polarizability 〈α〉 was determined by eqn (2).34
| 〈α〉 = 1/3(αxx + αyy + αzz) | (2) |
Analysis of the Gaussian output file yielded 10 hyperpolarizability tensors as oriented along the x, y, and z directions: βxxx, βxyy, βxzz, βyyy, βxxy, βyzz, βzzz, βxxz, βyyz, and βxyz. The magnitude of the total first hyperpolarizability (βtot) was calculated with the help of eqn (3).35
| βtotal = [(βxxx + βxyy + βxzz)2 + (βyyy + βyzz + βyxx)2 + (βzzz + βzxx + βxyz)2]1/2 | (3) |
The second hyperpolarizability was determined by utilizing eqn (4).33
| 〈γ〉 = 1/5[γxxxx + γyyyy + γzzzz +2(γxxxx + γyyyy + γzzzz)] | (4) |
Results and discussion
This quantum chemical study was performed for exploration of the NLO properties of non-fullerene chromophore (FH) and its derivatives. The reference chromophore consisted of an A–π–A architecture, and to a create strong push–pull mechanism in the derivatives one terminal acceptor unit was replaced with a donor moiety. By altering the axial donor (D) moieties while keeping the π-linkers and acceptor unit fixed, structural tailoring of all the designed compounds was performed. Therefore, the designed compounds consisted of: (a) (Z)-2-(2-ethylidene-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile as the acceptor, (b) 3,3,6,6,10,10-hexamethyl-2,3,6,7,8,10-hexahydro-1H-dicyclopenta[b,h]fluorine as the π-linker, and (c) various donor moieties. The different donor moieties used in designing the compounds were: (i) 9H-fluorene, (ii) triphenylamine, (iii) 1,2,2-trimethyl-1,2,3,4-tetrahydroquinoline, (iv) 10-methyl-10H-phenothiazine, (v) 10-methyl-10H-phenoxazine, and (vi) N,N-diphenylnaphthale-2-amine, as shown in Scheme 1. The optimized structures of the compounds are shown in Fig. S1.† | ||
| Scheme 1 Sketch map of the designed compounds with different donor moieties. | ||
The geometries of the chromophores were optimized with analyzing the vibrational frequencies employing the M06/6-311G (d,p) functional. The calculated bond lengths and bond angles are shown in Tables S8–S14.† For FH and FHD1–FHD6, the bond lengths of C1–C2, C1–C6, and C1–C15 were calculated as 1.413, 1.379, and 1.52 Å, respectively. These DFT values of C–C bond lengths were found to be in good agreement with the experimental values of C–C bond lengths.36 Moreover, the bond lengths between the functional group atoms, such as C42–O43 and C50–N51, in FH and FHD1–FHD6 were obtained as 1.213 and 1.156 Å, respectively. All these simulated values of the C–O and C–N bond lengths were in good agreement with the experimentally determined values.36 For the reference compound (FH), the bond angles of C29–C48–C49, C28–C42–O43, C7–C8–C58, and C20–C27–C34 were found to be 123°, 126°, 111°, and 135°, respectively. Moreover, the bond angles of C29–C48–C49, C28–C42–O43, and C7–C8–C58 were examined as 123°, 126°, and 111°, respectively, in all the designed compounds (FHD1–FHD6). The above-mentioned simulated bond angles were observed to be in good agreement with the literature values.37 DFT-based calculations were performed using the optimized geometries of FH and FHD1–FHD6 for exploration of their important quantum chemical properties, such as linear polarizability, first and second hyperpolarizabilities, UV-Vis analysis, frontier molecular orbital (FMO), natural bond orbital (NBO), density of states (DOS), transition density matrix (TDMs), and molecular electrostatic potential (MEP) (Fig. 1).
 | ||
| Fig. 1 The 2D structures of FH and its derivatives. | ||
Electronic structures
The frontier molecular orbital (FMO) theory is an effective tool for determination of the chemical stability, light absorbance, and optical and electronic properties of chromophores.27,38 The electron-accepting property is determined by the LUMO, while the electron-donating property is determined by the HOMO. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecules orbital (LUMO) analysis plays a vital role in determining the UV-Vis spectra and different types of reaction mechanisms. The parameters for prediction of the chemical reactivity and dynamic stability of molecules can be determined by the HOMO–LUMO energy gap.39 Molecules with a higher value of the HOMO–LUMO energy gap are regarded as chemically stable and may resist to chemical changes,40 while molecules with a higher degree of softness are considered to have less stability and exhibit a low HOMO–LUMO energy.41 Moreover, molecules with a lower HOMO/LUMO energy gap usually show high polarizability and as a consequence yield a promising NLO response.42 The EFMOs of FH and FHD1–FHD6 were calculated at the M06/6-311G (d,p) level of theory and their results are tabulated in Table 1. The other values, like EHOMO−1, ELUMO+1, EHOMO−2, and ELUMO+2, are collected in Table S15.†| Compounds | LUMO | HOMO | Band gap |
|---|---|---|---|
| a Units of EHOMO, ELUMO, and energy gaps are shown in eV. | |||
| FH | −3.37 | −5.930 | 2.560 |
| FHD1 | −3.214 | −5.583 | 2.369 |
| FHD2 | −3.211 | −5.425 | 2.214 |
| FHD3 | −3.212 | −5.417 | 2.205 |
| FHD4 | −3.216 | −5.533 | 2.317 |
| FHD5 | −3.212 | −5.420 | 2.208 |
| FHD6 | −3.208 | −5.515 | 2.307 |
The band gaps calculated for the reference chromophores was 2.560 eV with −5.930 and −3.37 eV, HOMO/LUMO energies. The data from Table 1 reveal that all the designed chromophores showed a comparable value of energy gap with the parent molecule. Nevertheless, the lowest energy difference between orbitals was seen in FHD3 (2.205 eV), which may be due to the 1,2,2-trimethyl-1,2,3,4-tetrahydroquinoline D moiety. The donor moiety in FHD3 has three electron-donating methyl (–CH3) groups along with the right conjugation (benzene ring) for creating a strong electron-donating effect with a powerful push–pull architecture (D–π–A), hence leading to a reduction in the band gap. The increasing band gap order of the compounds was FHD3 < FHD5 < FHD2 < FHD6 < FHD4 < FHD1 < FH. The accompanying energies of the orbitals and charge-transfer phenomenon between the orbitals for the studied chromophores is also explained by the FMO investigation, as shown in Fig. 2. In the reference chromophore, the charge density for the HOMO was concentrated largely over the π-bridge and a smaller portion over some atoms of the terminal acceptor unit, while the LUMO was located all over the molecule. Nevertheless, in the derivatives for the HOMO, the electron density mostly occurred on the donor moieties and partially over the π-conjugated linkers. In the LUMO case, the electron density was almost totally presented over the acceptor moieties and a little bit on the π-linkers, which indicated an excellent transfer of charge from D toward A by the π-spacer.
 | ||
| Fig. 2 HOMOs and LUMOs of the studied compounds. | ||
Density of states analysis
The delocalization of electrons exhibited in the HOMO and LUMO orbitals was also supported by a DOS investigation.43 For the studied chromophores, the DOS investigations were performed at the M06/6-311G (d,p) functional and the resulting graphs are illustrated in Fig. 3. As presented in Fig. 2 and 3, charge spreading of the electronic cloud was altered around the LUMO and HOMO because of the strong push and electron withdrawing architecture of the derivatives. As the reference molecule has A–π–A, to understand the DOS, FH was divided into two parts, namely the acceptor and π-linkers, while due to the D–π–A configuration of the derivatives, they were divided into three parts: donor, π-linkers, and acceptor, which are shown by green, blue, and red lines, respectively, in the DOS maps.44 | ||
| Fig. 3 DOS plots of the reference FH and designed compounds. | ||
In the reference compound FH, the acceptor contributes 24.6% to the HOMO and 62.7% for the LUMO. Similarly, π-linkers contributed 75.4% and 37.3% for the HOMO/LUMO, respectively. Among the derivatives, the acceptor contributions to the HOMO were 5.4%, 2.6%, 2.5%, 3.7%, 1.8%, and 3.9%, while the LUMO contributions were 62.7%, 49.6%, 65.9%, 65.8%, 62.3%, 66.0%, and 65.9% from FHD1–FHD6, respectively. In the same way, the π-linkers showed contributions of 68.2%, 40.6%, 40.4%, 68.1%, 50.2%, and 57.6% for the HOMO and 49.2%, 33.5%, 33.6%, 37.6%, 33.8%, and 33.9% for the LUMO from FHD1–FHD6, accordingly. These contributions show that the designing of different compounds by varying different efficient donor moieties play a significant role in the transmittance of the electronic charge in different ways. By these outcomes, it could be examined from the DOS graphs that the HOMOs of all the designed chromophores were largely concentrated on the donor and π-spacers as higher peaks of green and blue color, which were located at nearly −5.6 eV. Similarly, the LUMOs were significantly located on A and minorly on π-spacers in all the derivatives as higher peaks located near 6.5 eV, hence, these graphs significantly support the FMO diagrams (see Fig. 3 and 4). The DOS calculation showed that the charge was efficiently transferred from HOMO to LUMO in all the designed compounds from donor to acceptor through the π-spacers.
 | ||
| Fig. 4 TDM graphs of the studied compounds (FH and FHD1–FHD6). | ||
UV-visible analyses
TD-DFT computations were carried out using the conductor-like polarizable continuum model (CPCM) model34 in dichloromethane solvent at the M06 level with the 6-311G (d,p) basis set in order to have a better understanding of the absorption data.45 All the UV-Visible based parameters of FH and FHD1–FHD6 are collected in Table 2, while the absorption spectra are displayed in Fig. S8.†| Compounds | DFT λ (nm) | E (eV) | fos | MO contributions |
|---|---|---|---|---|
| a Units in eV. | ||||
| FH | 650.46 | 1.91 | 3.25 | H → L (92%) |
| FHD1 | 661.96 | 1.87 | 1.98 | H → L (88%), H-1 → L (8%) |
| FHD2 | 683.22 | 1.81 | 1.71 | H → L (83%), H-1 → L (13%) |
| FHD3 | 683.89 | 1.81 | 1.70 | H → L (82%), H-1 → L (14%) |
| FHD4 | 661.60 | 1.87 | 1.86 | H → L (78%), H-1 → L (16%) |
| FHD5 | 676.06 | 1.83 | 1.60 | H → L (75%), H-1 → L (20%) |
| FHD6 | 669.24 | 1.85 | 1.81 | H → L (80%), H-1 → L (13%) |
The absorption data from the above table indicated that all the studied chromophores showed absorption in the visible region of the spectrum. The λmax calculated for the reference molecule was 650.46 nm with 1.91 eV excitation energy. All the derivatives with D–π–A structures exhibited a higher value of the maximum absorption (661.60–683.89 nm) with a lower excitation energy (1.87–1.81 eV) than that of FH due to the strong push–pull configuration. Among the designed chromophores, the lowest values of λmax were observed for FHD1 and FHD4 as 661.95 and 661.60 nm, with 1.97 and 1.86 fos, respectively. These λmax values were further enhanced to 669.24 nm with 1.816 fos in FHD6 then 676.07 nm with 1.60 fos in FHD5. Table 2 shows that FHD2 and FHD3 had the maximum red-shifted values at 683.89 nm with 1.70 fos and 683.22 nm with 1.71 fos, respectively, among all the designed novel compounds. This might be due to the presence of the benzene ring and three electron-donating (methyl) groups as the D moiety in FHD3 and TPA, which acts as an internal donor unit in a number of hole-transporting amorphous materials due to its significant electron-donating ability in FHD2. These strong D moieties create a strong architecture of pull–push mechanism, which lowers the band gap and broadens the absorption spectra, as shown in Fig. S8† and Table 2. Increased values of the wavelengths were found in the following order: FH < FHD4 < FHD1 < FHD6 < FHD5 < FHD2 < FHD3. A graph was plotted between the wavelengths and molar absorptivity constant values of the studied chromophores and is shown in Fig. S8.† Throughout, this study shows that the absorption spectra of all the designed compounds were obtained reasonably broader as compared to FH, indicating that the D–π–A architecture created a strong pull–push mechanism in the derivatives, which lowered the band gap and allows them to be exploited as promising NLO-active compounds.
Global reactivity parameters
Numerous reactivity parameters were calculated using eqn (S1)–(S7)† and they were determined by FMO study (Table 3).46–49| Compounds | IP | EA | X | H | μ | ω | σ |
|---|---|---|---|---|---|---|---|
| a Units in Hartree (Eh). | |||||||
| FH | 0.217 | 0.123 | 0.170 | 0.047 | −0.170 | 0.310 | 10.629 |
| FHD1 | 0.205 | 0.118 | 0.162 | 0.043 | −0.161 | 0.300 | 11.486 |
| FHD2 | 0.199 | 0.118 | 0.159 | 0.040 | −0.158 | 0.309 | 12.291 |
| FHD3 | 0.199 | 0.118 | 0.159 | 0.041 | −0.158 | 0.310 | 12.340 |
| FHD4 | 0.203 | 0.118 | 0.161 | 0.042 | −0.161 | 0.303 | 11.744 |
| FHD5 | 0.199 | 0.118 | 0.159 | 0.041 | −0.159 | 0.310 | 12.324 |
| FHD6 | 0.202 | 0.117 | 0.160 | 0.042 | −0.160 | 0.303 | 11.795 |
The energy required to withdraw an electron from the HOMO orbital is equivalent to the ionization potential by which the electron-donating and -accepting abilities of an atom can be calculated.50 The electronegativity is a chemical property by which the tendency of interaction of the electronic cloud coming toward an atom is expressed.48 The chemical potential of a species helps in understanding the stability and reactivity of the compound. The above data revealed that all the studied chromophores exhibited higher values of ionization potential with lower electron-affinity values, which indicated their electron-acceptance nature. Further, the ionization potential, electron affinity, electronegativity, and global electrophilicity are also related to Egap. Among all the chromophores, FHD3 exhibit the lowest value of ionization potential, which supported its low Egap.
The global softness of a molecule refers to the extent of softness of a molecule and is inversely related to the bandgap, i.e. the smaller the bandgap, the softer the molecule will be in nature. Softness of the molecule can also be linked to its reactivity, as a lower bandgap is exhibited by soft molecules, hence their reactivity is greater.51 Polarizability can also be correlated to the softness, as soft molecules are considered as more polarized. Among the entire designed compounds, the largest value of the softness, i.e.12.340 Eh, was exhibited by FHD3, which showed maximum polarizability along its enhanced reactivity. This value of softness was reduced to 12.324 Eh in FHD5. Further drops in the value were observed in the case of FHD2, FHD6, and FHD4 of 12.291, 11.795, and 11.744 Eh, respectively. The bottom-most value of softness represents the least reactivity with less polarizability and was calculated in FHD1 (11.486 Eh). The decreased order of the softness as examined was: FHD3 > FHD5 > FHD2 > FHD6 > FHD4 > FHD1 > FH. Among all the compounds, the maximum value of hardness (0.047 Eh) was observed for FH, while the minimum value was found to be 0.040 Eh in FHD2. Similar to the hardness, the chemical potential value also seemed to decline in the same manner. Nevertheless, all the studied chromophores had a higher value of global softness (σ = 10.629–12.324 Eh) with the least global hardness (η = 0.040–0.042 Eh). These higher values of softness indicated the higher chemically reactivity of the studied molecules. From all this information, it can be interpreted that the designed chromophores might be active and have substantial NLO properties.
Natural bond orbital (NBO) analysis
Natural bonding orbital (NBO) analysis is frequently used to examine bond interactions and the intermolecular charge transfer (ICT) between orbitals.52 NBO is also used to predict the charge transfer between donor and acceptor moieties in D–π–A type organic compounds by calculating the natural charges.53 Therefore, NBO calculations were performed at the M06/6-311G (d, p) level using the optimized structures of FH and FHD1–FHD6 and their results in terms of natural charges for the donor, π-spacer, and acceptor segments are tabulated in Table 4.| Compounds | Donors | π-Linkers | Acceptors |
|---|---|---|---|
| FHD1 | 0.0836 | 0.0736 | −0.1572 |
| FHD2 | 0.0880 | 0.0707 | −0.1587 |
| FHD3 | 0.0871 | 0.0715 | −0.1587 |
| FHD4 | 0.0764 | 0.0790 | −0.1555 |
| FHD5 | 0.0898 | 0.0674 | −0.1572 |
| FHD6 | 0.1085 | 0.0516 | −0.1601 |
In FH, the acceptor, donor, and acceptor architecture exists, so the natural charges were calculated to be −0.1284, 0.2568, and −0.1284, respectively. The reference compound consisted of an A–D–A configuration, whereas the designed compounds were found to have an effective D–π–A configuration. From Table 4, it could be determined that the positive values of the donor moieties with regard to the NBO charges reveal that all the donors used for the studied compounds have excellent donor properties. Further, the acceptor moieties also have effective withdrawing capabilities in the studied compounds. Moreover, the π-conjugated linkers were determined to have positive values, which show their ability to facilitate charge transfer from the donor to acceptor moieties without any type of charge trapping. Among all the designed compounds, the highest positive value of donors was exhibited by FHD6 as 0.108508. This may be due to the π-electronic cloud and presence of naphthalene as a substituent on the D unit (amine, N,N-diphenylnaphthalen-2-amine). Furthermore, the detailed possible transitions were also calculated at the same level and the representative transitions of all the compounds are tabulated in Table 5.
| Compounds | Donor(i) | Type | Acceptor(j) | Type | E(2) |
|---|---|---|---|---|---|
| FH | C28–C30 | π | C32–C53 | π* | 31.51 |
| C70–N71 | π | C72–N73 | π* | 0.74 | |
| C32–H33 | σ | C30–S31 | σ* | 10.29 | |
| C30–S31 | σ | C30–C32 | σ* | 0.51 | |
| S31 | LP(2) | C28–C30 | π* | 22.51 | |
| S31 | LP(1) | C12–C21 | σ* | 1.05 | |
| FHD1 | C24–C25 | π | C31–C39 | π* | 32.47 |
| C50–N51 | π | C48–N49 | π* | 0.77 | |
| C31–H32 | σ | C24–S27 | σ* | 10.41 | |
| C86–H87 | σ | C86–C88 | σ* | 0.51 | |
| S27 | LP(2) | C18–C20 | π* | 29.92 | |
| S30 | LP(1) | C12–C21 | σ* | 0.95 | |
| FHD2 | C24–C25 | π | C31–C39 | π* | 32.52 |
| C50–N51 | π | C48–N49 | π* | 0.77 | |
| C31–H32 | σ | C24–S27 | σ* | 10.42 | |
| S30–C85 | σ | C22–C23 | σ* | 0.52 | |
| S27 | LP(2) | C | π* | 29.93 | |
| S30 | LP(1) | C12–C21 | σ* | 0.95 | |
| FHD3 | C24–C25 | π | C31–C39 | π* | 32.54 |
| C44–O45 | π | C31–C39 | π* | 0.74 | |
| C31–H32 | σ | C24–S27 | σ* | 10.44 | |
| C113–H114 | σ | C111–C113 | σ* | 0.50 | |
| S27 | LP(2) | C18–C20 | π* | 29.96 | |
| S30 | LP(1) | C12–C21 | σ* | 0.94 | |
| FHD4 | C24–C25 | π | C32–C40 | π* | 32.42 |
| C28–C30 | π | C87–C88 | σ* | 0.74 | |
| C32–H33 | σ | C24–S27 | σ* | 10.42 | |
| C84–N96 | σ | C97–H100 | σ* | 0.5 | |
| S27 | LP(1) | C2–C18 | σ* | 29.96 | |
| S101 | LP(2) | C83–C89 | π* | 0.89 | |
| FHD5 | C24–C25 | π | C32–C40 | π* | 32.48 |
| C49–N50 | π | C51–N52 | π* | 0.73 | |
| C32–H33 | σ | C24–S27 | σ* | 10.43 | |
| C19–C73 | σ | C73–H76 | σ* | 0.60 | |
| N97 | LP(1) | C84–C85 | π* | 36.44 | |
| O96 | LP(2) | C80–C81 | σ* | 0.68 | |
| FHD6 | C24–C25 | π | C32–C40 | π* | 32.62 |
| C51–N52 | π | C49–N50 | π* | 0.76 | |
| C32–H33 | σ | C24–S27 | σ* | 10.45 | |
| C96–H99 | σ | C77–N88 | σ* | 0.51 | |
| N88 | LP(1) | C80–C81 | π* | 41.21 | |
| N88 | LP(1) | C79–C80 | σ* | 0.51 |
The detailed NBO transitions of the reference and designed compounds are also shown in Tables S16–S22.†
Some dormant π→π* interactions can be noted like: π(C28–C30)→π*(C32–C53), π(C24–C25)→π*(C31–C39), π(C24–C25)→π*(C31–C39), π(C24–C25)→π*(C31–C39), π(C24–C25)→π*(C32–C40), π(C24–C25)→π*(C32–C40) and π(C24–C25)→π*(C32–C40) having greater stabilization energies at 31.51, 32.47, 32.52, 32.54, 32.42, 32.48, and 32.62 kcal mol−1 in FH–FHD6, respectively. Moreover, some other kinds of significant π→π* donor–acceptor intra-molecular charge-transfer (ICT) interplay were also studied, as shown in Tables S16–S22.† The lowest values of stabilization were determined as 0.74, 0.77, 0.77, 0.74, 0.74, 0.73, and 0.76 kcal mol−1 for π (C70–N71)→π*(C72–N73), π(C50–N51)→π*(C48–N49), π(C50–N51)→π*(C48–N49), π(C44–O45)→π*(C31–C39), π(C28–C30)→π*(C87–C88), π(C49–N50)→π*(C51–N52), and π(C51–N52)→π*(C49–N50) transitions in FH–FHD1, respectively (see Tables S16–S22†).
In σ→σ* transitions, larger values of stabilization energies were found to be 10.29, 10.41, 10.42, 10.44, 10.42, 10.43, and 10.45 kcal mol−1 for σ(C32–H33)→σ*(C30–S31), σ (C31–H32)→σ*(C24–S27), σ(C31–H32)→σ* (C24–S27), σ(C31–H32)→σ* (C24–S27), σ (C32–H33)→σ*(C24–S27), σ (C32–H33)→σ* (C24–S27), and σ (C32–H33)→σ* (C24–S27) interactions in FH–FHD6, respectively. Furthermore, some other transitions: σ(C30–S31)→σ*(C30–C32), σ(C86–H87)→σ*(C86–C88) σ(S30–C85)→σ*(C22–C23), σ(C113–H114)→σ*(C111–C113), σ(C84–N96)→σ*(C97–H100), σ(C19–C73)→σ*(C73–H76) and σ(C96–H99)→σ*(C77–N88) with the lowest stabilization energies of 0.51, 0.51, 0.52, 0.50, 0.50, 0.60, and 0.51 kcal mol−1, respectively, were also obtained for FH–FHD6, as shown in Tables S16–S22.† These least energy values were seen due to the emergence of weak interaction between the accepter and donor moieties.
Similar kinds of interaction were also investigated in accordance with resonance. LP2(S31)→π*(C28–C30), LP2(S27)→π*(C18–C20), LP2(S27)→π*(C18–C20), LP2(S27)→π*(C18–C20), LP1(S27)→σ*(C2–C18), LP1(N97)→π*(C84–C85) and LP1(N88)→ π* (C80–C81) with high stabilization energies, i.e. 22.51, 29.92, 29.93, 29.96, 29.96, 36.44, and 41.21 kcal mol−1 for FH–FHD6, respectively. On the other hand, LP1(S31)→σ*(C12–C21), LP1(S30)→σ*(C12–C2), LP1(S30)→σ*(C12–C2), LP1(S30)→σ*(C12–C2), LP2(S101)→ π*(C83–C89), LP2(O96)→σ*(C80–C81), and LP1(N88)→σ*(C79–C80) produced energies of 1.05, 0.95, 0.95, 0.95, 0.89, 0.68, and 0.51 kcal mol−1, respectively which exhibited the lowest electron-donating interaction energies in FH–FHD6 (Tables S16–S22†). The studied chromophores had the highest stability in the span of 32.62 to 0.17 kcal mol−1 for π→π* transitions owing to the extended hyper-conjugation. The increasing stability order of the compounds was noted as: FH < FHD4 < FHD1 < FHD5 < FHD2 < FHD3 < FHD6. All the designed compounds showed a greater stability than the reference compound (Table 5).
TDMs and exciton binding energy (Eb)
TDM analysis is used to determine the charge transfer from a donor to acceptor through a π-bridge.54 TDM analysis of the studied chromophores was performed to investigate the behavior of the transitions, essentially from the ground state (S0) to an excited state (S1) and the interaction between the donor and acceptor units. For this purpose, the atoms were divided into segments according to their role, i.e. donor (D), π-bridge (π), and acceptor (A). Hydrogen atoms were neglected by default as they do not have much involvement in charge transfer. The TDM heat maps for FH and FHD1–FHD6 are shown in Fig. 4. The DTM heat maps showed the efficient diagonal charge transfer coherence in all the studied chromophores. The larger portion of the electronic cloud was located over the π-bridge, which then successfully transferred through the π-bridge to the acceptor unit, without charge trapping. These heat maps also supported the DOS and FMO investigations, where the same phenomenon of charge transfer was examined. The binding energy is a functional key factor to determine the optoelectronic properties of compounds. The lower the binding energy, the lower the coulombic forces between the hole and electron, in which the excited state provides enhanced exciton dissociation. Actually the binding energy is calculated by the difference between the HOMO–LUMO energy gap and the first exciton energy. The binding energy is calculated by eqn (5) and the results are tabulated in Table 6.| Eb = EH–L − Eopt | (5) |
| Compounds | EH–L (eV) | Eopt (eV) | Eb (eV) |
|---|---|---|---|
| FH | 2.56 | 1.906 | 0.654 |
| FHD1 | 2.369 | 1.873 | 0.496 |
| FHD2 | 2.214 | 1.815 | 0.399 |
| FHD3 | 2.205 | 1.812 | 0.393 |
| FHD4 | 2.317 | 1.874 | 0.443 |
| FHD5 | 2.208 | 1.833 | 0.375 |
| FHD6 | 2.307 | 1.852 | 0.455 |
From Table 6, it is clear that the changing of the donor moieties plays a key role in the good binding energies. The binding energy of the reference compound was noted as 0.654 eV. The Eb values of compounds FHD1–FHD6 were 0.496, 1.497, 0.399, 0.393, 0.443, 0.375, and 0.455 eV, respectively. The binding energy values were noted to be in the increasing order of: FHD5 < FHD3 < FHD2 < FHD4 < FHD6 < FHD1 < FH. Note, molecules having an Eb lower than 1.9 eV are considered as perfect photonic materials with significant NLO responses. Interestingly, our studied chromophores exhibited binding energies smaller than 1.9 eV, illustrating them as perfect NLO materials.
Nonlinear optics (NLO)
NLO compounds are vastly used in the fields of optoelectronics and telecommunication. The optical properties of the molecules are characterized by their electronic analyses of the studied molecule, which supports the nonlinear response (hyperpolarizabilities:β and γ) and linear response (polarizability, α).55 Moreover, NLO compounds have been extensively used in optical switches, signal processing, optical memory devices, and communication technology.56 Here, the reference compound was reported originally with an A–D–A configuration, which showed a smaller NLO response compared to the designed compounds. The designed compounds (FHD1–FHD6) were found with D–π–A configuration, which was designed by the alteration of the donor moieties and exhibited an excellent push–pull architecture. The effective changes in the band gap and polarizability values were observed by the alternation of different donor moieties.The electronegativity difference is an effective factor to produce the dipole moment in a molecule. The dipole moment (μ) is directly related with the polarity of a system. The greater the electronegative difference in the molecules, the larger will be the dipole moment values. The μtotal value of the reference compound (FH) was reported as 6.558 D, with tensor values along the x, y, and z axes of 0.0004, 6.5575, and 0.0014 D, respectively. Data from Table 7 show that all the derivatives expressed larger dipole moment values, which indicated that these chromophores have greater polarity than their parent molecule. Among the derivatives, the least value of μtotal was found as 9.352 D in FHD4, which increased to 10.203 and 10.846 D in FHD5 and FHD1, respectively. Further enhancements in the dipole moment were noted as 11.3051, 11.5731, and 13.3253 D in FHD3, FHD2, and FHD6, respectively. The dipole moment values of the investigated compounds were found in decreasing order as: FHD6 > FHD5 > FHD2 > FHD3 > FHD1 > FHD4 > FH (Table 7). Similarly, the linear polarizability 〈α〉 parameter is also considered to be a key factor to determine the electronic properties of organic compounds. For the FH, the value of 〈α〉 was found to be 2.270 × 10−22 esu with 4.511 × 10−22, 1.723 × 10−22, and 5.755 × 10−23 esu along the αxx, αyy, and αzz, respectively. All the designed chromophores exhibited comparable values of linear polarizability in the range of (1.886 × 10−22–2.270 × 10−22 esu) with their parent compound. The FHD3 chromophore exhibited a larger value of linear polarizability [〈α〉 = 2.178 × 10−22 esu] among all the derivatives. The average polarizability in descending order of the investigated molecules was observed as: FH > FHD3 > FHD2 > FHD4 > FHD1 > FHD5 > FHD6.
| Compounds | μtotal | 〈α〉 × 10−22 | βtotal × 10−27 | 〈γ〉 × 10−32 |
|---|---|---|---|---|
| a Units of the dipole moment (μ) in Debye (D) while 〈α〉, (β) and 〈γ〉 are in esu. | ||||
| FH | 6.558 | 2.270 | 1.689 | 3.111 |
| FHD1 | 10.846 | 2.021 | 2.620 | 3.245 |
| FHD2 | 11.573 | 2.170 | 3.150 | 4.275 |
| FHD3 | 11.305 | 2.178 | 3.000 | 4.103 |
| FHD4 | 9.352 | 2.053 | 2.576 | 3.133 |
| FHD5 | 10.203 | 2.018 | 2.776 | 3.460 |
| FHD6 | 13.325 | 1.886 | 2.592 | 2.893 |
Dipole polarizabilities have been found to be a quantitative estimation for assessing the attractive NLO features of species. For instance, the βtotal and 〈γ〉 endorsed the estimation of the NLO response of the chromophores, which is then related with the ICT from the D moiety toward an A unit via π-linkers.53 Such kind of interactions for our compounds are displayed in Fig. 2. The interaction of the electron density with the external field increased the dipole moment, resulting in increased first-order hyperpolarizability. The reference compound (FH) exhibited a value of βtotal as 1.689 × 10−27 esu, with the major contributing tensor values of βxxx, βxxy, βyyz, βxzz, βyzz, and βzzz as 5.841 × 10−32, −1.852 × 10−28, −1.918 × 10−33, −5.946 × 10−35, 2.111 × 10−31, and −3.321 × 10−34 esu., respectively. Interestingly all the designed chromophores expressed larger responses for the second-order polarizabilities (β) as compared to their parent compound. This might be due to that all our derivatives have a D–π–A configuration with a strong pull–push architecture as compared to the parent chromophore, which exhibited an A–π–A structure. This pull–push architecture might decrease the band gap between the orbitals, increase the polarity, and enhance the ICT. Among all the derivatives, a larger value of βtotal was examined in FHD2 (3.150 × 10−27 esu), which then decreased to 3.000 × 10−27 esu in FHD3, and then declined to 2.776 × 10−27 esu in FHD5 and further reduced to 2.620 × 10−27 and 2.592 × 10−27 esu in FHD1 and FHD6, respectively. The minimum value of βtotal was studied in FHD4 as 2.576 × 10−27 esu among all the derivatives. The hyperpolarizability (β) with a descending order for the investigated molecules was observed as: FHD2 > FHD3 > FHD5 > FHD6 > FHD1 > FHD4 > FH. The detailed values of the dipole moment, linear polarizability, and hyperpolarizability of all the compounds along the x, y, and z axes are tabulated in Tables S30 and S31.†
The second-order hyperpolarizability (γ) also plays an important role in NLO compounds. The reference compound (FH) showed a value of second-order hyperpolarizability as 3.111 × 10−32 esu, with tensor values of 3.095 × 10−32, 1.452 × 10−34, and 6.125 × 10−36 esu along the γxx, γyy, and γzz axes, respectively. Among all the studied compounds, the highest second-order hyperpolarizability was observed in FHD2 with tensors γxx, γyy, and γzz as 4.259 × 10−32, 1.467 × 10−34, and 7.996 × 10−36 esu, respectively. The lowest value of second-order hyperpolarizability was noted in FHD6 with tensor values of 2.884 × 10−32, 7.584 × 10−35, and 9.571 × 10−36 esu along the γxx, γyy, and γzz axes, respectively. The values of second-order hyperpolarizability (γ) with descending order of molecules was: FHD2 > FHD3 > FHD5 > FHD1 > FHD4 > FH > FHD6. All the tensors values of the second-order hyperpolarizability (γ) values of all the compounds are collected in Table S30.† A literature review disclosed that the energy gap value influences the polarizability. A smaller bandgap and higher polarizability are clearly associated with stronger hyperpolarizabilities, as evidenced by the substantial NLO values.36 In our studied chromophores, an inverse relationship was observed between the bandgap and NLO response. Among the derivatives, larger values of first-order hyperpolarizability and second-order polarizability were examined in FHD5, FHD3, and FHD2 [(βtotal = 2.77 × 10−27, 3.00 × 10−27, and 3.150 × 10−27 esu) (〈γ〉 = 3.46 × 10−32, 4.103 × 10−32, and 4.275 × 10−32 esu)], which expressed a narrow band gap (2.208, 2.205, and 2.214, eV). The decrease in NLO values [(βtotal = 2.620 × 10−27, 2.576 × 10−27, and 2.592 × 10−27 esu) (〈γ〉 = 3.245 × 10−32, 3.133 × 10−32, and 2.893 × 10−32 esu)] was been noted as the band gap between orbitals was enhanced (2.369, 2.317, and 2.307 eV) in FHD1, FHD4, and FHD6, respectively.
Fantastically, it was also found that the designed chromophores exhibited a larger value of hyperpolarizability when compared with the standard para-nitroaniline (p-NA). The reported values for the first- and second-order hyperpolarizabilities were 6.46 × 10−30 and 7.92 × 10−36 esu, respectively.57 Interestingly, in all our chromophores, the effective NLO behavior, as three-order magnitude for βtotal and four-order magnitude for 〈γ〉, was investigated. This study concluded that by structural tailoring with efficient donor moieties, significant NLO materials could be obtained.
Molecular electrostatic potential (MEP)
MEP mapping is used to visualize the total electron density in a three dimensional framework of systems. The MEP plot performs quite well for its function to explain compound reactivity by predicting various electrophilic and nucleophile sites in chromophores.58 The scope of the electrostatic potential in MEP maps is described by different standard colors, namely white (neutral), red (negative), and blue (positive), to denote the electrostatic potential regions,59 and their pictorial demonstration is shown in Fig. 5. | ||
| Fig. 5 MEP diagrams of the reference FH and designed compounds FHD1–FHD6. | ||
In all the studied chromophores, a negative potential was centered over the oxygen atoms, which can be seen in Fig. 5. The blue color is mainly seen over carbon and hydrogen atoms, signaling the nucleophiles can approach these locations. The green area, which is localized across hydrogen atoms, indicates the average of two absolute values of electrostatic potential (red and blue) and is situated over the S atom in the investigated compounds.
Conclusion
In the present work, a series of chromophores (FHD1–FHD6) with a D–π–A framework was designed via varying the donor moieties consisting of promising electron-donating properties and a fixed π-bridge as well as acceptor groups. The obtained electronic properties showed that the donors have a positive effect on the D–π–A architectures, changing the intact properties of the designed molecules more than the synthetic compound. According to FMO analyses, HOMOs were found over the donor group and its neighboring π-linker, whereas LUMOs were found over the acceptors in all the compounds. The HOMO–LUMO energy gap was reduced from 2.369 eV to 2.205 eV for FHD1–FHD6, respectively. The HOMO–LUMO energy gap of FHD3 was found as 2.205 eV, which was the least among the designed derivatives. DOS and TDM investigations supported the FMO study as a significant ICT was found from the D moiety toward the A unit through the π-spacer. The NBO findings exploited a strong push–pull architecture in the designed chromophores as the donor and π-conjugates expressed positive values, while all the acceptors exhibited negative values. Moreover, the NBO study also revealed that the conjugation in the molecules led to a greater stability of the systems. Incredibly, lower binding energies (0.375–0.496 eV) with greater μtotal (6.558–13.325 D) and global softness (σ = 10.630–12.340 Eh) for the studied molecules were obtained, which predicted higher excitation, dissociation, and larger polarity. Interestingly, significant NLO findings as 〈α〉, βtot, and 〈γ〉 values were found as 2.170 × 10−22, 3.150 × 10−27, and 4.275 × 10−32 esu, respectively, for FHD2, which were greatest among all the studied chromophores. We can conscript the promising capabilities of our designed compounds for indicating a better NLO response. The FHD1–FHD6 derivatives may attract the attentions of researchers in terms of use in new synthetic approaches. Moreover, from our above-mentioned analyses, it can be concluded that the aforementioned D–π–A based organic NLO compounds may have promising applications in the NLO field.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was funded by Institutional Fund Projects under grant no (IFPRC-102-130-2020). Therefore, authors gratefully acknowledge technical and financial support from the Ministry of Education and King Abdul aziz University, Jeddah, Saudi Arabia.References
- Y. Wang, G. S. He, P. N. Prasad and T. Goodson, Ultrafast dynamics in multibranched structures with enhanced two-photon absorption, J. Am. Chem. Soc., 2005, 127, 10128–10129 CrossRef CAS PubMed.
- F.-X. Wang, M.-H. Chen, X.-Y. Hu, R.-R. Ye, C.-P. Tan, L.-N. Ji and Z.-W. Mao, Ester-modified cyclometalated iridium (III) complexes as mitochondria-targeting anticancer agents, Sci. Rep., 2016, 6, 1–15 CrossRef CAS PubMed.
- G. Walters, B. R. Sutherland, S. Hoogland, D. Shi, R. Comin, D. P. Sellan, O. M. Bakr and E. H. Sargent, Two-photon absorption in organometallic bromide perovskites, ACS Nano, 2015, 9, 9340–9346 CrossRef CAS PubMed.
- E. Walker and P. M. Rentzepis, A new dimension, Nat. Photonics, 2008, 2, 406–408 CrossRef CAS.
- R. Liu, M. Shu, J. Hu, S. Zhu, H. Shi and H. Zhu, Star-shaped D-π-A compounds with a 1, 3, 5-triazine core and N-aryl chromophore substituted fluorene arms: synthesis, aggregation induced emission and two-photon absorption, Dyes Pigm., 2017, 137, 174–181 CrossRef CAS.
- T.-C. Lin, Y.-H. Lee, B.-R. Huang, M.-Y. Tsai and J.-Y. Lin, Functionalized tetrafluorenylethylene-type chromophores: Synthesis, two-photon absorption and effective optical power-limiting properties in the visible-to-near IR region, Dyes Pigm., 2016, 134, 325–333 CrossRef CAS.
- P. Zhou, C. Zhong, X. Chen, J. Qin, I. s. Mariz and E. Maçôas, New kind of hyperbranched conjugated polymers containing alkyl-modified 2, 4, 6-Tris (thiophen-2-yl)-1, 3, 5-triazine unit for enhancing two-photon absorption, Macromolecules, 2014, 47, 6679–6686 CrossRef CAS.
- A. Qin, J. W. Lam, H. Dong, W. Lu, C. K. Jim, Y. Dong, M. Häussler, H. H. Sung, I. D. Williams and G. K. Wong, Metal-free, regioselective diyne polycyclotrimerization: Synthesis, photoluminescence, solvatochromism, and two-photon absorption of a triphenylamine-containing hyperbranched poly (aroylarylene), Macromolecules, 2007, 40, 4879–4886 CrossRef CAS.
- M. G. Vivas, D. L. Silva, J. Malinge, M. Boujtita, R. Zaleśny, W. Bartkowiak, H. Ågren, S. Canuto, L. De Boni and E. Ishow, Molecular structure–optical property relationships for a series of non-centrosymmetric two-photon absorbing push-pull triarylamine molecules, Sci. Rep., 2014, 4, 1–11 CAS.
- X. Liu, J. Liang, J. You, L. Ying, Y. Xiao, S. Wang and X. Li, Small molecular hole-transporting and emitting materials for hole-only green organic light-emitting devices, Dyes Pigm., 2016, 131, 41–48 CrossRef CAS.
- R. Chennoufi, H. Bougherara, N. Gagey-Eilstein, B. Dumat, E. Henry, F. Subra, S. Bury-Moné, F. Mahuteau-Betzer, P. Tauc and M.-P. Teulade-Fichou, Mitochondria-targeted triphenylamine derivatives activatable by two-photon excitation for triggering and imaging cell apoptosis, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed.
- B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, I.-Y. S. Lee and D. McCord-Maughon, Two-photon polymerization initiators for three-dimensional optical data storage and microfabrication, Nature, 1999, 398, 51–54 CrossRef CAS.
- Z.-M. Tang, T. Lei, J.-L. Wang, Y. Ma and J. Pei, Star-shaped donor-π-acceptor conjugated molecules: synthesis, properties, and modification of their absorptions features, J. Org. Chem., 2010, 75, 3644–3655 CrossRef CAS PubMed.
- Z. Zhou, H. W. Qiao, Y. Hou, H. G. Yang and S. Yang, Epitaxial halide perovskite-based materials for photoelectric energy conversion, Energy Environ. Sci., 2021, 14, 127–157 RSC.
- M.-S. Yuan, Q. Wang, W.-J. Wang, T.-B. Li, L. Wang, W. Deng, Z.-t. Du and J.-R. Wang, Symmetrical and asymmetrical (multi) branched truxene compounds: Structure and photophysical properties, Dyes Pigm., 2012, 95, 236–243 CrossRef CAS.
- S. A. Siddiqui, T. Rasheed, M. Faisal, A. K. Pandey and S. B. Khan, Electronic structure, nonlinear optical properties, and vibrational analysis of gemifloxacin by density functional theory, Spectroscopy, 2012, 27, 185–206 CrossRef CAS.
- L. Shi, M. Horn, S. Kobayashi and H. Mayr, Carbocationic n-endo-trig Cyclizations, Chem.–Eur. J., 2009, 15, 8533–8541 CrossRef CAS PubMed.
- F. Meyers, S. Marder, B. Pierce and J.-L. Bredas, Electric field modulated nonlinear optical properties of donor-acceptor polyenes: sum-over-states investigation of the relationship between molecular polarizabilities (alpha, beta, and gamma) and bond length alternation, J. Am. Chem. Soc., 1994, 116, 10703–10714 CrossRef CAS.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, Molecular optimization enables over 13% efficiency in organic solar cells, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- D. He, F. Zhao, J. Xin, J. J. Rech, Z. Wei, W. Ma, W. You, B. Li, L. Jiang and Y. Li, A fused ring electron acceptor with decacyclic core enables over 13.5% efficiency for organic solar cells, Adv. Energy Mater., 2018, 8, 1802050 CrossRef.
- C. Huang, X. Liao, K. Gao, L. Zuo, F. Lin, X. Shi, C.-Z. Li, H. Liu, X. Li and F. Liu, Highly efficient organic solar cells based on S, N-heteroacene non-fullerene acceptors, Chem. Mater., 2018, 30, 5429–5434 CrossRef CAS.
- Y. Chen, X. Wan and G. Long, High performance photovoltaic applications using solution-processed small molecules, Acc. Chem. Res., 2013, 46, 2645–2655 CrossRef CAS PubMed.
- W. Ni, M. Li, F. Liu, X. Wan, H. Feng, B. Kan, Q. Zhang, H. Zhang and Y. Chen, Dithienosilole-based small-molecule organic solar cells with an efficiency over 8%: investigation of the relationship between the molecular structure and photovoltaic performance, Chem. Mater., 2015, 27, 6077–6084 CrossRef CAS.
- S. Dai, F. Zhao, Q. Zhang, T.-K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu and W. You, Fused nonacyclic electron acceptors for efficient polymer solar cells, J. Am. Chem. Soc., 2017, 139, 1336–1343 CrossRef CAS PubMed.
- H. Feng, Y. Q. Q. Yi, X. Ke, J. Yan, Y. Zhang, X. Wan, C. Li, N. Zheng, Z. Xie and Y. Chen, New anthracene-fused nonfullerene acceptors for high-efficiency organic solar cells: energy level modulations enabling match of donor and acceptor, Adv. Energy Mater., 2019, 9, 1803541 CrossRef.
- J. Wang, J. Zhang, Y. Xiao, T. Xiao, R. Zhu, C. Yan, Y. Fu, G. Lu, X. Lu and S. R. Marder, Effect of isomerization on high-performance nonfullerene electron acceptors, J. Am. Chem. Soc., 2018, 140, 9140–9147 CrossRef CAS PubMed.
- T. Li, S. Dai, Z. Ke, L. Yang, J. Wang, C. Yan, W. Ma and X. Zhan, Fused Tris (thienothiophene)-Based Electron Acceptor with Strong Near-Infrared Absorption for High-Performance As-Cast Solar Cells, Adv. Mater., 2018, 30, 1705969 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. Khalid, H. M. Lodhi, M. U. Khan and M. Imran, Structural parameter-modulated nonlinear optical amplitude of acceptor–π–D–π–donor-configured pyrene derivatives: a DFT approach, RSC Adv., 2021, 11, 14237–14250 RSC.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, version 6.0. 16, Semichem Inc. Shawnee Mission KS, 2016 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- G. Zhurko and D. Zhurko, ChemCraft, version 1.6, URL: http://www.chemcraftprog.com, 2009 Search PubMed.
- C. Valverde, S. A. d. L. e Castro, G. R. Vaz, J. L. de Almeida Ferreira, B. Baseia and F. A. Osório, Third-order nonlinear optical properties of a carboxylic acid derivative, Acta Chim. Slov., 2018, 65, 739–749 CrossRef CAS.
- V. Barone and M. Cossi, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- Ö. Tamer, D. Avcı and Y. Atalay, Synthesis, X-Ray crystal structure, photophysical characterization and nonlinear optical properties of the unique manganese complex with picolinate and 1, 10 phenantroline: toward the designing of new high NLO response crystal, J. Phys. Chem. Solids, 2016, 99, 124–133 CrossRef.
- M. Khalid, A. Ali, M. F. U. Rehman, M. Mustaqeem, S. Ali, M. U. Khan, S. Asim, N. Ahmad and M. Saleem, Exploration of noncovalent interactions, chemical reactivity, and nonlinear optical properties of piperidone derivatives: a concise theoretical approach, ACS Omega, 2020, 5, 13236–13249 CrossRef CAS PubMed.
- M. Adnan, M. Y. Mehboob, R. Hussain and Z. Irshad, Banana-Shaped Nonfullerene Acceptor Molecules for Highly Stable and Efficient Organic Solar Cells, Energy Fuels, 2021, 11496–11506 CrossRef CAS.
- P.-H. Sung and T.-F. Hsu, Thermal stability of NLO sol—gel networks with reactive chromophores, Polymer, 1998, 39, 1453–1459 CrossRef CAS.
- M. I. Nan, E. Lakatos, G.-I. Giurgi, L. Szolga, R. Po, A. Terec, S. Jungsuttiwong, I. Grosu and J. Roncali, Mono-and di-substituted pyrene-based donor-π-acceptor systems with phenyl and thienyl π-conjugating bridges, Dyes Pigm., 2020, 181, 108527 CrossRef CAS.
- M. Yoshida and J.-i. Aihara, Validity of the weighted HOMO–LUMO energy separation as an index of kinetic stability for fullerenes with up to 120 carbon atoms, Phys. Chem. Chem. Phys., 1999, 1, 227–230 RSC.
- R. Hussain, F. Hassan, M. U. Khan, M. Y. Mehboob, R. Fatima, M. Khalid, K. Mahmood, C. J. Tariq and M. N. Akhtar, Molecular engineering of A–D–C–D–A configured small molecular acceptors (SMAs) with promising photovoltaic properties for high-efficiency fullerene-free organic solar cells, Opt. Quantum Electron., 2020, 52, 1–20 CrossRef.
- D. Dini and M. Hanack, Phthalocyanines as materials for advanced technologies: some examples, J. Porphyrins Phthalocyanines, 2004, 8, 915–933 CrossRef CAS.
- M. Khalid, I. Shafiq, M. Zhu, M. U. Khan, Z. Shafiq, J. Iqbal, M. M. Alam, A. A. C. Braga and M. Imran, Efficient tuning of small acceptor chromophores with A1-π-A2-π-A1 configuration for high efficacy of organic solar cells via end group manipulation, J. Saudi Chem. Soc., 2021, 25, 101305 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. U. Khan, M. Khalid, M. Ibrahim, A. A. C. Braga, M. Safdar, A. A. Al-Saadi and M. R. S. A. Janjua, First theoretical framework of triphenylamine–dicyanovinylene-based nonlinear optical dyes: structural modification of π-linkers, J. Phys. Chem. C, 2018, 122, 4009–4018 CrossRef CAS.
- M. Khalid, A. Ali, R. Jawaria, M. A. Asghar, S. Asim, M. U. Khan, R. Hussain, M. F. ur Rehman, C. J. Ennis and M. S. Akram, First principles study of electronic and nonlinear optical properties of A–D–π–A and D–A–D–π–A configured compounds containing novel quinoline–carbazole derivatives, RSC Adv., 2020, 10, 22273–22283 RSC.
- R. G. Parr, L. v. Szentpály and S. Liu, Electrophilicity index, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, Electronegativity: the density functional viewpoint, J. Chem. Phys., 1978, 68, 3801–3807 CrossRef CAS.
- N. Kovačević and A. Kokalj, Analysis of molecular electronic structure of imidazole-and benzimidazole-based inhibitors: a simple recipe for qualitative estimation of chemical hardness, Corros. Sci., 2011, 53, 909–921 CrossRef.
- R. G. Parr, L. v. Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- H. Chermette, Chemical reactivity indexes in density functional theory, Journal of computational chemistry, 1999, 20, 129–154 CrossRef CAS.
- S. Tariq, M. Khalid, A. R. Raza, S. L. Rubab, S. F. de Alcântara Morais, M. U. Khan, M. N. Tahir and A. A. C. Braga, Experimental and computational investigations of new indole derivatives: A combined spectroscopic, SC-XRD, DFT/TD-DFT and QTAIM analysis, J. Mol. Struct., 2020, 1207, 127803 CrossRef.
- M. Ahn, M.-J. Kim, D. W. Cho and K.-R. Wee, Electron Push–Pull Effects on Intramolecular Charge Transfer in Perylene-Based Donor–Acceptor Compounds, J. Org. Chem., 2020, 86, 403–413 CrossRef PubMed.
- G. Dennler, M. C. Scharber, T. Ameri, P. Denk, K. Forberich, C. Waldauf and C. J. Brabec, Design Rules for Donors in Bulk-Heterojunction Tandem Solar Cells Towards 15% Energy-Conversion Efficiency, Adv. Mater., 2008, 20, 579–583 CrossRef CAS.
- B. S. Mendis and K. N. de Silva, A comprehensive study of linear and non-linear optical properties of novel charge transfer molecular systems, J. Mol. Struct.: THEOCHEM, 2004, 678, 31–38 CrossRef.
- H. S. Nalwa, T. Watanabe, S. Miyata and J. Rabek, Optical second-harmonic generation in organic molecular and polymeric materials: measurement techniques and materials, Progress in Photochemistry and Photophysics, 1992, 5, 103–185 CAS.
- S. Muhammad, R. A. Shehzad, J. Iqbal, A. G. Al-sehemi, M. Saravanabhavan and M. Khalid, Benchmark study of the linear and nonlinear optical polarizabilities in proto-type NLO molecule of para-nitroaniline, J. Theor. Comput. Chem., 2019, 18, 1950030 CrossRef CAS.
- M. Akram, M. Adeel, M. Khalid, M. N. Tahir, M. U. Khan, M. A. Asghar, M. A. Ullah and M. Iqbal, A combined experimental and computational study of 3-bromo-5-(2, 5-difluorophenyl) pyridine and 3, 5-bis (naphthalen-1-yl) pyridine: Insight into the synthesis, spectroscopic, single crystal XRD, electronic, nonlinear optical and biological properties, J. Mol. Struct., 2018, 1160, 129–141 CrossRef.
- F. J. Luque, J. M. López and M. Orozco, Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”, Theor. Chem. Acc., 2000, 103, 343–345 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07183a |
| This journal is © The Royal Society of Chemistry 2022 |