
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAg2Mo2O7: an oxide solid-state Ag+ electrolyte†
Weixin Yana,
Dongmei Zhua,
Zhaofeng Wanga,
Yunhao Xiaa,
Dong-Yun Guib,
Fa Luoa and
Chun-Hai Wang *ac
*ac
aState Key Laboratory of Solidification Processing, Northwestern Polytechnical University, Xi'an, Shaanxi 710072, China. E-mail: chwang81@gmail.com
bInstitute of Functional Materials, College of Materials Science and Engineering, Xi'an University of Architecture and Technology, Xi'an, Shaanxi 710055, China
cSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia
First published on 26th January 2022
Abstract
Ag2Mo2O7 powders and micro-crystals were prepared at 400 °C for 24 h and 500 °C for 6 h using solid-state reactions. The Ag2Mo2O7 samples crystalized in a triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with the cell parameters a = 6.0972(1) Å, b = 7.5073(1) Å, c = 7.6779(2) Å, α = 110.43(1)°, β = 93.17(1)°, γ = 113.51(1)°, and V = 294.17(1) Å3 from Rietveld refinements. Ag2Mo2O7 powder is homogeneous with size of 2–8 μm and the ceramic pellets are in good sintering conditions with a relative density ∼93%. The indirect band gaps Eg(i) of Ag2Mo2O7 from reflectance measurements and DFT calculations are 2.63(1) and 1.80 eV. The vibrational modes of Ag2Mo2O7 were investigated by first-principles (DFT) calculations and Raman spectrum measurements with 24 of 33 predicted Raman modes recorded. According to DOS analyses, the valence bands (VB) of Ag2Mo2O7 are mainly constituted of O-2p and Ag-4d orbitals, while the conduction bands (CB) are mainly composed of Mo-4d and the O-2p orbitals. Regarding the impedance analysis, Ag2Mo2O7 is a silver oxide ion electrolyte with a conductivity of ∼5 × 10−4 S cm−1 at 450 °C. The carrier activation energy of Ag2Mo2O7 is 0.88(3) eV from the temperature dependent conductivity measurements.
space group with the cell parameters a = 6.0972(1) Å, b = 7.5073(1) Å, c = 7.6779(2) Å, α = 110.43(1)°, β = 93.17(1)°, γ = 113.51(1)°, and V = 294.17(1) Å3 from Rietveld refinements. Ag2Mo2O7 powder is homogeneous with size of 2–8 μm and the ceramic pellets are in good sintering conditions with a relative density ∼93%. The indirect band gaps Eg(i) of Ag2Mo2O7 from reflectance measurements and DFT calculations are 2.63(1) and 1.80 eV. The vibrational modes of Ag2Mo2O7 were investigated by first-principles (DFT) calculations and Raman spectrum measurements with 24 of 33 predicted Raman modes recorded. According to DOS analyses, the valence bands (VB) of Ag2Mo2O7 are mainly constituted of O-2p and Ag-4d orbitals, while the conduction bands (CB) are mainly composed of Mo-4d and the O-2p orbitals. Regarding the impedance analysis, Ag2Mo2O7 is a silver oxide ion electrolyte with a conductivity of ∼5 × 10−4 S cm−1 at 450 °C. The carrier activation energy of Ag2Mo2O7 is 0.88(3) eV from the temperature dependent conductivity measurements.
Introduction
The solid-state electrolyte (SSE) is a kind of solid or quasi-solid material with high ionic conductivity (and generally low electronic conductivity) at room or high temperatures.1–3 Solid-state electrolytes are widely used in battery, electrolysis and sensor applications. SSEs can be categorized into inorganic solid-state electrolytes, polymer solid-state electrolytes and composite solid-state electrolytes.4–6 Regarding the charge carriers, there are several well-known SSE materials, such as lithium ion SSEs (e.g. Li3N, Li3xLa(2/3)−xTiO3, Li7La3Zr2O12),7–9 sodium ion SSEs (e.g. NASICON, β-Al2O3),10,11 oxygen ion SSEs [e.g. yttria-stabilized zirconia (YSZ), gadolinium doped ceria (GDC), Bi4V2O11],12–14 proton SSEs (e.g. NAFION),15 and silver ion SSEs (e.g. AgI).16 Though Ag is a heavy element, the ionic radii and polarizable electron shells make Ag+ suitable for fast ion transport.17 More than 100 years ago, the very first Ag+ SSE silver iodide (α-AgI) was investigated by Turbandt and Lorenz.18 Owens and Argue observed the highest Ag+ ionic conductivity in KAg4I5 and RbAg4I5 with a conductivity σ of 0.21 cm−1 at 20 °C, respectively. They also reported that the conductivities are contributed by Ag+.19 To date, most Ag+ electrolytes investigated are limited in halides especially iodide. As oxides are very common in studies and shows significant chemistry difference with iodide, it is important to study the possible oxide solid-state Ag+ electrolyte to investigate the mechanism of oxide SSE and develop new SSE, which is critical in the study of related solid-state devices.Sliver dimolybdate, Ag2Mo2O7, was found in the fused salt Ag2MoO4–MoO3 using X-ray diffraction techniques by Gatehouse. Ag2Mo2O7 shows a triclinic symmetry (space group P) with cell parameters20 a = 6.11(2) Å, b = 7.49(2) Å, c = 7.66(2) Å, α = 109.8°, β = 93.5°, γ = 113.6°. Ag2Mo2O7 with an indirect band gap Eg of 2.65 eV,21 has been widely investigated in various applications, such as catalysts,22 antibacterial material,23 microwave applications24 and fuel cells.25 Zhang investigated Ag2Mo2O7 as an anode for lithium-ion batteries and observed a high specific capacity (discharge capacity of 825 mA h g−1 with capacity retention of 95% over 100 cycles) and superior rate capability (discharge capacity of 263 mA h g−1 at the high current density of 2 A g−1).26 Considering the crystal structure, Ag2Mo2O7 might be an oxide solid-state electrolyte in our experiences. To date, there is no studies reported on the conductivity and charge carries of Ag2Mo2O7 to our knowledge.
Generally, an ideal electrolyte is expected to give a high ionic conductivity and a low electronic conductivity, which should be an electronic insulator or semiconductor with ion migrations in physics. The electronic properties of solid-state materials are defined by electronic orbitals and band structures (including the density of state, DOS). The ion migration is confined by atomic vibrations and phonons. Thus, the study on the band structure (DOS and band gap) and phonons (vibrational modes) is important for the knowledge of solid-state electrolytes. To our knowledge, there is no studies reported on the band structures and vibrational modes of Ag2Mo2O7.
In this study, we synthesized Ag2Mo2O7 samples through solid-state reaction method and investigated the crystal structure of Ag2Mo2O7 by single crystal and powder X-ray diffractions and first-principle calculations. We also studied the vibrational modes, band structures, Raman spectra, diffuse reflectance, ionic conductivity and charge carriers of Ag2Mo2O7 in details.
Experimental procedures and calculation details
Synthesis
The Ag2Mo2O7 powder and microcrystal samples were prepared by the high temperature solid-state reaction method. Stoichiometric Ag2O and MoO3 (99.99%, Macklin, dried at 300 °C before use) were weighed and ground using an agate pestle and mortar. The mixture was then calcined at 400 °C for 24 h with an intermediate ground. After calcination, bright yellow Ag2Mo2O7 powder sample was synthesized according to the XRD analysis. The Ag2Mo2O7 powder was added with polyvinyl alcohol (PVA, 5 wt%) as binder and uniaxially pressed into pellets (thickness ∼1 mm) with a Φ15 mm die at about 120 MPa. The pressed pellets were heated at 400 °C for 1 h to drive off the PVA and sintered at 480 °C for 6 h with a ramp of 5 °C min−1 (both heat up and cool down). The sintered samples (Ag2Mo2O7 ceramics) were then carefully polished with sandpapers. The density of ceramics was determined using the Archimedes method. For the growth of Ag2Mo2O7 micro-crystals, Ag2Mo2O7 powder was heated at 500 °C for 6 h.Measurements
The phase of Ag2Mo2O7 samples was analyzed using powder X-ray diffraction (PXRD) (miniFlex 600, Rigaku, Cu-Kα) in 2θ range of 5–120° with a scanning rate 2° min−1 at room temperature. The single crystal X-ray diffraction (SXRD) data of a micro single crystal (20 × 10 × 10 μm) were collected by a Rigaku XtaLAB Synergy-R (Cu-Kα) diffractometer at 150 K (−123 °C). A total of 7725 frames were recorded at 7 s per frame. The crystal structure of Ag2Mo2O7was analyzed by Jana 2006![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 and TOPAS Academic (TA)28 and finally obtained by the Rietveld refinements against PXRD data measured. The crystal structure visualization was done using VESTA.29 The microstructure of the samples was observed by a scanning electron microscope (SEM, Tescan Vega3 SBH, Brno, Czech Republic). The Raman spectrum was collected on a pellet sample by a Raman spectrometric analyzer (Renishaw inVia, Britain) in 100–1200 cm−1 with the resolution ∼2 cm−1, excited by a 532 nm laser. The conductivity of Ag2Mo2O7 pellets was measured by an electrochemical workstation (CHI 660e, Chenhua) at different temperatures. The Ag electrode was prepared by heated the covered silver paste at 400 °C for 2 h and the Au electrode was made by ion sputter coating.
27 and TOPAS Academic (TA)28 and finally obtained by the Rietveld refinements against PXRD data measured. The crystal structure visualization was done using VESTA.29 The microstructure of the samples was observed by a scanning electron microscope (SEM, Tescan Vega3 SBH, Brno, Czech Republic). The Raman spectrum was collected on a pellet sample by a Raman spectrometric analyzer (Renishaw inVia, Britain) in 100–1200 cm−1 with the resolution ∼2 cm−1, excited by a 532 nm laser. The conductivity of Ag2Mo2O7 pellets was measured by an electrochemical workstation (CHI 660e, Chenhua) at different temperatures. The Ag electrode was prepared by heated the covered silver paste at 400 °C for 2 h and the Au electrode was made by ion sputter coating.
Calculations
The band structure and vibrational modes of Ag2Mo2O7 was calculated using the CASTEP software package based on the density functional theory (DFT).30 The plane wave basis was used for the expansion of the valence-electron wave functions based on generalized gradient approximation (GGA). The energy cutoff of plane wave basis was set to 1020 eV, and the criterion for self-consistency was set as eigen-energy within 10−10 eV per atom. The Brillouin zone sampling set (k-points) was 6 × 5 × 5 Monkhorst–Pack grid (reciprocal spacing ∼0.03 Å−1) and the pseudopotential (PBEsol) was constructed from the CASTEP database. Ag2Mo2O7 crystal structure obtained from the XRD analysis in this work was selected as original model and a further geometry optimization was run with convergence tolerance 10−9 eV per atom. The band structure and vibrational modes of the optimized Ag2Mo2O7 model were then calculated. The vibrational modes were calculated using linear response method31 with the convergence tolerance 10−5 eV Å−2.Results and discussions
Samples and microstructures
With the optimized synthesis described in the experimental section, desired Ag2Mo2O7 samples (powder, micro-crystal, and ceramics) were successfully synthesized. All the samples are bright yellow and there is no any impurity observed from XRD analysis. The microstructure of Ag2Mo2O7 powders, micro-crystals and ceramics (fractured surface) observed by SEM are shown in Fig. 1a–c. The fine Ag2Mo2O7 powder is obtained after the calcination at 400 °C with the particle size in 2–8 μm (Fig. 1a). The particles grew significantly to ∼20 μm (Fig. 1b) after heated at 500 °C for 6 h and most of the particles are single-domain micro-crystals from our single crystal diffraction analysis. The ceramic samples prepared are well sintered with a relative density of ∼93% and a grain size of 20–100 μm (Fig. 1c). Some pores distributed in the grains and grain boundaries should be caused by the fire out of the PVA binders. However, there is no any obvious voids or cracks in the sample, which is in consistent with the high relative density measured.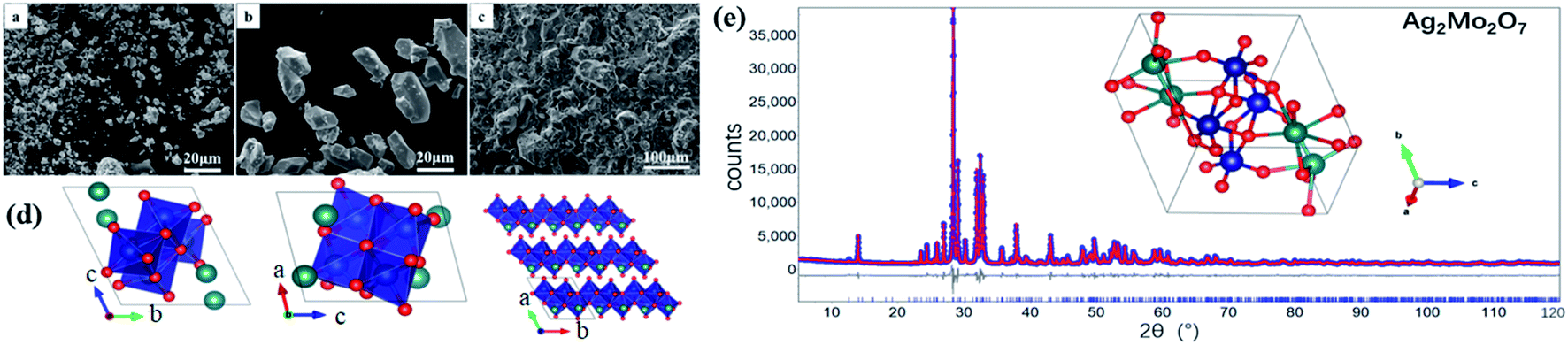 | ||
| Fig. 1 SEM images of Ag2Mo2O7 (a) powders, (b) micro-crystals, (c) fractured surface of a ceramic pellet samples; (d) representation of the crystal structure of Ag2Mo2O7 from a, b and c directions. Silver ions (green), molybdenum (navy), oxygen ions (red); (e) XRD patterns, Rietveld refinements and crystal structure of Ag2Mo2O7, circle points: observed, red solid line: calculated curve, solid line below the data: the difference curve, vertical tick marks: peak positions. | ||
Crystal structure of Ag2Mo2O7
The single-crystal X-ray diffraction data (SXRD) of Ag2Mo2O7 at 150 K were collected through a full reciprocal sphere with the resolution 0.8 Å to check the symmetry and space group. The observed reflections were well-indexed with the triclinic unit cell (P) a = 6.0907(4) Å, b = 7.4593(4) Å, c = 7.6668(5) Å, α = 110.370(5)°, β = 93.336(5)°, γ = 113.610(5)°, V = 291.18(4) Å3. By charge-flipping analysis and refinements, the crystal structure of Ag2Mo2O7 was obtained and a fairly good agreement was achieved with weighted residual factors (Rwp) of 0.054.
The XRD pattern of Ag2Mo2O7 powder (PXRD) sample at room temperature is shown in Fig. 1e. The pattern is in a reliable quality with the highest reflection >35000 counts. Rietveld refinements of the Ag2Mo2O7 structure model obtained from SXRD against the PXRD data were carried out to obtain a higher accuracy. In the refinements, background, zero shift, peak profile, the cell parameters, atomic coordinates, and atomic displacement parameters (ADPs) were refined. An excellent agreement between the diffraction data and the prediction from the structure model is achieved with the weighted residual factor (Rwp) ∼0.064. The refined profile is shown in Fig. 1d and the refined crystal structure is presented in Fig. 1e. Details of the refined structure is shown in Table 1 and given in the crystallographic information files (CIF) in the ESI.† All the reflections observed can be indexed in the refinements, which means the impurities is in a very low level. The cell parameters from the PXRD analysis are a = 6.0972(1) Å, b = 7.5073(1) Å, c = 7.6779(2) Å, α = 110.43(1)°, β = 93.17(1)°, γ = 113.51(1)°, V = 294.17(1) Å3, which are slight larger than that from SXRD analysis. This should be caused by the thermal expansions due to the different temperature in the measurements. The refined parameters here agree with the reported ones32 a = 6.095(1) Å, b = 7.501(1) Å, c = 7.681(1) Å, α = 110.4(1)°, β = 93.3(1)°, γ = 113.5(1)°. V = 294.37(4) Å3. In the crystal structure of Ag2Mo2O7, edge-shared MoO6 octahedra form chains along axis a and the Ag+ ions sit between the chains. The distance between neighboring Ag+ is ∼3.575(6) Å. Thus, it is possible for the Ag+ to migrate along axis a from crystallographic view and Ag2Mo2O7 should be a potential oxide Ag+ electrolyte.
| Space group | P (2) |
|---|---|
| PXRD | |
| a (Å) | 6.0972(1) |
| b (Å) | 7.5073(1) |
| c (Å) | 7.6779(1) |
| α (°) | 110.42(1) |
| β (°) | 93.16(1) |
| γ (°) | 113.50(1) |
| V (Å3) | 294.2(2) |
| dtheory (g cm−3) | 5.866(1) |
| x | y | z | Uiso (Å2) | |
|---|---|---|---|---|
| Ag1 | 0.7813(6) | 0.2741(5) | 0.2674(4) | 0.0066(2) |
| Ag2 | 0.2312(7) | 0.2397(5) | 0.0370(5) | 0.0073(2) |
| Mo1 | 0.6698(6) | 0.1706(5) | 0.7490(5) | 0.0005(2) |
| Mo2 | 0.7580(7) | 0.7710(6) | 0.4541(5) | 0.0003(2) |
| O1 | 0.603(3) | 0.505(3) | 0.278(3) | 0.0066(13) |
| O2 | 0.774(4) | 0.112(3) | 0.939(3) | 0.0066(13) |
| O3 | 0.362(4) | 0.140(3) | 0.766(3) | 0.0040(12) |
| O4 | 0.490(3) | 0.834(3) | 0.519(3) | 0.0015(11) |
| O5 | 0.921(4) | 0.115(3) | 0.600(3) | 0.0029(12) |
| O6 | 0.870(4) | 0.759(3) | 0.656(3) | 0.0074(13) |
| O7 | 0.843(4) | 0.429(3) | 0.821(2) | 0.0057(13) |
Raman spectrum
There are 22 atoms (Z = 2) in a Ag2Mo2O7 primitive cell, which gives 66 vibrational modes with 3 acoustic modes and 63 optical modes. According to group theory,33 the vibrational modes (acoustic modes Γacoustic and optical modes Γoptical) of Ag2Mo2O7 are| Γacoustic = 3Au | (1) |
| Γoptical = 33Ag + 30Au | (2) |
Of the optical modes, Ag are Raman active and Au are IR active. Thus, there are a total of 30 IR-active modes ΓIR = 30Au, and 33 Raman-active modes ΓRaman = 33Ag.
The Raman spectrum of Ag2Mo2O7 measured is shown in Fig. 2. 24 of the 33 predicted first-order Raman modes can be clearly recognized. By peak fitting using Voigt profile, the frequency, intensity, and peak width (full width at half maximum, FWHM) of recognized modes are obtained and listed in Table S1 which is shown in ESI.† The detailed fit profiles are also shown as Fig. S1 in ESI,† which shows excellent agreements from the fit.
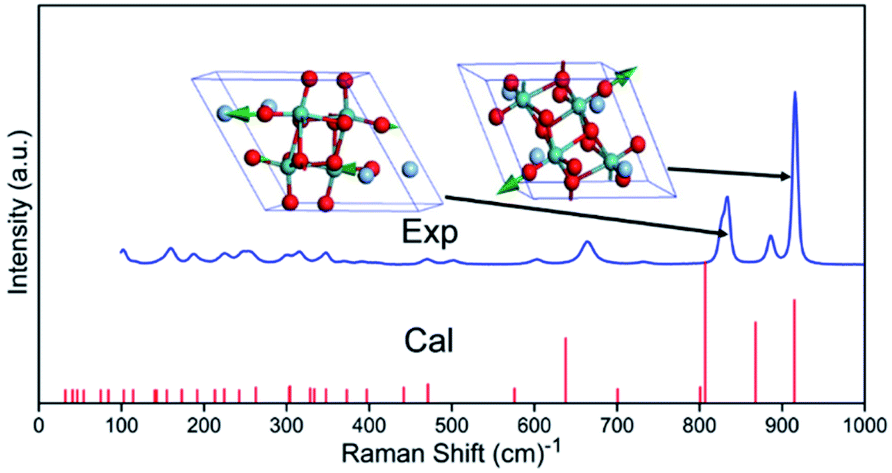 | ||
| Fig. 2 Raman spectra of Ag2Mo2O7. Exp.: measured, Cal.: calculated. Two Raman modes with highest intensity from DFT calculations are shown as inset. | ||
Diffuse reflectance
The optical band gap and absorption coefficient of semiconductor can be calculated by the equation34| (αhν)n = K(hν − Eg) |
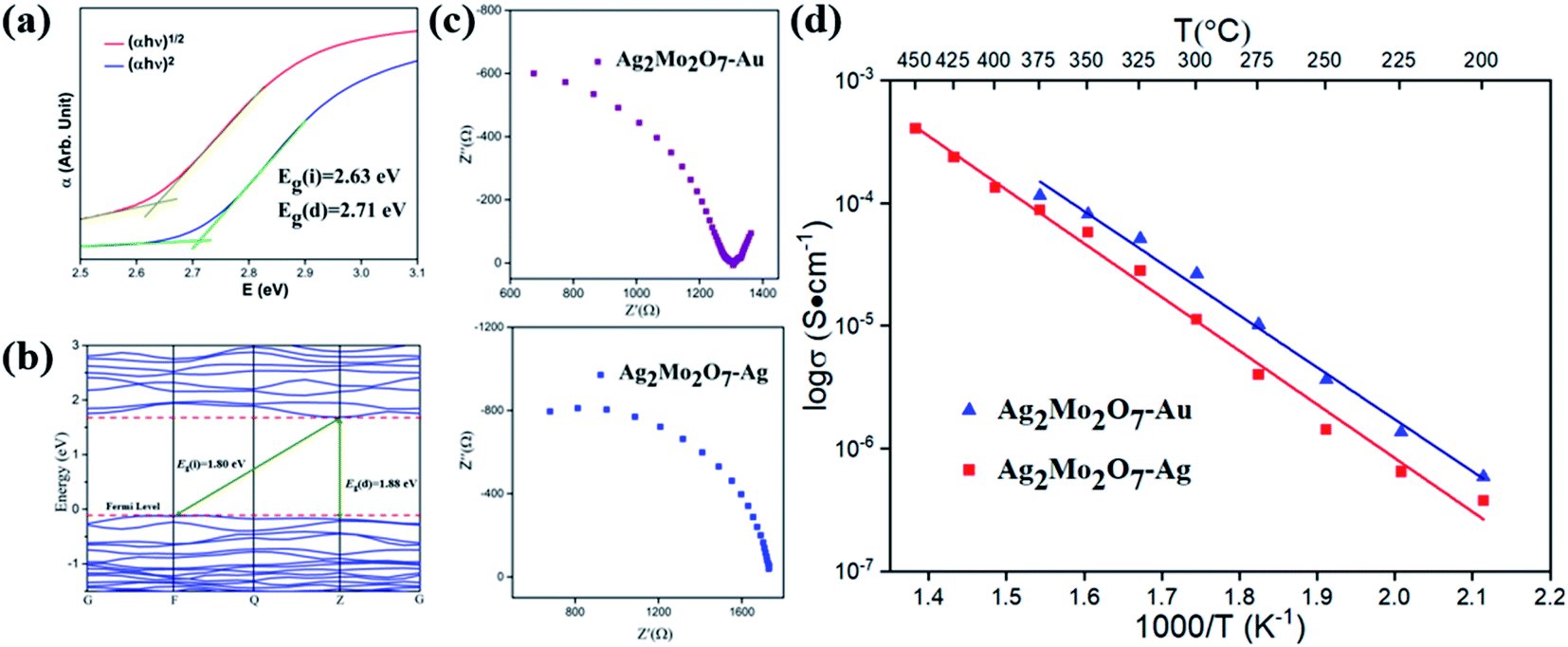 | ||
| Fig. 3 (a) (αhν)n − hν curve of Ag2Mo2O7 from diffuse reflectance measurements; (b) band structure and density of state (DOS) of Ag2Mo2O7. The Fermi level is set as EFemi = 0 eV; (c)Nyquist plot (impedance) of Ag2Mo2O7 with two different electrodes at 325 °C; (d) temperature dependent conductivity of Ag2Mo2O7 with two different electrodes. | ||
Band structures
After DFT geometry optimization, the cell parameters of the optimized structure show excellent agreements (difference of cell parameter <1%) with the one from our Rietveld refinements. The band structures of Ag2Mo2O7 calculated is plotted in Fig. 3b. Ag2Mo2O7 gives an indirect band gap of Eg(i) = 1.80 eV (F → Z) and a direct band gap of Eg(d) = 1.88 eV (Z → Z). This is slight smaller than the experimental results from diffuse reflectance analysis. In general, DFT calculations often underestimate the band gaps of solids, especially for semiconductors.35,36The partial density of state (PDOS) plots for different atoms and total density of state (DOS) of Ag2Mo2O7 are shown in Fig. S2 in ESI.† In the vicinity of the Fermi level, the main contribution to the valence bands (VB) is from the O-2p and Ag-4d orbitals, while the conduction bands (CB) are formed mainly by the Mo-4d and O-2p orbitals.
Vibrational modes
The eigenfrequency and intensity of the optical vibrational modes of Ag2Mo2O7 calculated are listed in Table S2 (Raman modes) and Table S3 (infrared modes) of ESI.† Refers to the theoretical intensity, we give the assignment of the observed Raman modes reliably, which are also given in Table S2.† For the Raman modes, the frequency of vibrational modes from DFT calculations (theoretical) is lower than that from measured spectra (experimental), which agrees with the general results from our previous studies.37 The vibration detail (vector of atomic motions) of five IR and Raman vibrational modes with high intensities is given as Fig. S3 and S4.†According to the atomic motion of vibrational modes, vibrations of O2− contribute to all the vibrational modes of Ag2Mo2O7. Generally, the vibrational modes with eigenfrequency f ≥ 195.52 cm−1 are dominated by the vibrations of O2− (light atom vibrations). The vibrations of Ag+ and Mo6+ mainly contribute to the vibrational modes at 126.84 cm−1 ≤ f ≤ 195.52 cm−1 and 32.01 cm−1 ≤ f ≤ 126.84 cm−1. The vibrational modes (phonons) of Ag+ give a rather low eigenfrequencies. As the frequency f of a vibration is determined by the restoring force k and the reduced mass m* (f ∝ k/m*). Thus, the restoring force k and the bonding of Ag+ in Ag2Mo2O7 is weak, which makes it easy to be activated for migration at a low or intermediate temperature.
Ionic conductivity
The conductivity of samples was determined by impedance analysis in the frequency range 0.1 Hz to 106 Hz. Two different electrodes (Au and Ag) were used to determine the species of charge carries in Ag2Mo2O7 and the Nyquist plots are shown in Fig. 3c. An obvious Warburg tail is observed for sample with Au electrodes, but disappeared for sample with Ag electrodes. This indicates the charge carries of Ag2Mo2O7 ceramics should be Ag+. As discussed in crystal structure of Ag2Mo2O7, the Ag+ sits between the Mo–O octahedra chains, which makes it possible for the migration of Ag+ in the structures. Resistance difference between gold electrode and sliver electrode is derived from the sample size and ceramic-electrode contact.For non-metal conductors, the conductivity σ of a non-metal system follows the Arrhenius equation38
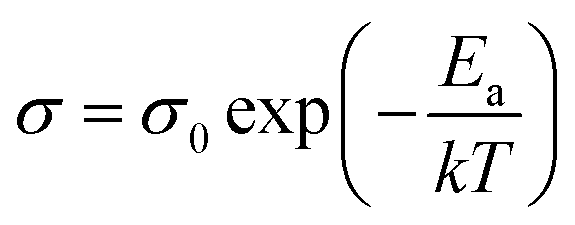
Conclusions
Ag2Mo2O7 powder with a particle size of 2–8 μm have been synthesized by conventional solid-state reaction. The crystal structure of Ag2Mo2O7 is in a triclinic P space group, with the cell parameters a = 6.0972(1) Å, b = 7.5073(1) Å, c = 7.6779(2) Å, α = 110.43(1)°, β = 93.17(1)°, γ = 113.51(1)°, V = 294.17(1) Å3. The band gap of Ag2Mo2O7 is estimated to be indirect band gap Eg(i) = 2.63(1) eV and direct band gap Eg(d) = 2.71(1) eV. The VB are mainly contributed by O-2p and Ag-4d orbitals, while the CB is dominated by the Mo-4d and the O-2p orbitals. 24 of the 33 predicted first-order Raman modes were recognized and assigned in the Raman spectrum. The vibrational modes with f ≥ 195.52 cm−1 are dominated by the vibrations of O2− (light atom vibrations). The Ag+ and Mo6+ mainly contributed to the vibrational modes at 126.84 cm−1 ≤ f ≤ 195.52 cm−1 and 32.01 cm−1 ≤ f ≤ 126.84 cm−1. Ag2Mo2O7 is a Ag+ solid state electrolyte with the activation energy Ea estimated to be 0.88(3) eV. The Ag+ conductivity of Ag2Mo2O7 is >5 × 10−4 S cm−1 at 450 °C.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22075229), and Natural Science Foundation of Shaanxi Province (2020JM-117). Thanks to Prof. Xiao-Ming Wang and Prof. Huan Jiao at School of Chemistry & Chemical Engineering, Shaanxi Normal University, for the single crystal XRD measurements.Notes and references
- Z. Zhang, Y. Shao, B. Lotsch, Y.-S. Hu, H. Li, J. Janek, L. F. Nazar, C.-W. Nan, J. Maier, M. Armand and L. Chen, Energy Environ. Sci., 2018, 11, 1945–1976 RSC.
- Z. G. G. Yang, J. W. Stevenson and P. Singh, Adv. Mater. Processes, 2003, 161, 34–37 CAS.
- O. Z. Sharaf and M. F. Orhan, Renew. Sust. Energ. Rev., 2014, 32, 810–853 CrossRef CAS.
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, Energy Environ. Sci., 2016, 9, 1602–1644 RSC.
- W. H. Kan, A. J. Samson and V. Thangadurai, J. Mater. Chem. A, 2016, 4, 17913–17932 RSC.
- J. B. Lu, Z. T. Zhang and Z. L. Tang, Rare Met. Mater. Eng., 2005, 34, 1177–1180 CAS.
- Y. Heng, W. Wang, Q. Li, X. Li, M. Dai, C. Yu, Z. Xue, J. Xia, G. Zhou and D. Jiang, J. Electroceram., 2021, 1–8 Search PubMed.
- Y. Ren, S. Yang, X. Ma, C. Zhang, B. Song, C. Sun, X. Tan and S. Sun, Appl. Surf. Sci., 2021, 567, 150746 CrossRef CAS.
- J. C. Verduzco, E. E. Marinero and A. Strachan, Integr. Mater. Manuf. Innov., 2021, 10, 299–310 CrossRef.
- M. Hou, X. Yang, F. Liang, P. Dong, Y. Chen, J. Li, K. Chen, Y. Dai and D. Xue, ACS Appl. Mater. Interfaces, 2021, 13, 33262–33271 CrossRef CAS PubMed.
- J. R. Walker and C. R. A. Catlow, J. Phys. C: Solid State Phys., 1982, 15, 6151–6161 CrossRef CAS.
- V. V. Kharton, A. V. Kovalevsky, A. P. Viskup, F. M. Figueiredo, A. A. Yaremchenko, E. N. Naumovich and F. M. B. Marques, J. Eur. Ceram. Soc., 2001, 21, 1763–1767 CrossRef CAS.
- V. Sharma, A. K. Shukla and J. Gopalakrishnan, Solid State Ionics, 1992, 58, 359–362 CrossRef CAS.
- M. J. Son, M. W. Kim, A. V. Virkar and H.-T. Lim, Electrochim. Acta, 2020, 353, 136450 CrossRef CAS.
- H. Gao and K. Lian, RSC Adv., 2014, 4, 33091–33113 RSC.
- M. C. R. Shastry and K. J. Rao, Solid State Ionics, 1992, 51, 311–316 CrossRef CAS.
- D. W. Kim, I.-S. Cho, S. Lee, S.-T. Bae, S. S. Shin, G. S. Han, H. S. Jung and K. S. Hong, J. Am. Ceram. Soc., 2010, 93, 3867–3872 CrossRef CAS.
- C. Tubandt and E. Lorenz, Z. Phys. Chem., 1914, 24, 513–543 CrossRef.
- B. B. Owens and G. R. Argue, Science, 1967, 157, 308–310 CrossRef CAS PubMed.
- B. M. Gatehouse and P. Leverett, J. Chem. Soc. D, 1970, 1, 484–496 Search PubMed.
- M. Hashim, C. Hu, Y. Chen, C. Zhang, Y. Xi and J. Xu, Phys. Status Solidi A, 2011, 208, 1937–1941 CrossRef CAS.
- S. A. Driscoll and U. S. Ozkan, Stud Surf Sci Catal, 1994, 82, 367–375 CrossRef CAS.
- H. Tang, A. Lu, L. Li, W. Zhou, Z. Xie and L. Zhang, Chem. Eng. J., 2013, 234, 124–131 CrossRef CAS.
- L. G. Vanuitert and S. Preziosi, J. Appl. Phys., 1962, 33, 2908–2909 CrossRef CAS.
- V. V. Kharton, F. M. B. Marques and A. Atkinson, Solid State Ionics, 2004, 174, 135–149 CrossRef CAS.
- M. Zhang, Y. Gao, N. Chen, X. Ge, H. Chen, Y. Wei, F. Du, G. Chen and C. Wang, Chem.–Eur. J., 2017, 23, 5148–5153 CrossRef CAS.
- V. Petricek, M. Dusek and L. Palatinus, Z. Krist-cryst. Mater., 2014, 229, 345–352 CrossRef CAS.
- A. A. Coelho, J. S. O. Evans, I. R. Evans, A. Kern and S. Parsons, Powder. Diffr., 2011, 26, S22–S25 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- V. Milman, K. Refson, S. J. Clark, C. J. Pickard and J. R. Yates, J. Mol. Struct.: THEOCHEM, 2010, 954, 22–35 CrossRef CAS.
- K. Refson, P. R. Tulip and S. J. Clark, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 155114 CrossRef.
- B. M. Gatehouse and P. Leverett, J. Chem. Soc., Dalton Trans., 1976, 1316–1320 RSC.
- D. M. P. Mingos, Nat. Phys. Sci., 1972, 236, 99–102 CrossRef CAS.
- T. J. Farrell, M. S. Patterson and B. Wilson, Med. Phys., 1992, 19, 879–888 CrossRef CAS.
- A. Seidl, A. Gorling, P. Vogl, J. A. Majewski and M. Levy, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 3764–3774 CrossRef CAS PubMed.
- H. Xiao, J. Tahir-Kheli and W. A. Goddard III, J. Phys. Chem. Lett., 2011, 2, 212–217 CrossRef CAS.
- C.-H. Wang, W. Shu, Y. Qing, F. Luo, D. Zhu and W. Zhou, J. Am. Ceram. Soc., 2021, 104, 1797–1805 CrossRef CAS.
- M. D. Archer, ACS Symp. Ser., 1989, 390, 115–126 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07097e |
| This journal is © The Royal Society of Chemistry 2022 |