
Large-Stokes-shifted yellow photoluminescence emission from an imide and polyimides forming multiple intramolecular hydrogen bonds†
Naiqiang
Liang
 ,
Shigeki
Kuwata
,
Ryohei
Ishige
and
Shinji
Ando
*
,
Shigeki
Kuwata
,
Ryohei
Ishige
and
Shinji
Ando
*
Department of Chemical Science & Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, Ookayama 2-12-1-E4-5, Meguro-ku, Tokyo 152-8552, Japan. E-mail: ando.s.aa@m.titech.ac.jp; Tel: +81-3-5734-2137
First published on 6th November 2021
Abstract
A novel imide compound (DH-MC) and polyimides (DH-PIs) that form multiple intramolecular hydrogen bonds (H-bonds) were synthesised from 2,2′-dihydroxybenzophenone-3,3′,4,4′-tetracarboxylic dianhydride (DHBA) to investigate the effects of distinct H-bond structures on the photoluminescence properties of these compounds. The DHBA moiety, which contains two proton donors and three proton acceptors, can form three types of H-bond structures (MC-0, MC-1, and MC-2). DFT calculations have predicted that the most energetically stable conformation is MC-1, forming an asymmetric H-bond structure, which is consistent with the FT-IR spectroscopy and single-crystal XRD analysis results. A colourless toluene solution of DH-MC exhibited orange fluorescence with a large Stokes shift (ν = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 905 cm−1), and DH-MC and the DH-PIs exhibited yellow fluorescence with a large ν of >10000 cm−1 in the solid state, with both originating from excited-state intramolecular proton transfer (ESIPT). In addition, these compounds exhibit small-Stokes-shifted fluorescence from the anionic form of the DHBA moiety, resulting in yellow coloration of the DH-PI film and the DH-MC powder. To reduce coloration, a polyimide copolymer (CoPI) film was prepared using DHBA and 4,4′-oxydiphthalic anhydride (ODPA) in which the molar ratio of DHBA was set at 3%. Owing to the dilution effect and efficient energy transfer from the blue-fluorescent ODPA to the DHBA moiety in the excited state, the colourless and transparent CoPI film exhibited prominent yellow fluorescence with a quantum yield of 0.14. The wavelength-converting spectrum demonstrated that the CoPI film absorbs a wide range of UV radiation from a xenon light source and significantly enhances the yellowish light component via ESIPT emission. The CoPI film is a promising candidate for solar spectral conversion applications.
905 cm−1), and DH-MC and the DH-PIs exhibited yellow fluorescence with a large ν of >10000 cm−1 in the solid state, with both originating from excited-state intramolecular proton transfer (ESIPT). In addition, these compounds exhibit small-Stokes-shifted fluorescence from the anionic form of the DHBA moiety, resulting in yellow coloration of the DH-PI film and the DH-MC powder. To reduce coloration, a polyimide copolymer (CoPI) film was prepared using DHBA and 4,4′-oxydiphthalic anhydride (ODPA) in which the molar ratio of DHBA was set at 3%. Owing to the dilution effect and efficient energy transfer from the blue-fluorescent ODPA to the DHBA moiety in the excited state, the colourless and transparent CoPI film exhibited prominent yellow fluorescence with a quantum yield of 0.14. The wavelength-converting spectrum demonstrated that the CoPI film absorbs a wide range of UV radiation from a xenon light source and significantly enhances the yellowish light component via ESIPT emission. The CoPI film is a promising candidate for solar spectral conversion applications.
1. Introduction
Polyimide (PI) is a high-performance polymer with excellent mechanical properties and outstanding thermal and chemical stability.1 In addition, PIs exhibit low dielectric constants, low thermal expansion and good flexibility, and PI films can be readily prepared. Therefore, PI films have been widely utilized for the production of microelectronics. Importantly, a variety of PIs derived from various dianhydrides and diamines have been reported and applied commercially. Thus, understanding the structure–property relationships of PIs is essential for their application.2 Over the past decade, the fluorescence properties of PI films have been extensively studied.3–7 Highly fluorescent PI films with excellent comprehensive performances are considered ideal wavelength-downshifting materials and are expected to be applied to luminescent solar concentrators, flat panel displays, crop cultivators, and photovoltaic devices.8–12 Due to their excellent thermal and chemical stability, fluorescent PIs can satisfy long-term use requirements even under extreme conditions, such as at elevated temperatures of >300 °C.Common aromatic PIs exhibit only weak photoluminescence. For example, Kapton, a commercialised PI synthesised from pyromellitic dianhydride (PMDA) and 4,4′-diaminodiphenyl ether (ODA), exhibits only weak fluorescence between 400 and 700 nm with a photoluminescence quantum efficiency (Φ) as low as 9.7 × 10−7.13,14 Time-dependent density functional theory (TD-DFT) calculations have indicated that this fluorescence originates from a charge-transfer (CT) transition. In this CT transition, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are located in the diamine and dianhydride moieties, respectively.3,15 Due to the small degree of overlap between the HOMO and LUMO, this CT transition gives rise to a low oscillator strength (f), which can explain the small Φ value of the Kapton film.
In addition to the CT transition, another transition occurring in PIs is the locally excited (LE) transition, which is observed in imide model compounds (MCs), which are small molecules that have the same structure as the repeating unit of a PI derived from cyclohexylamine.3 In such MCs, both the HOMO and LUMO are located in the dianhydride moieties with significant overlap, and the LE transition gives rise to a larger f value than the CT transition.3 However, conventional aromatic PIs predominantly show weak CT fluorescence due to the strong electron-donating and -accepting nature of the diamine and dianhydride moieties, respectively.
A series of highly fluorescent semi-aromatic PIs derived from aromatic dianhydrides and aliphatic diamines have been reported by our group.3–7,16 Aliphatic diamines, which are weakly electron donating, effectively inhibit the CT transition and enhance the LE transition. For example, a PI prepared from 1,4-bis(3,4-dicarboxyphenoxy)benzene dianhydride (HQDEA) and 4,4′-diaminodicyclohexylmethane (DCHM) gave rise to a high Φ value of 0.11.3 It is worth mentioning that the PIs synthesized from HQDEA, 4,4′-oxydiphthalic anhydride (ODPA), 3,3′,4,4′-biphenyltetracarboxylic dianhydride (s-BPDA), and 4,4′-(4,4′-isopropylidenediphenoxy)diphthalic anhydride (BPADA) in combination with DCHM displayed prominent fluorescence from the lowest LE (π–π*) transition due to the highly conjugated dianhydride moieties. Meanwhile, PIs derived from pyromellitic dianhydride (PMDA), 2,3,3′,4′-biphenyltetracarboxylic dianhydride (a-BPDA), 4,4′-(hexafluoroisopropylidene)diphthalic anhydride (6FDA) and 3,3′,4,4′-benzophenone-tetracarboxylic dianhydride (BTDA) with DCHM showed almost no fluorescence because the lowest energy LE (n–π*) transitions are forbidden.
To achieve intense and large-Stokes-shifted fluorescence, MCs and PIs exhibiting fluorescence originating from the excited-state intramolecular proton transfer (ESIPT) process have been developed.17–25 ESIPT is a photophysical process, whereby the molecule is excited in the enol form and fluorescence is emitted from the excited keto form.26–30 Because of the distinct configurations undergoing the excitation and emission processes, ESIPT can induce large-Stokes-shifted fluorescence. The introduction of phenolic groups adjacent to an imide ring is a facile strategy for obtaining PIs with ESIPT emission.17–19,31 For example, our group has reported that a PI derived from 1-hydroxypyromellitic dianhydride (PHDA, Chart 1) and DCHM exhibited large-Stokes-shifted orange photoluminescence via ESIPT. However, because of its planar structure, the PHDA moiety is highly likely to form aggregates. The aggregated form generates a competitive photophysical process via ESIPT. As a result, the Φ value of the PHDA/DCHM PI film is as low as 0.07.18,25
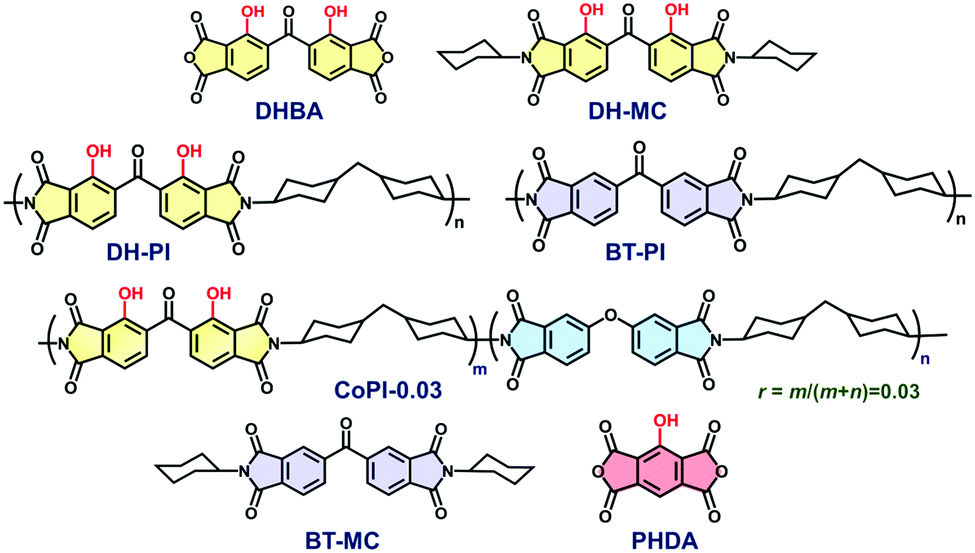 | ||
| Chart 1 Structures of dianhydrides (DHBA, PHDA), model compounds (DH-MC, BT-MC), and polyimides (DH-PI, BT-PI, and CoPI-0.03) derived from DHBA, ODPA, and BTDA dianhydrides and DCHM diamine. | ||
In this study, 2,2′-dihydroxybenzophenone-3,3′,4,4′-tetracarboxylic dianhydride (DHBA) was adopted as a new ESIPT dianhydride, and the optical properties of the PIs (DH-PI and CoPI) and MC (DH-MC) derived from DHBA are investigated (Chart 1). Compared with PHDA, the structure of DHBA is more flexible owing to the rotatable –CO– linkage, which may effectively suppress aggregation of the dianhydride moieties. Moreover, the MC and PI have two proton donors (–OH) adjacent to acceptors (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), enabling the formation of three different types of hydrogen (H)-bonded structures, which are expected to have distinct optical properties. For comparison, BT-PI and BT-MC (Chart 1) that do not emit ESIPT fluorescence were also prepared, and their optical properties were examined.
O), enabling the formation of three different types of hydrogen (H)-bonded structures, which are expected to have distinct optical properties. For comparison, BT-PI and BT-MC (Chart 1) that do not emit ESIPT fluorescence were also prepared, and their optical properties were examined.
2. Materials and methods
2.1 Materials
The reagents and purification procedures are listed in the ESI.†2.2 Synthesis
DHBA was synthesised as described in the literature with some modifications.32 The detailed synthetic procedure is illustrated in Scheme S1 in the ESI,† and the 1H-NMR spectra of the synthesised monomers are shown in Fig. S1 in the ESI.† The synthesis schemes and procedures for the model compounds (DH-MC, and BT-MC) and polyimides (DH-PI, BT-PI, and CoPI) are shown in Schemes S2–S5 (ESI†). The 1H and 13C NMR spectra of DH-MC are shown in Fig. S2 and S3 (ESI†). The synthesis and properties of BT-MC have been reported by Yamashita et al.33 The structures of the PIs were confirmed via their FT-IR spectra as shown in Fig. S4 (ESI†). The symmetric and anti-symmetric CO stretching vibrations in the imide ring (1778 and 1706 cm−1, respectively), and C–N stretching (1359 cm−1) at the imide–cyclohexane moiety are clearly observed, which can confirm the imide structure in the PIs. Due to the insolubility of the PIs in organic solvents, NMR spectra were not obtained for the PIs.
2.3 Measurements
 | (1) |
 | (2) |
2.4 Quantum chemical calculations
Density functional theory (DFT) calculations were conducted using Gaussian-16 software (Rev.C.01),35 as described in our previous studies.36,37 Geometry optimisation was independently conducted applying the B3LYP and CAM-B3LYP functionals with a 6-311G(d) basis set for the ground S0 and excited S1 states, respectively. The 6-311++G(d,p) basis set was used to calculate the vertical excitation wavelength and oscillator strength (f) for each of the S0 and S1 geometries. Each calculated transition was replaced by a Gaussian broadening function with a width of 0.10 eV, producing the shapes of the calculated spectra. The solvent effects in the ground and excited states were incorporated based on the polarizable continuum model (PCM) implemented in the Gaussian software.3. Results and discussion
3.1 Structure of H-bonds in DH-MC
The DH-MC molecule contains two phenolic groups as proton donors and three carbonyl groups as proton acceptors for H-bonding. Thus, DH-MC can form three types of conformers, MC-0, MC-1, and MC-2, which have distinct H-bond structures, as shown in Chart 2. The calculated total energy (E) values of these structures are listed in Table 1, where S0 and S1 denote the ground and excited states, respectively. Notably, the MC-2 conformer is energetically unstable in the S0 state, and geometry optimisation in the excited keto state, in which ESIPT occurs toward the central CO group, could not be achieved. By contrast, MC-1 showed the lowest E value among the enol forms, followed by MC-0, indicating that MC-1 is the most stable conformer in the ground state. Moreover, MC-1 also showed the lowest E value among the excited keto forms, indicating that MC-1 could be the most stable conformer in both the S0 and S1 states. As shown in Chart 2, intramolecular proton transfer occurs at the H-bond between the phenolic and imide CO groups in the excited state of MC-1, that is, the keto form. The proton-transferred keto conformer with the H-bond between the central CO and another phenolic group could not be optimised and suddenly relaxed to the enol form.
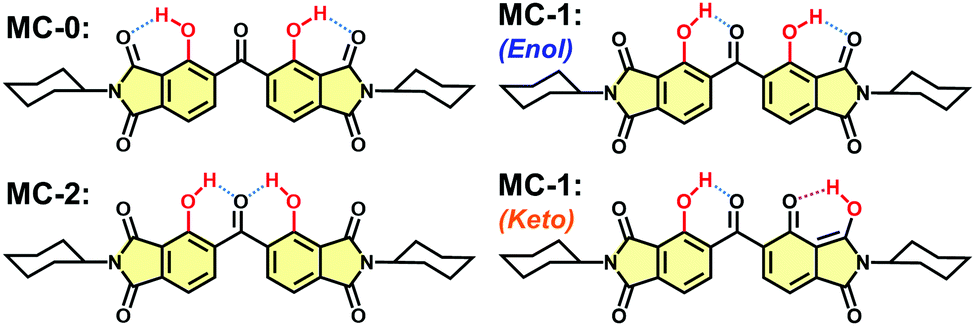 | ||
| Chart 2 Three possible H-bonding structures of DH-MC. The enol (S0) and keto (S1) conformers are shown for MC-1. | ||
The formation of H-bonds causes a shorter wavenumber shift in the stretching vibration of the carbonyl (>CO) group.38 For comparison, another MC that did not form intramolecular H-bonds was prepared using BTDA (BT-MC, Scheme S2, ESI†). Fig. 1 and Fig. S5 (ESI†) compare the FT-IR spectra of the DH-MC and BT-MC powders. For BT-MC, the symmetric and anti-symmetric stretching vibrations of the CO in the imide ring (1771 and 1703 cm−1, respectively), the symmetric stretching vibration of the central CO group (1663 cm−1), the aromatic CC stretching in the dianhydride moieties (1460 cm−1), and the C–N stretching in the main chain (1374 cm−1) were clearly observed. In DH-MC, only the stretching vibration of the central CO group has a short wavenumber shifted from 1663 to 1630 cm−1 due to H-bond formation. In addition, the peak at 1685 cm−1 is the short-wavenumber-shifted peak corresponding to the 1703 cm−1 peak (anti-symmetric stretching of CO in the imide ring), indicating the formation of H-bonds at the imide CO. The formation of H-bonds at two different CO groups was consistent with the MC-1 conformer. These experimental results agree well with the calculated FT-IR spectra of MC-1 (Fig. S6, ESI†).
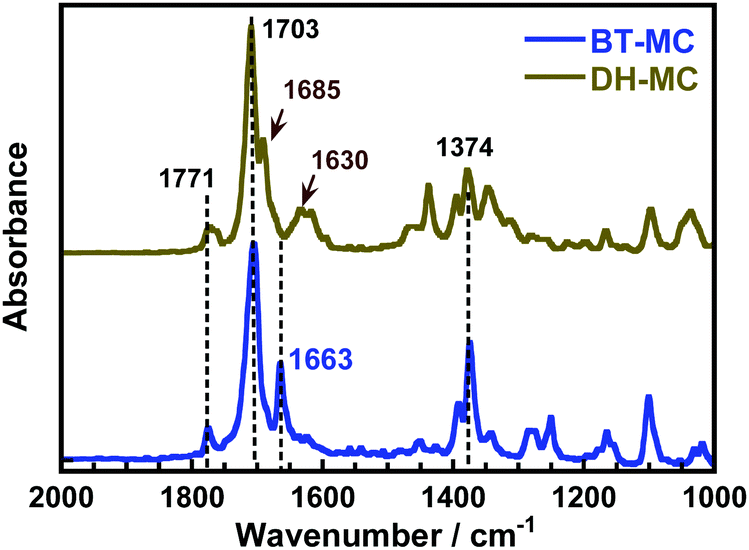 | ||
| Fig. 1 ATR FT-IR spectra of BT-MC and DH-MC powders. | ||
The precise conformation of DH-MC in the crystalline state was analysed using single-crystal XRD (Fig. 2), although complete structural refinements were hampered by the unsatisfactory quality of the crystals. Note that DH-MC only has the H-bonded asymmetric conformation represented by MC-1, which agrees well with the calculation results and the FT-IR spectrum. The torsion angles of O7–C29–C20–C19 are 70.9° in the crystal and 61.6° in the optimised geometry of the calculations (Fig. S7, ESI†). The intramolecular H-bond length of O3–O7 (2.612 Å) was considerably shorter than that of O6–O4 (3.002 Å). The planar structure of the phenolic and central CO groups is fixed by the short and strong H-bonds, and, interestingly, no double H-bonds are formed at the central CO group.
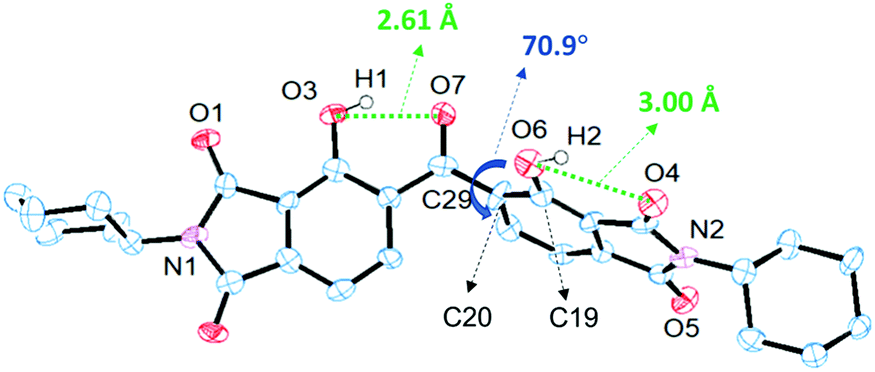 | ||
| Fig. 2 Crystal structure of DH-MC. | ||
Moreover, note that an obvious asymmetric structure is detected in the 13C NMR spectrum of DH-MC dissolved in CDCl3 (Fig. S3, ESI†). This can be attributed to a specific conformation in the solution induced by an asymmetric intramolecular hydrogen bond structure like MC-1. The 13C signals were assigned according to the calculated NMR spectra of MC-1 based on DFT.
Prior to evaluating the optical properties, the MC-1 electronic structures were examined. Table 2 lists the calculated values of the transition wavelengths, oscillator strengths (f), and dominantly contributing molecular orbitals (MOs) with assignments of one-electron transitions for the enol form of MC-1 in the optimised S0 geometry and those for the keto forms in the optimised S1 geometry. In addition, Fig. 3 and Fig. S8 (ESI†) illustrate the spatial distributions of the calculated MOs, where HOMO−m and LUMO+m denote the (m + 1)-th highest occupied MO and the (m + 1)-th lowest unoccupied MO, respectively. For the enol form of MC-1 in the ground state, the HOMO and HOMO−1 are located on the left-hand side of the dianhydride moiety, whereas the LUMO and LUMO+1 are uniformly distributed over the dianhydride moiety. Therefore, the HOMO → LUMO, HOMO → LUMO+1 and HOMO−1 → LUMO transitions are attributed to hybridised local and charge-transfer (HLCT) transitions.16,39–41 In addition, HOMO−2, HOMO−3, and HOMO−4 are predominantly located at the amine moieties. Thus, the transitions from these MOs to LUMO, LUMO+1, and LUMO+2 are attributable to CT (π–π*) transitions. Because of the significant overlap of the MOs, the oscillator strengths of the S0 → S1 (379 nm) and S0 → S3 (350 nm) transitions are larger than 0.1, which coincides well with the major absorptions in the experimental spectra.
| State | Transition wavelength (nm) | Oscillator strength | Orbitals | Assignment of transition | Contribution |
|---|---|---|---|---|---|
| Enol S1 | 379.28 | 0.1095 | HOMO–LUMO | HLCT (π–π*) | 0.47 |
| HOMO–LUMO+1 | HLCT (π–π*) | 0.01 | |||
| Enol-S2 | 370.77 | 0.0007 | HOMO−2–LUMO | CT (π–π*) | 0.24 |
| HOMO−2 – LUMO+1 | CT (π–π*) | 0.10 | |||
| Enol-S3 | 350.10 | 0.1450 | HOMO−1–LUMO | HLCT (π–π*) | 0.31 |
| Keto-S1 | 563.65 | 0.1924 | HOMO–LUMO | LE (π–π*) | 0.48 |
| Keto-S2 | 523.16 | 0.0247 | HOMO−1 – LUMO | CT (π–π*) | 0.49 |
| Keto-S3 | 494.28 | 0.0036 | HOMO−2–LUMO | CT (π–π*) | 0.26 |
| HOMO−4–LUMO | CT (π–π*) | 0.20 | |||
 | ||
| Fig. 3 Calculated molecular orbitals of the MC-1 (DH-MC) enol (S0) and keto (S1) forms. HOMO−m and LUMO+m denote the (m + 1)-th highest occupied orbital and the (m + 1)-th lowest unoccupied orbital, respectively. | ||
For the keto form of MC-1 in the excited state, the HOMO ← LUMO transition is attributable to the LE (π–π*) transition, and the HOMO and LUMO overlap spatially, in contrast to those of the enol form. Therefore, the oscillator strength of the S0 ← S1 transition increases considerably (f = 0.192), which indicates that MC-1 in the excited keto form is likely to exhibit strong emission. By contrast, TD-DFT calculations indicate that BT-MC exhibits almost no fluorescence because of the very low oscillator strengths for the S0 → S1 transition (370 nm, f = 0.0021) and the S0 ← S1 transition (443 nm, f = 0.0015) due to their LE (n–π*) nature (Fig. S9, Table S1, ESI†).
3.2 Optical properties of DH-MC
The steady-state UV-vis absorption and emission spectra of DH-MC in toluene solution (1 × 10−5 M) are displayed in Fig. 4. The three absorption peaks observed at 380, 370, and 340 nm are attributed to the CT (π–π*) transitions (MC-1, Table 2) of the S0 → S1, S0 → S2, and S0 → S3 states, respectively. Furthermore, prominent orange fluorescence was observed at 595 nm when DH-MC was excited at 350 nm, which was attributed to the large-Stokes-shifted (11905 cm−1) fluorescence originating from ESIPT. Note that the Φ value of this ESIPT fluorescence was as low as 0.01 in solution. One possible reason is that the vigorous rotational motion of DH-MC in solution enhances non-radiative deactivation.
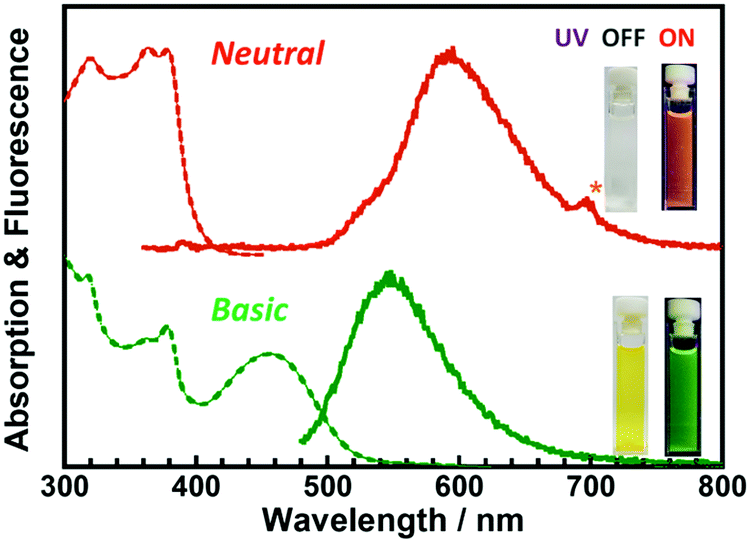 | ||
| Fig. 4 Steady-state UV-vis absorption and fluorescence spectra of DH-MC dissolved in toluene (λex = 350 nm; 10−5 M,) and toluene/DBU (λex = 460 nm; [DH-MC], 10−5 M; [DBU], 10−2 M). Insets are photos of DH-MC solutions under sunlight and UV light (375 nm). | ||
It has been reported that ESIPT molecules are sensitive to environmental acidity/basicity due to the presence of proton donors, such as –OH or –NH. Fig. 4 and Fig. S10 (ESI†) show the changes in the steady-state UV-vis absorption and emission spectra of DH-MC dissolved in toluene (1 × 10−5 M) before and after the addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU; 1 × 10−2 M), an organic base. Owing to deprotonation by DBU, anionic or dianionic species can be generated. Thus, new absorption and corresponding fluorescence emission peaks were observed at 460 and 530 nm, respectively. The DH-MC solution was colourless and exhibited orange fluorescence under neutral conditions, whereas the solution became yellow and exhibited green fluorescence under basic conditions, resulting from the anion/dianion of DH-MC. The excitation and emission spectra of DH-MC dissolved in toluene (λem = 525, 595 nm) are shown in Fig. S11 (ESI†). When the fluorescence is monitored at 525 nm, a small distinct peak is observed at 460 nm in the excitation spectrum. Since this peak position (460 nm) is the same as that of the absorption peak under basic conditions (Fig. 4), a small amount of anionic species generated by spontaneous deprotonation exists even in the neutral state.
Our group has reported that an ESIPT-MC having two phenolic groups at the pyromellitic dianhydride moiety appeared red or blue depending on the DBU concentration (10−3 M and 1 M) due to the successive formation of anions and dianions, respectively.17 Because DH-MC may also generate both anionic and dianionic species, a series of DH-MC/toluene solutions ([DH-MC] = 1 × 10−5 M) were prepared at DBU concentrations ranging from 0 to 1 M. However, as shown in Fig. S10 (ESI†), an increase in DBU concentration from 10−5 M to 10−1 M did not significantly affect the UV-vis absorption and emission spectra. This finding may indicate that both the anion and dianion of DH-MC show similar optical properties because the two phenolic groups are attached to different benzene rings, whereas the two phenolic groups are attached to the same benzene ring in our previously reported ESIPT-MC.17 The Stokes-shift of the anionic form is smaller (6830 cm−1) than that of the ESIPT emission.
Fig. 5 displays the steady-state excitation/emission spectra of DH-MC in the powder solid state. Although the sample was excited at 340 nm, close to the solution, the emission peak was observed at 540 nm, which is much shorter than the emission in the solution state (595 nm). This is due to the vigorous molecular motion in the solution state, wherein the averaged conformation probably resembles that optimised by the TD-DFT calculations. Thus, the emission wavelength in the solution was similar to the calculated result. However, in the solid-state, the conformation of DH-MC is fixed and differs from that in the solution. Furthermore, another fluorescence peak with λex = 450 nm and λem = 540 nm was observed for the DH-MC powder, which originates from the anionic form of DH-MC, and its emission peak overlapped with the prominent keto emission. Since imide compounds contain plural proton-accepting groups such as >CO and N–, spontaneous deprotonation of the phenolic –OH occurs in the solid state. By contrast, BT-MC dissolved in toluene presented neither prominent absorption over 350 nm (Fig. S12, ESI†) nor fluorescence, which is consistent with the TD-DFT calculation results for BT-MC.
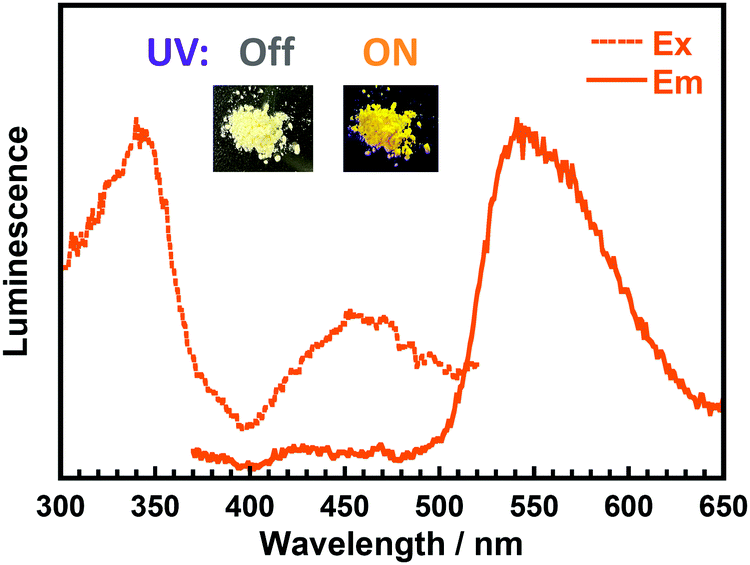 | ||
| Fig. 5 Steady-state excitation/emission spectra of DH-MC powder (λex = 340 nm, λem = 540 nm). Insets are photos of DH-MC powder under sunlight and under UV light (375 nm). | ||
3.3 Optical properties of DH-PI and CoPI
The DH-PI film demonstrates similar optical properties to those of the DH-MC powder. As shown in Fig. 6a, the DH-PI film exhibited two major absorption peaks at 340 and 470 nm. The former is attributable to the HLCT (π–π*) transition of the enol form, and the latter is attributable to the anionic form of the DHBA moiety. As shown in Fig. 6b, two adjacent fluorescence peaks were observed for the DH-PI film. The emission peak observed at 550 nm when the film was excited at 340 nm exhibited a large Stokes shift (v = 11230 cm−1), which originates from the ESIPT process. Another fluorescence peak at 540 nm when excited at 470 nm with a small Stokes shift (v = 2758 cm−1) originates from the anionic form of the DHBA moieties, and these peaks are comparable to those of the DHBA anions in toluene. As a result, the DH-PI film shows a yellowish colour and yellow fluorescence. As mentioned above, the yellowish colour of the DH-PI film is caused by the anionic form of DHBA, which is unfavourable for wavelength-converting applications. Moreover, the DH-PI film shows a relatively low Φ value of 0.04 due to the aggregation of the DHBA moieties. Comparably, a PI derived from BTDA (BT-PI) showed a very weak emission at ∼490 nm when excited at 440 nm (Fig. S13, ESI†). As this emission is not observed in the BT-MC solution, and the BT-PI film shows a relatively intense phosphorescence at ∼500 nm at lower temperatures (Fig. S14, ESI†) similar to that of the benzophenone crystal,42 this emission is attributable to room-temperature phosphorescence.
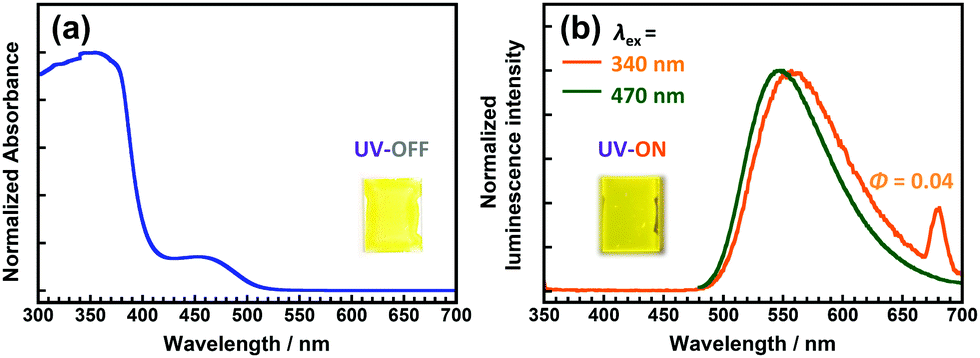 | ||
| Fig. 6 Steady-state (a) UV-vis absorption and (b) emission spectra of the DH-PI film. Insets are photos of the DH-PI film under sunlight and under UV light (375 nm). | ||
Although the DH-PI film exhibits large-Stokes-shifted yellow fluorescence (v = 10893 cm−1), it has an intense yellow appearance that originates from the anionic form generated by spontaneous deprotonation. Doping a small amount of sulfuric acid (H2SO4) in the poly(amic acid) solution effectively suppresses the characteristic absorption of the anionic form,25 but the addition of 2 equiv. of H2SO4 caused microphase separation, generating a haze due to light scattering (Fig. S15, ESI†). Another effective method for improving the colourlessness and transparency is copolymerization with a colourless PI.25,43 4,4′-Oxydiphthalic anhydride (ODPA) was chosen as the major component of the dianhydrides for preparing a copolyimide (CoPI) owing to the excellent colourlessness and intense blue fluorescence of ODPA/DCHM PI, which is attributable to the LE (π ← π*) transition of the ODPA moiety.3 The molar ratio of DHBA units in this CoPI was set to 0.03 (CoPI-0.03), while that of the ODPA units was 0.97. As shown in Fig. 7, the CoPI-0.03 film shows good colourlessness with a weak absorption peak at 470 nm originating from the anionic form of the DHBA moiety. Furthermore, this film exhibited weak blue fluorescence at 400 nm and intense yellow fluorescence at 542 nm when excited at 340 nm. The former originates from the LE (π ← π*) transition of the OPDA moiety, while the latter is assigned to the ESIPT fluorescence of the DHBA moiety. Although the molar fraction of DHBA was considerably lower than that of ODPA, prominent ESIPT fluorescence was observed, indicating that efficient energy transfer occurred from ODPA to DHBA in the PI chain.
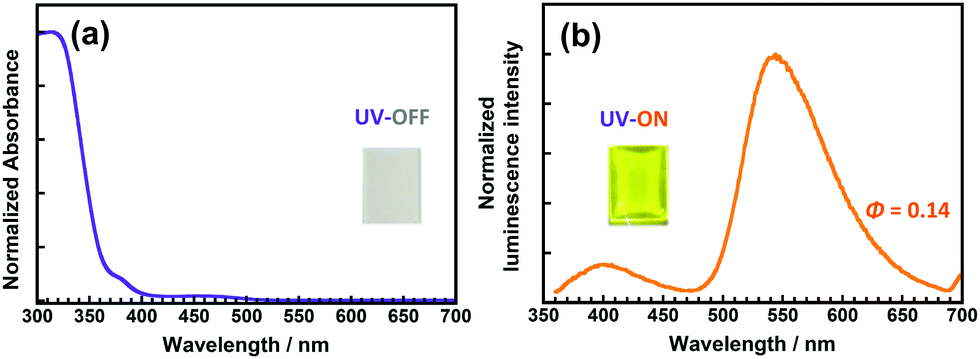 | ||
| Fig. 7 Steady-state (a) UV-vis absorption and (b) emission (λex = 340 nm) spectra of the CoPI-0.03 film. Insets are photos of the CoPI-0.03 film under sunlight and under UV light (375 nm). | ||
The energy-transfer efficiency (EFRET) can be estimated from the variation in the fluorescence lifetime τ of the energy donor (ODPA moiety),43,44 as follows:
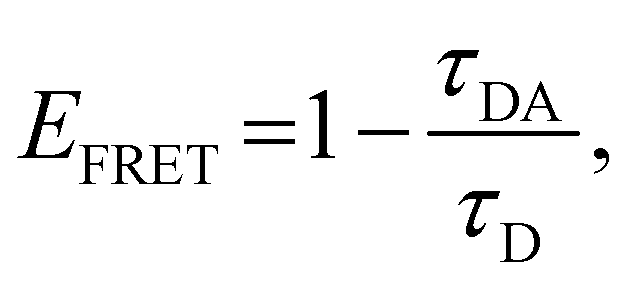 | (3) |
For practical applications that entail solar spectral conversion, sunlight (AM1.5) includes radiation with a wide range of wavelengths, from UV (∼300 nm) to near-infrared (∼1200 nm). Therefore, we generated a wavelength-converting spectrum (WCS), which plots the difference between the spectra obtained with and without the CoPI-0.03 film formed on fused silica substrates using a xenon light source, as we have previously reported.25Fig. 8 displays the WCS of CoPI-0.03, wherein the strong UV absorptions at 340 and 380 nm correspond to the absorptions of the ODPA and PHDA units, respectively. Note that the broad positive band observed at ∼540 nm agrees well with the ESIPT yellow fluorescence emitted from the DHBA unit. Accordingly, this CoPI film can function as an efficient solar spectral converter, which absorbs sunlight UV radiation and enhances the yellow light over a wide wavelength range without absorbing visible radiation.
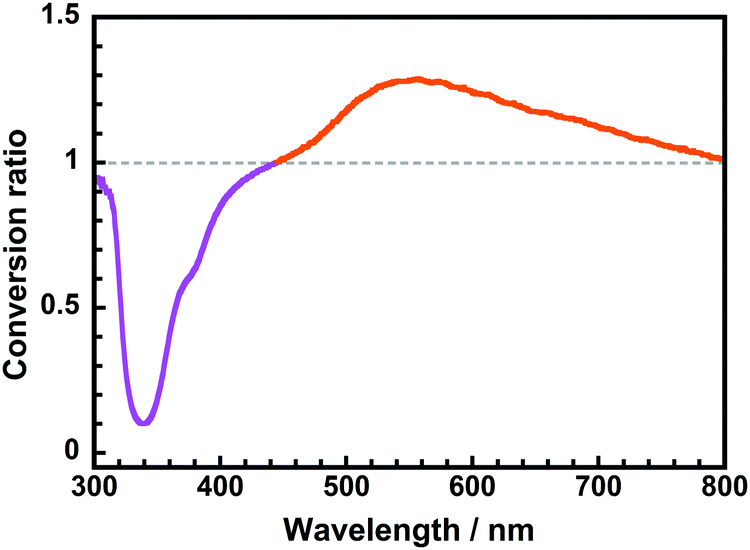 | ||
| Fig. 8 Wavelength-converting spectrum of the CoPI-0.03 film. | ||
4. Conclusions
2,2′-Dihydroxybenzophenone-3,3′,4,4′-tetracarboxylic dianhydride (DHBA) was synthesised, and the structures and optical properties of novel polyimide (DH-PI), copolyimide (CoPI-0.03), and a model compound (DH-MC) derived from DHBA were investigated. DH-MC can form three types of conformers (MC-0, MC-1, and MC-2) with distinct H-bond structures. DFT calculations show that MC-1 is the most stable conformer, and the results of FT-IR spectroscopy and single-crystal XRD analysis are consistent with the calculated results. In the solid-state, DH-MC and DH-PI showed large-Stokes-shifted (v > 10000 cm−1) fluorescence via the excited-state intramolecular proton transfer (ESIPT) process. The ESIPT process absorbs UV light at 340 nm and emits yellow light at 550 nm. Another fluorescence peak was observed at 540 nm using an excitation wavelength of 470 nm, originating from the anionic form of the DHBA moiety. Due to spontaneous deprotonation, DH-MC and DH-PI show yellow coloration in the solid state. The relatively low fluorescence quantum yield of the DH-PI film (Φ = 0.04) is attributable to the aggregation of the DHBA moieties.
To achieve highly fluorescent colourless PI films with high quantum yields, copolyimide (CoPI) was synthesised using DHBA and ODPA dianhydrides, whereby the molar ratio of DHBA was set to 0.03. Owing to efficient energy transfer (EFRET > 0.7) from the ODPA to the DHBA moieties, the CoPI-0.03 film exhibited prominent yellow ESIPT fluorescence from the DHBA moiety when excited at 340 nm. In addition, this CoPI film displays good colourlessness and a sufficiently high quantum yield (Φ = 0.14) owing to the suppression of DHBA aggregation, which enables the conversion of UV radiation from sunlight to yellow visible light, as evidenced in the wavelength-converting spectrum.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Ms Mayuko Nara and Ms Marina Doi at Tokyo Institute of Technology for their help with the synthesis and optical measurements. This work was financially supported by JSPS KAKENHI Grant Numbers 17H03112 and 21H01995. One of the authors (NL) thanks the China Scholarship Council (CSC).References
- C. E. Sroog, Polyimides, J. Polym. Sci., Macromol. Rev., 1976, 11, 161–208 CrossRef CAS.
- Y. Zhuang, J. G. Seong and Y. M. Lee, Polyimides containing aliphatic/alicyclic segments in the main chains, Prog. Polym. Sci., 2019, 92, 35–88 CrossRef CAS.
- J. Wakita, H. Sekino, K. Sakai, Y. Urano and S. Ando, Molecular Design, Synthesis, and Properties of Highly Fluorescent Polyimides, J. Phys. Chem. B, 2009, 113, 15212–15224 CrossRef CAS PubMed.
- K. Takizawa, J. Wakita, K. Sekiguchi and S. Ando, Variations in aggregation structures and fluorescence properties of a semialiphatic fluorinated polyimide induced by very high pressure, Macromolecules, 2012, 45, 4764–4771 CrossRef CAS.
- K. Kanosue, S. Hirata, M. Vacha, R. Augulis, V. Gulbinas, R. Ishige and S. Ando, A colorless semi-aromatic polyimide derived from a sterically hindered bromine-substituted dianhydride exhibiting dual fluorescence and phosphorescence emission, Mater. Chem. Front., 2019, 3, 39–49 RSC.
- Y. Zhuang, R. Orita, E. Fujiwara, Y. Y. Zhang and S. Ando, Colorless Partially Alicyclic Polyimides Based on Tröger’s Base Exhibiting Good Solubility and Dual Fluorescence/Phosphorescence Emission, Macromolecules, 2019, 52(10), 3813–3824 CrossRef CAS.
- E. Fujiwara, R. Orita, A. Vyšniauskas, M. Franckevičius, R. Ishige, V. Gulbinas and S. Ando, Ultrafast Spectroscopic Analysis of Pressure-Induced Variations of Excited-State Energy and Intramolecular Proton Transfer in Semi-Aliphatic Polyimide Films, J. Phys. Chem. B, 2021, 125, 2425–2434 CrossRef CAS PubMed.
- B. S. Richards, Enhancing the performance of silicon solar cells via the application of passive luminescence conversion layers, Sol. Energy Mater. Sol. Cells, 2006, 90, 2329–2337 CrossRef CAS.
- W. G. J. H. M. van Sark, K. W. J. Barnham, L. H. Slooff, A. J. Chatten, A. Büchtemann, A. Meyer, S. J. McCormack, R. Koole, D. J. Farrell, R. Bose, E. E. Bende, A. R. Burgers, T. Budel, J. Quilitz, M. Kennedy, T. Meyer, C. D. M. Donegá, A. Meijerink and D. Vanmaekelbergh, Luminescent Solar Concentrators - A review of recent results, Opt. Express, 2008, 16, 21773 CrossRef CAS PubMed.
- M. J. Currie, J. K. Mapel, T. D. Heidel, S. Goffri and M. A. Baldo, High-efficiency organic solar concentrators for photovoltaics, Science, 2008, 321, 226–228 CrossRef CAS PubMed.
- C. Haines, M. Chen and K. P. Ghiggino, The effect of perylene diimide aggregation on the light collection efficiency of luminescent concentrators, Sol. Energy Mater. Sol. Cells, 2012, 105, 287–292 CrossRef CAS.
- M. Rafiee, S. Chandra, H. Ahmed and S. J. McCormack, An overview of various configurations of Luminescent Solar Concentrators for photovoltaic applications, Opt. Mater., 2019, 91, 212–227 CrossRef CAS.
- G. Arjavalingam, G. Hougham and J. P. LaFemina, Emission mechanism in polyimide, Polymer, 1990, 31, 840–844 CrossRef CAS.
- M. Hasegawa and K. Horie, Photophysics, photochemistry, and optical properties of polyimides, Prog. Polym. Sci., 2001, 26, 259–335 CrossRef CAS.
- A. Demeter, T. Berces, L. Biczok, V. Wintgens, P. Valat and J. Kossanyi, Comprehensive model of the photophysics of W-phenylnaphthalimides: The role of solvent and rotational relaxation, J. Phys. Chem., 1996, 100, 2001–2011 CrossRef CAS.
- R. Orita, M. Franckevičius, A. Vyšniauskas, V. Gulbinas, H. Sugiyama, H. Uekusa, K. Kanosue, R. Ishige and S. Ando, Enhanced fluorescence of phthalimide compounds induced by the incorporation of electron-donating alicyclic amino groups, Phys. Chem. Chem. Phys., 2018, 20, 16033–16044 RSC.
- K. Kanosue, T. Shimosaka, J. Wakita and S. Ando, Polyimide and Imide Compound Exhibiting Bright Red Fluorescence with Very Large Stokes Shifts via Excited-State Intramolecular Proton Transfer, Macromolecules, 2015, 48, 1777–1785 CrossRef CAS.
- K. Kanosue, R. Augulis, D. Peckus, R. Karpicz, T. Tamulevičius, S. Tamulevičius, V. Gulbinas and S. Ando, Polyimide and Imide Compound Exhibiting Bright Red Fluorescence with Very Large Stokes Shifts via Excited-State Intramolecular Proton Transfer II. Ultrafast Proton Transfer Dynamics in the Excited State, Macromolecules, 2016, 49, 1848–1857 CrossRef CAS.
- J. Wakita, S. Inoue, N. Kawanishi and S. Ando, Excited-state intramolecular proton transfer in imide compounds and its application to control the emission colors of highly fluorescent polyimides, Macromolecules, 2010, 43, 3594–3605 CrossRef CAS.
- K. Kanosue and S. Ando, Fluorescence emissions of imide compounds and end-capped polyimides enhanced by intramolecular double hydrogen bonds, Phys. Chem. Chem. Phys., 2015, 17, 30659–30669 RSC.
- A. Tabuchi, T. Hayakawa, S. Kuwata, R. Ishige and S. Ando, Full-colour solvatochromic fluorescence emitted from a semi-aromatic imide compound based on ESIPT and anion formation, Mater. Adv., 2021, 2, 5629–5638 RSC.
- H. Okamoto, K. Itani, M. Yamaji, H. Konishi and H. Ota, Excited-state intramolecular proton transfer (ESIPT) fluorescence from 3-amidophthalimides displaying RGBY emission in the solid state, Tetrahedron Lett., 2018, 59, 388–391 CrossRef CAS.
- M. Fujii, M. Namba, M. Yamaji and H. Okamoto, Solvent-induced multicolour fluorescence of amino-substituted 2,3-naphthalimides studied by fluorescence and transient absorption measurements, Photochem. Photobiol. Sci., 2016, 15, 842–850 CrossRef CAS.
- L. Wang, M. Fujii, M. Namba, M. Yamaji and H. Okamoto, Fluorescence properties of amido-substituted 2,3-naphthalimides: Excited-state intramolecular proton transfer (ESIPT) fluorescence and responses to Ca2+ ions, Tetrahedron Lett., 2019, 60, 151189 CrossRef CAS.
- N. Liang, E. Fujiwara, M. Nara, R. Ishige and S. Ando, Colorless Copolyimide Films Exhibiting Large Stokes-Shifted Photoluminescence Applicable for Spectral Conversion, ACS Appl. Polym. Mater., 2021, 3, 3911–3921 CrossRef CAS.
- L. Chen, P. Fu, H. Wang and M. Pan, Excited-State Intramolecular Proton Transfer (ESIPT) for Optical Sensing in Solid State, Adv. Opt. Mater., 2021, 2001952 CrossRef.
- A. C. Sedgwick, L. Wu, H. Han, S. D. Bull, X. He, T. D. James and J. L. Sessler, Excited-state intramolecular proton-transfer (ESIPT) based fluorescence sensors and imaging agents, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- J. E. Kwon and S. Y. Park, Advanced organic optoelectronic materials: Harnessing excited-state intramolecular proton transfer (ESIPT) process, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS.
- V. S. Padalkar and S. Seki, Excited-state intramolecular proton-transfer (ESIPT)-inspired solid state emitters, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- A. P. Demchenko, V. I. Tomin and P. T. Chou, Breaking the Kasha Rule for More Efficient Photochemistry, Chem. Rev., 2017, 117, 13353–13381 CrossRef CAS PubMed.
- N. Liang, E. Fujiwara, M. Nara, R. Ishige and S. Ando, Photoluminescence properties of novel fluorescent polyimide based on excited state intramolecular proton transfer at the end groups, J. Photopolym. Sci. Technol., 2019, 32, 449–455 CrossRef CAS.
- G. J. Shin, J. C. Jung, J. H. Chi, T. H. Oh and J. B. Kim, Synthesis and micropatterning properties of a novel base-soluble, positive-working, photosensitive polyimide having ano-nitrobenzyl ether group, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 776–788 CrossRef CAS.
- T. Yamashita, T. Kudo, Y. Yoshida and S. Tagawa, Transient Absorption Spectra of Electron Beam Sensitive Polyimides and their Model Compounds, J. Photopolym. Sci. Technol., 2005, 18, 699–705 CrossRef CAS.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct, Chem., 2015, 71, 3–8 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Ando, T. Fujigaya and M. Ueda, DFT Calculations of Photoabsorption Spectra in the VUV Region for Design of Photoresist Materials for 157 nm Lithography, J. Photopolym. Sci. Technol., 2002, 15, 559–568 CrossRef CAS.
- S. Ando and M. Ueda, DFT Calculations of Photoabsorption Spectra for Alicyclic and Heterocyclic Compounds in the VUV Region, J. Photopolym. Sci. Technol., 2003, 16, 537–544 CrossRef CAS.
- M. A. Varfolomeev, D. I. Abaidullina, A. Z. Gainutdinova and B. N. Solomonov, FTIR study of H-bonds cooperativity in complexes of 1,2-dihydroxybenzene with proton acceptors in aprotic solvents: Influence of the intramolecular hydrogen bond, Spectrochim. Acta, Part A, 2010, 77, 965–972 CrossRef PubMed.
- L. Yao, Y. Pan, X. Tang, Q. Bai, F. Shen, F. Li, P. Lu, B. Yang and Y. Ma, Tailoring Excited-State Properties and Electroluminescence Performance of Donor-Acceptor Molecules through Tuning the Energy Level of the Charge-Transfer State, J. Phys. Chem. C, 2015, 119, 17800–17808 CrossRef CAS.
- T. Yoshihara, S. I. Druzhinin and K. A. Zachariasse, Fast intramolecular charge transfer with a planar rigidized electron donor/acceptor molecule, J. Am. Chem. Soc., 2004, 126, 8535–8539 CrossRef CAS PubMed.
- K. A. Zachariasse, S. I. Druzhinin, W. Bosch and R. Machinek, Intramolecular Charge Transfer with the Planarized 4-Aminobenzonitrile 1-tert-Butyl-6-cyano-1,2,3,4-tetrahydroquinoline (NTC6), J. Am. Chem. Soc., 2004, 126, 1705–1715 CrossRef CAS PubMed.
- J. L. Laporte, G. Nouchi and Y. Rousset, Phosphorescence of benzophenone crystals, J. Chem. Phys., 1972, 57, 1767–1769 CrossRef CAS.
- M. Nara, R. Orita, R. Ishige and S. Ando, White-Light Emission and Tunable Luminescence Colors of Polyimide Copolymers Based on FRET and Room-Temperature Phosphorescence, ACS Omega, 2020, 5, 14831–14841 CrossRef CAS.
- I. V. Gopich and A. Szabo, Theory of the energy transfer efficiency and fluorescence lifetime distribution in single-molecule FRET, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7747–7752 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of compounds with 1H NMR spectra; FT-IR spectra, TD-DFT-calculated FT-IR spectra; TD-DFT-calculated conformation of MC-1; calculated molecular orbitals of MC-1, calculated molecular orbitals of BT-MC; calculated electronic transition of BT-MC; steady-state UV-vis absorption spectra and emission spectra of DH-MC dissolved in toluene/DBU; steady-state UV-vis absorption spectra of BT-MC dissolved in toluene; steady-state UV-vis absorption spectra and excitation/emission spectra of BT-PI film; temperature-dependent fluorescence and phosphorescence spectra of the BT-PI film; steady-state UV-vis absorption spectra and emission spectra of DH-PI films with different H2SO4 concentrations; average photoluminescence lifetimes, energy transfer efficiencies and total quantum yields of ODPA/DCHM PI, DH-PI, and CoPI films. CCDC 2117075. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qm01280k |
| This journal is © the Partner Organisations 2022 |