
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient synthesis of thermoplastic elastomeric amorphous ultra-high molecular weight atactic polypropylene (UHMWaPP)†
Clement G.
Collins Rice
 ,
Jean-Charles
Buffet
,
Zoë R.
Turner
and
Dermot
O'Hare
*
,
Jean-Charles
Buffet
,
Zoë R.
Turner
and
Dermot
O'Hare
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: dermot.ohare@chem.ox.ac.uk
First published on 14th September 2022
Abstract
The selective and high-yielding synthesis of fully amorphous ultra-high molecular weight atactic polypropylene (UHMWaPP), with weight-average molecular weights (Mw) up to 2.0 MDa and narrow polydispersities is reported; permethylindenyl-phenoxy (PHENI*) titanium complexes immobilised on solid polymethylaluminoxane (sMAO) are remarkably efficient single-site catalysts for propylene polymerisation, with activities up to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kgPP molTi−1 h−1 bar−1. UHMWaPP was found to be a high-performance thermoplastic elastomer, exhibiting an ultimate tensile strength of 1.08 MPa (at 184% strain), and remarkably high ductility, with a tensile strain at break of >1900%. Excellent elastic recovery is observed with a set value as low as 7% in stress–strain hysteresis experiments. The mechanical properties reported are compared with commercial olefinic thermoplastic elastomers, demonstrating the potential of this technology to compete with commercial copolymers and composites as an elastic homopolymer.
000 kgPP molTi−1 h−1 bar−1. UHMWaPP was found to be a high-performance thermoplastic elastomer, exhibiting an ultimate tensile strength of 1.08 MPa (at 184% strain), and remarkably high ductility, with a tensile strain at break of >1900%. Excellent elastic recovery is observed with a set value as low as 7% in stress–strain hysteresis experiments. The mechanical properties reported are compared with commercial olefinic thermoplastic elastomers, demonstrating the potential of this technology to compete with commercial copolymers and composites as an elastic homopolymer.
Introduction
Commercially, polypropylene (PP) is predominantly synthesised using conventional Ziegler–Natta (ZN) catalyst technologies.1 The inherent chirality of the titanium centres on the TiCl3 crystal surface results in highly crystalline isotactic polymers.2 It is clear from detailed computational modelling of these mechanistically elusive systems that sterics play a key role in enforcing stereoselectivity, with the –CH3 group of the propylene interacting with chlorides adjacent to the metal centre.3 Tacticity and molecular weights (Mw) are highly coupled in these systems,4 with low Mw partially crystalline atactic PP (aPP) obtained as a by-product.5 The isotactic PP (iPP) produced in this manner has a non-uniform distribution of stereodefects, resembling a stereoblock microsctructure in contrast to the uniform polymers produced by single-site metallocene catalysts; it has been claimed that this is responsible for the difference in physical properties of these two classes of polymers.6 Group four metallocene and post-metallocene catalysts are typically used where independent control over polymer tacticity and molecular weight are desired and have been shown to be particularly effective in the synthesis of elastomeric high molecular weight aPP which is inaccessible using Ziegler–Natta technology.7,8 The relationship between the symmetry of single-site catalysts and resulting polypropylene tacticity is well understood,9,10 with complexes in the C2v and C2 point groups typically producing atactic and isotactic PP respectively, while asymmetric C1 complexes are reported to produce polymers with microstructures ranging from highly isotactic to atactic.9 Stereoselectivity in prochiral monomer insertion results principally from enantiomorphic site control with steric constraints enforcing a preference for either the re or si configuration of the monomer. C2-ansa-metallocenes are particularly effective, with the bridge requiring conformational rigidity of the ligand.11,12Amorphous high molecular weight aPP (HMWaPP, Mw > 200 kDa) is a material of growing industrial interest; beneficial elastic and optical properties lead to broad applications as adhesives, compatibilisers and as additives in a range of polymeric materials.5,7 There has been limited development of catalysts that are selective for the synthesis of such materials, with metallocene and post-metallocene complexes of titanium and zirconium receiving the most attention.8,13 The synthesis of ultra-high molecular weight aPP (UHMWaPP, Mw > 1 MDa) has been reported, though typically only in relatively low-yielding processes.13,14 Ultra-high molecular weight homopolymers of higher olefins from 1-butene to 1-octadecene have also been reported, typically in moderate yields or under forcing low-temperature conditions, with relatively limited specific catalyst development, exploration of polymer properties, and few proposed applications.15–19
Ultra-high molecular weight homopolypropylene elastomers, synthesised by Rieger et al. as stereodefective isotactic PP, were shown to have beneficial mechanical properties, including improved elastic recovery compared to lower Mw elastomeric PP.20 Homopolymer olefinic thermoplastic elastomers would offer beneficial processability and recyclability when compared with more conventional cross-linked rubbers.21,22 One such class of elastomer, ethylene propylene rubber (EPM), is commercially synthesised using highly active titanium cyclopentadienyl-amidinate complexes.23
Post-metallocene ansa-bridged complexes based on nitrogen and oxygen donors – such as the Constrained Geometry Complexes (CGCs),24,25 Phenoxy-Induced Complexes of Sumitomo (PHENICS),26–28 and our recently-reported permethylindenyl (C9Me6, Ind*, I*) analogue, PHENI*29 – have been shown to be highly active catalysts for ethylene polymerisation, with the latter producing substantially disentangled UHMWPE and demonstrating a remarkable 22-fold increase in activity over the analogous PHENICS complex. In the polyethylene case, solid polymethylaluminoxane (sMAO) has proven to be a highly competent dual-function activating catalyst support, forming strongly immobilised metallocene catalysts for use in heterogeneous slurry-phase reactions. These systems can produce polyolefins with single-site catalysis characteristics.30–32 Based on these results, we hypothesised that the PHENI* complexes would also be competent catalysts for UHMWPP.
In this work, we report the application of permethylindenyl-phenoxy (PHENI*) complexes in the homopolymerisation of propylene, both in solution-phase and sMAO-supported regimes. We demonstrate extremely high activities, excellent control, and the selective production of high and ultra-high molecular weight amorphous atactic polypropylene. The polypropylenes are shown to have excellent tensile and elastic properties, comparable to commercial engineering thermoplastic elastomers.
Results and discussion
Synthesis of complexes and pre-catalysts
Both CGCs and PHENICS complexes have been shown to have better catalytic activity than more traditional Kaminsky metallocenes utilising hydrocarbon ligands. Increased molecular weights are also observed in addition to more effective comonomer enchainment.24,33,34 The PHENICS catalysts are particularly notable for their very high temperature stability, with high activities maintained even at elevated temperatures (>200 °C).28 The permethylindenyl ligand has previously been shown to enhance the activity of Cp*-based ligands both in metallocene and CGC analogues.25,35–42 This is attributed to facile η5,η3-haptotropic ring slip – which lowers the barrier to associative addition;43–46 the steric kinetic stabilisation of the ligand;47 and the positive inductive effect of polyalkylation resulting from σ–π hyperconjugation.48 We have previously introduced the PHENI* ligand which is a development of PHENICS ligands, incorporating the many benefits of I*, and which shows higher activities both than the I* CGC analogues introduced by Williams et al. and the indenyl PHENICS analogue.25,29,43Titanium dichloride complexes 1–3, utilising PHENI* ligands, Me2SB(R1R2ArO,I*)TiCl2 ({(η5-C9Me6)Me2Si(2-R1-4-R2-C6H2O)}TiCl2; R1, R2 = Me, tBu, or CMe2Ph), were synthesised as previously reported.29 These complexes, and additionally the indenyl-PHENICS complex 4 (Me2SB(tBu,MeArO,I)TiCl2; {(η5-C9H6)Me2Si(2-tBu-4-Me-C6H2O)}TiCl2; I = C9H6), were then immobilised on sMAO following a standard procedure,49 with [AlsMAO]/[Ti] = 200, to afford 1sMAO–4sMAO (Chart 1).
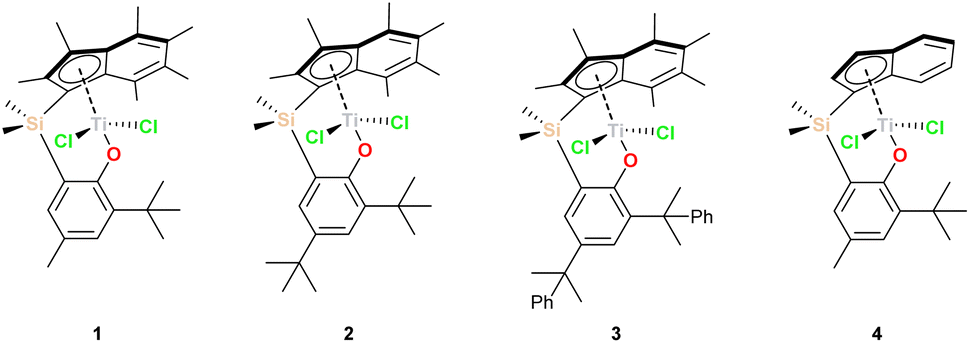 | ||
| Chart 1 Complexes used in this study. | ||
Polymerisation of propylene to afford ultra-high molecular weight polypropylene
Initial polymerisation studies using sMAO-supported solid catalysts were performed in 150 mL ampoules using 10 mg 1sMAO–4sMAO with 2 bar propylene in 50 mL hexanes for 30 minutes with MAO ([AlMAO]/[Ti] = 1000) acting as co-catalytic initiator and scavenger across a temperature of polymerisation (Tp) range of 60 ≤ Tp ≤ 80 °C (Table 1). As was found previously for ethylene polymerisation using supported catalysts,292sMAO displays the highest activity, 3200 kgPP molTi−1 h−1 bar−1 at 60 °C, with the trend 2sMAO > 1sMAO > 3sMAO. Comparing the indenyl and permethylindenyl analogues (1sMAO and 4sMAO) – a larger increase in activity is seen even than for ethylene polymerisation, being 26-fold greater for the I* complex at Tp = 60 °C. The weight-average molecular weight (Mw) of the resulting polypropylenes was determined by gel permeation chromatography (GPC) (Table 1 and Fig. 1, 2). The PHENI* sMAO supported catalyst 1sMAO produced aPP with a 5-fold higher Mw than the analogous PHENICS sMAO supported catalyst, 4sMAO, as well as having reduced polydispersity (PDI, Đ). sMAO supported catalyst 2sMAO produced the highest molecular weight polymers of the three PHENI* supported catalysts studied, up to 630 kDa at Tp = 60 °C.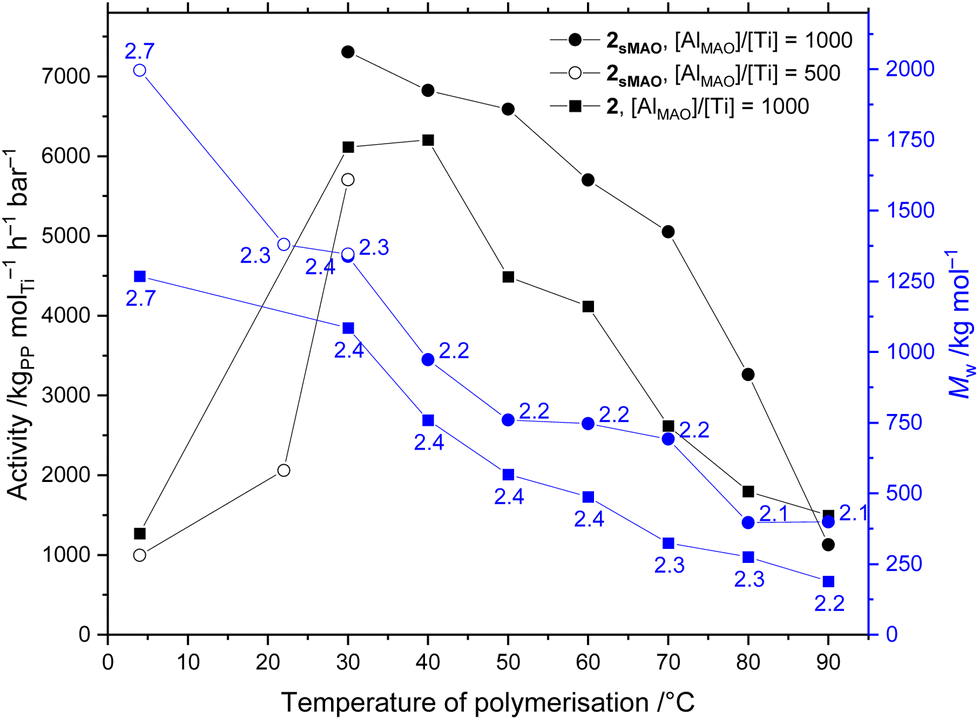 | ||
| Fig. 1 Propylene polymerisation activity and polypropylene molecular weight (polydispersity values (PDI, Đ = Mw/Mn) annotated) as a function of the temperature of polymerisation using homogeneous and sMAO-supported Me2SB(tBu2ArO, I*)TiCl2 (2, and 2sMAO respectively). Polymerisation conditions: 0.714 μmol Ti (10 mg solid catalyst, or 414 μg complex), 2 bar propylene, 50 mL hexanes, 10 minutes, and MAO ([AlMAO]/[Ti] = 500 or 1000). | ||
 | ||
| Fig. 2 Propylene polymerisation activity and polypropylene molecular weight (polydispersity values (PDI, Đ = Mw/Mn) annotated) as a function of [AlMAO]/[Ti] using homogeneous phase and sMAO-supported Me2SB(tBu2ArO, I*)TiCl2 (2, and 2sMAO respectively). Polymerisation conditions: 0.714 μmol Ti (10 mg solid catalyst, or 414 μg complex), 2 bar propylene, 50 mL hexanes, 10 minutes, and 30 °C. | ||
| Catalyst | T p/°C | Activitya | Productivityb | M w /kDa | PDIc |
|---|---|---|---|---|---|
| a kgPP molTi−1 h−1 bar−1. b kgpp gcat−1 h−1 bar−1. c Determined by GPC, PDI = Mw/Mn. | |||||
| 1sMAO | 60 | 2600 | 0.19 | 470 | 2.2 |
| 70 | 1400 | 0.10 | 370 | 2.3 | |
| 80 | 1100 | 0.080 | 230 | 2.3 | |
| 2sMAO | 60 | 3200 | 0.17 | 630 | 2.2 |
| 70 | 2200 | 0.16 | 430 | 2.2 | |
| 80 | 1600 | 0.11 | 350 | 2.1 | |
| 3sMAO | 60 | 1700 | 0.12 | 370 | 2.3 |
| 70 | 620 | 0.044 | 290 | 2.3 | |
| 80 | 640 | 0.047 | 200 | 2.2 | |
| 4sMAO | 60 | 100 | 0.0075 | 96 | 4.5 |
| 70 | 37 | 0.0026 | 63 | 3.2 | |
| 80 | 19 | 0.0013 | 75 | 3.5 | |
The formation of hexanes-soluble aPP led to an increasingly viscous reaction mixture during the course of the polymerisation. Runs were attempted as before with 2sMAO at Tp < 60 °C but the increased viscosity resulting from a greater polymer yield inhibited stirring. Thus, the reaction time was reduced to 10 minutes to allow complete thermal profiling of 2sMAO over a range of 4 ≤ Tp ≤ 90 °C (Fig. 1). As there was no advantage to be gained from a morphological templating effect of supported polymerisation, the homogeneous polymerisation activity of complex 2 was also investigated. Polymerisation conditions were as for runs using sMAO-supported solid catalysts, using 0.714 μmol 2 (414 μg, [Ti] = 14 μM, equal to the quantity supported on 10 mg 2sMAO). As anticipated, the activities of 2sMAO and 2 are broadly consistent, with the moderate increase in sMAO-supported activities largely attributed to the 20% increase in [Al] (from [AlMAO]/[Ti] = 1000 in solution-phase, to [Al(sMAO + MAO)]/[Ti] = 1200 with 2sMAO) which is shown vide infra to effect an increase in activity. While the per-moleTi activities were consistent it should be noted that the per-masscat productivities of the solution-phase system are far in excess of the supported catalysts.
A typical molecular weight temperature profile was observed using both 2sMAO and 2 (Fig. 1), with Mw increasing as polymerisation temperature decreases due to the increase in the rate of chain termination processes, including β-elimination and chain transfer, relative to propagation.50,51 With 2sMAO, Mw was found to be largely unaffected by the reduction in reaction time from 30 to 10 minutes, and continued to increase to 1.3 MDa at 30 °C. In solution-phase polymerisations with 2, polymer molecular weights were found to be consistently lower than when the catalyst is supported on sMAO though still affording UHMWaPP, up to 1.1 MDa at 30 °C, and 1.3 MDa at 4 °C. Polydispersities are low across this temperature range, with Mw/Mn ≤ 2.7, indicative of well-controlled single-site catalysis, which is maintained both in homogeneous phase and when supported on sMAO, showing that the supported system retains the benefits of homogeneous molecular catalysis. The production of UHMWaPP under ambient temperature conditions and moderate pressure is of potential industrial significance.
The effect of reaction scale
That polymerisation activity was found to be highest at 30 °C and decreases with increasing temperature – that is, with decreasing solubility of propylene in hexanes52 – is evidence that the catalytic activity of complex 2 in both homogeneous and supported regimes is limited principally by the availability of propylene in the system, with monomer diffusion being rate limiting. Further studies at higher pressures of propylene would be required to find the performance ceiling. This is evidenced by a 34% increase in activity of 2sMAO, to 9800 kgPP molTi−1 h−1 bar−1, when the solvent volume was increased to 250 mL in a larger reaction vessel leading to a more dilute, lower viscosity solution; more efficient stirring; and a reduced mass-transport barrier for the dissolution of propylene. In this large-scale regime, the quantity of MAO added was also increased five-fold to [AlMAO]/[Ti] = 5000 in order to maintain a consistent initial concentration of aluminium, [AlMAO] = 14 mM.To explore this further, the effects of reaction scale were investigated with 2, with higher catalyst dilutions leading to an increase in per-mole activity (see Fig. S1 in the ESI†). Activity appears to be proportional to the reciprocal concentration of catalyst, increasing to a value of 12000 kgPP molTi−1 h−1 bar−1 at one eighth relative dilution ([Ti] = 1.8 μM). The mass of MAO cocatalyst added was not changed, maintaining a concentration of [AlMAO] = 14 mM. The weight-average molecular weight of the resulting polypropylenes was found to increase from 1.1 to 1.5 MDa at one eighth dilution, showing a trend towards higher molecular weights at higher dilutions.
The effect of catalyst loading
The performance of both complex 2 and sMAO supported catalyst 2sMAO was investigated at Tp = 30 °C as a function of the ratio [AlMAO]/[Ti], ranging from 0 to 5000 (Fig. 2). In the absence of an aluminium cocatalyst, activity was found to be moderate with 2sMAO and no activity was recorded with 2. This demonstrates the dual-function nature of solid polymethylaluminoxane as an activating support.30 Increasing the MAO loading resulted in increased polymerisation activities up to a maximum value at 2500 equivalents of 12000 and 11000 kgPP molTi−1 h−1 bar−1 for 2sMAO and 2 respectively. This activity compares well with some of the most active polypropylene catalysts in literature: the TiCl4–Al/Mg system reported by Rishina et al. and the fluorenyl zirconocene reported by Resconi et al. have activities of 22000 and 29000 kgPP mol−1 h−1 respectively when used in liquid propylene,7,53 a far greater concentration of monomer than the 2 bar pressure used in this work.54 Runs were attempted using 2 and other alkyl aluminium cocatalysts – TMA, TEA, and TIBA – in place of MAO, but negligible activity was recorded in these cases. The catalyst system 2sMAO/TIBA by contrast did show moderate polymerisation activity (279 kgPP molTi−1 h−1 bar−1 at 60 °C), further demonstrating the activating nature of the sMAO support.
As expected, Mw was found to decrease with increasing MAO loading (Fig. 2) as a result of increased chain transfer to aluminium, with 2sMAO and 2 having maxima at [AlMAO]/[Ti] = 250 and 1000 respectively (1.4 and 1.1 MDa respectively). Polymerisations at 4 and 22 °C were performed with 2sMAO at [AlMAO]/[Ti] = 500 as a balance between high Mw and high activities, and at 4 °C with 2 and [AlMAO]/[Ti] = 1000 (Fig. 1). Activities were comparatively much reduced in these lower temperature conditions, though still classed as “very high”,55 and molecular weight was found to increase still further reaching 2.0 MDa at Tp = 4 °C with 2sMAO. In homogeneous polymerisation with 2, the increase in Mw at low temperature was less dramatic, reaching 1.3 MDa at 4 °C. The titanium phenoxy-imine and 2,2′-thiobis(6-tert-butyl-4-methylphenoxy) (TBP) complexes reported by Saito et al. and Miyatake et al. respectively were able to produce aPP with Mw > 8 MDa but with activities of only 48 kgPP molTi−1 h−1 bar−1 (20 °C, 1 bar) and 8900 kgPP molTi−1 h−1 (25 °C, liquid propylene) respectively,13,14 significantly lower than 2sMAO, reported in this work with activities up to 12000 kgPP molTi−1 h−1 bar−1 (30 °C, 2 bar).
Exploration of polymer properties; elastomeric UHMWaPP
Differential scanning calorimetry (DSC) confirmed the identity of the polymers as wholly amorphous aPP, with no melting endotherm observed. Glass transitions were observed between −12 and −3 °C, with ΔCp,Tg values in the region 0.4–0.6 J °C−1 g−1, consistent with the values reported by Wilkinson et al. for aPP and the predictions of Hirai and Eyring that, for a perfectly amorphous polyolefin, ΔCp,Tg should be 2.97 calories per degree per mole of chain atoms (corresponding to a value of 0.591 J °C−1 g−1 for PP).56–58 The PP synthesised by the indenyl complex 4sMAO has a Tg of −12 °C, in perfect agreement with literature values for aPP,56 with the PHENI* complexes 1sMAO–3sMAO producing PP with slightly elevated Tg values, presumably due to the higher molecular weights in these cases.13C{1H} Nuclear Magnetic Resonance (NMR) spectra were recorded at 298 K in chloroform-d and assigned according to the analysis of Miyatake et al. and are characteristic for atactic polypropylene.59–62 There was minimal evidence found for 2,1-insertion regioerrors in the 13C{1H} NMR spectra, particularly for aPP synthesised by 1sMAO, 2sMAO, and 2.63–65 This behaviour is typical for early transition metal post-metallocenes, where 2,1 regioerrors are usually rare <1 mol%.66 The presence of resonances at δ13C = 14–18 and 30–44 ppm in the NMR spectra of aPP synthesised by 3sMAO, and 4sMAO indicate an increased degree of 2,1-insertions, and thus poorer regiocontrol, in these cases.
The absence of Bragg reflections in the Wide-Angle X-ray Scattering (WAXS) pattern is also indicative of a fully amorphous material. The PHENI* complexes reported in this work are some of the highest activity catalysts in the literature, and to the authors’ knowledge the only such catalysts efficiently able to produce ultra-high molecular weight amorphous polypropylene (aPP) with low poly dispersity.
To facilitate mechanical characterisation, a batch of 33 g UHMWaPP (Mw = 1.2 MDa, Đ = 2.7) was cryogenically milled to a powder-like material and pressed at 180–200 °C to form an optically transparent plate. The bulk density was determined according to ISO1183, and was found to be 876 kg m−3.67 Optical transmissivity was found to be 85% across the visible light region (380–750 nm), approaching the values of more conventional transparent polyolefin block copolymers.68 Optical haze (Tdiff/Ttot) is calculated to be 50–60%, and total reflection measured at 7%, both quantities decreasing at longer wavelengths of incident light. The mechanical properties of the polymer were analysed according to ISO527.5A (Fig. 3) using type 5A test specimens.69 The Youngs modulus was found to be 2.05 MPa. The ultimate tensile strength was 1.08 MPa, at a strain of 184%, where a zero-slope condition is achieved demonstrating “yield-like” behaviour comparable to EPM copolymers (Fig. 3b).70
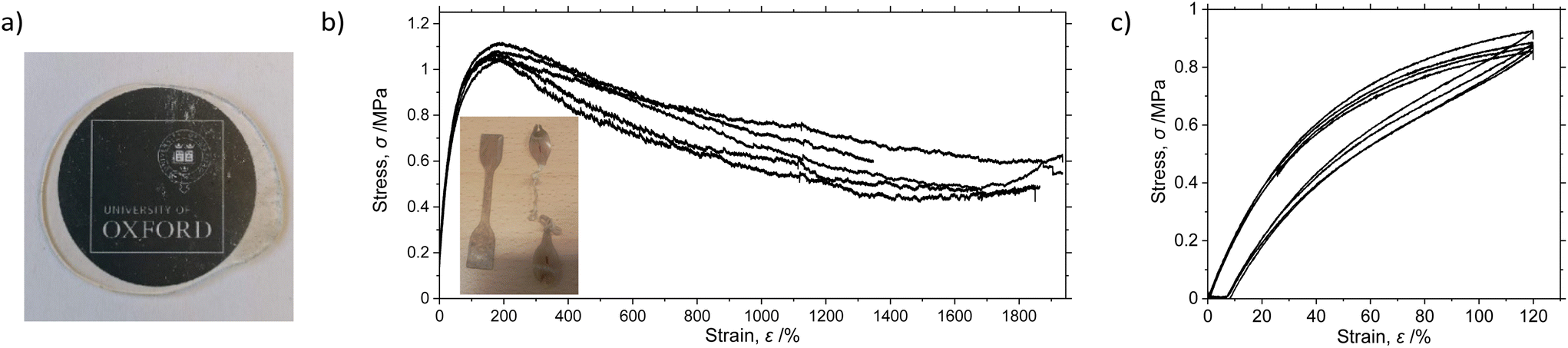 | ||
| Fig. 3 (a) Image of pressed transparent disc of UHMWaPP; (b) engineering stress–strain curves (inset: image of dog-bone samples before and after tensile testing), and (c) stress–strain hysteresis curves obtained from UHMWaPP samples. | ||
Break characteristics could not be determined; the elongation at break was higher than could be tested, but is at least 1900%, indicative of a tough, highly ductile material. Strain hardening was not observed. The “almost atactic” PP synthesised by Rishina et al. using Ti(OiPr)4 has Mw up to 98 kDa and exhibits an ultimate tensile strength of 2.3 MPa, and a strain at break of 550%; in this case, a yield point was not observed, with stress increasing nonlinearly to a limiting value.71
Elastic hysteresis curves were obtained by stress-relaxation from 120% strain (Fig. 3c) according to the ASTM D638 protocol used by Rieger et al.20 Excellent elastic recovery is observed with a set value of 7–8%. This is much less than for lower molecular weight aPPs, which suffer from large set values resulting from irreversible deformation typical of low crystallinity thermoplastic polyolefins. Behaviour is comparable to that reported by Rieger et al., who further noted the comparable performance with commercial thermoplastic elastomers such as the Kraton™ styrene–butadiene-styrene block copolymer.20
Ethylene–propylene copolymer rubbers are the most closely-related widely-studied materials, with the metallocene-catalysed materials produced by Faingol'd et al. having tensile strengths of 2.4–15.2 MPa and elongations at break up to 840% – slightly stronger but substantially less ductile than UHMWaPP.72 Unlike UHMWaPP, these polymers have a degree of crystallinity and exhibit mechanical strain hardening.
The elastomeric mechanical properties of UHMWaPP are postulated to be, at least in part, due to physical crosslinking made possible by the ultra-high molecular weights, which limits irreversible deformation by reducing chain mobility.73–75
These data compare favourably with commercial thermoplastic elastomers, such as the olefin block copolymers from Dow (e.g. INFUSE™ 9817), with a density of 877 kg m−3, a Youngs modulus of 2.31 MPa, an ultimate tensile strength of 7.00 MPa, an elongation at break of 1700%, and a compression set of 15%,76 or the olefinic thermoplastic elastomers from DuPont (e.g. MULTIFLEX™ TPO 50 D 41 81171) with a density of 890 kg m−3, and an elongation at break of 650%.77 By comparison the UHMWaPP reported in this work shows a stiffness of 2.05 MPa, elongation of >1900%, and a tension set of 7%. The initial mechanical properties of this UHMWaPP show highly promising results, suggesting that they may have to ability to encroach on the properties of more conventional materials. Such polymers, in particular UHMWaPP, warrant further study to determine commercial applicability.
Conclusions
Selective synthesis of UHMWaPP by an industrially viable high-yielding process has been described.
ansa-Permethylindenyl-phenoxy (PHENI*) complexes have proven to be highly active catalysts for the polymerisation of propylene in both solution-phase and when supported on solid polymethylaluminoxane (sMAO). We observe activities up to 12000 kgPP molTi−1 h−1 bar−1 at 2 bar propylene pressure, and this appears to be limited principally by reaction conditions. Furthermore, the product is a fully amorphous ultra-high molecular weight atactic polypropylene (UHMWaPP). Molecular weights were measured up to 2.0 MDa by GPC, among the highest values reported in the literature.
UHMWaPP demonstrates mechanical behaviour that is broadly comparable to EPM and other commercial thermoplastic elastomers, including remarkable ductility with a strain at break of >1900%, and a low elastic set of 7% in elastic recovery hysteresis experiments.
This work demonstrates the potential of PHENI* catalysts to synthesise efficiently homopolymeric UHMWaPP under mild conditions. The material properties of UHWMaPP are potentially competitive with commercial composite and copolymeric thermoplastic elastomers, with advantages in processability and recyclability, while offering optical transparency, comparable strength, and high ductility.
Data availability
Experimental details, polymerisation data and polymerisation characterisation data has been provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. G. C. R., J.-C. B. and Z. R. T. would like to thank SCG Chemicals Co., Ltd. (Thailand) for financial support. We also thank Ms Liv Thobru, Ms Sara Rund Herum, and Ms Rita Jenssen (Norner AS, Norway) for running GPC analysis; and Ms. Thea Høibjerg Glittum, and Ms. Heidi Nornes Bryntesen (Norner AS, Norway) for sample preparation and mechanical testing.References
- A. C. Shamiri, M. H. Chakrabarti, S. Jahan, M. A. Hussain, W. Kaminsky, P. V. Aravind and W. A. Yehye, Materials, 2014, 7, 5069–5108 CrossRef PubMed.
- P. Corradini, V. Barone, R. Fusco and G. Guerra, Eur. Polym. J., 1979, 15, 1133–1141 CrossRef CAS.
- N. Bahri-Laleh, A. Hanifpour, S. A. Mirmohammadi, A. Poater, M. Nekoomanesh-Haghighi, G. Talarico and L. Cavallo, Prog. Polym. Sci., 2018, 84, 89–114 CrossRef CAS.
- A. Shamiri, M. H. Chakrabarti, S. Jahan, M. A. Hussain, W. Kaminsky, P. V. Aravind and W. A. Yehye, Materials, 2014, 7, 5069–5108 CrossRef PubMed.
- Z. Zhang, R. Zhang, Y. Huang, J. Lei, Y.-H. Chen, J.-H. Tang and Z.-M. Li, Ind. Eng. Chem. Res., 2014, 53, 10144–10154 CrossRef CAS.
- C. De Rosa, F. Auriemma, C. Spera, G. Talarico and O. Tarallo, Macromol, 2004, 37, 1441–1454 CrossRef CAS.
- L. Resconi, R. L. Jones, A. L. Rheingold and G. P. A. Yap, Organometallics, 1996, 15, 998–1005 CrossRef CAS.
- Q. Wu, Q. Su, L. Ye, G. Li and Y. Mu, Dalton Trans., 2010, 39, 2525–2535 RSC.
- G. W. Coates, Chem. Rev., 2000, 100, 1223–1252 CrossRef CAS PubMed.
- L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253–1346 CrossRef CAS PubMed.
- J. A. Ewen, J. Am. Chem. Soc., 1984, 106, 6355–6364 CrossRef CAS.
- W. Spaleck, M. Antberg, J. Rohrmann, A. Winter, B. Bachmann, P. Kiprof, J. Behm and W. A. Herrmann, Angew. Chem., Int. Ed. Engl., 1992, 31, 1347–1350 CrossRef.
- J. Saito, M. Onda, S. Matsui, M. Mitani, R. Furuyama, H. Tanaka and T. Fujita, Macromol. Rapid Commun., 2002, 23, 1118–1123 CrossRef CAS.
- T. Miyatake, K. Mizunuma, Y. Seki and M. Kakugo, Makromol. Chem., Rapid Commun., 1989, 10, 349–352 CrossRef CAS.
- S. Segal, I. Goldberg and M. Kol, Organometallics, 2005, 24, 200–202 CrossRef CAS.
- K. Nomura, S. Pengoubol and W. Apisuk, Molecules, 2019, 24, 1634 CrossRef PubMed.
- K. C. Jayaratne and L. R. Sita, J. Am. Chem. Soc., 2000, 122, 958–959 CrossRef CAS.
- M. Fujita, Y. Seki and T. Miyatake, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1107–1111 CrossRef CAS.
- K. Nomura, S. Pengoubol and W. Apisuk, RSC Adv., 2016, 6, 16203–16207 RSC.
- B. Rieger, C. Troll and J. Preuschen, Macromol, 2002, 35, 5742–5743 CrossRef CAS.
- S. Basak, J. Macromol. Sci., Part A: Pure Appl.Chem., 2021, 58, 579–593 CrossRef CAS.
- D. Z. Pirityi and K. Pölöskei, Polymers, 2021, 13, 1116 CrossRef CAS PubMed.
- G. van Doremaele, M. van Duin, M. Valla and A. Berthoud, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2877–2891 CrossRef CAS.
- H. Braunschweig and F. M. Breitling, Coord. Chem. Rev., 2006, 250, 2691–2720 CrossRef CAS.
- T. J. Williams, J.-C. Buffet, Z. R. Turner and D. O'Hare, Catal. Sci. Technol., 2018, 8, 5454–5461 RSC.
- M. Nabika, H. Katayama, T. Watanabe, H. Kawamura-Kuribayashi, K. Yanagi and A. Imai, Organometallics, 2009, 28, 3785–3792 CrossRef CAS.
- H. Hanaoka, T. Hino, M. Nabika, T. Kohno, K. Yanagi, Y. Oda, A. Imai and K. Mashima, J. Organomet. Chem., 2007, 692, 4717–4724 CrossRef CAS.
- T. Senda, H. Hanaoka, S. Nakahara, Y. Oda, H. Tsurugi and K. Mashima, Macromol, 2010, 43, 2299–2306 CrossRef CAS.
- C. G. Collins Rice, J.-C. Buffet, Z. R. Turner and D. O'Hare, Chem. Commun., 2021, 57, 8600–8603 RSC.
- A. F. R. Kilpatrick, J.-C. Buffet, P. Nørby, N. H. Rees, N. P. Funnell, S. Sripothongnak and D. O'Hare, Chem. Mater., 2016, 28, 7444–7450 CrossRef CAS.
- T. A. Q. Arnold, J.-C. Buffet, Z. R. Turner and D. O'Hare, J. Organomet. Chem., 2015, 792, 55–65 CrossRef CAS.
- A. F. R. Kilpatrick, N. H. Rees, S. Sripothongnak, J.-C. Buffet and D. O'Hare, Organometallics, 2018, 37, 156–164 CrossRef CAS.
- V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–316 CrossRef CAS PubMed.
- J. Klosin, P. P. Fontaine and R. Figueroa, Acc. Chem. Res., 2015, 48, 2004–2016 CrossRef CAS PubMed.
- J. Tudor, S. Barlow, B. R. Payne, D. O'Hare, P. Nguyen, C. E. B. Evans and I. Manners, Organometallics, 1999, 18, 2281–2284 CrossRef CAS.
- P. Ransom, A. E. Ashley, N. D. Brown, A. L. Thompson and D. O'Hare, Organometallics, 2011, 30, 800–814 CrossRef CAS.
- J.-C. Buffet, T. A. Q. Arnold, Z. R. Turner, P. Angpanitcharoen and D. O'Hare, RSC Adv., 2015, 5, 87456–87464 RSC.
- J.-C. Buffet, Z. R. Turner and D. O'Hare, Chem. Commun., 2018, 54, 10970–10973 RSC.
- G. E. Hickman, C. M. R. Wright, A. F. R. Kilpatrick, Z. R. Turner, J.-C. Buffet and D. O'Hare, Mol. Catal., 2019, 468, 139–147 CrossRef CAS.
- J. V. Lamb, J.-C. Buffet, Z. R. Turner and D. O'Hare, Polym. Chem., 2019, 10, 1386–1398 RSC.
- J. V. Lamb, Synthesis and Characterisation of Unsymmetrical Group 4 Ansa-Metallocenes and their Applications as Polymerisation Catalysts, 2019 Search PubMed.
- J. V. Lamb, J.-C. Buffet, Z. R. Turner, T. Khamnaen and D. O'Hare, Macromol, 2020, 53, 5847–5856 CrossRef CAS.
- J. M. O'Connor and C. P. Casey, Chem. Rev., 1987, 87, 307–318 CrossRef.
- M. E. Rerek, L.-N. Ji and F. Basolo, J. Chem. Soc., Chem. Commun., 1983, 1208–1209 RSC.
- A. J. Hart-Davis and R. J. Mawby, J. Chem. Soc. A, 1969, 2403–2407 RSC.
- A. J. Hart-Davis, C. White and R. J. Mawby, Inorg. Chim. Acta, 1970, 4, 441–446 CrossRef CAS.
- D. C. Calabro, J. L. Hubbard, C. H. Blevins, A. C. Campbell and D. L. Lichtenberger, J. Am. Chem. Soc., 1981, 103, 6839–6846 CrossRef CAS.
- T. K. Miyamoto, M. Tsutsui and L.-B. Chen, Chem. Lett., 1981, 10, 729–730 CrossRef.
- J.-C. Buffet, N. Wanna, T. A. Q. Arnold, E. K. Gibson, P. P. Wells, Q. Wang, J. Tantirungrotechai and D. O'Hare, Chem. Mater., 2015, 27, 1495–1501 CrossRef CAS.
- M. Eskelinen and J. V. Seppala, Eur. Polym. J., 1996, 32, 331–335 CrossRef CAS.
- M. A. Parvez, M. Rahaman, M. A. Suleiman, J. B. P. Soares and I. A. Hussein, Int. J. Polym. Sci., 2014, 2014, 549031 Search PubMed.
- A. Dashti, S. H. Mazloumi, A. Bakhshi Ani and A. Akbari, J. Chem. Eng. Data, 2014, 59, 2258–2262 CrossRef CAS.
- L. A. Rishina, Y. V. Kissin, S. S. Lalayan and V. G. Krasheninnikov, J. Appl. Polym. Sci., 2019, 136, 47692 CrossRef.
- J. T. M. Pater, G. Weickert and W. P. M. van Swaaij, Chem. Eng. Sci., 2002, 57, 3461–3477 CrossRef CAS.
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed. Engl., 1999, 38, 428–447 CrossRef CAS.
- R. W. Wilkinson and M. Dole, J. Polym. Sci., 1962, 58, 1089–1106 CrossRef CAS.
- N. Hirai and H. Eyring, J. Appl. Phys., 1958, 29, 810–816 CrossRef CAS.
- N. Hirai and H. Eyring, J. Polym. Sci., 1959, 37, 51–70 CrossRef CAS.
- T. Miyatake, Y. Kawai, Y. Seki, M. Kakugo and K. Hikichi, Polym. J., 1989, 21, 809–814 CrossRef CAS.
- S.-N. Zhu, X.-Z. Yang and R. Chûjô, Polym. J., 1983, 15, 859–868 CrossRef CAS.
- V. Busico, R. Cipullo, G. Monaco, G. Talarico, M. Vacatello, J. C. Chadwick, A. L. Segre and O. Sudmeijer, Macromol, 1999, 32, 4173–4182 CrossRef CAS.
- V. Busico, R. Cipullo, G. Monaco, M. Vacatello and A. L. Segre, Macromol, 1997, 30, 6251–6263 CrossRef CAS.
- R. C. Klet, D. G. VanderVelde, J. A. Labinger and J. E. Bercaw, Chem. Commun., 2012, 48, 6657–6659 RSC.
- T. Asakura, Y. Nishiyama and Y. Doi, Macromol, 1987, 20, 616–620 CrossRef CAS.
- Z. Zhou, J. C. Stevens, J. Klosin, R. Kümmerle, X. Qiu, D. Redwine, R. Cong, A. Taha, J. Mason, B. Winniford, P. Chauvel and N. Montañez, Macromol, 2009, 42, 2291–2294 CrossRef CAS.
- V. Busico, R. Cipullo, J. C. Chadwick, J. F. Modder and O. Sudmeijer, Macromol, 1994, 27, 7538–7543 CrossRef CAS.
- ISO, ISO 1183: Plastics – Methods for determining the density of non-cellular plastics, 2019 Search PubMed.
- X. Song, L. Cao, R. Tanaka, T. Shiono and Z. Cai, ACS Macro Lett., 2019, 8, 299–303 CrossRef CAS PubMed.
- ISO, ISO 527: Plastics – Determination of tensile properties, 2019 Search PubMed.
- K. J. Wright and A. J. Lesser, Macromol, 2001, 34, 3626–3633 CrossRef CAS.
- L. A. Rishina, Y. V. Kissin, S. S. Lalayan, V. G. Krasheninnikov, E. O. Perepelitsina and T. I. Medintseva, Polym. Sci., Ser. B, 2016, 58, 152–162 CrossRef CAS.
- E. E. Faingol'd, S. L. Saratovskikh, A. N. Panin, O. N. Babkina, I. V. Zharkov, N. O. Garifullin, G. V. Shilov and N. M. Bravaya, Polymer, 2021, 220, 123559 CrossRef.
- C. Jourdan, J. Y. Cavaille and J. Perez, J. Polym. Sci., Part B: Polym. Phys., 1989, 27, 2361–2384 CrossRef CAS.
- P. Svoboda, R. Theravalappil, D. Svobodova, P. Mokrejs, K. Kolomaznik, K. Mori, T. Ougizawa and T. Inoue, Polym. Test., 2010, 29, 742–748 CrossRef CAS.
- M. Mihailov and L. Minkova, Colloid Polym. Sci., 1987, 265, 681–685 CrossRef CAS.
- Dow Chemical Company, “INFUSE™ 9817 Olefin Block Copolymer”, 400-00119899en, 29/09/2011.
- DuPont de Nemours, Inc., “MULTIFLEX™ TPO 50 D 41 81171 Polyolefin Thermoplastic Elastomer”, 08/11/2021.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, polymerisation data, polymer characterisation data. See DOI: https://doi.org/10.1039/d2py00708h |
| This journal is © The Royal Society of Chemistry 2022 |