
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular co-assembly of water-soluble nucleobase-containing copolymers: bioinspired synthetic platforms towards new biomimetic materials†
Laura Vasilica
Arsenie
a,
Mona
Semsarilar
 b,
Johannes C.
Brendel
cd,
Patrick
Lacroix-Desmazes
a,
Vincent
Ladmiral
*a and
Sylvain
Catrouillet
*a
b,
Johannes C.
Brendel
cd,
Patrick
Lacroix-Desmazes
a,
Vincent
Ladmiral
*a and
Sylvain
Catrouillet
*a
aICGM, University of Montpellier, CNRS, ENSCM, 34090 Montpellier, France. E-mail: sylvain.catrouillet@umontpellier.fr; vincent.ladmiral@enscm.fr
bIEM, University of Montpellier, CNRS, ENSCM, 34090 Montpellier, France
cLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, 07743 Jena, Germany
dJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, 07743 Jena, Germany
First published on 31st August 2022
Abstract
This study presents the development of co-assembled copolymer architectures at physiological pH (pH 7.4) formed via H-bonds between complementary nucleobase-containing copolymers. Well-defined hydrophilic copolymers were synthesised by RAFT polymerisation: statistical uracil- and thiomorpholine oxide-containing copolymers P(UrMAn-stat-THOXMAm) as well as diblock copolymers PEG112-b-P(AdMAn-stat-THOXMAm) composed of a PEG block and a second block of a copolymer of adenine- and thiomorpholine oxide-derived methacrylates. Binary mixtures of the resulting copolymers formed co-assembled nanoobjects in aqueous solution as a result of the H-bonds established between nucleobases. The influences of the polymer architecture (degree of polymerisation, co-monomer composition, length of the nucleobase-containing block), the ratio between complementary nucleobases, and the impact of H-bond competitors on the self-assembly properties were investigated. Light scattering techniques (SLS, DLS) and transmission electron microscopy (TEM) were used to characterise the co-assembled objects. This study demonstrates that the size of the resulting co-assemblies was mainly governed by the type and content of nucleobases, and by the length of the nucleobase block. Moreover, the in vitro evaluation of the nucleobase-containing polymers revealed that they were non-cytotoxic and hemocompatible. This study increases the understanding of nucleobase pairing in artificial copolymer architectures which are potential platforms for further use in biosciences.
Introduction
The structures of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) originate from self-assembly via H-bonds between complementary nucleobases such as adenine–thymine (in DNA) or adenine–uracil (in RNA).1 In the case of DNA, the complementary nucleobases match perfectly to pack in a double helix structure.1 RNA only has one strand, but could sometimes form secondary double helix structures as well.1 While the self-assembly via nucleobase pairing is well known in the cases of DNA and RNA, it has been barely explored in the case of synthetic copolymers containing nucleobases. Interesting attempts in this context were reported in the field of peptide nucleic acids (PNAs).2 The structure of PNAs consists of peptide-like backbones derived from 2-aminoethylglycine containing sequences of nucleobases able to form various supramolecular architectures with helicoidal morphologies mimicking the DNA. Numerous nucleobase-containing polymers reported so far were prepared by post-functionalisation of synthetic polymers such as PEG (polyethylene glycol), PCL (polycaprolactone) or PPG (polypropylene glycol).3–5 However, reaching high degrees of functionalization which would induce enhanced cooperativity by multiple H-bonds remains challenging. A solution is to polymerize vinyl monomers bearing nucleobases.This strategy was implemented to prepare nucleobase-containing copolymers by Ring-Opening Metathesis Polymerisation (ROMP)6,7 or Atom Transfer Radical Polymerisation (ATRP).8 Reversible addition fragmentation chain transfer (RAFT) polymerisation is arguably one of the most versatile polymerization techniques and it has been applied advantageously to prepare polymers incorporating a high density of nucleobases able to form well-defined self-assembled architectures.9–14
The use of RAFT allows the preparation of polymers with controlled molar masses, contents of nucleobases, architectures and chain-end functionality.15 Interactions between complementary nucleobase-containing polymers made by RAFT led to a variety of self-assembled architectures with tailored morphologies (spheres, vesicles, cylinders).9–14,16 Nevertheless, the hydrophobicity of the nucleobase moiety required the use of an organic solvent (DMF, DMSO, dioxane or chloroform) for the formation of self-assembled objects.9–14,16,17 For example, Kang et al. (2015)9 studied the self-assembly of poly(2-(2-(thymine-1-yl)acetoxyl)ethyl methacrylate)-b-poly(methyl methacrylate) diblock copolymers in different mixtures of CHCl3/dioxane and reported various morphological transitions, from long flexible cylinders (at 50% vol. CHCl3) to short worm-like structures (at 12.5% vol. CHCl3).
A few studies have reported the formation of self-assembled objects based on nucleobase polymers in an organic solvent/water mixture, using the solvent switch method.10,14,16,17 Representative examples by Hua et al. illustrated various morphological shapes such as spheres or cylinders obtained in DMF/water, by using block copolymers consisting of a hydrophilic poly(4-acryloyl morpholine) (PNAM) block and a hydrophobic poly(adenine propyl acrylamide) (PAAm) block.10 Therein, the self-assembly was mainly driven (in terms of size and morphologies) by hydrophobic interactions caused by the adenine heterocycle. Only a few examples of systems that form stacks of multiple supramolecular units (such as urea-containing toluidine heterocycles, peptides and nucleobases) are suitable to self-assemble in water. Their self-assembly is driven by both hydrogen bonds and hydrophobic interactions and is therefore rarely at the thermodynamic equilibrium in water.18–20 The hydrophobic interactions introduce a new parameter influencing the self-assembly that was poorly studied experimentally due to the synthetic difficulty of modulating the self-assembling moiety.21
The present study targets this problem and deals with hydrophilic nucleobase-containing copolymers able to co-assemble under physiological conditions. Polymethacrylate copolymers containing nucleobase (adenine or uracil) and hydrophilic thiomorpholine oxide were synthesized using the RAFT process. Various macromolecular structures were finely tailored in terms of degrees of polymerisation and contents of nucleobases. A binary mixture of uracil- and adenine-containing copolymers led to a wide range of co-assembled structures in water at physiological pH (pH 7.4). In contrast to previously described systems, H-bonds between complementary nucleobases rather than hydrophobic interactions were the main driving force of the assemblies. The influences of the number of nucleobases, the ratios between complementary nucleobases and the polymer architecture on the co-assembly were studied by SLS, DLS and TEM. In addition, in vitro investigations (cytotoxicity and compatibility assays on red blood cells) were carried out to test the potential of these nucleobase-containing copolymers for future biological applications.
Experimental section
Materials
Methacryloyl chloride (97% purity) was acquired from Fluka (Switzerland) and distilled (50 °C, 400 mbar) before use. 2-Bromoethanol (95% purity), adenine (Ad, 99% purity) and uracil (Ur, 98% purity) were purchased from Alfa Aesar (Germany). Thiomorpholine (98% purity) and 3-bromo-1-propanol (97% purity) were bought from Fluorochem (UK). Hydrogen peroxide (H2O2) solution (30 wt%) was received from Carlo Erba (France). 4-Dimethylaminopyridine (DMAP, 96% purity), triethylamine (TEA, 99% purity), 2-cyano-2-propyl benzodithioate (CTA, 97% purity), poly(ethylene glycol) methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate) (macro-CTA), disodium phosphate basic dodecahydrate (Na2HPO4·12H2O, 95% purity), potassium carbonate (K2CO3, 99.9% purity), sodium hydride (NaH, 90% purity) and deuterated solvents (deuterated chloroform, CDCl3, and hexadeuterodimethyl sulfoxide, DMSO-d6) were provided by Sigma Aldrich. 2,2′-Azobis(2-methylpropionitrile) (AIBN, 98% purity) was acquired from Sigma Aldrich (Germany) and recrystallised from methanol at 65 °C before use. Sodium chloride (NaCl) and citric acid monohydrate (C6H8O7 H2O, 99% purity) were obtained from VWR Chemical. Sodium bicarbonate (NaHCO3, 95% purity) was purchased from Fluka (France). Dimethylformamide (DMF, 99.8% purity) was acquired from Fisher Scientific (Belgium). HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffered saline solution (30 mM) was acquired from PromoCell (Germany). Dry solvents (dichloromethane, CH2Cl2, and acetonitrile, CH3CN) were purified using a PureSolv Micro solvent purification system purchased from Sigma Aldrich (USA). The dialysis membranes used for the purification of polymers (Spectra/Por 7 Pre-treated RC Dialysis Tubing, MWCO = 1 kDa, diameter 24 mm, 4.6 mL cm−1) were bought from Krackeler Scientific, USA. 2 mM L-glutamine 100 U mL−1 penicillin and 100 μg mL−1 streptomycin solutions were acquired from Biochrom. 10% fetal calf serum was obtained from FCS, Capricorn Scientific. PrestoBlue solution was acquired from Thermo Fisher, Germany. Human blood was provided by the Department of Transfusion Medicine from Jena University Hospital (Germany). Branched poly(ethylene imine) (bPEI) solution was purchased from Polysciences Inc.Instrumentation
1H and 13C-NMR spectra were recorded on a NMR Bruker Avance 400 MHz or NMR Bruker Avance III HD 400 MHz spectrometer using CDCl3 or DMSO-d6 as a deuterated solvent. The chemical shifts of protons were relative to tetramethylsilane (TMS) at δ = 0.Size exclusion chromatography (SEC) was performed in DMF containing 0.1 wt% LiCl, with a flow rate of 0.8 mL min−1 at 40 °C. Samples were filtered using TE36 Whatman PTFE-supported membrane filter paper (0.45 μm, 47 mm diameter) before the injection. The data were calibrated using polymethyl methacrylate (PMMA) standards.
∂n/∂c values in water were determined using a differential refractometer and were estimated from the integrated refractive index (RI)-signal knowing the polymer concentration. Light scattering measurements were performed using an LS spectrometer (from LS Instruments, Switzerland) incorporating a goniometer based multi-angle static light scattering (SLS) and dynamic light scattering (DLS) instrument. Transmission electron microscopy (TEM) analyses were conducted on a JEOL 1200 EXII-120 kV instrument.
L929 cells used for cytotoxicity tests were incubated by using 96 well plates from VWR, Germany. The fluorescence measurements used to determine the cell viability were assessed using an Infinite M200 PRO microplate reader from Tecan, Germany. The haemoglobin release measurements and the cell aggregation rates were obtained using a plate reader from Tecan, Männedorf, Switzerland.
Methods
Synthesis of 3-bromopropyl methacrylate and 2-bromoethyl methacrylate
To a solution of 4-(dimethylamino)pyridine (DMAP) (162.4 mg, 0.05 eq., 16.8 mmol) in CH2Cl2 (100 mL), 3-bromopropanol (2.4 mL, 1 eq., 26.54 mmol) and triethylamine (TEA) (7.4 mL, 2 eq., 53.2 mmol) were added under continuous stirring. Then, methacryloyl chloride (2.6 mL, 1 eq., 26.54 mmol) was added dropwise in an ice bath and under an inert (N2) atmosphere. The reaction mixture was kept at room temperature and under an inert atmosphere for 5 h. The resulting mixture was washed twice with a saturated NaHCO3 aqueous solution (2 × 100 mL) and then with distilled water (100 mL). The organic layer was collected, dried over magnesium sulfate and concentrated under vacuum to give a yellow oil (5 g, yield: 91%). 2-Bromoethyl methacrylate was obtained by a similar procedure to that of 3-bromopropyl methacrylate, following a published protocol.22Synthesis of 2-ethyl thiomorpholine oxide methacrylate (THOXMA)
2-Ethyl thiomorpholine oxide methacrylate was synthesized by oxidation of 2-ethyl thiomorpholine methacrylate, following a previously published procedure.23Synthesis of 3-(adenin-9-yl)propyl methacrylate (AdMA)
Adenine (1.5 g, 1 eq., 11.1 mmol) was dissolved in anhydrous DMF (100 mL) in a two-neck round-bottom flask and stirred for 1 h under an inert atmosphere at room temperature. Then, NaH (0.296 g, 1 eq., 11.1 mmol) was gently added and the mixture was kept under continuous stirring at room temperature for 30 min. Subsequently, freshly obtained 3-bromopropyl methacrylate (2.29 g, 1 eq., 11.1 mmol) was added and the flask containing the reaction mixture was immersed in an oil bath at 40 °C for 10 days. Then, the inorganic salts resulting from the reaction were removed by filtration. The resulting yellow filtrate was cryo-distilled to evaporate the organic solvent. A viscous yellow residue was formed after cryo-distillation. Anhydrous dichloromethane (100 mL) was added to the residue, and the resulting mixture was vigorously stirred for 30 min. Then, the mixture was filtered under vacuum and the filtrate was collected, dried with anhydrous magnesium sulphate and concentrated under vacuum to give a viscous yellow liquid (2 g, global yield: 68%). The monomer was characterised by 1H NMR (Fig. S1A†) and 13C NMR (Fig. S1B†) spectroscopy. 1H NMR, Fig. S1A† (400 MHz, DMSO-d6) δ (ppm) = 5.9 (d, CH2, denoted as a); 5.58 (d, CH2, denoted as a′); 1.87 (s, CH3, denoted as b); 4.27 (t, OCH2CH2, denoted as c); 2.20 (m, CH2CH2CH2, denoted as d); 4.10 (t, NCH2CH2, denoted as e); 7.26 (s, NH2, adenine heterocycle, denoted as g); 8.15 (s, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–N, adenine heterocycle, denoted as h); 8.13 (s, NCH–N, adenine heterocycle, denoted as i).
CH–N, adenine heterocycle, denoted as h); 8.13 (s, NCH–N, adenine heterocycle, denoted as i).
Synthesis of 3-(uracil-1-yl)propyl methacrylate (UrMA)
Uracil (1.5 g, 1 eq., 13.38 mmol) and anhydrous potassium carbonate (1.84 g, 1 eq., 13.38 mmol) were dissolved in anhydrous DMF (100 mL) in a two-neck round-bottom flask and stirred for 1 h under an inert atmosphere at room temperature. Then, 3-bromopropyl methacrylate (2.77 g, 1 eq., 13.38 mmol) was gently added using a syringe and the mixture was gently stirred at room temperature for 10 days. The mixture was filtered under vacuum and the filtrate was cryo-distilled to remove the organic solvent. After this step, dichloromethane (100 mL) was added and the resulting reaction mixture was stirred for another 15 min. Then, the mixture was filtered under vacuum to remove the inorganic salts. The filtrate was dried with anhydrous magnesium sulphate and then concentrated under vacuum, leading to a viscous pale yellow liquid (2 g, global yield: 63%). The product was characterised by 1H NMR (Fig. S2A†) and 13C NMR (Fig. S2B†) spectroscopy. 1H NMR, Fig. S2A† (400 MHz, DMSO-d6) δ (ppm) = 6.01 (d, CH2, denoted as a); 5.58 (d, CH2, denoted as a′); 1.87 (s, CH3, denoted as b); 4.11 (t, CH2CH2, denoted as e); 1.96 (m, CH2CH2CH2, denoted as d); 3.79 (t, CH2CH2, denoted as c); 7.62 (d, CHCH, uracil heterocycle, denoted as g); 5.53 (d, CHCH, uracil heterocycle, denoted as f); 11.5 (s, NH, uracil heterocycle, denoted as h).
Synthesis of poly((3-(uracil-1-yl) propyl methacrylate)-stat-(2-ethyl thiomorpholine oxide methacrylate)) P(UrMAn-stat-THOXMAm) by RAFT polymerisation
In a typical protocol, 2-cyano-2-propyl benzodithioate (CTA, 1 eq.), AIBN (0.25 eq.), uracil methacrylate (UrMA) (x eq.) and thiomorpholine oxide methacrylate THOXMA ((y − x) eq. where y is the targeted DP) were dissolved in a mixture of DMF/aqueous phosphate buffer (pH 4, CM = 4 M) in a volume ratio (DMF/buffer) of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The mixture was thoroughly degassed via 3 freeze–pump–thaw cycles, filled with nitrogen and immersed in an oil bath at 80 °C. At different times, an aliquot of the reaction mixture was taken and analysed by 1H-NMR and SEC. After 7 h, the reaction was stopped by exposure to air. The mixture was then dialysed against water (with a 1 kDa MWCO membrane) for 3 days, followed by lyophilisation for 2 days. The resulting pink polymer powder was analysed by 1H NMR in DMSO-d6 and DMF SEC. For example, to obtain P(UrMA8-stat-THOXMA34), the quantities of reagents used were CTA (1eq., 8 mg, 0.036 mmol), AIBN (0.25 eq., 1.5 mg, 0.009 mmol), uracil methacrylate UrMA (10 eq., 82 mg, 0.36 mmol), and thiomorpholine oxide methacrylate THOXMA (40 eq., 332 mg, 1.44 mmol), dissolved in DMF (2 mL)/aqueous buffer (pH 4, CM = 4 M, 60 eq., 0.54 mL). 1H NMR, Fig. S3† (400 MHz, DMSO-d6) δ (ppm) = 1.8 (s, CH2, polymerizable synthon, denoted as Ib); 1.8 (m, CH2, UrMA aliphatic linker, denoted as IIb); 3.43 (d, CH2, UrMA aliphatic linker, denoted as IIa and IIc); 3.43 (d, CH2, THOXMA aliphatic linker, denoted as IId and IIe); 2.62 (t, CH2CH2, thiomorpholine oxide cycle, denoted as IIIb); 3.87 (t, CH2CH2, thiomorpholine oxide cycle, denoted as IIIa); 0.87, 0.97, 1.1 (s, CH3, denoted as Ia); 7.57 (d, CHCH, uracil heterocycle, denoted as i); 5.87 (d, CHCH, uracil heterocycle, denoted as h); 11.5 (s, NH, uracil heterocycle, denoted as g).
:1. The mixture was thoroughly degassed via 3 freeze–pump–thaw cycles, filled with nitrogen and immersed in an oil bath at 80 °C. At different times, an aliquot of the reaction mixture was taken and analysed by 1H-NMR and SEC. After 7 h, the reaction was stopped by exposure to air. The mixture was then dialysed against water (with a 1 kDa MWCO membrane) for 3 days, followed by lyophilisation for 2 days. The resulting pink polymer powder was analysed by 1H NMR in DMSO-d6 and DMF SEC. For example, to obtain P(UrMA8-stat-THOXMA34), the quantities of reagents used were CTA (1eq., 8 mg, 0.036 mmol), AIBN (0.25 eq., 1.5 mg, 0.009 mmol), uracil methacrylate UrMA (10 eq., 82 mg, 0.36 mmol), and thiomorpholine oxide methacrylate THOXMA (40 eq., 332 mg, 1.44 mmol), dissolved in DMF (2 mL)/aqueous buffer (pH 4, CM = 4 M, 60 eq., 0.54 mL). 1H NMR, Fig. S3† (400 MHz, DMSO-d6) δ (ppm) = 1.8 (s, CH2, polymerizable synthon, denoted as Ib); 1.8 (m, CH2, UrMA aliphatic linker, denoted as IIb); 3.43 (d, CH2, UrMA aliphatic linker, denoted as IIa and IIc); 3.43 (d, CH2, THOXMA aliphatic linker, denoted as IId and IIe); 2.62 (t, CH2CH2, thiomorpholine oxide cycle, denoted as IIIb); 3.87 (t, CH2CH2, thiomorpholine oxide cycle, denoted as IIIa); 0.87, 0.97, 1.1 (s, CH3, denoted as Ia); 7.57 (d, CHCH, uracil heterocycle, denoted as i); 5.87 (d, CHCH, uracil heterocycle, denoted as h); 11.5 (s, NH, uracil heterocycle, denoted as g).
Synthesis of poly(ethylene glycol)-b-poly((3-(adenine-9-yl) propyl methacrylate)-stat-(2-ethyl thiomorpholine oxide methacrylate)) PEG112-b-P(AdMAn-stat-THOXMAm) by RAFT polymerisation
The PEG-macro chain transfer agent (macro-CTA PEG, 1 eq.), adenine methacrylate AdMA (x eq.) and thiomorpholine oxide methacrylate THOXMA ((y − x) eq. where y is the targeted DP) and AIBN (0.25 eq.) were dissolved in a mixture of DMF/aqueous phosphate buffer (pH 4, CM = 4 M) in a volume ratio (DMF/buffer) of 2:1. The mixture was degassed via 3 freeze–pump–thaw cycles, backfilled with nitrogen and then immersed in an oil bath at 80 °C for 7 hours. The conversion of the reaction was monitored each hour by 1H NMR and SEC. The reaction was then quenched by immersion in a liquid nitrogen bath and exposure to air. The final mixture was dialysed against water (with a 1 kDa MWCO membrane) for 3 days, followed by lyophilisation for 2 days. The resulting pale-yellow powder was analysed by 1H-NMR spectroscopy in DMSO-d6 and DMF SEC. For exemplification in the case of PEG112-b-P(AdMA30-stat-THOXMA70), the quantities of reagents used were macro-CTA PEG (1 eq., 20 mg, 0.0036 mmol), AIBN (0.25 eq., 0.15 mg, 9 × 10−4 mmol), adenine methacrylate AdMA (30 eq., 28.2 mg, 0.11 mmol), and thiomorpholine oxide methacrylate THOXMA (70 eq., 58.2 mg, 0.25 mmol), dissolved in DMF (1 mL)/aqueous buffer (pH 4, CM = 4 M, 130 eq., 0.12 mL). 1H NMR, Fig. S4† (400 MHz, DMSO-d6) δ (ppm) = 1.35 (s, CH2, polymerizable synthon, denoted as Ib); 1.35 (m, CH2, Ad aliphatic linker, denoted as IIb); 3.47 (m, CH2, PEG, denoted as IV); 3.96 (d, CH2, AdMA aliphatic linker, denoted as IIa and IIc); 3.96 (d, CH2, THOXMA aliphatic linker, denoted as IId and IIe); 4.44 (t, CH2CH2, thiomorpholine oxide cycle, denoted as IIIa and IIIb); 0.9 (s, CH3, denoted as Ia); 8.56 (s, NH2, adenine heterocycle, denoted as g); 8.97 (s, NCH–N, adenine heterocycle, denoted as h); 9.6 (s, NCH–N, adenine heterocycle, denoted as i).
Preparation of starting polymer solutions and self-assembled formulations
The starting polymer solutions (denoted as P1, P3-uracil polymer solutions and P2, P4 adenine polymer solutions see Table 2 and Table S1†) were prepared in HEPES buffer (pH 7.4) at a concentration of 5 g L−1 and stirred overnight at room temperature. Then, self-assembled formulations were obtained by slowly adding the uracil solution to the adenine solution, and the resulting mixture was then stirred for 2 days (Schemes 3 and S1†). For example, to prepare formulation A (P1 + P2, 1 mL), P1 solution (0.55 mL from a solution at a concentration of 5 g L−1, in HEPES buffer, calculated as stated using eqn (S15)†) was gently added using a micropipette (15 min, 200 rpm) to P2 solution (0.45 mL from a solution at a concentration of 5 g L−1, in HEPES buffer, calculated according to eqn (S16)†). Then, the solution mixture was stirred for 2 days (200 rpm, at room temperature). The solutions of starting polymers (P1, P2, P3, P4) were filtered through 0.2 μm pore size Waters filters (USA) prior to performing SLS, DLS and TEM characterization studies. The absence of the concentration variation due to filtration was checked. Dilutions were made by adding the filtered solvent (through 0.2 μm filters) and then stirring the solution for 5 min (Fig. S9 and S10†).| Polymer name | Experimental DPa | M n (g mol−1), by 1H-NMRb | M n (g mol−1), by SECc | Dispersity (Đ)c | Average number of nucleobases per polymer chain | M th (g mol−1) | Theoretical target DPd | |
|---|---|---|---|---|---|---|---|---|
| a Calculated by 1H-NMR performed in DMSO-d6, according to eqn (S4) and (S9).† b Calculated by 1H-NMR performed in DMSO-d6, according to eqn (S7) and (S14).† c SEC analysis performed in DMF containing 0.1% LiCl and by using PMMA standards. d Calculated using the following equation DPtarget = (([THOXMA]/[chain transfer agent]) × ConvTHOXMA) + (([AdMA or UrMA]/[chain transfer agent]) × ConvAdMA or UrMA). | ||||||||
| P1 | P(UrMA8-stat-THOXMA34) | 42 | 10000 |
10430 |
1.21 | 8 | 11840 |
50 |
| P2 | PEG112-b-P(AdMA30-stat-THOXMA70) | 104 | 30200 |
32100 |
1.32 | 30 | 29400 |
100 |
| P3 | P(UrMA22-stat-THOXMA19) | 41 | 10500 |
11200 |
1.1 | 22 | 11950 |
50 |
| P4 | PEG112-b-P(AdMA5-stat-THOXMA5) | 10 | 7900 | 9000 | 1.11 | 5 | 7700 | 15 |
| Formulation | Description | Aggregation number (Nagg)a | Particle hydrodynamic diameter (DH)b |
|---|---|---|---|
| a Evaluated by SLS. b Evaluated by DLS; formulations A, B, C, and D were prepared with a Ur/Ad molar ratio of 1/1. | |||
| A | P1 + P2 | 315 | 130 |
| B | P3 + P2 | 194 | 101 |
| C | P3 + P4 | 9.95 | 40 |
| D | P1 + P4 | 7.8 | 35 |
| Polymer solution | P1 | 1.6 | 11 |
| Polymer solution | P2 | 7.5 | 20 |
| Polymer solution | P3 | 3.3 | 13.2 |
| Polymer solution | P4 | 11.4 | 10.8 |
Static light scattering
Static light scattering (SLS) measurements were performed using an LS spectrometer operating with a vertically polarized laser of wavelength λ = 660 nm. All measurements (including dilutions) were done at room temperature (25 °C), collected from 30° to 90° with an interval of 5°, from 90° to 110° with an interval of 10°, and up to 150° with an interval of 20°. Prior to measurements, filtered toluene and filtered buffer (through a 0.2 μm pore size Waters filter membrane) were used as a reference and solvent, respectively.The Rayleigh ratio (Rθ) of the solution was determined following eqn (1).24,25
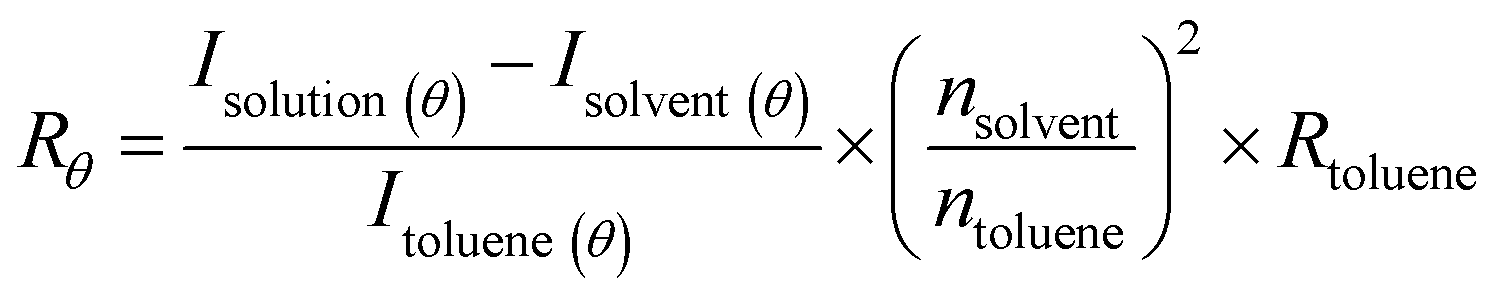 | (1) |
At a given concentration C, Rθ is related to the apparent weight average molar mass of the scatterers (or apparent molecular weight), Ma, and to the structure factor, S(q), which depends on the scattering wave vector, as shown using eqn (2).24,25 It is important to underline that Ma corresponds to the true molar mass (Mw) only in very dilute solutions, where the interactions between the scatterers can be neglected.25,26 At high concentrations, interactions cause Ma to differ strongly from Mw. For this reason, in order to accurately evaluate the true Ma of the self-assemblies, SLS measurements were performed for concentrations ranging between 5 g L−1 and 1 g L−1. Consequently, the curve representing each Ma corresponding to each tested concentration was fitted, in order to determine the real Mw of the self-assembled objects as the intercept.
| Rθ = K × C × Ma × S(q) | (2) |
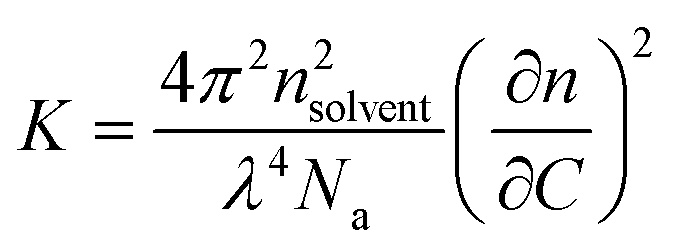 | (3) |
The aggregation number (Nagg) was expressed according to eqn (4):24,25
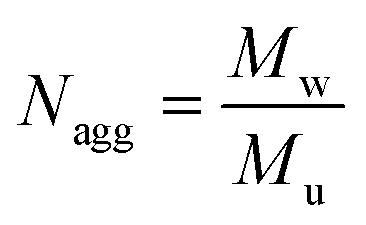 | (4) |
| Mu = wpolymer1 × Mpolymer1 + wpolymer2 × Mpolymer2 | (5) |
Dynamic light scattering
The particle size (DH, expressed as the hydrodynamic diameter of the particle) of co-assembled polymers was determined by dynamic light scattering (DLS). The DLS instrumentation consisted of the same LS spectrometer used for SLS experiments. DLS measurements were performed at 25 °C with a He–Ne 630 nm laser module, at a detection angle of 150°.Transmission electron microscopy
TEM analyses were performed using a JEOL 1400+ instrument equipped with a numerical camera, operating with an acceleration voltage of 120 kV at 25 °C. TEM samples were prepared by placing a drop (10.0 μL) of self-assembled polymer solution onto a carbon coated copper grid for 20 s, blotted with filter paper and then dried under ambient conditions. The experiments were performed at a concentration of 0.1% w/w.Cytotoxicity assays
Cytotoxic evaluation of nucleobase containing copolymers was assessed using the mouse fibroblast cell line L929 (400620, CLS), as recommended by ISO10993-5. L929 cells were cultured in Dulbecco's modified eagle's medium with 2 mM L-glutamine supplemented with 10% fetal calf serum, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C under a humidified 5% (v/v) CO2 atmosphere. To this, the L929 cells were firstly seeded at 103 cells per mL (104 cells per well) in a 96 well plate and then incubated for 24 h. No cells were seeded in the outer wells. The medium was changed to fresh cell culture medium 1 h prior to treatment. Cold nucleobase containing polymer solutions prepared in 20 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) were added to the cells at various concentrations (from 5 to 700 μg mL−1), and then the plates were incubated for 24 h. The control cells were incubated with fresh culture medium containing the same amount of HEPES as the treated cells. Moreover, the medium was replaced by a mixture of fresh culture medium and the resazurin-based solution PrestoBlue. After incubation for another 45 min at 37 °C under a humidified 5% (v/v) CO2 atmosphere, the fluorescence was measured at λex = 560 nm/λem = 590 nm with the gain set to optimal, with untreated cells on the same well plate serving as negative controls. The negative control was standardized as 0% of metabolism inhibition and referred to as 100% viability. Cell viability below 70% was considered to be an indication of cytotoxic behaviour. All experiments were performed in triplicate.Erythrocyte aggregation
Red blood cells from human blood were treated with nucleobase containing polymers at physiological pH (pH 7.4). Human blood was provided by the Department of Transfusion Medicine of the Jena University Hospital. Erythrocyte suspensions in PBS were prepared and mixed at a volume ratio of 1:1 with polymer solutions as described above. After incubation at 37 °C for 2 h, the erythrocyte aggregation was measured at 645 nm. As positive and negative controls, erythrocytes were treated with 50 μg mL−1 25 kDa branched poly(ethylene imine) (bPEI) solution or PBS buffer at pH 7.4. The aggregation activity of the polymer samples was expressed as the aggregation rate (eqn (6)): | (6) |
Hemolytic activity
The release of haemoglobin from erythrocytes was assessed in order to evaluate the damaging properties of the red blood cell membrane in the presence of nucleobase-containing copolymers. The blood was centrifuged at 4500g for 5 min. Subsequently, the pellets were washed three times with PBS (pH 7.4) by centrifugation at 4500g for 5 min. Furthermore, the erythrocytes were suspended in PBS at pH 7.4 to mimic the physiological conditions of blood/cytoplasm. Cold nucleobase containing polymer solutions were made in 20 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) at a stock concentration of 1 mg mL−1. Then the solutions were mixed at a volume ratio of 1:1 with cold erythrocyte suspensions and were incubated at 37 °C for 1 h. Erythrocyte suspensions were centrifuged at 2400g for 5 min. The release of hemoglobin in the supernatant was determined at 544 nm. The absorbance was measured using a plate reader. Additionally, determinations were conducted with washed erythrocytes either lysed with 1% Triton X-100 or suspended in PBS at pH 7.4 as a reference. The hemolytic activity of the polymers was calculated according to the following equation (eqn (7)):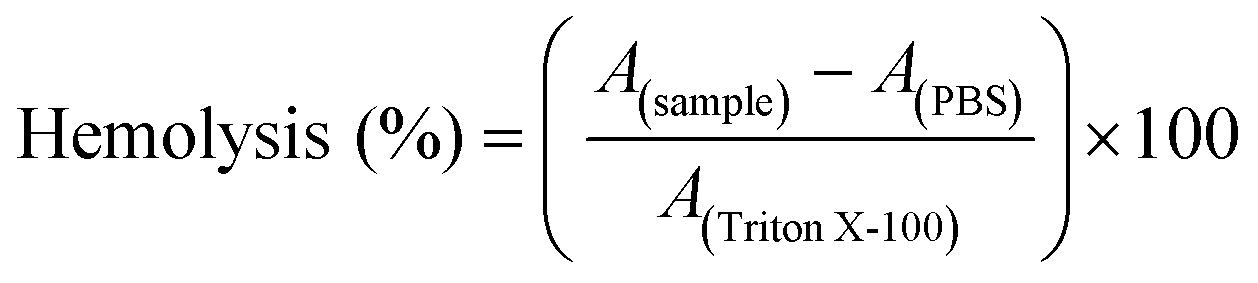 | (7) |
Results and discussion
Nucleobase-containing methacrylates
The synthesis of adenine- and thymine-containing methacrylates has already been reported by Kang et al.13,22,27 In this work, we adapted the previously reported synthetic routes to improve the yield and to reduce the synthetic efforts. Scheme 1 illustrates the synthesis of nucleobase-containing methacrylates used in the present study, called adenine methacrylate (AdMA) and uracil methacrylate (UrMA), which afforded the final monomer products with relatively high yields (above 63%) by straightforward techniques of organic chemistry.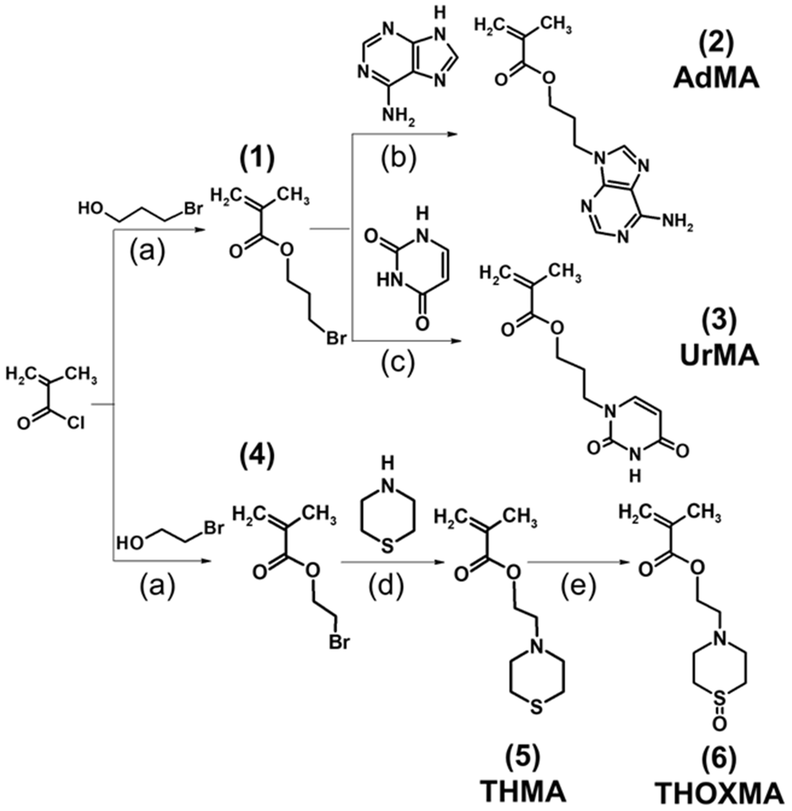 | ||
| Scheme 1 Syntheses of an adenine containing monomer (AdMA) (2), uracil methacrylate (UrMA) (3), thiomorpholine methacrylate (THMA) (5), and thiomorpholine oxide methacrylate (THOXMA) (6). (a): DMAP, TEA, CH2Cl2, RT, under N2, overnight; (b): NaH, DMF, 40 °C, 10 days; (c): K2CO3, DMF, RT, 10 days; (d) K2CO3, CH3CN, under N2, 40 °C, 6 days; (e): H2O2, under N2, RT, overnight. | ||
These adenine- and uracil-methacrylates were insoluble in water. Our previous study reported the synthesis and characterisation of a new hydrosoluble thiomorpholine oxide methacrylate. Using RAFT polymerisation, hydrophilic polymers with no cytotoxicity and high blood compatibility were synthesized.23 Ethyl-thiomorpholine oxide methacrylate (THOXMA) was thus used as a hydrophilic comonomer for the synthesis of nucleobase-containing copolymers to modulate their hydrophobicity.
Synthesis of nucleobase copolymers
Two families of nucleobase-containing copolymers were synthesised by RAFT polymerisation, as shown in Scheme 2: (A) diblock copolymers (PEG112-b-P(AdMAn-stat-THOXMAm)) composed of a hydrophilic block of PEG (with a constant DP = 112) and a second block driving the self-assembly prepared by the copolymerization of AdMA and THOXMA; (B) statistical copolymers of UrMA and THOXMA, denoted as P(UrMAn-stat-THOXMAm). In all structures, the nucleobases (adenine, uracil) are H-bond promoters. In the case of the adenine-containing block copolymer, PEG112-b-P(AdMAn-stat-THOXMAm), the PEG block was selected for its high hydrosolubility and the steric stabilisation it can provide to self-assembled structures in water,28 while THOXMA was introduced in the self-assembling moiety to modulate its hydrophobicity.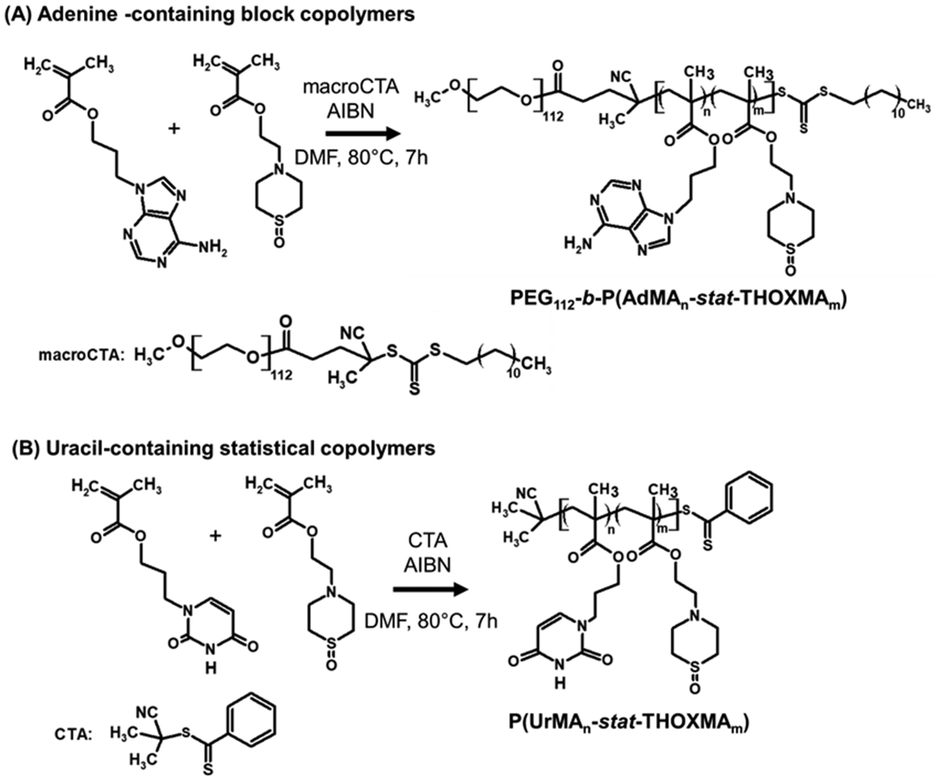 | ||
| Scheme 2 Syntheses of nucleobase-containing copolymers: (A) PEG112-b-P(AdMAn-stat-THOXMAm) diblock copolymers (P2, P4); (B) P(UrMAn-stat-THOXMAm) statistical copolymers (P1, P3). | ||
Both nucleobase copolymer families were synthesized by RAFT polymerization in a mixture of DMF/aqueous phosphate buffer (pH 4). The acidic buffer was used to prevent undesired aminolysis/hydrolysis of the dithioester and trithiocarbonate moieties of the chain transfer agents. The characterization data of these copolymers are summarized in Table 1. A good correlation between theoretical and experimental molar masses calculated by 1H NMR (Table 1) and comonomer ratios was observed (Table S3†). In addition, SEC analysis revealed a narrow molecular weight distribution (Đ ranging between 1.1 and 1.3, see Table 1) for all the copolymers, while the evolution of molar mass with conversion was linear, thus confirming that the polymerisation was controlled (Fig. S5 and S6†). These results indicate that the RAFT polymerisation led to the formation of well-defined nucleobase-containing copolymers with controlled molar masses and desired compositions.
Formation of co-assembled nucleobase polymer architectures at physiological pH
The obtained nucleobase copolymers were then dissolved in HEPES buffer (pH 7.4) and the properties of the resulting solutions were analysed (Scheme 3). First, starting filtered solutions of P1 and P2 were investigated by light scattering (SLS and DLS). The results are presented in Fig. 1, Table 2 and Table S2.† Low apparent molar mass (∼20000 g mol−1) and Nagg (∼1.6) were observed for the uracil-containing copolymer (P1) (Table S2†). This slight aggregation did not result in typical micellar structures observed in the case of previously reported nucleobase blocks. The analysis of the adenine-containing copolymer (P2) showed a slightly higher aggregation number Nagg (∼7.5) and apparent molar mass (Table S2†). Solutions of P3 and P4 showed similar behaviour but slightly higher aggregation numbers (3.3 and 11.4, respectively) likely caused by their higher molar contents in hydrophobic nucleobases (Table 2 and Table S2†). This slight aggregation is likely caused by hydrophobic interactions. Indeed the light scattering signatures of solutions of P1 or P2 were not affected by the addition of urea (Fig. 2).
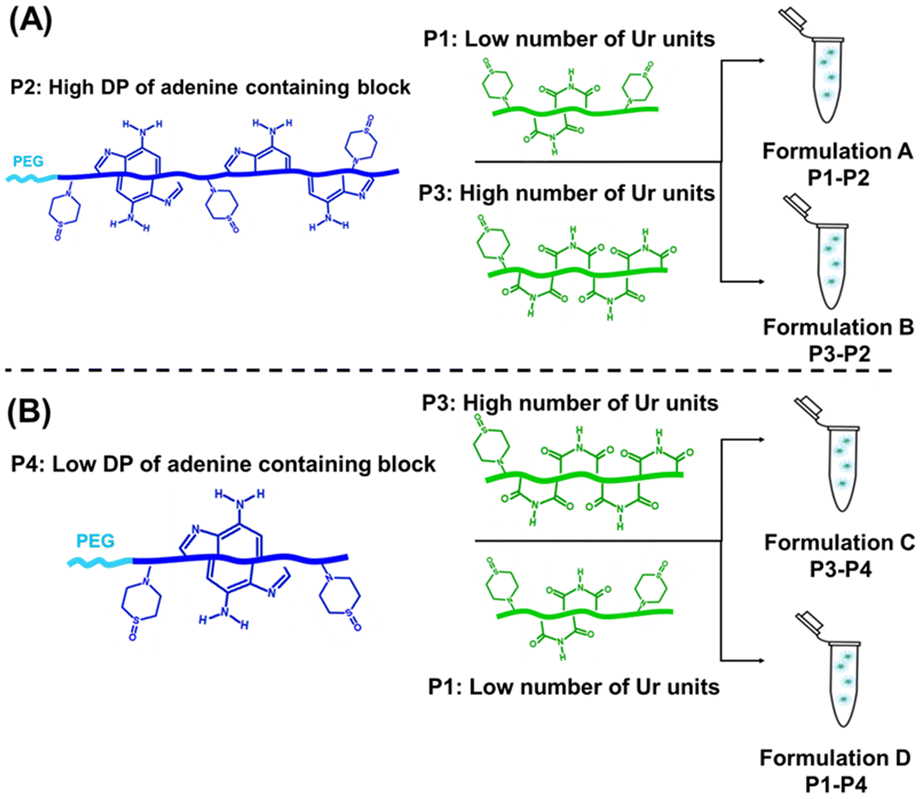 | ||
| Scheme 3 (A) Schematic representation of the composition of formulation A, made by adding a solution of P1 (with a low number of uracil units) to a solution of P2 (long DP of the adenine-containing block) and of the composition of formulation B, made by adding the solution of P3 (with a high number of uracil units) to a solution of P2 (long DP of the adenine-containing block); (B) schematic representation of the composition of formulation D, prepared by adding a solution of P1 (with a low number of uracil units) to a solution of P4 (short DP of the adenine-containing block) and of the composition of formulation C, formed by adding a solution of P3 (with a high number of uracil units) to a solution of P4 (short DP of the adenine-containing block). | ||
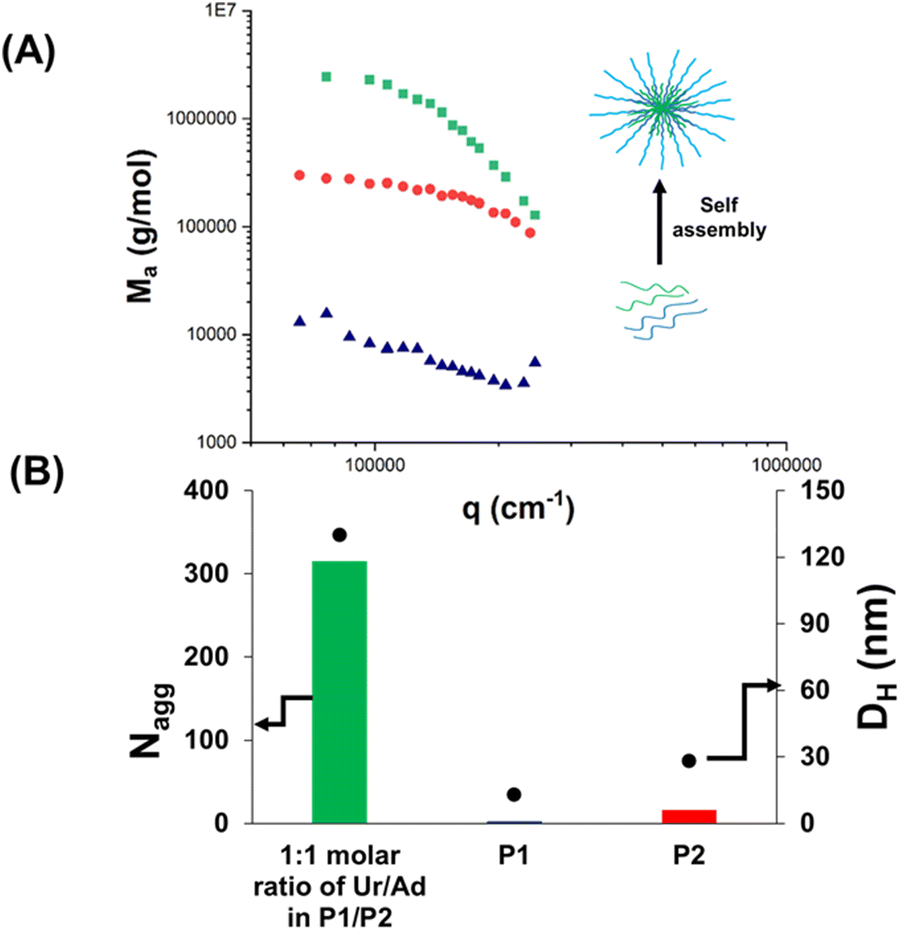 | ||
| Fig. 1 Co-assembly of P1 and P2 (formulation A): (A) apparent molar mass Ma evolution for individual polymer solutions (P1, P2) and formulation A, measured by SLS at a concentration of 5 g L−1; (B) Nagg (evaluated by SLS) and DH (evaluated by DLS) of individual polymer solutions and formulation A measured between 1 g L−1 and 5 g L−1. | ||
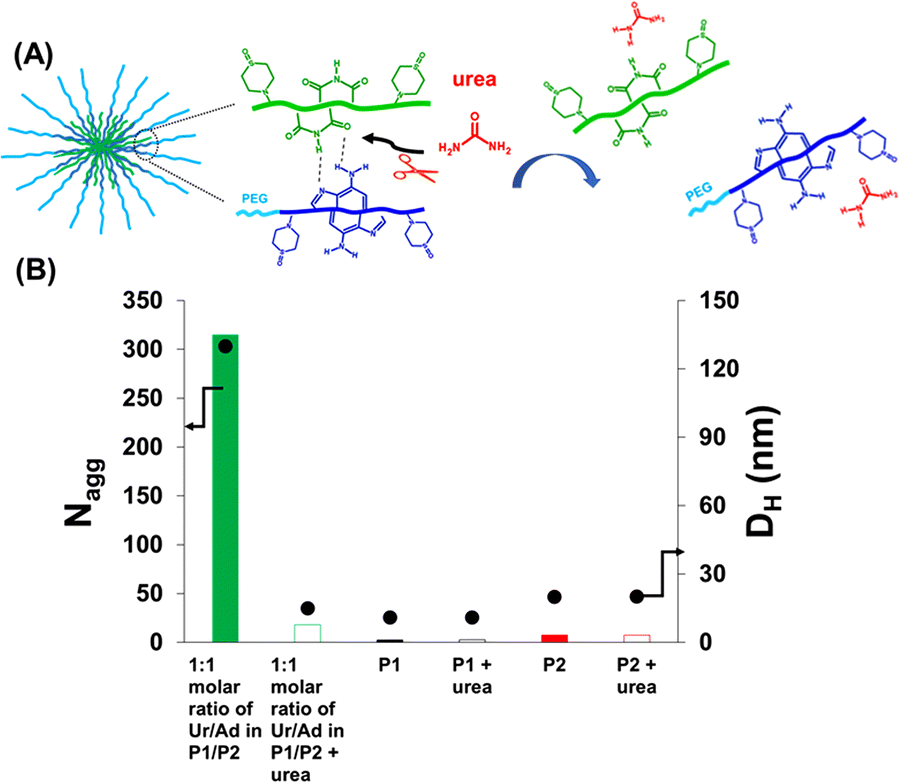 | ||
| Fig. 2 Disassembly of co-assembled nucleobase-containing copolymers P1 + P2 in HEPES buffer (pH = 7.4) in the presence of urea: (A) schematic representation of disassembly under urea influence; (B) Nagg (determined by SLS) and DH (measured by DLS) of formulation A, P1 and P2 before vs. after the addition of urea measured between 1 g L−1 and 5 g L−1. | ||
Overall, in contrast to the pure hydrophobic nucleobase-containing polymers reported so far, these results show that uracil-(P1) and adenine-containing (P2) copolymers have no tendency to form large aggregates (Nagg < 8 by SLS; particle hydrodynamic diameter DH < 20 nm, by DLS, as shown in Table 2).
Previous studies on DNA29 indicated that the co-association between complementary nucleobases is stronger than the uracil–uracil association or adenine–adenine association. Thus, it is expected that the co-assembly between uracil- and adenine-containing copolymers led to self-assembled objects with high Nagg values and large particle sizes, if the complementary H-bond interactions are strong enough to overcome the competition with water molecules.
For this reason, the next step consisted in the investigation of the co-assembly of P1 and P2 in aqueous solution at physiological pH. Compared to individual P1 or P2 solutions, formulation A (P1 + P2, Ur/Ad ratio of 1/1) featured a high increase of the apparent molar mass Ma (∼107 g mol−1) by SLS (Fig. 1A). This result proves the co-assembly of the complementary nucleobase-containing copolymers into aggregates with a high Nagg value (∼315). The formation of co-assembled aggregates was confirmed by DLS (Fig. 1B). Compared to P1 and P2, which showed low DHe values (particle hydrodynamic diameter < 20 nm), formulation A led to a DH value 6.5 times higher (130 nm) than those of the starting polymers. These results suggest that the co-assembly originates from strong interactions between complementary nucleobases.
Co-assembly in the presence of a H-bond competitor
Most of the self-assembled systems in water are driven by hydrophobic interactions. In order to prove unequivocally that the resulting co-assembly was mainly guided by H-bonds between complementary nucleobases instead of hydrophobic interactions, the effect of urea on the co-assembly was investigated. Urea is known as a strong competitor to H-bonds.30–33 A diluted solution of urea (0.01 M) was added to formulation A (P1 + P2, Ur/Ad ratio of 1/1), and the mixture was stirred for 1 h and then examined by SLS and DLS. As presented in Fig. 2, a significant decrease of the aggregation number was observed after the addition of urea. Small particles with an average hydrodynamic diameter of 15 nm (measured by DLS) were formed. The size and the Nagg value were close to those of individual P1 and P2 copolymers (Fig. 1, 2 and Table 2). Urea cleaves the H-bonds between complementary nucleobases of P1 and P2 thus preventing the formation of co-assemblies, and leading to the release of P1- and P2-urea structures.30 The dramatic effect of urea on formulation A (P1 + P2, Ur/Ad ratio of 1/1) confirms that H-bonds between complementary nucleobases are responsible for the co-assembly between P1 and P2.Impact of adenine and uracil nucleobases
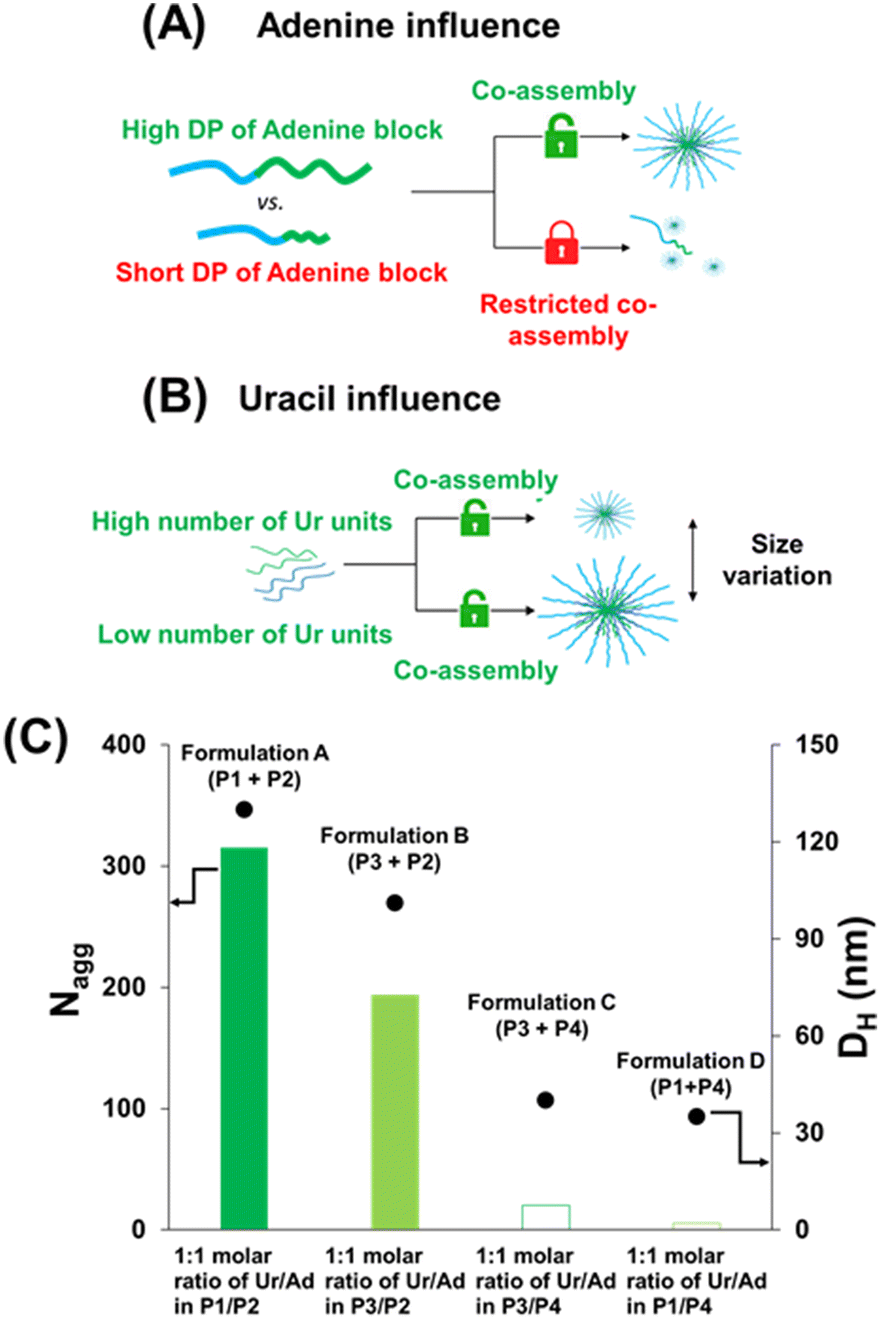
The effect of the length of the adenine-containing block and of the composition of the uracil containing polymer on the co-assembly was investigated. Four formulations (formulations A, B, C and D; Table 2 and Fig. 3) were compared. These formulations contained equimolar amounts of complementary nucleobases (ratio 1:1, eqn (S15) and (S16)†).
 | ||
| Fig. 3 Impact of adenine (A) and uracil (B) nucleobases on the properties of co-assembled structures; (C) variation of Nagg by SLS and particle size (DH) by DLS of formulations A, B, C and D. The experiments were performed at concentrations between 1 g L−1 and 5 g L−1. | ||
Formulations A and B were prepared using the same high length adenine-containing block copolymers (P2: PEG112-b-P(AdMA30-stat-THOXMA70), DP ∼ 100, 30 units of adenine) and two different uracil containing copolymers (P1, P3) with the same degree of polymerisation (DP ∼ 42) but different numbers of uracil units (P1: 8 units of Ur and P3: 22 units of Ur).
SLS and DLS data of formulations A and B displayed in Fig. 3 show that aggregates with high Nagg values (above 200) and particle sizes (above 100 nm) were formed when a long adenine-containing block (DP ∼ 100) was used (Table 2 and Fig. 3).
However, significant differences were noted depending on the uracil copolymer (P1 or P3) used in the co-assembled formulations A and B. Formulation A (P1 + P2), prepared with P1 that contains only 8 uracil units per chain, led to objects with a high Nagg values (∼315, by SLS). In contrast, formulation B (P2 + P3), in which P3 has 22 uracil units per chain, produced smaller co-assemblies with Nagg ∼ 194 (by SLS, Fig. 3). DLS measurements revealed a similar trend for the hydrodynamic particle diameters (DH) which reached 130 nm and 101 nm for formulation A and formulation B, respectively. Thus, increasing the number of uracil units per chain in the uracil-containing copolymer (from 8 to 22) led to smaller self-assembled objects. This might be because a smaller number of chains of the copolymer containing a higher density of uracil units (P3) is required to bind the complementary adenine containing copolymer (P2). This translates into a lower Nagg value and particle hydrodynamic diameter for formulation B than for formulation A. Spherical morphologies were observed by TEM in formulations A (∼150 nm) and B (∼100 nm) (Fig. 4), and their sizes were in agreement with the previous data obtained by DLS.
 | ||
| Fig. 4 TEM characterisation of co-assemblies (A) formulation A: P1 + P2 mixture (1:1); (B) formulation B: P3 + P2 mixture (1:1); (C) formulation C: P3 + P4 mixture (1:1); (D) formulation D: P1 + P4 mixture (1:1). The co-assemblies were prepared at an initial concentration of 1 g L−1. TEM experiments were performed at a concentration of 0.1% w/w. | ||
Formulations C and D were made using a shorter adenine containing block copolymer (P4: PEG112-b-P(AdMA5-stat-THOXMA5), DP ∼ 10, 5 units of adenine) and the same uracil containing copolymers (P1, P3).
Low Nagg values and hydrodynamic diameters were determined when short length adenine blocks (formulations C and D) were used. For formulation C (using P3, DP = 42, 22 units of uracil), a very low Nagg value (∼10, by SLS) and particle size (∼40 nm, by DLS) were observed. In addition, TEM analysis (Fig. 4) showed small spherical particles with a size of ca. 35 nm (formulation C, Fig. 4), in agreement with the DLS data. For formulation D (using P1, DP = 42, 8 units of Ur), Nagg (∼8) was calculated by SLS (Fig. 3). This low Nagg (∼8) obtained for formulation D in comparison to the results observed for formulation A shows that the use of polymers containing only small amounts and low density of adenine diminished the co-assembly ability to form large aggregates, likely because the H-bond cannot be formed in sufficiently high numbers.
Further investigation of the co-assembly in the presence of urea revealed a decrease of the particle hydrodynamic diameter and the number of aggregations in all formulations. The urea treatment leads to particles as small as 30 nm (Fig. S7†). This confirmed that the co-assemblies formed in the different formulations studied are driven by H-bonds between complementary nucleobase units rather than from hydrophobic interactions.
Influence of the molar ratios between Ur/Ad
Then, lower ratios of uracil and adenine equivalents (0.1/1 molar ratio of Ur/Ad) were studied on four formulations (formulations E, F, G, H), as presented in Table S1.† Co-assembled aggregates (Table S1 and Fig. S8A†) with an average hydrodynamic diameter of around 50 nm (corresponding to single populations in DLS) were formed. These results suggest that at such low stoichiometries, the amount of uracil was likely too low to completely complex (by H-bonds) with adenine.Moreover, high ratios between uracil and adenine equivalents (10/1 molar ratio of Ur/Ad) were investigated by preparing additional formulations (formulations I, J, K, L, Table S1 and Fig. S8B†). Co-assembled formulations with a moderate Nagg (∼100) and nanometric particle size (∼80 nm) were observed when long adenine-containing blocks (formulations I and J) were used, while a low Nagg value (below 40) and particle size (∼30 nm) were observed for the formulations based on short adenine-containing blocks (formulations K and L). These values were higher than those obtained for low ratios of Ur/Ad (i.e. Ur/Ad = 0.1/1), which is coherent since an increase in the number of complementary nucleobases increases the number of H-bonds which translates into the increase of Nagg and DH. However, compared to the 1/1 stoichiometry (Fig. 3), the increase of the stoichiometry to 10/1 led to an overall decrease of Nagg and DH. This behaviour was previously reported by Hua et al.10 who explained this phenomenon to be a consequence of a low energy barrier for chain exchange which occurs at high stoichiometry, since the system is oversaturated in one of the complementary nucleobase copolymers. Smaller spherical aggregates were, therefore, formed to reduce the increased corona-chain repulsion introduced through the insertion of the complementary hydrophobic copolymer.10 Applied to our system, the increase of the stoichiometry ratio increases the hydrophobicity of the system (by adding high amounts of the uracil containing copolymer) which led to more compact and smaller particles.
Globally, these results emphasize that structural parameters such as the copolymer architecture (DP, nucleobase number per polymer chain), the type of nucleobase, and the ratio between complementary nucleobases are important to control the size of the resulting co-assembled aggregates by H-bonds.
Biocompatibility of nucleobase copolymers
Since the obtained nucleobase-containing copolymers were soluble at physiological pH and might be interesting for further biomedical applications, their in vitro biocompatibility was evaluated. Cytotoxicity, blood aggregation rates, and hemolytic activity evaluation are summarized in Fig. 5. | ||
| Fig. 5 Cytotoxicity of nucleobase containing copolymers evaluated on the L929 mouse cell line, pH 7.4, using non-treated cells as a control (A); blood aggregation rate of copolymers evaluated at pH 7.4, on human blood (B); hemoglobin release of nucleobase copolymers tested at pH 7.4 on human blood (C). | ||
The cytotoxicity was assessed at pH 7.4 on the mouse fibroblast L929 cell line using the PrestoBlue assay (Fig. 5A).34 This test works as a cell health indicator which analyses the reducing ability of living cells to finally assess cellular viability.34 A cellular viability below 0.7 (or 70%) indicates a cytotoxic behavior.34 As shown in Fig. 5A, the nucleobase containing copolymers showed a cellular viability above 95% up to the highes tested concentration of 0.7 mg mL−1. Indeed, a slightly increased cell growth is observed for higher concentrations. A more detailed investigation of this phenomenon was however beyond the scope of this study.
These promising results led us to further evaluate the nucleobase containing copolymers towards blood compatibility, in terms of red blood cell aggregation activity (Fig. 5B) and hemoglobin release (Fig. 5C).
The aggregation activity was expressed by the aggregation rate and compared to a polycationic commercial polymer (i.e. polyethyleneimine PEI) which is known for its high aggregation rate (around 2.5).35 All the tested copolymers showed an aggregation rate below 1.2 which underlines that they do not provoke undesired cell aggregation. This result could be explained by the neutral character of the nucleobase copolymers at physiological pH which avoided cellular aggregation, a frequent phenomenon observed for cationic polymers. Moreover, we investigated the interaction of nucleobase copolymers with the red blood cell membrane. In this regard, the release of hemoglobin at physiological pH (pH 7.4) from the erythrocites was measured. As presented in Fig. 5C, the hemoglobin was released in low amounts (below 1%), which is consistent with the non-hemolytic activity.36 Overall, these results emphasize promising potential of neutral and hydrosoluble nucleobase-containing copolymers as alternatives to cationic polymers in biological applications.
Conclusions
A set of adenine- and uracil-containing statistical and block polymethacrylate copolymers have been successfully synthesized by RAFT polymerization. The resulting macromolecular structures were different from those previously reported in the literature, as a result of their hydrophilic character. The novelty of the developed co-assembled systems was that their preparation occurred exclusively in an aqueous buffer. The co-assembly was mainly driven by H-bonds between complementary adenine and uracil nucleobases, like in DNA. This approach is original compared to previously reported nucleobase polymethacrylate that self-assembled in organic solvents,5 because of their insolubility in water. A range of structural parameters (number of nucleobase units per polymer, length of blocks and ratio between complementary nucleobases) were investigated to examine their influence on the co-assembly behaviour and the physicochemical properties (aggregation number, particle size) of the resulting nano-objects. Overall, spherical co-assembled morphologies were observed by TEM. However, significant modifications were observed (by SLS, DLS) due to the variations in the length of the nucleobase-containing block as well as in the content of complementary nucleobases. A high DP of adenine blocks led to self-assembled architectures with high Nagg values, while a low DP of adenine blocks enabled the formation of small-sized spherical co-assembled objects. The uracil block had a significant contribution to the regulation of the size of the co-assemblies. Low contents in uracil groups led to co-assembled objects with high aggregation numbers and particle sizes, whereas higher contents of uracil resulted in smaller particles. Structural parameters (such as the number of nucleobases per polymer and the length of the nucleobase block) could thus lead to different co-assembly behaviour. We proposed that these results are a consequence of possible dynamic H-bond interchanges between the nucleobases present in the copolymers. Furthermore, individual uracil or adenine polymers showed no ability to self-assemble. This observation emphasized that complementary adenine–uracil H-bond interactions were at the core of co-assembly formation. The co-assembly by H-bonds was proven by competitive assays with urea which led to a significant decrease of the Nagg value and particle size of the co-assembled objects. Evaluation of the in vitro biological properties of the nucleobase copolymers showed that they are not cytotoxic and compatible with blood. This work emphasized huge potential for the use of nature inspired H-bond interactions between complementary nucleobases to develop novel co-assembled architectures which may find future applications in biological systems.Author contributions
Conceptualization, S. Catrouillet and V. Ladmiral; methodology, L. V. Arsenie, S. Catrouillet, and V. Ladmiral; validation, S. Catrouillet and V. Ladmiral; formal analysis, L. V. Arsenie; investigation, L. V. Arsenie and M. Semsarilar; resources, V. Ladmiral, P. Lacroix-Desmazes and J. C. Brendel; writing—original draft preparation, L. V. Arsenie; writing—review and editing, L. V. Arsenie, M. Semsarilar, J. C. Brendel, P. Lacroix-Desmazes, V. Ladmiral, and S. Catrouillet; supervision, S. Catrouillet and V. Ladmiral; project administration, S. Catrouillet and V. Ladmiral; and funding acquisition, V. Ladmiral and P. Lacroix-Desmazes. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors kindly thank Philippe Gonzales (IBMM, Montpellier, France) who performed dn/dc measurements, as well as Carolin Kellner (IOMC, Jena, Germany) who performed the biological investigations. This research was funded by DAAD (German Academic Exchange Service) Research Grants-Short Term Grants 2022 (Funding program number: 57588366) as well as by the French Ministry of higher education and research and the PHC Procope program (Project-ID: 48137ZE). JCB further thank the German Science Foundation (DFG) for generous funding within the Emmy-Noether Programme (Project-ID: 358263073).References
- F. Crick and J. Watson, Nature, 1953, 171, 737–738 CrossRef.
- C. Suparpprom and T. Vilaivan, RSC Chem. Biol., 2022, 3, 648–697 RSC.
- C. W. Huang, W. Ji and S. W. Kuo, Macromolecules, 2017, 50(18), 7091–7101 CrossRef CAS.
- I. Hong-Lin, C.-C. Cheng, C.-W. Huang, M.-C. Liang, J.-K. Chen, F.-H. Ko, C.-W. Chu, C.-F. Huang and F.-C. Chang, RSC Adv., 2013, 3, 12598–12603 RSC.
- C.-C. Cheng, I.-H. Lin, J.-K. Chen, Z.-S. Liao, J.-J. Huang, D.-J. Lee and Z. Xin, Macromol. Biosci., 2016, 16(3), 1415–1421 CrossRef CAS PubMed.
- H. S. Bazzi and H. F. Sleiman, Macromolecules, 2002, 35(26), 9617–9620 CrossRef CAS.
- H. Lu, J. Cai and K. Zhang, Polym. Chem., 2021, 12, 2193–2204 RSC.
- M. Garcia, K. Kempe, D. M. Haddleton, A. Khan and A. Marsh, Polym. Chem., 2015, 6, 1944–1951 RSC.
- Y. Kang, A. Pitto-Barry, H. Willcock, W.-D. Quan, N. Kirby, A. M. Sanchez and R. K. O'Reilly, Polym. Chem., 2015, 6, 106–117 RSC.
- Z. Hua, A. Pitto-Barry, Y. Kang, N. Kirby, T. R. Wilks and R. K. O'Reilly, Polym. Chem., 2016, 7, 4254–4262 RSC.
- Z. Hua, R. Keogh, Z. Li, T. R. Wilks, G. Chen and R. K. O'Reilly, Macromolecules, 2017, 50(9), 3662–3670 CrossRef CAS PubMed.
- Z. Hua, T. R. Wilks, R. Keogh, G. Herwig, V. G. Stavros and R. K. O'Reilly, Chem. Mater., 2018, 30(4), 1408–1416 CrossRef CAS.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 2(7), 581–586 CrossRef CAS PubMed.
- Y. Kang, A. Pitto-Barry, M. S. Rolph, Z. Hua, I. Hands-Portman, N. Kirby and R. K. O'Reilly, Polym. Chem., 2016, 7, 2836–2846 RSC.
- H. Yang and W. Xi, Polymers, 2017, 9(12), 666–690 CrossRef PubMed.
- S. Varlas, Z. Hua, J. R. Jones, M. Thomas, J. C. Foster and R. K. O'Reilly, Macromolecules, 2020, 53(22), 9747–9757 CrossRef CAS.
- G. B. Schuster, B. J. Cafferty, S. C. Karunakaran and N. V. Hud, J. Am. Chem. Soc., 2021, 143(25), 9279–9296 CrossRef CAS PubMed.
- S. C. Larnaudie, J. C. Brendel, I. Romero-Canélon, C. Sanchez-Cano, S. Catrouillet, J. Sanchis, J. P. C. Coverdale, J.-I. Song, A. Habtemariam, P. J. Sadler, K. A. Jolliffe and S. Perrier, Biomacromolecules, 2018, 19(1), 239–247 CrossRef CAS PubMed.
- E. Obert, M. Bellot, L. Bouteiller, F. Andrioletti, C. Lehen-Ferrenbach and F. Boué, J. Am. Chem. Soc., 2007, 129, 15601–15605 CrossRef CAS.
- S. Han, E. Nicol, F. Niepceron, O. Colombani, S. Pensec and L. Bouteiller, Macromol. Rapid Commun., 2018, 1800698–1800703 Search PubMed.
- D. Bochicchio, M. Salvalaglio and G. M. Pavan, Nat. Commun., 2017, 147(8), 1–11 Search PubMed.
- Y. Kang, A. Pitto-Barry, M. S. Rolph, Z. Hua, I. Hands-Portman, N. Kirby and R. K. O'Reilly, Polym. Chem., 2016, 7, 2836–2846 RSC.
- L. V. Arsenie, F. Hausig, C. Kellner, J. C. Brendel, P. Lacroix-Desmazes, V. Ladmiral and S. Catrouillet, Molecules, 2022, 27, 4233–4288 CrossRef CAS PubMed.
- K. E. B. Doncom, A. Pitto-Barry, H. Willcock, A. Lu, B. E. McKenzie, N. Kirby and R. K. O'Reilly, Soft Matter, 2015, 11, 3666–3676 RSC.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- R. Xu, Particuology, 2015, 18, 11–21 CrossRef.
- H. A. Spijker, F. L. van Delft and J. C. M. van Hest, Macromolecules, 2007, 40(1), 12–18 CrossRef CAS.
- J. Thevenot, A.-L. Troutier, L. David, T. Delair and C. Ladavière, Biomacromolecules, 2007, 8(11), 3651–3660 CrossRef CAS.
- S. Scott, C. Shaheen, B. McGuinness, K. Metera, F. Kouzine, D. Levens, C. J. Benham and S. Leslie, Nucleic Acids Res., 2019, 47(12), 6360–6368 CrossRef CAS PubMed.
- W. Zhang, M. Liu, C. Lee, B. J. Salena and Y. Li, Sci. Rep., 2018, 8, 3–10 CrossRef PubMed.
- W. K. Lim, J. Rosgen and S. Walter Englander, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(8), 2595–2600 CrossRef CAS PubMed.
- A. Bende, Theor. Chem. Acc., 2010, 125, 253–268 Search PubMed.
- F. Jia, H. Li, R. Chen and K. Zhang, Bioconjugate Chem., 2019, 30(7), 1880–1888 CrossRef CAS.
- M. Xu, D. J. McCanna and J. G. Sivak, J. Pharmacol. Toxicol. Methods, 2015, 71, 1–7 CrossRef CAS PubMed.
- A. Frère, M. Kawalec, S. Tempelaar, P. Peixoto, E. Hendrick, O. Peulen, B. Evrard, P. Dubois, L. Mespouille, D. Mottet and G. Piel, Biomacromolecules, 2015, 16(3), 769–779 CrossRef.
- I. Greco, N. Molchanova, E. Holmedal, H. Jenssen, B. D. Hummel, J. L. Watts, J. Hakansson, P. R. Hansen and J. Svenson, Sci. Rep., 2020, 10, 1–13 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00872f |
| This journal is © The Royal Society of Chemistry 2022 |