
A flow-based transition-metal-catalysed hydrogenolysis strategy to facilitate peptide side-chain deprotection†
Maria
Menti-Platten‡
 a,
Janice R.
Aldrich-Wright
ab and
Christopher P.
Gordon
*abc
a,
Janice R.
Aldrich-Wright
ab and
Christopher P.
Gordon
*abc
aSchool of Science, Western Sydney University, Locked Bag 1797, Penrith South DC, Australia. E-mail: c.gordon@westernsydney.edu.au
bNanoscale Organisation and Dynamics Group, Locked Bag 1797, Penrith South DC, Australia
cMolecular Medicine Research Group, Western Sydney University School of Medicine, Narellan Rd & Gilchrist Dr, 2560, Campbelltown, NSW, Australia
First published on 1st December 2021
Abstract
Orthogonal deprotection methodologies are an invaluable tool for the construction of site-specially modified peptides. Here, we report a facile 10% Pd/CaCO3-based procedure to selectively mediate Nβ-side-chain Cbz-lysis from extended peptide sequences in the presence of trityl and t-Butyl protecting groups.
Introduction
The compatibility of catalytic hydrogenolysis to facilitate side-chain and Nα-protecting group lysis on peptide-based molecules has long been recognised.1,2 For example, relative to the standard acidic and alkaline-driven cleavage strategies, hydrogenolysis is less prone to inducing epimerisation or degradation of sensitive residues.3 Further, as the post-cleavage by-products are typically gaseous or volatile in nature, the employment of hydrogenolysable protecting groups can expedite workup and isolation processes.3 Nevertheless, presumably as consequences of traditionally proving incompatible with ‘on-resin’ transformations,4,5 and further, the susceptibility of platinum-group metals to sulfur- and nitrogen-induced deactivation, the employment of metal-catalysed hydrogenolysis within the context of peptide synthesis remains relatively limited.6–9Notwithstanding these limitations, metal-catalysed hydrogenolysis has been most successfully employed within the construction of a number of natural and non-natural peptidyl-based products such as Callipeltin E (1, Fig. 1),10 Alterobactin A (2, Fig. 1),11 and Schizopeptin 791 (3, Fig. 1)12 Hence despite the aforementioned limitations, metal-catalysed hydrogenolysis displays a significant value for mediating off-resin late-stage of synthesis modifications. Moreover, as a number of standard protecting functionalities such as tert-butyloxycarbonyl (Boc), trityl (Trt), 2-trimethylsilylethyl (TMSE) and 4-methyltrityl (Mtt) are reported to remain intact under hydrogenation conditions, metal-catalysed hydrogenolysis presents the potential for applications in the development of site-specific side-chain modification protocols.2
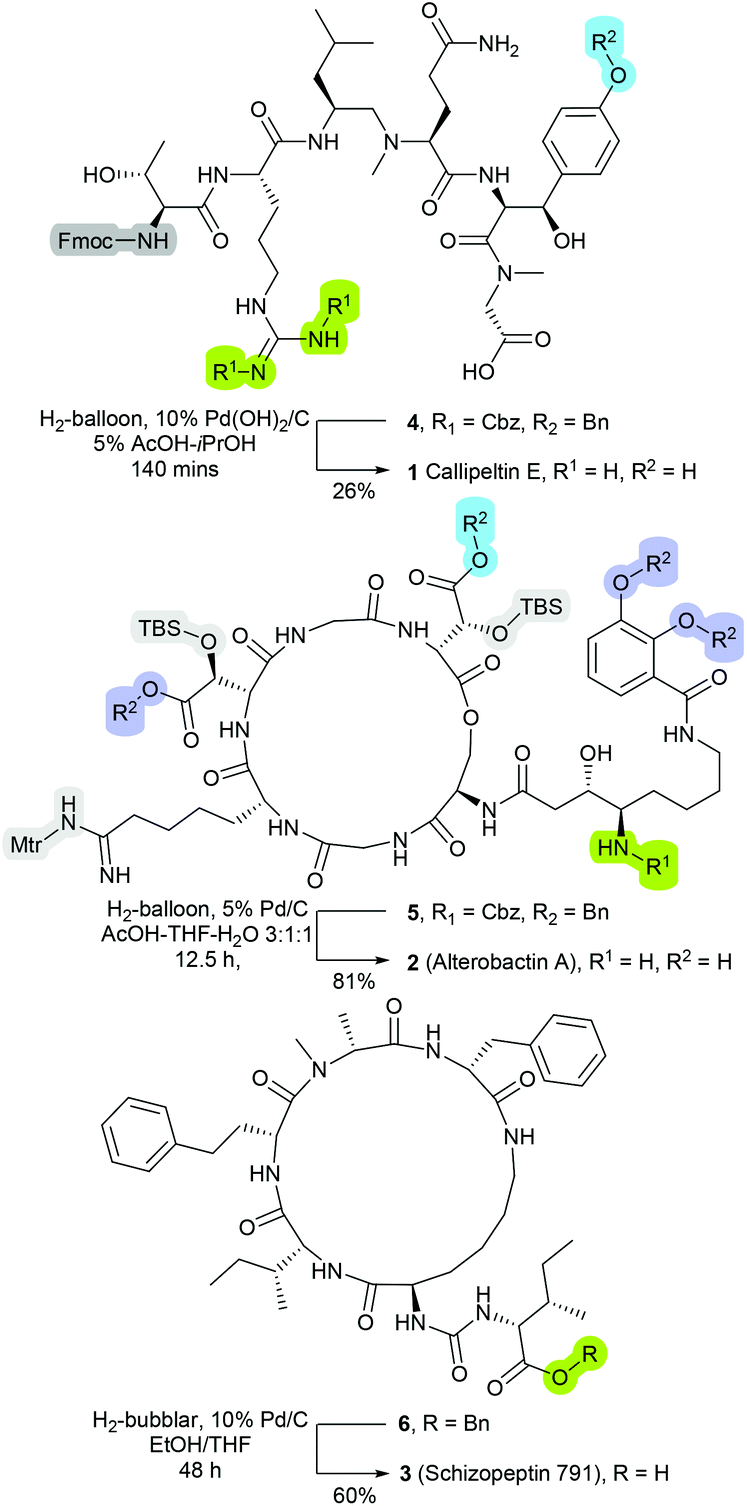 | ||
| Fig. 1 Structures of as Callipeltin E (1),10 Alterobactin A (2),11 and Schizopeptin 791 (3), in which were synthesised through protocols that incorporated a catalytic hydrogenolysis deprotection transformation.12 | ||
Our interest in exploring transition-metal-catalysed hydrogenolysis pertained to the synthesis of Gram-positive bacterial quorum sensing autoinducing peptides (AIPs).13 Typically, AIPs produced by Gram-positive bacteria, such as Staphylococcus aureus (S. aureus) are constructed around a side-chain-to-tail penta-residue macrocyclic scaffold. To date, we have primarily relied on synthetic strategies which are underpinned by a combination of a hyper-acid liable resin-linker and side-chain protecting moiety (Scheme 1a) to derive AIP analogues.14 Whilst this approach has broadly proven adequate-for-purpose, affording cyclisations of ≈80%, we speculate that the carry-through of stoichiometric quantities of post-cleavage methoxytrityl-species released from the concomitant resin and side-chain cleavage step is deleterious (i.e. stage 2, Scheme 1a). Of particular concern is the affordance of trityl-peptide reattachment adducts and unintentional protecting group lysis through Lewis acid-mediated pathways.15
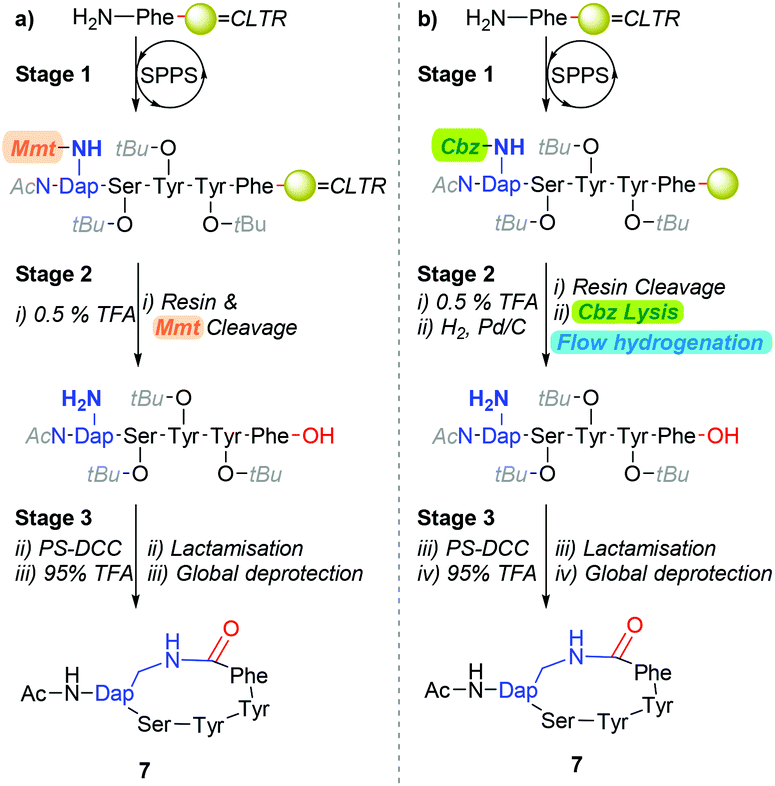 | ||
| Scheme 1 a) The traditional cyclisation pathway, with hyper-acid labile Mtt as a protecting group. (b) Alternative approach to synthesise lactam-based AIP analogues using catalytic hydrogenation to afford the free side-chain amine. | ||
Accordingly, we were eager to examine the potential of incorporating transition-metal-catalysed hydrogenolysis into the polystyrene-supported-carbodiimide (PS-DCC) mediated cyclisation reaction sequence (i.e.Scheme 1a) as an alternative approach to facilitate the lysis of a β-amino-Cbz moiety from an intra-sequence diaminopropionic acid (Dap) residue (Scheme 1b). To this end, although traditional batch and shaker-type approaches have proven most adequate,3,10–12,16,17 the established advantages of conducting hydrogenations under flow conditions,18,19 which include enhancing catalyst longevity, prompted our interest in exploring the practicability of performing reductive chemistry on peptides under flow conditions.20–22
Although outwardly seeming plausible, literature precedent for mediating amino-side-chain protecting group lysis under flow conditions is seemingly limited to the naphthoyl-cbz-protected lysine derivative (Table 1, entry 1).20 Indeed, it appears that this is representative of only three examples whereby amino side-chain deprotection has been effected under metal-catalysed flow hydrogenation.20 The remainder of examples describe Nα-Cbz lysis from either single amino acid derivatives (Table 1, entry 2), a small number of dipeptides, a tripeptide, and a pentapeptide (Table 1, entries 3–5).
| Entry | Substrates |
|---|---|
a Reagents and conditions: 10% Pd/C, EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH (1:1), 1.0 mL min−1, 80 °C, ref. 20.
b Reaction performed on 3 g scale; 30% AcOH was added to the solvent ref. 20.
c 10% Pd/C, EtOAc:EtOH (1:1), 0.5 mL min−1, 60 °C, ref. 21.
d 10% Pd/C, EtOAc:EtOH (1:1), 1.0 mL min−1, 60 °C, ref. 22. :EtOH (1:1), 1.0 mL min−1, 80 °C, ref. 20.
b Reaction performed on 3 g scale; 30% AcOH was added to the solvent ref. 20.
c 10% Pd/C, EtOAc:EtOH (1:1), 0.5 mL min−1, 60 °C, ref. 21.
d 10% Pd/C, EtOAc:EtOH (1:1), 1.0 mL min−1, 60 °C, ref. 22.
|
|
| 1a |
|
| 2a |

|
| 3a,b |

|
| 4a,c |
|
| 5d |
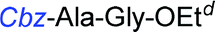
|
| 6c |

|
Results and discussion
Across the aforementioned examples in which peptidyl-based deprotection has been reported under flow, each has been performed using comparable reaction conditions.20–22 Specifically, with the exception of flow rate and temperature, hydrogenations were accomplished by passaging a 0.05 M solution of the peptidyl–substate in a solvent system of 1:1 ethanol in ethyl acetate through a fixed bed of 10% Pd/C (Table 1). Whilst these conditions were reported to promote near quantitative conversion, with respect to the proposed cyclisation pathway (Scheme 1b), both EtOH and EtOAc have proven incompatible with PS-DCC mediated coupling. Hence, given the well-established compatibility of chloroform (CHCl3) with polystyrene-based resin matrices, we initially focused on exploring whether CHCl3 could serve as a viable reaction medium to facilitate lysis of hydrogen-labile protecting groups from peptide-based substrates. Whilst cognisant that CHCl3 could be considered an atypical solvent for the intended purpose, we noted that previous studies have demonstrated that CHCl3 and dichloromethane (DCM) can absorb sufficient H2 to mediate the Pd/C catalysed reduction of α,β-unsaturated through to di-substituted olefinic substrates.23,24
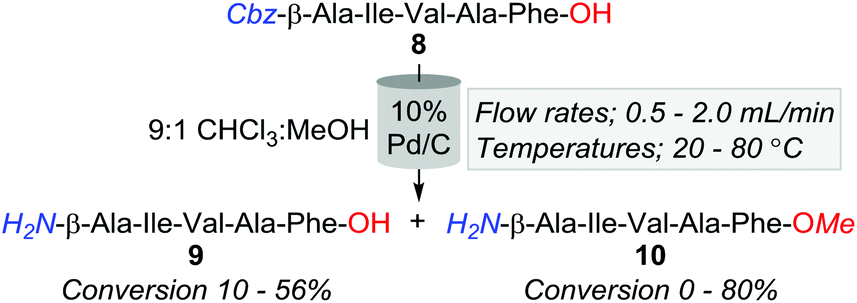
However, an initial series of trial reactions in which various permeations of catalyst bed temperature (40–100 °C), flow rate (0.5–3.0 mL min−1), and substrate concentration (≈4–0.06 mM, i.e. ≈2.5–0.04 mg L−1) were explored, revealed that limited product solubility precluded the use of neat CHCl3 as a reaction medium. For instance, with the use of 10% Pd/C and the CHCl3 soluble Cbz-β-Ala-Ile-Val-Ala-Phe-OH (8); whilst trace amounts of the desired Cbz-lysed sequence H2N-β-Ala-Ile-Val-Ala-Phe-OH (9) were observed, precipitation of product, both within and directly downstream of the catalyst bed, triggered pump over pressurisation failures.
Nonetheless, a subsequent screen of alternative solvent combinations ascertained that a 9:1 CHCl3:MeOH reaction solution alleviated the complications associated with precipitation. However, the employment of this cosolvent mixture promoted the formation of the methyl ester adduct, 10, with conversions ranging from 0 to 80% (Table 2). Although we speculate that trace amounts of HCl within the solvent were a causative factor of methyl esterification, the afforded quantities of 10 were inversely proportionate to temperature and flow rate (Table 2). That is, at 80 °C and a flow rate of 0.5 mL min−1, 10 was obtained in an 80% conversion, whilst only 5% of 8 was converted to the desired product, 9. In contrast, 40 °C and 1.0 mL min−1 offered lower O-methylation, with 50% and 30% of 8 converted to 9 and 10, respectively. Although milder conditions increased the production of 8; in accordance with a handful of previous batch reports, Pd/C afforded unidentified higher polarity by-products, which we speculate are resultant of amide-bond reductions due to the well-documented inconstancies associated with Pd/C catalysts.9,25
|
|
|||||
|---|---|---|---|---|---|
| Entry | Flow rate (mL min−1) | Temp. (°C) | 8 (%) | 9 (%) | 10 (%) |
| a Percent conversions determined by HPLC analysis at 214 nm. | |||||
| 1 | 0.5 | 20 | 70 | 25 | 0 |
| 2 | 1.0 | 20 | 18 | 45 | 7 |
| 3 | 2.0 | 20 | 45 | 35 | 0 |
| 4 | 0.5 | 40 | 5 | 45 | 36 |
| 5 | 1.0 | 40 | 5 | 50 | 30 |
| 6 | 2.0 | 40 | 10 | 56 | 15 |
| 7 | 0.5 | 60 | 0 | 10 | 55 |
| 8 | 1.0 | 60 | 0 | 50 | 50 |
| 9 | 2.0 | 60 | 0 | 63 | 32 |
| 10 | 0.5 | 80 | 0 | 5 | 80 |
| 11 | 1.0 | 80 | 0 | 10 | 54 |
| 12 | 2.0 | 80 | 0 | 10 | 60 |
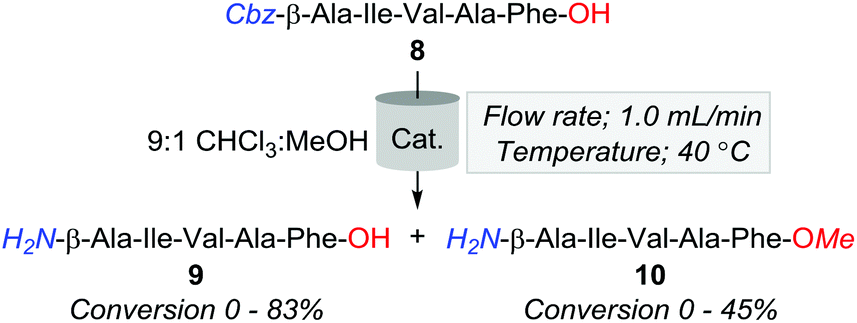
Hence, in a bid to optimise the production of 9, whilst minimising the formation of by-products, we surveyed a series of heterogeneous (Table 3, entries 1–8) and polymer immobilised catalysts (Table 3, entries 9 and 10). Of the catalysts screened, 10% Pd/CaCO3 proved most effective, affording an 83% and 17% conversion of 8 to 9 and 10, respectively. Additionally, 20% Pd(OH)2/C was observed to convert 55% and 45% of 8 to 9 and 10, respectively. Despite this improvement, methylation was still evident across several conditions, and as such, attention turned to examining the effects of manipulating the percent H2 solvation in reducing the formation of the O-methylated by-product.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | 8 (%) | 9 (%) | 10 (%) |
| Reagents and conditions: H-Cube Pro, 8 in 9:1 CHCl3:MeOH, 1 atm, full H2, 40 °C, 1.0 mL min−1.a Percent conversions determined by HPLC analysis at 214 nm. |
||||
| 1 | 10% Pd/C | 5 | 50 | 30 |
| 2 | CuO/Al2O3 | >95 | 0 | 0 |
| 3 | 10% Pd/CaCO3 | 0 | 83 | 17 |
| 4 | 20% Pd(OH)2/C | 0 | 55 | 45 |
| 5 | Nickel sponge | >95 | 0 | 0 |
| 6 | RaCopper | >95 | 0 | 0 |
| 7 | RaNi | >95 | 0 | 0 |
| 8 | Rh(COD)(dppf) | >95 | 0 | 0 |
| 9 | Fibrecat 1001 | >95 | 0 | 0 |
| 10 | Pd(II) Encat | >95 | 0 | 0 |
Accordingly, the subsequent series of trials were performed using a flow rate of 1.0 mL min−1 and employed either 10% Pd/CaCO3 or 20% Pd(OH)2/C heated to 40 °C, and examined the effect of incrementally increasing percent solvent H2 saturation from 25 to 100%. As outlined in Fig. 2, O-methylation decreased with a reduction of available H2 gas; however, the production of 10 was still significantly higher across all amounts of H2 saturation under 20% Pd(OH)2/C in comparison to using 10% Pd/CaCO3. However, whilst conditions of 40 °C, 1.0 mL min−1, and 25% H2 using 10% Pd/CaCO3 appeared optimal for the desired purpose, it was apparent that these conditions lead to rapid catalyst inactivation as, upon increasing reaction scale to 100 mg, conversion of 8 to 9 was observed to decrease as a function of time with an overall ≈30% conversion of 8 to 9 obtained.
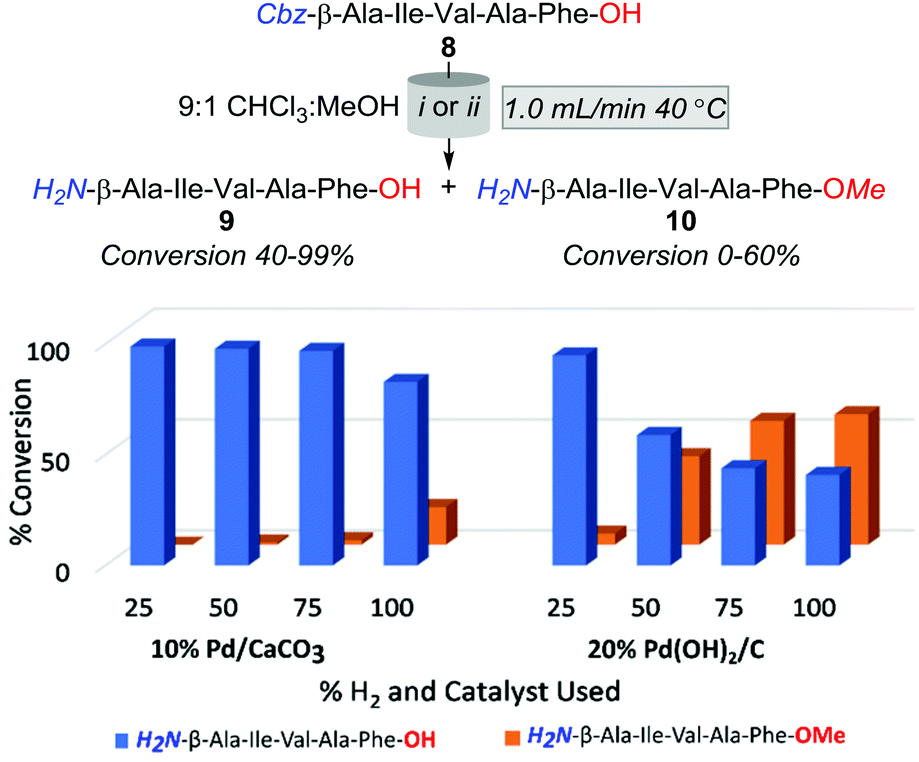 | ||
| Fig. 2 Optimisation trials altering H2 saturation. (i) 10% Pd/CaCO3, (ii) 20% Pd(OH)2/C. | ||
Whilst for N-hydrogenolysis, catalyst poisoning is nearly-inevitable due to the high coordination affinity of amines with transition metals; in this instance, the onset of deactivation appeared exceptionally rapid. To demonstrate, in the aforementioned 100 mg trial, near-complete catalyst deactivation was observed after the turnover of only 46 μmoles of the substrate. In contrast, Ley et al. reported turning over 8070 μmoles of Cbz-Pro-tetrazole-OH in quantitative conversion under comparable conditions.20 Furthermore, no discernible increases in catalyst longevity were observed in our subsequent up-scaling attempts, which employed recognised catalyst preservation strategies of solvent acid doping and intermittent catalyst washing (ESI†).20
Consequently, as the employment of a chloroform-based solvent system remained the most obvious point of difference between the current and previously reported scaling-up protocols, we explored several studies examining hydrodechlorination for the treatment of CHCl3 contaminated groundwater. Such studies describe supported Pd catalysts as highly efficient effectors of hydrodechlorination under ambient conditions.26,27 Further, Xu et al. proposed that the conversion pathway of CHCl3 to methane entailed the generation of several radical atomic species wherein C* and Cl* served as potential site-blocking species.28
Accordingly, whilst contrary to the overall objective of developing an end-to-end single solvent flow sequence, we turned to an assessment of potential non-halogenated solvent systems to effect Cbz-lysis from 8. Whilst 1:1 EtOAc:EtOH was unsuccessful in solubilising 8, following the traditional orthodoxy that hydrogenation rates are dependent on the solvent dielectric constant (ε), and dipole moment (μ),29–32 we turned to an examination of alcoholic solvents. From a quantitative assessment of the solubility of 8 within isopropyl t-butyl alcohol, methanol to ethanol through to n-propanol and n-hexanol inclusively, MeOH presented as the most viable solvent alternative. Indeed, upon reattempting Cbz-hydrogenolysis of 1530 μmoles of 8 utilising MeOH as a reaction medium and the afore-described conditions of 40 °C, 1.0 mL min−1, and 25% H2 using 10% Pd/CaCO3, 9 was afforded in quantitative conversion with no observable evidence of 10.
Although improving catalyst longevity and mitigating O-methylation (i.e. formation of 10), the exclusion of CHCl3 afforded minimal improvement in previously trialled catalysts. For instance, upon reattempting Cbz-lysis of 8 in MeOH at 40 °C, under 25% H2 solvent saturation, and a flow rate of 1.0 mL min−1, CuO/Al2O3, RaCopper, RaNi, Fibercat1001, and Pd(II) Encat remained ineffectual. Nevertheless, the use of a methanolic solution at a flow rate of 1.0 mL min−1 with a 10% Pd/C catalyst bed heated to 40 °C, proved viable for the mediation of Nβ Cbz-lysis from β-Ala (i.e.8, Fig. 3 & 11, Fig. 4) and Dap residues (i.e.13, 15, 17 & 19, Fig. 4). Here, in addition to conservation of both t-Bu (i.e.12, 14, 16, 18, & 20) and Trt (i.e.18) protecting groups, these mild conditions imparted a degree of Fmoc preservation with >85% of 19 converted to 20 (ESI†). Additionally, initial investigations indicated that the aforementioned 10% Pd/CaCO3 based protocol remained effective for the lysis of the O-Bn moiety (i.e.21→22, Fig. 4).
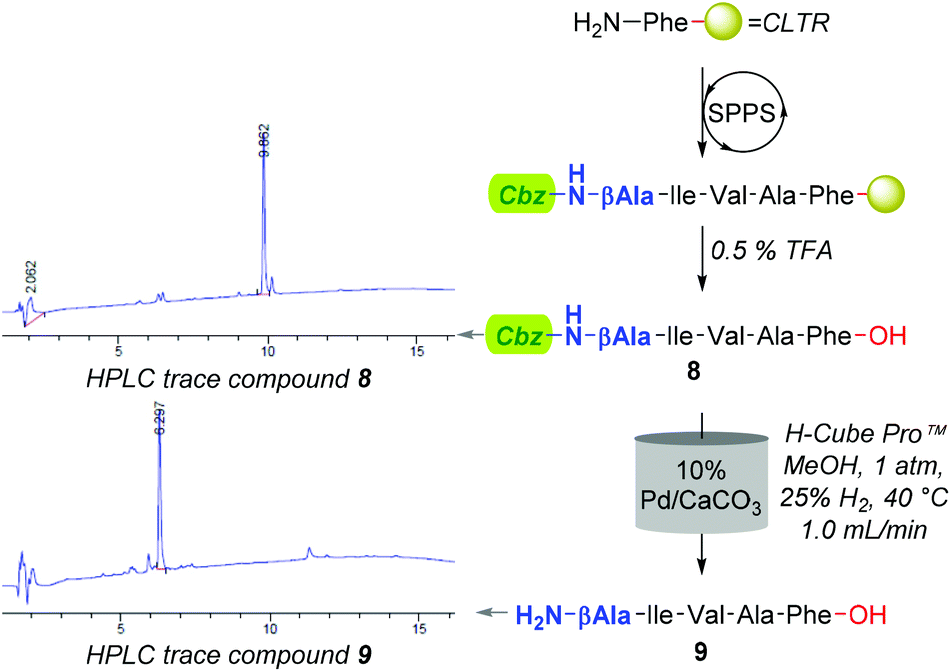 | ||
| Fig. 3 Overall synthetic sequence to access compound 9 with HPLC traces acquired for compounds 8 and 9 to provide an illustrative example of the purities afforded for the crude reaction material prior (i.e.8) and post (i.e.9) hydrogenation. | ||
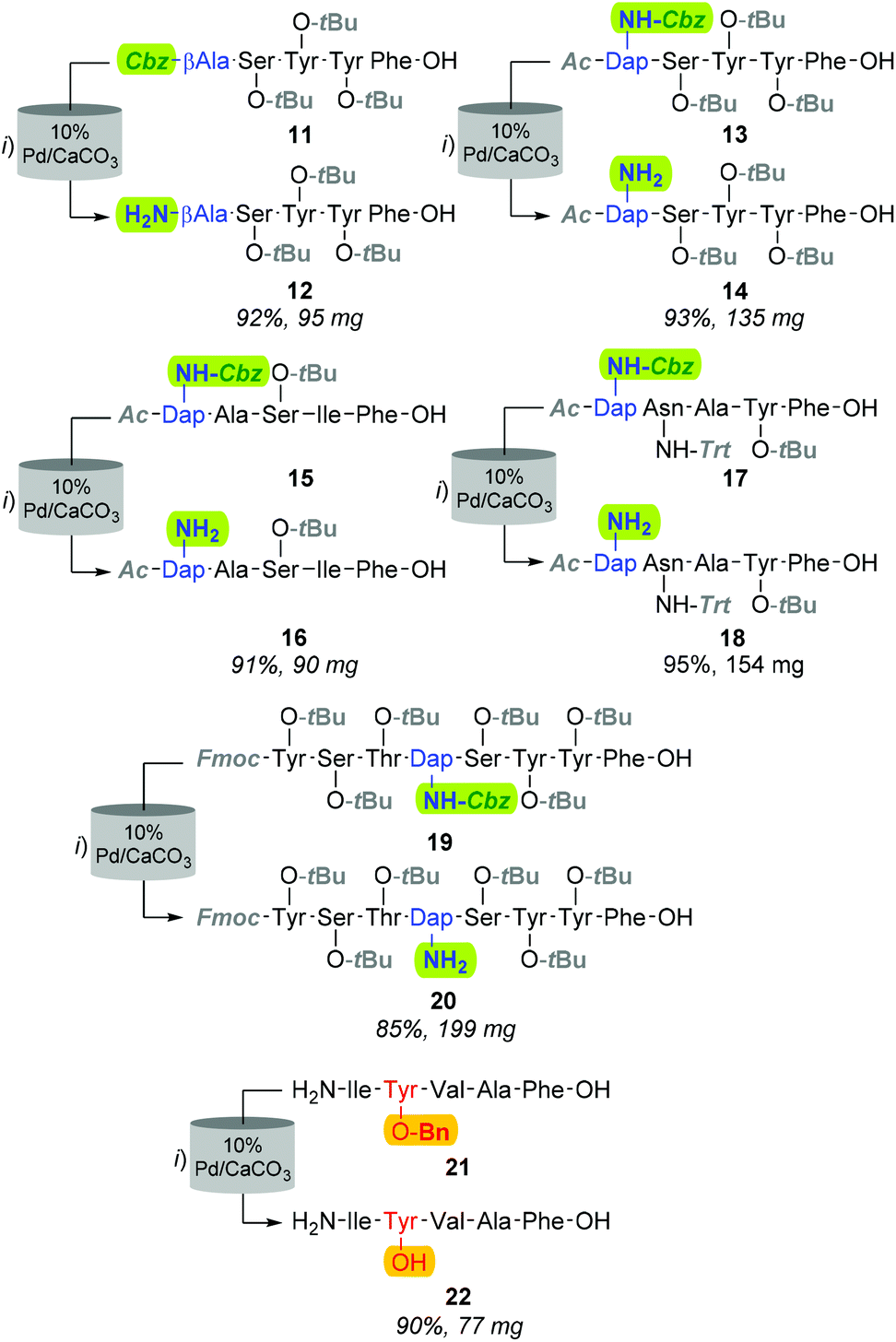 | ||
| Fig. 4 Structures of all Cbz-protected and Cbz-deprotected peptide sequences in addition to the Bn-protected (21) and Bn-deprotected (22) derivative. Reagents and conditions: (i) H-Cube Pro, MeOH, 10% Pd/CaCO3, 1 atm, 25% H2, 40 °C, 1.0 mL min−1. | ||
Although the observed catalyst deactivating properties of CHCl3 diminished the potential of end-to-end single solvent synthesis, it appeared that the absence of carry-through post-deprotection trityl species proved advantageous. Here, following in vacuo MeOH evaporation and CHCl3 re-solvation of the hydrogenated eluent, in each instancePS-DCC induced side-chain-to-tail lactamisation of the partially deprotected sequences (i.e. compound 12, 14, 16, 18 & 20), followed by global deprotection, affording the desired macrocyclic adducts 7 and 23–26, in discernibly higher purities than previously observed with alternative acid-lability-dependent approaches (Fig. 5).
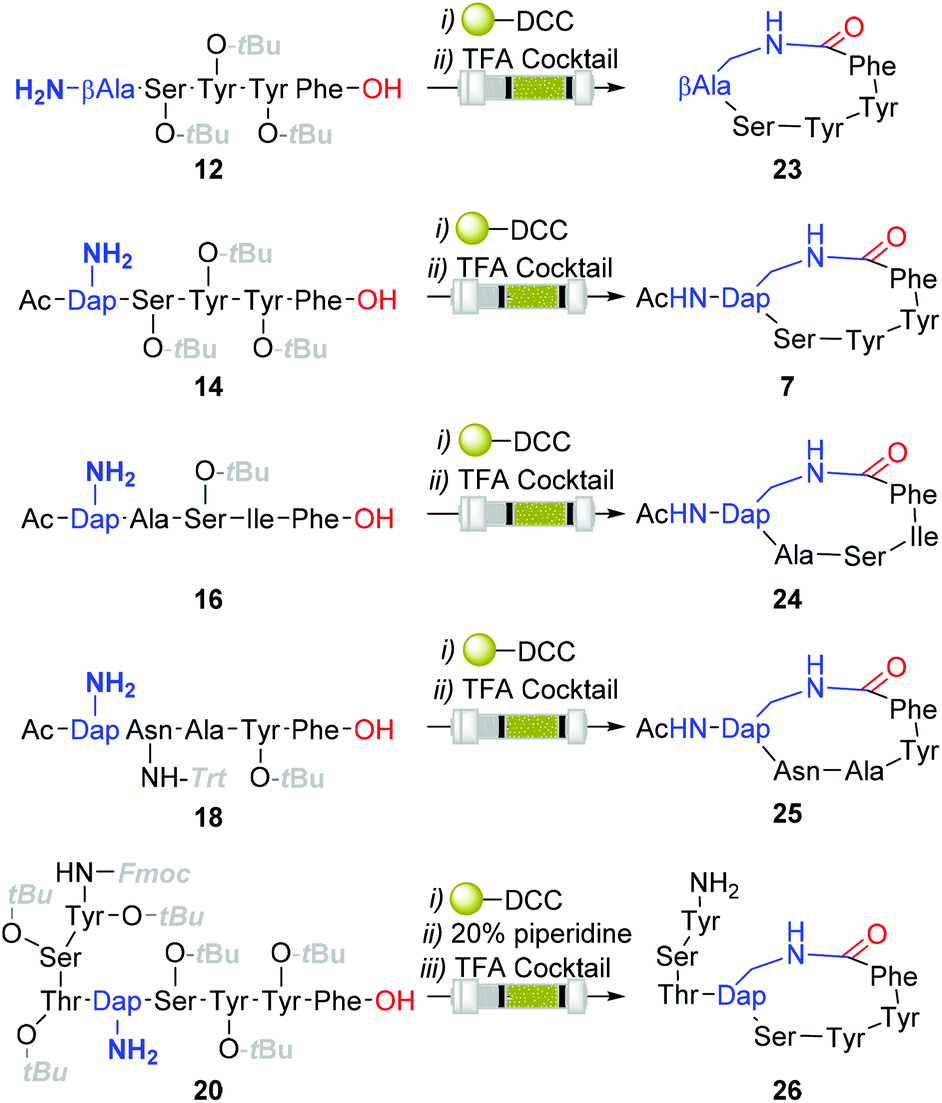 | ||
| Fig. 5 Structure of all Cbz-deprotected and final macrocyclic structures. Reagents and conditions (i) PS-DCC:HOAt (5:1 equiv.), CHCl3, 72 h; (ii) TFA:H2O:Et3SiH:EtSMe (90:5:3:2), 1 h, purification by RP-HPLC. | ||
Conclusions
Whilst CHCl3-associated catalyst deactivation precluded the overarching objective of devising a single solvent continuous flow protocol to access macrocyclic peptides, a facile 10% Pd/CaCO3-based procedure to effect Nβ-Cbz-deprotection from extended peptide sequences was delineated. In addition, from preliminary evaluations, it appears that the amenability of the 10% Pd/CaCO3 based conditions extend to the facilitation of O-debenzylation (i.e.21→22, Fig. 4). Perhaps the broader significance of the technique pertains to its applications within the field of artificial peptide modifications, which continue to underpin an enormity of developing peptide–drug conjugates as therapeutic strategies. As such, considering the combination of hyper-acid-labile, in addition to the considered application of orthogonal allyl-based deprotection-based methods,33,34 hydrogen-labile protecting groups could provide access to site-specific diamino functionalised peptides. Therefore, further development of peptide-based flow hydrogenation represents a valuable addition to the peptide chemist's toolbox.Experimental
General considerations
Protected amino acids, PS-2ClTrt-Cl (loading 0.9 mmol g−1) and (2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylammonium hexafluorophosphate) (HCTU) were purchased from Auspep and used as received to synthesise the linear peptide sequences as previously published. All other reagents and reactants were purchased from standard suppliers and utilised without purification unless otherwise stated. PS-carbodiimide was purchased from biotage (loading 1.27 mmol g−1). All solvents were purchased from ChemSupply and distilled under reduced pressure, with the exception of HPLC grade solvents being used as received. HPLC solvents were degassed before use. Deuterated solvents were purchased from Cambridge Isotope Laboratories and stored in a desiccator.All flow hydrogenation methods were carried out on a ThalesNano H-Cube Pro™ continuous flow reactor. The choice of catalyst (ThalesNano CatCarts®), and reaction parameters including temperature (°C) H2 pressure (bar), flow rate (mL min−1), H2 production (%), and number of cycles are defined in each individual experiment.
All 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded at 25 °C on a Varian ‘Mercury’ 300 MHz spectrometer or a Bruker ‘Avance’ 400 MHz spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) and are measured in reference to the designated deuterated solvent. Coupling constants (J) are recorded in Hz and significant multiplicities described by singlet (s), doublet (d), doublet of doublets (dd), doublet of triplets (dt), triplet (t), quadruplet (q), broad (br s), multiplet (m). Spectra were assigned using appropriate gCOSY sequences.
Analytical RP-HPLC analysis was performed using an Agilent instrument comprising six modules; 1260 Bin Pump, 1260 HIP Degasser, 1260 ALS, 1260 TCC, 1260 DAD and 1260 FC-AS. Analytical RP-HPLC was performed using Phenomenex Onyx Monolithic reversed-phase C18 column (4.6 × 100 mm). A: 0.06% TFA in H2O and solvent B: 0.06% TFA in CH3CN:H2O (9:1), flow rate of 1.0 mL min−1, gradient 10–100 (%B), curve = 6, over 15.0 min, and detection at 214 and 254 nm. For peptide purification semi-preparative RP-HPLC was performed using a Waters 2525 binary gradient pump equipped with a water 2487 dual λ absorbance detector and a Chromolith®SemiPrep RP-18e 100–10 mm column. A flow rate of 10 mL min−1 was used with solvent A: 0.06% TFA in water and solvent B: 0.06% TFA in CH3CN:H2O (90:10). Gradient 10–75 (%B) over 15 min, curve = 6, with UV detection at 214 nm and 254 nm.
Mass spectrometry data was obtained at the Western Sydney University Mass Spectrometry Facility. Low-resolution mass spectrometry (LRMS) experiments were performed using a Waters ‘Xevo TQ Triple Quadrupole’ Mass Spectrometer via the ESI method. Spectra were recorded in positive ion mode from analyte solutions injected into a 0.1% formic acid in methanol solution flowing at 0.1 mL min−1. A capillary voltage of 3.0 kV, cone voltage of 30 V, desolvation temperature of 300 °C and desolvation flow rate of 500 L h−1. Spectra were recorded over 1 min with an m/z range of 100–2000. High resolution mass spectra (HRMS) were obtained on a Waters ‘Xevo Quadrupole-Time of Flight’ spectrometer via the ESI method and were within 5.00 ppm of the acceptable limit. The instrument was fitted with an ESI probe and mass-corrected sample spectra were recorded in positive mode, using leucine encephalin (200 pg mL−1 in 50% aqueous acetonitrile + 0.1% formic acid) as a lockmass reference.
General methods
:1 DMF:piperidine, 5.0 mL min−1) until the detector reached maximum absorption and returned to <0.1 AU. The resin was then washed with solution A (2 min, 5.0 mL min−1). The amino acid solution (Fmoc-AA-OH 4 eq., HCTU 4 eq., DIPEA 8 eq., DMF 1.0 mL) was injected into the sample loop of the reactor and flowed through the Omnifit™ column (2.0 mL min−1) until the UV detector reached maximum absorbance and subsequently returned to <0.1 AU. This was followed by Fmoc-deprotection with solution B until the detector reached maximum detection and returned to <0.1 AU. The resin was then washed with solution A (2 min, 5.0 mL min−1). The above steps of AA solution injection (step 1), Fmoc-deprotection (step 2) and washing (step 3) were repeated for all additional AA's in sequence. Once the final Fmoc-deprotection was achieved, the column was removed from the heating block and the resin was washed with DCM (2 × 5 mL), MeOH (2 × 5 mL), and Et2O (2 × 10 mL) and then dried in vacuo.
:DCM). Following the injection of coupling solution into the continuous DCM stream (0.5 mL min−1) the column eluent was monitored via UV and upon absorbance detection, the reaction stream was collected. Collection of the column eluent continued until UV absorbance of the solvent stream returned to baseline.
Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
The authors acknowledge the facilities and technical assistance provided by the WSU Biomedical Magnetic Resonance Facility (BMRF) and the WSU Mass Spectrometry Facility. Additionally, the authors thank WSU School of Science for facilities access and financial support. MM thanks WSU for an award of an MRes scholarship.Notes and references
- M. Bergmann and L. Zervas, Ber. Dtsch. Chem. Ges., 1932, 65, 1192–1201 CrossRef.
- A. Isidro-Llobet, M. Alvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS PubMed.
- A. J. Pallenberg, Tetrahedron Lett., 1992, 33, 7693–7696 CrossRef CAS.
- J. M. Schlatter, R. H. Mazur and O. Goodmonson, Tetrahedron Lett., 1977, 2851–2852, DOI:10.1016/s0040-4039(01)83091-4.
- O. Kanie, G. Grotenbreg and C.-H. Wong, Angew. Chem., Int. Ed., 2000, 39, 4545–4547 CrossRef CAS.
- H. Sajiki and K. Hirota, Tetrahedron, 1998, 54, 13981–13996 CrossRef CAS.
- H. Sajiki and K. Hirota, Chem. Pharm. Bull., 2003, 51, 320–324 CrossRef CAS PubMed.
- H. Sajiki, H. Kuno and K. Hirota, Tetrahedron Lett., 1998, 39, 7127–7130 CrossRef CAS.
- F.-X. Felpin and E. Fouquet, Chem. – Eur. J., 2010, 16, 12440–12445 CrossRef CAS PubMed.
- S. Calimsiz, A. I. Morales Ramos and M. A. Lipton, J. Org. Chem., 2006, 71, 6351–6356 CrossRef CAS PubMed.
- J. Deng, Y. Hamada and T. Shioiri, J. Am. Chem. Soc., 1995, 117, 7824–7825 CrossRef CAS.
- C. Bérubé, A. Borgia and N. Voyer, Org. Biomol. Chem., 2018, 16, 9117–9123 RSC.
- A. R. Horswill and C. P. Gordon, J. Med. Chem., 2020, 63, 2705–2730 CrossRef CAS PubMed.
- C. P. Gordon, Org. Biomol. Chem., 2020, 18, 379–390 RSC.
- G. Pallikonda and M. Chakravartya, J. Org. Chem., 2016, 81, 2135–2142 CrossRef CAS PubMed.
- K. F. McClure and M. Z. Axt, Bioorg. Med. Chem. Lett., 1998, 8, 143–146 CrossRef CAS.
- H. Hilpert, Tetrahedron, 2001, 57, 7675–7683 CrossRef CAS.
- M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316 CrossRef CAS PubMed.
- P. J. Cossar, L. Hizartzidis, M. I. Simone, A. McCluskey and C. P. Gordon, Org. Biomol. Chem., 2015, 13, 7119–7130 RSC.
- K. R. Knudsen, J. Holden, S. V. Ley and M. Ladlow, Adv. Synth. Catal., 2007, 349, 535–538 CrossRef CAS.
- K. R. Knudsen, M. Ladlow, Z. Bandpey and S. V. Ley, J. Flow Chem., 2014, 4, 18–21 CrossRef.
- I. R. Baxendale, S. V. Ley, C. D. Smith and G. K. Tranmer, Chem. Commun., 2006, 4835–4837, 10.1039/B612197G.
- K. C. H. Tijssen, B. J. A. van Weerdenburg, H. Zhang, J. W. G. Janssen, M. C. Feiters, P. J. M. van Bentum and A. P. M. Kentgens, Anal. Chem., 2019, 91, 12636–12643 CrossRef CAS PubMed.
- M. Soto, R. G. Soengas and H. Rodriguez-Solla, Adv. Synth. Catal., 2020, 362, 5422–5431 CrossRef CAS.
- S. A. Yakukhnov and V. P. Ananikov, Adv. Synth. Catal., 2019, 361, 4781–4789 CrossRef CAS.
- F. J. Urbano and J. M. Marinas, J. Mol. Catal. A: Chem., 2001, 173, 329–345 CrossRef CAS.
- W.-X. Zhang, C.-B. Wang and H.-L. Lien, Catal. Today, 1998, 40, 387–395 CrossRef CAS.
- L. Xu, S. Bhandari, J. Chen, J. Glasgow and M. Mavrikakis, Top. Catal., 2020, 63, 762–776 CrossRef CAS.
- C. Reichaedt, Solvents and Solvent Effects in Organic Chemistry, 3rd edn, 2003 Search PubMed.
- X. Ma, Y. Liu, X. Li, J. Xu, G. Gu and C. Xia, Appl. Catal., B, 2015, 165, 351–359 CrossRef CAS.
- R. A. Rajadhyaksha and S. L. Karwa, Chem. Eng. Sci., 1986, 41, 1765–1770 CrossRef CAS.
- R. L. Augustine and P. Techasauvapak, J. Mol. Catal., 1994, 87, 95–105 CrossRef CAS.
- A. M. Giltrap, L. J. Dowman, G. Nagalingam, J. L. Ochoa, R. G. Linington, W. J. Britton and R. J. Payne, Org. Lett., 2016, 18, 2788–2791 CrossRef CAS PubMed.
- N. Thieriet, J. Alsina, E. Giralt, F. Guibe and F. Albericio, Tetrahedron Lett., 1997, 38, 7275–7278 CrossRef CAS.
- L. K. Spare, M. Menti, D. G. Harman, J. R. Aldrich-Wright and C. P. Gordon, React. Chem. Eng., 2019, 4, 1309–1317 RSC.
- L. K. Spare, V. Laude, D. G. Harman, J. R. Aldrich-Wright and C. P. Gordon, React. Chem. Eng., 2018, 3, 875–882 RSC.
- C. Dankers, J. Tadros, D. G. Harman, J. R. Aldrich-Wright, T. V. Nguyen and C. P. Gordon, ACS Comb. Sci., 2020, 22, 255–267 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, HPLC chromatograms, HRMS spectra. See DOI: 10.1039/d1ob02179f |
| ‡ Present address; School of Chemistry & Molecular Bioscience, University of Wollongong, Wollongong, NSW, 2522, Australia. |
| This journal is © The Royal Society of Chemistry 2022 |