
Copper(I)-catalysed intramolecular hydroarylation-redox cross-dehydrogenative coupling of N-propargylanilines with phosphites†
Guangzhe
Li
 *a,
Guo
Yu
b,
Chengdong
Wang
c,
Taiki
Morita
b,
Xuhai
Zhang
a and
Hiroyuki
Nakamura
*b
*a,
Guo
Yu
b,
Chengdong
Wang
c,
Taiki
Morita
b,
Xuhai
Zhang
a and
Hiroyuki
Nakamura
*b
aState Key Laboratory of Fine Chemicals, Department of Pharmaceutical Sciences, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China. E-mail: liguangzhe@dlut.edu.cn
bLaboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, Yokohama 226-8503, Japan. E-mail: hiro@res.titech.ac.jp
cDepartment of Nuclear Medicine, First Affiliated Hospital of Dalian Medical University, Dalian 116024, China
First published on 23rd November 2021
Abstract
Intramolecular hydroarylation-redox cross-dehydrogenative coupling of N-propargylanilines with phosphite diesters proceeded in the presence of Cu(I)-catalysts (20 mol%) to selectively give 2-phosphono-1,2,3,4-tetrahydroquinolines in good yields with 100% atomic utilization. P–H and two C–H bonds are activated at once and these hydrogen atoms are trapped by a propargylic triple bond in the molecule.
α-Aminophosphonic acids (α-aminophosphonates) are structural isosteres of α-amino acids (esters) in which the planar carboxylic acid group has been replaced by a tetrahedral phosphonate group. Owing to the tetrahedral configuration of the phosphorus atom, the α-aminophosphonic acid functional group can mimic the transition state of the hydrolysis of peptides in a stable manner and inhibit the enzymes involved in peptide metabolism, which endows α-aminophosphonic acid (ester) with a wide range of important physiological activities,1 such as antibacterial,2 antioxidant,3 anti-tumour,4 and enzyme inhibitor5 activities (Fig. 1).
 | ||
| Fig. 1 Typical bioactive α-amino phosphonic acid derivatives. | ||
Because of these attractive biological properties in medicinal chemistry, numerous efficient synthesis protocols have been developed for the synthesis of α-aminophosphonates in the last decade.6
The biological activities and synthesis methods of cyclic α-aminophosphonates have also attracted extensive attention due to the characteristics of their restricted conformations.1b,7 However, despite the developments and application potential of the cyclic α-amino phosphonate possessing 1,2,3,4-tetrahydroquinoline (2-phosphonotetrahydroquinoline),8 which is the conformationally constrained analog of pipecolic acid, the convenient and effective synthesis of 2-phosphonotetrahydroquinoline has remained a challenge. To date, two types of protocols have been reported for the synthesis of 2-phosphonotetrahydroquinolines: (1) the direct addition of phosphorylating reagents to quinolines (Scheme 1-1)8a–d and (2) multi-step reactions in which quinolines react with the acid chlorides to generate N-acylquinolinium intermediates, followed by the addition of phosphorus nucleophiles and subsequent hydrogenation (Scheme 1-2).8e,f These protocols are non-selective and have limitations for multi-substituted tetrahydroquinoline synthesis.
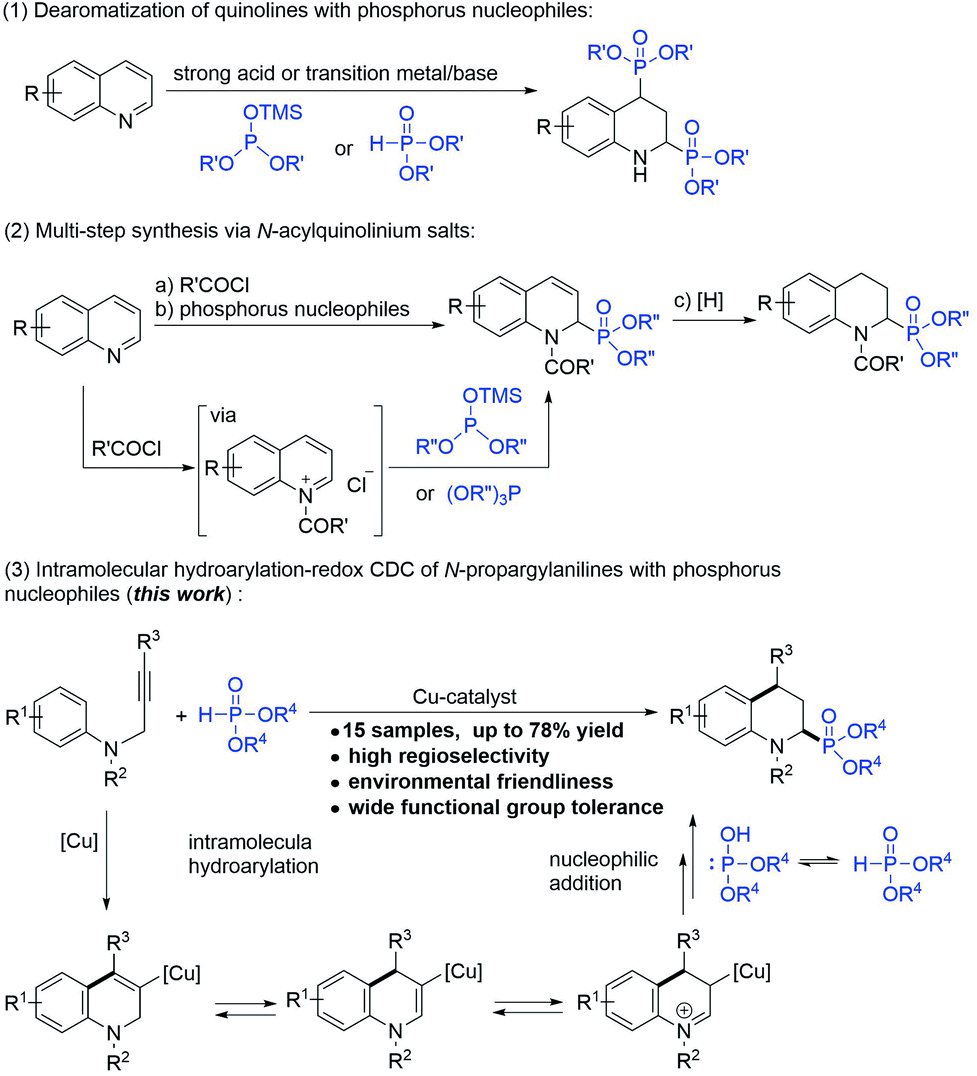 | ||
| Scheme 1 Synthetic strategies to 2-phosphono-1,2,3,4-tetrahydroquinolines. | ||
The intramolecular hydroarylation–redox cross-dehydrogenative-coupling (IH-Redox CDC) reaction of the N-propargyl aromatic amine with different carbon nucleophiles, such as indoles (sp2 carbon nucleophiles), nitromethane (sp3 carbon nucleophiles), and phenylacetylenes (sp carbon nucleophiles), was first reported by our group.9 Notably, the alkynyl group of the N-propargyl aromatic amine acted as an internal oxidant that scavenged hydrogen, eliminating the need for external oxidants other than metal catalysts (copper or zinc) and achieving 100% atomic utilization via a green process. Based on our previous observations, phosphorus nucleophiles (H-phosphonates), as alternatives to carbon nucleophiles, were subjected to the IH-Redox CDC reaction of N-propargyl aromatic amines. In this paper, as an alternative synthesis method to the methods derived from quinolines, we report the simple and site-selective synthesis of 2-phosphonotetrahydroquinolines, in which tetrahydroquinoline was constructed by a cyclization reaction and the phosphoric acid (ester) group was introduced to the C2-position at the same time (Scheme 1-3).
First, N-benzyl-N-(2-propargyl)-aniline (1a) and dimethyl phosphite (2) were chosen as template compounds to conduct the IH-Redox CDC reaction. The 2-phosphono tetrahydroquinoline product (3aa) was obtained with only a 20% yield (Table 1, entry 1) in the presence of ZnBr2 (20 mol%) at 120 °C for 12 h in dichloroethane (DCE). Meanwhile, 2,4-diphosphono tetrahydroquinoline (4aa) was also obtained with a 7% yield as a by-product.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Solvent | Yielda of 3aa (%) | Yielda of 4aa (%) | Recoverya of 1a (%) |
Reaction conditions: 1a (0.25 mmol, 1.0 equiv.), 2a (3.0 equiv.) and catalyst (20 mol%) were heated in DCE (0.25 ml) in a sealed vial tube at 120 °C for 12 h.a The yield was estimated by 1H nuclear magnetic resonance (NMR) with 1,1,2-trichloroethane as internal standard.b Isolated yield.c The ratio of M (major configuration) to m (minor configuration) for 4aa is 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.8e DCE: dichloroethane; DCM: dichloromethane; THF: Tetrahydrofuran; Tf: trifluoromethanesulfonyl; N.D.: not detected; Quan.: quantitative. :1.8e DCE: dichloroethane; DCM: dichloromethane; THF: Tetrahydrofuran; Tf: trifluoromethanesulfonyl; N.D.: not detected; Quan.: quantitative. |
|||||
| 1 | ZnBr2 | DCE | 20 | 7 | 71 |
| 2 | ZnI2 | DCE | 7 | 6 | 55 |
| 3 | ZnCl2 | DCE | 22 | 2 | 50 |
| 4 | Zn(OAc)2 | DCE | 5 | 2 | 71 |
| 5 | Cu(OAc)2 | DCE | 21 | 8 | Trace |
| 6 | CuBr2 | DCE | 50 | 10 | Trace |
| 7 | CuSO4 | DCE | 36 | 9 | N.D. |
| 8 | Cu(OTf)2 | DCE | 16 | 6 | N.D. |
| 9 | CuOTf | DCE | 31 | 5 | N.D. |
| 10 | CuBr | DCE | 51 | 16 | Trace |
| 11 | CuCl | DCE | 50 | 15 | Trace |
| 12 | CuI | DCE | 75 (70)b,c | 12 | Trace |
| 13 | CuI | MeOH | 46 | 14 | N.D. |
| 14 | CuI | DCM | 61 | 11 | N.D. |
| 15 | CuI | THF | 45 | 16 | N.D. |
| 16 | CuI | Acetone | 31 | 20 | N.D. |
| 17 | CuI | 1,4-Dioxane | 67 | 10 | N.D. |
| 18 | Pd(OAc)2 | DCE | 40 | 14 | Trace |
| 19 | PdCl2 | DCE | N.D. | N.D. | Quan. |
| 20 | AgNO3 | DCE | N.D. | N.D. | 43 |
| 21 | AgOAc | DCE | N.D. | N.D. | 60 |
| 22 | AgBF4 | DCE | N.D. | N.D. | 27 |
Other zinc metal catalysts that were sequentially investigated led to unsatisfactory results (Table 1, entries 2–4). It was found that the yield of CuI was greatly promoted from 22% to 75% (Table 1, entry 12) after several copper salts were examined. In addition, palladium (Table 1, entries 18 and 19) and silver (Table 1, entries 20–22) catalysts were also employed. However, the catalytic effects were not satisfactory.
Since CuI was found to give the best results in DCE, the effect of the solvent on the Redox CDC reaction was next investigated. However, other solvents, such as MeOH, dichloromethane (DCM), tetrahydrofuran (THF), acetone, and 1,4-dioxane, gave lower yields of 3aa than DCE (Table 1, entries 13–17). Meanwhile, the equivalent ratio of phosphite ester (2a) was also examined, and no further improvement was observed above three equivalents (Table S1, entries 1–5†).
In addition, the reaction yields were promoted when the reaction time was extended from 4 to 12 h (entry 12 in Table 1vs. entries 6 and 7 in Table S1†). Prolonging the reaction time continuously to 24 h reduced the reaction yield, and increasing the reaction time further to 36 h resulted in increasing generation of 4aa (Table S1, entries 8 and 9†). Furthermore, other influential conditions, including the applicable temperature of the reaction, were also explored. The yield of 3aa was greatly increased from 44% to 75% as the temperature was raised from 80 to 120 °C (entry 12 in Table 1vs. entries 11 and 12 in Table S1†). However, when the reaction was carried out at 140 °C (Table S1, entry 10†), neither the products (3aa and 4aa) nor the starting material 1a were detected.
Finally, the concentration of the starting material (1a, Table S1, entries 13–16†) was investigated, and it was found that a 1 M concentration of 1a was optimal. As a result, the target product 3aa was obtained optimally with a 75% yield (nuclear magnetic resonance (NMR) yield, isolated yield 70% in Table 1, entry 12) in the presence of CuI (20 mol%) at 120 °C for 12 h in DCE.
With the optimised reaction conditions of the IH-Redox CDC reaction, the reactions of various types of N-(2-propargyl)aniline with phosphite ester were investigated. The results are summarized in Scheme 2. The N-(2-propargyl)-aniline (R2 = Bn, R3 = H) bearing different substituents at R1, such as a hydrogen atom (1a) or a methoxy group (1b), chloro group (1c), methyl group (1d), and fluoro group (1e), underwent the IH-Redox CDC reaction with dimethyl phosphite (2a) smoothly to provide the corresponding N-substituted-1,2,3,4-tetrahydroquinolines with good yields (Scheme 2, 3aa–3ea). N-(2-Propynyl)anilines (R1 = H, R3 = H) with various functional groups at R2, such as methyl propionate (1f), phenyl (1g), allyl (1h), and methyl (1i) groups, were also compatible with the reaction to provide the corresponding 2-substituted N-benzyl-1,2,3,4-tetrahydroquinolines (Scheme 2, 3fa–3ia) with moderate yields.
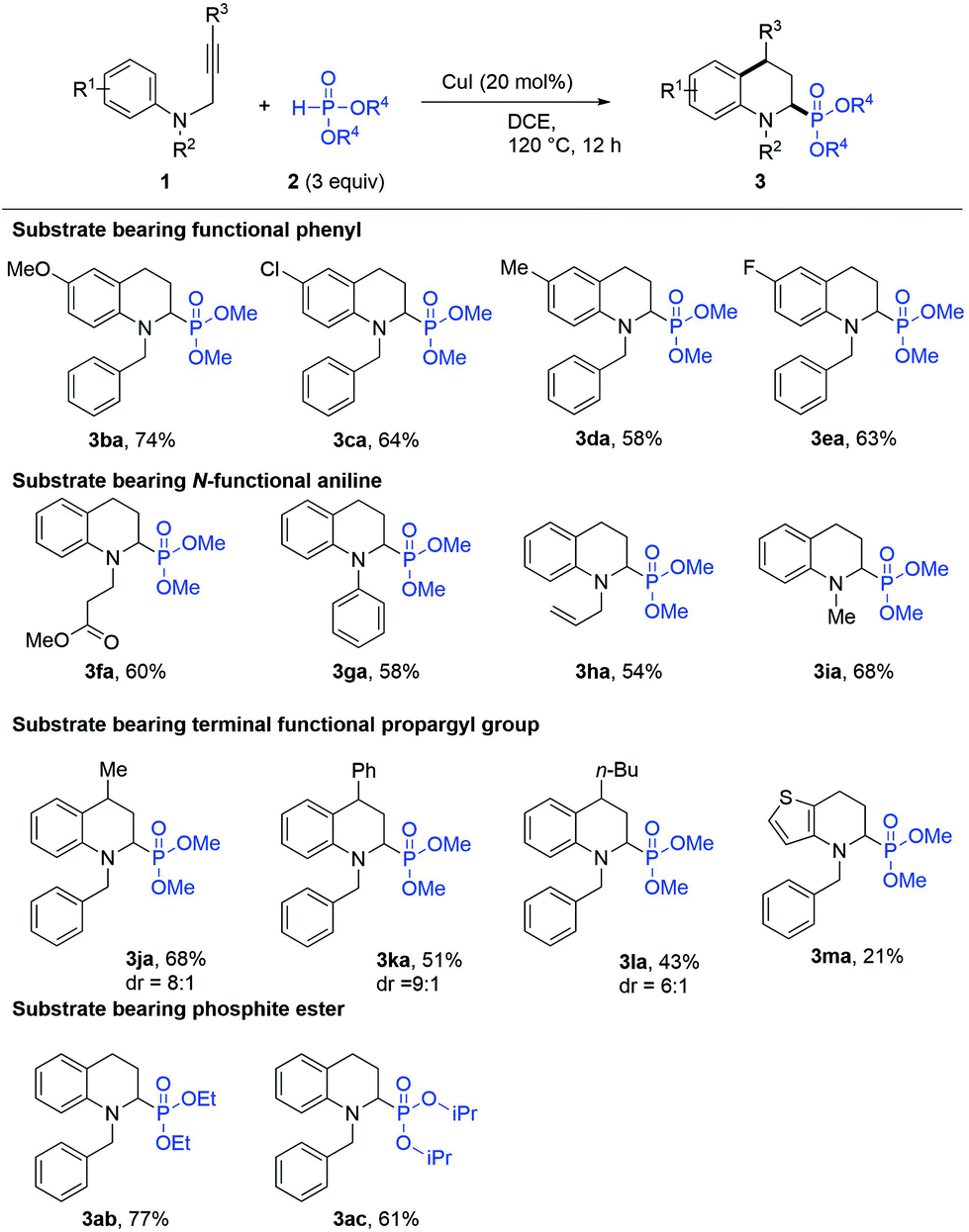 | ||
| Scheme 2 Exploration of substrate scope of 2-phosphono tetrahydroquinolines. Reaction conditions: 1 (0.25 mmol, 1.0 equiv.), 2 (3.0 equiv.), and CuI (20 mol%) were heated in DCE (0.25 mL) in a sealed vial tube at 120 °C for 12 h. | ||
In addition, the IH-Redox CDC reaction of N-propargylanilines bearing substituents at R3, such as methyl (1j), phenyl (1k), and n-butyl (1l) groups, with dimethyl phosphite (2a) were examined to obtain products with moderate to good yields and higher trans-diastereoselectivity8d (Scheme 2, 3ja–3la). Moreover, the phosphite esters with ethyl (2b) and i-propane (2c) groups at the R4 position underwent the IH-Redox CDC reaction with N-(2-propargyl)-aniline (1a) to obtain the 2-phosphate-substituted tetrahydroquinolines 3ab and 3ac with 77% and 61% yields, respectively. It was inferred that the large steric hindrance of the substituents at the R4 position eventually inhibited the reactivity. Finally, the aromatic compound N-(2-propargyl)thiophen-3-amine (1m) undergoing the IH-Redox CDC reaction provided dimethyl (4-benzyl-4,5,6,7-tetrahydrothieno[3,2-b]pyridin-5-yl)phosphonate (3ma) with a 21% yield, suggesting that the strong electron donating heterocyclic structure adversely affected the reaction progress.
Thus, various tetrahydroquinoline derivatives with phosphorus substituents at the C2 position were obtained in high yields via the established IH-Redox CDC coupled reaction of N-(2-propargyl)aniline with phosphite esters.
Meanwhile, the demand for the tetrahydroquinolines with phosphonic groups at the C2 and C4 positions can also be met. Thus, the formation mechanism of 2,4-diphosphono tetrahydroquinoline (4aa) was explored. We assumed that there were two possible processes for generating 4aa: (1) dimethyl phosphite (2a) cross-coupled with compound 1a at the terminal position of propargyl group to yield compound 5,10 and then compound 5 underwent IH-Redox CDC reaction with dimethyl phosphite to afford product 4aa; (2) compound 1a underwent the IH-Redox CDC reaction with dimethyl phosphite to yield mono-phosphono tetrahydroquinoline (3aa), and then 3aa was cross-coupled with 2a to afford 4aa as the target product (Scheme 3, eqn (1)).
 | ||
| Scheme 3 Control experiments for the study of the reaction mechanism. | ||
To confirm our hypothesis, we conducted two kinds of reactions (Scheme 3, eqn (2) and (3)). Compound 6 was subjected to the IH-Redox CDC reaction with 2a to verify this possibility through path 1. Since the desired product, 7 or 7′, was not detected (Scheme 3, eqn (2)), the generation of 5 would not be involved in the reaction pathway.
Furthermore, to confirm the possibility of the reaction via path 2, 3aa was used as the starting material to react with dimethyl phosphite (2a) under the established conditions of the IH-Redox CDC reaction (Scheme 3, eqn (3)), and 2,4-diphosphono tetrahydroquinoline (4aa) was afforded with a 33% yield. However, when 0.25 equivalents of TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) was added to the reaction as a radical scavenger, 4aa was not generated. Based on these observations, it is speculated that the 2,4-diphosphono product(4aa) was formed via the radical reaction of the mono-phosphono product (3aa) with dimethyl phosphite (2a).
In conclusion, we have developed the IH-Redox CDC reaction of N-propargylanilines with phosphites to prepare 2-phosphate-substituted tetrahydroquinoline derivatives with 100% atomic utilization. CuI was found to be essential for an effective IH-Redox CDC reaction with a wide range of substrates. Due to the conformational similarity between the phosphate groups and the carboxyl groups, the development of biologically active 2-substituted tetrahydroquinoline alkaloids is of great significance, and our present results provide a convenient and efficient protocol for the synthesis of 2-phosphate (ester group)-substituted tetrahydroquinolines as an alternative to the methods derived from quinolines.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22178049) and the Fundamental Research Funds for the Central Universities (DUT20YG114, DUT18LK11).Notes and references
- (a) A. Maestro, E. M. D. Marigorta, F. Palacios and J. Vicario, Asian J. Org. Chem., 2020, 9, 538 CrossRef CAS; (b) A. Mucha, P. Kafarski and L. Berlicki, J. Med. Chem., 2011, 54(17), 5955 CrossRef CAS; (c) L. Berlicki and P. Kafarski, Curr. Org. Chem., 2005, 9, 1829 CrossRef CAS; (d) P. Kafarski and B. Lejczak, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 301 CrossRef CAS.
- (a) M. R. Sivala, S. R. Devineni, M. Golla, V. Medarametla, G. K. Pothuru and N. R. Chamarthi, J. Chem. Sci., 2016, 128, 1303 CrossRef CAS; (b) S. A. Dake, D. S. Raut, K. R. Kharat, R. S. Mhaske, S. U. Deshmukh and R. P. Pawar, Bioorg. Med. Chem. Lett., 2011, 21, 2527 CrossRef CAS PubMed; (c) F. R. Atherton, C. H. Hassall and R. W. Lambert, J. Med. Chem., 1986, 29(1), 29 CrossRef CAS.
- (a) R. Damiche and S. Chafaa, J. Mol. Struct., 2017, 1130, 1009 CrossRef CAS; (b) K. U. M. Rao, S. Swapna, D. M. Manidhar, K. M. K. Reddy and C. S. Reddy, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190(2), 232 CrossRef CAS.
- (a) R.-Z. Huang, C.-Y. Wang, J.-F. Li, G.-Y. Yao, Y.-M. Pang, M.-Y. Ye, H.-S. Wang and Y. Zhang, RSC Adv., 2016, 6, 62890 RSC; (b) Q. Wang, L. Yang, H. Ding, X. Chen, H. Wang and X. Tang, Bioorg. Chem., 2016, 69, 132 CrossRef CAS; (c) G.-Y. Yao, M.-Y. Ye, R.-Z. Huang, Y.-J. Li, Y.-J. Pan, Q. Xu, Z.-X. Liao and H.-S. Wang, Bioorg. Med. Chem. Lett., 2014, 24, 501 CrossRef CAS PubMed; (d) A. K. Bhattacharya, D. S. Raunt, K. C. Rana, I. K. Polanki, M. S. Khan and S. Tram, Eur. J. Med. Chem., 2013, 66, 146 CrossRef CAS PubMed; (e) S. A. Dake, D. S. Raut, K. R. Kharat, R. S. Mhaske, S. U. Deshmukh and R. P. Pawar, Bioorg. Med. Chem. Lett., 2011, 21, 252 CrossRef PubMed.
- (a) Y. Feng, J. Park, S. Li, R. Boutin, P. Viereck, M. A. Schilling, A. M. Berghuis and Y. S. Tsantrizos, J. Med. Chem., 2019, 62, 9691 CrossRef CAS; (b) M. Sienczyk, L. Winiarski, P. Kasperkiewicz, M. Psurski, J. Wietrzyk and J. Oleksyszyn, Bioorg. Med. Chem. Lett., 2011, 21, 1310 CrossRef CAS PubMed.
- A. Amira, Z. Aouf, H. K'Tir, Y. Chemam, R. Ghodbane, R. Zerrouki and N. Aouf, ChemistrySelect, 2021, 6, 6137–6149 CrossRef CAS.
- K. Moonen, I. Laureyn and C. V. Stevens, Chem. Rev., 2004, 104, 6177 CrossRef CAS PubMed.
- (a) Q. Q. Zhang, D.-D. Wei, X. L. Cui, D. Zhang, H. Wang and Y. J. Wu, Tetrahedron, 2015, 71, 6087 CrossRef CAS; (b) Y. X. Gao, H. G. Deng, S. S. Zhang, W. H. Xue, Y. L. Wu, H. W. Qiao, P. X. Xu and Y. F. Zhao, J. Org. Chem., 2015, 80, 1192 CrossRef CAS PubMed; (c) A. D. Blieck, S. Catak, W. Debrouwer, J. Drabowicz, K. Hemelsoet, T. Verstraelen, M. Waroquier, V. V. Speybroeck and C. V. Stevens, Eur. J. Org. Chem., 2013, 1058 CrossRef; (d) A. D. Blieck, K. G. R. Masschelein, F. Dhaene, E. R. Sokolowska, B. Marciniak, J. Drabowiczc and C. V. Stevens, Chem. Commun., 2010, 46, 258 RSC; (e) H. N. Sun, G. Chen, Y. L. Zhu, B. Liu, W. Zhou and Z. Q. Shao, Chem. – Eur. J., 2017, 23, 5983 CrossRef; (f) M. Ordóñez, A. Arizpe, F. J. Sayago, A. I. Jiménez and C. Cativiela, Molecules, 2016, 21, 1140 CrossRef.
- (a) G. Li, C. Wang, Y. Li, K. Shao, G. Yu, S. Wang, X. Guo, W. Zhao and H. Nakamura, Chem. Commun., 2020, 56, 7333 RSC; (b) G. Li and H. Nakamura, Angew. Chem., Int. Ed., 2016, 55, 6758 CrossRef CAS PubMed.
- (a) X. Liu, Z. S. Wang, T. Y. Zhai, C. Luo, Y. P. Zhang, Y. B. Chen, C. Deng, R. S. Liu and L. W. Ye, Angew. Chem., Int. Ed., 2020, 59, 17984 CrossRef CAS PubMed; (b) Q. Yuan, W. D. Rao, S. Y. Wang and S. J. Ji, J. Org. Chem., 2020, 85, 1279 CrossRef CAS; (c) P. Sun, J. J. Yang, Z. Song, Y. Cai, Y. J. Liu, C. Chen, X. Chen and J. Peng, Synthesis, 2020, 52, 75 CrossRef CAS; (d) R. M. Gorman, T. E. Hurst, W. F. Petersen and R. J. K. Taylor, Tetrahedron, 2019, 75, 130711 CrossRef; (e) K. Sun, X. L. Chen, S. J. Li, D. H. Wei, X. C. Liu, Y. L. Zhang, Y. Liu, L. L. Fan, L. B. Qu, B. Yu, K. Li, Y. Q. Sun and F. Y. Zhao, J. Org. Chem., 2018, 83, 14419 CrossRef CAS PubMed; (f) R. Li, X. Chen, X. R. Song, H. X. Ding, P. Y. Wang, Q. Xiao and Y. M. Liang, Adv. Synth. Catal., 2017, 359, 3962 CrossRef CAS; (g) M. J. Xiong, X. Xie and Y. H. Liu, Org. Lett., 2017, 19, 3398 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob02091a |
| This journal is © The Royal Society of Chemistry 2022 |