
Construction of sulfur-containing compounds with anti-cancer stem cell activity using thioacrolein derived from garlic based on nature-inspired scaffolds†
Taichi
Yoneda
a,
Naoto
Kojima
a,
Takahiro
Matsumoto
 a,
Daisuke
Imahori
a,
Tomoe
Ohta
b,
Tatsusada
Yoshida
b,
Tetsushi
Watanabe
a,
Hisashi
Matsuda
a and
Seikou
Nakamura
*a
a,
Daisuke
Imahori
a,
Tomoe
Ohta
b,
Tatsusada
Yoshida
b,
Tetsushi
Watanabe
a,
Hisashi
Matsuda
a and
Seikou
Nakamura
*a
aKyoto Pharmaceutical University, 1 Misasagi-Shichono-cho, Yamashina-ku, Kyoto 607-8412, Japan. E-mail: naka@mb.kyoto-phu.ac.jp
bFaculty of Pharmaceutical Sciences, Nagasaki International University, 2825-7 Huis Ten Bosch-Cho, Sasebo, Nagasaki 859-3298, Japan
First published on 2nd December 2021
Abstract
Sulfur-containing compounds, such as cyclic compounds with a vinyl sulfane structure, exhibit a wide range of biological activities including anticancer activity. Therefore, the development of efficient strategies to synthesize such compounds is a remarkable achievement. We have developed a unique approach for the rapid and modular preparation of nature-inspired cyclic and acyclic sulfur-containing compounds using thioacrolein, a naturally occurring chemically unstable intermediate. We constructed thiopyranone derivatives through the regioselective sequential double Diels–Alder reaction of thioacrolein produced by allicin, a major component in garlic, and two molecules of silyl enol ether as the diene partner. The cytotoxicity toward cancer stem cells of the thiopyranones was equal to or higher than that of (Z)-ajoene (positive control) derived from garlic, and the thiopyranones had higher chemical stability than (Z)-ajoene.
Introduction
Ajoene (1) is one of the major sulfur-containing compounds in garlic (Allium sativum) (Scheme 1).1,2 It has a vinyl sulfane structure and is a mixture of geometric Z and E isomers (1a and 1b). That 1 has attracted attention for its pharmacological activity underscores the importance of the vinyl sulfane structure in the development of new therapeutic drugs. It has been reported that (Z)-ajoene (1a) exhibits an inhibitory effect on cancer stem cells (CSCs).3 This is a very important finding because of the paucity of anti-CSC substances. However, 1 has low chemical stability and is therefore not used in clinical practice. The percent residue was 19.9% when 1a was heated in ethyl acetate at 60 °C for 3 days.4 Therefore, it is necessary to develop sulfur-containing compounds that have improved chemical stability and are able to maintain their pharmacological activity.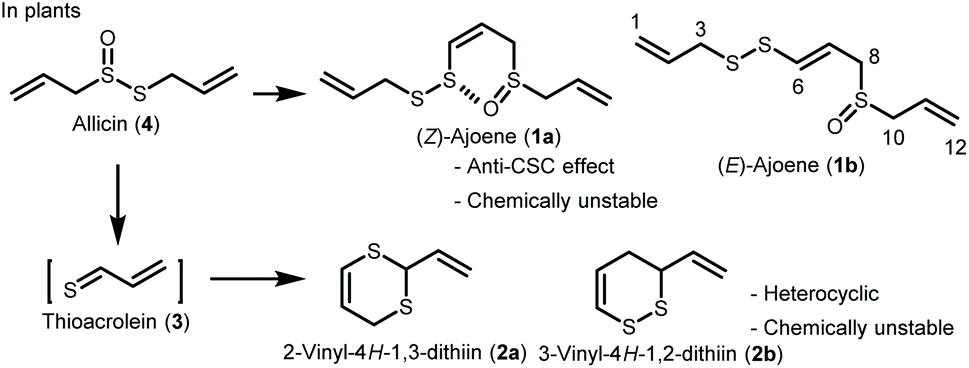 | ||
| Scheme 1 Structures of ajoene (1) and vinyldithiin (2). | ||
It has been reported that (Z)-ajoene (1a) has more potent biological activities than (E)-ajoene (1b), including anticancer,5–7 neuroprotective,8 antibacterial,9 antifungal,10 and anti-inflammatory activities.11 We hypothesize that the Z-form (1a) would bind more readily to the target molecule because its molecular conformation is fixed by the S–O interaction between sulfur and oxygen atoms in the (Z)-ajoene (1a) molecule.
In this study, we focused on 2-vinyl-4H-1,3-dithiin (2a) and 3-vinyl-4H-1,2-dithiin (2b),1,12 which are the major cyclic sulfur-containing components in garlic. Compounds 2a and 2b are produced by the Diels–Alder reaction of thioacrolein (3), which is generated by the decomposition of allicin (4) obtained by an enzymatic reaction from cysteine sulfoxide, an amino acid component in garlic. Compounds 2a and 2b exhibit diverse biological activities, but, like 1, are chemically unstable and therefore not used in clinical practice. Here, we assumed that thioacrolein (3), the biosynthetic precursor of 2a and 2b, could be used to construct new cyclic sulfur-containing compound 5 with anticancer activity, instead of 2 (Scheme 2). In the present study, we developed a facile regioselective synthesis of thiopyranone derivative 5 using thioacrolein (3) obtained from allicin (4), evaluated the inhibitory effects of 5 on CSCs, and compared the biological activity and chemical stability of 5 with those of (Z)-ajoene (1a).
 | ||
| Scheme 2 Outline of thiopyranone synthesis. | ||
Results and discussion
Synthesis of thiopyranone derivatives
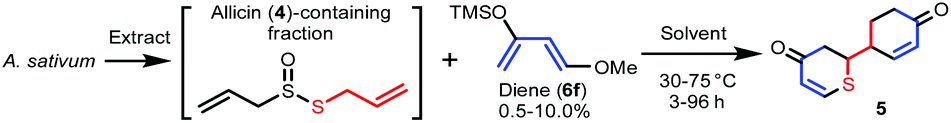
A biogenetically inspired synthetic process to furnish a thiopyranone derivative is outlined in Scheme 2. Using thioacrolein (3) as the key building block, we drew up a plan to synthesize the objective thiopyranones through the generation of four new covalent bonds and a new bicyclic ring system using a two-fold Diels–Alder reaction. To initiate our study, we tried to synthesize and isolate 3, but were unable to do so. To the best of our knowledge, isolation of 3 has not been reported in the literature, the reason being that 3 is highly reactive and therefore somewhat prone to Diels–Alder dimerization. As a second approach to 5, we investigated the use of 3 generated by the decomposition of a fraction containing allicin (4), which was obtained from garlic. First, we examined the extraction conditions for 4. Water, MeOH, acetone, toluene, ethyl acetate, and diethyl ether were considered as the extraction solvent (Fig. S1 and S2†). Polar solvents, such as water, MeOH, and acetone, extracted not only 4 but various garlic components as well. Therefore, the allicin (4)-containing fractions obtained using these solvents were considered inappropriate for use in the synthesis of 3. On the other hand, 4 was selectively extracted by non-polar solvents, such as toluene, ethyl acetate, and diethyl ethers, and therefore these solvents were considered excellent extraction solvents to yield the allicin (4)-containing fraction. We decided to use diethyl ether, which has excellent 4 extraction efficiency, as the extraction solvent. From thus-obtained allicin (4), thioacrolein (3) was prepared in situ and simultaneously subjected to the sequential double Diels–Alder reaction with two molecules of silyl enol ether as the diene partner.When silyl enol ethers 6a–e [(((2Z,4E)-hexa-2,4-dien-3-yl)oxy)trimethylsilane, (E)-trimethyl((5-methylhexa-1,3-dien-2-yl)oxy)silane, (E)-trimethyl(octa-1,3-dien-2-yloxy)silane, (E)-trimethyl((4-phenylbuta-1,3-dien-2-yl)oxy)silane, and (E)-4-(3-((trimethylsilyl)oxy)buta-1,3-dien-1-yl)phenol] were used as the diene, the reaction did not proceed at all (Fig. S3†). Therefore, a more active diene, (E)-((4-methoxybuta-1,3-dien-2-yl)oxy)trimethylsilane (Danishefsky's diene, 6f)13 was used, and desired compound 5 was obtained (Scheme 3). On the other hand, when (E)-3-((tert-butyldimethylsilyl)oxy)-N,N-dimethylbuta-1,3-dien-1-amine (Rawal's diene, 6g)14 was used, 5 was not obtained. Instead, a variety of sulfur-containing compounds were obtained, including different types of acyclic sulfur-containing compounds.
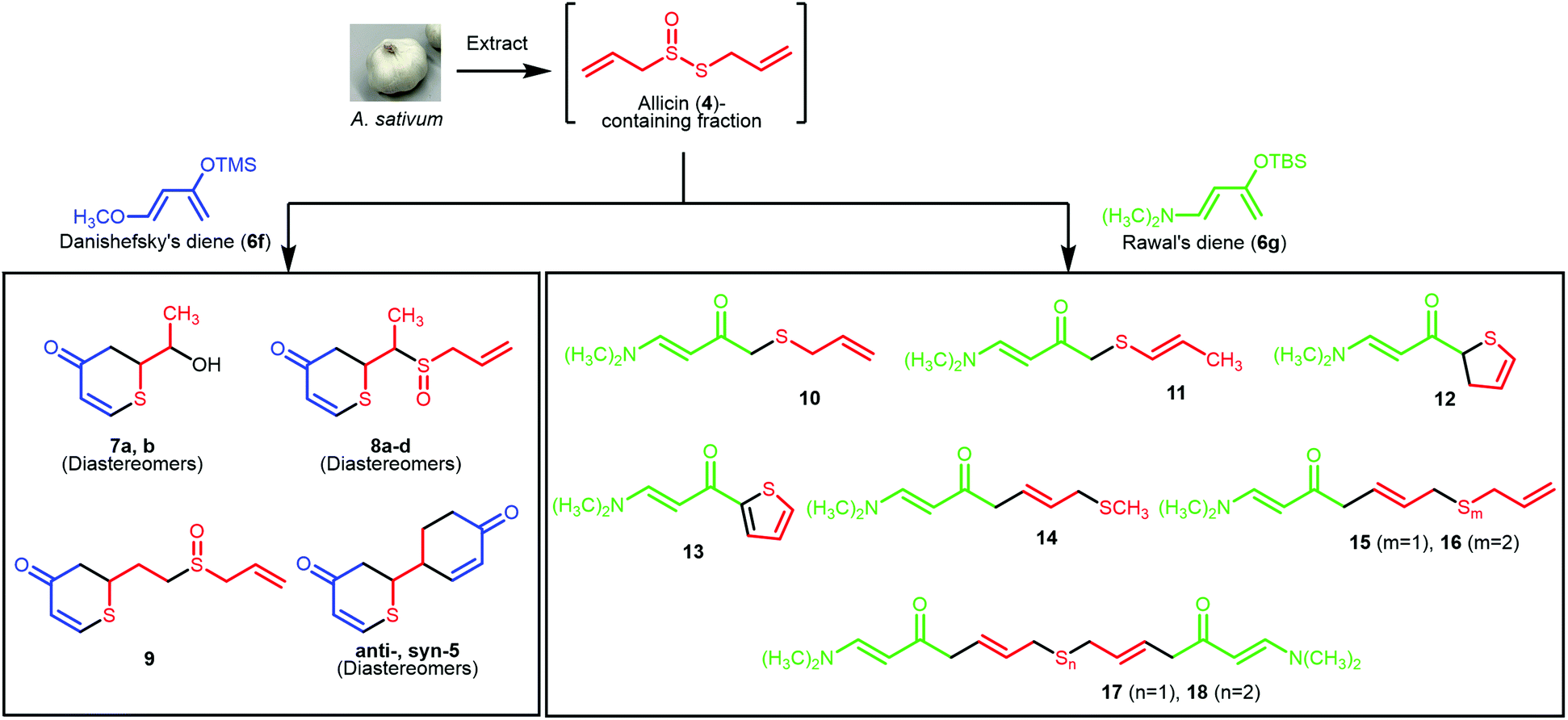 | ||
| Scheme 3 Synthesized compounds. | ||
2-(4-Oxocyclohex-2-en-1-yl)-2,3-dihydro-4H-thiopyran-4-ones (5) were synthesized as a yellow oil. The molecular formula (C11H12O2S) was determined using HRMS and NMR spectroscopy. Quasimolecular ion peaks were observed using ESIMS (m/z 231 [M + Na]+). The 1H and 13C NMR (CDCl3) spectra recorded for anti-5 and syn-5 show signals corresponding to three methylenic hydrogens, two methinic hydrogens, four olefinic hydrogens, and two carbonyl groups. These spectroscopic data suggest that the planar structures of anti-5 and syn-5 are the same. The positions of the functional groups described above were determined on the basis of the double quantum filter correlation spectroscopy (DQF COSY) experiment on 1 indicated the presence of partial structures written in bold lines (Fig. 1), and in the heteronuclear multiple bond connectivity spectroscopy (HMBC) experiment, long-range correlations were observed between the following protons and carbon pairs: anti-5: H-2 and C-4, 6 and 2′; H-3 and C-4; H-5 and C-3; H-6 and C-2 and 4; H-2′ and C-2 and 4′; H-5′ and C-1′ and 4′; syn-5: H-2 and C-4 and 2′; H-3 and C-4 and 1′; H-5 and C-3; H-6 and C-2 and 4; H-2′ and C-4′ and 6′; H-5′ and C-1′ and 4′; H-6′ and C-2′ and 4′ (Fig. 1). Furthermore, stereoisomers anti-5a, anti-5b, syn-5a, and syn-5b with optical rotations (anti-5a: [α]21D −132.9, anti-5b: [α]21D +118.6, syn-5a: [α]21D −136.0, syn-5b: [α]21D +153.3 in MeCN) could be obtained by separating compounds anti-5 and syn-5 on a chiral column, respectively (Fig. S5 and S6†). The absolute configuration was elucidated from the calculated ECD spectrum (Fig. 1, S10 and S11†). The calculated ECD spectra of anti-5a (2R,1′S), anti-5b (2S,1′R), syn-5a (2R,1′R), and syn-5b (2S,1′S) were identical to those observed experimentally, respectively. On the basis of all these lines of evidence, the chemical structures of anti-5a, anti-5b, syn-5a, and syn-5b were determined and are shown in Fig. 1.
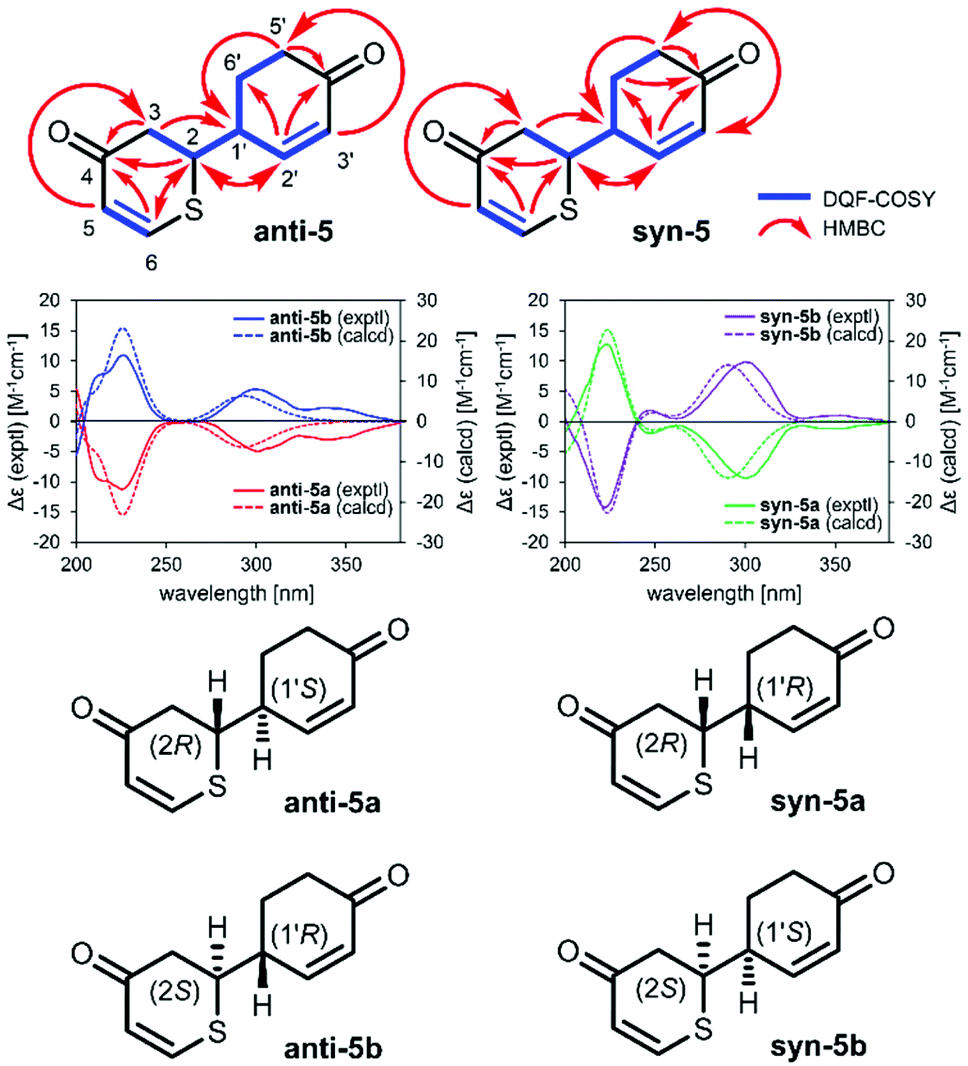 | ||
| Fig. 1 Experimental and calculated ECD spectra of 5. | ||
The chemical stability of the obtained compounds was also investigated (Table 1). The compounds were dissolved in DMSO-d6 or CD3OD and stored at 60 °C for 1 week. By quantifying the concentrations of the compounds in the solution before and after storage using the internal standard method in NMR, the amount of residue was determined. The results showed that both anti-5 and syn-5 had more residue than (Z)-ajoene (1a), meaning that obtained cyclic compounds anti-5 and syn-5 were more chemically stable than 1a.
| Compound | Solvent | |
|---|---|---|
| DMSO-d6 | CD3OD | |
| Each yield was quantified by NMR. | ||
| syn-5 | 99% | 96% |
| anti-5 | 96% | 83% |
| 1a | 77% | 81% |
A proposed synthetic pathway for the obtained compounds is shown in Scheme 4. In step 1, the enzymatic reaction proceeds by pre-extraction processing, and active sulfur components such as allicin (4) are produced in garlic. After extraction with ether, the active sulfur components are arbitrarily degraded into the important intermediate thioacrolein (3) and intermediates i–iv.15–17 When the Diels–Alder reaction between thioacrolein (3) and two molecules of Danishefsky's diene (6f) proceeds, target thiopyranone 5 is synthesized. Compounds 7–9 are synthesized by the oxidation of, or the addition of intermediate i![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 to, the product of the Diels–Alder reaction of 3 and a single molecule of 6f. On the other hand, Rawal's diene (6g) does not undergo the Diels–Alder reaction with 3. It is proposed that compounds 10 and 11 are synthesized by the radical addition reaction of intermediates ii and iii. This is supported by the isolation of compounds 12 and 13 and the fact that sulfur is prone to radical generation and stabilizes radicals.19,20 Compounds 14–18, which are compounds with an extended carbon chain, are thought to have been synthesized using compound 19, which is isolated at the same time, as an intermediate. Compound 19 is synthesized by the aldol condensation of 6g and acrolein (iv). The iv-based carbon chain extension reaction is sometimes reported as a biosynthetic process for compounds derived from Allium plants,21,22 and the aldol reaction of 6g is also known.23 Compounds 14–16 are synthesized by subjecting 19 to a thio-Michael addition reaction with a thiol, such as methyl mercaptan or intermediate ii.24 Finally, it is estimated that compounds 17 and 18 are synthesized by the dimerization of compounds 14–16.
18 to, the product of the Diels–Alder reaction of 3 and a single molecule of 6f. On the other hand, Rawal's diene (6g) does not undergo the Diels–Alder reaction with 3. It is proposed that compounds 10 and 11 are synthesized by the radical addition reaction of intermediates ii and iii. This is supported by the isolation of compounds 12 and 13 and the fact that sulfur is prone to radical generation and stabilizes radicals.19,20 Compounds 14–18, which are compounds with an extended carbon chain, are thought to have been synthesized using compound 19, which is isolated at the same time, as an intermediate. Compound 19 is synthesized by the aldol condensation of 6g and acrolein (iv). The iv-based carbon chain extension reaction is sometimes reported as a biosynthetic process for compounds derived from Allium plants,21,22 and the aldol reaction of 6g is also known.23 Compounds 14–16 are synthesized by subjecting 19 to a thio-Michael addition reaction with a thiol, such as methyl mercaptan or intermediate ii.24 Finally, it is estimated that compounds 17 and 18 are synthesized by the dimerization of compounds 14–16.
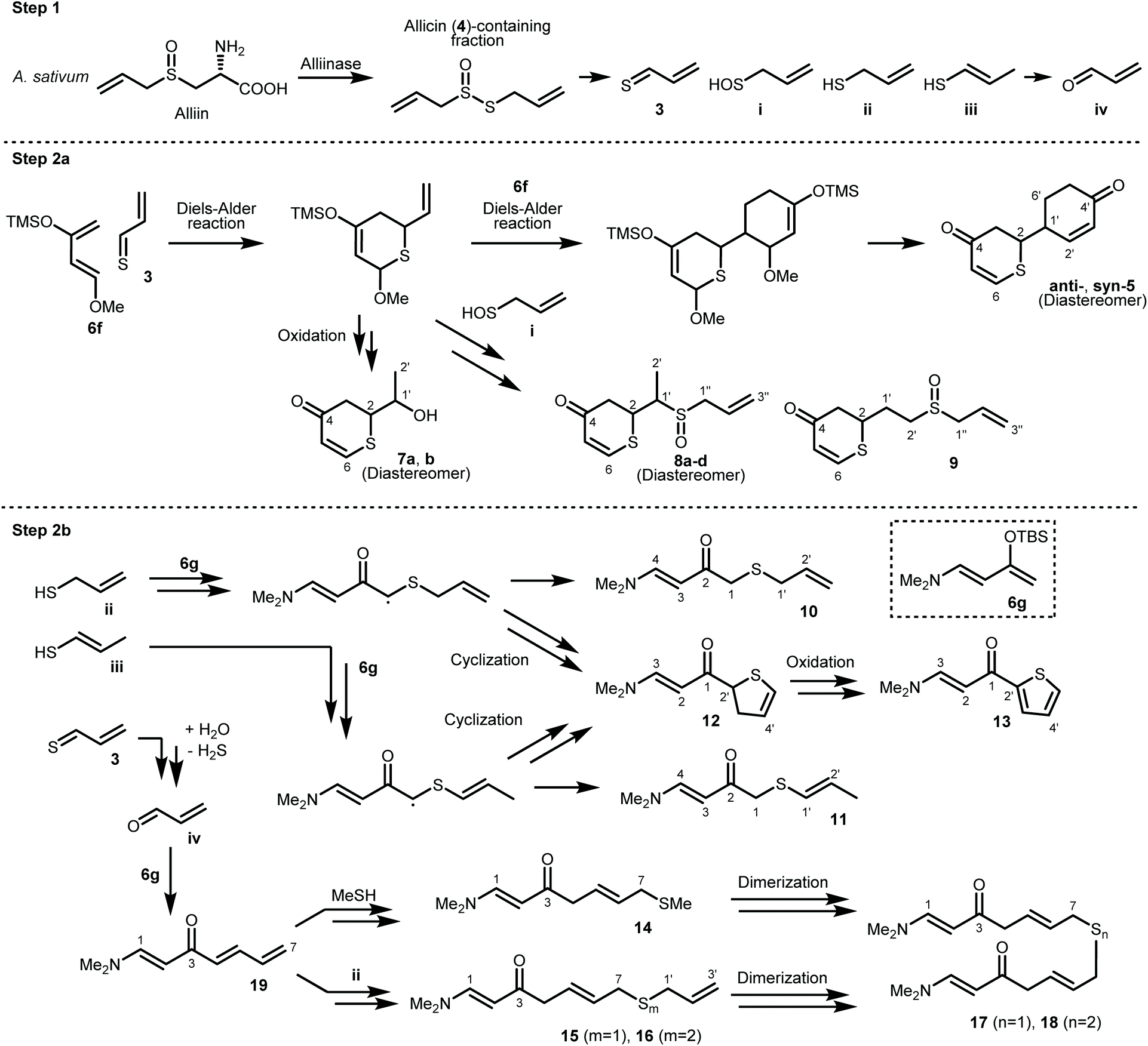 | ||
| Scheme 4 Proposed synthetic pathway for compounds 7–19. | ||
Examination of regioselectivity of Diels–Alder reaction
In principle, the cycloaddition reaction of 3 and 2 mol of 6f could lead to four regioisomers 5, 20, 21, and 22 (Scheme 5 and Fig. S7†). However, 20, 21, and 22 were not obtained this time, and only regioisomer 5 was selectively synthesized. In order to gain further insight into this reaction, we performed DFT calculations using the Gaussian 09_E25 and Reaction Plus Pro programs26 (B3LYP/6-31G(d,p),27 298 K, 1 atm).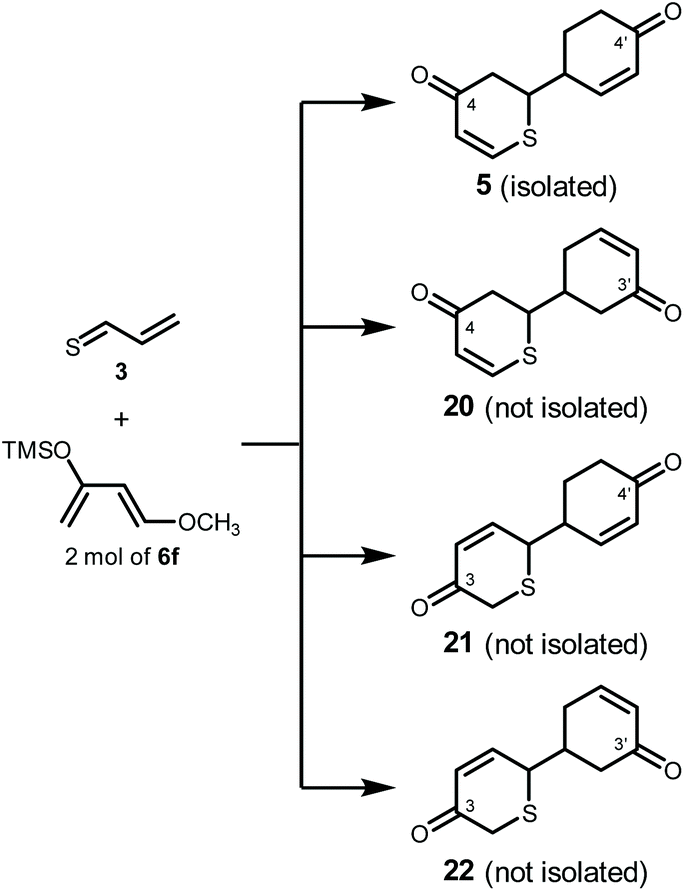 | ||
| Scheme 5 Four regioisomers theoretically obtained by the Diels–Alder reaction of 3 and 2 mol of 6f. | ||
Thioacrolein (3) as a dienophile had two reaction sites, a thiocarbonyl group and a vinyl group. However, it is considered that the reaction of the thiocarbonyl group is prioritized from the calculation in previous studies.28,29 Furthermore, experimentally, thiopyrans 7–9 without the cyclohexanone skeleton were obtained. This supported the hypothesis that the thiocarbonyl group reacted first.
Thus, because it was found that the thiocarbonyl group of 3 reacted first, the energy profiles of the reaction pathways for the thiocarbonyl moiety of 3 and a single molecule of 6f were first examined to understand the experimentally observed regioselectivity (Fig. 2 and S8†). In Fig. 2, the free energy profile of the observed thio-Diels–Alder reaction leading to the formation of thiopyranes (products1-I to IV) is outlined. In this reaction, 3 had four possible ways to approach 6f (transition states via TS1-I, TS1-II, TS1-III, and TS1-IV). These pathways should lead to the formation of four diastereomers (product1-I, product1-II, product1-III, and product1-IV). TS1-I, TS1-II, TS1-III, and TS1-IV were identified as energy transition states located at 35.3, 40.9, 60.9, and 66.5 kJ mol−1, respectively. Intrinsic reaction path (IRC) calculations30,31 did not show a shoulder on the path to products1-I to IV, that is, no intermediate could be located, suggesting an asynchronous concerted mechanism. All reactions were exothermic (ΔE (kJ mol−1) = product1-I: −88.4; product1-II: −74.1; product1-III: −72.9; and product1-IV: −36.6). Due to the large energetic barriers of the backward reactions (>103.1 kJ mol−1), the final product distribution was most likely to be kinetically controlled. In this regard, product1-I was favored by 5.6, 25.6, and 31.2 kJ mol−1 over products1-II, III, and IV, respectively. According to the Boltzmann equation, such an energy difference should not give product1-III and product1-IV, and the product distribution of product1-I and product1-II should correlate with 9:1, in agreement with experimental observations (21 and 22 were not isolated).
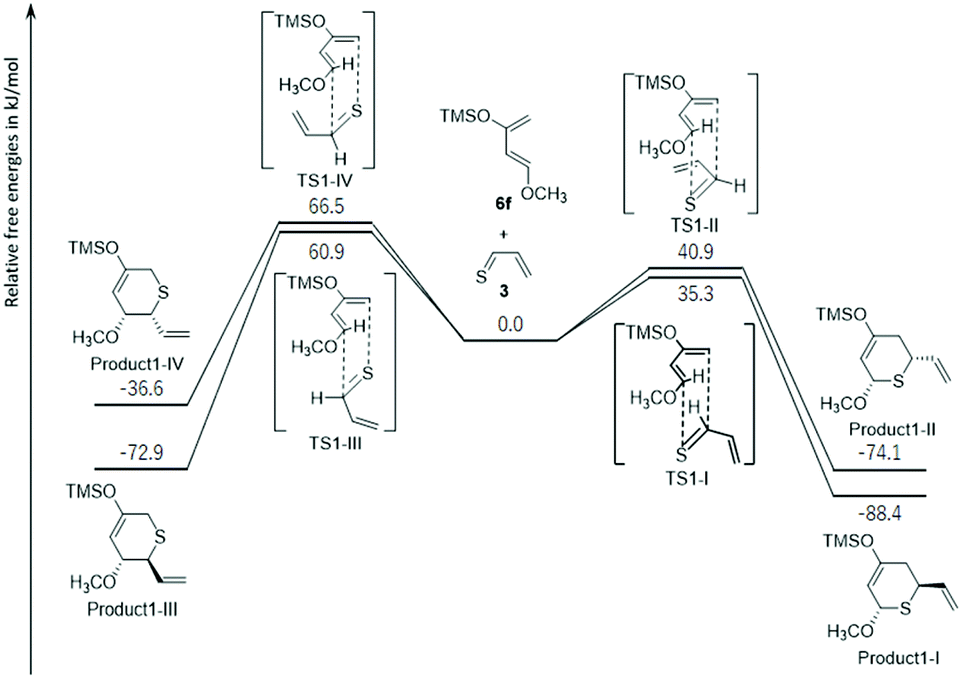 | ||
| Fig. 2 DFT computed relative free energies [ΔG (kcal mol−1), 298 K, 1 atm] and transition-state structures for the thio-Diels–Alder reactions of thioacrolein (3) and Danishefsky's diene (6f). | ||
Therefore, because product1-I was preferentially synthesized in the first-stage reaction, the energy profiles of the reaction pathways for the second molecule of 6f and product1-I were investigated (Fig. 3 and S9†). In this reaction, product1-I had eight possible ways to approach 6f (transition states via TSs2-I to VIII). They were also directly linked to cyclic products (products2-I to VIII) in IRC. Due to the large energy barrier of the reverse reaction (>209.6 kJ mol−1), the second-stage reaction was also most likely to be kinetically controlled. The most preferred products, product2-I and product2-II, were precursor compounds of syn-5 and anti-5, consistent with experimental results.
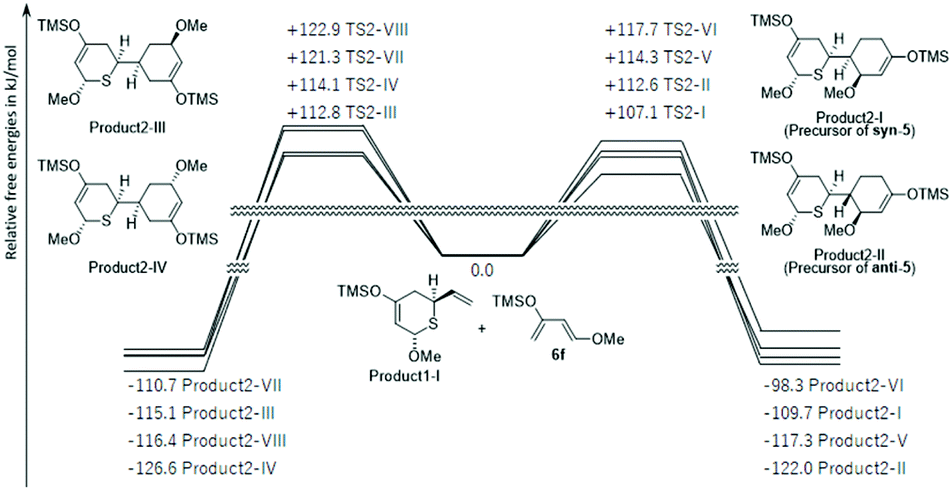 | ||
| Fig. 3 DFT computed relative free energies [ΔG (kcal mol−1), 298 K, 1 atm] and product structures for the Diels–Alder reactions of product1-I and 6f. | ||
Optimization of reaction conditions
Now that desired thiopyran 5 was obtained, we attempted to optimize the reaction conditions with the aim of obtaining a higher yield. First, the effects of changing the amount of diene 6f added and the reaction time under the reaction condition of heating to 75 °C in toluene were investigated (Table 2, entries 1–6). The amount of 6f added was determined on the basis of the fresh weight of A. sativum whole bulb, and using HPLC, the yield was calculated on the basis of the amount of allicin (4) contained in garlic reported in the literature.32,33 In the toluene study, we were able to improve the yields of compound 5 to 7.4% (entry 5), but we re-examined solvent conditions with the aim of further improving yield (entries 7–11). A study of solvent conditions suggested that CH2Cl2 was most suitable for the synthesis of 5 (entry 11). Then, the amount of diene added and the reaction time were assessed (entries 12–21). As a result of the examination, 5 was obtained in 9.8% yield in the reaction shown in entry 16, in which the reaction solvent was CH2Cl2, the reaction temperature was 30 °C, the amount of diene 6f added was 1.0%, and the reaction time was 72 h. In this case, the yield of 5 determined on the basis of the fresh weight of A. sativum whole bulb was 0.027%, and that determined on the basis of the dry weight of A. sativum whole bulb was 0.068%.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solv. | Temp. (°C) | Dienea (w/w%) | Time (h) | Mol% yield from 4b | Ratio from entry 1c |
| a Calculated from the fresh weight of garlic whole bulb. b Quantification using HPLC based on the theoretical value of allicin (4) contained in A. sativum.32,33 c The ratio of the yield when the yield of 5 from 4 in entry 1 (2.1%) is 1. d The yields of compounds 7, 8 and 9 were 1.6%, 2.1% and 0.5%, respectively. | ||||||
| 1 | Toluene | 75 | 1.0 | 3 | 2.1 | 1.0 |
| 2 | Toluene | 75 | 1.0 | 24 | 2.7 | 1.3 |
| 3 | Toluene | 75 | 1.0 | 48 | 2.2 | 1.0 |
| 4 | Toluene | 75 | 1.0 | 72 | 2.2d | 1.0 |
| 5 | Toluene | 75 | 1.5 | 48 | 7.4 | 3.6 |
| 6 | Toluene | 75 | 2.0 | 48 | 4.2 | 2.0 |
| 7 | Toluene | 30 | 1.0 | 48 | 3.8 | 1.8 |
| 8 | Benzene | 30 | 1.0 | 48 | 4.0 | 1.9 |
| 9 | Neat | 30 | 1.0 | 48 | 4.8 | 2.3 |
| 10 | CH3CN | 30 | 1.0 | 48 | 5.7 | 2.8 |
| 11 | CH2Cl2 | 30 | 1.0 | 48 | 9.4 | 4.6 |
| 12 | CH2Cl2 | 30 | 0.5 | 24 | 0.8 | 0.4 |
| 13 | CH2Cl2 | 30 | 0.5 | 48 | 1.9 | 0.9 |
| 14 | CH2Cl2 | 30 | 1.0 | 3 | 1.2 | 0.6 |
| 15 | CH2Cl2 | 30 | 1.0 | 24 | 6.5 | 3.2 |
| 16 | CH2Cl2 | 30 | 1.0 | 72 | 9.8 | 4.7 |
| 17 | CH2Cl2 | 30 | 1.0 | 96 | 8.0 | 3.9 |
| 18 | CH2Cl2 | 30 | 1.5 | 24 | 3.7 | 1.8 |
| 19 | CH2Cl2 | 30 | 2.0 | 24 | 4.9 | 2.4 |
| 20 | CH2Cl2 | 30 | 5.0 | 48 | 2.6 | 1.3 |
| 21 | CH2Cl2 | 30 | 10.0 | 48 | 5.0 | 2.4 |
On the other hand, it was reported that 110 mg of vinyldithiin (2) was isolated from 600 g of A. sativum.1 This signified that 2 was synthesized from garlic-derived 4 in 19.6% yield. The fact that 5 was obtained in 9.8% yield indicated that the biosynthetic pathway from 3 to 2 was efficiently switched to the synthesis of 5 (Scheme 1 and Fig. S4†).
Examination of anti-CSC effect
CSCs are present in small numbers in various cancer cells, and possess both self-renewal ability and pluripotency to produce many types of cancer cells.34 In addition, unlike more differentiated cancer cells, CSCs are often in the mitotic phase. Therefore, CSCs are resistant to existing anticancer drugs and are involved in cancer recurrence after chemotherapy.35 There is a pressing need to develop a therapeutic drug that specifically targets CSCs. The cytotoxic activities of the 16 synthesized compounds (7–14, 16, and 18) on U-251 MG cells and their CSCs were evaluated using (Z)-ajoene (1a) as positive control. The CSCs were generated using the sphere formation assay as described previously,36 and the formation of spheres was confirmed from cell morphology. Cell viability was determined using the CellTiter-Glo® 3D cell viability assay that is based on the quantification of cellular ATP level.Compound syn-5 showed significant cytotoxic activity on both U-251 MG non-CSCs and CSCs at 50 μM. However, other sulfur-containing compounds including anti-5, the diastereomer of syn-5, showed no significant cytotoxic activity on U-251 MG non-CSCs and CSCs at 50 μM.
To obtain further knowledge of CSC toxicity, the cytotoxic activities of 5 and 1 on MDA-MB-231 non-CSCs and their CSCs were evaluated (Table 3). Compounds syn-5a, syn-5b, 1a, and 1b exhibited cytotoxic activities on both MDA-MB-231 non-CSCs and CSCs, as were observed in the case of U-251 MG cells. The cytotoxic activities on CSCs of syn-5a and syn-5b were the same as or higher than that of positive control 1a. On the other hand, the cytotoxic activities on non-CSCs of syn-5a and syn-5b were slightly lower than that of 1a. The cytotoxicity of 1b toward non-CSCs was lower than those of syn-5a, syn-5b, and 1a. On the other hand, compounds anti-5a and anti-5b at 50 μM did not show cytotoxic activities on MDA-MB-231 CSCs as in the case of U-251 MG CSCs and non-CSCs. anti-5 and syn-5 are diastereomers, and it is interesting that one diastereomer has a cytotoxic effect whereas the other diastereomer has no effect. The differences in the three-dimensional molecular structures of these compounds might contribute to the pharmacological activity.
| U-251 MG CSCs | U-251 MG nonCSCs | MDA-MB-231 CSCs | MDA-MB-231 nonCSCs | |
|---|---|---|---|---|
| a Each value represents the mean ± S.E.M. (N = 3). b No effect at 50 μM. | ||||
| Cytotoxic activity (IC50, μM)a | ||||
| syn-5a (2R,1′R) | 39 ± 3 | 26 ± 5 | 43 ± 1 | 32 ± 2 |
| syn-5b (2S,1′S) | 30 ± 7 | 32 ± 5 | 40 ± 2 | 24 ± 5 |
| anti-5a (2R,1′S) | N. E.b | N. E.b | N. E.b | 47 ± 3 |
| anti-5b (2S,1′R) | N. E.b | N. E.b | N. E.b | 47 ± 1 |
| (Z)-Ajoene (1a, positive control) | 39 ± 1 | 19 ± 1 | 43 ± 1 | 15 ± 1 |
| (E)-Ajoene (1b) | 40 ± 2 | 40 ± 2 | 43 ± 1 | 36 ± 2 |
| Adriamycin (positive control) | 0.051 ± 0.014 | 0.040 ± 0.009 | 0.48 ± 0.08 | 0.20 ± 0.02 |
Conclusions
In 2013, Hergenrother and co-workers reported the “Complexity-to-Diversity” (CtD) strategy for creating complex natural product-like derivatives via the controlled application of ring distortion reactions on natural products.37–39 This CtD strategy was inspired by nature's proclivity to produce complex natural products from naturally occurring common intermediates. In this study, we used a naturally occurring chemically unstable intermediate, thioacrolein (3), for the construction of cyclic sulfur-containing compounds with anticancer effects. We showed for the first time that thioacrolein (3) produced by allicin (4), a major component in garlic, undergoes regioselective sequential double Diels–Alder reactions with two molecules of silyl enol ether as the diene partner. In addition, several cyclic and acyclic sulfur-containing compounds were obtained in the reactions.Surprisingly, whereas thiopyranones syn-5a and syn-5b exhibited significant toxicity toward CSCs, their diastereomers anti-5a and anti-5b showed no such activity. The toxicity toward CSCs of syn-5a and syn-5b was equal to or higher than that of (Z)-ajoene (1a) derived from garlic. Moreover, syn-5a and syn-5b had higher chemical stability than 1a.
Experimental
General experimental procedures
The following instruments were used to obtain physical data: specific rotation, Horiba SEPA-300 digital polarimeter (l = 5 cm); IR spectra, JASCO FT/IR-4600 Fourier Transform Infrared Spectrometer; ESIMS, Agilent Technologies Quadrupole LC/MS 6130; HR-ESI-MS, SHIMADZU LCMS-IT-TOF; EIMS and HREIMS, JEOL JMS-GCMATE mass spectrometer; 1H NMR spectra, JEOL JNM-ECA 600 (600 MHz) spectrometer; 13C NMR spectra, JEOL JNM-ECA 600 (150 MHz) spectrometer with tetramethylsilane as the internal standard; MPLC, a YMC (Kyoto, Japan) Multiple Preparative HPLC LC-Forte/R. YMC-DispoPack AT SIL-25 (12 g) column was used for analytical and preparative purposes. The following materials were used for chromatography: normal-phase silica gel column chromatography (CC), silica gel BW-200 (Fuji Silysia Chemical, Ltd, 150–350 mesh); reversed-phase silica gel CC, Chromatorex ODS DM1020T (Fuji Silysia Chemical, Ltd, 100–200 mesh); TLC, precoated TLC plates with silica gel 60F254 (Merck, 0.25 mm) (normal phase) and silica gel RP-18 F254S (Merck, 0.25 mm) (reversed phase); reversed-phase HPTLC, precoated TLC plates with silica gel RP-18 WF254S (Merck, 0.25 mm). Detection of compounds was achieved by UV irradiation and by spraying with 1% Ce(SO4)2–10% aqueous H2SO4 followed by heating.Plant material
Fresh garlic (A. sativum bulb) were obtained as commercial products in Kyoto, Japan in 2018–2021. The plants were identified by the author (S. Nakamura).Measurement of dry weight loss of A. sativum
A flat weighing bottle was dried for 1 h at 110 °C in a drying oven. After drying, it was transferred to a desiccator and allowed to cool for 30 min. After cooling, the weighing bottle was weighed, a portion of A. sativum (weight A: 2.2263 g, 2.4467 g, 2.5166 g) was placed in the weighing bottle respectively, and the whole was weighed again. The weighed A. sativum sample was heated in a 500 W microwave oven for 30 s and then transferred to a drying oven set at 110 °C for 5 h. After drying, the A. sativum sample was placed in a desiccator and allowed to cool for 30 min. After cooling, dried weight was measured (weight B: 0.8143 g, 1.0339 g, 0.9687 g). From these results, the dry weight of the dry A. sativum was calculated by considering the dry weight loss (61%; calculated by (1 − [total of weight B]/[total of weight A]) × 100) of fresh A. sativum.Isolation of (E/Z)-ajoenes (1)
(E/Z)-Ajoenes (1) were obtained by partially modifying the method reported by Block et al.1 The allicin (4)-containing fraction was extracted by chopping fresh Allium sativum (370 g) whole bulb and blending with ether for 3 minutes in a mixer. Plant fibers were removed by filtration, and the allicin (4)-containing fraction (2.5 g, 0.7 w/w% from fresh A. sativum whole bulb) was obtained by concentrating the filtrate under reduced pressure. The allicin (4)-containing fraction (2.5 g) was suspended in acetone–H2O (3:2, v/v) and reflux for 4 h. After the reaction, the solvent was evaporated in vacuo to obtain a residue (2.3 g). The residue (2.3 g) was subjected to normal-phase MPLC {mobile phase: n-hexane–ethyl acetate (70:30 → 0:100 gradient 50 min, v/v) [YMC-DispoPack AT SIL-25 (40 g)]} to give 1a (72.5 mg, 0.020%) and 1b (146.8 mg, 0.040%).
Syntheses of compounds 5, 7–9
The allicin (4)-containing fraction was extracted by chopping fresh Allium sativum (195.2 g) whole bulb and blending with ether for 3 minutes in a mixer. Plant fibers were removed by filtration, and the extract was dried over sodium sulfate. The allicin (4)-containing fraction (890.3 mg, 0.5 w/w% from fresh A. sativum whole bulb) was obtained by concentrating the filtrate under reduced pressure. The allicin (4)-containing fraction (890.3 mg) and Danishefsky's diene (1.75 mL, 9 mmol) were suspended in toluene (3 mL) and stirred at 75 °C for 72 h. After the reaction, the solvent was evaporated in vacuo to obtain a residue (962.4 mg). The residue (962.4 mg) was subjected to reversed-phase MPLC {mobile phase: H2O–MeOH (90:10 → 0:100 gradient 50 min, v/v) [YMC-DispoPack AT ODS-25 (40 g)]} to give five fractions [Fr. 1 (59.0 mg), Fr. 2 (63.0 mg), Fr. 3 (62.3 mg), Fr. 4 (99.4 mg), and Fr. 5 (238.3 mg)]. Fr. 2 (63.0 mg) was purified by HPLC {mobile phase: H2O–MeOH (75:25, v/v) [5C18-MS-II (250 × 10 mm i.d.)]} to give 7a (2.6 mg, 0.016 mmol, 0.0013% from A. sativum), 7b (3.6 mg, 0.023 mmol, 0.0018%), 8a (2.7 mg, 0.012 mmol, 0.0014%), 8b (4.1 mg, 0.018 mmol, 0.0021%), 9 (2.6 mg, 0.011 mmol, 0.0013%), 8c (2.6 mg, 0.011 mmol, 0.0013%), and 8d (2.3 mg, 0.010 mmol, 0.0012%). Fr. 3 (62.3 mg) was purified by HPLC {mobile phase: H2O–MeOH (75:25, v/v) [5C18-MS-II (250 × 10 mm i.d.)]} to give a racemic mixture of anti-5 (4.5 mg, 0.022 mmol, 0.0023%) and a racemic mixture of syn-5 (6.9 mg, 0.033 mmol, 0.0035%). The racemic mixture of anti-5 (1.6 mg) was purified by HPLC {mobile phase: n-hexane–2-propanol (60:40, v/v) [CHIRAL PAK AD-H (250 × 4.6 mm i.d.)]} to give anti-5a (0.8 mg) and anti-5b (0.7 mg). The racemic mixture of syn-5 (1.9 mg) was purified by HPLC {mobile phase: n-hexane–2-propanol (50:50, v/v) [CHIRAL PAK AD-H (250 × 4.6 mm i.d.)]} to give syn-5a (1.0 mg) and syn-5b (0.6 mg).
syn-5a (2R,1′R) and syn-5b (2S,1′S): yellow oil; syn-5a: [α]21D −136.0 (c = 1.0, CH3CN); ECD (CH3CN) λmax (Δε) [223 nm (+12.8), 300 nm (−9.4)]; syn-5b: [α]21D +153.3 (c = 1.0, CH3CN); ECD (CH3CN) λmax (Δε) [222 nm (−14.2), 300 nm (+9.8)]; IR (ATR) νmax = 2952, 1667, 1574, 1548 cm−1; HRMS (EI) m/z: [M]+ calcd for C11H12O2S 208.0558; found 208.0561; LRMS (ESI) m/z: [M + Na]+ 231; 1H NMR (CDCl3, 400 MHz): δ 7.47 (H-6, d, J = 10.1), 6.88 (H-2′, dd-like, J = 10.2), 6.24 (H-5, d, J = 10.1), 6.13 (H-3′, d, J = 10.2), 3.77 (H-2, ddd, J = 12.4, 5.7, 3.7), 2.83 (H-1′, m), 2.82 (H-3a, dd, J = 16.2, 3.7), 2.74 (H-3b, dd, J = 16.2, 12.4), 2.60 (H-5′a, dt-like, J = 16.9, 4.4), 2.40 (H-5′b, ddd, J = 16.9, 13.3, 4.9), 2.17 (H-6′a, ddd-like, J = 14.5, 4.9, 1.7), 2.04 (H-6′b, dddd-like, J = 14.5, 13.3, 10.0, 4.4); 13C NMR (CDCl3, 100 MHz): δ 198.4 (C-4′), 193.8 (C-4), 148.9 (C-2′), 145.3 (C-6), 131.5 (C-3′), 123.8 (C-5), 46.5 (C-2), 41.8 (C-3), 36.6 (C-5′), 25.4 (C-6′).
7b: yellow oil, high resolution ESI-MS: calcd for (C7H10O2S + Na)+: 181.0294. Found: 181.0291. IR (ATR): 2973, 2894, 2161, 2018, 1649, 1545 cm−1. ESI-MS: m/z 181 (M + Na)+. 1H NMR (CDCl3, 500 MHz): δ 7.42 (H-6, d, J = 10.2), 6.19 (H-5, d, J = 10.2), 4.03 (H-1′, quin-like, J = 6.2), 3.53 (H-2, ddd, J = 11.2, 5.7, 4.0), 2.87 (H-3a, dd, J = 16.2, 4.0), 2.78 (H-3b, dd, J = 16.2, 11.2), 1.34 (H-2′, d, J = 6.2). 13C NMR (CDCl3, 125 MHz): δ 194.3 (C-4), 145.0 (C-6), 123.5 (C-5), 68.6 (C-1′), 49.2 (C-2), 39.2 (C-3), 20.5 (C-2′).
8b: yellow oil, high resolution ESI-MS: calcd for (C10H14O2S2 + Na)+: 253.0327. Found: 253.0324. IR (ATR): 2932, 1656, 1548 cm−1. ESI-MS: m/z 253 (M + Na)+. 1H NMR (CDCl3, 500 MHz): δ 7.42 (H-6, d, J = 10.2), 6.23 (H-5, d, J = 10.2), 5.82 (H-2′′, dddd, J = 17.1, 10.3, 8.0, 7.0), 5.45 (H-3′′a, d-like, J = 10.3), 5.42 (H-3′′b, d-like, J = 17.1), 3.80 (H-2, ddd, J = 9.5, 7.9, 4.7), 3.56 (H-1′′a, dd-like, J = 13.0, 7.0), 3.43 (H-1′′b, dd-like, J = 13.0, 8.0), 2.97 (H-3a, d-like, J = 4.7), 2.96 (H-3b, d-like, J = 9.5), 2.91 (H-1′, m), 1.45 (H-2′, d, J = 6.8). 13C NMR (CDCl3, 125 MHz): δ 192.8 (C-4), 144.2 (C-6), 125.5 (C-2′′), 124.0 (C-5), 123.9 (C-3′′), 54.3 (C-1′), 54.1 (C-1′′), 44.5 (C-2), 41.9 (C-3), 7.9 (C-2′).
8c: yellow oil, high resolution ESI-MS: calcd for (C10H14O2S2 + Na)+: 253.0327. Found: 253.0330. IR (ATR): 2968, 2933, 2348, 1657, 1548 cm−1. ESI-MS: m/z 253 (M + Na)+. 1H NMR (CDCl3, 400 MHz): δ 7.45 (H-6, d, J = 10.2), 6.21 (H-5, d, J = 10.2), 5.97 (H-2′′, dddd, J = 17.0, 10.1, 8.2, 6.6), 5.51 (H-3′′a, d-like, J = 10.1), 5.43 (H-3′′b, d-like, J = 17.0), 4.27 (H-2, dt, J = 11.7, 3.8), 3.66 (H-1′′a, dd, J = 13.5, 6.6), 3.30 (H-1′′b, dd, J = 13.5, 8.2), 3.07 (H-1′, qd, J = 7.1, 3.8), 2.92 (H-3a, dd, J = 16.4, 3.8), 2.78 (H-3b, dd, J = 16.4, 11.7), 1.25 (H-2′, d, J = 7.1). 13C NMR (CDCl3, 100 MHz): δ 193.1 (C-4), 145.0 (C-6), 124.9 (C-2′′), 124.3 (C-3′′), 123.8 (C-5), 57.1 (C-1′), 52.9 (C-1′′), 40.9 (C-2), 39.1 (C-3), 9.9 (C-2′).
8d: yellow oil, high resolution ESI-MS: calcd for (C10H14O2S2 + Na)+: 253.0327. Found: 253.0322. IR (ATR): 2967, 2916, 1706, 1656, 1548 cm−1. ESI-MS: m/z 253 (M + Na)+. 1H NMR (CDCl3, 400 MHz): δ 7.45 (H-6, dd, J = 10.2, 1.0), 6.22 (H-5, d, J = 10.2), 5.97 (H-2′′, dddd, J = 16.9, 10.2, 8.4, 6.4), 5.52 (H-3′′a, d-like, J = 10.2), 5.42 (H-3′′b, d-like, J = 16.9), 4.29 (H-2, ddd, J = 10.9, 4.8, 3.3), 3.69 (H-1′′a, dd, J = 13.6, 6.4), 3.28 (H-1′′b, dd, J = 13.6, 8.4), 3.03 (H-1′, qd, J = 7.0, 3.3), 2.80 (H-3a, dd, J = 16.4, 10.9), 2.74 (H-3b, dd, J = 16.4, 4.8), 1.24 (H-2′, d, J = 7.0). 13C NMR (CDCl3, 100 MHz): δ 193.4 (C-4), 145.2 (C-6), 124.9 (C-2′′), 123.9 (C-3′′), 124.0 (C-5), 58.2 (C-1′), 52.7 (C-1′′), 42.1 (C-3), 41.5 (C-2), 9.7 (C-2′).
Syntheses of compounds 10–19
The allicin (4)-containing fraction was extracted by chopping fresh Allium sativum (69.42 g) whole bulb and blending with ether for 3 minutes in a mixer. Plant fibers were removed by filtration, and the extract was dried over sodium sulfate. The allicin (4)-containing fraction (453.4 mg, 0.65 w/w% from fresh A. sativum whole bulb) was obtained by concentrating the filtrate under reduced pressure.The allicin (4)-containing fraction (453.4 mg) and Rawal's diene (0.75 mL, 660 mg, 2.902 mmol) was suspended in toluene (3 mL) and stirred at 75 °C for 3 h. After the reaction, the solvent evaporated in vacuo to obtain a residue (880.7 mg). The residue (880.7 mg) was subjected to reversed-phase MPLC {mobile phase: H2O–MeOH (90:0 → 0:100 gradient 50 min, v/v) [YMC-DispoPack AT ODS-25 (40 g)]} to give six fractions [Fr. 1 (142.1 mg), Fr. 2 (9.6 mg), Fr. 3 (25.6 mg), Fr. 4 (35.2 mg), Fr. 5 (104.9 mg), and Fr. 6 (122.0 mg)]. Fr. 3 (25.6 mg) was purified by HPLC {mobile phase: H2O–MeOH (65:35, v/v) [5C18-MS-II (250 × 10 mm i.d.)]} to give 12 (1.0 mg, 0.00546 mmol, 0.0014% from A. sativum), 13 (1.4 mg, 0.00772 mmol, 0.0020%), 10 (1.3 mg, 0.00702 mmol, 0.0019%), 11 (0.2 mg, 0.00108 mmol, 0.00029%), and 14 (0.7 mg, 0.00351 mmol, 0.0010%). Fr. 4 (35.2 mg) was purified by HPLC {mobile phase: H2O–MeOH (45:55, v/v) [5C18-MS-II (250 × 10 mm i.d.)]} to give 19 (1.6 mg, 0.0106 mmol, 0.0023%), 17 (1.9 mg, 0.00565 mmol, 0.0027%), 15 (1.4 mg, 0.00621 mmol, 0.0020%), 18 (1.2 mg, 0.00326 mmol, 0.0017%), and 16 (1.0 mg, 0.00388 mmol, 0.0014%).
Computational methods
Biological evaluation
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.21 software. The statistical analysis was conducted using one-way analysis of variance (ANOVA) followed by a Tukey–Kramer or Dunnett test to analyze differences between treatment groups. The differences were considered significant when *P < 0.05, **P < 0.01, or ***P < 0.001.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant No. 20K07109 (S. N.) and 20H03397 (S. N., T. M., T. O., T. W.).References
- E. Block, S. Ahmad, M. K. Jain, R. W. Crecely, R. Apitz-Castro and M. R. Cruz, J. Am. Chem. Soc., 1984, 106, 8295–8296 CrossRef CAS.
- S. Nakamura, S. Nakashima and H. Matsuda, J. Asian Assoc. Sch. Pharm., 2021, 10, 1–8 Search PubMed.
- Y. Jung, H. Park, H. Y. Zhao, R. Jeon, J. H. Ryu and W. Y. Kim, Mol. Cells, 2014, 37, 547–553 CrossRef PubMed.
- M. T. Naznin, M. Akagawa, K. Okukawa, T. Maeda and N. Morita, Food Chem., 2008, 106, 1113–1119 CrossRef CAS.
- D. W. Lee, M. Y. Lee, B. Ku, S. H. Yi, J. H. Ryu, R. Jeon and M. Yang, Arch. Toxicol., 2014, 88, 283–290 CrossRef CAS PubMed.
- C. H. Kaschula, R. Hunter, N. Stellenboom, M. R. Caira, S. Winks, T. Ogunleye, P. Richards, J. Cotton, K. Zilbeyaz, Y. Wang, V. Siyo, E. Ngarande and M. I. Parker, Eur. J. Med. Chem., 2012, 50, 236–254 CrossRef CAS.
- R. Hunter, C. H. Kaschula, I. M. Parker, M. R. Caira, P. Richards, S. Travis, F. Taute and T. Qwebani, Bioorg. Med. Chem. Lett., 2008, 18, 5277–5279 CrossRef CAS.
- D. Y. Yoo, W. Kim, S. M. Nam, M. Yoo, S. Lee, Y. S. Yoon, M. H. Won, I. K. Hwang and J. H. Choi, Food Chem. Toxicol., 2014, 72, 1–7 CrossRef CAS.
- R. Ohta, N. Yamada, H. Kaneko, K. Ishikawa, H. Fukuda, T. Fujino and A. Suzuki, Antimicrob. Agents Chemother., 1999, 43, 1811–1812 CrossRef CAS.
- C. F. Williams, A. R. Vacca, L. Dunham, D. Lloyd, M. P. Coogan, G. Evans, M. Graz and J. Cable, Mol. Biochem. Parasitol., 2016, 206, 20–28 CrossRef CAS.
- D. Y. Lee, H. Li, H. J. Lim, H. J. Lee, R. Jeon and J. H. Ryu, J. Med. Food, 2012, 15, 992–999 CrossRef CAS.
- E. Block, S. Ahmad, J. L. Catalfamo, M. K. Jain and R. Apitz-Castro, J. Am. Chem. Soc., 1986, 108, 7045–7055 CrossRef CAS.
- S. Danishefsky and T. Kitahara, J. Am. Chem. Soc., 1974, 96, 7807–7808 CrossRef CAS.
- S. A. Kozmin and V. H. Rawal, J. Org. Chem., 1997, 62, 5252–5253 CrossRef CAS.
- M. Fukaya, S. Nakamura, R. Nakagawa, S. Nakashima, M. Yamashita and H. Matsuda, Org. Lett., 2018, 20, 28–31 CrossRef CAS PubMed.
- M. Fukaya, S. Nakamura, H. Hayashida, D. Noguchi, S. Nakashima, T. Yoneda and H. Matsuda, Front. Chem., 2020, 8, 282 CrossRef CAS PubMed.
- T. Yoneda, S. Nakamura, S. Okui, S. Okazaki, S. Nakashima and H. Matsuda, Heterocycles, 2021, 102, 2168 CrossRef.
- R. Kubec, I. Štefanová, M. Moos, P. Urajová, M. Kuzma and J. Zápal, J. Agric. Food Chem., 2018, 66, 8783–8794 CrossRef CAS PubMed.
- R. Castarlenas, A. Di Giuseppe, J. J. Pérez-Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2013, 52, 211–222 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- R. Kubec, P. Curko, P. Urajová, J. Rubert and J. Hajšlová, J. Agric. Food Chem., 2017, 65, 10615–10620 CrossRef CAS.
- R. Kubec, P. Urajová, O. Lacina, J. Hajšlová, M. Kuzma and J. Zápal, J. Agric. Food Chem., 2015, 63, 10192–10199 CrossRef CAS.
- M. De Rosa and A. Soriente, Tetrahedron, 2011, 67, 5949–5955 CrossRef CAS.
- Y. Liu, Z. Lai, P. Yang, Y. Xu, W. Zhang, B. Liu, M. Lu, H. Chang, T. Ding and H. Xu, RSC Adv., 2017, 7, 43104–43113 RSC.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- HPC Systems Inc., http://www.hpc.co.jp/chem/react1.html (written in Japanese).
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2013.
- G. M. Li, S. Niu, M. Segi, K. Tanaka, T. Nakajima, R. A. Zingaro, J. H. Reibenspies and M. B. Hall, J. Org. Chem., 2000, 65, 6601–6612 CrossRef CAS PubMed.
- E. Vedejs, D. A. Perry, K. N. Houk and N. G. Rondan, J. Am. Chem. Soc., 1983, 105, 6999–7001 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- P. Rose, M. Whiteman, P. K. Moore and Z. Z. Yi, Nat. Prod. Rep., 2005, 22, 351–368 RSC.
- E. Block, S. Naganathan, D. Putman and S.-H. Zhao, J. Agric. Food Chem., 1992, 40, 2410–2430 Search PubMed.
- M. F. Clarke, J. E. Dick, P. B. Dirks, C. J. Eaves, C. H. M. Jamieson, D. L. Jones, J. Visvader, I. L. Weissman and G. M. Wahl, Cancer Res., 2006, 66, 9339–9344 CrossRef CAS.
- H. D. Hemmati, I. Nakano, J. A. Lazareff, M. Masterman-Smith, D. H. Geschwind, M. Bronner-Fraser and H. I. Kornblum, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15178–15183 CrossRef CAS.
- T. Takada, K. Takata and E. Ashihara, J. Physiol. Sci., 2016, 66, 387–396 CrossRef CAS PubMed.
- R. W. Huigens, K. C. Morrison, R. W. Hicklin, T. A. Timothy, M. F. Richter and P. J. Hergenrother, Nat. Chem., 2013, 5, 195–202 CrossRef CAS.
- J. J. Ciardiello, H. L. Stewart, H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Bioorg. Med. Chem., 2017, 25, 2825–2843 CrossRef CAS.
- V. Srinivasulu, S. M. Sieburth, M. A. Khanfar, I. A. Abu-Yousef, A. Majdalawieh, M. Ramanathan, A. Sebastian and T. H. Al-Tel, J. Org. Chem., 2021, 86, 12872–12885 CrossRef CAS.
- Spartan '10, Wavefunction Inc., Irvine, CA, 2010.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Pecitelli, SpecDis, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- (a) H. Jónsson, G. Mills, K. W. Jacobsen and B. J. Berne, in Classical and Quantum Dynamics in Condensed Phase Simulations, ed. G. Ciccotti and D. F. Coker, World Scientific, Singapore, 1998, p. 385 Search PubMed; (b) G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob01992a |
| This journal is © The Royal Society of Chemistry 2022 |