
Organocatalytic synthesis of (Het)biaryl scaffolds via photoinduced intra/intermolecular C(sp2)–H arylation by 2-pyridone derivatives†
Tapas Kumar
Das
ab,
Mrinalkanti
Kundu
 *a,
Biswajit
Mondal
ab,
Prasanjit
Ghosh
*b and
Sajal
Das
*b
*a,
Biswajit
Mondal
ab,
Prasanjit
Ghosh
*b and
Sajal
Das
*b
aTCG Lifesciences Pvt. Ltd., BN-7, Sector V, Salt Lake City, Kolkata-700091, India. E-mail: mrinal.kundu@tcgls.com
bDepartment of Chemistry, University of North Bengal, Darjeeling, 734013, India. E-mail: sajal.das@hotmail.com; prasanjitmeister@yahoo.com
First published on 30th November 2021
Abstract
A unique N,O-bidentate ligand 6-oxo-1,6-dihydro-pyridone-2-carboxylic acid dimethylamide (L1) catalyzed direct C(sp2)–H (intra/intermolecular) arylation of unactivated arenes has been developed to expedite access to (Het)biaryl scaffolds under UV-irradiation at room temperature. The protocol tolerated diverse functional groups and substitution patterns, affording the target products in moderate to excellent yields. Mechanistic investigations were also carried out to better understand the reaction pathway. Furthermore, the synthetic applicability of this unified approach has been showcased via the construction of biologically relevant 4-quinolone, tricyclic lactam and sultam derivatives.
Introduction
Synthetic chemists have always paid substantial attention to the pursuit of newer protocols to expedite the synthesis of (Het)biaryl scaffolds due to their widespread prevalence in drugs and natural products and use as ligands in cross-coupling reactions.1 Undeniably, the transition-metal catalyzed direct C–H bond arylation technique offers the most promising and straightforward route to construct biaryl derivatives although it is associated with significant disadvantages such as the requirement of elevated temperature, high economic cost and the possible contamination of the final target compounds with transition-metal impurities, including drug-like substances.2 Therefore, the discovery of sustainable and environmentally benign approaches continues to see a great surge of interest and the concept of organocatalyzed direct C(sp2)–H arylation has revamped the traditional cross-coupling reaction over the past few decades. In this regime, the C–C cross-coupling between unactivated arenes and aryl halides using the combination of metal tert-butoxides3 and basic amino group containing ligands such as quinoline-1-amino-2-carboxylic acid,4 an amino-linked nitrogen heterocyclic carbene,5N,N′-dimethylethylenediamine and other diamines,6 a phenalenyl-based ligand,7N,N-bidentate nitrogen heterocycles like 1,10-phenanthroline and its derivatives,8etc. is the most relevant and viable alternative to transition metals in terms of environmental benignity, cost effectiveness and non-hazardousness. Initially, these organic promoters strongly coordinated with the metal ion of the base used, e.g. t-BuOK, to form five-membered ring structures and thus initiated the single-electron-transfer (SET) from metal tert-butoxides to ligands under heating3a or in the presence of a high catalyst loading, sometimes with the help of an additive6b or under light9 or with a combination of DMSO under microwave irradiation.3c Interestingly, photoinduced C–C coupling reactions have only been investigated for intermolecular arylations either using the t-BuOK–DMSO system9a or in the presence of a sub-stoichiometric amount (50 mol%)9b of a ligand, e.g. 1,10-phenanthroline. In the last decade, photoredox catalysis has emerged as an exciting approach for the activation of organic molecules by translating visible light energy via SET, mostly under catalysis of transition metals, e.g. silver, iridium, and ruthenium.10 After the seminal research using organometallic photocatalysts, visible-light-mediated organic transformations have become promising strategies because of the increasing need to develop sustainable reaction conditions and to access novel radical intermediates and pathways.11 Thus, UV-visible light mediated organic transformations have been increasingly appreciated as an abundant, renewable, and clean energy source.On the other hand, 4-quinolone and polyfused lactam derivatives represent an important class of potent privileged structural frameworks found in many alkaloids and pharmaceutically active compounds having widespread applications as anti-cancer, anti-HIV and anti-malarial agents.12 Some of the biologically active molecules are shown in Fig. 1.
 | ||
| Fig. 1 A few examples of pharmaceutically important quinolone and tricyclic lactam derivatives. | ||
In our research works, we are actively engaged in discovering new N,N and N,O-bidentate directing groups which are effective for C–H functionalizations and we have identified suitably substituted 2-pyridone units having potential applications in chelation assisted unactivated C(sp2)–H and C(sp3)–H activation reactions under metal catalysis and also as suitable organocatalysts in asymmetric Michael addition reactions with >99% diastereoselectivity and enantioselectivity.13 Monodentate pyridone derivatives have also been used in the past as ligands to accelerate the (non)directed transition metal-catalyzed coupling reactions of arenes at elevated temperature.14 Recently, bidentate [2,2′-bipyridin]-6(1H)-one has also been used with the Pd catalyst at high temperature (Fig. 2).15 Thus, in continuation of our studies on C–H functionalization by 2-pyridone derivatives,13 herein we describe 6-oxo-1,6-dihydro-pyridone-2-carboxylic acid dimethylamide (L1, Scheme 1) as a weak N,O-coordinated ligand whose deprotonated form can effectively help in transition metal-free catalytic C(sp2)–H arylation, including the synthesis of biologically relevant 4-quinolone and tricyclic lactam scaffolds in the presence of t-BuOK as the base under UV light irradiation at room temperature (Fig. 2).
 | ||
| Fig. 2 Cross coupling reactions: relevant previous works vs. our development. | ||
 | ||
| Scheme 1 C(sp2)–H arylation using different ligands under UV irradiation. | ||
Results and discussion
To test our hypothesis, we utilized three 2-pyridone-based compounds (L1–L3) and one imidazopyridine derivative L4 (Scheme 1). The trials were conducted with 4-iodotoluene (1a, 0.5 mmol) in benzene (2.5 mL) in the presence of t-BuOK (3 equiv.) as the base under UV irradiation (λ ≤ 310 nm) at room temperature for 24 h using a catalytic amount (15 mol%) of the ligands L1–L4 respectively.Pleasingly, all the arylation reactions were successful albeit with varying yields, and amongst the ligands, L1 was found to be the best, resulting in 82% yield of the desired product 3a. It is also worth noting that the most commonly used N,N-coordinated ligand phenanthroline afforded 3a in a moderate yield (45%) under the same conditions. Based on these observations, we started our investigation on the reaction optimization using L1 and Table 1 shows the results. Changing the base from t-BuOK to either t-BuONa or Cs2CO3 or Ag2CO3 resulted in a decrease in the yields of 3a (Table 1, entries 2 and 3).
|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yield of 3a (%) |
| a Reaction conditions: 4-iodotoluene (0.5 mmol), benzene (5 mL), t-BuOK (3 equiv.), L1 (15 mol%), N2 atmosphere, UV light irradiation (λ ≤ 310 nm). b Under thermal heating (oil bath). | ||
| 1 | None | 82 |
| 2 | t-BuONa | 50 |
| 3 | Cs2CO3/Ag2CO3 | 38/40 |
| 4 | Without ligand | <10 |
| 5b | 120 °C | 50 |
| 6 | No base | 0 |
| 7 | Without UV light | 0 |
| 8 | 10 mol% of ligand | 28 |
| 9 | 1.5 equiv. of t-BuOK | 31 |
| 10 | 16 h | 38 |
| 11 | 4-Br toluene | 32 |
| 12 | 4-Cl toluene | 20 |
| 13 | In the presence of visible light | 0 |
| 14 | In the presence of blue LED light (8 W) | 0 |
| 15 | λ ≤ 254 nm (UV light) | 70 |
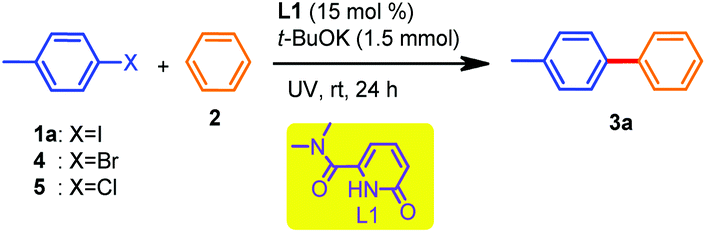
When the reaction was conducted without ligand L1 (keeping other parameters the same), a meager conversion was observed, suggesting the crucial role of this ligand in the C–C coupling reaction (Table 1, entry 4). The reaction was attempted under thermal heating conditions (oil bath) at 120 °C, and this resulted in a reduction in the yield (Table 1, entry 5). The absence of t-BuOK was absolutely detrimental (Table 1, entry 6) which was in accordance with the previous reports where t-BuOK was proposed to facilitate the plausible radical pathway due to the increased availability of the butoxide anion in a low polar solvent like benzene when complexed with an organocatalyst.3a,16 Notably, the role of UV irradiation was significant, as was evident from the trial mentioned in entry 7, where no product was formed. Performing the same reaction either with a lower equivalent of L1 or t-BuOK or in a shorter reaction time provided detrimental results (Table 1, entries 8–10). The use of either 4-bromotoluene or 4-chlorotoluene resulted in much lower yields of 3a (Table 1, entries 11 and 12). Moreover, further attempts to perform the reaction in the presence of both visible and blue LED (8 W) light resulted in deleterious outcomes (Table 1, entries 13 and 14). Replacing UV irradiation (λ ≤ 310 nm) with λ ≤ 254 nm reduced the efficiency of the current catalytic transformation (Table 1, entry 15). Therefore, from our optimization studies, the combination of L1 (15 mol%) as an organocatalyst and t-BuOK as a suitable base under ambient conditions in the presence of UV irradiation (λ ≤ 310 nm) was found to be optimal to achieve the direct C(sp2)–H arylation of unactivated arene (benzene).
With the optimized conditions in hand, the substrate scope of the C(sp2)–H arylation reactions was explored next with various (hetero)aryl iodides (Table 2). In general, the reaction scope was quite broad and both electron-withdrawing and electron-donating groups performed well under the current catalytic conditions. 4-Substituted aryl iodides showed good reactivity towards coupling reactions. For instance, methoxy, –t-Bu, –CF3 or cyano substituted iodo compounds, all afforded the desired products 3b–3e in high isolated yields (78–88%). As noted above in Table 1, the iodo derivative was more reactive compared to the bromo derivative; therefore we judiciously used 1-bromo-4-iodobenzene, leading to the product 3f in 64% yield. Interestingly, 2,4-dichloro iodobenzene led to a moderate yield of 3h (Table 2, 3h, 45%), presumably due to steric hindrance, which was also observed in the case of product 3i, whereas meta-substituted iodobenzenes afforded compounds 3j and 3k in decent yields (62% and 85%, respectively). Heteroaryl iodides like 3-iodothiophene and 4-iodo-1-methyl pyrazole were also amenable, affording the target compounds 3m and 3n in 67% and 81% yields, respectively.
| a Reaction conditions: (hetero)aryl iodides (0.5 mmol), benzene (3 mL), t-BuOK (3 equiv.), L1 (15 mol%), N2 atmosphere, UV (λ ≤ 310 nm), rt, 24 h. b 6-Iodoquinolone and derivatives (0.25 mmol), benzene (3 mL), DMSO (0.5 mL), t-BuOK (3 equiv.), L1 (15 mol%), N2 atmosphere, UV (λ ≤ 310 nm), rt, 40 h. c From GCMS (not isolated). All other yields reported are isolated yields after column chromatography. |
|---|

|
As discussed above, the 4-quinolone scaffold has drawn significant attention in medicinal chemistry for designing new molecular entities such as HIV-1 integrase inhibitors (Fig. 1) for which the aryl group at the 6-position of the quinolone ring was introduced by a transition metal-catalyzed C–C cross coupling reaction. Herein, we report the synthesis of quinolone derivatives 3p–3r following our methodology (DMSO was added due to the meager solubility of 6-iodoquinolones in benzene and the reactions were conducted for 40 h) and to the best of our knowledge, these are the first examples of this kind. The reactions with two other heteroaryl iodides 1s and 1t afforded the requisite products 3s and 3t in low to moderate yields. Conversely, cyclohexyl iodide showed a downward trend in reactivity under the standard reaction conditions and the presence of both DMSO as the solvent and DMAP as the additive did not have any conspicuous effect on the yield of the final product.
Gratifyingly, the scope of this direct C–H functionalization strategy was extended to pyridine instead of benzene. When 4-iodotoluene reacted with pyridine under the optimized conditions, the desired coupling product 7a was obtained in 74% yield as an inseparable mixture of 1-/2-/3-isomers in a 2.7/1.4/1 ratio (from 1H NMR of the crude reaction mixture), as has been reported in the previous literature.9 All individual isomers were further isolated through column purification and characterized by spectroscopic analysis (see the Experimental section). Moreover, 4-iodoanisole and 4-iodobenzotrifluoride also afforded the corresponding C–H arylated products 7b and 7c in satisfactory yields as a mixture of regioisomers.
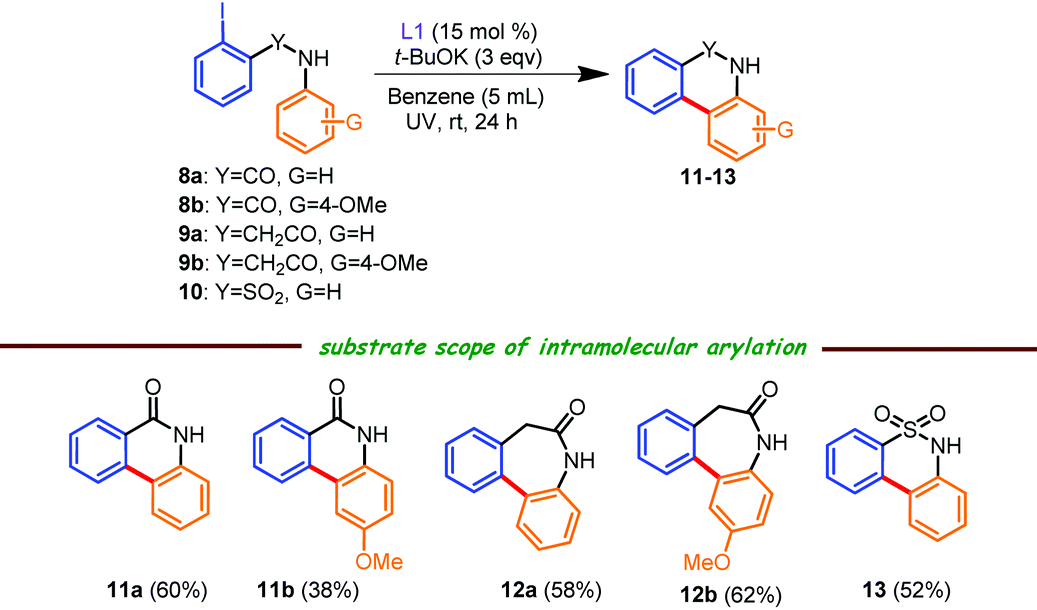
To portray the synthetic versatility of this protocol, we next envisaged the synthesis of tricyclic lactams and a sultam analogue through intramolecular C–C cross coupling reactions (Table 3), which have advantages over the previously published report,6b where different N,N- or N,O-coordinating ligands afforded a meager yield of the tricyclic compound under conventional heating and in the presence of low to even significantly high catalyst loadings. All the reactions shown in Table 3 were performed using benzene as a solvent16 as the starting materials were solid in nature and t-BuOK in THF (1 M) was used due to the partial solubility of iodo compounds in benzene. Of note, these intramolecular C–C coupling reactions carried out with a low catalyst loading at room temperature are, to the best of our knowledge, reported for the very first time. As an example, phenanthridinone 11a was isolated in 60% yield from 2-iodobenzanilide 8a under our reaction conditions, which has advantages over the previously published literature method6b (<30% of the desired product was formed at elevated temperature using 10–40% of a variety of catalysts). Next, when amide 8b was judiciously used in this protocol, a mixture of 6-exo (Table 3, 11b) and 5-endo (11c, see the Experimental section and the ESI†) regioisomers were isolated in an ∼2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (57% overall yield), which was in agreement with previous reports.6b,16 The structure of 11b was confirmed by 2D experiments (see the ESI†). Furthermore, phenylacetamides 9a and 9b smoothly participated under the current catalytic conditions, resulting in the seven-membered lactams 12a (58% isolated yield; ∼25% of the des-iodo product was also formed; data not shown) and 12b (62% isolated yield; obtained as an exclusive isomer). 2-Iodo-N-phenylbenzenesulfonamide 10 was also suitable, affording the tricyclic sultam derivative 13 in a reasonable yield.
:1 ratio (57% overall yield), which was in agreement with previous reports.6b,16 The structure of 11b was confirmed by 2D experiments (see the ESI†). Furthermore, phenylacetamides 9a and 9b smoothly participated under the current catalytic conditions, resulting in the seven-membered lactams 12a (58% isolated yield; ∼25% of the des-iodo product was also formed; data not shown) and 12b (62% isolated yield; obtained as an exclusive isomer). 2-Iodo-N-phenylbenzenesulfonamide 10 was also suitable, affording the tricyclic sultam derivative 13 in a reasonable yield.
To shed light on the reaction pathway, a competitive intermolecular C(sp2)–H arylation experiment was conducted by employing two electronically dissimilar iodoarenes (4-iodoanisole and 4-iodotrifluoromethyl benzene) as coupling partners under the optimized conditions (Scheme 2). The corresponding coupling products 3b and 3v were obtained in 27% and 25% isolated yields (after column chromatography), indicating that the electronic effects of an aryl iodide did not have a major impact on the relative yields of the corresponding products. Rather, this observation suggested the involvement of a radical intermediate in the catalytic cycle. In addition, we also used 4-iodonitrobenzene as a coupling unit under the standard conditions. This did not result in any improved outcome. This may be attributed to the fact that the catalytic activity of 2-pyridone was inhibited through the formation of the intermolecular hydrogen bonding network with 4-iodonitrobenzene. Radical quenching experiments in the presence of over stoichiometric amount of TEMPO/BHT/1,1-diphenylethylene/benzoquinone (as the radical scavenger) had unfavorable outcomes, reinforcing the involvement of the SET (single electron transfer) mechanism. Moreover, we conducted some traditional-cross coupling reactions with ArB(OH)2 and ArMgBr, which resulted in satisfactory yields of only 3a instead of trace amounts of any of the cross-coupled products. These observations further confirmed the importance of the single-electron transfer mechanism (SET). In addition, KIE (kH/kD) values of 2.62 were obtained from a parallel experiment, indicating that the C–H bond cleavage step from complex 20 was the rate-limiting step.
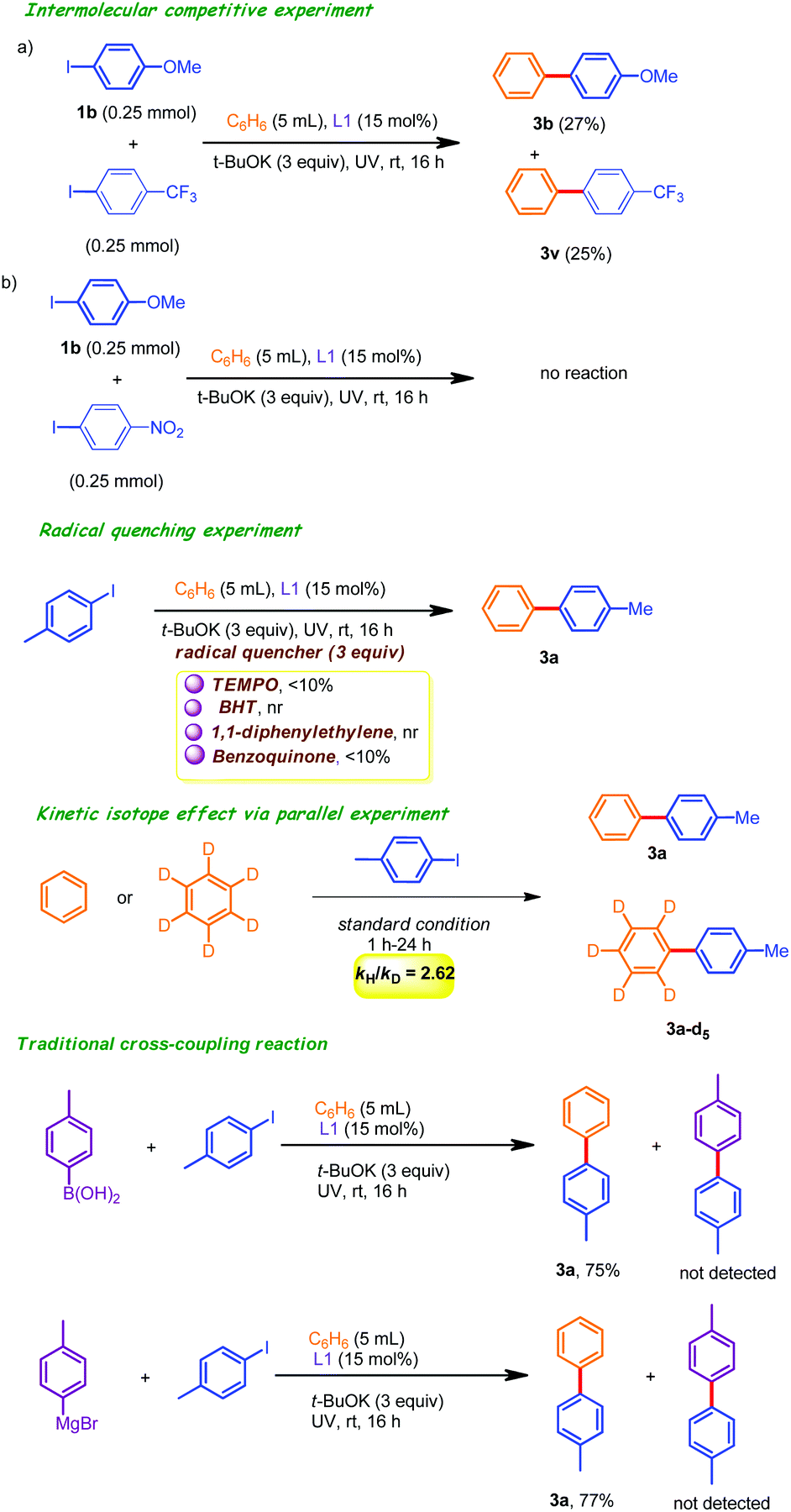 | ||
| Scheme 2 Control experiments. | ||
From the control experiments (Scheme 2) and previous literature precedence,3d,8b a plausible mechanistic pathway is outlined for the intermolecular C(sp2)–(hetero)arylations (Scheme 3). At the outset, we proposed that the deprotonated form of the pyridone catalyst L1 will form the N,O-coordinated 5-membered ring (14) with potassium. This complex initiates the SET process in the presence of light through the transfer of an electron to (hetero)aryl iodide (1), thereby generating the radical anion 17 which spontaneously dissociates into the (hetero)aryl radical 18 (and an iodide ion). The (hetero)aryl radical was trapped by benzene to furnish Int-19 and the single electron oxidation of Int-19 by the complex radical cation Int-16 led to the generation of the (hetero)aryl substituted cation Int-20. Finally, cation Int-20 underwent a base-assisted deprotonation followed by aromatization, delivering the desired products 3 and t-BuOH. In addition, the cationic metal species Int-21 reacted with t-BuOK to regenerate the active metal-chelated complex Int-14 for the continuation of the next catalytic cycle.
 | ||
| Scheme 3 Plausible mechanistic pathway for photoinduced L1 catalyzed intermolecular (hetero)arylation reactions. | ||
Conclusions
In summary, we disclose an environmentally benign approach for transition-metal free intermolecular and intramolecular C(sp2)–H(hetero)arylation reactions at room temperature under UV-light irradiation (λ < 310 nm) using a catalytic amount of 6-oxo-1,6-dihydro-pyridone-2-carboxylic acid dimethylamide (L1) as a unique N,O-bidentate ligand. The crucial effects of temperature, reagent equivalence and substrates having a variety of functional groups were investigated during the reaction optimization which shows the advantage of this method that uses the weak N,O-coordinated catalyst over the existing methods, wherein either elevated temperature or sub-stoichiometric catalyst loading along with the presence of an external additive in some cases has been used. Moreover, the present methodology has been implemented to synthesize pharmaceutically relevant 4-quinolones and tricyclic amides. Investigations on the exact reaction mechanism including the synthesis of new molecular entities and their various biological studies are currently under progress in our laboratory and these observations will be forthcoming.Experimental
General remarks
All reactions were conducted in oven-dried glassware fitted with rubber septa under an atmosphere of nitrogen using freshly distilled solvents, unless specified otherwise. Stainless steel needles were oven-dried, cooled in desiccators and flushed with nitrogen prior to the handling of dry solvents and reagents. Commercial reagents were used as received. DMF, DMSO, benzene, and pyridine (anhydrous) were commercially available and were purchased from Aldrich, Alfa Aesar or Fluorochem. Before performing the reactions, the moisture content in all reagents and solvents was checked. A reaction temperature above 25 °C refers to oil bath temperature. Thin layer chromatography was performed on Merck silica gel 60 F-254 precoated plates (0.25 mm) and visualized by UV irradiation using KMnO4, anisaldehyde or (NH4)2Ce(NO3)6 solutions. Hexanes and ethyl acetate (ACS grade) were used as received from vendors. All the compounds were purified by using RediSep® normal-phase silica Flash columns in a Teledyne ISCO CombiFlash system. Melting points were recorded on a digital melting point apparatus. 1H, 13C{1H} and 19F NMR spectra were recorded on Bruker 400 MHz UltraShield spectrometers with 13C and 19F NMR operating frequencies of 100 MHz and 376 MHz, respectively. Chemical shifts (in δ units, ppm) were referenced to SiMe4 (1H and 13C) and CFCl3 (19F) and values are reported in ppm relative to the residual solvent CDCl3 signal (δ = 7.24 for 1H NMR and δ = 77.0 for 13C NMR) and DMSO-d6 signal (δ = 2.50 for 1H NMR and δ = 39.4–40.6 for 13C NMR). Chemical shifts (δ) are reported in ppm and multiplicities are indicated by s (singlet), d (doublet), q (quartet), t (triplet) m (multiplet), and br (broad). Coupling constants J are reported in hertz (Hz). The spectral data were recorded at 297.8 K unless otherwise noted. 1H–13C HSQC, HMBC and NOE experiments were used to help with the signal assignments.Synthesis of ligands L1–L3
The pyridone derivatives L117 and L218 were synthesized according to exact reports available, whereas L3 is a new compound and the synthesis was carried out following the previous literature.19 The imidazopyridine derivative L4 and phenanthroline are commercially available.
General procedure for photoinduced intermolecular C(sp2)–H (hetero)arylation (synthesis of 3a–o, 3s, 3t, 7a–c)
6-Hydroxy-N,N-dimethylpicolinamide (L1) (12.5 mg, 0.075 mmol) and t-BuOK (168 mg, 1.5 mmol) were charged in a 20 mL oven-dried test tube with a rubber septum under a nitrogen atmosphere. Benzene (5 mL) and iodo compounds (0.5 mmol) were added subsequently. The reaction mixture was sonicated for 5 min and degassed with N2 for 10 min. The reaction mass was then placed in a UV light set-up instrument and irradiated (λ ≤ 310 nm) for 24 h at room temperature (25 °C). The reaction mass was diluted with dichloromethane (∼30 mL) and filtered to separate the insoluble parts. The filtrate was concentrated under reduced pressure and the crude products were purified by silica gel CombiFlash column chromatography using an ethyl acetate/hexane mixture as the eluent to obtain the arylated products.m-(7a)9b. 1H NMR (400 MHz, CDCl3): δ = 8.83 (s, 1H), 8.56 (d, J = 5.3 Hz, 1H), 7.85 (d, J = 7.9 Hz, 1H), 7.48 (d, J = 8.0 Hz, 2H), 7.34 (dd, J = 7.9, 4.8 Hz, 1H), 7.28 (d, J = 7.8 Hz, 2H), 2.40 (s, 3H); 13C NMR (100 MHz, CDCl3) δ = 148.1, 148.1, 138.0, 136.5, 134.9, 134.1, 129.8, 126.9, 123.5, 21.1.
p-(7a). 1H NMR (400 MHz, CDCl3) δ = 8.63 (d, J = 5.1 Hz, 2H), 7.54 (d, J = 7.9 Hz, 2H), 7.49 (d, J = 5.2 Hz, 2H), 7.29 (d, J = 8.1 Hz, 2H), 2.41 (s, 3H); 13C NMR (100 MHz, CDCl3) δ = 150.1, 148.0, 140.1, 135.1, 129.6, 126.6, 121.2, 21.0.
m-(7b). 1H NMR (400 MHz, CDCl3): δ = 8.80 (bs, J = 1.5 Hz, 1H), 8.53 (d, J = 4.0 Hz, 1H), 7.83 (d, J = 7.9 Hz, 1H), 7.50 (d, J = 8.7 Hz, 2H), 7.31 (dd, J = 7.8, 4.8 Hz, 1H), 6.99 (d, J = 8.6 Hz, 2H), 3.85 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 159.8, 148.0, 147.9, 136.2, 133.8, 130.3, 128.2, 123.5, 114.5, 55.3.
p-(7b). 1H NMR (400 MHz, CDCl3): δ = 8.61 (d, J = 5.4 Hz, 2H), 7.60 (d, J = 8.7 Hz, 2H), 7.46 (d, J = 5.6 Hz, 2H), 7.01 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 160.5, 150.2, 147.8, 130.4, 128.1, 121.1, 114.6, 55.4.
m-(7c). 1H NMR (400 MHz, CDCl3) δ = 8.86 (d, J = 1.6 Hz, 1H), 8.65 (d, J = 3.5 Hz, 1H), 7.89 (d, J = 7.9 Hz, 1H), 7.77–7.65 (m, 4H), 7.40 (dd, J = 7.9, 4.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 149.3, 148.3, 141.3, 135.2, 134.4, 130.6 (q, J = 32.3 Hz), 127.4, 126.0 (q, J = 3.8 Hz), 125.4 (q, J = 270.4 Hz), 123.6.
p-(7c). 1H NMR (400 MHz, CDCl3): δ = 8.70 (d, J = 5.6 Hz, 2H), 7.74 (s, 4H), 7.50 (d, J = 6.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 150.5, 146.9, 141.7, 131.5 (q, J = 32.2 Hz), 127.4, 126.1(d, J = 3.8 Hz), 125.3 (d, J = 270.6 Hz), 121.7.
General procedure for 6-arylation of iodoquinoline derivatives 3p and 3q
6-Iodo-4(1H)-quinolone (1p) is commercially available. Compound 1q28 was synthesized following the literature procedure. Both the iodo compounds were then used for intermolecular arylation following the protocol given here.
6-Hydroxy-N,N-dimethylpicolinamide (L1) (6.25 mg, 0.0375 mmol) and t-BuOK (84 mg, 0.75 mmol) were charged in a 20 mL oven-dried test tube with a rubber septum under a nitrogen atmosphere. Benzene (3 mL) and a mixture of iodo compounds (0.25 mmol) in 0.5 mL DMSO were added. The reaction mixture was sonicated for 5 min and degassed with N2 for 10 min. The reaction mass was then placed in a UV light set-up instrument and irradiated (λ ≤ 310 nm) for 40 h at room temperature (25 °C). The reaction was quenched with water and the organic parts were extracted with ethyl acetate. The organic part was separated, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by reverse phase preparative HPLC (70% MeCN, 0.1% NH4HCO3) followed by lyophilization to obtain pure arylated products.
:10) to obtain ethyl 1-(2-acetoxyethyl)-6-iodo-4-oxo-1,4-dihydroquinoline-3-carboxylate (1r) as an off white solid. Yield: 70%. M.p.: 173–175 °C. 1H NMR (400 MHz, DMSO-d6): δ = 8.64 (s, 1H), 8.49 (d, J = 1.8 Hz, 1H), 8.07 (dd, J = 8.9, 1.9 Hz, 1H), 7.70 (d, J = 8.9 Hz, 1H), 4.63 (t, J = 4.7 Hz, 2H), 4.35 (t, J = 4.7 Hz, 2H), 4.20 (q, J = 7.1 Hz, 2H), 1.90 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ = 171.4, 169.8, 164.1, 150.1, 140.6, 138.4, 134.7, 129.7, 119.5, 110.4, 90.2, 60.9, 59.8, 51.1, 20.3, 14.2.
Step 2: 6-Hydroxy-N,N-dimethylpicolinamide (L1) (6.25 mg, 0.0375 mmol) and t-BuOK (84 mg, 0.75 mmol) were charged in a 20 mL oven-dried test tube, fitted with a rubber septum under a nitrogen atmosphere. Then benzene (3 mL) and a mixture of ethyl 1-(2-acetoxyethyl)-6-iodo-4-oxo-1,4-dihydroquinoline-3-carboxylate (1r) (107 mg, 0.25 mmol) in 0.5 mL DMSO were added. The reaction mixture was sonicated for 5 min and degassed with N2 for 10 min. After that, the reaction mass was placed in a UV light set-up instrument and irradiated (λ ≤ 310 nm) for 40 h at room temperature (25 °C). The reaction was quenched with water and the organic parts were extracted with ethyl acetate. The combined organic phase was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was then purified by reverse phase preparative HPLC (70% MeCN, 0.1% HCOOH) followed by lyophilization to afford pure 1-(2-hydroxyethyl)-4-oxo-6-phenyl-1,4-dihydroquinoline-3-carboxylic acid (3r) as an off-white solid. Yield: 62% (47 mg). M.p.: 195–197 °C. 1H NMR (400 MHz, DMSO-d6): δ = 15.24 (s, 1H), 8.92 (s, 1H), 8.61 (d, J = 2.2 Hz, 1H), 8.28 (d, J = 10.2 Hz, 1H), 8.18 (d, J = 9.0 Hz, 1H), 7.83 (d, J = 7.5 Hz, 2H), 7.55 (t, J = 7.6 Hz, 2H), 7.46 (t, J = 7.3 Hz, 1H), 5.06 (t, J = 5.6 Hz, 1H), 4.68 (t, J = 5.1 Hz, 2H), 3.81 (q, J = 5.1 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ = 177.7, 166.1, 150.1, 138.7, 138.0, 137.7, 132.4, 129.2, 128.2, 126.9, 125.9, 122.8, 119.1, 107.1, 58.4, 55.9.
General procedure for intramolecular C–C cross-coupling reactions (synthesis of 11a, 11b, 12a, 12b and 13)
The iodo compounds 8a and 8b,319a and 9b,32 and 1033 were synthesized as per literature reports and used for the intramolecular C–C couplings reactions following the procedure as described here.
6-Hydroxy-N,N-dimethylpicolinamide (L1) (12.5 mg, 0.075 mmol) and t-BuOK, 1 M in THF (1.5 mL, 1.5 mmol; THF solution was used to make the reaction mixture homogeneous as the iodo compounds were partially soluble in benzene) were charged in a 20 mL oven-dried test tube with a rubber septum under a nitrogen atmosphere. Then benzene (5 mL) and iodo compounds (0.5 mmol) were added. The reaction mixture was sonicated for 5 min and degassed with N2 for 10 min. After that, the reaction mass was placed in a UV light set-up instrument and irradiated (λ ≤ 310 nm) for 24 h at room temperature (25 °C). The reaction mass was diluted with dichloromethane (∼30 mL) and filtered to separate the insoluble parts. The filtrate was concentrated under reduced pressure and the crude products were purified by silica gel CombiFlash column chromatography using an ethyl acetate/hexane mixture as the eluent to obtain the arylated products.
Experimental details for the KIE study (synthesis of the mixture of 3a and 3a–d5)
6-Hydroxy-N,N-dimethylpicolinamide (L1) (5 mg, 0.03 mmol) and t-BuOK (50 mg, 0.45 mmol) were charged in a 10 mL oven-dried test tube with a rubber septum under a nitrogen atmosphere. Benzene (1.5 mL) or benzene-d6 (1.5 mL) and 4-iodo toluene (33 mg, 0.15 mmol) were added subsequently. The reaction mixture was sonicated for 5 min and degassed with N2 for 10 min. The reaction mass was then placed in a UV light set-up instrument and irradiated (λ ≤ 310 nm) for 24 h at room temperature (25 °C). The reaction mixture was monitored by GCMS/GCFID in four intervals. The reaction mass was diluted with dichloromethane (∼10 mL) and filtered to separate the insoluble parts. The filtrate was concentrated under reduced pressure and the crude products were purified by silica gel CombiFlash column chromatography using ethyl acetate/hexane mixture as the eluent to obtain the two products.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 5.
Colorless liquid, 1H NMR (400 MHz, CDCl3): δ 2.39 (s, 3H), 7.24 (d, J = 7.2 Hz, 2H), 7.48(d, J = 8.0 Hz, 2H).
5.
Colorless liquid, 1H NMR (400 MHz, CDCl3): δ 2.39 (s, 3H), 7.24 (d, J = 7.2 Hz, 2H), 7.48(d, J = 8.0 Hz, 2H).
General procedure for the traditional coupling reactions with ArB(OH)2 and ArMgBr
6-Hydroxy-N,N-dimethylpicolinamide (L1) (5 mg, 0.03 mmol) and t-BuOK (50 mg, 0.45 mmol) were charged in a 10 mL oven-dried test tube with a rubber septum under a nitrogen atmosphere. Benzene (1.5 mL), 4-iodo toluene (0.15 mmol) and 4-methylphenyl boronic acid (0.15 mmol)/p-tolylmethyl magnesium bromide, 1 M in THF (0.15 mmol) were added subsequently. The reaction mixture was sonicated for 5 min and degassed with N2 for 10 min. The reaction mass was then placed in a UV light set-up instrument and irradiated (λ ≤ 310 nm) for 16 h at room temperature (25 °C). The reaction mass was diluted with dichloromethane (∼10 mL) and filtered to separate the insoluble parts. The filtrate was concentrated under reduced pressure and the crude products were purified by silica gel CombiFlash column chromatography using an ethyl acetate/hexane mixture as the eluent.Author contributions
This manuscript was prepared with contribution from all the authors. TKD carried out the synthesis of all compounds at TCGLS. MK, SD and PG conceptualized the key studies. TKD and BM analyzed the spectral data and wrote the ESI.† MK and SD jointly supervised TKD and BM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TKD gratefully acknowledges TCGLS and the University of North Bengal for giving the opportunity to pursue his PhD work and is thankful to the analytical team at TCGLS for the structural elucidation and discussion of the spectral data.Notes and references
-
(a) G. Bringmann, C. Günther, M. Ochse, O. Schupp and S. Tasler, Fortschr. Chem. Org. Naturst., 2001, 82, 1–249 CrossRef CAS
; (b) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC
-
(a) Y. Wang, D. Leow and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 13864–13867 CrossRef
-
(a) For recent reviews, see: G. Nocera and J. A. Murphy, Synthesis, 2020, 52, 327–336 CrossRef CAS
- Y. Qiu, Y. Liu, K. Yang, W. Hong, Z. Li, Z. Wang, Z. Yao and S. Jiang, Org. Lett., 2011, 13, 3556–3559 CrossRef CAS PubMed
- W.-C. Chen, Y.-C. Hsu, W.-C. Shih, C.-Y. Lee, W.-H. Chuang, Y.-F. Tsai, P. P.-Y. Chen and T.-G. Ong, Chem. Commun., 2012, 48, 6702–6704 RSC
-
(a) W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS
- A. Banik, R. Paira, B. K. Shaw, G. M. Vijaykumar and S. K. Mandal, J. Org. Chem., 2018, 83, 3236–3244 CrossRef CAS PubMed
-
(a) C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X. Y. Lu, K. Huang, S. F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed
-
(a) M. E. Buden, J. F. Guastavino and R. A. Rossi, Org. Lett., 2013, 15, 1174–1177 CrossRef CAS PubMed
-
(a) W. Wu, E. Cui, Y. Zhang, C. Zhang, F. Zhu, C.-H. Tung and Y. Wang, ACS Catal., 2019, 9, 6335–6341 CrossRef CAS
-
(a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC
-
(a) M. Hadjeri, E.-L. Peiller, C. Beney, N. Deka, M. A. Lawson, C. Dumontet and A. Boumendjel, J. Med. Chem., 2004, 47, 4964–4970 CrossRef CAS
-
(a) B. Mondal, P. Ghosh, M. Kundu, T. K. Das and S. Das, Org. Biomol. Chem., 2021, 19, 360–364 RSC
-
(a) P. Wang, G.-C. Li, P. Jain, M. E. Farmer, J. He, P.-X. Shen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14092–14099 CrossRef CAS PubMed
- V. Salamanca, A. Toledo and A. C. Albeniz, J. Am. Chem. Soc., 2018, 140, 17851–17856 CrossRef CAS PubMed
- B. S. Bhakuni, A. Kumar, S. J. Balkrishna, J. A. Sheikh, S. Konar and S. Kumar, Org. Lett., 2012, 14, 2838–2841 CrossRef CAS PubMed
- T. P. Tran, P. B. Mullins, C. W. am End and M. Pettersson, Org. Lett., 2013, 15, 642–645 CrossRef CAS
-
(a) T. Takahashi and A. Koshiro, Chem. Pharm. Bull., 1961, 9, 426–432 CrossRef CAS
- Y. Suna, Y. Himeda, E. Fujita, J. T. Muckerman and M. Z. Ertem, ChemSusChem, 2017, 10, 4535–4543 CrossRef CAS
- C.-C. Chiu, H.-T. Chiu, D.-S. Lee and T.-J. Lu, RSC Adv., 2018, 8, 26407–26415 RSC
- S. Wu, N. Ding, P. Jiang, L. Wu, Q. Feng, L. Zhao, Y. Wang, Q. Su, H. Zhang and Q. Yang, Tetrahedron Lett., 2020, 61, 152656 CrossRef CAS
- F. Mou, Y. Sun, W. Jin, Y. Zhang, B. Wang, Z. Liu, L. Guo, J. Huang and C. Liu, RSC Adv., 2017, 7, 23041–23045 RSC
- L. Candish, M. Freitag, T. Gensch and F. Glorius, Chem. Sci., 2017, 8, 3618–3622 RSC
- J. Duczynski, A. N. Sobolev, S. A. Moggach, R. Dorta and S. G. Stewart, Organometallics, 2020, 39, 105–115 CrossRef CAS
- F. Mäsing, H. Nüsse, J. Klingauf and A. Studer, Org. Lett., 2018, 20, 752–755 CrossRef
- M. J. West and A. J. B. Watson, Org. Biomol. Chem., 2019, 17, 5055–5059 RSC
- K. Kiriyama and E. Shirakwa, Chem. Lett., 2017, 46, 1757–1759 CrossRef CAS
- X. Dr. Ji, D. Li, Z. Wang, M. Tan, H. Huang and G.-J. Deng, Asian J. Org. Chem., 2018, 7, 711–714 CrossRef
- P. Bichovski, T. M. Haas, D. Kratzert and J. Streuff, Chem. – Eur. J., 2015, 21, 2339–2342 CrossRef CAS PubMed
-
(a) S. Pasquini, C. Mugnaini, C. Tintori, M. Botta, T. Alejandro, R. K. Arvela, M. Larhed, M. Witvrouw, M. Michiels, F. Christ, Z. Debyser and F. Corelli, J. Med. Chem., 2008, 51, 5125–5129 CrossRef CAS
- B. Yao, C. Jaccoud, Q. Wang and J. Zhu, Chem. – Eur. J., 2012, 18, 5864–5868 CrossRef CAS
- W. Du and D. P. Curran, Org. Lett., 2003, 10, 1765–1768 CrossRef PubMed
- K. S. Etse, B. Dassonneville, G. Zaragoza and A. Demonceau, Tetrahedron Lett., 2017, 58, 789–793 CrossRef CAS
- Q.-F. Hu, T.-T. Gao, Y.-J. Shi, Q. Lei and L.-T. Yu, RSC Adv., 2018, 8, 13879–13890 RSC
- V. H. Thorat, J.-C. Hsieh and C.-H. Cheng, Org. Lett., 2020, 22, 6623–6627 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob01798e |
| This journal is © The Royal Society of Chemistry 2022 |