
Design of novel tripyridinophane-based Eu(III) complexes as efficient luminescent labels for bioassay applications†
Nadine
Leygue
a,
Claude
Picard
 a,
Pamela
Faure
b,
Emmanuel
Bourrier
b,
Laurent
Lamarque
*b,
Jurriaan M.
Zwier
b and
Chantal
Galaup
*a
a,
Pamela
Faure
b,
Emmanuel
Bourrier
b,
Laurent
Lamarque
*b,
Jurriaan M.
Zwier
b and
Chantal
Galaup
*a
aSPCMIB, UMR5068 CNRS-Université Paul Sabatier-Toulouse III, 118 Route de Narbonne, F-31062 Toulouse, France. E-mail: galaup@chimie.ups-tlse.fr
bCisbio Bioassays, BP 84175, 30200 Codolet, France. E-mail: Laurent.Lamarque@perkinelmer.com
First published on 25th November 2021
Abstract
In this work, the development of highly luminescent europium(III) complexes in water solution is reported, including their syntheses, analyses of their photophysical properties and applications in bioassays. Three Eu(III) complexes are derived from new ligands based on a tripyridinophane platform. There are four distinct sections in the structure of these ligands: an 18-membered polyaminocarboxylic macrocycle to bind efficiently lanthanide ions in aqueous solutions, three chromophoric subunits (4-(phenylethynyl)pyridine moieties) to effectively sensitize the emission of the metal, two peripheral moieties to solubilise the complex in aqueous media (sulfonate, sulfobetaine or glucose groups) and a free NH2 group available for grafting or bioconjugation. In our synthetic procedure, a pivotal macrocyclic platform is obtained with a high yield in the crucial macrocyclization step due to a metal template ion effect (74% yield). In Tris aqueous buffer (pH 7.4), the Eu(III) complexes show a maximum excitation wavelength at 320 nm, a suitable overall quantum yield (14%), a relatively long lifetime (0.80 ms) and a one-photon brightness in the range of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1. Importantly, these photophysical properties are retained at dilute concentrations, even in the presence of a very large excess of potentially competing species, such as EDTA or Mg2+ ions. Furthermore, we report the bioconjugation of a Eu(III) complex labelled by an N-hydroxysuccinimide ester reactive group with an antibody (anti-glutathione-S-transferase) and the successful application of the corresponding antibody conjugate in the detection of GST-biotin in a fluoroimmunoassay. These new complexes provide a solution for high sensitivity in Homogeneous Time-Resolved Fluorescence (HTRF®) bioassays.
000 M−1 cm−1. Importantly, these photophysical properties are retained at dilute concentrations, even in the presence of a very large excess of potentially competing species, such as EDTA or Mg2+ ions. Furthermore, we report the bioconjugation of a Eu(III) complex labelled by an N-hydroxysuccinimide ester reactive group with an antibody (anti-glutathione-S-transferase) and the successful application of the corresponding antibody conjugate in the detection of GST-biotin in a fluoroimmunoassay. These new complexes provide a solution for high sensitivity in Homogeneous Time-Resolved Fluorescence (HTRF®) bioassays.
Introduction
In the large family of fluorescent probes for biological applications, lanthanide-based emitters now have their place alongside organic fluorophores, transition metal-based dyes, quantum dots or native fluorescent proteins. The numerous and various applications of lanthanide reporters such as detection of endogenous ions and biological or bioactive molecules by luminescence spectroscopy and in vitro imaging by luminescence microscopy have been emphasized in recent reviews.1 In a number of these applications, luminescent Ln(III) complexes participate in a non-radiative energy process analogous to FRET (Förster Resonance Energy Transfer) which is often referred to as time-resolved FRET (TR-FRET) or luminescence/lanthanide resonance energy transfer (LRET). In the TR-FRET approach, lanthanide luminophores serve as energy donors to active common acceptors such as organic dyes, fluorescent proteins, or quantum dots.2 Today, Homogeneous Time-Resolved Fluorescence (HTRF®) technology is used in immunoassays to detect events such as DNA or RNA hybridization, enzyme activity, and various drug discovery applications.3 As a consequence, several commercial bioassays based on this technology were developed by different companies. The key feature of lanthanide luminescence that makes it particularly attractive for these applications is that lanthanide ions have long emission lifetimes which can extend into the millisecond range for Eu(III) and Tb(III) complexes. This property mitigates the interference of background fluorescence originating from the biological media (which is in the nanosecond range), greatly improving the signal-to-noise ratio and increasing the overall probe sensitivity through the time-resolved luminescence technique. In TR-FRET bioassays, this allows temporal discrimination between direct acceptor emission and acceptor emission due to FRET. However, exploitation of this luminescence requires that the lanthanide ion is coordinated by an organic ligand containing an aromatic structure (chromophore) able to strongly absorb photons and transfer the collected energy to the emissive level of the metal, in order to balance the inherently weak metal-centered absorption bands (luminescence sensitization or antenna effect).4The detection sensitivity that luminescent Ln(III) probes can provide largely depends on the chemical and photophysical properties of the chromophore structure. Among those, the potential donor level of the antenna (singlet, triplet, intra-ligand charge transfer or ligand-to-metal charge transfer) should lie above the resonance energy level of the Ln(III) ion. As far as Eu(III) and Tb(III) complexes are concerned, a phenomenological rule suggests that the ligand donor level energy should be located between 2500 and 5000 cm−1 above the Ln(III) receiving levels to provide fast energy transfer and avoid thermal energy back transfer.5 It is also desirable that the sensitizer should be located close to the Ln(III) ion, in order to maximize its energy transfer rate. A sensitizer with suitable coordination site(s) to directly bind the Ln(III) ion is therefore needed. This can be illustrated by the extensive use of N-donor aromatic chromophores as Ln(III) sensitizers.6 The excitation wavelength of the antenna should be above 300 nm so as to minimize the strong absorption of nucleic acids and the aromatic amino acids in proteins (primary inner filter effect).7 However, the upper limit for excitation depends on the nature of the lanthanide ion, i.e. their excited emissive levels. Taking into account the conventional sensitization pathway through the ligand triplet excited state, Steemers et al. estimated that the long wavelength absorption edge of the antenna cannot be much above 346 nm for Tb(III) or above 385 nm for Eu(III) complexes.8 On the other hand, ligand structures enabling efficient excitation of the europium ion at wavelengths higher than 400 nm were reported. For example, Yuan et al. described an Eu(III) complex with a maximum excitation wavelength at 415 nm and a luminescence quantum yield of 42% in aqueous Tris buffer.9 Another essential requirement is that the antenna possesses a large molar absorption cross-section. The detectability of a luminescent (fluorescent) label is directly related to its brightness parameter which represents the product of the molar extinction coefficient at the excitation wavelength and the luminescence quantum yield (B = ελexc × Φ). In this regard, a common strategy is to incorporate several antennae around a single Ln(III) ion in a low molecular weight ligand, thereby facilitating high absorption of the incident radiation. This approach is exemplified with trisbipyridyl cryptands developed by Lehn and Mathis and more recent probes containing three or four same chromophores around the metal.10 The increasing number of the same chromophores in a Ln(III) complex increases the overall brightness, but can also lead to a decrease in the luminescence quantum yield, connected to a decrease in the energy transfer efficiency from the ligand to metal.11 Attention should also be paid to the water solubility of complexes bearing several polyaromatic or conjugated aromatic groups.
In addition to governing the Ln(III) sensitization process by incorporating in its structure chromophore(s), the ligand should also (i) fill entirely the first coordination sphere of the Ln(III) ion (the use of multidentate chelates, CN = 8–10), to protect it from water interactions, which are responsible for non-radiative deactivation processes, (ii) endow the complex with high stability in aqueous solutions at subnanomolar concentrations, and (iii) possess a suitable reactive chemical functionality for attachment to targeting vectors or for biomolecule labeling, e.g. carboxylic acid or primary amine group.
We have recently reported the efficient synthesis of an 18-membered tripyridinophane macrocycle, tpptac (Fig. 1), and the promising photophysical properties of the corresponding Eu(III) and Tb(III) complexes.12 In this ligand, the three intracyclic pyridine rings serve both as chelating subunits and as antennae chromophores and the three aminoacetate moieties ensure the stability of Ln(III) complexes in aqueous solutions. This nonadentate ligand matches well with the large coordination number of lanthanide ions (8–10 in aqueous solutions). Consequently, the thermodynamic stability constants of the Eu(III) and Tb(III) complexes are relatively high (logKLnL = 18.7 and 18.0, respectively), with excellent selectivity over the endogenous cations Ca2+, Cu2+and Zn2+ (logKML = 9.3–14.4), thus preventing transmetallation reactions. At physiological pH (7.4), the Eu(III) complex has a conditional stability constant (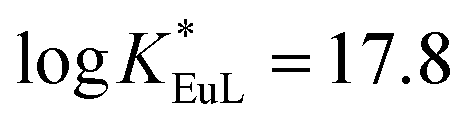 ), higher than that of Eu-DOTA (
), higher than that of Eu-DOTA (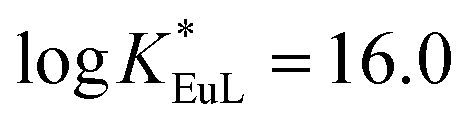 ).13 Interestingly, the use of this tripyridinophane platform results also in more stable lanthanide complexes in aqueous solutions than those based on a tris-picolinate-triazacyclononane core (
).13 Interestingly, the use of this tripyridinophane platform results also in more stable lanthanide complexes in aqueous solutions than those based on a tris-picolinate-triazacyclononane core (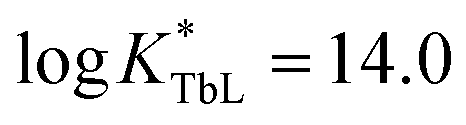 at pH 7.4).14 At the maximum excitation wavelength, the Eu(III) and Tb(III) complexes display calculated Φ × ε values of 570 and 3600 M−1 cm−1 in water solution, respectively, above the lower limit of 300 M−1 cm−1, required for emissive lanthanide complexes acting as tags for bioassays or optical probes.15 However, the major limitation of these lanthanide complexes is the excitation wavelength which corresponds to the absorption band of a pyridine ring (λmax = 267 nm). Lower excitation energy is needed to minimize potential damage to biological media and the inner filter effect. To overcome this weakness, we have set out to prepare a ligand based on the pyridinophane core tpptac where the 4-position of the three pyridine rings is substituted by a phenylethynyl group. From the literature data, the 4-(phenylethynyl)pyridine moiety is a valuable chromophore for photosensitizing the europium ion. This antenna has been introduced in various linear ligands like EDTA16 and dipicolinic acid17 or macrocyclic ligands like cyclen (1,4,7,10-tetraazacyclododecane),18 tacn (1,4,7-triazacyclononane)19 or more recently pyclen (1,4,7,10-tetraaza-2,6-pyridinophane)20 platforms bearing pendant picolinate arms. In this work, we report the incorporation of such an antenna in the tpptac platform. These ligands are shown in Fig. 1. Two antennae are decorated at their extremity by anionic (sulfonate moieties) or neutral (zwitterionic sulfobetaine or glucose moieties) hydrosolubilizing fragments, in order to mask the hydrophobic nature of the three aryl-alkynyl chromophores. The overall charge of the complex plays an important role in limiting non-specific interactions with biological surroundings or ensuring cellular permeabilization. Moreover, a reactive amino functionality was incorporated at the extremity of the third antenna for conjugation purposes. In the final labeling complex, the activation of this amino functionality can be achieved by a direct transformation to the isothiocyanato group or by an indirect transformation to maleimide or N-hydroxysuccinimide-ester with the use of heterobifunctional reagents.
at pH 7.4).14 At the maximum excitation wavelength, the Eu(III) and Tb(III) complexes display calculated Φ × ε values of 570 and 3600 M−1 cm−1 in water solution, respectively, above the lower limit of 300 M−1 cm−1, required for emissive lanthanide complexes acting as tags for bioassays or optical probes.15 However, the major limitation of these lanthanide complexes is the excitation wavelength which corresponds to the absorption band of a pyridine ring (λmax = 267 nm). Lower excitation energy is needed to minimize potential damage to biological media and the inner filter effect. To overcome this weakness, we have set out to prepare a ligand based on the pyridinophane core tpptac where the 4-position of the three pyridine rings is substituted by a phenylethynyl group. From the literature data, the 4-(phenylethynyl)pyridine moiety is a valuable chromophore for photosensitizing the europium ion. This antenna has been introduced in various linear ligands like EDTA16 and dipicolinic acid17 or macrocyclic ligands like cyclen (1,4,7,10-tetraazacyclododecane),18 tacn (1,4,7-triazacyclononane)19 or more recently pyclen (1,4,7,10-tetraaza-2,6-pyridinophane)20 platforms bearing pendant picolinate arms. In this work, we report the incorporation of such an antenna in the tpptac platform. These ligands are shown in Fig. 1. Two antennae are decorated at their extremity by anionic (sulfonate moieties) or neutral (zwitterionic sulfobetaine or glucose moieties) hydrosolubilizing fragments, in order to mask the hydrophobic nature of the three aryl-alkynyl chromophores. The overall charge of the complex plays an important role in limiting non-specific interactions with biological surroundings or ensuring cellular permeabilization. Moreover, a reactive amino functionality was incorporated at the extremity of the third antenna for conjugation purposes. In the final labeling complex, the activation of this amino functionality can be achieved by a direct transformation to the isothiocyanato group or by an indirect transformation to maleimide or N-hydroxysuccinimide-ester with the use of heterobifunctional reagents.
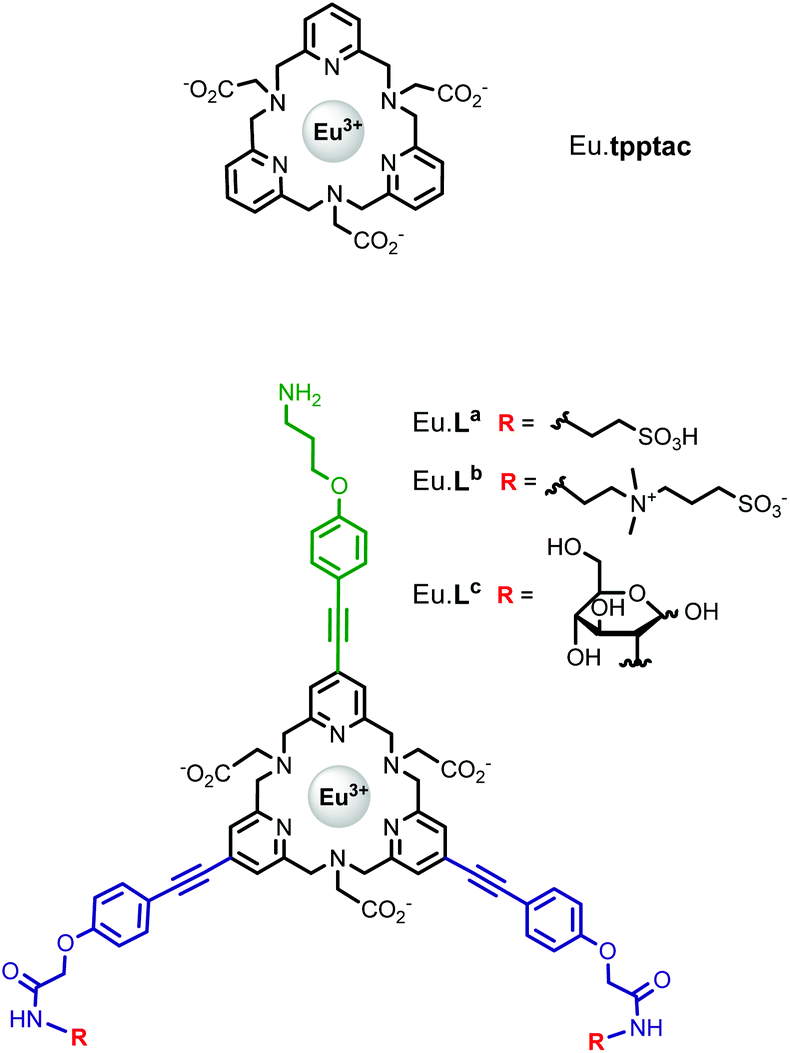 | ||
| Fig. 1 Structures of the studied Eu·tpptac and europium(III) complexes Eu·La, Eu·Lb, and Eu·Lc. | ||
In this contribution, we report the synthesis route of three new bifunctional chelates and the luminescence properties and relative stability of their Eu(III) complexes in aqueous solutions. We also describe the bioconjugation of a Eu(III) complex labeled by an NHS reactive group with an antibody and the application of the corresponding conjugate in the detection of GST-biotin in a HTRF® assay.
Results and discussion
Synthesis
The synthesis of target ligands La, Lb and Lc requires that one of the phenyl alkynyl moieties can be distinguished from the other two. In this regard, our synthetic pathway used for the preparation of the parent tpptac ligand is well adapted.21 This strategy involves the synthesis of a key multifunctionalized 18-membered macrocycle and the prior preparation of monopyridine and dipyridine building blocks as shown in Scheme 1.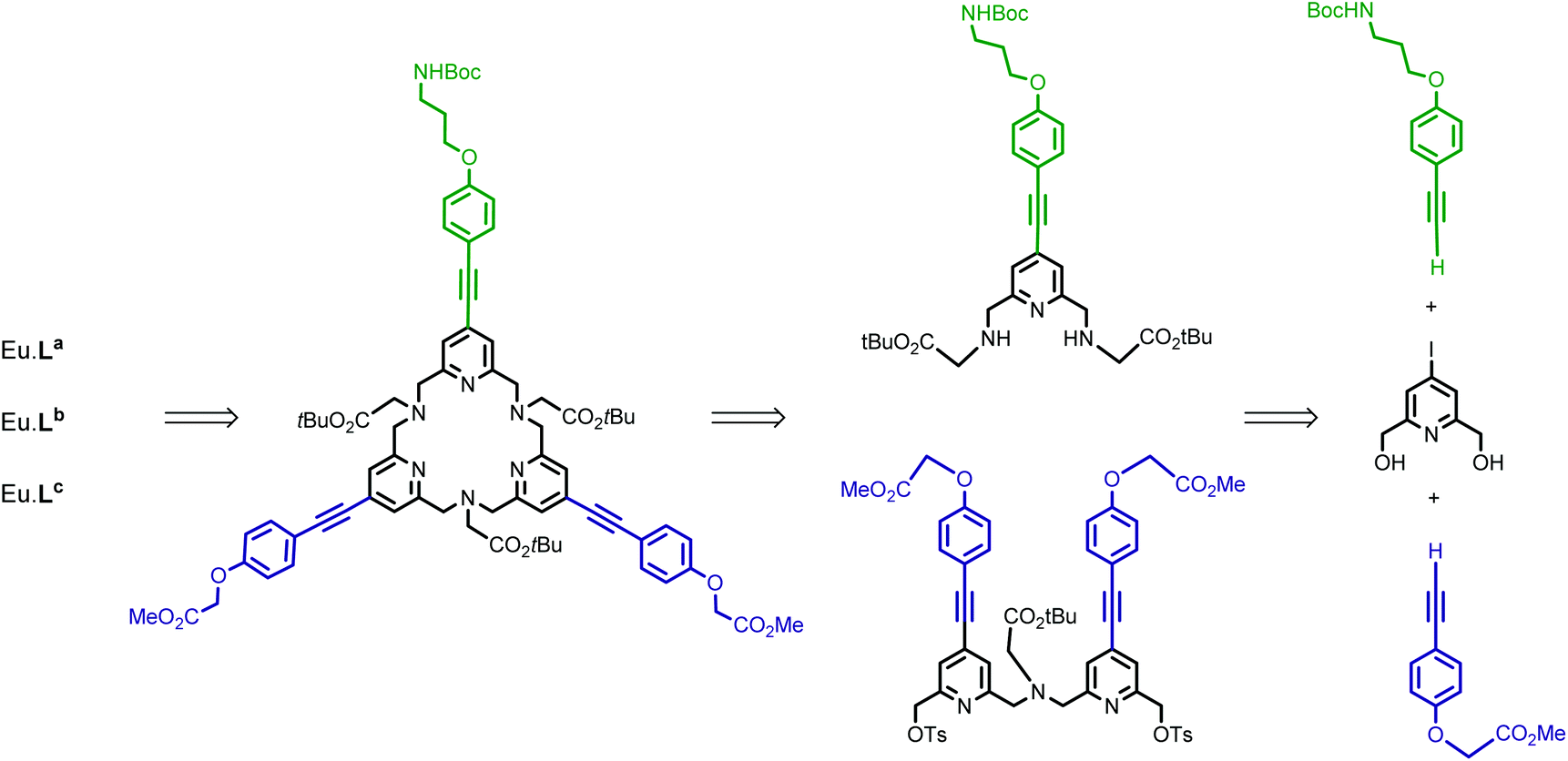 | ||
| Scheme 1 Retrosynthesis of target europium complexes Eu·La, Eu·Lb, and Eu·Lc. | ||
The 4-iodo-2,6-pyridinedimethanol derivative 1 is the common starting material for the preparation of the two building blocks 6 (Scheme 2) and 13 (Scheme 3) and is readily available on a multigram scale and in three steps by following literature procedures (47% overall yield from commercially available chelidamic acid).22 In compound 1, alcohol functionalities can be converted into more reactive groups as bromide or sulfonate ester and the 4-position of pyridine carries an iodo leaving group, well suitable for the introduction of an aryl alkynyl moiety by a Pd-catalyzed cross-coupling reaction.
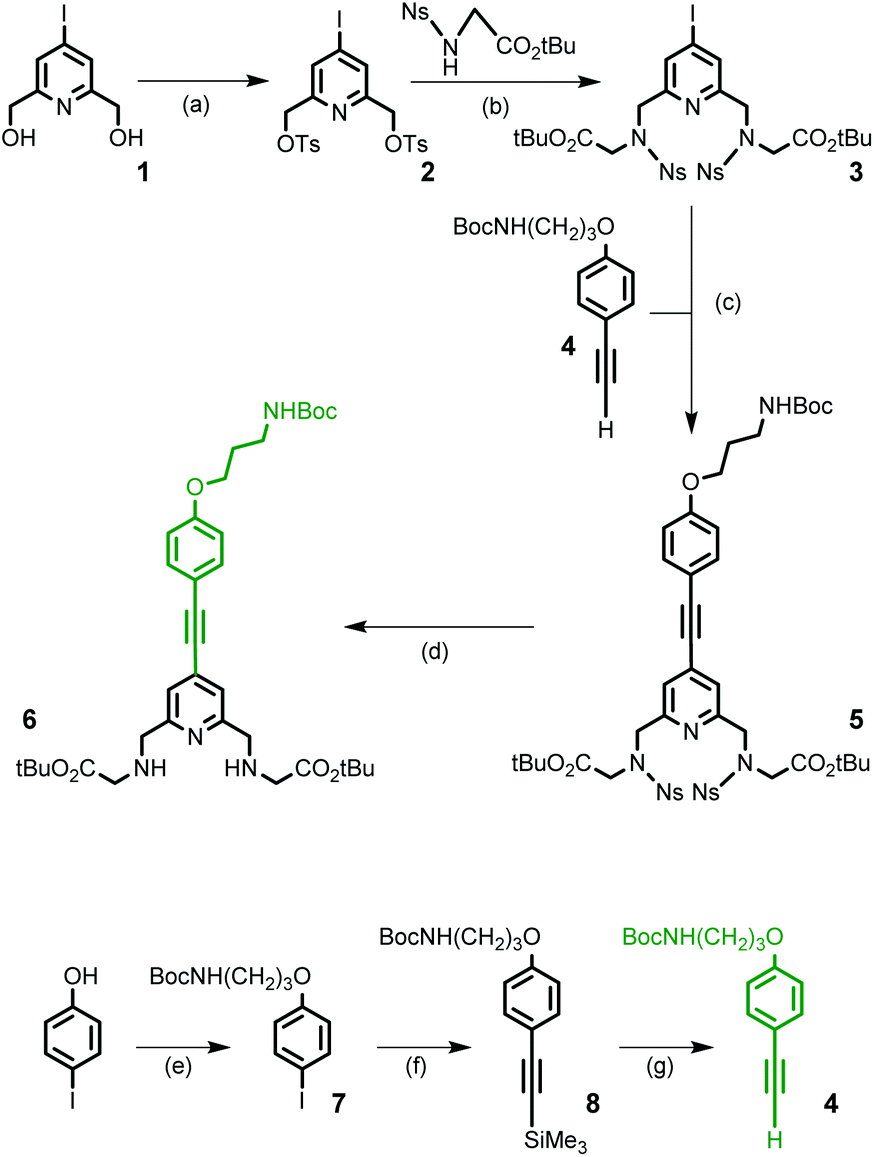 | ||
| Scheme 2 Synthesis of building block 6 and derivative 4. Reagents and conditions: (a) TsCl, NaOH, H2O, THF, rt, 20 h, 84%; (b) Na2CO3, MeCN, reflux, 72 h, 89%; (c) Pd(dppf)Cl2, CuI, Et3N, THF, 60 °C, 20 h, 57%; (d) HOCH2CH2SH, DBU, MeCN, rt, 45 min, 55%; (e) Br(CH2)3NHBoc, Cs2CO3, NaI, CH3CN, 60 °C, 24 h, 85%; (f) ethynyltrimethylsilane, Pd(PPh3)Cl2, CuI, Et3N, THF, MW (100 W), 30 min, 99%; and (g) K2CO3, MeOH, rt, 24 h, 98%. | ||
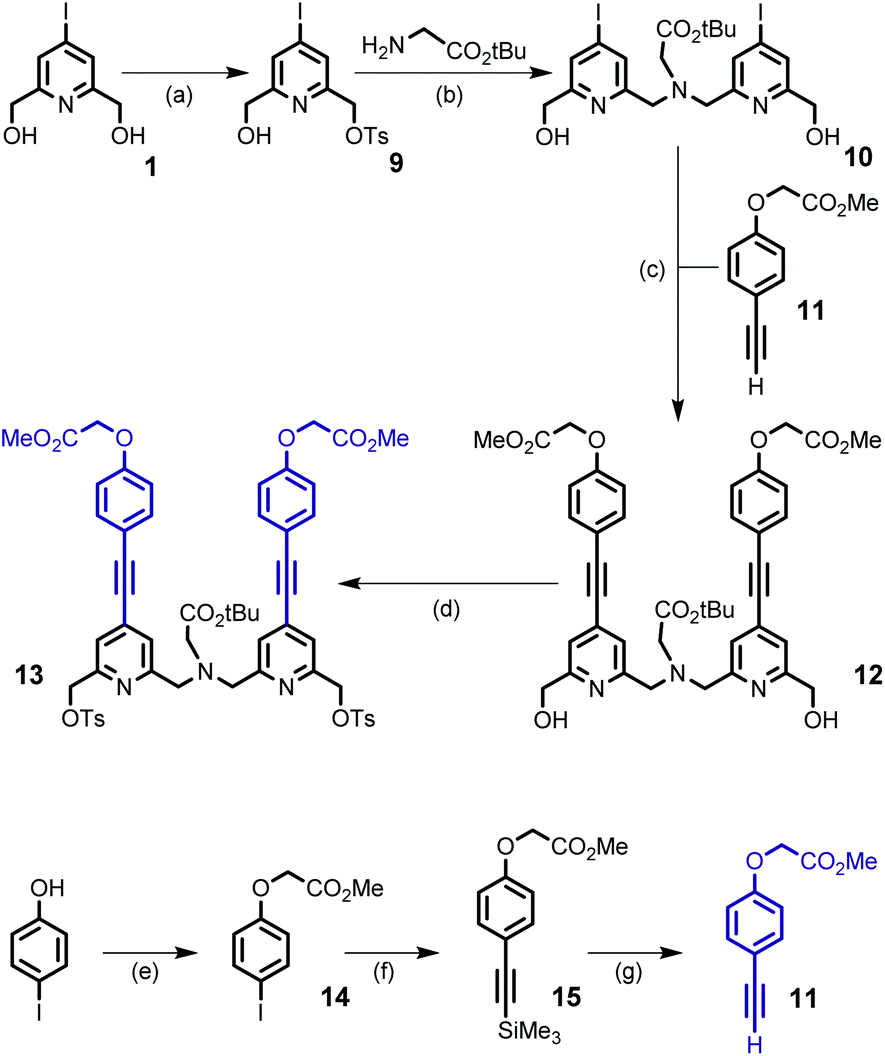 | ||
| Scheme 3 Synthesis of building block 13 and derivative 11. Reagents and conditions: (a) TsCl, Ag2O, KI, CH2Cl2, rt, 16 h, 44%; (b) K2CO3, CH3CN, 60 °C, 20 h, 90%; (c) Pd(dppf)Cl2, CuI, THF, Et3N, MW (100 W), 30 min, 71%; (d) TsCl, Ag2O, KI, CH2Cl2, rt, 16 h, 88%; (e) BrCH2CO2Me, K2CO3, acetone, 50 °C, 12 h, 97%; (f) ethynyltrimethylsilane, Pd(dppf)Cl2, CuI, THF, Et3N, 65 °C, 12 h, 65%; and (g) K2CO3, CH2Cl2, MeOH, rt, 1 h, 83%. | ||
For the synthesis of the monopyridine building block 6, a four-step procedure was devised (Scheme 2). Ditosylation of diol 1 followed by alkylation of the nosyl derivative of glycine tert-butyl ester (2.2 equivalents) in the presence of Na2CO3 as a base led to the formation of the 4-iodo-pyridyl compound 3 in good yield. In parallel, the (4-substituted)phenylethynyl fragment 4, containing a masked primary amino group that will ensure the conjugation process, was prepared in three steps from 4-iodo-phenol. Using a series of O-alkylation reaction, Sonogashira coupling and deprotection of the intermediate silylated alkyne, compound 4 was obtained in an overall yield of 82% and on a multigram scale. A Sonogashira cross-coupling reaction was then performed between 3 and 4. This reaction was carried out in degassed THF and in the presence of the classic catalytic system (Pd(dppf)Cl2/CuI) and afforded the alkyne derivative 5 in 57% yield. The cleavage of nosyl groups was then achieved by using 2-mercaptoethanol in the presence of DBU at room temperature to give the target compound 6 in moderate 55% yield. As a matter of fact, two side-products were isolated during this deprotection step and identified by NMR and mass spectrometry data as thioalkene derivatives. These side reactions are time-dependent. This is the result of nucleophilic monoaddition of the thiol functionality of 2-mercaptoethanol on the acetylenic bond. The thiol–yne reaction catalyzed by the base DBU at room temperature has been previously reported.23
The preparation of the second building block 13 is depicted in Scheme 3. Monotosylation of symmetrical diol 1 was performed by the use of the TsCl/Ag2O/KI system and without the use of an excess of diol as described for the non-halogenated diol.21 The conjugated dipyridine derivative 12 was then obtained by N,N-dialkylation of glycine tert-butyl ester with monotosylate ester 9 (2 equivalents) followed by a double Sonogashira coupling reaction with the alkyne derivative 11 containing an ester group as the starting point for the introduction of hydrosolubilizing moieties. Compound 11 was obtained from 4-iodo-phenol and in a similar sequence to that of 4 (overall yield 52%). For the double Sonogashira coupling reaction, the use of microwave activation afforded the coupling product 12 in 30 min and in an appropriate yield (71%). After purification by column chromatography on silica gel, we can note that the target compound 12 was isolated in two separate forms: free species 12 and copper complex Cu·12 which gave 12 after treatment with an aqueous solution of EDTA. The initial experiments revealed that diol 12 could not be properly and efficiently transformed into its dibromide counterpart, either in the presence of PBr3 or NBS/PPh3 (yield < 20%). So, we turned our attention to the preparation of the ditosylate derivative 13 which was isolated in 88% yield after purification by column chromatography.
The two units 6 and 13 were then assembled on the basis of our recently published macroring process implementing an efficient sodium template ion effect (Scheme 4).21 The crucial macrocyclization step was carried out in CH3CN under reflux in the presence of sodium carbonate (10 equivalents) as a base and NaI (0.1 equivalent) in a batchwise procedure (reagent concentration = 2 × 10−3 M). The sodium complex [Na·16]+ was evidenced by NMR and MS analyses as the major species of the crude reaction mixture and the free macrocycle 16 was obtained in 74% yield after purification by column chromatography, as a result of alumina-mediated dissociation. This could be expected because of the soft character of N-donor atoms towards sodium ions. Besides, it is worth noting that treatment of the free ligand 16 with Na2CO3 in CH3CN under reflux afforded a mixture of the free ligand and Na·16 complex. It should also be noted that these sodium and free species can be readily distinguished by their 1H NMR spectra (see the Experimental section). The next step was a selective cleavage of the methyl ester groups appended on the pyridine subunits. This was performed by base-catalyzed hydrolysis using lithium hydroxide in a mixture of CH3CN/H2O. The following syntheses were carried out on a small scale (μmol range) and therefore the purity of compounds 18a, 18b, and 18c and complexes Eu·La, Eu·Lb and Eu·Lc was established on the basis of HPLC analysis and their structures by high-resolution mass spectrometry (HRMS) analysis.
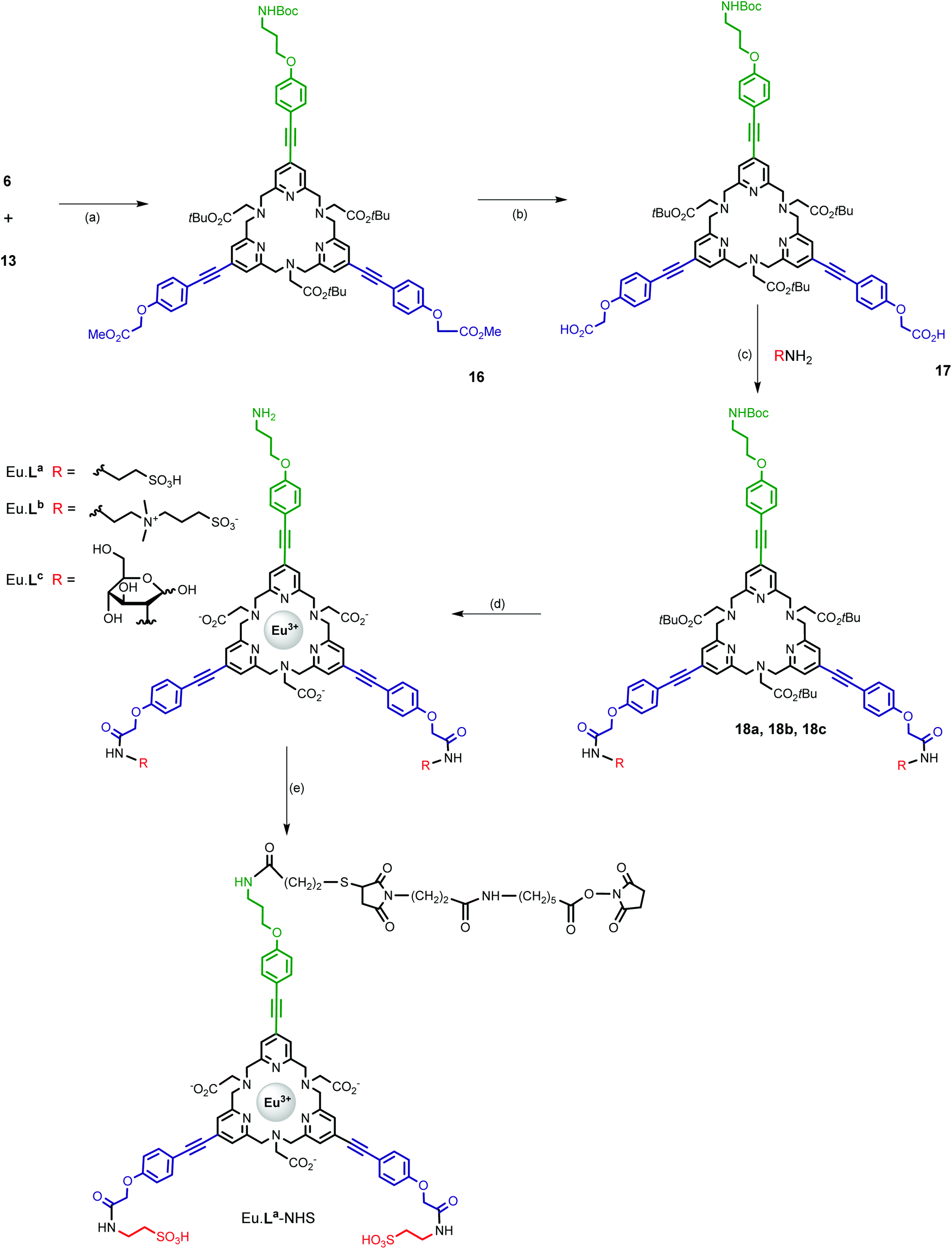 | ||
| Scheme 4 Synthesis of complexes Eu·La, Eu·Lb, Eu·Lc and Eu·La-NHS. Reagents and conditions: (a) Na2CO3, NaI, MeCN, reflux, 40 h, 74%; (b) LiOH (1 M), CH3CN, rt, 1 h, 60%; (c) HATU, DIPEA, RNH2, DMSO, rt, 10–40 min, 76% (18a), 60% (18b), 87% (18c); (d) (i) TFA, CH2Cl2, rt, 1 h, (ii) EuCl3·6H2O, aqueous NaOH pH 6, rt, 12 h, 38% (Eu·La), 41% (Eu·Lb), 18% (Eu·Lc); and (e) (i) SPDP, DIPEA, DMSO, rt, 1 h, 100%, (ii) TCEP, TEAA, H2O, rt, 1 h, 100%, and (iii) SMPH, TEAA, DMSO, rt, 30 min, 34%. | ||
The different hydrosolubilizing moieties (RNH2, Scheme 4) were then introduced by the formation of amide bonds using the HATU/DIPEA system in DMSO with acceptable yields (60–85%), after purification by preparative HPLC. The final europium complexes were prepared in two subsequent steps: (i) hydrolysis of the tert-butyl ester functionality of the tpptac core and deprotection of the BOC group by TFA and (ii) complexation of the corresponding triacid compounds with EuCl3·6H2O salt in aqueous solution at room temperature and pH 6. These complexes were isolated by preparative HPLC. They exhibit good water solubility allowing the determination of their photophysical properties in various aqueous buffers.
In order to conjugate these Eu(III) complexes with biological materials, we investigate the conversion of Eu·La into the Eu·La-NHS derivative, bearing an N-hydroxysuccinimide ester moiety, reactive towards the amino groups of ε-lysine residues present in proteins and antibodies. Our synthetic pathway involved the use of two heterobifunctional reagents that contain at opposite sites NHS ester and pyridyl disulfide functionalities (SPDP) or maleimide and NHS ester reactive groups (SMPH), and thus capable of two distinct sequential reactions. Eu·La-NHS was obtained in a three step protocol: (i) amidation of the amino group of Eu·La with SPDP, (ii) reduction of the disulfide bond with TCEP, and (iii) introduction of an NHS ester moiety by the formation of a thioether bond with SMPH. For each step, the product was purified by RP-HPLC and an overall yield of 34% was obtained for Eu·La-NHS.
Photophysical properties
First, the photophysical properties of the europium complex, Eu·La, were evaluated in hydrogenated and deuterated aqueous buffer (air equilibrated Tris buffer, 50 mM, pH 7.4) and are presented in Table 1| Solventa,b | λ max (nm) | τ (ms) |
Φ
ovd (%)c |
I F2/IF1e | |
|---|---|---|---|---|---|
| a Subscripts H and D refer to hydrogenated and deuterated media, respectively. b Tris (50 mM, pH 7.4), phosphate (10 mM, pH 7.4), and HEPES (50 mM, pH 7.4). c Determined by excitation into the lowest-energy ligand centred absorption band and recording the intensity of the 5D0 → 7F2 transition (620 nm). d Overall quantum yields were determined under ligand excitation. e Intensity ratio of the 5D0 → 7F2 and 5D0 → 7F1 transitions of the corrected emission spectrum. f Data from ref. 12. | |||||
| Eu·La | TrisH | 322 | 0.68 | 14.0 | 5.7 |
| TrisD | 322 | 1.58 | 30.0 | 5.8 | |
| TrisD (77 K) | 1.59 | ||||
| Phosphate | 320 | 0.65 | 12.5 | 5.75 | |
| HEPES | 321 | 0.68 | 14.5 | 5.8 | |
| H2O | 320 | 0.66 | 14.0 | 5.7 | |
| CH3OH | 322 | 0.97 | 21.0 | 5.1 | |
| CH3CN | 317 | 1.69 | 42.0 | 4.3 | |
| DMF | 324 | 1.38 | 44.0 | 5.3 | |
| Eu·Lb | TrisH | 321 | 0.69 | 13.0 | 5.7 |
| TrisD | 1.59 | 40 | |||
| Eu·Lc | TrisH | 321 | 0.71 | 14.7 | 5.7 |
| TrisD | 1.62 | 38 | |||
| Eu·tpptacf |
TrisH | 267 | 0.78 | 4.8 | 3.0 |
| TrisD | 267 | 1.99 | 11.8 | ||
In this buffer, the UV-vis absorption spectrum of the Eu·La complex shows a large absorption band centred at 322 nm (λcut-off = 355 nm) characterized by a high molar absorption coefficient (ε = 72000 M−1 cm−1) and no shoulder was observed at lower energy (Fig. 2). By comparison with the parent pyridine complex, Eu·tpptac, the substitution of the three pyridine sensitizing moieties with phenyl-ethynyl groups results in a significant redshift of the maximal absorption band (Δλ = 55 nm) and cut-off wavelength (Δλ = 70 nm), and an increase of the epsilon value by a factor of 6. No support of the assignment of this band at 322 nm as an ILCT transition was evidenced by a solvatochromic study. We do not observe a significant variation of this band with solvents of decreasing polarity: λ(H2O) = 320 nm and λ(DMF) = 324 nm (Table 1). In this direction, Parker et al. reported a hypochromic shift of 24 nm from H2O to DMF, in relation to an absorption band of a similar antenna with ILCT character.24
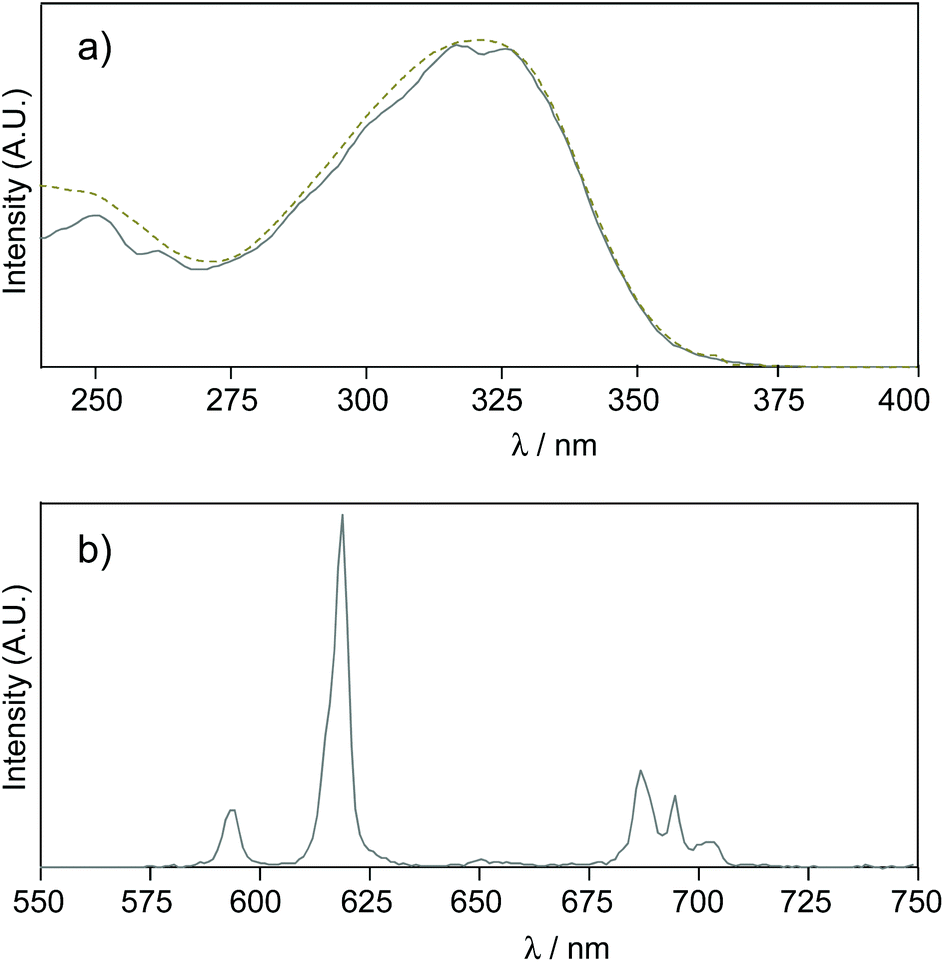 | ||
| Fig. 2 (a) Normalized UV-vis (solid line) and corrected excitation (dashed line) spectra (λem = 619 nm) of Eu·La. (b) Corrected emission spectrum of Eu·La (λexc = 322 nm, excitation and emission bandpasses: 5 nm). These spectra are recorded in Tris buffer (50 mM, pH 7.4) at 298 K at a concentration of 10 μM. | ||
At room temperature, the emission spectrum of the complex shows typical narrow-band europium luminescence in the 580–720 nm range after excitation into the ligand absorption band at 322 nm (Fig. 2). The emission spectrum is characterized by five bands due to 5D0 → 7FJ (J = 0–4) transitions, with relative intensities of 0.03, 1, 5.7, 0.3 and 3.1 using the J = 1 magnetic dipole transition as the internal reference. The hypersensitive 5D0 → 7F2 transition centred at 619 nm and the sensitive 5D0 → 7F4 transition at around 690 nm are dominant and account for ∼56% and 31% of the total emission intensity, respectively. Interestingly, these two electric dipole transitions are considered to be efficient donating transitions in TR-FRET experiments, while the magnetic dipole transition (5D0 → 7F1) is not regarded as the resonance partner.25 On the other hand, the introduction of three aryl-alkynyl groups in Eu·La perturbs the coordination environment around the Eu(III) center. The intensity ratio of the hypersensitive 5D0 → 7F2 transition and the magnetic dipole 5D0 → 7F1 transition (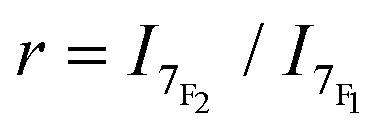 ) is higher in Eu·La (r = 5.7) than in Eu·tpptac (r = 3.0). This ratio is highly dependent on both the dipolar polarizabilities of the ligand atoms or groups and coordination geometry (symmetry on the coordination sphere of the europium ion).26 The excitation spectrum monitored at each of the emission bands is located on the absorption range of the complex and has a similar shape as the absorption spectrum, consistent with an antenna effect.
) is higher in Eu·La (r = 5.7) than in Eu·tpptac (r = 3.0). This ratio is highly dependent on both the dipolar polarizabilities of the ligand atoms or groups and coordination geometry (symmetry on the coordination sphere of the europium ion).26 The excitation spectrum monitored at each of the emission bands is located on the absorption range of the complex and has a similar shape as the absorption spectrum, consistent with an antenna effect.
The luminescence lifetime of the complex was measured at both ambient (298 K) and low (77 K) temperatures. The decay curves of the luminescence from the 5D0 state were well described by fitting them to a single exponential decay equation. In TrisH buffer, the lifetime is relatively long (τ = 0.68 ms) but shorter than in TrisD buffer (τ = 1.58 ms), reflecting the quenching contribution of proximate OH oscillators (inner- and outer-sphere water molecules). Using both the Horrocks methodology and Supkowski refined equation taking into account the quenching contribution of outer-sphere water molecules, the number of water molecules in the primary coordination sphere of Eu(III) is estimated to be q = 0.58.27 Given the typical error related to this approach (±0.2), this non-integer value points to a hydration equilibrium between mono- and non-hydrated species. A similar non-integer value (q = 0.52) was also obtained for the parent non-functionalized europium complex, Eu·tpptac, which presents a hydration equilibrium involving a monohydrated species (q = 1) with a coordination number of 10 and a 9-coordinated nonhydrated (q = 0) species.12 Similar hydration equilibrium was also reported for some europium complexes derived from pyclen ligands bearing pendant picolinate arms.20a,28 The lack of any temperature dependence of the lifetime in TrisD buffer indicates that no back energy transfer between the ligand donor excited state and the acceptor excited state on the europium ion or alternately no quenching via the low LMCT state, commonly observed for Eu(III) complexes, occurs. Sensitized emission quantum yields, Φov = 14% in TrisH buffer and Φov = 30% in TrisD buffer, were also determined at pH 7.4 using [Ru(bpy)3]Cl2 as a reference. The higher quantum yield in TrisD buffer confirms that the quenching process due to O–H vibrations is operative in this complex. These quantum yields are higher than those of Eu·tpptac and in the range of those reported in aqueous solutions (Φ = 6–31%) for europium complexes derived from tris-picolinate TACN ligands featuring a similar alkoxy phenyl ethynyl substituent on the 4-pyridyl ring.19a,29 The Eu·La complex is thus characterized by a significant brightness following excitation at 330 nm (B = 9600 M−1 cm−1), an excitation wavelength compatible with a N2 laser commonly used in HTRF assays. The photophysical properties of Eu·La were also examined in organic solvents (Table 1). The increase of τ and Φov values going from H2O to MeOH is in line with the lower efficiency of MeOH in inducing nonradiative deactivation processes via O–H groups. This is based on the assumption that MeOH behaves like half a water molecule. As expected, the lifetime and overall quantum yields are higher in CH3CN and DMF. To understand the limitation and the advantage of a ligand to photosensitize a lanthanide ion, two parameters of interest to be determined are the intrinsic quantum yield (ΦEu) and the sensitization efficiency (ηsens), which take into account the stepwise pathway of the antenna effect. The intrinsic quantum yield represents the efficiency of the radiative deactivation of the lanthanide ion relative to all its deactivations. The sensitization efficiency represents both the efficiency with which the donor state of the ligand is populated by the initially excited state and the efficiency of the energy transfer from the donor state to the accepting Ln(III) ion.
The combined efficiency of these two steps corresponds to the overall quantum yield of the complex (Φov = ΦEu × ηsens). The parameter ΦEu was determined according to a well-established protocol and by using the following equations:30
| ΦEu = kr/(kr + ∑knr) = τobs/τR | (1) |
| 1/τR = kr = 14.65 × n3 × (I∑Fj/IF1) | (2) |
| Eu·La (TrisH) | Eu·La (TrisD) | Eu·tpptaca (TrisH) |
|
|---|---|---|---|
| a Data from ref. 12. b Experimentally measured parameters. c For calculated parameters, see the text. d ∑knr = kobs − kr. | |||
|
I
∑Fj/IF1b |
10.1 | 10.3 | 6.7 |
| k r (s−1)c | 349 | 354 | 230 |
| k obs (s−1)b | 1470 | 633 | 1282 |
| Φ Eu (%)c | 24 | 56 | 18 |
| η sens (%)c | 59 | 54 | 27 |
| Φ ov (%)b | 14 | 30 | 4.8 |
| ∑knr (s−1)c,d | 1121 | 279 | 1052 |
We have also determined the photophysical properties of Eu·Lb and Eu·Lc complexes bearing neutral sulfobetaine and glucoside moieties (Table 1; see also Fig. S36, ESI†). These data indicate that variation of the peripheral solubilising groups does not perturb optical parameters (λ, ε, τ and Φov) and the hydration state value (q) of the europium complex. Similar metal centered luminescence and sensitization efficiencies (ΦEu and ηsens) are also calculated (see Table S1, ESI†).
Relative stability
The emission properties of Eu·La remained unchanged in aerated Tris buffer after several days at room temperature, highlighting its kinetic inertness in this medium. Similar photophysical properties (Table 1) and kinetic stability were observed in 50 mM HEPES (pH 7.3) and 10 mM phosphate (pH 7.4) buffers, indicating that Eu·La is also operative in these buffers that are often used in biological applications. We can thus note that the presence of phosphate groups, which are ubiquitous in biological media and also have strong affinity for Ln(III) ions, does not interfere with the luminescence properties of this complex at ratios up to 106:1. Nevertheless, the evaluation of a new europium complex as a potential luminescent bioprobe should also focus on its behaviour in different environments, for example, in the presence of cations, anions or competing ligands. This sort of study is often overlooked in reports claiming that the described lanthanide complexes are effective luminescent probes for biological applications. In this direction, we turned to luminescence to check the relative stability of the Eu·La complex with respect to common physiological ions (Na+, K+, Mg2+and Ca2+) or metal ions needed for certain types of enzyme activity (Mn2+). In the experiments (see Fig. 3), the complex was present at 1 nM concentration in HEPES buffer (pH 7.4), containing 0.1% bovine serum albumin, which corresponds to the experimental conditions appropriate for bioassays, and a great excess of disturbing species was used (≥20 mM).
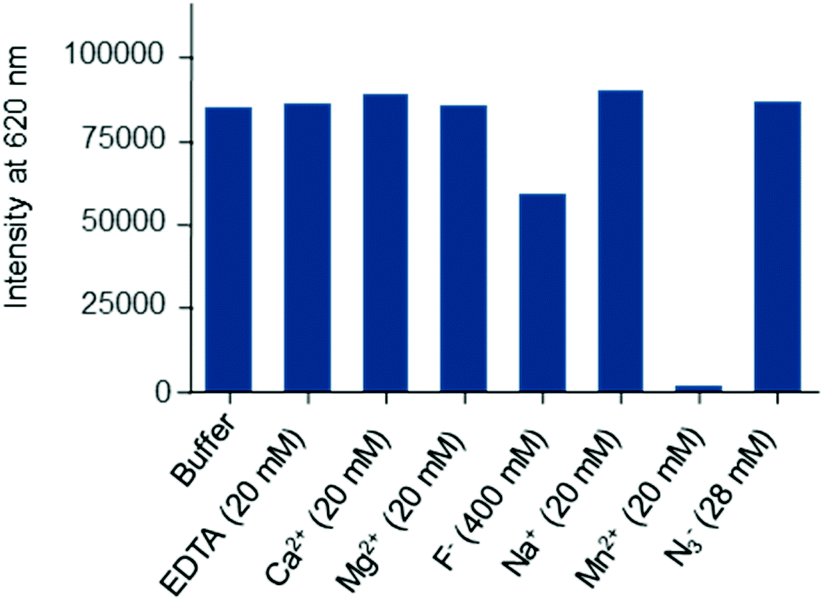 | ||
| Fig. 3 Stability assessment: changes of the europium intensity of Eu·La (recorded at λem = 620 nm) after 18 h incubation in the presence of the stated additives. The complex was used at a concentration of 1 nM in 0.05 M HEPES and 0.1% BSA, pH 7.4. | ||
Except for Mn2+, these ions do not have any effects on Eu·La luminescence. When Eu·La was challenged with Mn2+, a complete luminescence quenching was observed. Given that the parent ligand tpptac showed affinity for Mn2+several orders of magnitude lower than for Eu3+ (logKMn = 13.0 and logKEu = 18.7), we can assume that this quenching is not due to a transmetallation process. The quenching effect is probably related to the dynamic energy transfer or possibly oxidation of Mn2+ ions by the excited state of the chromophore.11b,31 In parallel, the sensitivity of Eu·La to a fluoride ion was assessed, since this hard anion may have a strong influence on lanthanide luminescence.32 In the presence of 0.4 M KF, we observe some perturbations in the excited state lifetime and shape of the emission spectrum, but no change in the overall luminescence intensity. The lifetime was characterized by a 65% increase, but does not reach the value obtained in D2O, and the intensity at 619 nm by a 30% decrease. This may arise from a partial substitution of coordinated water molecules by fluoride ions and a subsequent change of the symmetry of the complex. We observed a significant ratio change between the 613 and 619 nm bands within the 5D2 manifold. Scission of the 5D1 band is also observed upon binding with F− (Fig. S37, ESI†). On the other hand, Eu·La is not susceptible to quenching by sodium azide. This salt is commonly used as a bacteriostatic agent in biological solutions and the quenching effect of N3− anions is well documented.33 Finally, the chelator EDTA was chosen as a ligand competitor for the transchelation experiment, since this common chelator binds strongly to Eu3+ and is largely used in bioscreening assays to stop the enzymatic activity and to minimize the disturbing metal ion concentration. After overnight incubation, no change in the photophysical parameters was observed, consistent with a high kinetic inertness of this complex with respect to dissociative ligand exchange. These results highlight the potential of these europium complexes in biological applications.
HTRF immunoassay
To assess whether this new series of europium chelates can be used in time-gated HTRF assays, we chose to evaluate the N-hydroxysuccinimide (NHS)-ester of [Eu·La] in a ubiquitous assay format to detect the glutathione-S-transferase (GST)-protein. The GST-protein is used in recombinant protein production and purification and can subsequently be used for instance to design protein–protein or protein–ligand in vitro assays using it as a tag. The presence of the protein of interest can then be detected using dye labeled anti-GST antibodies. In the sandwich assay designed here (Fig. 4), we wanted to detect biotinylated-GST using an anti-GST labeled [Eu·La] and a cyanine d2 labeled streptavidin (SA-d2).3b,34 The labelling of biotinylated-GST with Eu·La-NHS was performed following the standard protocol described in the Experimental section. An average of 12 complexes per conjugate was estimated from the relative intensity of antibody and complex absorptions.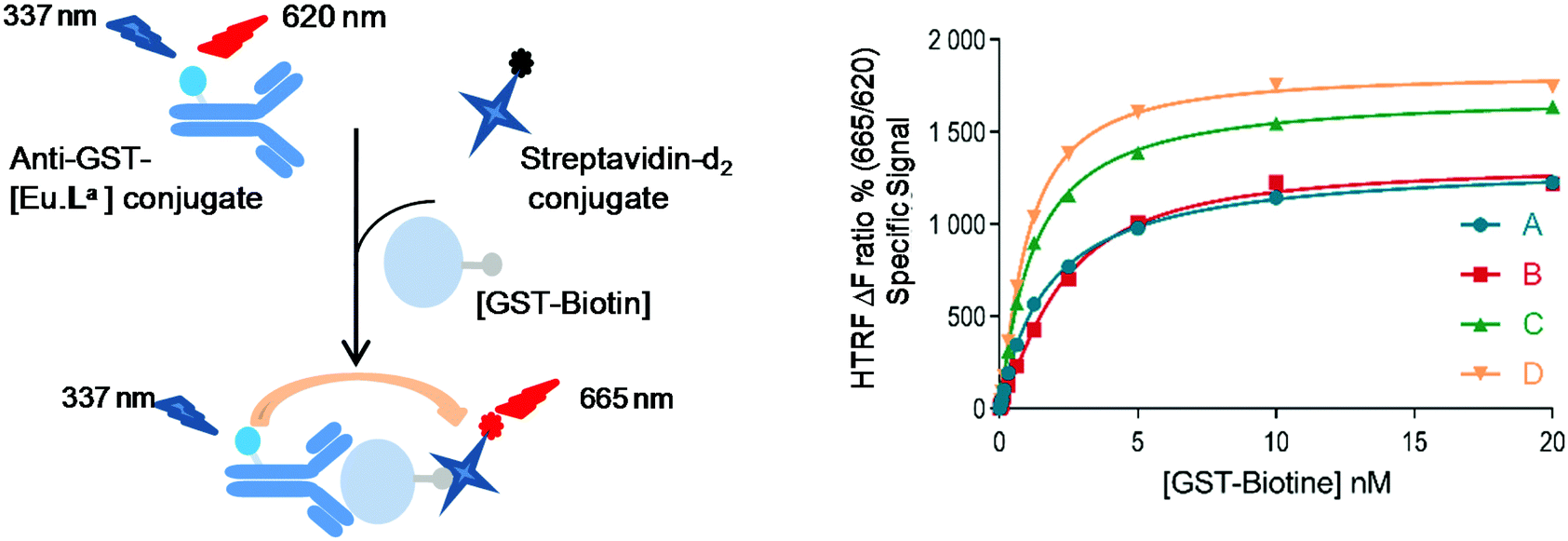 | ||
| Fig. 4 HTRF detection of GST-biotin using anti-GST[Eu·La] and SA-d2 conjugates in 0.05 M HEPES 0.1% BSA, pH 7.4 at (A) 0.5 h, (B) 0.5 h (+0.4 M KF), (C) 20 h, and (D) 20 h (+0.4 M KF) of incubation. ΔF% = [(ratio sample − ratio negative control)/ratio negative control] × 100, with ratio = I665 nm/I620 nm × 104. | ||
In Fig. 4, the assay performance of anti-GST-[Eu·La] is shown in the presence or not of 0.4 M of KF at 30 min and 20 h at different GST-biotin concentrations using 0.25 nM anti-GST-[Eu·La] and 5 nM streptavidin-d2. The assay response behaves linearly up to a concentration of ca. 5 nM and then due to the limited concentration of SA-d2 saturates. The curves are fitted with four-parameter logistic (4PL) regression. In both the presence and absence of F−, the analyte is detected from 40 pM upwards in accord with other HTRF assays. A maximum assay window up to 18 at 10 nM GST-biotin in the presence of 0.4 M KF is observed which is slightly higher than that in the absence of KF. The assay equilibrium is not reached at 30 min under both conditions, because the assay performance especially at lower concentrations is enhanced overnight. The slight difference of the response at concentrations of GST-biotin >2.5 nM of curve C compared to curve D is most probably due to a significant change in ionic force. This can change the antibody–antigen affinity, leading to a change in the TR-FRET response.
Conclusions
To satisfy as much as possible the numerous requirements necessary to obtain a europium probe suitable for fluorescent biological assays, we report the study of new multidentate ligands based on a tripyridinophane platform. To that end, the prototype macrocyclic ligand tpptac was functionalized with three π-conjugated pyridine donors decorated with hydrosolubilizing end groups and a bioconjugatable handle. The first part of this work emphasizes the utility of our synthetic approach in more complex structures than those previously reported. In particular, this synthetic pathway is highly versatile and provides high yield in the crucial macrocyclization step. These ligands form anionic or neutral Eu(III) complexes that are soluble in aqueous solutions and their sensitizing properties have been investigated by luminescence spectroscopy in Tris buffer (pH 7.4). The Eu(III) complexes display an efficient energy transfer process (ηsens ≈ 60%) with luminescence lifetimes in the millisecond range and significant overall quantum yields. They also show a brightness in the range of 10000 M−1 cm−1 at 330 nm, a wavelength compatible with the use of flash lamps or nitrogen laser excitation sources, commonly used in bioassays. Under neutral conditions and at common assay concentrations, these complexes are kinetically inert in biological buffers (Tris, HEPES, and phosphate) and in the presence of certain buffer components (EDTA and sodium azide) or most common physiological ions. A reactive handle NHS was used to graft Eu·La to an antibody detecting GST, and the proof of concept of a HTRF immunoassay with these complexes was successfully performed for the detection of GST-biotin in a HTRF® assay. It should be possible to further modify the ligands to optimize or to mix antenna moieties and to incorporate new chemical properties (exclusion of coordinated solvent molecules). This work is currently in progress.
Experimental section
Details of instrumentation, analytical and preparative HPLC systems, and general methods of analysis are given in the ESI.† The syntheses of compounds 4 and 11, NMR and HRMS spectra, and HPLC chromatograms of selected compounds, and luminescence spectra and photophysical data of Eu·Lb and Eu·Lc complexes are also given in the ESI.†Compound 2
To a solution of 4-iodo-2,6-pyridinedimethanol (3.0 g, 11.3 mmol) in THF (10 mL) was added 10 mL of an aqueous solution of NaOH (1.36 g, 34.0 mmol) at room temperature. After cooling to 0 °C, a solution of tosyl chloride (6.47 g, 34.0 mmol) in THF (10 mL) was added and the mixture was stirred for 20 h at room temperature. To this mixture were added CH2Cl2 (100 mL) and brine (50 mL). After decantation, the aqueous layer was extracted with CH2Cl2 (2 × 100 mL). The combined organic layers were dried over Na2SO4, filtered off, and then concentrated under vacuum. The residue was subjected to purification by column chromatography on silica gel (eluent: cyclohexane/CH2Cl2 gradient from 50/50 to 0/100) to yield compound 2 (5.41 g, 84%) as a white solid. Mp. 143.5–144.5 °C. 1H NMR (CDCl3, 400 MHz) δ 7.81 (4H, d, J = 8.2 Hz), 7.64 (2H, s,), 7.36 (4H, d, J = 8.2 Hz), 5.01 (4H, s), 2.47 (6H, s) ppm. 13C NMR (CDCl3, 100 MHz) δ 154.2, 145.4, 132.6, 130.4, 130.0, 128.1, 107.0, 70.4, 21.7 ppm. HRMS (ESI+) m/z calcd for C21H21INO6S2 [M + H]+ = 573.9855, found 573.9853.Compound 3
To a solution of N-(o-nosyl)glycine tert-butylester21 (1.83 g, 5.75 mmol) in dry CH3CN was added dry Na2CO3 (2.77 g, 26.2 mmol), and then the mixture was stirred at 80 °C for 1 h and cooled to room temperature. Ditosylate 2 (1.5 g, 2.62 mmol) was added to the mixture and stirred at 80 °C for 72 h. After cooling to room temperature, the mixture was filtered off and the filtrate was concentrated under vacuum. The crude oil was purified by column chromatography on silica gel (eluent: EtOAc/MeOH from 98/02 to 95/05) to yield compound 3 (2.0 g, 89%) as a yellow oil. 1H NMR (CDCl3, 400 MHz) δ 8.08 (2H, dd, J = 7.6 Hz, 1.6 Hz), 7.75–7.66 (6H, m), 7.60 (2H, s), 4.63 (4H, s), 4.15 (4H, s), 1.39 (18H, s) ppm. 13C NMR (CDCl3, 100 MHz) δ 167.4, 156.4, 147.8, 133.8, 133.5, 131.8, 130.85, 130.45, 124.2, 107.35, 82.6, 52.8, 49.2, 28.0 ppm. HRMS (ESI+) m/z calcd for C31H37IN5O12S2 [M + H]+ = 862.0925, found 862.0922.Compound 5
A solution of compound 4 (2.58 g, 9.37 mmol) and compound 3 (7.36 g, 8.54 mmol) in a mixture of triethylamine (42 mL) and THF (42 mL) was degassed with argon by bubbling for 15 min. To this solution were added Pd(dppf)Cl2·CH2Cl2 (1.39 g, 1.70 mmol) and CuI (0.49 g, 2.57 mmol). The mixture was stirred at 60 °C for 20 h, cooled to room temperature and filtered off over Celite. The filtrate was concentrated under vacuum. The crude product was purified by column chromatography on silica gel (eluent: cyclohexane/CH2Cl2/EtOAc gradient from 50/20/30 to 20/50/30) to yield compound 5 (4.90 g, 57%) as a yellow foam. 1H NMR (CDCl3, 400 MHz) δ 8.10 (2H, dd, J = 7.6 Hz, 1.7 Hz), 7.75–7–65 (6H, m), 7.49 (2H, d, J = 8.7 Hz), 7.32 (2H, s), 6.92 (2H, d, J = 8.7 Hz), 4.76 (1H, bs), 4.68 (4H, s), 4.17 (4H, s), 4.08 (2H, t, J = 6.0 Hz), 3.36 (2H, m), 2.03 (2H, m), 1.47 (9H, s), 1.38 (18H, s) ppm. 13C NMR (CDCl3, 100 MHz) δ 167.4, 159.8, 156.1, 155.8, 147.9, 133.9, 133.65, 133.60, 131.8, 130.9, 124.2, 122.8, 114.7, 113.9, 95.3, 85.5, 82.5, 79.35, 65.9, 53.1, 49.2, 37.9, 29.6, 28.4, 27.9 ppm. HRMS (ESI+) m/z calcd for C47H57N6O15S2 [M + H]+ = 1009.3323, found 1009.3322.Compound 6
To a solution of compound 5 (0.1 g, 0.099 mmol) in CH3CN (4.3 mL) were added 2-mercaptoethanol (76 μL, 1.09 mmol) and 1,8-diazabicyclo[5-4-0]undec-7-ene (DBU, 67 μL, 0.445 mmol), and the reaction mixture was stirred at room temperature. The progress of the reaction was monitored by TLC and after 45 min the mixture was concentrated under vacuum. The residue was purified by column chromatography on silica gel (EtOAc/MeOH, 98/02 and 96/04) to yield compound 6 (35 mg, 55%) as a beige oil. 1H NMR (CDCl3, 300 MHz) δ 7.44 (2H, d, J = 8.8 Hz), 7.29 (2H, s), 6.86 (2H, d, J = 8.8 Hz), 4.80 (1H, bs), 4.02 (2H, t, J = 6.0 Hz), 3.90 (4H, s), 3.36 (4H, s), 3.30 (2H, m), 2.42 (2H, bs), 1.97 (2H, m), 1.45 (18H, s), 1.42 (9H, s) ppm. 13C NMR (CDCl3, 75 MHz, J Mod) δ 171.35 (Cq), 159.4 (Cq), 159.0 (Cq), 156.0 (Cq), 133.4 (CH), 132.6 (Cq), 122.0 (CH), 114.6 (CH), 114.3 (Cq), 93.7 (Cq), 86.1 (Cq), 81.2 (Cq), 79.2 (Cq), 65.8 (CH2), 54.3 (CH2), 51.1 (CH2), 37.8 (CH2), 29.45 (CH2), 28.3 (CH3), 28.1 (CH3) ppm. HRMS (ESI+) m/z calcd for C35H51N4O7[M + H]+ = 639.3758, found 639.3784.Compound 9
To a mixture of diol 1 (3 g, 11.3 mmol), Ag2O (3.93 g, 17.0 mmol) and KI (0.37 g, 2.2 mmol) in dry CH2Cl2 (50 mL) cooled at −20 °C was added portionwise tosyl chloride (2.37 g, 12.4 mmol). The mixture was stirred for 16 h at room temperature, then filtered off over Celite and the filtrate was concentrated under vacuum. The crude oil was purified by column chromatography on silica gel (eluent: CH2Cl2) to yield compound 9 (2.1 g, 44%) as a white solid. Mp. 97–98 °C. 1H NMR (CDCl3, 400 MHz) δ 7.84 (2H, d, J = 8.4 Hz), 7.64 (1H, s), 7.62 (1H, s), 7.38 (2H, d, J = 8.4 Hz), 5.10 (2H, s), 4.67 (2H, s), 3.20 (1H, bs), 2.48 (3H, s) ppm. 13C NMR (CDCl3, 100 MHz) δ 160.0, 153.4, 145.4, 132.7, 130.0, 129.5, 129.4, 128.1, 106.9, 70.6, 63.5, 21.7 ppm. HRMS (ESI+) m/z calcd for C14H15INO4S [M + H]+ = 419.9766, found 419.9759.Compound 10
A suspension of glycine tert-butyl ester hydrochloride (0.76 g, 4.5 mmol) and dry K2CO3 (1.88 g, 13.6 mmol) in dry CH3CN (76 mL) was heated at 55 °C for 30 min. After cooling to room temperature, monosulfonate ester 9 (3.77 g, 9.0 mmol) was added as a solid in one portion, and the mixture was stirred at 60 °C for 20 h. After cooling to room temperature, the mixture was filtered and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: EtOAc/MeOH from 97/03 to 90/10) to yield compound 10 (2.53 g, 90%) as a white foam. 1H NMR (CDCl3, 400 MHz) δ 7.79 (2H, s), 7.53 (2H, s), 4.70 (4H, s), 3.96 (4H, s), 3.86 (2H, bs), 3.40 (2H, s), 1.53 (9H, s) ppm. 13C NMR (CDCl3, 100 MHz) δ 170.1, 159.6, 158.9, 131.05, 128.3, 106.9, 81.6, 63.6, 59.2, 56.7, 28.3 ppm. HRMS (ESI+) m/z calcd for C20H26I2N3O4 [M + H]+ = 626.0013, found 626.0015.Compound 12
A solution of compound 10 (1.64 g, 2.62 mmol) and compound 11 (1.5 g, 7.89 mmol) in a mixture of THF (7.5 mL) and triethylamine (7.5 mL) was degassed with argon by bubbling for 15 min. To this mixture were added Pd(dppf)Cl2·CH2Cl2 (0.64 g, 0.78 mmol) and CuI (0.3 g, 1.57 mmol). The mixture was irradiated with microwaves (100 W) for 30 min, then cooled to room temperature and filtered off over Celite. The filtrate was concentrated under vacuum and the residue was purified by column chromatography on silica gel (CH2Cl2/EtOAc/MeOH gradient from 100/0/0, 50/50/0, and 0/100/0 to 0/90/10) to give a mixture of the copper complex and free ligand. This mixture was dissolved in THF (80 mL) and a 15% aqueous solution of EDTA (150 mL, pH 7.3) and stirred overnight at room temperature to give the desired compound 12 (1.39 g, 71%) as a beige foam. 1H NMR (CDCl3, 400 MHz) δ 7.51 (2H, s), 7.48 (4H, d, J = 8.7 Hz), 7.19 (2H, s), 6.87 (4H, d, J = 8.7 Hz), 4.75 (4H, s), 4.68 (4H, s), 4.16 (2H, bs), 4.03 (4H, s), 3.84 (6H, s), 3.43 (2H, s), 1.53 (9H, s) ppm. 13C NMR (CDCl3, 100 MHz) δ 170.4, 168.9, 158.6, 158.3, 158.0, 133.6, 132.7, 123.7, 120.6, 115.4, 114.8, 93.7, 86.2, 81.4, 65.2, 63.95, 59.5, 56.5, 52.4, 28.25 ppm. HRMS (ESI+) m/z cacld for C42H44N3O10 [M + H]+ 750.3027, found 750.3022.Compound 13
To a solution of diol 12 (250 mg, 0.33 mmol) in CH2Cl2 (8 mL) were added KI (23 mg, 0.14 mmol) and Ag2O (231 mg, 1 mmol). To this mixture cooled to −5 °C was added as a solid in one portion tosyl chloride (190 mg, 1 mmol). The resulting reaction mixture was stirred at room temperature for 16 h. The mixture was then filtered and the filtrate was concentrated under vacuum. The crude product was purified by column chromatography on silica gel (eluent: CH2Cl2 and CH2Cl2/MeOH from 99/01 to 98/02 with 0.2% increase) to yield compound 13 (307 mg, 88%) as a beige oil. 1H NMR (CDCl3, 300 Hz) δ 7.81–7.76 (4H, m), 7.52 (2H, d, J = 1.5 Hz), 7.47–7.42 (4H, m), 7.31–7.27 (4H, m), 7.28 (2H, d, J = 1.5 Hz), 6.86–6.81 (4H, m), 5.07 (4H, s), 4.65 (4H, s), 3.87 (4H, s), 3.80 (6H, s), 3.32 (2H, s), 2.39 (6H, s), 1.46 (9H, s) ppm. 13C NMR (CDCl3, 75 MHz, J Mod) δ 170.1 (Cq), 168.8 (Cq), 159.0 (Cq), 158.3 (Cq), 153.1 (Cq), 145.0 (Cq), 133.5 (CH), 133.0 (Cq), 132.6 (Cq), 129.8 (CH), 128.0 (CH), 124.2 (CH), 121.6 (CH), 115.1 (Cq), 114.7 (CH), 94.1 (Cq), 85.9 (Cq), 81.2 (Cq), 71.3 (CH2), 65.0 (CH2), 59.5 (CH2), 56.0 (CH2), 52.3 (CH3), 28.1 (CH3), 21.5 (CH3) ppm. HRMS (ESI+) m/z calcd for C56H56N3O14S2 [M + H]+1058.3204, found 1058.3241.Compound 16
To a solution of ditosylate compound 13 (312 mg, 0.295 mmol) and diamine 6 (188 mg, 0.295 mmol) in anhydrous acetonitrile (140 mL) were added Na2CO3 (312 mg, 2.95 mmol) and NaI (4.4 mg, 0.0295 mmol) and the mixture was stirred under reflux for 40 h. The reaction mixture was cooled at room temperature, the inorganic salts were filtered off and the filtrate was concentrated under vacuum. The residue was subjected to purification by column chromatography on alumina (CH2Cl2/MeOH 98:02 and 97:03) to give the free macrocycle 16 as a beige oil (297 mg, 74%). IR (film) ṽmax 3428 (NH), 3072, 3054, 2975, 2929, 2871, 2210 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 1761 (C=O), 1734 (C=O), 1596, 1546, 1510, 1437, 1367, 1248, 1211, 1174 cm−1. 1H NMR (CDCl3, 300 MHz) δ 7.34–7.28 (6H, m), 7.22 (2H, s), 7.20–7.15 (4H, m), 6.70–6.62 (6H, m), 4.89 (1H, bs), 4.59 (4H, s), 3.98 (2H, t, J = 6.0 Hz), 3.91–3.82 (12H, m), 3.81 (6H, s), 3.41 (2H, s), 3.39 (4H, s), 3.32 (2H, q, J = 6.2 Hz), 1.98 (2H, m), 1.48 (27H, s), 1.44 (9H, s) ppm. 13C NMR (CDCl3, 75 MHz, J Mod) δ 170.6 (Cq), 168.9 (Cq), 159.1 (Cq), 158.48 (Cq), 158.45 (Cq), 158.4 (Cq), 157.8 (Cq), 156.0 (Cq), 133.43 (CH), 133.41 (CH), 132.0 (Cq), 131.7 (Cq), 123.0 (CH), 122.97 (CH), 122.8 (CH), 115.8 (Cq), 114.65 (Cq), 114.4 (CH), 114.3 (CH), 92.9 (Cq), 92.4 (Cq), 86.6 (Cq), 86.4 (Cq), 81.1 (Cq), 79.2 (Cq), 65.7 (CH2), 65.0 (CH2), 60.3 (CH2), 60.2 (CH2), 60.1 (CH2), 59.0 (CH2), 58.75 (CH2), 52.3 (CH3), 37.7 (CH2), 29.4 (CH2), 28.4 (CH3), 28.2 (CH3) ppm. HRMS (ESI+) m/z calcd for C77H90N7O15 [M + H]+1352.6495, found 1352.6521.
C), 1761 (C=O), 1734 (C=O), 1596, 1546, 1510, 1437, 1367, 1248, 1211, 1174 cm−1. 1H NMR (CDCl3, 300 MHz) δ 7.34–7.28 (6H, m), 7.22 (2H, s), 7.20–7.15 (4H, m), 6.70–6.62 (6H, m), 4.89 (1H, bs), 4.59 (4H, s), 3.98 (2H, t, J = 6.0 Hz), 3.91–3.82 (12H, m), 3.81 (6H, s), 3.41 (2H, s), 3.39 (4H, s), 3.32 (2H, q, J = 6.2 Hz), 1.98 (2H, m), 1.48 (27H, s), 1.44 (9H, s) ppm. 13C NMR (CDCl3, 75 MHz, J Mod) δ 170.6 (Cq), 168.9 (Cq), 159.1 (Cq), 158.48 (Cq), 158.45 (Cq), 158.4 (Cq), 157.8 (Cq), 156.0 (Cq), 133.43 (CH), 133.41 (CH), 132.0 (Cq), 131.7 (Cq), 123.0 (CH), 122.97 (CH), 122.8 (CH), 115.8 (Cq), 114.65 (Cq), 114.4 (CH), 114.3 (CH), 92.9 (Cq), 92.4 (Cq), 86.6 (Cq), 86.4 (Cq), 81.1 (Cq), 79.2 (Cq), 65.7 (CH2), 65.0 (CH2), 60.3 (CH2), 60.2 (CH2), 60.1 (CH2), 59.0 (CH2), 58.75 (CH2), 52.3 (CH3), 37.7 (CH2), 29.4 (CH2), 28.4 (CH3), 28.2 (CH3) ppm. HRMS (ESI+) m/z calcd for C77H90N7O15 [M + H]+1352.6495, found 1352.6521.
Compound 17
To a solution of compound 16 (110 mg, 81 μmol) in CH3CN (3 mL) was added an aqueous solution of 1 M lithium hydroxide (810 μL, 810 μmol) and the solution was stirred at room temperature for 1 h. The solution was purified by preparative HPLC (system A, see the ESI†) to give compound 17 (64 mg, 60%). Analytical HPLC (system B, see the ESI†): tR13.95 min. 1H NMR (CD3OD, 500 MHz) δ 7.54–7.46 (6H, m), 7.39–7.32 (6H, m), 7.02–6.95 (6H, m), 4.45 (4H, s), 4.10–3.92 (14H, m), 3.29–3.20 (8H, m), 2.01–1.93 (2H, m), 1.45 (18H, s), 1.31 (9H, s), 1.30 (9H, s) ppm. 13C NMR (CD3OD, 125 MHz) δ 175.9, 172.8, 161.5, 161.4, 159.77, 159.74, 159.72, 158.6, 135.1, 135.0, 134.6, 134.5, 125.4, 125.35, 125.33, 116.2, 115.95, 115.0, 114.9, 96.3, 96.1, 86.24, 86.16, 82.9, 80.0, 68.4, 66.8, 61.4, 61.3, 61.2, 56.72, 56.69, 38.35, 30.6, 28.8, 28.5, 28.3 ppm. HRMS (ESI+) m/z calcd for C75H86N7O15 [M + H]+ 1324.6182, found 1324.6183.Compound 18a
To a solution of compound 17 (34 mg, 25.7 μmol) in anhydrous DMSO (1275 μL) were added taurine (12.85 mg, 102.7 μmol), diisopropylethylamine (26.4 μL, 154 μmol), and HATU (39 mg, 102.7μmol). The solution was stirred at room temperature for 40 min and purified by preparative HPLC (system C, see the ESI†) to give 18a (30 mg, 76%). Analytical HPLC (system B, see the ESI†): tR 11.33 min. HRMS (ESI+) m/z calcd for C79H97N9O19S2 [M + 2H]2+ 769.8171, found 769.8164.Compound 18b
To a solution of compound 17 (15 mg, 11.3 μmol) in anhydrous DMSO (562 μL) were added 3-((2-aminoethyl)dimethylammonio)propane-1-sulfonate35 (15 mg, 71.58 μmol), diisopropylethylamine (11.6 μL, 67.9 μmol), and HATU (17.2 mg, 45.3 μmol). The solution was stirred at room temperature for 10 min and purified by preparative HPLC (system D, see the ESI†) to give 18b (11.6 mg, 60%). Analytical HPLC (system B, see the ESI†): tR 11.14 min. HRMS (ESI+) m/z calcd for C89H119N11O19S2 [M + 2H]2+ 854.9062, found 854.9055.Compound 18c
To a solution of compound 17 (15 mg, 11.3 μmol) in anhydrous DMSO (562 μL) were added glucosamine (9.77 mg, 45.3 μmol), diisopropylethylamine (11.6 μL, 67.9 μmol), and HATU (17.2 mg, 45.3 μmol). The solution was stirred at room temperature for 10 min and purified by preparative HPLC (system D, see the ESI†) to give 18c (16.3 mg, 87%). Analytical HPLC (system B, see the ESI†): tR 11.62 min. HRMS (ESI+) m/z calcd for C87H108N9O23 [M + H]+ 1646.7558, found 1646.7554.General procedure for the preparation of europium complexes
To compound 18a, 18b or 18c (19.5 μmol) was added CF3COOH (500 μL) and the solution was stirred at room temperature for 2 h and then concentrated under vacuum. The residue was dissolved in a mixture of H2O (6 mL) and CH3CN (2 mL) and the pH was adjusted to 7 with a 3 M aqueous solution of NaOH. To this solution were added EuCl3·6H2O (72 μmol) and a 3 M aqueous solution of NaOH to adjust the pH to 6. The solution was stirred at room temperature for 16 h and the complex was purified by preparative HPLC (system E, see the ESI†).The number of complexes per antibody was calculated by UV-visible spectroscopy and using the following equations:
| Labelling ratio = [Complex]/[Conjugate] |
| [Complex] = ODλmax/ελmax |
| [Conjugate] = (OD280 − ODλmax/Rz)/ε280(antibody) with Rz = ελmax/ε280(complex) |
| ΔF (%) = ((Ratio Sample − Ratio Negative Control)/Ratio Negative Control) × 100 |
| With Ratio = (I665 nm/I620 nm) × 104. |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from University Paul Sabatier-Toulouse III and Centre National de la Recherche Scientifique is gratefully acknowledged.References
- For recent references, see: (a) J. H. S. K. Monteiro, Molecules, 2020, 25, 2089 CrossRef CAS PubMed; (b) U. Cho and J. K. Chen, Cell Chem. Biol., 2020, 27, 921–936 CrossRef CAS; (c) R. Zhang and J. Yuan, Acc. Chem. Res., 2020, 53, 1316–1329 CrossRef CAS PubMed; (d) S. H. Hewitt and S. J. Butler, Chem. Commun., 2018, 54, 6635–6647 RSC; (e) K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS; (f) M. L. Aulsebrook, B. Graham, M. R. Grace and K. L. Tuck, Coord. Chem. Rev., 2018, 375, 191–220 CrossRef CAS.
- (a) P. R. Selvin, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 275–302 CrossRef CAS PubMed; (b) P. R. Selvin, Nat. Struct. Biol., 2000, 7, 730–734 CrossRef CAS.
- (a) J. M. Zwier and N. Hildebrandt, Time-gated FRET detection for multiplexed biosensing, in Reviews in fluorescence 2016, ed. C. D. Geddes, Springer International Publishing, Cham, 2017, pp. 17–43 Search PubMed; (b) J. M. Zwier, H. Bazin, L. Lamarque and G. Mathis, Inorg. Chem., 2014, 53, 1854–1866 CrossRef CAS; (c) A. K. Hagan and T. Zuchner, Anal. Bioanal. Chem., 2011, 400, 2847–2864 CrossRef CAS PubMed; (d) D. Geißler and N. Hildebrandt, Curr. Inorg. Chem., 2011, 1, 17–35 CrossRef.
- N. Sabbatini, M. Guardigli and J.-M. Lehn, Coord. Chem. Rev., 1993, 123, 201–228 CrossRef CAS.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J.-C. Rodriguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- (a) A. De Bettencourt-Dias, P. S. Barber and S. Viswanathan, Coord. Chem. Rev., 2014, 273–274, 165–200 CrossRef CAS; (b) E. Brunet, O. Juanes and J.-C. Rodriguez-Ubis, Curr. Chem. Biol., 2007, 1, 11–39 CAS.
- J. Kimball, J. Chavez, L. Ceresa, E. Kitchner, Z. Nurekeyev, H. Doan, M. Szabelski, J. Borejdo, I. Gryczynski and Z. Gryczynski, Methods Appl. Fluoresc., 2020, 8, 033002 CrossRef CAS PubMed.
- F. J. Steemers, W. Verboom, D. N. Reinhoudt, E. B. van der Tol and J. W. Verhoeven, J. Am. Chem. Soc., 1995, 117, 9408–9414 CrossRef CAS.
- L. Tian, Z. Dai, Z. Ye, B. Song and J. Yuan, Analyst, 2014, 139, 1162–1167 RSC.
- (a) H. Sund, K. Blomberg, N. Meltola and H. Takalo, Molecules, 2017, 22, 1807 CrossRef; (b) J. W. Walton, A. Bourdolle, S. J. Butler, M. Soulie, M. Delbianco, B. K. McMahon, R. Pal, H. Puschmann, J. M. Zwier, L. Lamarque, O. Maury, C. Andraud and D. Parker, Chem. Commun., 2013, 49, 1600–1602 RSC; (c) J. Xu, T. M. Corneillie, E. G. Moore, G.-L. Law, N. G. Butlin and K. N. Raymond, J. Am. Chem. Soc., 2011, 133, 19900–19910 CrossRef CAS; (d) B. Alpha, V. Balzani, J.-M. Lehn, S. Perathoner and N. Sabbatini, Angew. Chem., Int. Ed. Engl., 1987, 26, 1266–1267 CrossRef; (e) B. Alpha, J.-M. Lehn and G. Mathis, Angew. Chem., Int. Ed. Engl., 1987, 26, 266–267 CrossRef.
- (a) N. Alzakhem, C. Bischof and M. Seitz, Inorg. Chem., 2012, 51, 9343–9349 CrossRef CAS; (b) D. Parker, J. W. Walton, L. Lamarque and J. M. Zwier, Eur. J. Inorg. Chem., 2010, 3961–3966 CrossRef CAS.
- N. Leygue, C. Galaup, A. Lopera, E. Delgado-Pinar, R. M. Williams, H. Gornitzka, J. M. Zwier, E. Garcia-Espana, L. Lamarque and C. Picard, Inorg. Chem., 2020, 59, 1496–1512 CrossRef CAS.
- pKa and logKEu values for DOTA from: A. Pasha, G. Tircso, E. T. Benyo, E. Brucher and A. D. Sherry, Eur. J. Inorg. Chem., 2007, 4340–4349 CrossRef CAS PubMed.
- G. Nocton, A. Nonat, C. Gateau and M. Mazzanti, Helv. Chim. Acta, 2009, 92, 2257–2273 CrossRef CAS.
- G. Mathis and H. Bazin, Stable Luminescent Chelates and Macrocyclic Compounds, in Lanthanide luminescence-Photophysical, Analytical and Biological Aspects, ed P. Hänninen and H. Härmä, Springer, 2011, vol. 7, pp. 47–88 Search PubMed.
- H. Takalo, E. Hanninen and J. Kankare, Helv. Chim. Acta, 1993, 76, 877–883 CrossRef CAS.
- A. D'Aléo, A. Picot, A. Beeby, J. A. G. Williams, B. Le Guennic, C. Andraud and O. Maury, Inorg. Chem., 2008, 47, 10258–10268 CrossRef PubMed.
- (a) A. T. Bui, M. Beyler, Y.-Y. Liao, A. Grichine, A. Duperray, J.-C. Mulatier, B. L. Guennic, C. Andraud, O. Maury and R. L. Tripier, Inorg. Chem., 2016, 55, 7020–7025 CrossRef CAS; (b) Z. Liang, C.-F. Chan, Y. Liu, W.-T. Wong, C.-S. Lee, G.-L. Law and K.-L. Wong, RSC Adv., 2015, 5, 13347–13356 RSC.
- (a) M. Starck, J. D. Fradgley, R. Pal, J. M. Zwier, L. Lamarque and D. Parker, Chem. – Eur. J., 2021, 27, 766–777 CrossRef CAS PubMed; (b) H. Sund, Y.-Y. Liao, C. Andraud, A. Duperray, A. Grichine, B. Le Guennic, F. Riobé, H. Takalo and O. Maury, ChemPhysChem, 2018, 19, 3318–3324 CrossRef CAS; (c) S. J. Butler, M. Delbianco, L. Lamarque, B. K. McMahon, E. R. Neil, R. Pal, D. Parker, J. W. Walton and J. M. Zwier, Dalton Trans., 2015, 44, 4791–4803 RSC; (d) M. Delbianco, V. Sadovnikova, E. Bourrier, G. Mathis, L. Lamarque, J. M. Zwier and D. Parker, Angew. Chem., Int. Ed., 2014, 53, 10718–10722 CrossRef CAS; (e) H. Takalo, I. Hemmila, T. Sutela and M. Latva, Helv. Chim. Acta, 1996, 79, 789–802 CrossRef CAS.
- (a) N. Hamon, A. Roux, M. Beyler, J.-C. Mulatier, C. Andraud, C. Nguyen, M. Maynadier, N. Bettache, A. Duperray, A. Grichine, S. Brasselet, M. Gary-Bobo, O. Maury and R. Tripier, J. Am. Chem. Soc., 2020, 142, 10184–10197 CrossRef CAS PubMed; (b) N. Hamon, M. Galland, M. Le Fur, A. Roux, A. Duperray, A. Grichine, C. Andraud, B. Le Guennic, M. Beyler, O. Maury and R. L. Tripier, Chem. Commun., 2018, 54, 6173–6176 RSC.
- N. Leygue, A. Perez e Iñiguez de Heredia, C. Galaup, E. Benoist, L. Lamarque and C. Picard, Tetrahedron, 2018, 74, 4272–4287 CrossRef CAS.
- (a) G. Chessa, L. Canovese, F. Visentin, C. Santo and R. Seraglia, Tetrahedron, 2005, 61, 1755–1763 CrossRef CAS; (b) A. Picot, C. Feuvrie, C. Barsu, F. Malvolti, B. Le Guennic, H. Le Bozec, C. Andraud, L. Toupet and O. Maury, Tetrahedron, 2008, 64, 399–411 CrossRef CAS; (c) O. Storm and U. Luning, Eur. J. Org. Chem., 2002, 3680–3685 CrossRef CAS.
- V. X. Truong and A. P. Dove, Angew. Chem., Int. Ed., 2013, 52, 4132–4136 CrossRef CAS.
- E. R. Neil, A. M. Funk, D. S. Yufit and D. Parker, Dalton Trans., 2014, 43, 5490–5504 RSC.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- (a) R. M. Supkowski and W. D. W. Horrocks Jr., Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef CAS; (b) W. D. W. Horrocks Jr. and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334–340 CrossRef.
- M. Le Fur, E. Molnar, M. Beyler, O. Fougere, D. Esteban-Gomez, O. Rousseaux, R. Tripier, G. Tircso and C. Platas-Iglesias, Inorg. Chem., 2018, 57, 6932–6945 CrossRef CAS PubMed.
- (a) M. Starck, R. Pal and D. Parker, Chem. – Eur. J., 2016, 22, 570–580 CrossRef CAS; (b) V. Placide, D. Pitrat, A. Grichine, A. Duperray, C. Andraud and O. Maury, Tetrahedron Lett., 2014, 55, 1357–1361 CrossRef CAS.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542–1548 RSC.
- B. C. Barja, A. Remorino, M. J. Roberti and P. F. Aramendia, J. Argent. Chem. Soc., 2005, 93, 81–96 CAS.
- (a) G. Mathis, Clin. Chem., 1993, 39, 1953–1959 CrossRef CAS PubMed; (b) N. Sabbatini, S. Perathoner, G. Lattanzi, S. Dellonte and V. Balzani, J. Phys. Chem., 1987, 91, 6136–6139 CrossRef CAS.
- S. Lis, T. Kimura and Z. Yoshida, J. Alloys Compd., 2001, 323–324, 125–127 CrossRef CAS.
- F. Degorce, A. Card, S. Soh, E. Trinquet, G. P. Knapik and B. Xie, Curr. Chem. Genomics, 2009, 3, 22–32 CrossRef CAS PubMed.
- A. Natrajan, D. Sharpe and D. Wen, Org. Biomol. Chem., 2012, 10, 1883–1895 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of instrumentation, analytical and preparative HPLC systems, and general methods of analysis. Synthesis of compounds 4 and 11, NMR and HRMS spectra, and HPLC chromatograms of selected compounds, and luminescence spectra and photophysical data of Eu·Lb and Eu·Lc complexes. See DOI: 10.1039/d1ob02092g |
| This journal is © The Royal Society of Chemistry 2022 |