
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePurely electrical SARS-CoV-2 sensing based on single-molecule counting†
Xander F.
van Kooten‡
 ,
Yana
Rozevsky‡
,
Yulia
Marom
,
Efrat
Ben Sadeh
and
Amit
Meller§
*
,
Yana
Rozevsky‡
,
Yulia
Marom
,
Efrat
Ben Sadeh
and
Amit
Meller§
*
Department of Biomedical Engineering, Technion-IIT, Haifa 32000, Israel. E-mail: ameller@technion.ac.il
First published on 7th March 2022
Abstract
The majority of RNA based COVID-19 diagnostics employ enzymatic amplification to achieve high sensitivity, but this relies on arbitrary thresholding, which complicates the comparison of test results and may lead to false outcomes. Here we introduce solid-state nanopore sensing for label-free quantification of SARS-CoV-2 RNA in clinical nasal swab samples. This PCR-free method involves reverse transcribing a target gene on the viral RNA before enzymatically digesting all but the resulting dsDNA. Ratiometric quantification of RNA abundance is achieved by single-molecule counting and length-based nanopore identification of dsDNA from a SARS-CoV-2 gene and a human reference gene. We graded nasal swab samples from >15 subjects and find that the SARS-CoV-2 ratiometric nanopore index correlates well with the reported RT-qPCR threshold cycle for positive classified samples. Remarkably, nanopore analysis also reports quantitative positive outcomes for clinical samples classified as negative by RT-qPCR, suggesting that the method may be used to diagnose COVID-19 in samples that may evade detection. We show that the sample preparation workflow can be implemented using a compact microfluidic device with integrated thermal control for semi-automated processing of extremely small sample volumes, offering a viable route towards automated, fast and affordable RNA quantification in a small and portable device.
1. Introduction
The outbreak of the COVID-19 pandemic in late 20191 has sparked a global interest in diagnostic methods that would enable accurate, rapid, and affordable on-site detection of viral infections, thus preventing transmission and reducing unnecessary isolation.2 Currently, reverse transcription quantitative polymerase chain reaction (RT-qPCR), a well-established nucleic acid amplification test, remains the most widespread, as it offers high sensitivity, specificity and throughput.3–6 RT-qPCR relies on enzymatic amplification to produce a detectable number of DNA copies after initially reverse transcribing a part of the viral genome.4,7 However, the lack of a universal quantitative molecular benchmark for RT-qPCR diagnostics has made it challenging to assess and compare reports of day-to-day variations in threshold cycle for the same patient8,9 as well as false-negative10–13 and false positive1,14 rates. Furthermore, the non-linear amplification process complicates the quantification and comparison of viral load between cohorts, a crucial step towards developing an understanding of disease progression and spread among the wider population.12,15Single-molecule sensing can overcome some of the limitations of amplification-based detection, provided that it offers sufficient specificity and sensitivity, and preferably avoids non-linear and error-prone amplification steps.16–18 Nanopores are an emerging class of single-molecule biosensors, developed primarily for single-molecule DNA sequencing. Owing to their high sensitivity, in recent years nanopore biosensors have been adapted for sensing clinical biomarkers in biofluids, such as circulating DNAs and even proteins.19–22 Specifically, solid-state nanopores (ssNPs) can be used to quantify specific RNA and mRNA molecules with high accuracy and sensitivity by converting them to double-stranded DNA using a lossless biochemical assay.23 Reverse transcription quantitative nanopore sensing (RT-qNP) allows direct quantification of gene expression, bypassing the need for PCR amplification.23 To date, however, the use of ssNP biosensors for quantitative molecular detection of SARS-CoV-2 in clinical samples has not been reported. Here we present a proof-of-principle study to assess the ability of ssNPs to quantify multiple RNA types directly extracted from clinical patient samples. We show that the nanopore can distinguish between viral RNA and a human reference gene, thus allowing direct counting of the two genes in a rapid and label-free molecular test. Importantly, the ratio of the two RNA events can serve as a quantitative measure for the viral load in the sample, showing good correlation with the Ct value obtained by RT-qPCR run in a certified diagnostic lab. Remarkably, our method reports quantitative RNA loads in clinical samples classified as negative SARS-CoV-2 by RT-qPCR, in some cases indicating a clear positive result.
The purely additive library preparation of the sensing method described here lends itself for liquid handling in a microfluidic device, as it does not involve any centrifugation or purification steps. This may open the possibility for fully automated and fast sample processing and subsequent nanopore analysis. To demonstrate this, we introduce a design for a device capable of preparing clinical sample volumes for purely electrical analysis by ssNPs, hence permitting future development of a fully portable compact sensing technology. The fluidic device performs all the necessary functions for reverse transcription of RNA and digestion of the undesired analytes, using pressure-driven flow with capillary synchronization valves in a disposable lab-on-chip format. We use this device to process both analytical samples containing SARS-CoV-2 RNA and clinical samples from confirmed COVID-19 patients, and show that nanopore quantification of these samples yields results in line with those processed in a standard well-plate format.
2. Results and discussion
2.1. Method validation using synthetic RNA samples
Quantitative nanopore sensing eliminates molecular amplification and instead involves ratiometric, single-molecule counting of well-defined molecular species (dsDNA of two different lengths). As described schematically in Fig. 1a, unlabeled dsDNA molecules are counted by analyzing their distinct electrical signature, in this case the dwell time tD of their translocation through a nanopore, and the normalized amplitude of current blockage IB = iB/io, where iB is the mean blocked ion current level during the passage of the molecule and io is the open pore current level. A typical ssNP device is shown in Fig. 1b. The measurement cell includes two Teflon (PTFE) parts, forming two small chambers (‘cis’ and ‘trans’) separated by the silicon chip (Fig. 1c). A voltage bias is applied using the two wire electrodes also used to measure the ionic current through the ssNP, which serves as the only passage from cis to trans. Typical sets of translocation events of the short (I) and long (II) dsDNA are shown in Fig. 1d.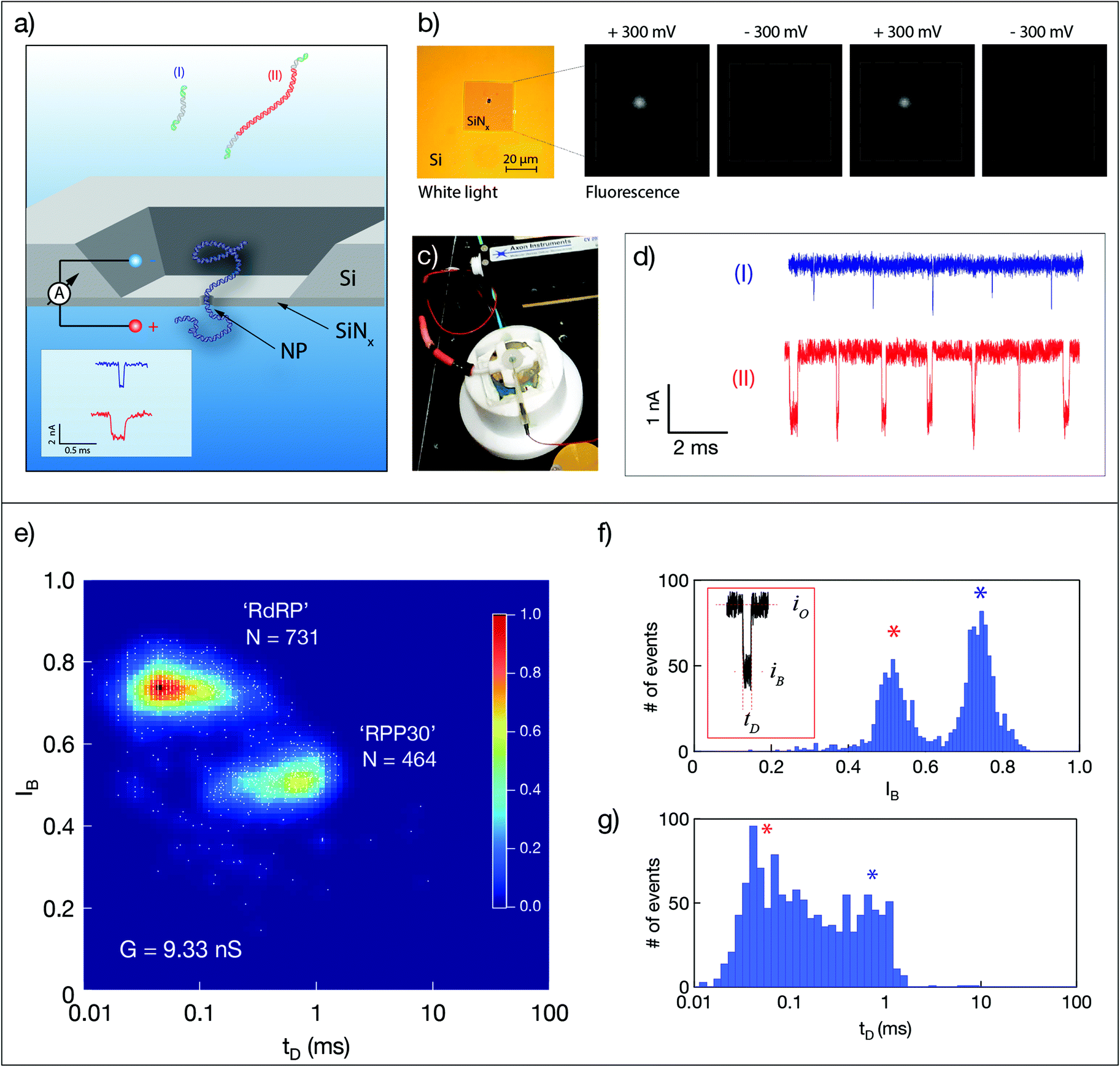 | ||
| Fig. 1 Solid-state nanopore-based sensing of SARS-CoV-2 RNA validation using synthetic RNA samples. (a) Schematic illustration of the method. RNA molecules corresponding to the two target genes (I and II) are first reverse transcribed. The resulting dsDNA molecules are then sensed using a ssNP. The dsDNA strands of the two target genes are designed to be 107 bp and 758 bp, respectively, enabling single-molecule counting using a ∼5 nm ssNP. (b) ssNPs are fabricated in a 25 μm square silicon nitride membrane. Wide-field fluorescence image of a typical ssNP using under ±300 mV and calcium indicator dye, RHOD-2. (c) The measurement apparatus consists of two small fluidic chambers, filled with an electrolyte solution and connected by Ag/AgCl wire electrodes to a current amplifier. (d) Typical ion-current blockade events associated with RPP30 dsDNA (human reference gene, blue) and RdRP dsDNA (viral gene, red). (e) A characteristic event density diagram for the two genes produced from synthetic RNA sources and analysed separately using the same ssNP (G = 9.33 nS). The two distinct event populations are distinguished by (f) their fractional blockade level (IB) and (g) typical event dwell times (tD). | ||
In this study we count the ratio of dsDNA molecules reverse transcribed from two genes: SARS-CoV-2 RdRP, and the human housekeeping gene RPP30. The two dsDNA fragments are produced in the same reaction by including specific reverse primers for the second strand synthesis, resulting in 107 bp and 758 bp long molecules for the two genes, respectively. This design allows straightforward discrimination between the two strands using ssNPs ranging from 3.5 nm to roughly 6 nm in diameter. Fig. 1e shows typical dsDNA translocations corresponding to the two target genes reverse transcribed from synthetic RNAs sources (see ESI Fig. S1 and S2†). The event diagram heat map shows the density of single-molecule events, each represented by its IB and tD values. Notably, in this method validation step, results were obtained in two consecutive experiments using the same nanopore device (G = 9.33 nS, corresponding to a ∼4 nm pore). The device was washed thoroughly with clean buffer in between measurement until no events were observed for at least 10 minutes. The two dsDNA lengths produced two distinct clusters of translocation events, where the longer RPP30 produced longer and deeper amplitude events, as expected. Double-Gaussian fitting of the histograms yields a clear separation of peaks for IB (Fig. 1f), but less so for tD, due to the broader distribution of events (Fig. 1g). A good separation of populations could nevertheless be obtained by using a Gaussian Mixture Model, which uses two-dimensional information to cluster events. Importantly, the ability to serially perform independent analyses of the two synthetic and pure dsDNA components using the same ssNP, establishes the ability of the nanopore to quantify multiple components for a ratiometric approach.
2.2. Analysis of SARS-CoV-2 from clinical samples
Next, we evaluated our method for the analysis of clinical nose/throat swabs, collected as part of the COVID-19 testing program at Rambam Medical Center (Haifa, Israel). After virus inactivation in lysis buffer, 5 μL of each sample was taken for reverse transcription of the SARS-CoV-2 gene RdRP and the human reference gene RPP30, followed by enzymatic digestion using DNAse, RNAse and a broad-spectrum protease (see Fig. 2a). The samples were then analyzed using solid-state nanopores, with translocation data undergoing GMM clustering to annotate the events in two distinct groups.23,24 Arrival times of the translocation events yielded the event rates for the two target genes. A detailed description of the protocol can be found in Methods.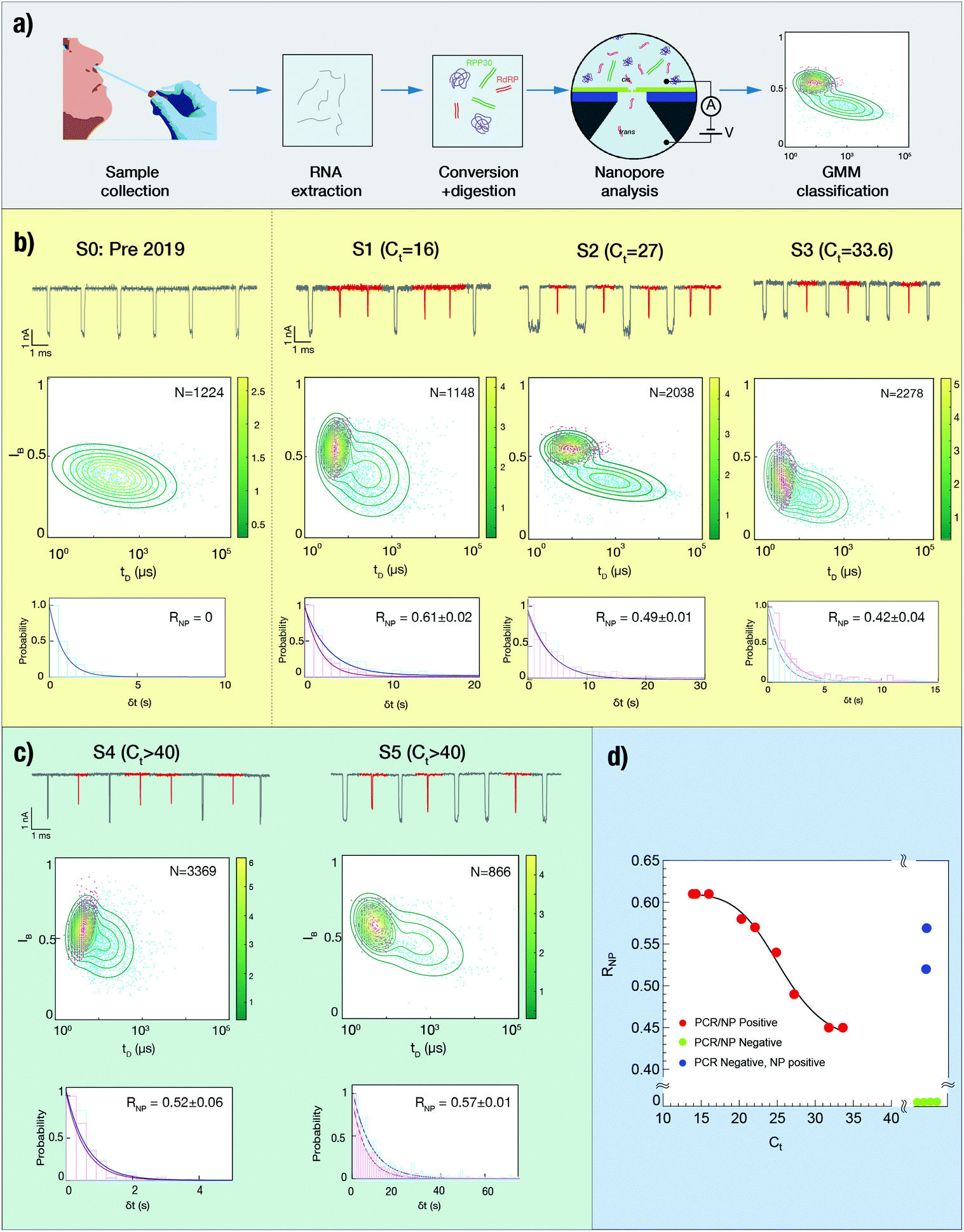 | ||
| Fig. 2 Single-molecule analysis of clinical SARS-Cov-2 samples. (a) A general flow of the sample treatment and ssNP-based sensing for clinical samples. Nasal swabs collected using standard protocols are immediately suspended in virus-inactivated lysis buffer, followed by RNA extraction (on site). RNA to DNA conversion is done in a single step using specific oligonucleotide primers for the target genes, as well as RT and DNA polymerase for second strand synthesis. Then all single-stranded nucleic acids and proteins are enzymatically digested, and the sample is analyzed using an ssNP of roughly 4 nm diameter. The dsDNA translocation events are clustered in two groups representing the abundance of the two target genes in sample using GMM. (b) Four clinical samples with increasing Ct value, analysed consecutively using ssNPs with an average conductance of 13.8 ± 1 nS. S0 is a Pre-Covid-19 sample, used as a true negative control. Upper panels: typical events traces in which the events assigned to RdRP gene fragments are marked in red, and the events assigned to RPP30 are in grey. Middle panels: The two distinct populations of the SARS-CoV-2 RdRP gene (magenta) and human reference gene RPP30 (cyan) are classified using a two-dimensional GMM algorithm. Lower panels: the annotated translocation events are used to calculate the event rate for each gene, from which the SARS-CoV-2 ratiometric nanopore index is calculated. (c) Analysis of two SARS-Cov-2 clinical samples for which RT-qPCR results were classified as negative. In both cases the nanopore index is moderate, suggesting that these samples are likely false negatives. (d) Comparison of ssNP analysis and RT-qPCR for nine clinical COVID-19 samples. The data set of positive samples (red points) is empirically fit using a non-linear Hill function (solid line). Green points represent samples that were negative (undetermined) in RT-qPCR as well as in the nanopore analysis. Blue points represent samples that were found negative (undetermined) by RT-qPCR, but positive in the nanopore analysis. The expected Ct values of these samples are estimated by the non-linear Hill function. | ||
The events capture rate (R), which is proportional the dsDNA concentration in the cis chamber, depends on the size and shape of the nanopore used,25 as well as on the efficiency of the sample collection and preparation. As such, the events rate of RdRP alone is an unsuitable metric for comparing between samples analyzed using different nanopore devices. Instead, the reference gene, which acts as an internal calibrator for the event rate, should be used to calculate a normalized metric. Accordingly, we define the SARS-CoV-2 ratiometric nanopore index as:
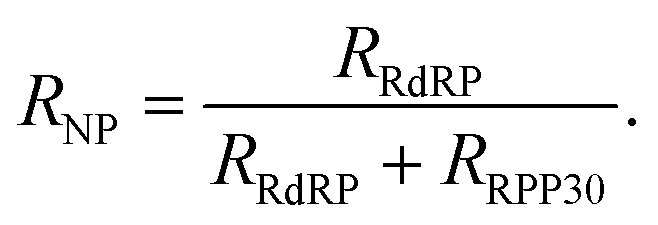 | (1) |
Fig. 2b displays the analysis of 4 clinical samples, using 4 different nanopore devices. We start by analyzing a pre-COVID-19 nasal swab taken before the outbreak of the pandemic, serving as a true negative clinical sample (S0). As expected, despite the presence of both primer sets in the reaction, this sample displays a single population of events with relatively low IB value of 0.4, which we attribute to the human RPP30 gene. We note that the other reaction byproducts, such as digested ssDNA or digested proteins, do not produce additional signals. The measurement was repeated using a different nanopore device, yielding the same result (see ESI Fig. S10†). In contrast, samples S1, S2 and S3, with decreasing SARS-CoV-2 RNA abundance as indicated by their RT-qPCR cycle threshold (Ct) values, exhibit two distinct populations of events. We ascribe long, low-amplitude events to RdRP (labelled in red) and longer, deeper events to RPP30. The upper part of each panel displays typical sets of events, the middle part displays the 2D GMM clustering of the events and the lower part shows the arrival time histogram for both populations. We find that while the event distributions may vary among different nanopore devices, the GMM classification is able to differentiate between the two populations in each experiment. This highlights the importance of co-analyzing the reference gene as an internal control for sample integrity and for nanopore benchmarking.
About a dozen additional clinical samples were analyzed in a similar fashion. We find that in nine of the samples analyzed, as the Ct value increased from 16 to nearly 34, the corresponding ratiometric nanopore index (eqn (1)) decreased from 0.61 to 0.42 (Fig. 2d, red circles). Histograms of IB and tD for all samples are provided in ESI Fig. S3–S6.† We find that the nanopore results and the relative RNA expression levels measured by single-molecule counting can be related by a non-linear Hill function: 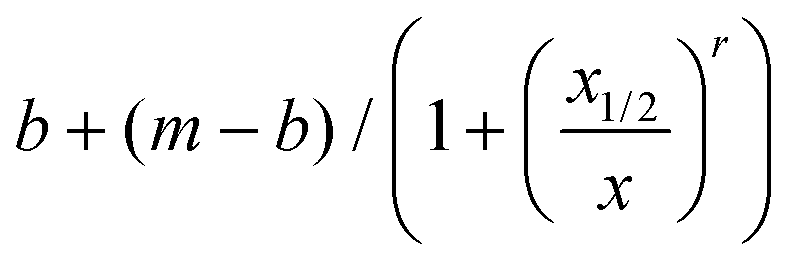 with the following parameters: b = 0.43 ± 0.01; m = 0.611 ± 0.005; r = −8.37 ± 1.57; x1/2 = 25.6 ± 0.7. Here x1/2 represents the RT-qPCR cycle number at which the nanopore identifies equal abundance of the viral gene (RdRP) and the reference human gene (RPP30). The strong non-linear factor r is likely related to the fact that we compare an exponential PCR amplification process to unamplified molecule-by-molecule counting, which is strictly linear. Notably, at both extremes, namely at very high viral load (Ct < 15 cycles) and very low viral load (Ct > 35), the results appear to be less dependent on the number of cycles. This is a consequence of the RT-qPCR process and is likely not due to the nanopore sensitivity limits, as these limit values correspond to RNP values of 0.75 and 1.57 for the base (b) and maximum (m) values, respectively. In a typical translocation batch, one would then have to misclassify roughly 25% to 34% events, which is highly unlikely in these experiments.24
with the following parameters: b = 0.43 ± 0.01; m = 0.611 ± 0.005; r = −8.37 ± 1.57; x1/2 = 25.6 ± 0.7. Here x1/2 represents the RT-qPCR cycle number at which the nanopore identifies equal abundance of the viral gene (RdRP) and the reference human gene (RPP30). The strong non-linear factor r is likely related to the fact that we compare an exponential PCR amplification process to unamplified molecule-by-molecule counting, which is strictly linear. Notably, at both extremes, namely at very high viral load (Ct < 15 cycles) and very low viral load (Ct > 35), the results appear to be less dependent on the number of cycles. This is a consequence of the RT-qPCR process and is likely not due to the nanopore sensitivity limits, as these limit values correspond to RNP values of 0.75 and 1.57 for the base (b) and maximum (m) values, respectively. In a typical translocation batch, one would then have to misclassify roughly 25% to 34% events, which is highly unlikely in these experiments.24
Next, we analyzed six additional samples that were classified as negative, as they produced no detectable signal in RT-qPCR after 40 cycles. The analysis of each sample was repeated at least twice using independent ssNP devices (ESI Fig. S3, S5 and S6†). Four out of these six samples yielded a single population of events, and were therefore assigned a value of RNP = 0 (Fig. 2d, green circles). However, two other samples (ESI Fig. S3, S5,† and Fig. 2c) returned a positive outcome in three repeats despite being classified as negative by RT-qPCR. These samples are marked as blue circles in Fig. 2d. We estimated the expected Ct values for each false negative case by the Hill function, corresponding to moderate SARS-CoV-2 abundance (Ct = 23–25). The data used for the quantitative analysis is summarized in Table 1 and ESI Fig. S3–S5.†
| Sample | C t value | R RPP30 (s−1) | R RdRP (s−1) | R NP |
|---|---|---|---|---|
| S0 | — | 1.27 ± 0.02 | 0 | 0 |
| S1 | 16.03 | 0.36 ± 0.02 | 0.57 ± 0.02 | 0.61 ± 0.02 |
| S2 | 27.22 | 0.26 ± 0.01 | 0.25 ± 0.01 | 0.49 ± 0.01 |
| S3 | 33.62 | 0.87 ± 0.02 | 0.63 ± 0.03 | 0.42 ± 0.04 |
| S4 | N.D. | 1.60 ± 0.04 | 1.75 ± 0.05 | 0.52 ± 0.06 |
| S5 | N.D. | 0.12 ± 0.01 | 0.16 ± 0.01 | 0.57 ± 0.01 |
| S6 | 24.86 | 0.15 ± 0.01 | 0.18 ± 0.01 | 0.54 ± 0.01 |
| S7 | N.D. | 5.25 ± 0.06 | 0 | 0 |
| S8 | N.D. | 0.35 ± 0.03 | 0 | 0 |
| S9 | N.D. | 0.74 ± 0.03 | 0 | 0 |
| S10 | N.D. | 0.48 ± 0.01 | 0 | 0 |
| S11 | 20.28 | 0.81 ± 0.04 | 1.13 ± 0.04 | 0.58 ± 0.04 |
| S12 | 31.77 | 0.37 ± 0.01 | 0.30 ± 0.02 | 0.45 ± 0.02 |
| S13 | 14.3 | 0.07 ± 0.01 | 0.11 ± 0.01 | 0.61 ± 0.01 |
| S14 | 13.89 | 0.29 ± 0.03 | 0.46 ± 0.03 | 0.61 ± 0.03 |
| S15 | 22.07 | 0.14 ± 0.01 | 0.19± 0.01 | 0.57 ± 0.01 |
2.3. On-chip clinical sample preparation for solid-state nanopore analysis
Microfluidic library preparation is advantageous for the ssNP method as it offers repeatable sample processing and enables the use of small sample volumes. The ssNP sensing workflow (Fig. 2) is well-suited for integration on a fluidic device as the method avoids any purification steps or sample centrifugation. Fig. 3a shows the concept of our disposable microfluidic device containing mixing zones and incubation chambers atop a thermoelectric heater. Liquids are introduced in the sample reservoir and three reagent reservoirs, and the inlet reservoirs are sealed to allow hands-off flow and temperature control via a LabVIEW interface (Fig. 3b). The sample and reagent channels lead to merging junctions between the incubation chambers. At the first junction, 1 μL reverse transcriptase (RT) and second-strand synthesis (SSS) mix (reservoir 1) is added to 1 μL of sample (reservoir 2) and incubated at 60 °C for 10 min. At the second junction, 1.2 μL exonuclease and RNase (digestion mix, reservoir 3) is added to the reverse transcribed sample and incubated at 37 °C for 30 min. Finally, 3.2 μL protease (degradation mix, reservoir 4) is added to the sample and incubated at 50 °C for 20 min. The sequence for controlling valves, pressures and temperatures is shown in Fig. 3c. At the end of the sample preparation workflow, the sample is transferred to the nanopore device. While sample delivery to an in-line nanopore sensor has been shown,26,27 here we manually transferred the processed sample to enable a direct comparison with the fully-manual workflow.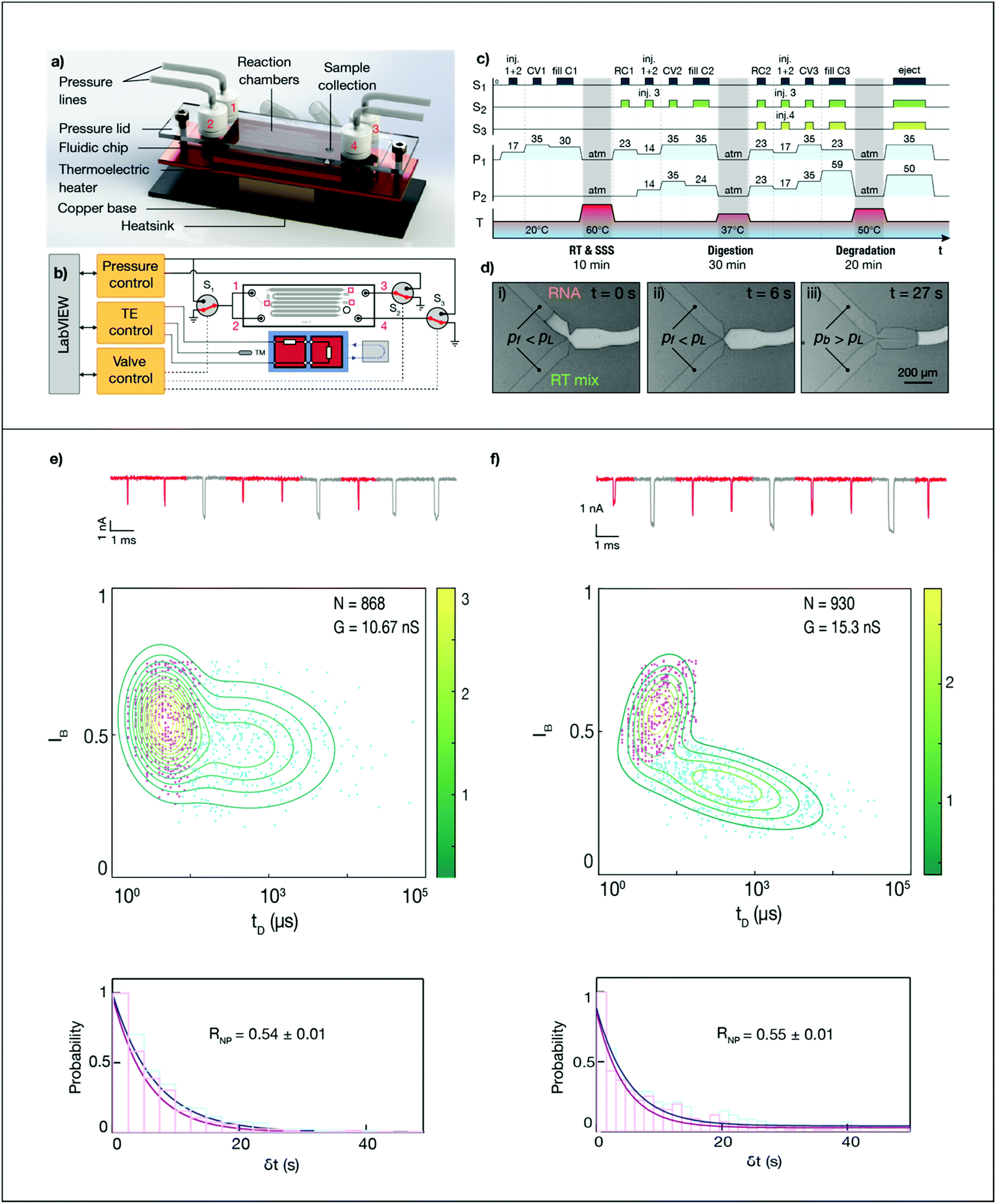 | ||
| Fig. 3 Nanopore sensing of SARS-CoV-2 using on-chip sample preparation. (a) Overview of the fluidic device, showing pressure lines connected to sealed reservoirs, incubation chambers and a closed-loop thermoelectric heater for three consecutive incubation steps (reverse transcription and second-strand synthesis, nuclease digestion, and enzymatic degradation). After incubation, the sample is transferred from the sample collection port to a nanopore device for single-molecule sensing. (b) Injection, mixing and incubation of reagents is controlled on-chip using pressurized reservoirs, capillary valves and thermoelectric (TE) heating. The process is automated using LabView-based control code. S: pressure selector valve; TM: thermistor. Red squares mark capillary valve junctions. Inlets 1–4 are respectively loaded with sample, RT mix, nuclease mix and proteinase. (c) Timing diagram showing pressures (P1, P2, mbar), selector valve signals (S1, S2, S3, on/off) and temperature control in the microfluidic device. CV: capillary valve, RC: reaction chamber retention valve; C: incubation chamber. Note that the time axis is not to scale. (d) Synchronized merging of flows at a capillary valve. Two reagents are introduced under a filling pressure pf, which is less than the Laplace pressure pL of the pinned interface. Once the applied pressure exceeds the Laplace pressure, the valve bursts and the flows merge. (e and f) Nanopore analysis of the same patient sample, S6 (Ct = 25) processed using the conventional workflow in a vial (e) and on the fluidic chip (f). The conductance of the nanopores used was respectively 10.67 nS and 15.3 nS. Top figure: concatenated ionic current traces, with shorter events corresponding to the SARS-CoV-2 RdRP gene. Middle panel: event diagram, with GMM-classified translocation events. Bottom panel: arrival time histogram, exponentially fitted to yield event rates for the RdRP and RPP30 gene. | ||
To ensure controlled mixing with minimal dispersion of incubated sample, the liquids are sequentially introduced into an initially empty (air-filled) hydrophobic channel. However, this approach is likely to trap air bubbles,28 which expand dramatically at elevated temperatures and can push liquids back to the reservoirs. To overcome this, we implemented capillary valves at each junction, which synchronize interfaces arriving at different times by retaining them until the applied pressure exceeds the Laplace (‘burst’) pressure, which is defined by the geometry.28–30 The operation of a capillary synchronization junction is shown in Fig. 3d, where a filling pressure Pf is initially applied to the reservoir containing the RNA sample and RT mix. Once both interfaces have reached the junction (ii), a pressure Pb, which exceeds the Laplace pressure, is applied, causing the valve to burst and the flows to merge (iii). Apart from preventing bubbles and allowing temperature-sensitive reagents to be stored away from the ‘hot zone’ of the chip, capillary valves offer a way towards full automation, by presenting incoming flows with a ‘soft’ block that can withstand an applied pressure. A similar constriction geometry is added as a retention valve at the end of every incubation chamber, to pin the fluid front and prevent capillary creeping during incubation.
To demonstrate the performance of the microfluidic device for nanopore quantification of viral samples, we compared ssNP sensing of an additional clinical sample (S6, Ct = 25) after sample preparation in the microfluidic device and in a vial. Fig. 3 and ESI Fig. S7–S9† show the nanopore results for samples processed using the conventional and microfluidic workflows (Fig. 3e and f, respectively). Remarkably, despite being run on pores with a different conductance, the microfluidic and conventional workflow yield nearly identical relative CoV-2 rates (RNP = 0.55 ± 0.01 and 0.54 ± 0.01, respectively). This demonstrates the feasibility of microfluidic sample processing and highlights the sensitivity and robustness of the nanopore-based workflow.
2.4. Discussion
We developed a ratiometric nanopore-based RNA quantification method based on single-molecule and label-free counting optimized for clinical SARS-CoV-2 samples. This method circumvents PCR amplification, hence reducing the reliance on specialized lab equipment while cutting down test time and conserving or improving accuracy. Towards full field implementation of our method we further introduced a custom microfluidic device capable of performing the sample processing workflow, starting from just 1 μl of extracted RNA mix (about 8-fold less than RT-qPCR) and producing a nanopore-ready solution. In this device, capillary synchronization valves enable bubble-free reagent injection and merging of flows and provide a pressure-based stop valve that is compatible with timed automation without the need for feedback.A fully autonomous device would further require integration of an in-line nanopore sensor to eliminate the manual sample transfer,33 and should support pre-loading of reagents in e.g. thermoelectrically cooled reservoirs. Furthermore, an investigation of reaction efficiency in each stage (currently totaling 60 min) is expected to show that incubation times can be substantially reduced, making the microfluidic workflow suitable for fast-turnaround testing scenarios.
Our studies open an avenue towards a lab-on-chip device for biomedical and clinical needs, demonstrated in the context of COVID-19, but potentially also relevant to other pathogens as well as cancer diagnostics based on RNA biomarkers. Here, we illustrated the strength of the technique by diagnosing a range of SARS-CoV-2 positive and negative clinical samples. In some cases, our results point to false negative RT-qPCR classification (Fig. 2 and ESI Fig. S3, S5†). While the disparity between nanopore and RT-qPCR results in these cases remains to be investigated, false negative RT-qPCR outcomes are known to occur, perhaps most notably in samples taken during disease progression.10,11
We believe that the accuracy of the nanopore-based test can be largely attributed to the fact that both the viral and human gene are co-processed and co-analyzed, and the results are always presented as a ratio of the two. This cancels out sample-to-sample variations due to e.g. sample source concentration, enzymatic process efficiency and sampling errors. Furthermore, the lack of amplification eliminates any potential amplification bias or error. In the nanopore test, true negative samples (pre-COVID-19 nasal swabs) clearly display a single event population corresponding to the human reference gene RPP30 (see Fig. 2 and ESI Fig. S6†). The positive signal of RPP30 provides a powerful confirmation that the clinical sample was acquired effectively. In contrast, the appearance of two event populations in samples with significant relative counts is strong evidence of the presence of the viral gene. This translates to high nanopore accuracy, as shown in Fig. 2 and ESI Fig. S3–S5.†
The workflow shown here is expected to be compatible with various RNA sampling approaches, including direct detection from saliva without any extraction31 or even from sewage.32 The combination of a microfluidic processing device with nanopore sensing has an important impact towards establishing rapid lab-on-chip devices for medical diagnostics in general.
3. Experimental
3.1. Sample processing for single-molecule analysis
Positive control samples from a mixture of synthetic SARS-CoV-2 RNA and extracted hRPP30: either 50 ng of DNaseI-treated RNA, extracted from HCT116 cells using GeneJet RNA Purification Kit (Thermo Fisher scientific), or 0.5 × 106 copies of synthetic SARS-CoV-2 RNA (control 2; MN908947.3; Twist Biosciences) were reversed transcribed with specific primers (ESI Table 1†) for human RPP30 cDNA or for two amplicons within the RdRP open reading frame of SARS-CoV-2. The reaction contained the following ingredients: 20 mM Tris–HCl, 10 mM (NH4)2SO4, 10 mM KCl, 0.1% Triton® X-100 (pH 8.8 @ 25 °C), 6 mM MgSO4, 1.4 mM dNTPs, 0.2 mM of each primer, 3 U WarmStart RTx (NEB) and 6 U Bst 2.0 WarmStartTM DNA polymerase (NEB). The reaction was carried out at 62 °C for 10 min. Afterwards, cDNA samples were treated with 20 U of Exonuclease I (NEB) at 37 °C for 15 min, followed by 2.5 U RNase I (Thermo Fisher Scientific) at 50 °C for 20 min, and finally 0.2 U of ProK (NEB). For gel electrophoresis verification, cDNA was PCR amplified using Kapa HiFi polymerase (Roche). PCR was performed under the following conditions: 95 °C for 3 min followed by 30 cycles of 98 °C for 20 s, 62 °C for 15 s and 72 °C for 30 s.Extracted full NA clinical SARS-CoV-2 samples were provided by Rambam Medical Center (Haifa, Israel). All samples were approved by Rambam Health Corporation Ethics Committee (Haifa, Israel) and were diagnosed as positive or negative, based on RT-qPCR results (Ct values). Nasal swabs taken both from nostrils and throat were collected by healthcare providers. The samples were mixed with lysis buffer and then whole nucleic acids (NA) were extracted using automated NA instruments, STARMag 96 × 4 Universal Cartridge kit’ (Seegene Inc., South Korea) or magLEAD (Precision System Science). RT-qPCR was performed on the E, RdRP, and N genes of SARS-CoV-2 (Sarbeco probes).3,34 Reactions with 8 μl of samples were heated to 50 °C for 30 minutes for reverse transcription, denatured in 95 °C for 10 minutes, and then 40 cycles of amplification were carried out at 95 °C for 15 seconds and 55 °C for 32 seconds. Fluorescence was measured using the FAM parameters. As a result, the cycle threshold (Ct) was determined except for the undetermined cases in which no signal was observed for 40 PCR cycles or more (classified as “negative”). Information on Ct values is presented in Table 1. The clinical samples were divided into 5 μl aliquots and stored at −80 °C.
For the Nanopore-based analysis 5 μl aliquots of the same NA extracted samples used for the RT-qPCR, were diluted to final volume of 10 μl, with the following buffer: 20 mM Tris–HCl 10 mM (NH4)2SO4, 10 mM KCl, 0.1% Triton® X-100 (pH 8.8 @ 25 °C), 6 mM MgSO4, 1.4 mM dNTPs, 0.2 mM of each primer, 3 U WarmStart® RTx (NEB) and 6 U Bst 2.0 WarmStart™ DNA polymerase (NEB). No loop primers were used in the reaction to avoid amplification. The reaction was carried out at 62 °C for 10 min. A positive control sample composed of 0.5 × 106 copies synthetic SARS-CoV-2 and 50 ng of extracted total RNA from HCT116 cells, was run in parallel. Afterwards, cDNA samples were treated with 20 U of Exonuclease I (NEB) at 37 °C for 15 min, followed by 2.5 U RNase I (Thermo Fisher Scientific) at 50 °C for 20 min, and finally 0.2 U of ProK (NEB). The sample was diluted 40-fold in nanopore buffer.
Clinical sample processing for the microfluidic workflow: the clinical samples were prepared using the same concentrations as detailed above, but only 1 μl of each sample was introduced into the device. After processing the samples with the fluidic device, the sample was diluted 20-fold in nanopore buffer.
3.2. Nanopore fabrication
Nanochip fabrication was performed as previously described.35 Briefly, nanopore devices were fabricated on a 100 mm double-side polished silicon wafer, coated on both sides with 500 nm SiO2 and 50 nm low-stress SiNx. The silicon nitride on the front side of the wafer was thinned to 8–12 nm in 2 μm circular regions, using CF4/O2 reactive ion etching (RIE). The nitride and oxide on the back side of the wafer were patterned by photolithography and etched using RIE and buffered oxide etch (BOE) to open a hard mask for subsequent anisotropic Si etching in 33% KOH. Finally, the remaining SiO2 was etching in BOE to release the free-standing nitride membranes.Chips were cleaved from the wafer and cleaned using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of H2SO4/H2O2. They were then glued to a Teflon insert using Ecoflex 5 (Smooth-ON) and immersed in buffer (1 M KCl, 40 mM Tris–HCl, 1 mM EDTA). An Ag/AgCl electrode was placed in both the cis and trans reservoir and nanopores were drilled using dielectric breakdown (DB).36 The locally thinned SiNx ensured that the pore was formed at the center of the membrane. DB voltage pulses with an amplitude of 8–9 V and a duration of 225 ms were applied at 1 s intervals, and the ionic current was probed at 300 mV between pulses. Once the current exceeded 0.4 nA, the pulse amplitude was reduced to 1–3 V until the desired nanopore conductance was reached.
:1 solution of H2SO4/H2O2. They were then glued to a Teflon insert using Ecoflex 5 (Smooth-ON) and immersed in buffer (1 M KCl, 40 mM Tris–HCl, 1 mM EDTA). An Ag/AgCl electrode was placed in both the cis and trans reservoir and nanopores were drilled using dielectric breakdown (DB).36 The locally thinned SiNx ensured that the pore was formed at the center of the membrane. DB voltage pulses with an amplitude of 8–9 V and a duration of 225 ms were applied at 1 s intervals, and the ionic current was probed at 300 mV between pulses. Once the current exceeded 0.4 nA, the pulse amplitude was reduced to 1–3 V until the desired nanopore conductance was reached.
Afterward, the nanopores were kept under a low probing voltage (0.15 to 0.3 V) in a buffer solution (1 M KCl, 40 mM Tris–HCl, 1 mM EDTA, pH 7.5) to obtain a stable open-pore current. During the experiment, translocation events were monitored using an Axon 200B amplifier, filtered at 100 kHz, and acquired using a custom LabVIEW software (National Instruments). After collecting the data, we performed offline analysis using a custom LabVIEW program to extract the dwell time (tD), current blockage (IB), and arrival time (ta) of each translocation event according to an electrical threshold. Collisional events (short current spikes due to unsuccessful translocations) could be filtered by setting a current threshold in the analysis of IB < 0.8.
Optical monitoring of the nanopores was performed as follows: 500 mM CaCl2 was added to the Cis chamber, 500 nM of RHOD-2 (Molecular Probes®) and 10 mM EGTA (Ca2+ chelator) were added to the trans chamber. When negative potential bias is applied, Ca2+ ions driven to the pore and binds to RHOD-2, releasing strong fluorescence signal in the pore location. The signal is radially declines as Ca2+ ions bind to EGTA. A custom wide field microscope with a collimated 560 nm laser (iFlex-Viper, PointSource) coupled through a single-mode optical fiber is focused on a high NA objective (Olympus Plan Apochromat 60×/1.45) and expanded to illuminate the membrane position at the image plane. The emitted light is collected by the same objective and focused onto an EMCCD camera (Andor iXon 887).36
3.3. Microfluidic device
We fabricated microchannels in PDMS by soft lithography. A 40 μm layer of SU-8 3050 was spin-coated on a 100 mm silicon wafer, patterned, developed in PGMEA and hard baked at 150 °C for 2 min. We then vapor-coated the mold with trichlorosilane in a desiccator and cast Sylgard 184 polydimethylsiloxane (PDMS) in a 10:1 ratio of monomer base to crosslinker onto the wafer. The PDMS was cured for 2 hours at 80 °C, peeled off the mold and cut. We then punched 3 mm reservoirs and irreversibly bonded the PDMS to a glass slide after 30 s oxygen plasma treatment.
The PDMS/glass device was placed on the copper heat diffuser of two thermoelectric heaters (Melcor CPI-4-31-045L) wired in series and controlled in a closed loop (3040, Newport). The cold side was bonded with thermal grease to a heat sink which was cooled using a chiller (PolyScience). After pipetting the reagents into the reservoirs, we connected the pressure lines. The pressure in the reservoirs was set by a controller (MFCS-EZ, Fluigent). Each reservoir was connected to two parallel on/off valves (075-T2NC12-32 M, Bio-Chem) acting as a two-way selector valve. The valves were controlled by a 6-channel valve controller (VC-6, Warner Instrument Corp.). For visualization purposes, the device was imaged with an upright stereomicroscope (AZ100, Nikon).
3.4. GMM analysis
We generated histograms of the nanopore data for each axis (x: dwell time, y: blockage amplitude) and determined the amplitude, mean and covariance matrix of each peak. The maximum values are used as an initial estimate of the concentration ratio of the two populations, which are in turn used as initial conditions for a GMM algorithm that clusters events into two populations. The posterior probability is calculated to assign the likelihood of an event belonging to a specific population. All graphs and fits were generated with Igor Pro 6 (Wavemetrics, Lake Oswego, OR) and data analysis was performed in MATLAB (MathWorks, Natick, MA).Author contributions
X. van Kooten and Y. Rozevsky co-led the work and performed critical sample preparation, nanopore and nanofluidic fabrication and data analysis. Y. Marom and E. Ben Sadeh assisted in the data acquisition. A. Meller supervised the research and assisted in data analysis. All authors contributed to the manuscript writing and approved its final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Ronit Almog and the staff in the epidemiology unit at the Rambam Medical center (Haifa, IL) for fruitful, ongoing collaboration. We thank Dr Diana Huttner for assistance in the sample processing; Navneet Verma and Neeraj Soni for assistance in optical microscopy. This project has received funding from the European Research Council (ERC) No. 833399 (NanoProt-ID) and ERC-PoC No. 966824 (OptiPore) both under the European Union's Horizon 2020 research and innovation programme grant agreements.References
- P. Zhou, X.-L. Yang, X. G. Wang, B. Hu, L. Zhang, W. Zhang, H. R. Si, Y. Zhu, B. Li, C. L. Huang, H. D. Chen, J. Chen, Y. Luo, H. Guo, R. Di Jiang, M. Q. Liu, Y. Chen, X. R. Shen, X. Wang, X. S. Zheng, K. Zhao, Q. J. Chen, F. Deng, L. L. Liu, B. Yan, F. X. Zhan, Y. Y. Wang, G. F. Xiao and Z. L. Shi, Nature, 2020, 579, 270 CrossRef CAS PubMed.
- B. Gates, N. Engl. J. Med., 2020, 382, 1677 CrossRef CAS PubMed.
- V. M. Corman, O. Landt, M. Kaiser, R. Molenkamp, A. Meijer, D. K. Chu, T. Bleicker, S. Brünink, J. Schneider, M. L. Schmidt, D. G. Mulders, B. L. Haagmans, B. van der Veer, S. van den Brink, L. Wijsman, G. Goderski, J.-L. Romette, J. Ellis, M. Zambon, M. Peiris, H. Goossens, C. Reusken, M. P. Koopmans and C. Drosten, Eurosurveillance, 2020, 25, 2000045 Search PubMed.
- I. A. Mattioli, A. Hassan, O. N. Oliveira and F. N. Crespilho, ACS Sens., 2020, 5, 3677 Search PubMed.
- M. A. C. Huergo and N. T. K. Thanh, Analyst, 2021, 146, 382 RSC.
- B. D. Kevadiya, J. Machhi, J. Herskovitz, M. D. Oleynikov, W. R. Blomberg, N. Bajwa, D. Soni, S. Das, M. Hasan, M. Patel, A. M. Senan, S. Gorantla, J. McMillan, B. Edagwa, R. Eisenberg, C. B. Gurumurthy, S. P. M. Reid, C. Punyadeera, L. Chang and H. E. Gendelman, Nat. Mater., 2021, 20, 593 CrossRef CAS PubMed.
- H. D. VanGuilder, K. E. Vrana and W. M. Freeman, BioTechniques, 2008, 44, 619 CrossRef CAS PubMed.
- X. He, E. H. Y. Lau, P. Wu, X. Deng, J. Wang, X. Hao, Y. C. Lau, J. Y. Wong, Y. Guan, X. Tan, X. Mo, Y. Chen, B. Liao, W. Chen, F. Hu, Q. Zhang, M. Zhong, Y. Wu, L. Zhao, F. Zhang, B. J. Cowling, F. Li and G. M. Leung, Nat. Med., 2020, 26, 672 CrossRef CAS.
- L. Zou, F. Ruan, M. Huang, L. Liang, H. Huang, Z. Hong, J. Yu, M. Kang, Y. Song, J. Xia, Q. Guo, T. Song, J. He, H. L. Yen, M. Peiris and J. Wu, N. Engl. J. Med., 2020, 382, 1177 CrossRef PubMed.
- L. M. Kucirka, S. A. Lauer, O. Laeyendecker, D. Boon and J. Lessler, Ann. Intern. Med., 2020, 173, 262 CrossRef PubMed.
- Y. Li, L. Yao, J. Li, L. Chen, Y. Song, Z. Cai and C. Yang, J. Med. Virol., 2020, 92, 903 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, Y. Chen and Q. Qin, J. Med. Virol., 2020, 92, 568 CrossRef CAS PubMed.
- Y. Wang, H. Kang, X. Liu and Z. Tong, J. Med. Virol., 2020, 92, 538 CrossRef CAS PubMed.
- A. Borst, A. T. A. Box and A. C. Fluit, Eur. J. Clin. Microbiol. Infect. Dis., 2004, 23, 289 CrossRef CAS.
- S. Bustin and T. Nolan, Eur. J. Clin. Invest., 2017, 47, 756 CrossRef PubMed.
- T. Gilboa, P. M. Garden and L. Cohen, Anal. Chim. Acta, 2020, 1115, 61 CrossRef CAS PubMed.
- S. Cai, T. Pataillot-Meakin, A. Shibakawa, R. Ren, C. L. Bevan, S. Ladame, A. P. Ivanov and J. B. Edel, Nat. Commun., 2021, 12, 1 CrossRef PubMed.
- N. Burck, T. Gilboa, A. Gadi, M. Patkin Nehrer, R. J. Schneider and A. Meller, Clin. Chem., 2021, 67, 753 CrossRef PubMed.
- K. J. Freedman, L. M. Otto, A. P. Ivanov, A. Barik, S.-H. Oh and J. B. Edel, Nat. Commun., 2016, 7, 1 Search PubMed.
- Y. Wang, K. Tian, R. Shi, A. Gu, M. Pennella, L. Alberts, K. S. Gates, G. Li, H. Fan, M. X. Wang and L.-Q. Gu, ACS Sens., 2017, 2, 975 CrossRef CAS.
- K. Tian, X. Chen, B. Luan, P. Singh, Z. Yang, K. S. Gates, M. Lin, A. Mustapha and L. Q. Gu, ACS Nano, 2018, 12, 4194 CrossRef CAS PubMed.
- A. K. Thakur and L. Movileanu, ACS Sens., 2019, 4, 2320 CrossRef CAS.
- Y. Rozevsky, T. Gilboa, X. F. van Kooten, D. Kobelt, D. Huttner, U. Stein and A. Meller, ACS Nano, 2020, 14, 13964 CrossRef CAS.
- A. H. Squires, E. Atas and A. Meller, PLoS One, 2015, 10, 1 Search PubMed.
- M. Wanunu, W. Morrison, Y. Rabin, A. Y. Grosberg and A. Meller, Nat. Nanotechnol., 2010, 5, 160 CrossRef CAS.
- N. Varongchayakul, J. S. Hersey, A. Squires, A. Meller and M. W. Grinstaff, Adv. Funct. Mater., 2018, 28, 1804182 CrossRef PubMed.
- R. Tahvildari, E. Beamish, K. Briggs, S. Chagnon-Lessard, A. N. Sohi, S. Han, B. Watts, V. Tabard-Cossa and M. Godin, Small, 2017, 13, 1602601 CrossRef PubMed.
- J. Melin, N. Roxhed, G. Gimenez, P. Griss, W. Van Der Wijngaart and G. Stemme, Sens. Actuators, B, 2004, 100, 463 CrossRef CAS.
- M. Zimmermann, P. Hunziker and E. Delamarche, Microfluid. Nanofluid., 2008, 3, 395 CrossRef.
- H. Cho, H. Y. Kim, J. Y. Kang and T. S. Kim, J. Colloid Interface Sci., 2007, 306, 379 CrossRef CAS PubMed.
- T. Baier, T. E. Hansen-Hagge, R. Gransee, A. Crombé, S. Schmahl, C. Paulus, K. S. Drese, H. Keegan, C. Martin, J. J. O'Leary, L. Furuberg, L. Solli, P. Grønn, I. M. Falang, A. Karlgård, A. Gulliksen and F. Karlsen, Lab Chip, 2009, 9, 3399 RSC.
- G. Xun, S. T. Lane, V. A. Petrov, B. E. Pepa and H. Zhao, Nat. Commun., 2021, 12, 2905 CrossRef CAS PubMed.
- K. Yaniv, E. Ozer, M. Shagan, S. Lakkakula, N. Plotkin, N. S. Bhandarkar and A. Kushmaro, Environ. Res., 2021, 201, 111653 CrossRef CAS.
- I. Yelin, N. Aharony, E. S. Tamar, A. Argoetti, E. Messer, D. Berenbaum, E. Shafran, A. Kuzli, N. Gandali, O. Shkedi, T. Hashimshony, Y. Mandel-Gutfreund, M. Halberthal, Y. Geffen, M. Szwarcwort-Cohen and R. Kishony, Clin. Infect. Dis., 2020, 71, 2073 CrossRef CAS PubMed.
- A. Squires, E. Atas and A. Meller, Sci. Rep., 2015, 5, 11643 CrossRef CAS PubMed.
- A. Zrehen, T. Gilboa and A. Meller, Nanoscale, 2017, 9, 16437 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr07787b |
| ‡ Equally contributing authors. |
| § Current address: The WYSS Institute, Harvard University, Boston, MA 02115 USA. |
| This journal is © The Royal Society of Chemistry 2022 |