
1D alignment of Co(II) metalated porphyrin–napthalimide based self-assembled nanowires for photocatalytic hydrogen evolution†
Botta
Bhavani
ab,
Nageshwarrao
Chanda
ab,
Vishal
Kotha
 c,
Govind
Reddy
a,
Pratyay
Basak
ab,
Ujjwal
pal
ab,
Lingamallu
Giribabu
*ab and
Seelam
Prasanthkumar
*ab
c,
Govind
Reddy
a,
Pratyay
Basak
ab,
Ujjwal
pal
ab,
Lingamallu
Giribabu
*ab and
Seelam
Prasanthkumar
*ab
aPolymer & Functional Materials Division, CSIR-Indian Institute of Chemical Technology (IICT), Tarnaka, Hyderabad-500007, Telangana, India. E-mail: prasanth@iict.res.in; giribabu@iict.res.in
bAcademy of Scientific and Innovation Research (AcSIR), Ghaziabad-201 002, India
cDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai-400076, Maharastra, India
First published on 22nd November 2021
Abstract
The splitting of water into hydrogen and oxygen under visible light is an emerging phenomenon in green energy technology. Nevertheless, selecting an appropriate photocatalyst is rather significant to enhance hydrogen production on a large scale. In this context, organic photocatalysts have received considerable attention owing to their larger surface area, control in diffusion adsorption, nanostructures and electronic properties. Herein, we have developed five either free base or transition metalated porphyrin–napthalimide based donor–acceptor systems (PN1–PN5) and studied their morphology, electronic properties and catalytic behaviour. Detailed studies suggest that the Co(II) substituent D–A system (PN2) displayed a well-aligned one-dimensional (1D) nanowire with high electrical conductivity promoting remarkable photocatalytic hydrogen production rate (18 mM g−1 h−1) when compared to that of porphyrin-based derivatives reported until now. Thus, these results propose to investigate diverse metalated π-conjugated materials as photocatalysts for hydrogen production.
 Seelam Prasanthkumar | Seelam Prasanthkumar received his PhD degree in organic chemistry from CSIR-National Institute of Interdisciplinary Science and Technology (NIIST), Trivandrum, India in 2012. Afterwards, he joined as a postdoctoral fellow in the group of Prof. Takuzo Aida, RIKEN, Japan. Subsequently, he was selected for DST-Inspire faculty award and joined as a Inspire faculty fellow in CSIR-Indian Institute of Chemical Technology (IICT), Hyderabad, India. Currently, he is working as a Scientist, Assistant Professor (AcSIR) in Polymer & Functional Materials Division, CSIR-IICT, India. His research interest includes developing the stimuli responsive materials for sensors, solar cells and hydrogen production. |
Introduction
Light-stimulated water splitting into hydrogen has received significant attention for the cleanest energy production and storage.1 In this context, several inorganic semiconductors were utilized as photocatalysts for water splitting towards hydrogen evolution.2 However, lack of control on the surface area of nanostructures and band gap alignments has resulted in finding alternative semiconductors. Hence, semiconductors with fine-tuning of their critical parameters such as crystallinity, band gap, stability of transient species and exciton migration are rather important. In this regard, organic semiconductors are considered alternative catalysts for H2 evolution.1 Organic semiconductors with low-lying HOMO levels are highly desirable to improve the catalytic process under visible light and also to overcome the kinetic barrier of the process. Thus, so far, very few examples are reported on organic photocatalysts such as carbon nitrides, porous and linear conjugated polymers and organic frameworks.3–8 However, exciton recombination, low surface area and poor charge separation direct us to develop small π-conjugated materials with high photocatalytic activity.9,10 Viewing this, the porphyrin-based π-conjugated system has attracted much attention in organic electronics due to its high ionization potential, electron affinity, planarity and photo/thermal stability. The extended π-conjugated structure of porphyrin promotes self-assembled nanostructures via supramolecular interactions.11 On the other hand, porphyrins have been employed as a photosensitizer in third-generation solar cells, however, their photocatalytic performance is not much explored. Herein, we have developed porphyrin comprised donor–acceptor systems in which porphyrin acts as an electron donor and napthalimide as an electron acceptor. Napthalimide (NMI) is a well-known electron acceptor having miscellaneous optoelectronic applications. Additionally, the NMI structure possesses rigidity and planarity, which facilitates self-assembly by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯π and π–π interactions.12 Indeed, porphyrin and NMI individually exhibit excellent well-defined nanostructures, and designing the combination of porphyrin–NMI in π-conjugated structure can serve as a donor–acceptor (D–A) system, which aid to improve the electronic properties through the self-assembly.
O⋯π and π–π interactions.12 Indeed, porphyrin and NMI individually exhibit excellent well-defined nanostructures, and designing the combination of porphyrin–NMI in π-conjugated structure can serve as a donor–acceptor (D–A) system, which aid to improve the electronic properties through the self-assembly.
Thereby, we have designed five porphyrin–NMI based D–A derivatives (PN1–PN5) with a free base (PN1), and incorporated transition metals (PN2–PN5): cobalt (Co), nickel (Ni), copper (Cu) and zinc (Zn) (Fig. 1). As is known, the energy structure of porphyrin is non-planar and shows S4 symmetry, while transition metal-doped porphyrins exhibit planarity which results in the enhancement of symmetry to D4h. On the other hand, transition metalated porphyrins depict high ionization potential, binding energy, electronic affinity and high chemical hardness. Among four transition metals, Co(II) metalated porphyrin exhibits high stability and poor reactivity due to low electron affinity, high chemical hardness and binding energy between the nitrogen complex with cobalt metal.13 These molecules were synthesized using sonogashira coupling reactions, followed by metalation. Subsequently, these molecules were characterized by using spectroscopic techniques. These five derivatives showed broad range absorption from ultraviolet to the visible region and their redox nature facilitates plausible electron transfer from the donor to acceptor. Subsequently, these molecules form self-assembled one-dimensional nanostructures using supramolecular interactions resulting in high bulk conductivity, however, PN2 is remarkable amongst them. Theoretical calculations also reveal that the PN2 exhibits low lying HOMO and LUMO energy levels suggest that the desirable catalyst for hydrogen production. Consequently, PN2 implies photocatalytic hydrogen production with a high rate promotes transition metalated linear π-conjugated D–A systems are highly enviable photocatalysts for future cleanest energy production and storage applications.
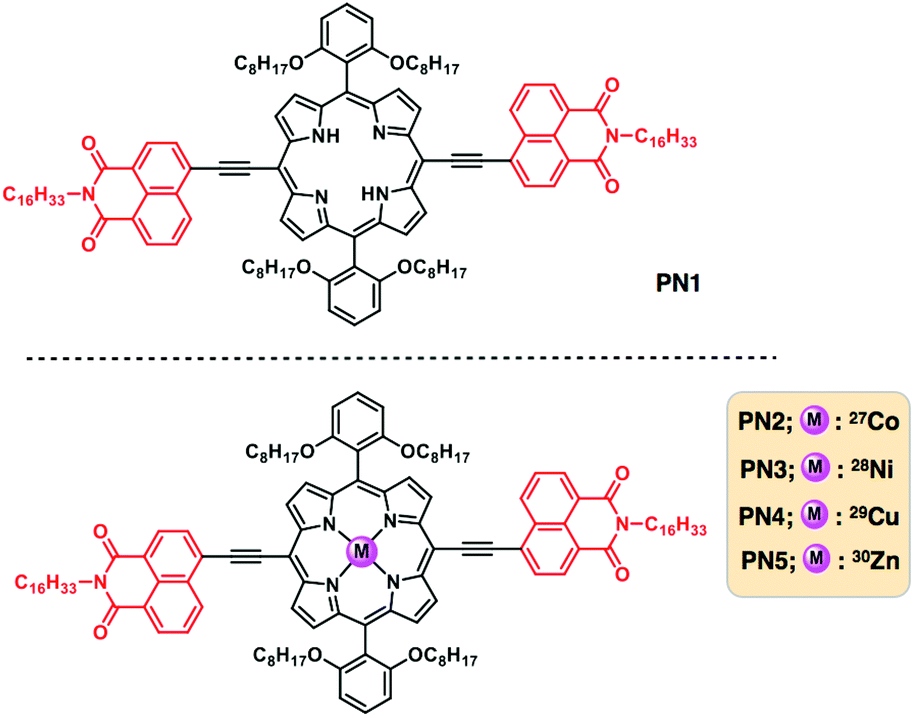 | ||
| Fig. 1 Chemical structures of the free base and transition metalated porphyrin–napthlimide series and represented PN1 for the free base, PN2–PN5 for Co(II), Ni(II), Cu(II) and Zn(II) metalated derivatives. | ||
Results and discussion
UV-visible absorption spectroscopic analysis of freebase and metalated porphyrin-based D–A systems were carried out in solution state at a concentration of 1 × 10−4 M. Fig. 2a represents the absorption spectra of PN1–PN5 in chloroform showed prominent absorption bands at 400–500 nm and 570–750 nm, respectively. The typical absorption spectrum of porphyrin exhibits two significant bands at 400 nm and 550 nm, which correspond to strong and weak electronic transitions from S0 to S2 and S0 to S1. These absorption bands indicate the Soret band or B-band and Q-bands, which arise due to π–π* transitions of the porphyrin moiety. Whereas in PN series (PN1–PN5), the spectral bands show bathochromic shift compared to the typical porphyrin core suggesting that the PN series consisting of porphyrin-linked naphthalimide facilitates extended π-conjugation, which promotes red shift absorption towards the visible region (Table S1†). However, transition metalated derivatives (PN2–PN5) exhibit a blue shift at the Q-band region compared to the free base (PN1), indicating that the vibronic states were stabilized upon metal binding in the porphyrin core. On the other hand, PN2 and PN3 showed a significant hypsochromic shift of ∼20–30 nm than that of PN4 and PN5, suggesting that Co, Ni metalated PN series has low lying ground state energy levels and higher stability relatively. Thus, the ground state spectra of the PN series showed a broad absorption from UV to visible range and also considerable spectral variations in the free base and metallated derivatives.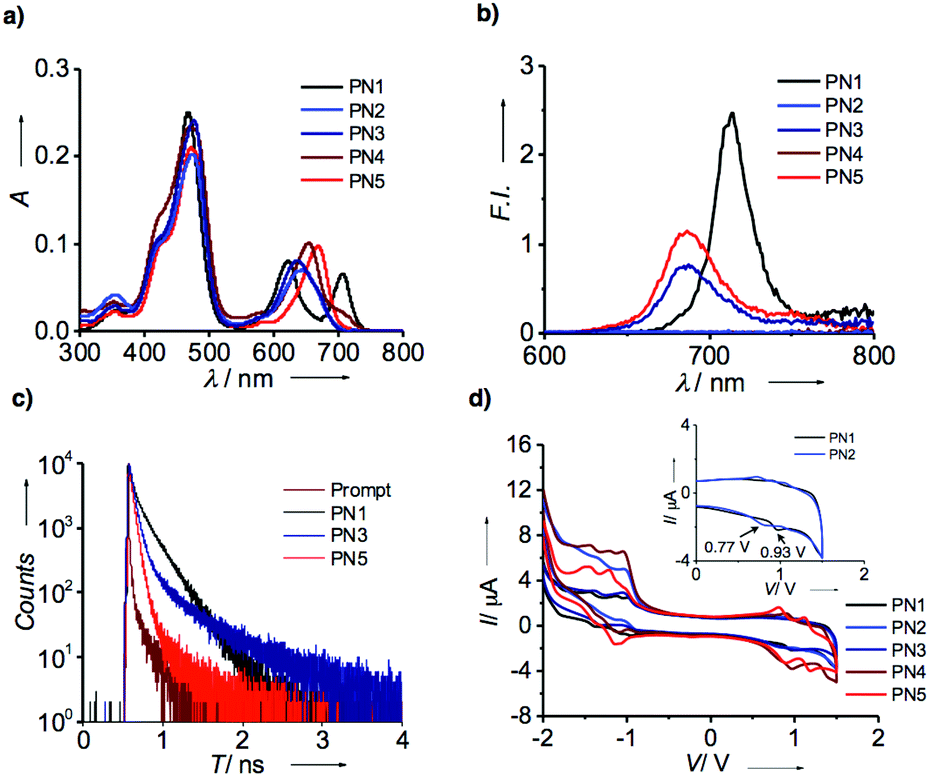 | ||
| Fig. 2 (a) Ultraviolet-visible absorption spectra of PN series (PN1–PN5) in CHCl3 at a concentration of 1 × 10−4 M. (b) Fluorescence spectra of PN Series at an excitation wavelength of 470 nm. (c) Time correlated single photon counting spectra of PN1, PN3 and PN5 using an excitation wavelength of 440 nm. (d) Cyclic voltammetry of PN series in CHCl3 at a scan rate of 200 mV s−1 (inset represents the oxidation potentials of PN1 and PN2 at 0–2 V). | ||
Subsequently, emission spectral data on the PN series in chloroform were recorded upon excitation using the wavelength of 470 nm with an optical density of 0.15 (Fig. 2b). Fluorescence spectra of PN1 showed the emission maximum at 712 nm, while PN3 and PN5 at 685 nm with significant quenching of the emission intensity suggest the possibility of electron transfer in metalated derivatives. On the contrary, PN2 and PN4 did not show emission intensity due to the paramagnetic nature of Co and Cu substituent's influence on the loss of excited-state energy by passing through the intersystem crossing. Furthermore, lifetime analysis of PN1, PN3 and PN5 were performed to confirm the electron transfer mechanism using the time-correlated single-photon counting technique with an excitation wavelength of 440 nm. The resultant spectral data revealed that three derivatives displayed a biexponential decay profile with significant differences in lifetime values suggesting that the electron transfer is more favoured in metalated derivatives than the free base (Fig. 2c). As aforementioned, PN2 and PN4 did not show fluorescence thereby lifetime values were not observed. The ground and excited-state electronic properties confirmed that porphyrin and NMI appended PN series depict extended visible absorption and efficient electron transfer phenomenon in metalated derivatives.
Later, the redox-active nature of the PN series was analyzed by cyclic voltammetry (CV) at a scan rate of 200 mV s−1. To examine their electrochemical properties, we measured each sample from the PN series in 0.1 M tetrabutylammonium hexafluorophosphate (NBu4PF6) as a supporting electrolyte in chloroform followed by the three electrode system; saturated calomel, glassy carbon, platinum (Pt) wire as a reference, working and counter electrode, respectively. Fig. 2d represents the redox spectra of PN1–PN5 showed quasi-reversible oxidation and reduction potentials (Table S1†). Freebase PN1 displays oxidation potentials of 0.934 V, while PN2 exhibits 0.77 V (inset Fig. 1b). Likewise, other derivatives PN3–PN5 perform relatively similar to each other (Table S1†). Detailed CV analysis revealed that PN2 showed lower oxidation potentials compared to that of free base and other metalated PN series indicating that Co(II) substituent plays a vital role to ease of oxidation among other metal substituents, which entails an efficient electron transfer from the donor to acceptor.
Having confirmed that the optical and electrochemical properties of metalated derivatives had prominent ground/excited state electron properties and superior redox potentials prompts us to investigate their surface morphology. As mentioned above, porphyrins possess planarity and symmetry aid to form hierarchical self-assembled nanostructures. Whereas, in the PN series, tethering of porphyrin and NMI enhances conjugation leading to extended π-structure perhaps promoting long axial growth of well-defined nanostructures when compared to individual porphyrin. Thus, we have prepared aggregates of PN series by the methanol vapour diffusion approach mechanism. Subsequently, each of PN series were taken in 1 ml chloroform solution separately in a vial and placed in methanol for 24 h, resulting in green-coloured aggregates. These aliquots were analyzed using optical, X-ray diffraction and electron microscopic techniques. Initially, UV-visible absorption studies of aggregates were recorded in methanol at 25 °C. Fig. 3a represents absorption spectra of PN1 and PN2 showing the hypsochromic shift at the Soret band Q-band when compared to solution-state, indicating that these molecules exist as H-aggregates (Fig. 3a). Likewise, aggregates of PN3–PN5 also performed in methanol and showed similar trends (Fig. S15†). Therefore, absorption spectra of the PN series suggest that these five molecules are arranged in head-to-head stacking, however, variations in a blue shift of each PN derivative might differ in molecular packing whilst undergoing self-assembly.
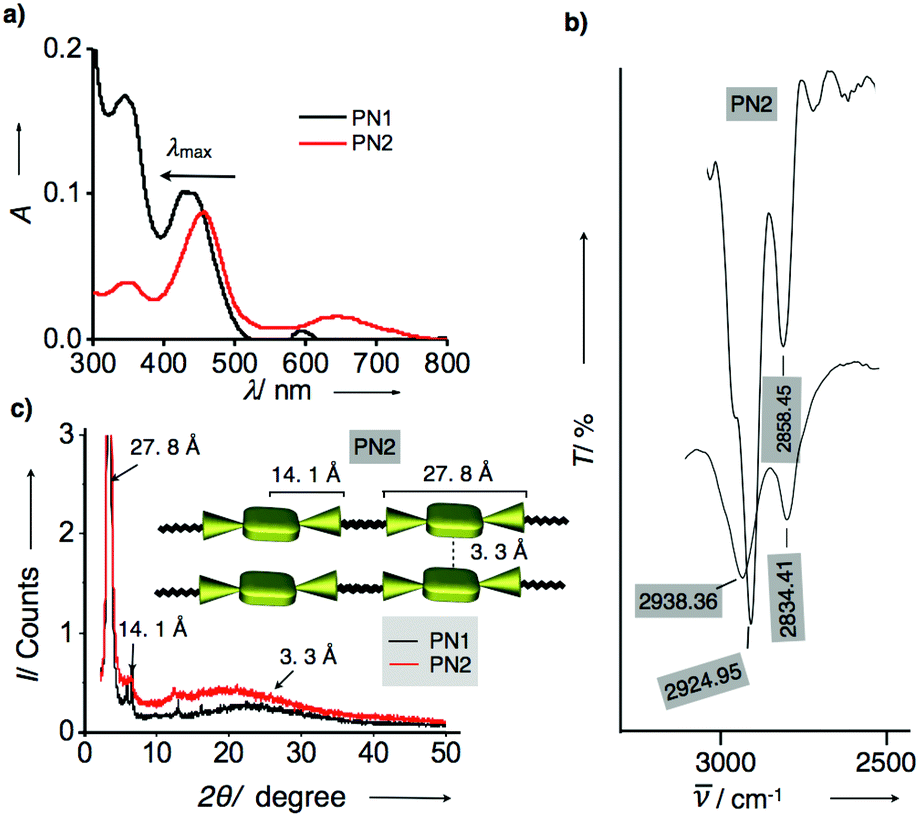 | ||
| Fig. 3 (a) UV-visible absorption spectra of PN1 and PN2 aggregates in methanol at 25 °C. (b) FT-IR spectra of PN2 in solution and aggregate state recorded in chloroform and methanol. (c) Powder X-ray diffraction analysis of PN1 (black line) and PN2 (red line) aggregates and denoted d-spacing values for PN2. Inset shows the schematic illustration of molecular stacking in PN2 aggregates and represented d-spacing values for each length of end-to-end, center to end and π–π stacking. | ||
Furthermore, Fourier transform infrared spectroscopic analysis (FT-IR) of PN series were carried out in the solution and aggregate state using chloroform and methanol to ensure the stretching of long alkyl chains whilst aggregation (Fig. S15†). In chloroform, the CH2 stretching frequencies of PN2 were observed at 2924.95 and 2858.45 cm−1, respectively. Whereas, CH2 stretching frequencies of aggregated samples have shown a significant shift to 2938.36 and 2834.41 cm−1 as methanol solvent interference in CH2 stretching frequency results in the shift of asymmetric and symmetric methylene stretching frequencies. Thus, FT-IR analysis revealed that the presence of hexadecyl chains in PN2 are stretched confirmation, which facilitated interdigitation with other molecules whilst under self-assembly (Fig. 3b and S15†).14 Subsequently, the other four derivatives PN1, PN3–PN5 were also studied, and their spectral characteristics resembled PN2. However, minor changes in the stretching frequencies indicate that the presence of distinct metals and freebase in the PN series directs variation in molecular packing, which defines divergent nanostructures from one to another. Furthermore, aggregates were analyzed using the powder X-ray diffraction method to confirm the molecular arrangement and their crystallinity whilst self-assembly at 25 °C. Fig. 3c represents that PN1 and PN2 exhibit sharp diffraction peaks at small and wide-angle regions signifying that molecules exhibit crystalline nature. PN2 showed a broad peak observed at a small angle region, which corresponds to the d-spacing values of 3.3 Å, suggesting intermolecular π–π stacking between molecules. However, other diffraction peaks with d-spacing values of 27.8 Å and 14.1 Å represent the end-to-end length of the NMI–porphyrin–NMI and metal centre to NMI reveals that the rigidity of the π-conjugated structure is extending over supramolecular nanostructures (Inset Fig. 3c). Along with that, hexadecyl chains also play a vital role in aggregation via van der Waal's interactions. On the other hand, PN1, PN3–PN5 also showed similar diffraction peaks similar to PN2 but considerable differences in d-spacing values suggest that stacking patterns differ from each other (Fig. S16†). Thus, the resultant UV-visible absorption, IR analysis and PXRD depict that the PN series undergoes H-aggregation leading to extended supramolecular nanostructures via π–π stacking and van der Waals interactions. Nevertheless, the variation in molecular packing of each PN series is still unclear, thereby, microscopic visualization and their electron diffraction pattern are considered as a significant approach to studying the molecular stacking, alignment and supramolecular structures at the nanoscopic level.
Furthermore, we measured the surface morphology of aggregates of the PN series to check their self-assembled nanostructures and alignment by using electron microscopic and diffraction techniques. Scanning electron microscopy of PN1 revealed nanorod-shaped structures with an average width of 0.5–1 micrometer and 3–4 μm in length (Fig. 4a). Subsequently, transmission electron microscopy studies have been conducted by drop-casting the suspension of PN1 in methanol at 25 °C. The TEM image of PN1 displayed nanorod morphology with their dimensions similar to the SEM image (Fig. 4b). Eventually, we have performed electron diffraction analysis on the area same as in the TEM image and found that PN1 did not show the precise molecular alignment in nanorod morphology (Fig. 4c). In contrast, the SEM image of PN2 depicts mesh wreath-type morphology wherein thin nanowires were aligned and joined together (Fig. 4d). In addition, small pores were observed in this morphology that showed a greater advantage when applied to photocatalytic hydrogen production. Interestingly, TEM images revealed well-aligned nanowires with an average width of 10 nm and length of 50–300 nm (Fig. 4e). Whereas, high-resolution TEM images show that these nanowires are unidirectionally aligned and connected to each other (Fig. 4g and h). Afterwards, diffraction analysis confirmed that the molecules undergo co-facial stacking, which was aligned in a specific direction, resulting in an ordered one-dimensional nanowires (Fig. 4f and i). Likewise, the morphology of three derivatives PN3–PN5 was also studied and PN3 showed nanorod formation with an average dimension of 0.5–1 μm and several micrometers in length. In some places, these bundles of nanorods were wrapped together forming supramolecular nanoropes. Whereas, PN4 exhibits nanowire morphology with several micrometers in length, which is in contrast to PN2. Subsequently, PN5 was also studied and it showed nanorods shaped structures similar to PN1 and PN3 (Fig. S17†). Also, we performed the electron diffraction analysis on PN3–PN5, and their corresponding images reveal a significant alignment of molecular arrangement in supramolecular nanostructures when compared to PN1, but relatively less than that of PN2. Thus, microscopic analysis suggests that the PN2 ascertain well-aligned molecular arrangement leading to uni-directionality among 1D nanostructures (Fig. 4j). While PN series showed significant molecular alignment in 1D nanostructures, prompting us to perform their electronic characterization for organic electronic applications.
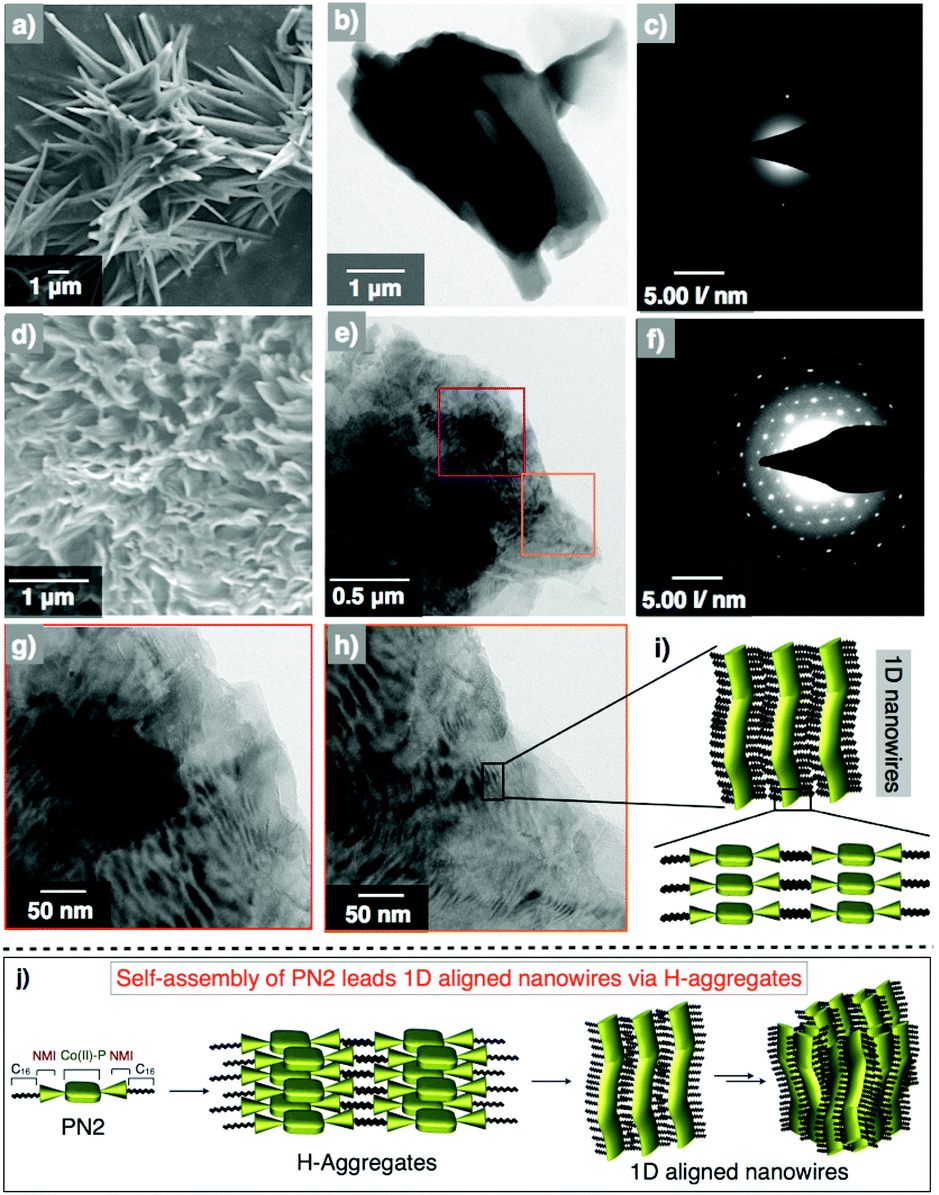 | ||
| Fig. 4 (a, b) SEM and TEM images of an air-dried suspension of PN1 at a concentration of 1 × 10−4 M at 25 °C. (c) The corresponding diffraction pattern of the PN1 image. (d, e) SEM and TEM of an air-dried suspension of PN2. Purple- and orange-coloured boxes denote the are for high-resolution imaging. (f) Diffraction pattern recorded on the TEM images of PN2. (g, h) High-resolution TEM images of PN2 from the selected area of image-e. (i) Proposed scheme of 1D nanowires from the molecular aggregates. (j) Schematic illustration of PN2 undergoes self-assembly leading to 1D nanowires via H-aggregates. | ||
Hence, the electrical conductivity of the PN series was analyzed using electrochemical impedance studies (Fig. 5, S18–S22 and Table S2†). To assess the conductivity behavior of these assemblies, we coated the aggregates of each PN series on indium tin oxide glass plate suitably etched to provide 1 × 1 cm cell dimensions and recorded the spectra. Fig. 5a represents Nyquist plots of PN1–PN5 at 25 °C that revealed typical depressed semi-circular curves characteristic of contributions from both bulk resistance (Rb) and capacitance (Cb) when in a parallel combination. Amongst all, Co(II) metalated PN2 showed the least bulk resistance (Rb) of 400 MΩ, which corresponds to a specific conductivity of ∼33 mS cm−1. Whereas, other PN derivatives displayed slightly higher resistance in the range of 500–2000 MΩ that relates to specific conductivities in the range of 3–23 mS cm−1, respectively (Fig. 5d and Table S2†). Although metalated PN series have achieved remarkably higher bulk conductivity than PN1, Cu(II) substituent PN4 yielded lower conductivity possibly to poor Ohmic contacts at the electrode interface with irregular and entangled nanowires. Subsequently, the temperature-dependent behavior of the PN series was recorded as a function of temperature from 25 °C to 65 °C that displayed a steady rise in the resistance of bulk (Fig. 5b and S18–S22†). This steady increase in the resistance against temperature is indicative of the predominant contribution of electronic conduction within the bulk (Fig. S18†). Furthermore, the bulk capacitance of PN2 was estimated from the significant parameters at the apogee of the semicircular arc where the condition ωRCbulk = 1 is satisfied. From the radial peak frequency (ωpeak = 2πfpeak) obtained from the spectroscopic plots of imaginary impedance (Z′′) against log (f) the bulk relaxation time (τb), of PN2 and rest of the samples were estimated (Fig. 5c and Table S2†). Consequently, the calculated capacitance of PN2 is 3.7 nF at 25 °C. Likewise, studies on PN1, PN3–PN5 were also conducted and the bulk capacitance values were calculated with the variation of temperature from 25 °C to 65 °C (Fig. S19–S22 and Table S2†). Overall, these results confirmed that PN2 exhibits the highest bulk electrical conductivity and capacitance values among the PN series.
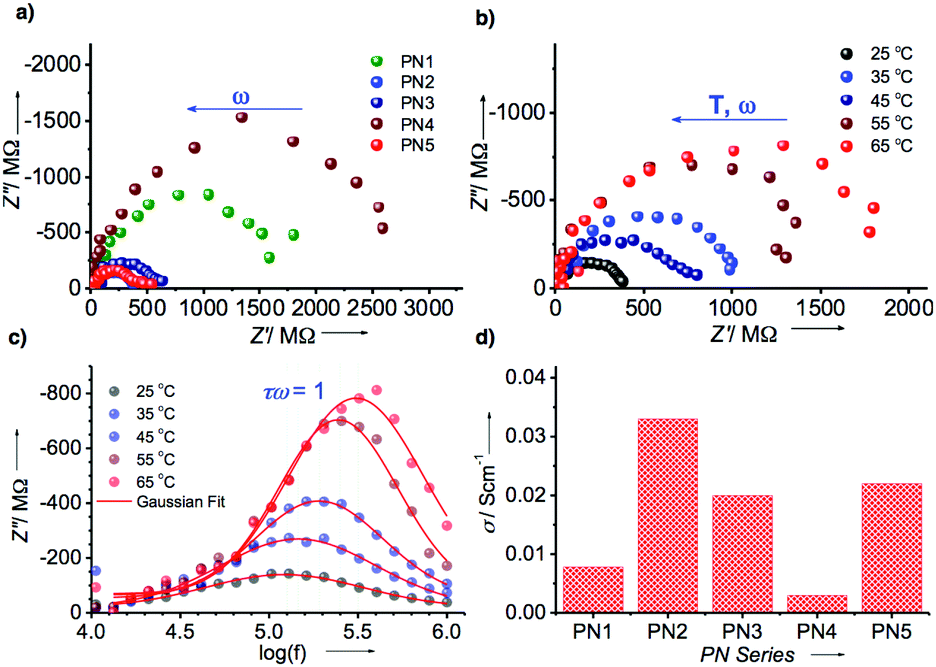 | ||
| Fig. 5 (a) Nyquist plot of PN1–PN5 derivatives. (b) Temperature-dependent Nyquist plot of PN2 with ranging from 25 °C–65 °C. (c) Temperature-dependent logarithmic frequency (log (f)) against imaginary impedance of PN2. (d) The bar diagram of the PN series vs. specific conductivity at 25 °C. | ||
As mentioned above, microscopic and impedance analysis of PN2 displayed well-aligned 1D nanowires with high electrical conductivity among other transition metal substituted PN series, prompting us to utilize PN2 as a catalyst for hydrogen evolution. Alongside, we also performed the density functional theory (DFT) calculations to check the appropriate energy levels of the PN series with the TiO2 electrode, are a foremost criterion to be considered for the catalyst. Thereby, theoretical calculations of the PN series were studied using Gaussian 09 package with a functional basis set of the B3LYP/6-31G(d,p) level. The optimized structure of PN1 shows the electron density distribution on the highest occupied molecular orbital (HOMO) on the porphyrin moiety and the lowest unoccupied molecular orbital (LUMO) on the porphyrin–NMI and the resultant values were 5.03 and 2.98 eV, respectively (Fig. 6). On the other hand, electron density allocation of HOMO−1 and LUMO+1 on porphyrin and napthalimide exhibits unequivocal discrimination (Table S3†). The calculated band gap of PN1 from the HOMO and LUMO yielded 2.05 eV. Subsequently, values for transition metalated PN series (PN2–PN5) were also calculated and the resultant HOMO and LUMO values were lower than PN1 (Fig. 6 and Table S3†). Electrostatic potentials (ESP) of the PN series displayed similar electron density distribution in five derivatives (Table S3†). However, low lying HOMO levels and band gap variations suggest that the metalated derivatives were more suitable with TiO2 nanoparticles than freebase (PN1). Amongst, Co(II) substituent PN2 exhibits low lying HOMO and LUMO energy levels, hence PN2 is a potential candidate to apply as a catalyst for hydrogen production.
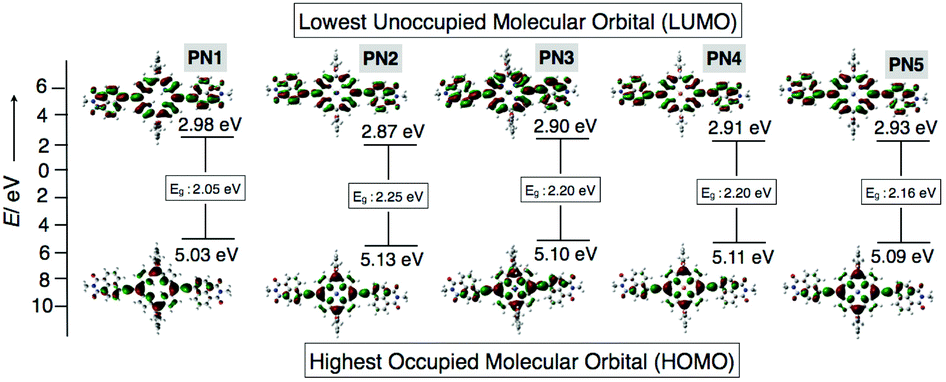 | ||
| Fig. 6 Optimized structures of PN series (PN1–PN5) from DFT calculations. The corresponding HOMO/LUMO energy levels and band gap values. | ||
Thus, photocatalytic hydrogen evolution experiments were performed using PN2 as a catalyst with aq. triethanolamine (TEOA) solution (20 vol%) as a sacrificial agent and platinum as the cocatalyst for titanium oxide (TiO2) electrode. Fig. 7a represents hydrogen yields of PN2 recorded under visible light irradiations (λ = 420 nm) up to 4 h at neutral, acidic and basic pH conditions. At neutral pH, hydrogen yields exhibit 15 mmol g−1 until 4 h irradiation. Later, acidic and basic pH values were also recorded by controlled addition of concentrated HCl which results in higher H2 yield in basic media when compared to acidic and neutral pH. Subsequently, the H2 production rate was calculated and those values were remarkably higher than the previous porphyrin-based D–A systems reported until now.15 The resultant H2 rate of PN2 is 17.5 mmol g−1 h−1 (pH = 10), 15.7 mmol g−1 h−1 (pH = 7) and 4.0 mmol g−1 h−1 (pH = 3) (Fig. 7b). In addition, turnover number (TON) and TOF were calculated for PN2 to determine the number of reacting molecules in comparison with the active sites whilst irradiation time. The resultant values are 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 790.84 and 16197.71, respectively. Nevertheless, the stability of the photocatalyst is rather significant for hydrogen production. Thereby, we conducted the stability studies of PN2 by extending the irradiation time from 4 h to 16 h revealed that the H2 production rate remains constant still for a longer time (Fig. 7c). Furthermore, we recorded photocurrent against time under the light on/off conditions suggesting that the photocurrent of Pt/TiO2–PN2 is higher than that of Pt/TiO2 alone and stable until 550 s (Fig. 7d). Overall, Co(II) substituent PN2 showed a higher H2 production rate and stability owing to the LUMO energy levels close to the conduction band of TiO2, which implies efficient electron transfer from the dye to the TiO2 electrode (Fig. 7e). Therefore, a well-defined 1D molecular orientation of Co(II) substituted PN2 with high electrical conductivity leading to photocatalytic H2 production facilitates enhancing the performance of organic electronic devices.
790.84 and 16197.71, respectively. Nevertheless, the stability of the photocatalyst is rather significant for hydrogen production. Thereby, we conducted the stability studies of PN2 by extending the irradiation time from 4 h to 16 h revealed that the H2 production rate remains constant still for a longer time (Fig. 7c). Furthermore, we recorded photocurrent against time under the light on/off conditions suggesting that the photocurrent of Pt/TiO2–PN2 is higher than that of Pt/TiO2 alone and stable until 550 s (Fig. 7d). Overall, Co(II) substituent PN2 showed a higher H2 production rate and stability owing to the LUMO energy levels close to the conduction band of TiO2, which implies efficient electron transfer from the dye to the TiO2 electrode (Fig. 7e). Therefore, a well-defined 1D molecular orientation of Co(II) substituted PN2 with high electrical conductivity leading to photocatalytic H2 production facilitates enhancing the performance of organic electronic devices.
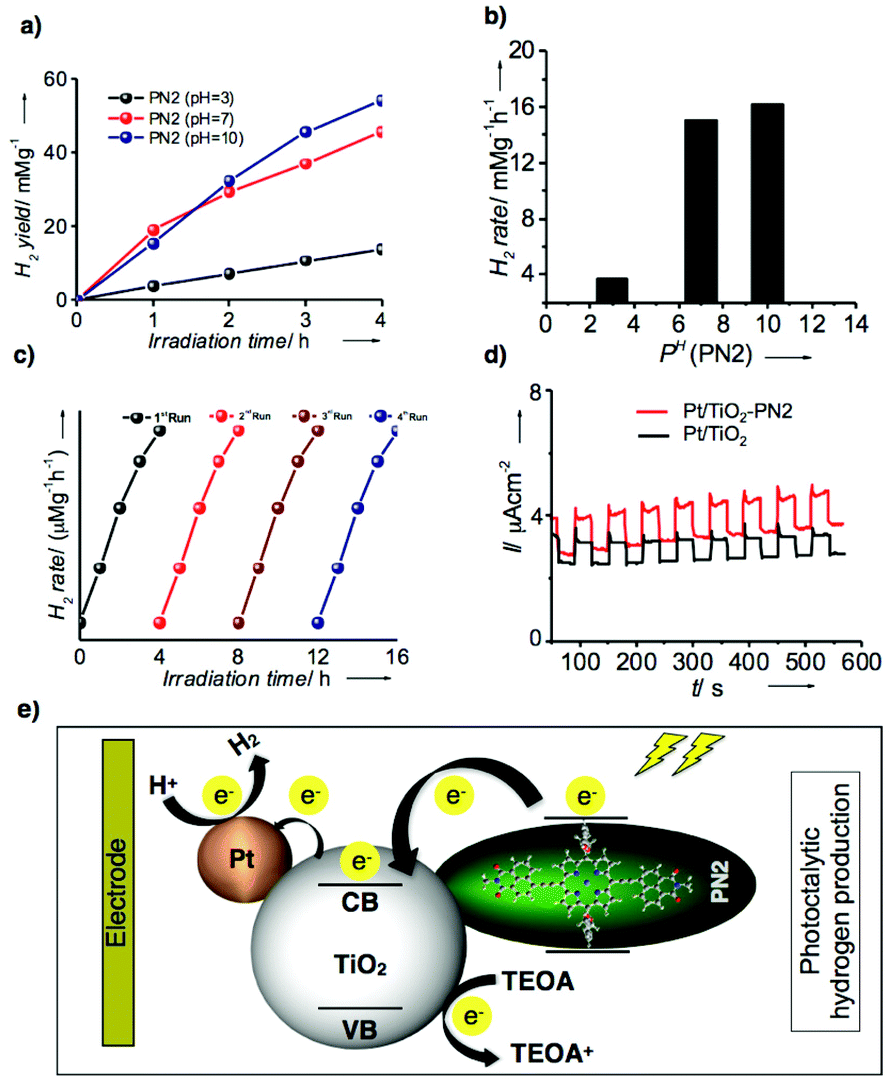 | ||
| Fig. 7 (a) Photocatalytic activity of PN2 at different PH variations (acidic, neutral and basic conditions). (b) Bar diagram of H2 rate against PH of PN2. Stability studies: (c) hydrogen production rate against the time variation from 0 h to 16 h. (d) Photocurrent response vs. time (seconds) between Pt/TiO2–PN2 and Pt/TiO2. (d) Schematic representation of possible electron transfer mechanism and hydrogen evolution under visible light. | ||
Conclusions
In conclusion, we have demonstrated the five porphyrin–napthelimide (PN1–PN5) as donor–acceptor systems in which PN2 showed well-aligned molecular arrangements directing 1D nanowires, which exhibit high electrical conductivity and photocatalytic hydrogen production. The optical and electrochemical properties revealed that metalated derivatives displayed efficient intramolecular electron transfer from the donor to acceptor than free base. Microscopic data and diffraction analysis suggested that the precise molecular packing of Co(II) substituent PN2 exclusively formed aligned nanowires among other derivatives. Hence, it showed remarkable bulk electrical conductivity and accomplished high hydrogen production rate under visible light. Consequently, transition metal substituted macrocyclic-based D–A systems to be explored in detail for realistic challenges in clean and green technology.Author contributions
S. P., B. B. and L. G. designed the experiment, analysis of data and wrote the manuscript. B. B. and G. R. executed the synthesis, characterization, spectroscopic measurements of the PN series. N. C. and U. P. performed the photocatalytic hydrogen production analysis. P. B. conducted electrochemical impedance studies. V. K. measured the microscopic and electron diffraction analyses. All authors participated in the discussion of results and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. P. thanks to the Department of Science and Technology and Inspire programme (DST/INSPIRE/04/2015/001457) for funding support. B. B., N. C., and V. K. acknowledge DST-Inspire, UGC and CSIR for fellowship. We thank Director, CSIR-IICT for his support (IICT/pubs./2021/308).Notes and references
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS; Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201 CrossRef PubMed; C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Adv. Mater., 2020, 32, 1907296 CrossRef PubMed; Y. Bai, K. Hippalgaonkar and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 16222 RSC; F. Liu, M. Wang, X. Liu, B. Wang, C. Li, C. Liu, Z. Lin and F. Huang, Nano Lett., 2021, 21, 1643 CrossRef; K. Kondo, Y. Watanabe, J. Kuno, Y. Ishii, S. Kawasaki, M. Kato, G. Kalita, Y. Hattori, O. Mashkov, M. Sytnyk and W. Heiss, Energy Technol., 2021, 9, 2100123 CrossRef.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC; M. G. Kibria, F. A. Chowdhury, S. Zhao, B. Alotaibi, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2015, 6, 6797 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed; J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970 CrossRef PubMed.
- L. Li, Z. Cai, Q. Wu, W. Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681 CrossRef CAS.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712 CrossRef CAS PubMed; J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef; C. Shu, C. Han, X. Yang, C. Zhang, Y. Chen, S. Ren, F. Wang, F. Huang and J.-X. Jiang, Adv. Mater., 2021, 33, 2008498 CrossRef PubMed.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265 CrossRef CAS PubMed; Y. S. Kochergin, D. Schwarz, A. Acharjya, A. Ichangi, R. Kulkarni, P. Eliášová, J. Vacek, J. Schmidt, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2018, 57, 14188 CrossRef; R. S. Sprick, C. M. Aitchison, E. Berardo, L. Turcani, L. Wilbraham, B. M. Alston, K. E. Jelfs, M. A. Zwijnenburg and A. I. Cooper, J. Mater. Chem. A, 2018, 6, 11994 RSC; Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063 CrossRef PubMed.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632 Search PubMed; P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423 CrossRef CAS PubMed; K. C. Ranjeesh, R. Illathvalappil, V. C. Wakchaure, G. Gouda, S. Kurungot and S. S. Babu, ACS Appl. Energy Mater., 2018, 1, 6442 CrossRef; K. C. Ranjeesh, L. George, A. Maibam, S. Krishnamurty and S. S. Babu, ChemCatChem, 2021, 13, 1717 CrossRef.
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400 CrossRef CAS PubMed; S. Ghosh, A. Nakada, M. A. Springer, T. Kawaguchi, K. Suzuki, H. Kaji, I. Baburin, A. Kuc, T. Heine, H. Suzuki, R. Abe and S. Seki, J. Am. Chem. Soc., 2020, 142, 9752 Search PubMed.
- Y. Zhong, J. F. Wang, R. F. Zhang, W. B. Wei, H. M. Wang, X. P. Lu, F. Bai, H. M. Wu, R. Haddad and H. Y. Fan, Nano Lett., 2014, 14, 7175 CrossRef CAS PubMed; Y. Z. Chen, A. X. Li, Z. H. Huang, L. N. Wang and F. Y. Kang, Nanomaterials, 2016, 6, 51 CrossRef PubMed; N. Zhang, L. Wang, H. M. Wang, R. H. Cao, J. F. Wang, F. Bai and H. Y. Fan, Nano Lett., 2018, 18, 560 CrossRef PubMed; Q. Li, N. N. Zhao and F. Bai, MRS Bull., 2019, 44, 172 CrossRef.
- A. G. Mojarrad and S. Zakavi, ACS Appl. Mater. Interfaces, 2020, 12, 46190 CrossRef CAS; G. B. Bodedla, J. Huang, W. Wong and X. Zhu, ACS Appl. Nano Mater., 2020, 3, 7040 CrossRef; F. Zeng, J. Liao, M. Ding and G. Ou, Dyes Pigm., 2021, 192, 109430 CrossRef; V. Nikolaou, G. Charalambidis and A. G. Coutsolelos, Chem. Commun., 2021, 57, 40554058 RSC; Y. Kobayashi, A. Muranaka, K. Kato, A. Saeki, T. Tanaka, M. Uchiyama, A. Osuka, T. Aida and T. Sakurai, Chem. Commun., 2021, 57, 120 Search PubMed; E. Nikoloudakis, M. Pigiaki, M. N. Polychronaki, A. Margaritopoulou, G. Charalambidis, E. Serpetzoglou, A. Mitraki, P. A. Loukakos and A. G. Coutsolelos, ACS Sustainable Chem. Eng., 2021, 9, 7781 CrossRef.
- M. Mrinalini, B. S. P. Achary, S. Ghosh, D. Koteshwar, S. Prasanthkumar and L. Giribabu, J. Phys. Chem. C, 2018, 122, 10255 CrossRef CAS; M. Mrinalini, S. S. Pathak, B. S. Achary, L. S. Panchakarla and S. Prasanthkumar, Chem. – Asian J., 2019, 14, 537 CrossRef PubMed; M. Mrinalini, M. Naresh, S. Prasanthkumar and L. Giribabu, J. Porphyrins Phthalocyanines, 2021, 25, 382 CrossRef; B. Bhavani, M. Mrinalini, J. V. S. Krishna, P. Basak, L. Giribabu and S. Prasanthkumar, ACS Appl. Electron. Mater., 2021, 3, 176 CrossRef.
- D. L. Reger, J. J. Horger and M. D. Smith, Chem. Commun., 2011, 47, 2805 RSC; J. A. Kitchen, P. N. Martinho, G. G. Morgan and T. Gunnlaugsson, Dalton Trans., 2014, 43, 6468 RSC; S. Pagidi, N. K. Kalluvettukuzhy and P. Thilagar, Organometallics, 2018, 37, 1900 CrossRef CAS; N. Meher and P. K. Iyer, Angew. Chem., Int. Ed., 2018, 57, 8488 CrossRef PubMed; S. Sahin Un, S. Zehra Topal and Y. Zorlu and, J. Lumin., 2017, 190, 23 CrossRef; G. B. Bodedla, L. L. Li, Y. Y. Che, Y. J. Jiang, J. Huang, J. Z. Zhao and X. J. Zhu, Chem. Commun., 2018, 54, 11614 RSC; G. B. Bodedla, G. Tang, J. Zhao and X. A. Zhu, Sustainable Energy Fuels, 2020, 4, 2675 RSC.
- D. R. Roy, E. V. Shah and S. M. Roy, Spectrochim. Acta, Part A, 2018, 190, 121 CrossRef CAS; E. V. Shah, V. Kumar, B. K. Sharma, K. Rajput, V. P. Chaudhary and D. R. Roy, J. Mol. Model., 2018, 24, 239 CrossRef PubMed.
- W. Jin, Y. Yamamoto, T. Fukushima, N. Ishii, J. Kim, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 9434 CrossRef CAS PubMed.
- P. Y. Ho, M. F. Mark, Y. Wang, S. C. Yiu, W. H. Yu, C. L. Ho, D. W. McCamant, R. Eisenberg and S. Huang, ChemSusChem, 2018, 11, 2517 CrossRef CAS PubMed; Z. Chen, J. Wang, S. Zhang, Y. Zhang, J. Zhang, R. Li and T. Peng, ACS Appl. Energy Mater., 2019, 2, 5665 CrossRef; L. Y. Huang, J. F. Huang, Y. Lei, S. Qin and J. M. Liu, Catalysts, 2020, 10, 656 CrossRef; N. Chanda, K. Devulapally, S. Gonuguntla, S. Bojja, U. Pal and L. Giribabu, Mater. Adv., 2021, 2, 4762 RSC; P. S. Gangadhar, S. S. Gonuguntla, S. Madanaboina, N. Islavath, U. Pal and L. Giribabu, J. Photochem. Photobiol., A, 2020, 392, 112408 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr06961f |
| This journal is © The Royal Society of Chemistry 2022 |