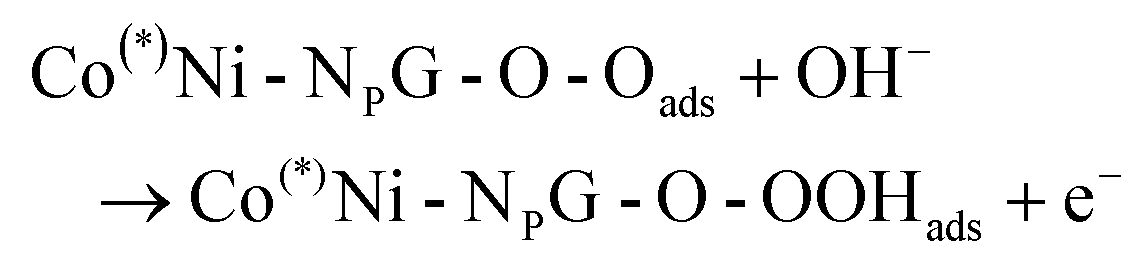DOI:
10.1039/D1NR06334K
(Paper)
Nanoscale, 2022,
14, 187-195
Electronic properties of double-atom catalysts for electrocatalytic oxygen evolution reaction in alkaline solution: a DFT study†
Received
26th September 2021
, Accepted 19th November 2021
First published on 19th November 2021
Abstract
In alkaline solution, the electrocatalytic oxygen evolution reaction (OER) of dual transition metal atom (2TM) nitrogen-decorated graphene as a double-atom catalyst (DAC) has received special attention. Here, using density functional theory (DFT) calculations, the OER electrocatalysis of 2TM-pyridine/amino-nitrogen-decorated graphene (2TM-NPAG and 2TM-NPG. 2TM represents FeCo, FeNi, Conti) is studied. The electrocatalytic OER mechanism is that 2TM-NPG acts as the pre-catalyst, while the real catalysts are 2TM-NPAG and 2TM-NPG-O. In particular, CoNi-NPAG and CoNi-NPG-O exhibit higher OER activity compared to state-of-the-art RuO2 at pH = 14. It is confirmed that the potential-determining step is also the rate-determining step. Amino-nitrogen is the main accepter of electrons from CoNi atoms and pyridine-nitrogen is the main acceptor of electrons from nearby C atoms. The role of different N coordination continues to influence the entire electrocatalytic OER process of CoNi-NG. Simultaneously, the overpotential of CoNi-NG is in a volcano-shaped relationship with the electronic properties (oxidation state or d-band center) of the catalytic site of Co. Moreover, CoNi-NPAG and CoNi-NPG-O are the closest to the center of the OER overpotential (a function of the d-band center and oxidation state) contour plot, implying that they exhibit the best catalytic activity among all the CoNi-NG materials. The optimal electronic properties of CoNi-NPAG and CoNi-NPG-O contribute towards their excellent OER performance, and provide a new breakthrough in developing high-performance DACs.
1. Introduction
As the oxidative half reaction of electrochemical water splitting, the oxygen evolution reaction (OER) is the key reaction in electrochemical technology.1–4 The OER process with four-electron-transfer has a greater kinetic barrier than the thermodynamic equilibrium potential. A considerable overpotential is required to drive the electrochemical reaction, which hinders the large-scale application of water electrolysis.5–8 The reaction pathway of OER is related to the pH value. The electrocatalytic OER occurs mostly under alkaline conditions, experimentally.9 The following elementary steps (eqn (1)–(4)) are dominant in the OER process under alkaline conditions, where * refers to the active site on the electrocatalyst surface.10| | | OH* + OH− → O* + H2O(l) + e− | (2) |
| | | OOH* + OH− → * + O2(g) + H2O(l) + e− | (4) |
The research on electrocatalytic OER is still in its infancy. The demand for developing clean energy electrocatalysts with excellent activity for the OER process has increased over recent years. Owing to the synergistic effect of dual transition metal atom (2TM) active sites, and the special coordination configurations between 2TM and the substrate,11,12 double-atom catalysts (DACs) exhibit enhanced OER catalytic performances in comparison with their single-atom catalyst (SAC) counterparts.3 Therefore, 2TM nitrides have the intrinsic advantages of high conductivity and tunable electronic structure. It is expected that they will be next-generation OER electrocatalysts in alkaline electrolyte.13,14 The OER applications of 2TM nitrides (Table S1†) are still at the initial stage and the theoretical computations are still relatively limited.15 Recently, 2TM-pyridine/amino-nitrogen-decorated graphene has been shown to exhibit significant activity and durability for the OER under alkaline conditions.14 Hu and coworkers16 prepared atomically-dispersed binary Co–Ni sites in nitrogen-doped hollow carbon nanocubes for the OER at pH = 14, in which the synergistic effect of neighboring Co–Ni dual metal atoms ultimately promoted the electrocatalysis of the OER. Lu et al.17 reported the harnessing of the interplay of Fe–Ni atoms embedded in nitrogen-doped carbon for bifunctional oxygen electrocatalysis at pH = 14. These structures of dual transition metal atoms embedded in pyridine nitrogen-doped graphene are universally named as 2TM-NPG. However, the OER catalytic mechanisms involved are inconsistent in the theoretical computations in the above reports. In addition, Zhu et al.18 also reported another neighboring Zn/Co monomer configuration. We named this structure of dual transition metal atoms embedded in pyridine and amino nitrogen-doped graphene as 2TM-NPAG. The catalytically active sites can be mainly ascribed to Fe/Co/Ni metal atoms with variable valence electrons in 3d orbitals, while the Ti/V/Mn/Cu/Zn metal atoms usually play a supplementary role in the OER activity.4 Thus, a fundamental understanding of the mechanism of Fe/Co/Ni based electrocatalysts is an important step in the developing of principles for OER catalyst design.
As is known, the catalytic state is essentially far from the equilibrium state, which complicates the direct observation of this process. In May 2021, Chueh et al.19 found that particles swelled to form an α-CoO2H1.50.5H2O-like structure-produced at pre-catalytic voltages, in which the oxidation number of cobalt was +2.5. Upon increasing the voltage to drive oxygen evolution, contracted β-CoOOH particles containing Co3+ species were formed, which significantly improved the OER catalytic activity. In June 2021, Yao et al.20 reported a type of structurally stable bimetallic FeNi-metal–organic framework (MOF_ nanoarray with self-optimized electrocatalytic activities in the OER. Such a unique dynamic phenomenon is related to the gradual valence increments of Fe ions in MOFs, which triggers a continuous performance improvement before the optimal steady state is reached. Thus, quantitative charge transfer at the catalytic sites significantly improves the OER activity. At the same time, on 16 June 2021, Zhou et al.21 demonstrated a volcano-shaped relationship for a NiFe-MOF between its d-band center, eg filling and OER activity, and found that it has an optimized energy level and electronic structure situated close to the volcano summit. Therefore, the study of electronic properties, such as the oxidation state and d-band center of the catalytic site, will help aid the design of novel electrocatalysts with a stable structure and excellent oriented catalytic properties for electrocatalytic OER.
In this work, we systematically investigated the catalytic activities of 2TM-pyridine/amino-nitrogen-decorated graphene (2TM-NG) as an OER electrocatalyst via density functional theory (DFT) calculations. The 2TM-NG includes six systems of 2TM-NPAG and 2TM-NPG, in which 2TM represents FeCo, FeNi or CoNi. The OER activities of 2TM-NG under alkaline conditions (pH = 14) demonstrate that CoNi-NG exhibits the best OER activity. Then, the catalytic mechanism of the OER at pH = 14 was proposed by analyzing the catalytic pathway of CoNi-NG in detail. Finally, based on the Bader charge analysis, charge density differences, spin-polarized density of states and d-band center, the origin of the excellent activity of CoNi-NG was further revealed.
2. Research methods
2.1 Computational details
All of the DFT calculations were performed using the first principles-based Vienna ab initio simulation package (VASP).22–24 The projector augmented wave (PAW) potentials25 and Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional26 were adopted in the calculations with a plane-wave cut-off energy of 520 eV. Spin polarization, dipole moment and van der Waals corrections were taken into consideration. The energy convergence criterion was set to 10−5 eV while the force converged to less than 0.02 eV Å−1 on each ion. A 2 × 2 × 1 Monkhorst–Pack k-point mesh was used to sample the first Brillouin zone for structural optimizations, while 7 × 7 × 1 sampling was used for the spin-polarized density of states calculations. Gaussian smearing with a width of σ = 0.05 eV was used. A 7 × 7 supercell with a periodic boundary condition along the x–y plane was employed to model the infinite graphene sheet. A vacuum layer greater than 15 Å in the z direction was used, so that the interaction among periodic images along the z direction could be neglected. The transition states (TS) were located by using the climbing image nudged elastic band method (CI-NEB), where the force converged to less than 0.05 eV Å−1 on each ion. An implicit solvation model was calculated using VASPsol to illustrate the solute-water solvent interactions in different electrochemical systems.27 The d-band center of the d-states was calculated from the spin-up and spin-down d-states.
2.2 Catalyst models
Based on the previous computational and experimental studies, graphene samples with pyridine- or amino-nitrogen decorative vacancies are generally stable, thus, the configurations of 2TM-NPAG and 2TM-NPG (Fig. S1†) were constructed.16–18 As shown in Fig. 1, there are four pyridine-N atoms and two amino-N atoms in the 2TM-NPAG unit cell, and six pyridine-N atoms in the 2TM-NPG unit cell.
 |
| | Fig. 1 The geometric structures of (a) 2TM-NPAG and (b) 2TM-NPG. | |
According to the calculations, 2TM (FeCo, FeNi or CoNi) can adopt an optimal coordination configuration (2TM-NPAG) with formation energies (Ef) of −3.77, −4.01 or −2.89 eV, respectively, which are more negative than the corresponding energies of 2TM-NPG (−2.47, −3.22 or −2.84 eV) (Fig. S2†). This indicates that for each 2TM combination the formation probability of 2TM-NPAG is higher than that of the corresponding 2TM-NPG. In particular, the Ef values of CoNi-NPAG (−2.89 eV) and CoNi-NPG (−2.84 eV) are relatively approximate, which means that the possibility of forming these two structures is analogous.
The binding energies of 2TM-NPAG and 2TM-NPG were calculated as descriptors for the bimetal atom stability against metal atom aggregation. As shown in Fig. S3,† the binding energies of 2TM (FeCo, FeNi, and CoNi) in NPAG are −17.60, −16.57 and −16.60 eV respectively, which are lower than those of −12.49, −11.98 and −12.74 eV in NPG. Thus the ability of NPAG to trap 2TM is higher than that of NPG. The binding energies are lower than the cohesive energy Ecoh (eqn (s3)†) (−4.91 eV for Fe, −4.97 eV for Co, and −5.06 eV for Ni). Thus, the FeCo, FeNi and CoNi atoms being embedded in the substrates is energetically more favorable than metal atom aggregation.
3. Results and discussion
3.1 OER activity under alkaline conditions
The OH radical adsorption on the electrocatalysts (OH*) is the first step of the OER process under alkaline conditions, as shown in Fig. S4 and S5.† Its initial adsorption configuration and the adsorption energy (Eads) play important roles in the subsequent O and OOH radical adsorption on the electrocatalysts (O* and OOH*).28 We added the calculation of optimized OOH* adsorption sites,29 generally for the OER potential-determining step, and found that the active sites are the same as those for OH*. 2TM-NG is expected to achieve superior OER activity. The OER performances of 2TM-NPAG and 2TM-NPG under alkaline conditions were systematically studied.
Fig. 2(a–c) show the Gibbs free energies of the different steps on different sites under a potential of U = 0 V and equilibrium potential (U = 0.40 V) at pH = 14. It is known that the overpotential (η) is determined by the largest energy difference between two adjacent steps, which is labeled in green. As shown in Fig. 2(a), for FeCo-NPAG, the first step (OH− → OH*) is the potential-determining step, with η = 0.42 V. However, for FeCo-NPG, the overpotential (1.41 V) in the third step (O* → OOH*) is three times higher than that of FeCo-NPAG. The strong interaction of the O radical with the substrate leads to the high energy required for the step from O* to OOH*. According to a study by Jin,30 the transition metal atoms are readily oxidized to metal oxides that can serve as real active sites for OER catalysis. Therefore, FeCo-NPG may act as a pre-catalyst for the final conversion to the corresponding oxides (FeCo-NPG-O). Then, for FeCo-NPG-O, the overpotential of the potential-determining step (O* → OOH*) is reduced from 1.41 V to 0.98 V, in which the OER catalytic site is the Fe atom. In addition, for FeNi-NPAG and CoNi-NPAG (Fig. 2(b and c)), the third step (O* → OOH*) is the potential-determining step with an overpotential of 0.65 or 0.38 V, respectively. For the other pre-catalysts FeNi-NPG and CoNi-NPG, the third step (O* → OOH*) is also the potential-determining step. The overpotential values are as high as 1.49 and 1.43 V, respectively, while the overpotential values of the real catalysts FeNi-NPG-O and CoNi-NPG-O for OER catalysis are reduced to 0.91 and 0.31 V, respectively. Among all the DACs shown in Fig. 2, the overpotential values of the real catalysts CoNi-NPAG and CoNi-NPG-O are as low as 0.38 and 0.31 V, respectively. They are much lower than those of noble metal catalysts (0.42 V for the OER on RuO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31) by 26.19%, clarifying the perfect performance of CoNi-NG in terms of its OER activity.
31) by 26.19%, clarifying the perfect performance of CoNi-NG in terms of its OER activity.
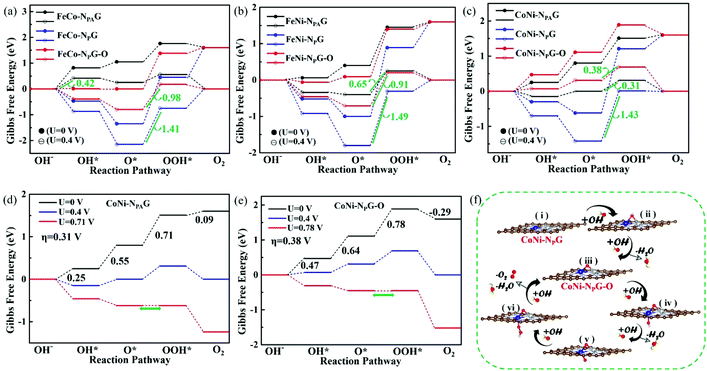 |
| | Fig. 2 Gibbs free energy for the OER pathway for the (a) FeCo, (b) FeNi and (c) CoNi electrocatalysts. Gibbs free energy for the OER pathway at an applied potential of (d) U = 0.71 V for CoNi-NPAG and (e) U = 0.78 V for CoNi-NPG-O. (f) Mechanism process of the OER on CoNi-NPG (pH = 14). | |
To more intuitively show the better OER activity of the real catalysts, CoNi-NPAG and CoNi-NPG-O, as shown in Fig. 2(d and e), a variety of applied potentials (U) were set to surmount the energy barrier. For CoNi-NPAG (Fig. 2(d) and Fig. S6 (a, b)†), almost all the steps are uphill at U = 0. When the applied potential is increased to 0.71 V, all of the elementary steps are downhill. Therefore, the overall OER catalytic mechanism of CoNi-NPAG was proposed as in eqn (5)–(8):
| | | CoNi-NPAG + OH− → Co(*)Ni-NP4A2G-OHads + e− | (5) |
| |  | (6) |
| | | Co(*)Ni-NPAG-Oads + OH− → Co(*)Ni-NPAG-OOHads + e− | (7) |
| |  | (8) |
where Co
(*) denotes the catalytically active metal site at the surface.
For CoNi-NPG (Fig. S6(c and d)†), the step of the OH radical adsorbed on substrate (OH*) and the step of OH* → O* present negative Gibbs free energies (−0.30 eV for ΔGOH*, and −0.32 eV for ΔGO*) at U = 0 V, implying that the reactions are exothermic. While the overpotential is as high as 1.43 V in the step of O* → OOH*, suggesting that it is difficult to catalyze O* to OOH*. Therefore, the real catalyst CoNi-NPG-O (Fig. 2(e)) was used for OER. Similar to CoNi-NPAG, when the applied potential is increased to 0.78 V, the OER pathway on CoNi-NPG-O starts to occur spontaneously. Moreover, based on the high OER activity of CoNi-NPG-O, starting from the adsorption of an OH radical on CoNi-NPG, the overall OER catalytic mechanism was proposed as in eqn (9)–(14):
| | | CoNi-NPG + OH− → Co(*)Ni(*)-NPG-OHads + e− | (9) |
| |  | (10) |
| | | CoNi-NPG-Oads + OH− → Co(*)Ni-NPG-O-OHads + e− | (11) |
| |  | (12) |
| |  | (13) |
| |  | (14) |
where Co
(*) and Ni
(*) denote catalytically active metal sites at the surface. Note: the OH radical was inclined to adsorb on Co
(*)Ni
(*) dual sites of CoNi-N
PG and the Co
(*) single site of CoNi-N
PG-O.
The above discussion was considered from a thermodynamic point of view. In general, the potential-determining step should also be the rate-determining step in one electrocatalytic reaction.32 Thus, to give more insight into the optimal OER catalysts CoNi-NG, their dynamics need to be studied. As shown in Fig. 3, we calculated the energy barrier for each reaction step using the CI-NEB for CoNi-NPAG, with the best catalytic activity, as an example. It can be seen that the step of OH radical adsorption onto CoNi-NPAG-Oads (Fig. S7†), i.e., O* → OOH*, requires an energy barrier of 0.64 eV. It is both the rate-determining step and the potential-determining step. In addition, the energy barriers for the potential-determining step (O* + OH− → OOH* + e) of CoNi-NPG and CoNi-NPG-O were also calculated (Fig. S8†). The energy barrier of CoNi-NPG is 1.35 eV and that of CoNi-NPG-O is 0.68 eV. Surprisingly, the trends in the energy barriers and potential barriers are consistent for the different CoNi-NPAG, CoNi-NPG and CoNi-NPG-O catalysts.
 |
| | Fig. 3 Dynamic reaction pathway of the OER for CoNi-NPAG. Inset: the dynamics data of the rate-determining steps of CoNi-NPG (pink) and CoNi-NPG-O (green). | |
In addition, the thermal stability of CoNi-NG was demonstrated by ab initio molecular dynamics (AIMD) within the NVT canonical ensemble at 500 K with an overall time scale of 10 ps. As shown in Fig. S9,† the total energies oscillate within a small range, and the structures show no significant geometrical distortion, indicating the stable configuration of the CoNi-NG catalysts.
E
solv is the solvation energy of the catalytic reaction intermediates, defined as: Esolv = Esol − Evac. Esol and Evac are the energies of the intermediates under vacuum and the implicit solvation model, respectively.27 The solvation energies are shown in Table 1. It can be seen that the implicit solvation model results in more stable reaction intermediates in real catalysts CoNi-NPAG and CoNi-NPG-O. η/vac and η/sol are overpotential values calculated under vacuum and the implicit solvation model, respectively. There is a subtle difference between the overpotentials of the CoNi electrochemical systems with and without the implicit solvation model. In FeCo-NG and FeNi-NG (Table S2†), after adding the implicit solvation model for the Gibbs free energy calculations, the OER catalytic mechanism was found to be consistent with that under vacuum. Meanwhile, both structures of the CoNi-based catalysts exhibit better OER catalytic activity, relative to those of the FeCo- and FeNi-based catalysts. Taken together, we will still systematically discuss the origin of the OER catalytic activity of the CoNi-based catalysts in the next section.
Table 1 Solvation energies (Esolv) of the catalytic reaction intermediates, η/vac and η/sol for the OER reactions of CoNi-NPAG, CoNi-NPG and CoNi-NPG-O
| |
CoNi-NPAG |
CoNi-NPG |
CoNi-NPG-O |
|
E
solv (eV) |
OH* |
−0.06 |
−0.08 |
−0.28 |
| O* |
−0.10 |
0.00 |
−0.23 |
| OOH* |
−0.11 |
−0.02 |
−0.30 |
|
η/vac (eV) |
0.31 |
1.43 |
0.38 |
|
η/sol (eV) |
0.30 |
1.41 |
0.31 |
3.2 Electronic properties
3.2.1 Oxidation state and d-band center.
In order to gain more insight into the real catalyst, the Bader charge analysis, charge density differences and spin-polarized density of states for 2TM-NPAG, 2TM-NPG and 2TM-NPG-O were calculated. The values of charge transfer were obtained from the Bader charge analysis,33–35 where the positive (+) and negative (−) charge indicate the decrease and increase of total valence electrons. The charge density differences of 2TM-NPAG and 2TM-NPG help to visualize the charge transfer between the metal atoms and the substrate, which is defined as eqn (15):36| | | Δρ(r) = ρ(2TM-NG) − ρ(TM1) − ρ(TM2) − ρ(NG) | (15) |
where ρ(2TM-NG) is the charge of 2TM-NG and ρ(TM1), ρ(TM2) and ρ(NG) are the charges of the independent TM1, TM2, and NG, respectively.
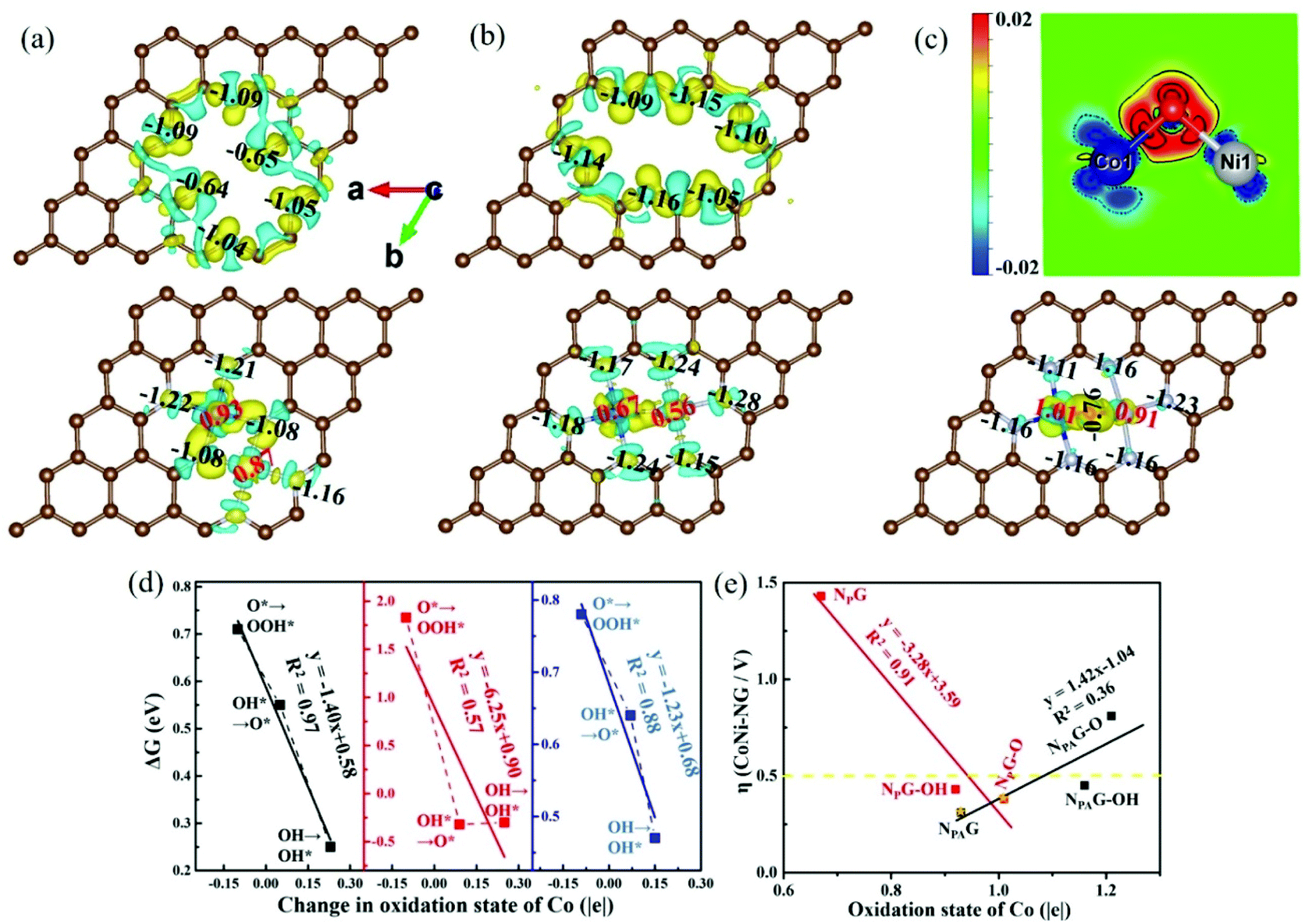
As shown in Fig. 4(a and b), there is a substantial amount of electrons relocation to N atoms from the Co, Ni and nearby C atoms. The Co/Ni atom electron decrease of CoNi-NPAG (+0.93|e| for Co, +0.87|e| for Ni) is greater than that of CoNi-NPG (+0.67|e| for Co, +0.56|e| for Ni). When CoNi atoms are embedded in NG, for CoNi-NPAG and CoNi-NPG, the number of electron changes in the N atoms are −1.35|e| and −0.57|e| electrons, respectively, while there are −0.60|e| and −1.32|e| in nearby C atoms, respectively (Table S3†). Therefore, the N atoms, especially in NA, are the main acceptors of electrons from the CoNi atoms, which leads to a higher oxidation state of CoNi atoms in CoNi-NPAG. For CoNi-NPG, NP is the main acceptor of electrons from nearby C atoms. Thus, the competition between the nearby C atoms and CoNi atoms for providing electrons results in a lower oxidation state of the CoNi atoms. This result can be directly illustrated by the 2D charge density differences (differences between an interacting system and the superposition of atomic charge densities) in Fig. S10.† And it can also be seen from the distance between the Co/Ni and N atoms, i.e., the shortest distance of Co/Ni-NA in CoNi-NPAG (Fig. S1†).
 |
| | Fig. 4 The Bader charge analysis and the 3D charge density differences for (a) NPAG and CoNi-NPAG, (b) NPG and CoNi-NPG and (c) CoNi-NPG-O (bottom) and the 2D plots (top) of the clipping plane corresponding to CoNi-NPG-O crossing the three atoms of Co, O and Ni. The yellow (solid contour line) and blue (dashed contour line) colors correspond to the accumulation and depletion of total valence electrons, respectively. The isosurface value is 0.008 e per bohr3. (d) The relationship between the ΔG of CoNi-NPAG (black line), CoNi-NPG (red line), CoNi-NPG-O (blue line) and change in the oxidation state of the catalytic site Co. (e) Relationship between the overpotential of CoNi-NG and the oxidation state of the catalytic site Co. | |
For CoNi-NPAG, the electron decrease of the Co atom (+0.93|e|) is greater than that of the Ni atom (+0.87|e|). The same behavior occurs on CoNi-NPG. Since the OH− reactant is negative, the metal site (Co) that has fewer electrons and becomes more positive more readily adsorbs OH−.37 Therefore, for CoNi-NG, the OH radical is more likely to adsorb at the Co atom site. For FeCo/FeNi-NG (Fig. S11 and S12†), the catalytic site is the Fe atom site. Obviously, the changing trend of the charge transfer is consistent with that of the element electronegativity (1.83 for Fe, 1.88 for Co, 1.91 for Ni). In order to show the charge change after the second step of the OER catalysis (OH* → O*) of 2TM-NPG, the Bader charge analysis and charge density differences (Δρ(r) = ρ(2TM-NPG-O) − ρ(2TM-NPG) − ρ(O)) were investigated and the corresponding 2D plot of the clipping plane crossing the three atoms of TM1, O and TM2 (Fig. 4(c) and Fig. S11, S12†) was constructed. It can be seen that the OER catalytic site remains unchanged and its valence electrons decrease to +1.01|e|.
To understand the role of different N coordination in the entire process of the electrocatalytic OER of CoNi-NG, the oxidation states of Co/Ni atom were calculated and are shown in Table S4.† And the relationships between the ΔG of CoNi-NPAG, CoNi-NPG, CoNi-NPG-O and change in the oxidation state of the catalytic site Co are shown in Fig. 4(d). With CoNi-NPG as the catalyst (red line), the slope is as high as 6.25, which indicates that the O* intermediate is too stable, resulting in a large ΔG for the third step (O* → OOH*). However, when CoNi-NPG-O is used as the catalyst (blue line), the O atom gains some of the electrons from the CoNi atoms, which weakens the binding strengths of the reaction intermediates to CoNi-NPG-O and thus improves the stability of the reaction.38 In CoNi-NPAG (black line), the N atoms gain more electrons than CoNi-NPG from the CoNi atoms, and the reason for its better electrocatalytic OER performance is similar to that of CoNi-NPG-O. In addition, for CoNi-NPAG-OH, O* gains electrons from the Co atom with a high oxidation state, making the reaction, which occurs on the other side different from the O atom, difficult to carry out. So, the potential-determining step shifts to OH* → O*, and the overpotential becomes large. For CoNi-NPAG-O, the oxidation state of the Co atom is too high, meaning that O* has to get electrons from the Ni atom, and the competition with the NA atoms becomes greater, leading to a rapid rise in the overpotential. The potential-determining step is also OH* → O*. In summary, the overpotentials of CoNi-NG show a volcano-like relationship with the oxidation state of Co (Fig. 4(e)), indicating that the more suitable oxidation state of the catalytic site, the better the electrocatalytic OER of the catalyst. This also further illustrates the feasibility of the oxidation state of the catalytic site in tuning the OER activity.
According to Fig. S13† and previous calculations,39 N-doped graphene is nonmagnetic, and the spin magnetic moments for NPAG and NPG are 0.00μB. However, for CoNi-NPAG and CoNi-NPG-O (Fig. 5), it is obvious that the spin-up and spin-down PDOS of the Co and Ni 3d orbitals are asymmetric, which indicates the spin magnetic moment properties of the two structures (0.76μB for Co atom, 0.13μB for Ni atom in CoNi-NPAG; −0.85μB for Co atom, −0.06μB for Ni atom in CoNi-NPG-O). For CoNi-NPG, the asymmetry is not obvious, thus, the PDOS of Co/Ni 3d and N 2p split orbitals are given (Fig. S14†). Due to the asymmetry of the Co/Ni 3d split orbitals, especially for the dz2 orbital of the Co atom, CoNi-NPG also generates spin magnetic moments (−1.10μB for the Co atom, −0.04μB for the Ni atom). To show the spin magnetic moments more intuitively, the spin-charge density was also calculated (Fig. S15†). Karim et al.40 concluded that some magnetic materials have variable valence electrons, and obviously promote the catalytic process. Y. Sun et al.32 further concluded that a good OER catalyst requires a moderate spin magnetic moment.
 |
| | Fig. 5 Spin-polarized density of states for (a) CoNi-NPAG, (b) CoNi-NPG and (c) CoNi-NPG-O. (d) Corresponding values and the correlation between the overpotential (η) of CoNi-NG and the d-band center (εd) of the catalytic site Co. (e) Contour plot of the overpotential as a function of the d-band center and oxidation state for the catalytic site Co of CoNi-NG. | |
We also studied the spin-polarized density of states to gain more understanding about the selection of catalytic site selectivity. As shown in Fig. 5(a–c), when Co and Ni atoms are trapped by nitrogen-doped vacancies, they make some contribution towards the TDOS of CoNi-NPAG and CoNi-NPG near to the Fermi level (EF). The N 2p orbitals are strongly hybridized with the C 2p orbitals between the −8 and 0 eV energy levels, which contributes towards catalyst stability. For CoNi-NPAG, there are some couplings between the N 2p and Co 3d orbitals near to the Fermi level, in contrast to that of the electron of the Ni atom, which is rarely distributed near to the Fermi level. This indicates that the Co atom plays a more crucial role in adsorbing and activating the incoming OH radical than the Ni atom. For CoNi-NPG, there is hybridization between the N 2p, Co 3d and Ni 3d orbitals around the Fermi level. However, the Ni 3d orbital peaks are lower than the Co 3d orbitals in CoNi-NPG-O. Both the Co 3d and Ni 3d orbitals have an effect on the adsorption of the OH radical, but the Co 3d orbitals play a major role in CoNi-NPG-O. This result is consistent with the above discussion of the Bader charge analysis and charge density differences, that is, the catalytic site may be a CoNi dual site for CoNi-NPG and a Co single site for CoNi-NPAG and CoNi-NPG-O.
From the above discussion, it can be seen that the oxidation state of the catalytic site Co will affect the activity of the OER. According to Nørskov et al., the d-band center (εd) as an electronic property of transition metals can also be used to describe their reactivity.41 It is well known that the binding between an adsorbate and 2TM-NG catalyst is due to the coupling between the oxygen 2p orbitals of oxygen-containing intermediates (such as O*, OH*, OOH*) and the 3d orbitals of the transition metal atoms,42 which leads to the formation of filled bonding states and partially occupied anti-bonding states. An increase in the proportion of unoccupied anti-bonding states leads to an increase in adsorption-surface interactions.21 We carefully studied the correlation between the OER activity of CoNi-NG and different types of d-band center, such as the catalytic site Co (Fig. 5(d)), the averaged d-band center of dual transition metal atoms CoNi (Fig. S116†), and the supplementary transition metal Ni (Fig. S17†). It is worth noting that the overpotential of CoNi-NG is in a volcano-shaped relationship with the d-band center of the catalytic site Co, which further illustrates the feasibility of the catalytic site Co in tuning the OER activity. For the averaged d-band center of dual transition metal atoms CoNi, a similar volcano-shaped relationship was also obtained (Fig. S16†) due to the contribution from the catalytic site Co atom. In contrast, there is no clear relationship between the overpotential and the values of the d-band center obtained from the supplementary transition metal Ni (Fig. S17†). These results indicate that, similar to the oxidation state, the d-band center of the catalytic site is one of the key parameters that affects the OER activity.
As shown in Fig. S18,† the linear relationship between the oxidation state and the d-band center indicates that the physical mechanism for tuning the OER activity is similar. At the same time, a contour plot of the OER overpotential as a function of the d-band center and the oxidation state of CoNi-NG was obtained (Fig. 5(e)). The value of the overpotential gradually increases from the red area to the green area, and the blue area has the highest overpotential. It can be seen that CoNi-NPAG and CoNi-NPG-O are closest to the red center, indicating that they have the lowest overpotential. Due to the optimal electronic properties (oxidation state and d-band center) of CoNi-NPAG and CoNi-NPG-O, their OER catalytic activity is the best.
3.2.2 OER catalytic process.
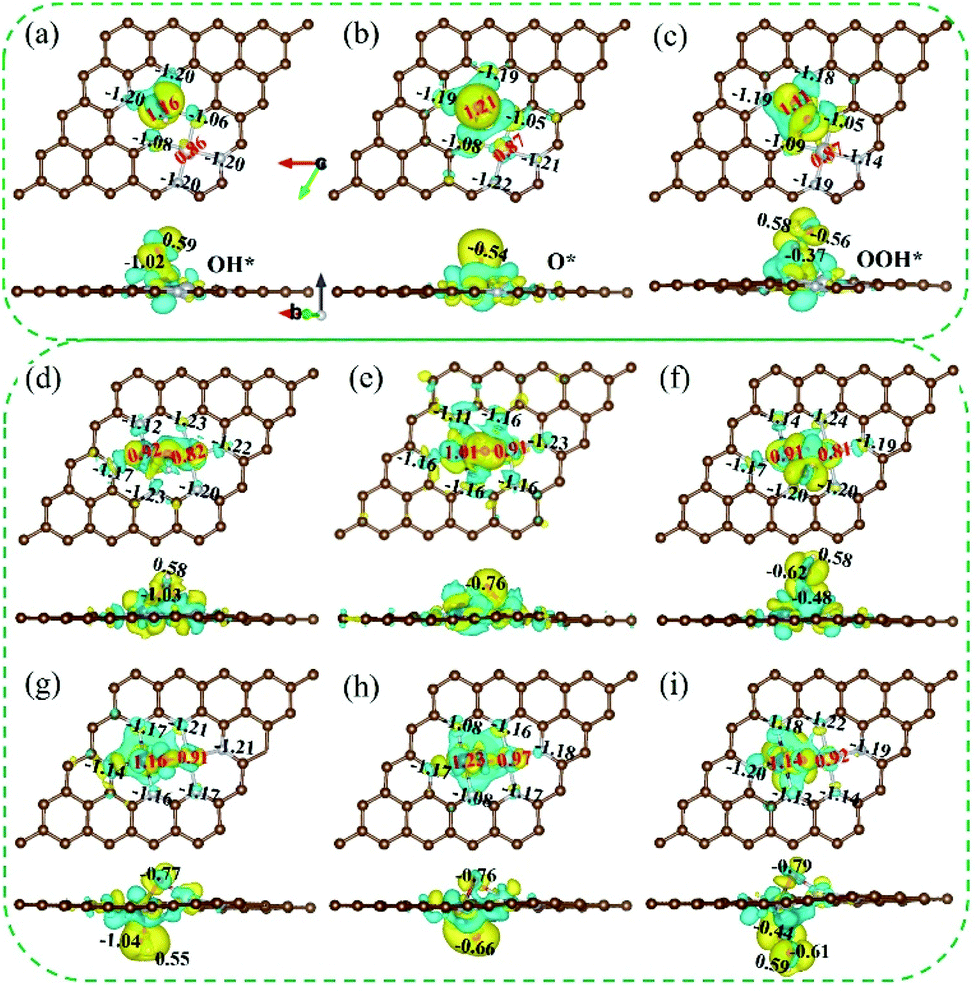
To further understand the OER catalytic process for the real catalysts CoNi-NPAG, CoNi-NPG-O and pre-catalyst CoNi-NPG at pH = 14, the Bader charge analysis, charge density differences and spin polarized density of states of the adsorbed intermediates OH*, O* and OOH* were calculated.
For the adsorbed intermediate OH* of CoNi-NPAG and CoNi-NPG-O (Fig. 6(a and g)), the electron depletion area near to the Co site (+1.16|e|) is more obvious than that of the Ni atom (+0.86–+0.91|e|), which indicates that the electrons are more readily transferred from the Co site to the O atom. In addition, the electron accumulation region on the N atom stabilizes the hydrated cations through non-covalent interactions in alkaline electrolyte, which further enhances the interaction between OH− and the catalyst surface.43 At the same time, effective charge of −1.02 to −1.04|e| and +0.55 to +0.59|e| are generated on the O and H atoms of the OH*, respectively. The electron decrease of the CoNi atoms is greater than that of the H atom, because the electronegativities of the Co and Ni atoms are much lower than that of the H atom. Therefore, the O atom of OH* tends to receive electrons from the CoNi atoms rather than from the H atom, which weakens the interaction between the O and H atoms. The positive charge of the CoNi atoms is considered to be the possible reason for the easy dissociation of the bond in O–H. For CoNi-NPG (Fig. 6(d)), the OH radical is simultaneously adsorbed on the two sites of the Co and Ni atoms. The oxidation state of the CoNi sites is increased from +1.23|e| (Fig. 4(b)) to +1.74|e|. This indicates that the interaction between the OH radical and NiCo-NPG is stronger (Fig. S5†) and that the O–H bond more readily dissociates. Next, after the second step of the OER catalytic process (OH* → O*), the adsorbed intermediate O* is obtained. As shown in Fig. 6(b, e and h), the enhanced charge transfer between the CoNi and O atom indicates a strong interaction between them. In addition, compared to the OH* system, the distance between the O and Co atoms is shortened in the O* system. Finally, the depletion of the total valence electrons of the CoNi atoms in the OOH* system (+1.98|e| for CoNi-NPAG, +2.06|e| for CoNi-NPG-O) decreases, compared with the O* system (+2.08|e| for CoNi-NPAG, +2.20|e| for CoNi-NPG-O) (Fig. 6(c and i)). That is, the decrease in the total valence electron depletion of CoNi-NPAG is 0.10|e|, and that of CoNi-NPG-O is 0.14|e|. This result indicates that the third step (O* → OOH*) of CoNi-NPAG requires a lower applied potential than that of CoNi-NPG-O. The effective electron transfer between the CoNi atoms and the nearest O atom is reduced. The interaction between them becomes weaker, resulting in an extension of the Co–O bond (1.69 Å for O* system, 1.87–1.88 Å for OOH* system). For CoNi-NPG (Fig. 6(f)), the electron transfer between the CoNi atoms and the nearest O atom is also reduced and the decrease in the total valence electron depletion is 0.20|e|, indicating that it is more difficult for OH radicals to continue to adsorb on the O* system. To sum up, it is thought that O* → OOH* is the potential/rate-determining step, which is consistent with the above discussion in 3.1.
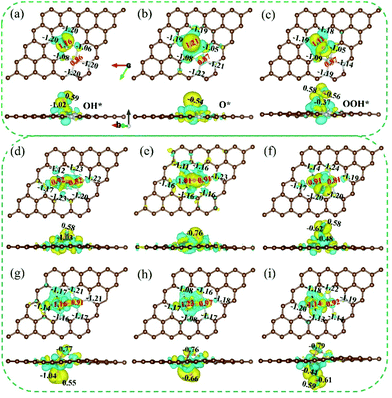 |
| | Fig. 6 Bader charge analysis and the 3D charge density differences for the adsorption intermediates OH*, O*, and OOH* of (a–c) CoNi-NPAG, (d–f) CoNi-NPG, (g–i) CoNi-NPG-O. Yellow and blue correspond to accumulation and depletion of total valence electrons, respectively. The isosurface value is 0.0035 e per bohr3. | |
The TDOS and PDOS are within the energy range from −8 to 8 eV (Fig. S19–S21†). For the adsorbed intermediate OH*, there are some hybridizations between the Co 3d orbitals and the O 2p orbitals, indicating the strong interaction between the Co site and the O atom. However, for the adsorbed intermediate O*, the hybridization between the Co 3d orbitals and the O 2p orbitals is stronger compared with OH*. This result also shows that the O* system is stable and more readily formed during the OER catalytic process. For the adsorbed intermediate OOH*, the spin-up and spin-down PDOS of the various atoms in CoNi-NPAG and CoNi-NPG-O are generally symmetrical, and the spin magnetic moment of the CoNi atoms is 0.00μB. In contrast, for CoNi-NPG, the Co 3d orbitals and Ni 3d orbitals are asymmetric, suggesting the generation of magnetic moments (0.78μB for the Co atom, 0.62μB for the Ni atom). The results show that there are no additional catalytic sites on Co*Ni-NPAG-OOH and Co*Ni-NPG-O-OOH, and the magnetic moment on Co*Ni*-NPG-OOH indicates that it is not a complete OER catalytic process or that it may also be a site for another OER catalytic process.
4. Conclusions
Under alkaline conditions, the OER electrocatalytic mechanisms were found to be that 2TM-NPG acts as a pre-catalyst, and that the real catalysts are 2TM-NPAG and 2TM-NPG-O. CoNi-NG exhibits perfect OER catalytic performances (η = 0.31 eV for CoNi-NPAG, and η = 0.38 eV for CoNi-NPG-O). The potential-determining step also being the rate-determining step was confirmed from CI-NEB dynamics calculations. Then, the role of different N coordination was analyzed. For CoNi-NPG, NA is the main acceptor of electrons from CoNi atoms, this leads to a higher oxidation state of CoNi atoms of CoNi-NPAG. For CoNi-NPG, NP is the main accepter of electrons from nearby C atoms, which leads to the lower oxidation state of the CoNi atoms. This role continues to influence their OER activity. In addition, the overpotentials of CoNi-NG show a volcano-like relationship with the oxidation state of Co. This further illustrates the feasibility of the oxidation state of the catalytic site in tuning the OER activity. At the same time, the overpotential of CoNi-NG is also in a volcano-shaped relationship with the d-band center of the catalytic site Co, indicating that the d-band center of the catalytic site is one of the key parameters that determines the OER activity. The linear relationship between the oxidation state and d-band center indicates that the physical mechanism for tuning the OER activity is similar. Finally, according to the OER overpotential (a function of the d-band center and oxidation state) contour plot, CoNi-NPAG and CoNi-NPG-O have the lowest overpotentials. The electronic properties, such as the oxidation state and d-band center can be extended to other DAC systems. It is thus believed that the content reported may provide a way to further develop new electrocatalysts for clean energy applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number 11474123) and the Scientific Research Foundation of the Education Department of Jiangxi Province (grant number GJJ203114).
References
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Y. Mo, S. P. Ong and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 205446 CrossRef.
- W. H. Lee, Y. J. Ko, J. Y. Kim, B. K. Min, Y. J. Hwang and H. S. Oh, Chem. Commun., 2020, 56, 12687–12697 RSC.
- K. Wang, X. Wang, Z. Li, B. Yang, M. Ling, X. Gao, J. Lu, Q. Shi, L. Lei and G. Wu, Nano Energy, 2020, 77, 105162 CrossRef CAS.
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef PubMed.
- X. Cheng, Z. Pan, C. Lei, Y. Jin, B. Yang, Z. Li, X. Zhang, L. Lei, C. Yuan and Y. Hou, J. Mater. Chem. A, 2019, 7, 965–971 RSC.
- J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2019, 141, 693–705 CrossRef CAS.
- J. Cao, C. Lei, J. Yang, X. Cheng, Z. Li, B. Yang, X. Zhang, L. Lei, Y. Hou and K. K. Ostrikov, J. Mater. Chem. A, 2018, 6, 18877–18883 RSC.
- W. Peng, Y. Feng, X. Yan, F. Hou, L. Wang and J. Liang, Adv. Sustainable Syst., 2021, 5, 2000213 CrossRef CAS.
- C. Yang, Z. D. Yang, H. Dong, N. Sun, Y. Lu, F. M. Zhang and G. Zhang, ACS Energy Lett., 2019, 4, 2251–2258 CrossRef CAS.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J. P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 RSC.
- G. Zheng, L. Li, Z. Tian, X. Zhang and L. Chen, J. Energy Chem., 2021, 54, 612–619 CrossRef.
- C. Cui, X. Hu and L. Wen, J. Semicond., 2020, 41, 091705 CrossRef CAS.
- C. Huang, Y. Li, N. Wang, Y. Xue, Z. Zuo, H. Liu and Y. Li, Chem. Rev., 2018, 118, 7744–7803 CrossRef CAS.
- Y. Wang, C. Xie, D. Liu, X. Huang, J. Huo and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 18652–18657 CrossRef CAS.
- X. Han, X. Ling, D. Yu, D. Xie, L. Li, S. Peng, C. Zhong, N. Zhao, Y. Deng and W. Hu, Adv. Mater., 2019, 31, 1905622 CrossRef CAS.
- X. Zhu, D. Zhang, C. J. Chen, Q. Zhang, R. S. Liu, Z. Xia, L. Dai, R. Amal and X. Lu, Nano Energy, 2020, 71, 104597 CrossRef CAS.
- W. Zhu, L. Zhang, S. Liu, A. Li, X. Yuan, C. Hu, G. Zhang, W. Deng, K. Zang and J. Luo, Angew. Chem., Int. Ed., 2020, 59, 12664–12668 CrossRef CAS PubMed.
- J. T. Mefford, A. R. Akbashev, M. Kang, C. L. Bentley, W. E. Gent, H. D. Deng, D. H. Alsem, Y. S. Yu, N. J. Salmon, D. A. Shapiro, P. R. Unwin and W. C. Chueh, Nature, 2021, 593, 67–73 CrossRef CAS PubMed.
- C. P. Wang, Y. Feng, H. Sun, Y. Wang, J. Yin, Z. Yao, X. H. Bu and J. Zhu, ACS Catal., 2021, 11, 7132–7143 CrossRef CAS.
- J. Zhou, Z. K. Han, X. K. Wang, H. Y. Gai, Z. K. Chen, T. Guo, X. B. A. Hou, L. L. Xu, X. J. Hu, M. H. Huang, S. V. Levchenko and H. Q. Jiang, Adv. Funct. Mater., 2021, 31, 2102066 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. R. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef.
- M. Lei, Y. Zhang, M. Wang, W. Yang and Z. Gao, Chem. Eng. J., 2021, 421, 129747 CrossRef CAS.
- C. Tang, L. Chen, H. Li, L. Li, Y. Jiao, Y. Zheng, H. Xu, K. Davey and S. Z. Qiao, J. Am. Chem. Soc., 2021, 143, 7819–7827 CrossRef CAS.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- Y. Sun, J. Wang, Q. Liu, M. Xia, Y. Tang, F. Gao, Y. Hou, J. Tse and Y. Zhao, J. Mater. Chem. A, 2019, 7, 27175–27185 RSC.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- H. M. Mu, H. X. Yu, D. R. Zhu, S. N. Zhao and X. C. Wang, Appl. Surf. Sci., 2019, 498, 143823 CrossRef CAS.
- H. X. Yu, H. M. Mu, D. R. Zhu, Y. Zhang, X. C. Wang and S. X. A. Zhang, Int. J. Hydrog. Energy, 2019, 44, 19920–19928 CrossRef CAS.
- X. C. Wang, Q. B. Lin, R. Q. Li and Z. Z. Zhu, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 245404 CrossRef.
- Y. Liu, X. Xie, G. Zhu, Y. Mao, Y. Yu, S. Ju, X. Shen and H. Pang, J. Mater. Chem. A, 2019, 7, 15851–15861 RSC.
- Z. Zeng, L. Y. Gan, H. B. Yang, X. Su, J. Gao, W. Liu, H. Matsumoto, J. Gong, J. Zhang, W. Cai, Z. Zhang, Y. Yan, B. Liu and P. Chen, Nat. Commun., 2021, 12, 4088 CrossRef CAS.
- M. Wu, C. Cao and J. Z. Jiang, Nanotechnology, 2010, 21, 505202 CrossRef CAS PubMed.
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Nature, 2017, 541, 68–71 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- C. Wei, Y. M. Sun, G. G. Scherer, A. C. Fisher, M. Sherburne, J. W. Ager and Z. C. Xu, J. Am. Chem. Soc., 2020, 142, 7765–7775 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr06334k |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Yang
Wu
a,
Yuxiu
Wang
b,
He-Na
Zhang
a,
Liang-Hui
Zhu
a and
Xiao-Chun
Wang
a,
Yang
Wu
a,
Yuxiu
Wang
b,
He-Na
Zhang
a,
Liang-Hui
Zhu
a and
Xiao-Chun
Wang