
Alkyl selenol reactivity with common solvents and ligands: influences on phase control in nanocrystal synthesis†
Eric A.
Ho‡
 ,
Antony R.
Peng
and
Janet E.
Macdonald
*
,
Antony R.
Peng
and
Janet E.
Macdonald
*
Department of Chemistry, Vanderbilt University, Nashville, Tennessee 37235, USA. E-mail: janet.macdonald@vanderbilt.edu
First published on 6th December 2021
Abstract
This study develops mechanistic understanding of the factors which control the phase in syntheses of copper selenide nanocrystals by investigating how the chemistry of the dodecylselenol reactant is altered by the ligand and solvent environment. 1H NMR and 77Se NMR were used to study how commonly used solvents (octadecene and dioctylether) and ligands (oleylamine, oleic acid, stearylamine, stearic acid and trioctyl phosphine) change the nature of the dodecylselenol reactant at 25 °C, 155 °C and 220 °C. Unsaturations were prone to selenol additons, carboxylates underwent selenoesterification, amines caused the release of H2Se gas, and the phosphine formed phosphine selenide. Adventitious water caused oxidation to didodecyldiselenide. NMR studies were correlated with the phases that resulted in syntheses of nanocrystalline copper selenides, in which berzalianite, umangite or a metastable hexagonal phase were produced as identified by X-ray diffraction, depending on the ligand and solvent environemnts. Formation of the rare hexagonal Cu2−xSe phase could be assigned to cases that included DD2Se2 as a reactive intermediate, or strong L-type ligation of amines which was dependant on alkyl chain length.
 Janet E. Macdonald | Janet Macdonald received her BS (Honors) in Chemistry from McGill University where she had her first whiff of organo-chalcogenide chemistry in the laboratory of David Harpp. Her PhD in nanocrystal synthesis was conducted under Jonathan Veinot at the University of Alberta, followed by post-doctoral research with Uri Banin at the Hebrew University of Jerusalem. She is currently an Associate Professor at Vanderbilt University. Her group is particularly interested in discovering transcendent principles for phase control in nanocrystal synthesis, but also tackles challenges in charge transfer phenomena from nanocrystals and nanostructures as pertains to solar energy capture. |
1. Introduction
The geologic record displays a diverse array of binary metal compounds of varying composition and crystal structures. The directed and controlled synthesis of nanocrystals in these phases represents a significant challenge.1,2 For example, there are several instances of phase-control in transition-metal chalcogenide nanoparticles,1,3–5 but these are for the most part developed serendipitously, with insufficient mechanistic understanding of the reactions to accurately synthesize other target phases on demand. The ability to selectively synthesize desirable phases will enable us to leverage their unique properties in applications.Ligand environment has been identified as a key component in phase control. For example, phase control between hexagonal wurtzite and cubic zinc blende CdSe has been established based on ligand choice in the synthesis.6 More recently, the Schimpf group developed conditions for tuning both the phase and size of WSe2 nanocrystals through the ratio of two ligands.7 It is not often understood why specific ligand environments give particular phases.
One significant hurdle to achieving directed phase control in nanocrystal synthesis is that there is an incomplete mechanistic understanding of how molecular precursors break down and change in the progress of a reaction. Several studies have tackled this problem8–11 but have only covered a few reactants and reaction conditions. Furthermore, very rarely interrogated are the effects of ligands and solvents on the rates and mechanism of precursor decomposition, which in turn can influence the trapping of kinetic phases. Work by the Krauss group8 uncovering active selenium precursors in quantum dot syntheses was influential, bridging the gap between nanomaterials and organic chemistry with proposed mechanisms for the release of Se and Cd into solution. The Hogarth group found that amine ligands changed the nature of the dithiocarbamate sulfur precursor and thereby influenced the phase of nickel sulfide produced.12,13 These works emphasize the importance of understanding the role of ligand environment on molecular processes in nanocrystal synthesis, beyond simply surface passivation.
Particularly intriguing are conditions where unexpected and new polytypes and phases form, because these give rise to new material properties. Metastable polytypes previously unobserved in nature have been synthesized using organoselenium precursors;1,5 however, the metastable phases only seem to form under very specific reaction and ligand conditions, otherwise forming thermodynamic phases. The structure of the selenium precursors, such as alkyl-, phenyl-, or benzyl-selenols and diselenides have significant effects on the resultant phase in nanoparticle synthesis.14–17 The rate of precursor decomposition and corresponding release of Se into a nucleating crystal is suggested to be a determining factor for phase control in the copper selenide model system, resulting in a variety of phases such as hexagonal CuSe, cubic Cu2−xSe, and hexagonal Cu2Se.3,5 Phase control via precursor structure is therefore sensitive to reactions of organoselenium precursors with the ligands and solvent environment, which can result in alternative product phases.16–18 We are particularly interested in the conditions under which the rare and newly discovered polytype of hexagonal Cu2Se5,19 forms, because understanding of this selectivity may lead to the discovery of more undiscovered phases across other material systems.
In the case of polytypism, two dominant theories on the nucleation and phase determination of nanoparticles have formed the basis for synthetic reasoning since the late 1800's and have been applied most thoroughly by the nanocrystal community to the synthesis of CdSe quantum dots. The first argues that phase selection is controlled kinetically; it is summarized by Ostwald's rule of stages,20 which proposes that the kinetic phase forms first, and then undergoes a transition to the thermodynamically more stable phase. Slow reaction kinetics in CdSe nanocrystal synthesis can produce large, 15 nm metastable zinc blende nanocrystals that lack the structural defects which catalyze the transformation to the thermodynamic wurtzite phase, and so the crystals become kinetically trapped in the metastable phase.21
Alternatively, metastable phases can be captured through thermodynamic stabilization. When only a few unit cells comprise a crystal, surface energy dominates over subtle enthalpic differences between the polytypes,22 and even fluctuating structure can occur.23–25 Carefully chosen surface ligands can stabilize one polytype over another. Strong X-type ligands favor the cubic zinc blende phases because of the eight charged [111] surfaces. In contrast, L-type donors prefer to bind to the cations on neutral surfaces presented by [100] surfaces of hexagonal wurtzite phases.6,26
The evidence for both the kinetic and thermodynamic stabilization of CdSe nanocrystalline polytypes underscores the need to be cognizant of both factors in the synthesis of other nanocrystalline products. Ligands do not necessarily have singular, well defined roles in nanoparticle syntheses because they can change both the kinetics and thermodynamics of the reaction. With multiple possible roles, what are the most important factors that ligands have in phase control?
Here we study closely the chemistry of a selenium precursor used for metal selenide syntheses, dodecyl selenol.5,27,28 Alkylselenols are important precursors because they react at temperatures considered low for nanocrystal synthesis (below 200 °C) which gives opportunity for metastable phases to be trapped. By extensively employing 77Se and 1H NMR, the reactions of this precursor with common ligands and solvents were identified and tracked, revealing highly varied chemistry that fundamentally changes the nature of the precursor in situ. We correlate these chemistries with the resulting phase in a model nanocrystal system of copper selenides. These experiments illuminate the contrasting roles that ligands can play in phase determination, indicating that both kinetic and thermodynamic contributions must be considered.
2. Experimental
2.1 Materials
Whatman lead acetate indicator paper was purchased from GE Healthcare Life Sciences. Copper(II) acetylacetonate (Cu(acac)2, ≥98%) was purchased from Strem. All other reagents were purchased from Aldrich. Trioctylphosphine (TOP, 97%) was used without further purification. 1-Octadecene (ODE, 90%, technical grade), dioctyl ether (DOE, 99%), oleic acid (90%, technical grade), stearic acid (99%), dodecylamine (95%), tetradecylamine (95%), hexadecylamine (90%), and stearylamine (90%) were heated at 120 °C under vacuum for 16 h and stored in the glovebox or used directly. Oleylamine (70%, technical grade) was purified by vacuum distillation before use and stored in a glovebox. Dodecyl selenol (DDSeH) was synthesized following a previously-reported procedure and stored in an N2 glovebox freezer at −35 °C until use.292.2 Reactions of dodecyl selenol with ligands
DDSeH (0.1 mmol) and ligand (0.2 mmol; TOP, ODE, oleylamine, oleic acid, stearylamine, stearic acid, or DOE) were combined in glass NMR tubes in a nitrogen-filled glovebox. DDSeH oxidizes readily to DD2Se2 under atmospheric conditions, necessitating the use of air-free techniques. After export, each tube was fitted with a balloon filled with inert gas (either N2 or Ar) through a needle adapter to allow for safe gas expansion during heating. Three sets of vessels were prepared for each ligand, and were either held at room temperature or heated to 155 °C or 220 °C for 90 min and allowed to cool. Each tube was injected with a standard solution of 0.1 mmol dioxane in 500 μL CDCl3 for analysis. When testing for H2Se formation, the NMR scale experiments were prepared and performed as described. In addition, a strip of lead acetate indicator paper was placed into each tube such that the bottom of the paper was just above the level of the reactants. After cooling, the lead acetate paper was removed and analysed by powder X-ray diffraction (pXRD).2.3 Synthesis of Cu2−xSe nanocrystals
Cu(acac)2 (0.09 mmol) and ligand (1.4 mmol; TOP, ODE, oleylamine, oleic acid, stearylamine, stearic acid, or DOE) were combined in a 1-dram vial in a nitrogen-filled glovebox and sealed with a septum. DDSeH oxidizes readily to DD2Se2 under atmospheric conditions, necessitating the use of air-free techniques. Separately, DDSeH (0.2 mmol) and ligand (1.4 mmol) were combined in a 5 mL pear-shaped flask in the glovebox and sealed with a septum. After export, the flask was fitted with a balloon filled with inert gas (either N2 or Ar).The flask was heated in a bath of silicone oil to a temperature of either 155 °C, 200 °C or 220 °C for 1 h with strong stirring, while the vial was warmed separately on a hot plate to melt its contents. At the 1-h mark, the contents of the vial were rapidly injected into the flask. In some cases, warming the glass syringe was necessary to ensure the ligand remained liquid throughout the transfer. The reaction was allowed to continue for an additional 1 h in the oil bath.
Upon cooling, ∼0.5 mL chloroform was injected into each flask to suspend the particles. Warm isopropanol (∼20 mL, ∼60 °C) was added, and the particles were separated through centrifugation (8700 rpm, 5 min) and the supernatant discarded. Twice more, the particles were further resuspended in ∼0.5 mL chloroform and precipitated with ∼20 mL acetone.
2.5 Characterization
1H and 77Se nuclear magnetic resonance (1H and 77Se NMR) spectra were acquired on a Bruker AV-400 console with a 9.4 Tesla Oxford magnet and 5 mm Z-gradient broadband (BBFO) probe, tuned to 400 MHz. Full instrumental parameters for NMR experiments are available in Table S1.† CDCl3 was used as a standard NMR solvent unless otherwise noted, in which case C6D6 was used. Dioxane (1H NMR δ = s, 3.71 ppm) was used as an internal standard. Powder X-ray diffraction (XRD) measurements were performed with a Rigaku SmartLab powder X-ray diffractometer with a Cu Kα (λ = 0.154 nm) radiation source set to 40 kV and 44 mA and a D/teX Ultra 250 1D silicon strip detector. XRD patterns were acquired using a step size of 0.1° at 1° min−1 (nanocrystals) or 15° min−1 (lead acetate paper). Transmission electron microscopy images (TEM) were acquired using an FEI Tecnai Osiris TEM operated at 200 keV.3. Results and discussion
3.1 NMR Studies
In this work, the chemistry of dodecylselenol (DDSeH) (1) was studied under common conditions for nanocrystal synthesis, specifically targeting conditions used in existing preparations of copper selenide from DDSeH.5,28 Several solvents and ligand environments were tested, featuring amine, carboxylic acid, phosphine, alkene and ether groups. 1-Octadecene (ODE) (2) is often considered a “benign” solvent for nanocrystal synthesis and is most often used in conjunction with stronger ligands,30 yet it is known to polymerize31 and act as a reducing agent.32 Furthermore, ODE contains weakly coordinating unsaturation at the 1-position. Dioctyl ether (DOE) (8), was included in the study as a representative poorly-coordinating solvent with suitable physical properties and without the reactive unsaturation of ODE. Oleylamine33 (3) and oleic acid (4) are frequently selected for use in nanoparticle synthesis as L- and X-type ligands, respectively, due to their convenient physical properties.34–36 Both have high boiling points that can tolerate high-temperature reaction conditions, and are liquids at room temperature due to mid-chain cis-unsaturation in contrast to their saturated counterparts stearylamine (5) and stearic acid (6).There is additional literature precedent for the use of Se-amine solutions in nanoparticle syntheses. Yang et al., report the dissolution of Se powder in oleylamine at room temperature enabled by a reducing agent such as a thiol.17 Phosphines are another common ligand type used in nanocrystal synthesis, and trioctylphosphine (TOP) (7) is known to react with elemental selenium to produce TOP:Se, an organoselenium precursor commonly used in preparations of CdSe QDs.8
NMR-scale reactions were performed containing DDSeH and each ligand studied at 25 °C, 155 °C or 220 °C. While some information was gleaned from 1H NMR, 77Se NMR proved to be highly informative and readily interpreted, as 77Se is spin ½ with ∼7% natural abundance. The 77Se NMR clearly showed the changes in soluble selenium species before and after reaction. Lead acetate paper was placed in the head-space of the reaction to detect the evolution of any gaseous H2Se, identifiable by the conversion of lead acetate to lead selenide.
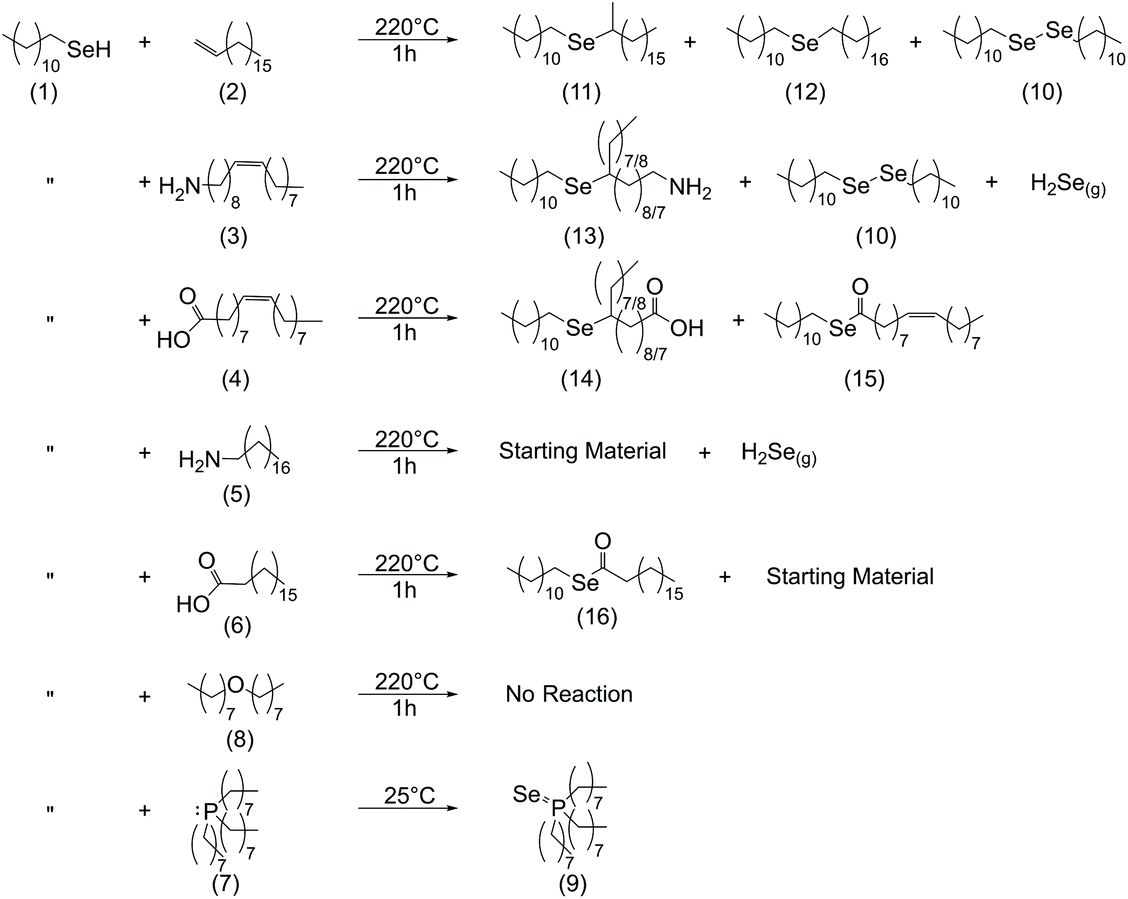
At room temperature, neither 1H nor 77Se NMR indicated that there had been a reaction between DDSeH (1) (77Se NMR: δ = −13.8 ppm)16 and any of the solvents or ligands tested (Fig. S1–S25†). The exception was TOP, where even at room temperature DDSeH was completely consumed to form TOP:Se (9) identified by a doublet (δ = −382.6 ppm, 1JSe-P = 3.56 kHz) in the 77Se NMR spectrum. A doublet results from coupling with the adjacent phosphorus (Fig. 1A and Scheme 1).8,10
 | ||
| Fig. 1 (A) 77Se NMR of reaction mixture between DDSeH and various ligands at 155 °C. Major products are listed numerically and match Scheme 1. H2Se evolution was detected qualitatively using Pb(OAc)2 strips. *TOP reacts at room temperature with DDSeH and was not heated. ODE, oleylamine, stearylamine, DOE, and TOP in CDCl3 solvent. Oleic acid and stearic acid in C6D6 (B) XRD of nanocrystalline products when copper precursor was included. Dominant product phases are listed. Complete XRD data for Pb(OAc)2 tests are available in Fig. S26.† Hexagonal Cu2Se reference pattern from Manna et al.,19 cubic (ICSD: 181661), umangite (ICSD: 16949). | ||
 | ||
| Scheme 1 Summary of identified products in the reactions of dodecylselenol with common ligands and solvents used in nanocrystal synthesis. | ||
ODE showed no reaction with DDSeH at 155 °C (Fig. 1A), but did react at 220° which was readily apparent by both 77Se NMR (Fig. 2A) and 1H NMR (Fig. S3†). The starting DDSeH (1) was nearly completely consumed and replaced with signals in regions belonging to selenoethers (R–Se–R) and dialkyldiselenides (RSeSeR). The 77Se NMR signal at δ = 307.7 ppm can be assigned to didodecyldiselenide (DD2Se2)5 (10). Similarly, a new peak appears in the 1H NMR at δ = 2.92 ppm which can be assigned to the α protons of DD2Se2.5 Experimental rigor was used to specifically prevent oxidation by air or water, so the formation of the diselenide must be a product of some other chemical process that occurs in the presence of ODE. No elemental Se was detected in the post-reaction mixture.37
 | ||
| Fig. 2 (A) 77Se NMR of reaction mixture between DDSeH and various ligands at 220 °C. Major products are listed numerically and match Scheme 1. H2Se evolution was detected qualitatively using Pb(OAc)2 strips. ODE, oleylamine, stearylamine, DOE, and TOP in CDCl3 solvent. OA and SA in C6D6. *TOP reacts at room temperature with DDSeH and was not heated. (B) XRD of nanocrystalline products when copper precursor was included. Dominant product phases are listed. Complete XRD data for Pb(OAc)2 tests are available in Fig. S27.† Hexagonal Cu2Se reference pattern from Manna et al.,19 cubic (ICSD: 181661), umangite (ICSD: 16949). | ||
The signals of the vinylic protons of ODE disappeared (1H NMR δ = 5.82 and 4.96 ppm) and were replaced by a multiplet at δ = 5.4 ppm suggesting isomerization of the 1-alkene into a 2-alkene (Fig. S3†). Such isomerization of the alkene matches a previous report on the reaction of ODE with elemental selenium,16 and suggests that the vinylic protons of ODE are interacting with the selenol. In addition to the isomerization of the double bond, the overall vinylic signal decreased in intensity, suggesting a further chemical reaction between the selenol and ODE.
The Raston group studied the reaction between ODE and elemental Se and proposed two structural families as potential products.16 First is a family of products formed by cross-linkage of ODE olefins with bridging Se chains of various lengths. Second is a family of episelenides, an epoxide analogue, formed by adduction of Se to the olefin. However, Seppelt et al., reported that their attempts to synthesize episelenides were unsuccessful.38 The episelenide species remained stable only as a cationic adduct, not as a neutral molecule, which instead yielded more stable diselenides. Therefore, in the reaction between ODE and DDSeH studied here, an episelenide is unlikely.
Instead, in the reaction between DDSeH and ODE, the selenol adds across the double bond in a reaction analogous to thiolene addition. The signal at 77Se NMR δ = 161.0 ppm correlates well with a long chain di-n-alkyl selenide,39,40 and the largest product with its 77Se NMR signal at 266 ppm matches those of structurally similar 1°–2° (at the 2-positon) asymmetric alkyl selenide.40 These two signals are from Markovnikov (11) and anti-Markovnikov (12) additions respectively, and it is unsurprising that there is such a mixture given the elevated temperature. Both Lewis-acid-catalyzed and radical pathways are known for the corresponding thiolene reaction, but the concomitant formation of DD2Se2 (10) (77Se NMR δ = 307 ppm) is suggestive of a radical pathway (vide infra) (Scheme 1).
Oleylamine and DDSeH also showed little change in the 77Se NMR after reaction at 155 °C (Fig. 1A), but did react and completely consumed the DDSeH at 220 °C (Fig. 2A). The signal at 1H NMR δ = 5.38 ppm changed shape and is ascribed to the cis-to-trans isomerization of the alkene (Fig. S6†).41 It also decreased in intensity, indicating chemical reduction. The 77Se NMR signal at δ = 227 ppm can be assigned to a selenol addition across the double bond of oleylamine producing a mid-chain selenoether at either the 9- or 10-position of the oleyl chain (13), similar to that seen for ODE. A similar signal was seen for syntheses with oleic acid, suggesting similar addition products (14) (Scheme 1). No direct 77Se NMR standard was found in the literature; however, a 40 ppm decrease in chemical shift was observed between R-SePh at the 2-position of an alkyl chain vs. the 4-position.42 Here we observe a similar 39 ppm decrease between the 2-position addition of ODE and the 9-position addition of oleylamine, and find selenol addition across the oleylamine alkene to be a likely mechanism.
DD2Se2 (77Se δ = 307 ppm) is a major product in the reaction of oleylamine and DDSeH at 220 °C. DD2Se2 does not form in the presence of saturated stearylamine (Fig. 2A and Scheme 1), so it must occur as a product of the reaction with the alkene, and likely forms from coupling of two DDSe˙ radicals. Therefore, the reduction of unsaturation by DDSeH begins with radical abstraction of the selenol H by the alkene. The remaining DDSe˙ radical can be captured by the radicalized oleylamine to make the selenoether or can couple to make DD2Se2. Saturated octadecane and stearic acid from two successive additions of H˙ are likely unidentified products of these radical processes.
Dushkin et al., have previously observed H2Se formation from similar mixtures of amines and selenols under conditions in excess of 300 °C,14 though there are no previous reports of H2Se generation at milder temperatures. Under the conditions studied here, DDSeH produced H2Se only when amines were present. With stearylamine, H2Se was observed to from at 155 °C and 220 °C. With oleylamine, H2Se was only observed at 220 °C. The other radical chemistry of oleic acid, described above, actively competes with the H2Se formation pathway when unsaturation is present (Fig. 1A, 2A and Scheme 1).
When the two carboxylic acids, oleic acid and stearic acid, were heated in the presence of DDSeH, the resultant NMR expressed that a reaction had occurred at 155 °C but it was difficult at first to identify the product. In CDCl3 solvent, the signal from the methylene protons α to the Se (1H NMR δ = 2.57 ppm) in DDSeH had decreased in intensity. At 220 °C, this signal was further decreased for stearic acid (Fig. S15†) and completely absent for oleic acid (Fig. S19†). The 77Se NMR also showed a decrease in the intensity of the starting material, yet did not show a significant new selenium-containing product between −100 and 1500 ppm (Fig. 2 and Fig. S25†).
It was noticed that in the CDCl3 solvent, an amber-colored precipitate settled over the course of the NMR experiment. Changing the NMR solvent to deuterated benzene, which has increased solubility over chloroform for long chain fatty esters,43 revealed a new signal in the 77Se NMR (oleic acid δ = 463 ppm, stearic acid δ = 473 ppm) (Fig. 2A). Direct esterification between selenols and carboxylic acids and is known to occur at 160 °C.44 The new NMR signals are from the dodecyl selenoester of oleic acid (15) or stearic acid (16), making a very nonpolar, insoluble C30 chain with only a mid-chain selenoester (and one degree of unsaturation for oleic acid) (Scheme 1). The relative insolubility suggests that the selenoesters will be a stubborn impurity in nanocrystal products, especially if chloroform is used rather than toluene or benzene in purification steps.
DOE was chosen as another common solvent used in nanocrystal synthesis. When heated with DDSeH to 155 °C or 220 °C, only starting material was observed. Under the conditions studied here, it was the only true “benign” solvent that had no chemical reaction with the selenol at high temperatures (Fig. 1A, 2A and Scheme 1).
3.2 Nanocrystal syntheses
The NMR studies showed that DDSeH has active chemistries with many common functional groups seen in nanocrystal synthesis - especially unsaturation, amines and carboxylic acids. Therefore, it is important to identify how the resultant changes in the nature of the selenium reactant might influence phase in a chemical reaction to produce metal chalcogenide nanocrystals. Ligands of course also passivate the surfaces of growing particle nuclei, changing the thermodynamic landscape. With such synthetic complexity, what are the most important factors in phase control?The copper selenides were chosen as model synthetic targets because there are several phases of differing stoichiometry, and several polytypic pairs, including hexagonal/cubic Cu2−xSe phases. The metastable hexagonal Cu2−xSe phase has only been achieved through colloidal techniques and so understanding the nuances of how and why this phase is selected in certain nanocrystal syntheses may lead to the discovery of new metastable phases in other metal chalcogenide systems. The hexagonal/cubic Cu2−xSe polytypic pair provides an interesting comparator to the existing literature on phase control in hexagonal/cubic CdSe because the hexagonal phase is the thermodynamic phase in CdSe and metastable in Cu2Se.
Ostwald's rule of stages suggests that the metastable phases always form first and then are transformed into the thermodynamic phase. With this in mind, here we presume that the hexagonal Cu2−xSe phase is always present at the nucleation step and is then transformed into the thermodynamic cubic phase. Previous work has suggested that the metastable to thermodynamic transformation of nanocrystals can be prevented through slow reaction kinetics that create more prefect crystals, lacking the defects that catalyze the needed structural transformations for relaxation to the thermodynamic phase.21
In the case of Cu2−xSe, the existing direct synthesis to hexagonal Cu2−xSe is the result of slow reaction kinetics provided by the DD2Se2 precursor.5 At first glance, it might be expected that if DD2Se2 was produced in reaction with the ligands, the hexagonal phase might result in a comparative nanocrystal synthesis. Yet previous work has shown that nanocrystals of the hexagonal phase transitioned to the cubic phase at 151 °C in a heated XRD experiment.5 Going into the phase control experiments, a prediction can be made that reactions at 220 °C will only yield the thermodynamic cubic phase, even if DD2Se2 forms as a product of the ligands chemistry with unsaturation, because the synthesis temperature is well above the phase transition temperature.
The phase transition for hexagonal Cu2−xSe at 151 °C was measured under a very weak ligation environment. Strong ligands, by way of lowering the surface energy, can be active in stabilizing one polytypic phase over the other, either hindering or promoting the phase transformation as has been seen with metastable cubic CdSe.6,26 Ligand stabilization of the hexagonal/cubic phases of Cu2−xSe has yet to be reported, and may be observed here, especially for L-type ligands (amines, phosphines and unsaturation) that stabilize hexagonal phases.
DDSeH was heated in 7 eq. of solvent/ligand at 155 °C or 220 °C for 1 h. A similarly prepared solution of Cu(acac)2 in the same solvent/ligand was then injected. The reactant pre-heat procedure was chosen to amplify the effect of selenol chemistry on nanocrystal synthesis. The resultant copper selenides were isolated and characterized with XRD.
When TOP was employed, no particles could be isolated either at 155 °C or 220 °C (Fig. 1B and 2B). CdSe syntheses with TOP:Se are typically performed in excess of 300 °C, but there indications that TOP:Se can decompose at temperatures as low as 170 °C.45 Other studies indicate that CdSe nanocrystals are nucleated by more reactive dioctylphophineselenide impurities.10 Here, the lack of particle nucleation may be speculatively explained by Cu2+ being less reactive than Cd2+ towards TOP:Se, and the use of neat TOP in the synthesis, in contrast to its typical use paired with other weaker-coordinating solvents such as OLAM35,46 or dichloromethane.34
When carboxylic acid ligands were employed, the drying/degassing step was critical in determining the phase of copper selenide that formed. Under non-rigorous conditions, the resultant nanocrystals were a mixture of cubic and hexagonal Cu2−xSe (Fig. S28†), but there was often the concomitant formation of a blue precipitate, which was presumed to be Cu(OH)2 from dissolved water. Oleic acid stored under ambient conditions can contain hundreds to thousands of ppm of water,11 and adventitious water present in stearic acid has been found to oxidize DDSeH to DD2Se2 at 155 °C (Fig. S26†), which is already known to produce the hexagonal phase.5 When rigorous degassing/drying steps of the ligand were employed (120 °C, 16 h under vacuum) the blue precipitate did not form and the resultant copper selenide was cubic berzelianite for both acids at 155 °C (Fig. 1B) and at 220 °C (Fig. 2B).
In addition to ambient water, the formation of the selenoesters, which was observed at 220 °C, will also produce an equivalent of water. This was probably driven off during the long pre-soak heating period, however, it could be speculated that variations in heating rates and injection times could cause variable results in phase in nanocrystal synthesis.
Carboxylic acids are X-type ligands and are known to stabilize cubic phases with their many charged [111] surfaces. However, here the slow reaction kinetics of DD2Se2 must override the surface stabilization factor, since some hexagonal phase could be formed under the wet conditions.
Under rigorously dry conditions, berzelianite forms at 155 °C (when the mixture is DDSeH and carbocyclic acid) and at 220 °C, (when the mixture is stearic/oleic-didodecylselenoester and carbocyclic acid). The selenoester must also be an active selenizing reactant at 220 °C. Particles produced in the presence of oleic acid and steric acid often were contaminated by a crystalline material with low-angle reflections (Fig. 1B, stearic acid as an example). This organic by-product is likely unreacted selenoesters which were found to be very insoluble and difficult to separate from the nanocrystalline product.
When oleylamine or stearylamine was employed, the product at 155 °C was the metastable hexagonal phase (Fig. 1B). Umangite Cu1.5Se is also observed for oleylamine, which reportedly forms from hexagonal phase in the presence of excess amine.19 The production of the hexagonal phase contrasts with a previously reported synthesis at this temperature without an amine present that gave the cubic phase,5 and so the amine is an active part in phase determination. A change in the identity of the active reagent was considered. To avoid oxidation by water to DD2Se2, the amines underwent rigorous drying; the oleylamine was vacuum distilled and stored in the glovebox, and the stearylamine was degassed under vacuum for 1.5 h at 120 °C. Similarly, the NMR studies indicated the chemistry to produce DD2Se2 from radical processes was only active for oleylamine or was prevalent at only 155 °C (Fig. 1A). Therefore, DD2Se2 was not the culprit for the formation of metastable hexagonal Cu2−xSe in the presence of amines. H2Se was also considered. While H2Se was formed by DDSeH and stearylamine at 155 °C, it was not formed for oleylamine at this temperature and yet the hexagonal phase formed for both (Fig. 1A). Therefore, a changing reactant identity was rejected as an explanation.
Surface ligation by amines was therefore identified as the reason for the formation of the hexagonal phase. As L-type donors, amines stabilize the predominantly neutral surfaces ([100]-type) of hexagonal crystals and direct to these phases.
At 220 °C, stearylamine continued to give predominantly hexagonal product, suggesting that the amine can stabilize the hexagonal phase to high temperatures well above the 151 °C transition temperature seen previously (prepared from DD2Se2 in ODE). However, oleylamine was not able to stabilize the hexagonal particles at 220 °C and instead, a cubic phase was identified by XRD after 1 h (Fig. 2B). Oleylamine cannot provide the necessary surface stabilization for the hexagonal phase to persist, because it cannot pack as tightly as stearylamine due to the mid-chain kink caused by the unsaturation.
As further evidence that amines provide surface stabilization of hexagonal Cu2−xSe surfaces, reactions of DDSeH and Cu(acac)2 were performed in the presence of 14 equivalents of amines of increasing chain length: C12H15NH2, C14H29NH2, C16H33NH2 and C18H37NH2 (stearylamine) at 200 °C (Fig. 3). The shortest chain gave only cubic berzelianite, but increasing the chain length increased the proportion of hexagonal phase, in a linear trend up to ∼70% for stearylamine. Increased van der Waals forces between the amine chains47 increase the stabilization of the hexagonal phase.
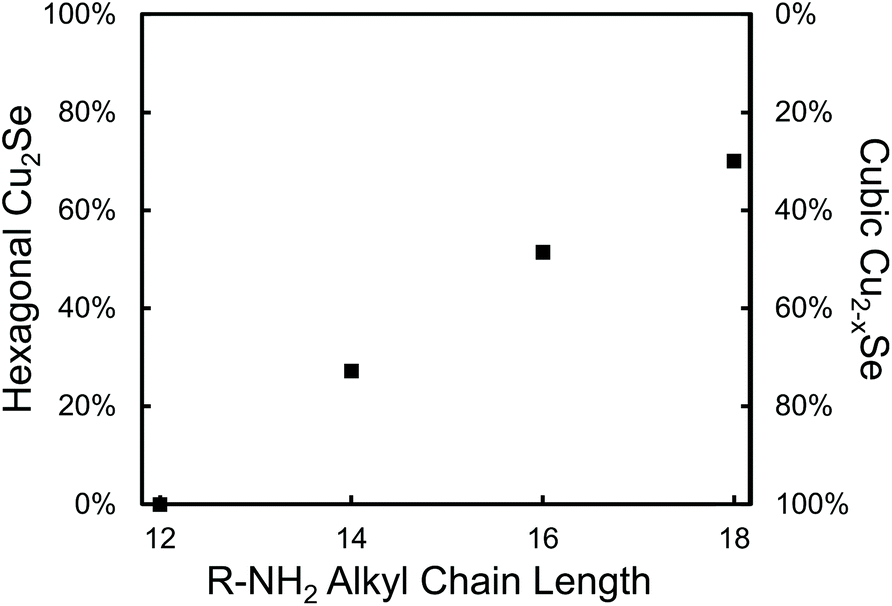 | ||
| Fig. 3 Fractions of nanocrystalline product phases for saturated amines of increasing length. Percentages determined by Rietveld refinement with X2 < 3. | ||
When ODE and DOE were employed, which are both common solvents in nanocrystals synthesis, berzelianite was the universal product (Fig. 1B and 2B). While both are technically L-type, neither solvent is strongly coordinating. Like oleylamine and short-chain amines, ODE and DOE do not provide adequate surface stabilization to prevent relaxation of hexagonal nuclei to the cubic phase. This is notable since, like oleylamine, ODE changes the nature of the DDSeH reactant to a combination of selenoethers and DD2Se2. In the case of ODE and oleylamine, at 220 °C, the effect of poor surface stabilization overrides the “slow kinetics” provided by the less reactive precursors. If this is the case, then the hexagonal phase should be expected if ODE is preheated with DDSeH to give DD2Se2 but then reacted with copper at a lower temperature. As expected, the hexagonal phase formed after a pre-heat at 220 °C, followed by a reaction with Cu(acac)2 at 155 °C (Fig. S30†).
As a final note, this last experiment was conducted with 4× DDSeH compared to the standard experiment in order to obtain enough nanocrystalline product for XRD analysis. While DD2Se2 is known to react at 155 °C, the other seleno-ether byproducts of the reaction of DDSeH and ODE are likely not reactive at this modest temperature. The reaction yields suffered accordingly without the additional reagent. Measuring reaction yields precisely throughout these experiments was not facile. The different sizes of particles and the presence of ligands prevented reliable mass comparisons. Using the Beer–Lambert law and the excitation coefficients well above the band gap is complex because of the presence of multiple phases. Therefore, the complex molecular chemistry of DDSeH with common ligands and solvents may have an unrecognized effect on yields.
4. Conclusions
Commonly-used ligands and solvents are active in the organoselenium chemistry that precedes nanocrystal formation. In the case of the reactant dodecylselenol, the chemistry is varied and extensive (Scheme 1). Changing the nature of the reactant in situ can consequently change the phase of the nanocrystalline product (Scheme 2), which can contrast or confound expectations from surface ligation arguments.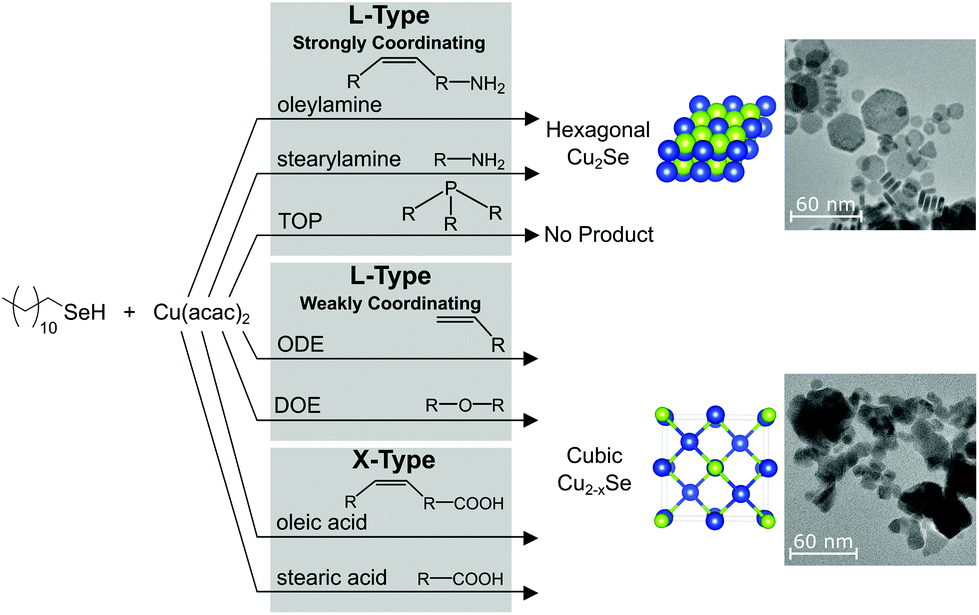 | ||
| Scheme 2 Summary of ligand classes, which predominantly produce one of two phases of copper selenide nanoparticles at 155 °C. | ||
Employing trioctylphosphine with DDSeH causes the formation of TOP:Se at room temperature. Unsaturation in a solvent such as 1-octadecene, or ligand such as oleylamine and oleic acid is subject to addition of the selenol across the double bonds through radical mechanisms, which also leads to the concomitant formation of diselenide. Carboxylic acids undergo selenoesterification. All of these chemistries did not occur at 155 °C but were extensive and often consumed the selenol reactant at 220 °C. Amine functional groups, in ligands such as oleylamine and stearylamine, lead to the formation of H2Se at 155 °C and 220 °C, but this chemistry did not consume a significant fraction of the reactant. The only truly “benign” commonly-used nanocrystal solvent tested was dioctyl ether, which did not react with the selenol up to 220 °C.
Since the alkylselenium reactant is so fundamentally changed in each of these ligand conditions, one would expect both the mechanism and kinetics of release of selenium to be affected in nanocrystal synthesis.
Here we studied the formation of copper selenide as a model material. It was found that three factors were most important in phase control: in situ formation of DD2Se2, reaction temperature and its relation to the phase transition temperature, and strong L-type ligation.
In most cases, the expected thermodynamic product cubic berzelianite Cu2−xSe formed, yet in very specific cases, the rare hexagonal polymorph formed. With carboxylic acid ligands, the hexagonal phase could be attributed to adventitious water which oxidized the selenol to the dialkyldiselenide. The diselenide reactant is known to give the hexagonal polymorph because of its slow kinetics of releasing selenium into solution. Dialkyldiselenide formation was also observed when unsaturation was present. The required temperatures of 220 °C for this radical chemistry were above the activation energy for the phase transition temperature from hexagonal to cubic Cu2Se. Therefore, only the cubic phase was seen.
In contrast, amines also promoted the formation of the hexagonal polymorph, but in this case, phase selection can be attributed to the strong L-type donation of amines which stabilize the predominantly neutral surfaces of hexagonal crystallites. Neat TOP, while also an L-type donor, caused the formation of TOP:Se and prevented nucleation of copper selenides altogether.
In summary, the conditions most favorable for producing the metastable hexagonal Cu2Se include using DD2Se2 as a reagent, long chain saturated L-type amine ligands, and keeping the synthetic temperature to as low as possible. In contrast, in order to produce the thermodynamic cubic Cu2−xSe phase, weak L-type ligation provided by ODE, DOE, or short chain amines or oleylamine could be used at higher synthetic temperatures of 220 °C. Carboxylic acids should be avoided as they produce insoluble selenoesters, and if not perfectly dry will cause a mixture of phases. Equally, TOP should be avoided as it produces TOP:Se which can be prepared by more conventional means with Se metal. In general, pre-heating DDSeH with ligands should be minimized, especially ones containing amines, acids, and unsaturation. The changes in the molecular nature of the precursor will alter the kinetics and reproducibility of the nanocrystal synthesis as well as the yields.
The kinetic trapping of metastable phases, with their new material properties, is one of the great advantages of colloidal nanocrystal synthesis. Carefully examining the molecular transformations in solution that occur before nucleation is one of the keys to predicting phase outcomes in nanocrystal synthesis. With such information in hand, it will be possible to rationally target metastable phases, possibly ones yet to be discovered.
Author contributions
E. A. H. designed experimental methodology, conducted the investigation, visualized data, and co-wrote the original draft. A. R. P. assisted with the investigation, validated results, and reviewed and edited the manuscript. J. E. M. acquired funding, administered and supervised the project, and co-wrote the original draft.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
The authors thank the U.S. National Science Foundation (CHE 1905265) for financial support. They also thank D. F. Stec for assistance acquiring 77Se NMR spectra.Notes and references
- B. A. Tappan, G. Barim, J. C. Kwok and R. L. Brutchey, Chem. Mater., 2018, 30, 5704–5713 CrossRef CAS.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 364 CrossRef CAS PubMed.
- Y. Wu, I. Korolkov, X. Qiao, X. Zhang, J. Wan and X. Fan, J. Solid State Chem., 2016, 238, 279–283 CrossRef CAS.
- S. C. Riha, D. C. Johnson and A. L. Prieto, J. Am. Chem. Soc., 2011, 133, 1383–1390 CrossRef CAS PubMed.
- E. A. Hernández-Pagán, E. H. Robinson, A. D. La Croix and J. E. Macdonald, Chem. Mater., 2019, 31, 4619–4624 CrossRef.
- B. Mahler, N. Lequeux and B. Dubertret, J. Am. Chem. Soc., 2010, 132, 953–959 CrossRef CAS PubMed.
- J. Q. Geisenhoff, A. K. Tamura and A. M. Schimpf, Chem. Commun., 2019, 55, 8856–8859 RSC.
- L. C. Frenette and T. D. Krauss, Nat. Commun., 2017, 8, 1–8 CrossRef CAS PubMed.
- M. P. Hendricks, M. P. Campos, G. T. Cleveland, I. Jen-La Plante and J. S. Owen, Science, 2015, 348, 1226–1230 CrossRef CAS PubMed.
- C. M. Evans, M. E. Evans and T. D. Krauss, J. Am. Chem. Soc., 2010, 132, 10973–10975 CrossRef CAS PubMed.
- J. E. Macdonald, C. J. Brooks and J. G. C. Veinot, Chem. Commun., 2008, 3777–3779 RSC.
- A. Roffey, N. Hollingsworth, H. U. Islam, M. Mercy, G. Sankar, C. R. A. Catlow, G. Hogarth and N. H. De Leeuw, Nanoscale, 2016, 8, 11067–11075 RSC.
- N. Hollingsworth, A. Roffey, H. U. Islam, M. Mercy, A. Roldan, W. Bras, M. Wolthers, C. R. A. Catlow, G. Sankar, G. Hogarth and N. H. De Leeuw, Chem. Mater., 2014, 26, 6281–6292 CrossRef CAS.
- G. G. Yordanov, H. Yoshimura and C. D. Dushkin, Colloid Polym. Sci., 2008, 286, 813–817 CrossRef CAS.
- J. Jasieniak, C. Bullen, J. Van Embden and P. Mulvaney, J. Phys. Chem. B, 2005, 109, 20665–20668 CrossRef CAS PubMed.
- C. Bullen, J. van Embden, J. Jasieniak, J. E. Cosgriff, R. J. Mulder, E. Rizzardo, M. Gu and C. L. Raston, Chem. Mater., 2010, 22, 4135–4143 CrossRef CAS.
- Y. Liu, D. Yao, L. Shen, H. Zhang, X. Zhang and B. Yang, J. Am. Chem. Soc., 2012, 134, 7207–7210 CrossRef CAS PubMed.
- B. C. Walker and R. Agrawal, Chem. Commun., 2014, 50, 8331–8334 RSC.
- G. Gariano, V. Lesnyak, R. Brescia, G. Bertoni, Z. Dang, R. Gaspari, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2017, 139, 9583–9590 CrossRef CAS PubMed.
- W. Z. Ostwald, Phys. Chem., 1897, 22, 289–330 CAS.
- A. L. Washington, M. E. Foley, S. Cheong, L. Quffa, C. J. Breshike, J. Watt, R. D. Tilley and G. F. Strouse, J. Am. Chem. Soc., 2012, 134, 17046–17052 CrossRef CAS PubMed.
- Y. Gao and X. Peng, J. Am. Chem. Soc., 2014, 136, 6724–6732 CrossRef CAS PubMed.
- T. J. Pennycook, J. R. McBride, S. J. Rosenthal, S. J. Pennycook and S. T. Pantelides, Nano Lett., 2012, 12, 3038–3042 CrossRef CAS PubMed.
- J. R. McBride, T. J. Pennycook, S. J. Pennycook and S. J. Rosenthal, ACS Nano, 2013, 7, 8358–8365 CrossRef CAS PubMed.
- U. Soni, V. Arora and S. Sapra, CrystEngComm, 2013, 15, 5458–5463 RSC.
- J. Huang, M. V. Kovalenko and D. V. Talapin, J. Am. Chem. Soc., 2010, 132, 15866–15868 CrossRef CAS PubMed.
- K. H. Low, C. H. Li, V. A. L. Roy, S. S. Y. Chui, S. L. F. Chan and C. M. Che, Chem. Sci., 2010, 1, 515–518 RSC.
- A. C. Berends, W. Van Der Stam, Q. A. Akkerman, J. D. Meeldijk, J. Van Der Lit and C. de Mello Donega, Chem. Mater., 2018, 30, 3836–3846 CrossRef CAS PubMed.
- V. S. Dilimon, G. Fonder, J. Delhalle and Z. Mekhalif, J. Phys. Chem. C, 2011, 115, 18202–18207 CrossRef CAS.
- Q. Dai, D. Li, H. Chen, S. Kan, H. Li, S. Gao, Y. Hou, B. Liu and G. Zou, J. Phys. Chem. B, 2006, 110, 22951 CrossRef CAS.
- E. Dhaene, J. Billet, E. Bennett, I. Van Driessche and J. De Roo, Nano Lett., 2019, 19, 7411–7417 CrossRef CAS PubMed.
- Y. A. Yang, H. Wu, K. R. Williams and Y. C. Cao, Angew. Chem., Int. Ed., 2005, 44, 6712–6715 CrossRef CAS PubMed.
- F. Jiang, L. T. Peckler and A. J. Muscat, Cryst. Growth Des., 2015, 15, 3565–3572 CrossRef CAS.
- D. W. Houck and B. A. Korgel, Chem. Mater., 2018, 30, 8359–8367 CrossRef CAS.
- Z. Yang and K. J. Klabunde, J. Organomet. Chem., 2009, 694, 1016–1021 CrossRef CAS.
- A. Pein, M. Baghbanzadeh, T. Rath, W. Haas, E. Maier, H. Amenitsch, F. Hofer, C. O. Kappe and G. Trimmel, Inorg. Chem., 2011, 50, 193–200 CrossRef CAS PubMed.
- B. Bureau, C. Boussard-ple, M. Lefloch, J. Troles, J. Lucas, D. Rennes and D. Beaulieu, Society, 2005, 33, 6130–6135 Search PubMed.
- H. Poleschner and K. Seppelt, Chem. – Eur. J., 2021, 27, 649–659 CrossRef CAS PubMed.
- N. P. Luthra, A. M. Boccanfuso, R. B. Dunlap and J. D. Odom, J. Organomet. Chem., 1988, 354, 51–62 CrossRef CAS.
- A. N. Hancock, S. Lobachevsky, N. L. Haworth, M. L. Coote and C. H. Schiesser, Org. Biomol. Chem., 2015, 13, 2310–2316 RSC.
- B. Hou, D. Benito-Alifonso, N. Kattan, D. Cherns, M. C. Galan and D. J. Fermín, Chem. – Eur. J., 2013, 19, 15847–15851 CrossRef CAS PubMed.
- H. Duddeck, P. Wagner and B. Rys, Magn. Reson. Chem., 1993, 31, 736–742 CrossRef CAS.
- R. S. Sedgwick, C. W. Hoerr and H. J. Harwood, J. Org. Chem., 1952, 17, 327–337 CrossRef CAS.
- W. H. H. Günther, J. Org. Chem., 1966, 31, 1202–1205 CrossRef.
- R. K. Čapek, K. Lambert, D. Dorfs, P. F. Smet, D. Poelman, A. Eychmüller and Z. Hens, Chem. Mater., 2009, 21, 1743–1749 CrossRef.
- Y. Hu, M. Afzaal, M. A. Malik and P. O'Brien, J. Cryst. Growth, 2006, 297, 61–65 CrossRef CAS.
- G. Menagen, J. E. Macdonald, Y. Shemesh, I. Popov and U. Banin, J. Am. Chem. Soc., 2009, 131, 17406–17411 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) availableAdditional instrumental parameters, additional 1H NMR and 77Se NMR, representative electron microscopy. See DOI: 10.1039/d1nr06282d |
| ‡ Currently at Department of Chemistry, Emory University, Atlanta, Georgia 30322, USA. |
| This journal is © The Royal Society of Chemistry 2022 |