
Non-invasive transferrin targeted nanovesicles sensitize resistant glioblastoma multiforme tumors and improve survival in orthotopic mouse models†
Puja
Sandbhor
a,
Jayant
Goda
*b,
Bhabani
Mohanty
ca,
Pradip
Chaudhari
bc,
Shilpee
Dutt
 d and
Rinti
Banerjee
*a
d and
Rinti
Banerjee
*a
aDepartment of Biosciences and Bioengineering, Indian Institute of Technology, Bombay, Powai, Mumbai, India. E-mail: rinti@iitb.ac.in
bDepartment of Radiation Oncology ACTREC, Tata Memorial Center, Kharghar Navi-Mumbai, India. E-mail: jgoda@actrec.gov.in
cDepartment of Comparative Oncology and Small Animal Imaging Facility, ACTREC, Tata Memorial Center, Kharghar Navi-Mumbai, India
dDepartment Shilpee Lab/DNA Repair and Cellular Oncology Lab, ACTREC, Tata Memorial Center, Kharghar Navi-Mumbai, India
First published on 13th December 2021
Abstract
The blood–brain barrier (BBB) and tumor heterogeneity have resulted in abysmally poor clinical outcomes in glioblastoma (GBM) with the standard therapeutic regimen. Despite several anti-glioma drug delivery strategies, the lack of adequate chemotherapeutic bioavailability in gliomas has led to a suboptimal therapeutic gain in terms of improvement in survival and increased systemic toxicities. This has paved the way for designing highly specific and non-invasive drug delivery approaches for treating GBM. The intranasal (IN) route is one such delivery strategy that has the potential to reach the brain parenchyma by circumventing the BBB. We recently showed that in situ hydrogel embedded with miltefosine (HePc, proapoptotic anti-tumor agent) and temozolomide (TMZ, DNA methylating agent) loaded targeted nanovesicles prevented tumor relapses in orthotopic GBM mouse models. In this study, we specifically investigated the potential of a non-invasive IN route of TMZ delivered from lipid nanovesicles (LNs) decorated with surface transferrin (Tf) and co-encapsulated with HePc to reach the brain by circumventing the BBB in glioma bearing mice. The targeted nanovesicles (228.3 ± 10 nm, −41.7 ± 4 mV) exhibited mucoadhesiveness with 2% w/v mucin suggesting their potential to increase brain drug bioavailability after IN administration. The optimized TLNs had controlled, tunable and significantly different release kinetics in simulated cerebrospinal fluid and simulated nasal fluid demonstrating efficient release of the payload upon reaching the brain. Drug synergy (combination index, 0.7) showed a 6.4-fold enhanced cytotoxicity against resistant U87MG cells compared to free drugs. In vivo gamma scintigraphy of 99mTc labeled LNs showed 500- and 280-fold increased brain concentration post 18 h of treatment. The efficacy of the TLNs increased by 1.8-fold in terms of survival of tumor-bearing mice compared to free drugs. These findings suggested that targeted drug synergy has the potential to intranasally deliver a high therapeutic dose of the chemotherapy agent (TMZ) and could serve as a platform for future clinical application.
1. Introduction
Glioblastoma multiforme (GBM) is the most lethal and a highly aggressive primary brain tumor causing 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths and 25000 new cases every year in the United States.1 Despite advances in the contemporary management of GBM, the survival rates are abysmally low with only 5% patients surviving for 5 years.2 The current treatment modalities have been abstemiously successful in reducing the tumor burden. This has been attributed to the presence of the blood–brain barrier (BBB) that limits the permeability of systemically administered chemotherapeutics across the brain resulting in decreased drug bioavailability in the brain. Tumor heterogeneity, diffusely infiltrating nature often leading to suboptimal resections, pseudo palisading of actively migrating tumor cells, hypoxia, and angiogenesis are responsible for its aggressive behaviour.3 Moreover, the diffuse topography of GBM has been regarded as a forerunner to frequent tumor recurrences.4 Over the past few years, several drug delivery approaches such as Convection Enhanced Delivery (CED) which operate on the principle of pressure gradient to actively deliver drugs at the targeted site have been tried.5 CED is associated with off-target spread of the therapeutic agent as a result of intrinsic backflow and challenges in achieving an optimal ratio of the volume of the infusion (Vi) to the volume of distribution (Vd).6 Direct delivery of the drug in the tumor bed can be considered an efficient mechanism which bypasses the BBB; for instance carmustine loaded polymeric implants (Gliadel® wafers) are directly placed into the tumor bed after surgical resection. However, these approaches are not without limitations, such as Gliadel wafers have issues like limited drug diffusion (∼2–3 mm), and heterogeneous drug concentration in the brain leading to a sub-therapeutic effect. In point of fact, these delivery strategies have resulted in only a modest improvement in survival and at exorbitant costs.7 Temozolomide (TMZ), an oral alkylating agent, was found to be successful in treating GBM as its bioavailability in the brain was high and soon became the standard chemotherapy regimen used concurrently with radiation therapy.8 However, TMZ is associated with excessive myelosuppression abrogating the tumoricidal benefits on gliomas.9
000 deaths and 25000 new cases every year in the United States.1 Despite advances in the contemporary management of GBM, the survival rates are abysmally low with only 5% patients surviving for 5 years.2 The current treatment modalities have been abstemiously successful in reducing the tumor burden. This has been attributed to the presence of the blood–brain barrier (BBB) that limits the permeability of systemically administered chemotherapeutics across the brain resulting in decreased drug bioavailability in the brain. Tumor heterogeneity, diffusely infiltrating nature often leading to suboptimal resections, pseudo palisading of actively migrating tumor cells, hypoxia, and angiogenesis are responsible for its aggressive behaviour.3 Moreover, the diffuse topography of GBM has been regarded as a forerunner to frequent tumor recurrences.4 Over the past few years, several drug delivery approaches such as Convection Enhanced Delivery (CED) which operate on the principle of pressure gradient to actively deliver drugs at the targeted site have been tried.5 CED is associated with off-target spread of the therapeutic agent as a result of intrinsic backflow and challenges in achieving an optimal ratio of the volume of the infusion (Vi) to the volume of distribution (Vd).6 Direct delivery of the drug in the tumor bed can be considered an efficient mechanism which bypasses the BBB; for instance carmustine loaded polymeric implants (Gliadel® wafers) are directly placed into the tumor bed after surgical resection. However, these approaches are not without limitations, such as Gliadel wafers have issues like limited drug diffusion (∼2–3 mm), and heterogeneous drug concentration in the brain leading to a sub-therapeutic effect. In point of fact, these delivery strategies have resulted in only a modest improvement in survival and at exorbitant costs.7 Temozolomide (TMZ), an oral alkylating agent, was found to be successful in treating GBM as its bioavailability in the brain was high and soon became the standard chemotherapy regimen used concurrently with radiation therapy.8 However, TMZ is associated with excessive myelosuppression abrogating the tumoricidal benefits on gliomas.9
The blood–brain barrier (BBB), considered to be a major detriment in CNS penetration of drugs, can be circumvented by various approaches which include transient opening of the BBB by ultrasound, chemical methods,10,11 or receptor mediated transport of therapeutics using specific ligands (e.g. receptors like transferrin [Tf], insulin-like growth factor [IGF], diphtheria toxin, and vascular endothelial growth factor [VEGF]) that are over-expressed on the endothelial cells of the BBB and rapidly infiltrating and proliferating tumor cells.12,13 However, these approaches are limited by the progressive loss of therapeutic activity in vivo due to gradual reduction in the expression of receptors over a period of time resulting in drug resistance. Moreover, accumulation of the payload at non-cranial sites also brings down its therapeutic efficacy.14,15 Consequently, a more specific and rational strategy is required to treat these aggressive tumors.
Intranasal (IN) administration of drugs is an attractive alternative route for CNS delivery of drugs as it is more effective in circumventing the BBB than systemic administration. The intranasally administered drug reaches the parenchymal space of the brain or cerebrospinal fluid (CSF) through trigeminal and olfactory nerves using axonal or endocytic pathways, and intra- or extra-neuronal pathways respectively.16 Several studies have reported the advantages of the IN route such as non-invasiveness, improved brain uptake and favourable pharmacodynamic and pharmacokinetic profiles of CNS drugs.17,18 The perceived advantage of the IN route is further augmented by using nano delivery platforms like lipid based and polymer based systems entrapped with the chemotherapeutic drug or their active metabolites that improve the therapeutic efficacy of the drug in high-grade gliomas especially grade III & IV gliomas (anaplastic astrocytomas & GBM) by increasing the permeability across the blood–brain barrier and blood–brain tumor barrier through the Enhanced Permeation and Retention (EPR) effect and controlled release of the payload at the designated site within the brain.19–21
In this study, we utilized the non-invasive IN route for targeted delivery of synergistic nanovesicles in the orthotopic GBM induced mouse model (standardized by micro-CT and bioluminescence imaging). This study demonstrated the ability of the intranasal route to enhance drug delivery to the brain by overcoming nasal mucosal barriers, due to the biocompatibility, and mucoadhesive property of nanovesicles, thereby improving the brain bioavailability of the incorporated drug. Furthermore, selective targeting of the orthotopic GBM bearing mouse using transferrin receptor modified intranasal nanovesicles represents the abilities of theragnostic cargos for various CNS malignancies in the future.
2. Results and discussion
The non-invasive IN route is a novel, alternative, and expedited approach that overcomes the BBB, considered a significant bottleneck in GBM management.22 Furthermore, precise targeting of the overexpressed Tf receptors on the surface of these tumors allows for ligand modification of the drug delivery system and results in improved cellular uptake and internalization of the drug within the tumor.23 Given the presence of BBB and the overexpression of Tf receptors on GBM cells, we sought to exploit Tf as a targeting moiety against glioma, decorated on synergistic drugs loaded IN nanovesicles. Currently, TMZ is reported to be the only effective chemotherapeutic drug against malignant gliomas.24 Although the oral formulation of TMZ free drug is used as a standard formulation for treating gliomas due to its high CNS bioavailability, its efficacy as an anti-glioma agent is tempered with high systemic toxicities, especially severe thrombocytopenia due to myelosuppression compromising the eventual clinical outcome of survival in these patients.9 Consequently, alternative drug delivery strategies are entailed to bring about a paradigm shift in managing these tumors.25 In this regard, a non-invasive intranasal approach with combinatorial targeted drug delivery could enhance retention and receptor-mediated internalization of the drugs into glioblastoma cells to achieve an ideal anti-glioma effect (Fig. 1).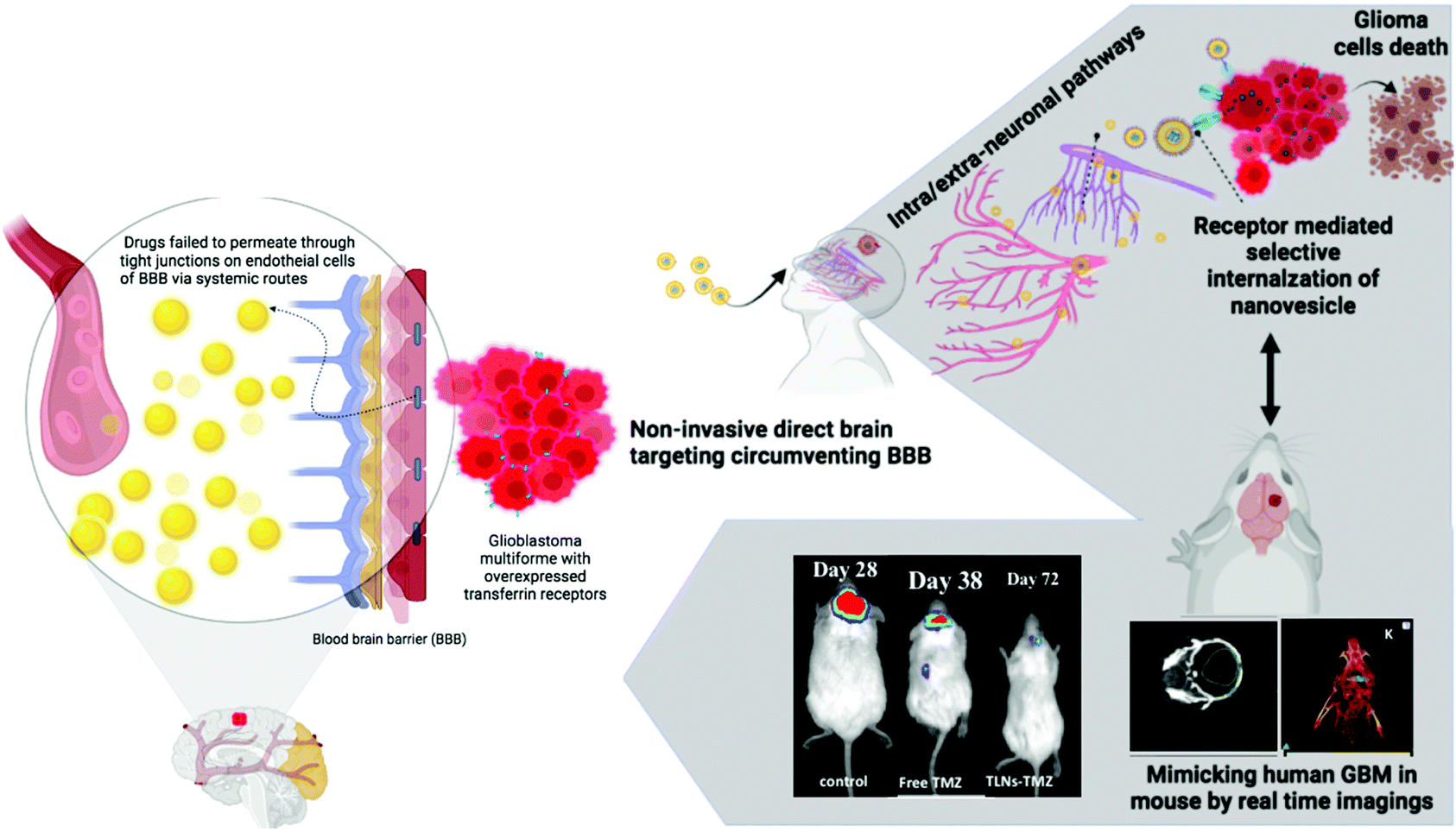 | ||
| Fig. 1 Schematic illustration of the non-invasive GBM targeting approach and motivation for developing a synergistic drug-loaded transferrin-targeted nanovesicle system reaching the brain by circumventing the blood–brain-barrier (BBB) through neurological pathways. The nanovesicles appear to modulate the BBB using intra/extra-neuronal channels to reach the brain. The transferrin coupling to the nano delivery vehicle carrying drug synergy effectively targets the infiltrating glioma cells via transferrin receptor-mediated endocytosis pathway and selectively homes the nanovesicles to an orthotopic GBM induced mouse model. Image created by Bio-render software. | ||
2.1. Intranasal targeted nanovesicle proof-of-concept evaluations
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of amide-I (1610–1690 cm−1) and N–H stretching of amide-II (1520–1620 cm1) of the protein.27 The amide-I and amide-II shifts indicated the interaction between Tf and nanovesicles; thus, it was considered a proof-of-concept evaluation for successful Tf conjugation to nanovesicles for further studies.
O stretching of amide-I (1610–1690 cm−1) and N–H stretching of amide-II (1520–1620 cm1) of the protein.27 The amide-I and amide-II shifts indicated the interaction between Tf and nanovesicles; thus, it was considered a proof-of-concept evaluation for successful Tf conjugation to nanovesicles for further studies.
Moreover, Tf embedded in the periphery of the nanovesicles was determined by UV-Visible spectroscopy28 by scanning the Tf specific absorbance from 200–400 nm using pure Tf (control), non-conjugated conjugated nanovesicles. UV-Visible spectra of the pure holo-transferrin exhibited absorption maxima at ∼280 nm, whereas complete recovery (represented by a decrease in the protein peak) of the total absorbance at 280 nm did not occur, probably due to reduced Tf absorptivity as it was conjugated with nanovesicles (Fig. S1c†). The coupling efficiency of Tf to nanovesicles was found to be 995.6 ± 76.8 μg of Tf per μmole of phospholipids by the BCA protein assay kit.
Drug-loaded nanovesicles had unimodal size distribution. The hydrodynamic size was 115.8 ± 13.2 nm, and the zeta potential was −30.6 ± 2.1 mV under pH 7.4, whereas post Tf modification size and zeta potential increased to 228.3 ± 10 nm and −41.7 ± 3.9 mV, respectively. This was probably due to the coupling of negatively charged protein to the vesicles. We observed that the size range was suitable for efficient mucosal penetration and their prospect for an ideal nose to brain drug delivery.29 We did not observe any discernible change in the stability of the nanovesicles following Tf modification, as revealed by the unchanged polydispersity index, encapsulation efficiency, and drug loading percent (Fig. 2a). Furthermore, high-resolution transmission electron microscopy (HR-TEM) and cryo-field emission gun scanning electron microscopy (cryo-FEG-SEM) image analysis showed spherical particles free from aggregations supporting the evidence that the nanovesicles were stable even after Tf modification (Fig. 2b and c).
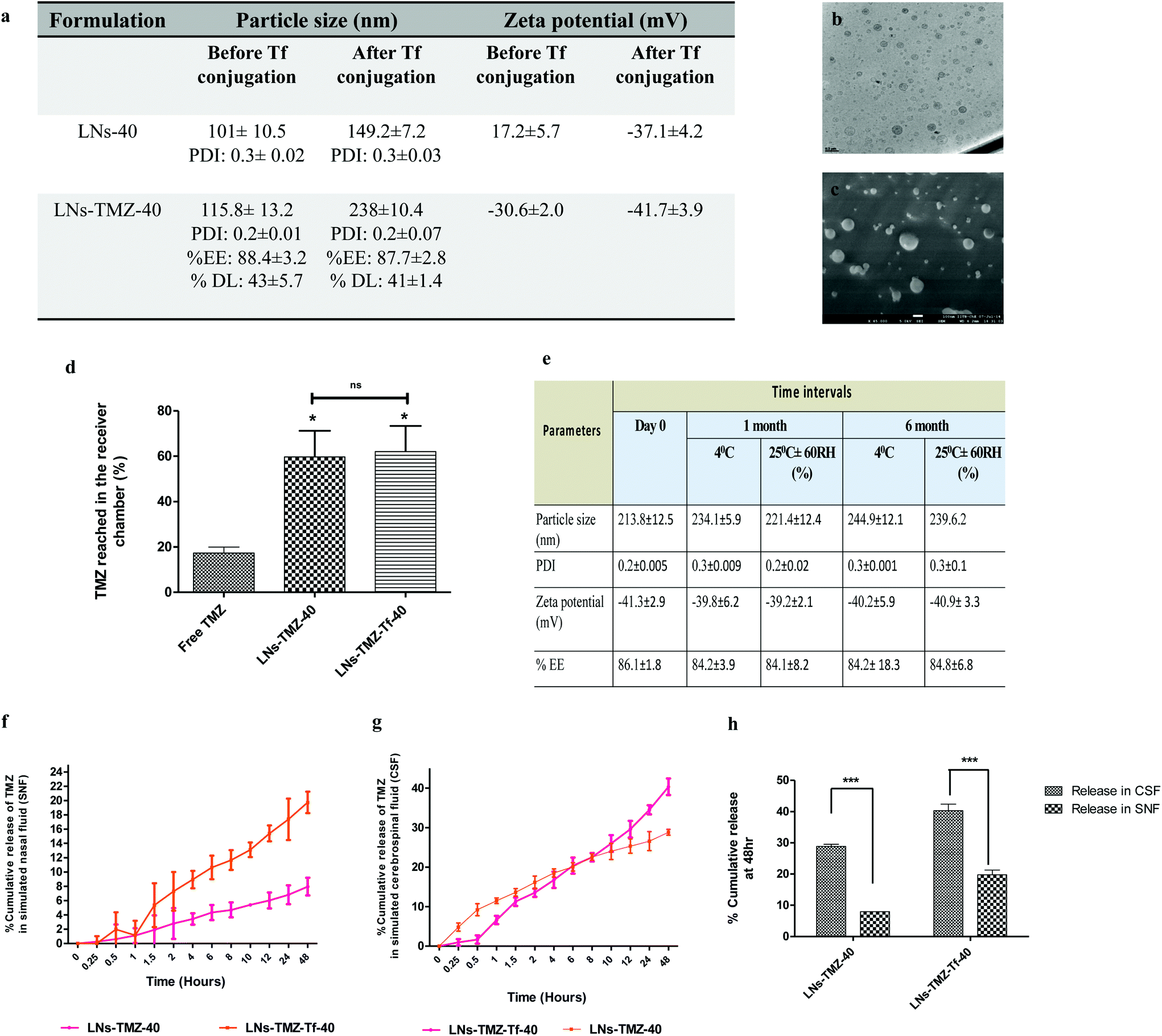 | ||
| Fig. 2 In vitro characterization of intranasal nanovesicles. (a) Physicochemical characterization of blank (LNs-40) and temozolomide (LNs-TMZ-40) encapsulated nanovesicles before and after transferrin (Tf) coupling showing particle size (nm), surface charge (mV), polydispersity index (PDI), percentage encapsulation efficiency (% EE) and drug loading (%DL) determined by dynamic light scattering, ZetaPALS analyzer and mass spectroscopy for three replicate measurements. (b and c) Representative high-resolution transmission electron microscopy (HR-TEM) and cryo-field emission gun scanning electron microscopy (Cryo-FEG-SEM) images of nanovesicles (scale bar: 200 nm) (d) ex vivo Parallel artificial membrane permeability assay (PAMPA) showing percentage of TMZ reached in the receiver chamber after 18 h of incubation using Franz's diffusion cells. All the results are expressed as mean ± SEM. Statistical significance was calculated using One way ANOVA indicating the significant difference at nsp ≥ 0.05, **p ≤ 0.01 vs. free TMZ, (n = 3) (e) stability profile of LNs-TMZ-Tf-40 as per International Council for Harmonization (ICH) under different storage conditions for six months (f and g) in vitro release kinetics of LNs-TMZ-40 and LNs-TMZ-Tf-40 in (f) simulated nasal fluid (SNF) (g) simulated cerebrospinal fluid (CSF) at 37 °C and (h) comparative evaluation of release profile of LNs at 48 h (mean ± SEM, at significant difference, ***p ≤ 0.001, *p ≤ 0.05 nsp ≥ 0.05, (n = 3)). | ||
000), which was characterized by atomic force microscopy (AFM), and cryo-field emission gun scanning electron microscopy analysis techniques (Fig. S2c†). The free TMZ, targeted/non-targeted TMZ loaded nanovesicles were screened according to their transport permeability across that membrane. As shown in Fig. 2d, the percent TMZ accumulated in the receiver chamber was significantly high for targeted (63 ± 7%) and non-targeted (60 ± 8.1%) nanovesicles compared to the free drug (29.2 ± 4.1%) at 18 h (p ≤ 0.05). Improved permeability of nanovesicles across the brain lipid barrier might be attributed to its lipid composition mainly, the surface-active nature of soya PC and miltefosine,34 and also the ability of miltifosine to interact with cholesterol35 one of the major components of the BBB, thereby fluidizing it and resulting in the transport of the encapsulated drugs across it. The above observations show the ability of the IN nanovesicle to permeate through the BBB.
2.2. In vitro human chemoresistant U87MG cell cytotoxicity and cellular uptake of intranasal nanovesicles
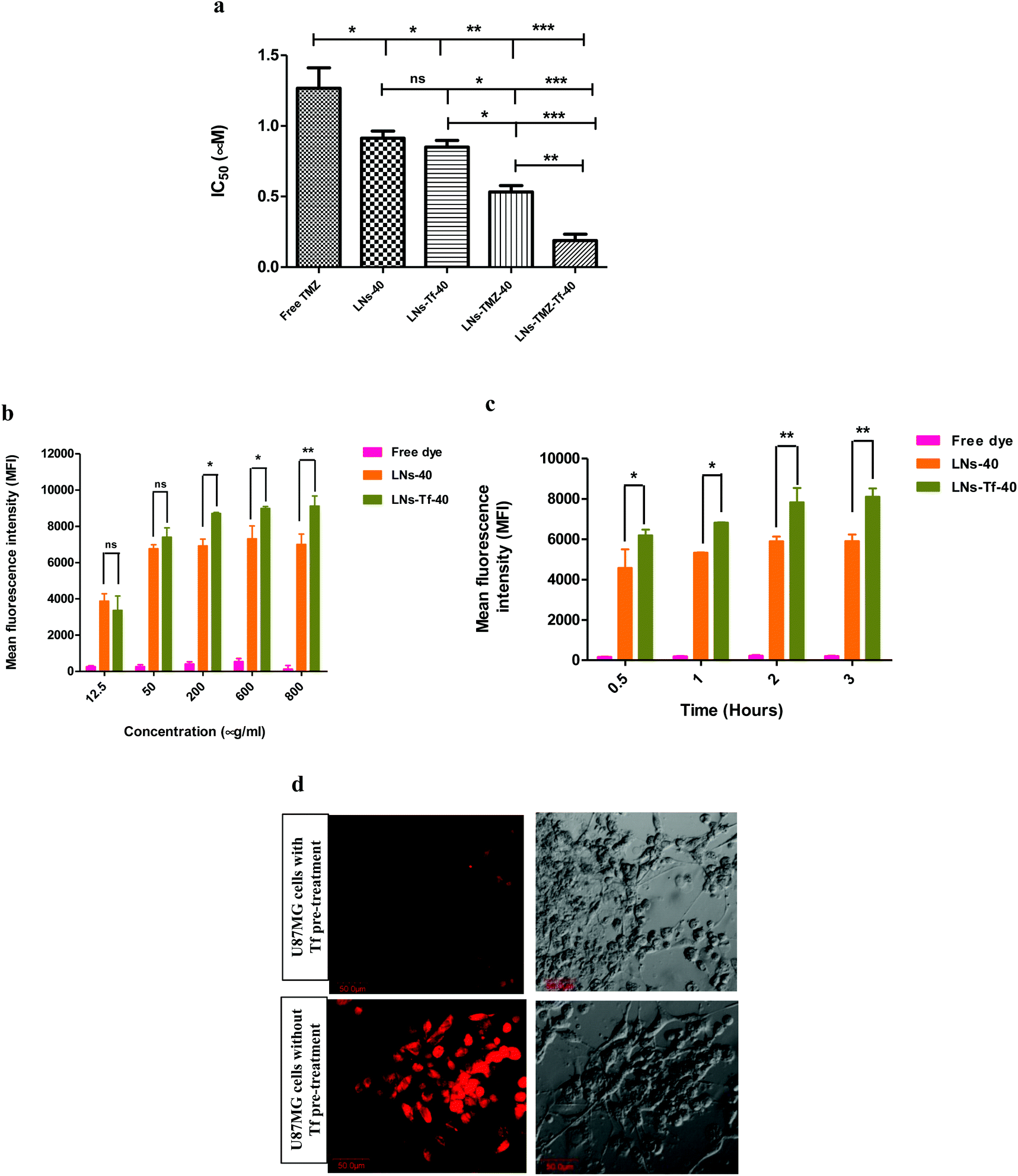 | ||
| Fig. 3 In vitro cytocompatibility, cytotoxicity, and cellular uptake studies. (a) MTT assay showing IC50 values of different nanovesicles on U87MG cells. Values represented as mean ± SEM, at significant difference, ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01 & *p ≤ 0.05 vs. free TMZ, (n = 3). (b and c) Flow cytometry quantitative results of in vitro cellular uptake of the free dye, rhodamine-6G loaded LNs-40 and LNs-Tf-40 at concentrations incubated for one h at 37 °C, and different time intervals at an equal dose (200 μg mL−1) (d) in vitro cellular uptake showing competitive inhibition in the presence of free ligand (transferrin) for rhodamine-6G labeled targeted nanovesicles. | ||
2.3. Intranasal route triggered an increased intracerebral uptake of targeted nanovesicles in vivo
To overcome the BBB barrier, we aimed to find an alternative delivery strategy against GBM. Hence, TMZ loaded Tf modified/non-modified nanovesicles and free TMZ were intranasally administered to find out the intracerebral concentration of TMZ. To understand the potential benefits of LNs-TMZ-Tf-40 administered by this non-invasive route, we investigated its brain accumulation and the pharmacokinetics and pharmacodynamic profiles by non-compartmental analysis and compared them with free TMZ using the HPLC technique. At predetermined time points, TMZ was extracted from the plasma and tissue homogenate of Sprague Dawley Rats after acetone treatment (Fig. 4a). Its concentration was determined using acetonitrile and formic acid (0.5%) in water (86:14% v/v) as a mobile phase. After standardization of the extraction efficiency, the recovery and limit of detection (LOD) were observed to be >89.3% and 0.04 μM, respectively.
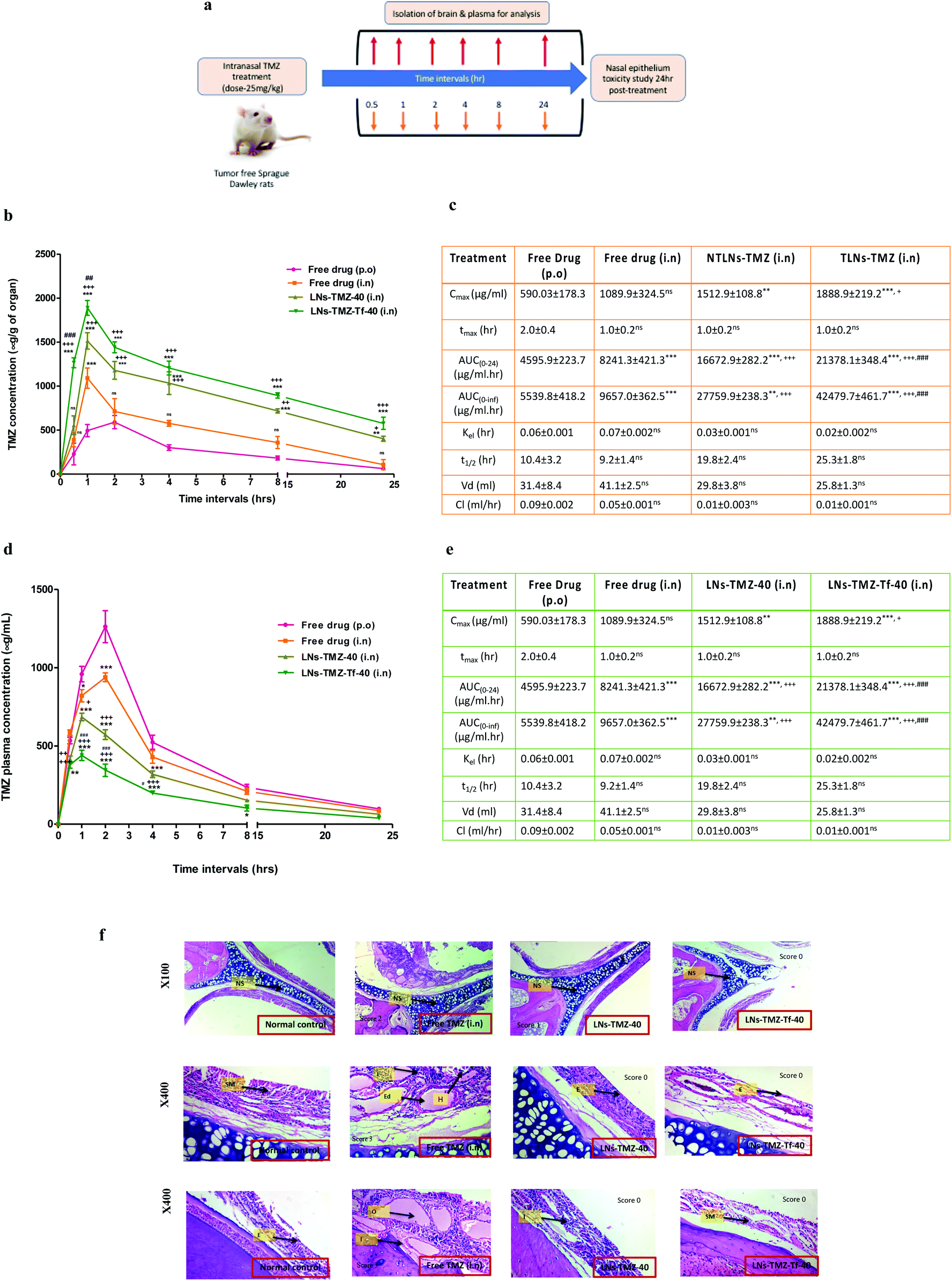 | ||
| Fig. 4 In vivo brain uptake and pharmacokinetic evaluation of intranasal nanovesicles. (a) Schematic of intranasal dosing regimen of drug-loaded lipid nanovesicle formulations in tumor-free animals. (b and c) Brain concentration vs. time profile and brain pharmacokinetics of different formulation & free drug in tumor-free animals (d and e) plasma concentration vs. time profile and pharmacokinetics of free drug & TMZ loaded different formulation in tumor-free animals. Results are expressed as mean ± SEM and significant difference at ***p ≤ 0.001, **p ≤ 0.01, vs. free drug (p.o), +++p ≤ 0.001, +p ≤ 0.05 vs. free drug (i.n) and ###p ≤ 0.001, for LNs-TMZ-Tf-40 vs. LNs-TMZ-40, (n = 6) and (f) photomicrographs of normal control (NC, saline-treated), free TMZ (i.n), TMZ loaded non-targeted (LNs-TMZ-40) and targeted (LNs-TMZ-Tf-40) lipid nanovesicles after 24 h post-treatment, 25 mg kg−1 (×100 & ×400 magnification, (n = 4) {NS: nasal septum; H: hyperplasia of nasal epithelium: I: submucosal infiltration of inflammatory cells; Ed: submucosal edema}. | ||
The plasma concentration-time profile for targeted/non-targeted nanovesicles and free TMZ is illustrated in Fig. 4d. The plasma pharmacokinetic parameters are summarized in Fig. 4e. We observed that the concentration of TMZ in plasma was negligible in the targeted nanovesicle group compared to free TMZ groups. High peak plasma concentration (Cmax, 1262.0 ± 121.03 μg ml−1 and 938.6 ± 87.4 μg ml−1) was observed when free TMZ was administered orally or by the IN route, respectively. LNs-TMZ-Tf-40 achieved a 2.8-fold reduction in Cmax. Similarly, free drugs exhibited a significantly (P ≤ 0.001) higher AUC as compared to TLNs-TMZ. However, other plasma pharmacokinetics parameters such as drug clearance, t1/2 and tmax among all the groups were observed to be statistically non-significant (p ≥ 0.05).
Brain pharmacokinetic parameters of targeted/non-targeted nanovesicles and free TMZ are summarized in Fig. 4c. Relative augmentation in Cmax and AUClast after IN administration of LNs-TMZ-Tf-40 was 2.5-fold and 4.6-fold higher than that in oral free TMZ, respectively. The prompt and enhanced appearance of brain tmax after IN dosing of TMZ from TLNs may be due to direct nose to brain targeting and a shorter path that the drug needs to traverse to reach the brain parenchyma. After IN dosing, the early tmax value for the brain (1.0 ± 0.2 h) was higher than that for oral administration. This could be due to superior transport from nose to brain assisting speedy uptake of nanovesicles. The brain levels of the intranasally treated LNs-TMZ-Tf-40 group were substantially higher than those of the oral TMZ group. The intranasal targeted TMZ loaded nanovesicle treated group showed a peak concentration of ∼30% at one hour and ∼9% of the administered dose in the brain post 24 h compared to the oral free TMZ treated group (i.e. 7% at one hour and 1% at 24 h). This could be due to a longer residence time in the nasal cavity (mucoadhesiveness). This resulted in higher penetration of the drug payload into the brain parenchyma as opposed to higher systemic absorption when administered through the oral route. These findings imply that intranasally administered targeted nanovesicles result in higher accumulation of the drug in the brain with lower plasma concentration and thereby can be used as a potential platform for direct brain targeting through the nose to treat CNS malignancies.
2.4. Intranasal route enhanced in vivo brain retention of nanovesicles and mitigating off-target toxicity
Radioisotopes have been extensively used in the development of multimodality imaging agents. To determine TMZ retention in the brain and other vital organs, we radiolabeled TMZ loaded targeted/non-targeted nanovesicles and free TMZ with 99mTc (∼2 mCi) using stannous chloride. The labeling efficiency was found to be >93.4%. In vivo biodistribution of IN formulations and free TMZ was performed in tumor-free nude mice and analysed by the gamma scintigraphy technique (Fig. 5a). Biodistributions of 99mTc-labeled LNs-TMZ-Tf-40, LNs-TMZ-40, and free TMZ were expressed as injected dose per g of tissue/organs (% ID per gram of tissue).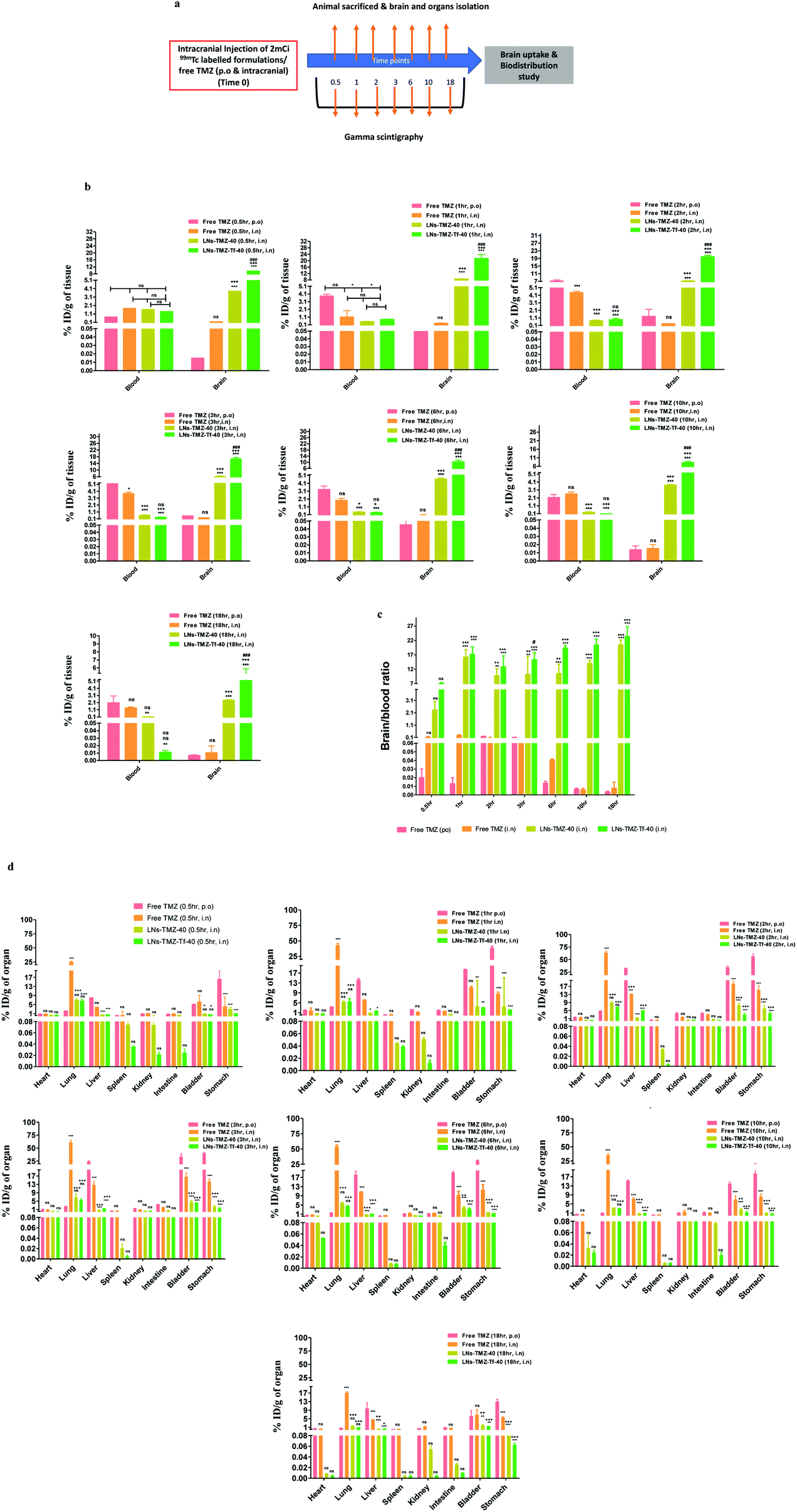 | ||
| Fig. 5 In vivo brain retention of 99mTc-labeled intranasal nanovesicles by gamma scintigraphy technique in nude mice. (a) Schematic of organ isolation for brain and biodistribution, and gamma scintigraphy analysis of nude mice at different time points and (b) brain and blood distribution of radiolabeled free TMZ and TMZ loaded nanovesicles at different time intervals after intranasal (i.n)/oral (p.o) administration in nude mice (c) brain/blood ratio of radiolabeled free TMZ and TMZ loaded nanovesicles at different time intervals after intranasal/oral administration in nude mice and (d) biodistribution of radiolabeled free TMZ and TMZ loaded lipid-nanovesicles at different time intervals after oral and intranasal administration in nude mice. All the results are expressed as mean ± SEM and statistical significance was calculated using One way ANOVA indicating the significant difference at ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05 and nsp ≥ 0.05 free drug (p.o), +++p ≤ 0.001, ++p ≤ 0.01, +p ≤ 0.05 vs. free drug (i.n) and ###p ≤ 0.001, ##p ≤ 0.01 for LNs-TMZ-Tf-40 vs. LNs-TMZ-40, (n = 4, TMZ dose: 25 mg kg−1). | ||
We observed that the intracerebral distribution of nasally administered 99mTc labeled LNs-TMZ-Tf-40 and LNs-TMZ-40 was significantly higher over free TMZ (p ≤ 0.001) for all the time points measured. Although ligand anchoring had shown a comparatively higher brain uptake of the modified nanovesicles than the unmodified nanovesicles (Fig. 5b). We measured the brain to blood ratio of TMZ and observed a significantly longer retention (p ≤ 0.001) and higher drug concentration in the brain than in the blood of LNs-TMZ-Tf-40 compared to free TMZ post 18 h treatment (Fig. 5c). However, considering the 6 h half-life of 99mTc, decay of the isotope enabled follow-up of the nanovesicle treated mice for a maximum time of 18 h. We noted the significant amount of 99mTc-labeled TMZ in the liver and bladder from the free drug treated group compared to targeted/non-targeted nanovesicle treated groups (p ≤ 0.05). However, we observed that the accumulation of LNs-TMZ-Tf-40 in liver (probably due to lower expression of transferrin receptor-2 (TfR2) in liver for iron homeostasis) was comparatively higher (p ≤ 0.01) than LNs-TMZ-40 which could be attributed to the presence of Tf receptors in the liver. These findings suggested that transferrin modification enables the nanovesicle uptake by liver to be metabolized and eliminated via urine/faeces.40,41 Moreover, insignificant (p ≤ 0.01) uptake of LNs-TMZ-Tf-40 was observed in other vital organs such as the spleen, lungs, intestine, stomach, kidneys, and heart at 18 h post-treatment (Fig. 5d).
2.5. Intranasal nanovesicles improved the anti-glioma efficacy in the orthotopic GBM induced mouse model
The following demonstration of the efficacy of Tf conjugated LNs to deliver TMZ into the glioma cells; studies were addressed to investigate whether the developed nanovesicles would be efficient in delivering the encapsulated payloads to the intracranial tumor when administered via the intranasal route. For this purpose, we developed and standardized the orthotopic GBM induced mouse model that mimicked human GBM by employing real-time imaging techniques (bioluminescence imaging). The anti-glioma efficacy, safety, and tolerability of nanovesicles containing the anti-glioma agents were evaluated by measuring the tumor burden utilizing longitudinal bioluminescence imaging and calculating the total flux intensity, the whole body weight, and survival time of the U87MG glioblastoma bearing mice, respectively (Fig. 6a). | ||
| Fig. 6 In vivo anti-glioma efficacy evaluation of intranasal nanovesicles in orthotopic GBM induced mice. (a) Schematic of treatment and imaging regimen of the orthotopic GBM mouse model (TMZ dose 25 mg kg−1) and (b) standardization of the orthotopic GBM mouse model showing (A–E) bioluminescence image analysis in U87MG Luc expressed mice on different days and (F–K) micro CT analysis of tumor volume and their 3D view at pre-determined time intervals (c) orthotopic GBM mouse model showing micro CT analysis of tumor and their 3D view (A–C) coronal view (D–F) right sagittal view (G–I) left sagittal view and (J–L) axial view at pre-determined time intervals (d) quantification of total flux (p/s) in the orthotopic GBM induced mouse model. Values expressed as mean ± SEM and significant difference at $$$p ≤ 0.001 for free TMZ (per oral) vs. free TMZ (i.n), and nanovesicle formulations, @@p ≤ 0.01 and @@@p ≤ 0.001 for free TMZ (i.n) vs. LNs-40 and LNs-Tf-40, LNs-TMZ-40, and LNs-TMZ-Tf-40 (i.n) respectively, (n = 12) (e) body weight change of different groups post-treated with TMZ in the orthotopic GBM induced mouse model showing a significant difference at ***p ≤ 0.001, **p ≤ 0.01 for control vs. treatment groups, (n = 12) (f) evaluation of anti-glioma efficacy by Kaplan Meier survival analysis shows a significant difference at ***p ≤ 0.001 for control vs. treatment groups, (n = 8) (g) representative luciferase images from mice treated with free TMZ and TMZ loaded different formulations in orthotopic GBM induced mouse model and (h) histopathological analysis showing the toxicity profile of mice treated with free TMZ, different nanovesicle formulations loaded with TMZ and control mice (saline treated) in the orthotopic glioblastoma induced mouse model (n = 4). | ||
3. Conclusions
Glioblastoma (GBM) is the most common adult brain tumour with aggressive biology and is associated with poor prognosis and high relapse rates. This is due to resistance to conventional treatment, tumor heterogeneity, and limited permeability of chemotherapeutics across the blood–brain barrier (BBB). Temozolomide (TMZ) is one of the most effective and commonly used drugs to treat GBM. However, there is still an unmet need for delivering the drug specifically at the tumour site to overcome the systemic toxicities and therapeutic limitations of TMZ. Subsequently, better modification of the existing drugs or identifying novel chemical entities with enhanced anti-glioma efficacy and low systemic side effects is constantly creditable. Therefore, we developed a novel approach for enhanced brain retention of TMZ across the BBB through an alternative route (i.e., intranasal) using lipid-based targeted synergistic nano-drug delivery to improve the anti-glioma treatment efficacy. The present study offered a rational, non-invasive, selectively targeted combinatorial approach to treat chemorefractory tumors like GBM and provided a feasible translational approach to eradicate glioma cells in the brain through ligand (transferrin) modification on the surface of the nanovesicles.Transferrin (Tf) targeting to the nanovesicles facilitated the selective internalization of the payload via receptor-mediated endocytosis in in vitro studies. The targeted TMZ nanovesicles imparted significantly higher release kinetics in CSF and mucoadhesiveness with 2% w/v mucin, reflecting their suitability as an ideal candidate for an effective nose to brain drug delivery to treat GBM. The targeted TMZ nanovesicles showed 6.4-fold higher cytotoxic effects against chemoresistant human U87MG cells compared to free drugs. Combining HePc (a proapoptotic agent) with TMZ conferred a synergistic cytotoxic effect against U87MG cells.
The results from in vivo biodistribution studies divulged a significantly higher brain-to-blood ratio of intranasally administered targeted nanovesicles than free drugs given by either oral or IN routes. Importantly, significant improvement and prolonged survival of orthotopic GBM bearing mice treated with IN targeted nanovesicles indicated the enhanced anti-glioma efficacy over clinically approved systemic TMZ. In addition, we anticipate that targeted nanovesicles will be better tolerated than free TMZ when combined with HePc and reduce off-target organ toxicities often observed in the clinic. The present study could lead to a better anti-glioma efficacy in GBM in comparison with conventional systemic administration routes, achieving higher TMZ levels of brain concentration and mitigate systemic drug exposure. On the other hand, the developed technology could improve patient compliance with minimal psychological stress under chronic use through the non-invasive route of administration. Conclusively, our preclinical experiments have shown that these smart delivery platforms have the potential to be translated to clinics and significantly augment the therapeutic indices of anti-glioma chemotherapy by overcoming the current biological barrier commonly encountered in pre-clinical drug discovery and development.
4. Experimental section
4.1. Materials
1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine and hexadecylphosphocholine (HePc) or miltefosine (purity > 99%) were obtained from Avanti Polar Lipids (Alabaster, AL, USA) and Lipoid GmbH (Germany), respectively. Temozolomide (purity > 99%) was purchased from Ark Pharma. Inc. (China). Soya phosphatidylcholine (SPC), dialysis membrane of molecular wt cut-off 12000–14000, N-hydroxysuccinimide (NHS) and 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide (EDC) were procured from HiMedia Laboratories Pvt Ltd, Mumbai (India). Human holo-transferrin with purity >99% was purchased from Sigma Aldrich (India). Fluorescein isothiocyanate (FITC), rhodamine-6G, and Nile red were procured from Anaspec Inc. (USA). The bicinchoninic acid protein assay kit (BCA), 2-(N-morpholino) ethanesulfonic acid (MES), and high-pressure liquid chromatography (HPLC) grade acetonitrile, chloroform, and methanol were obtained from Thermo Scientific (USA), Merck (India), respectively. Milli-Q water with 18.2 MΩ cm resistivity was used in all studies using the Millipore (USA) system.
4.2. Cell line
The human U87MG glioblastoma multiforme cell line was procured from National Centre for Cell Science (India). Fetal bovine serum (FBS), minimum essential medium (MEM), Dulbecco's modified Eagle's medium (DMEM), antibiotic-antimycotic solution, Dulbecco's phosphate-buffered saline (PBS), trypsin-ethylene diamine tetra-acetic acid (EDTA) solutions, Hank's balanced salt solution, and bovine serum albumin (BSA) were procured from HiMedia Laboratories Pvt. Ltd (India). The cell culture flasks (25 cm2 and 75 cm2) and plates (96 and 24 well-plates) were purchased from Corning, California (USA).4.3. Fabrication of intranasal nanovesicles
Transferrin (Tf) targeted, and non-targeted HePc/TMZ loaded nanovesicles (LNs-Tf and LNs-TMZ-Tf) were prepared by a thin film hydration method26 with slight modification and using different HePc contents ranging from 10 mol% to 40 mol% to soya PC and covalent coupling of Tf to nanovesicles using EDC-NHS based carbodiimide chemistry (Fig. S1a†). The non-targeted drug/blank loaded nanovesicles were labeled as LNs-TMZ-10/LNs-10, LNs-TMZ-20/LNs-20, LNs-TMZ-30/LNs-30, and LNs-TMZ-40/LNs-40 depending on the preselected molar ratio of HePc to soya PC The purified targeted nanovesicles were labeled as LNs-TMZ-Tf-10/LNs-Tf-10, LNs-TMZ-Tf-20/LNs-Tf-20, LNs-TMZ-Tf-30/LNs-Tf-30, and LNs-TMZ-Tf-40/LNs-Tf-40.4.4. Characterization of intranasal nanovesicles and controlled drug release in simulated fluids
We analysed the polydispersity index (PDI) and size distribution of nanovesicles by photon correlation spectroscopy using a laser particle analyser (BI200SM, Brookhaven Instrument Corporation, USA). Charge on the nanovesicles surface was analysed with a Zeta potential analyser using ZetaPALS, Brookhaven Instrument Corporation, (USA). Analysis was carried out in triplicate, and results were presented as a mean value ± SEM. Furthermore, we evaluated the encapsulation efficiency,42 Tf content and its binding efficiency to nanovesicles, and phospholipid (PL) concentration in the nanovesicles after Tf coupling43 according to the protocols described previously. We used a high-resolution transmission electron microscope (HR-TEM, model JEM 2100 ultra, at 200 kV) to evaluate the morphology of the nanovesicles. Next, we additionally performed UV-Visible spectroscopy28,44 to validate Tf embedded in the periphery of nanovesicles and Fourier transform infrared (FTIR) spectroscopy to confirm the interaction between Tf and nanovesicles after coupling as proof-concept studies. Briefly, the presence of Tf specific absorbance was observed via scanning the wavelength range from 200–400 nm using pure holo-Tf (standard) blank nanovesicles before and after Tf conjugation.To investigate the mucoadhesiveness of nanovesicles loaded with TMZ before and after Tf modification, the sessile drop technique was employed using optical contact angle meter (CAM-100 from KSV Instruments (Finland) as reported previously.45 The parallel artificial membrane permeability assay (PAMPA) based on porcine brain lipid extract (PBLE) was carried out as described previously33 with slight modifications. Briefly, 1 g of wet porcine brain tissue was homogenized in 5 ml of 0.25% acetic acid for 1 min and then kept undisturbed for 15 min at room temperature. It was then centrifuged at 3000 rpm for 5 min followed by decantation of the supernatant. The pellet was slurried with 40 ml of chloroform and methanol (1:1 v/v) and kept for 5 min aside and then subsequently homogenized at a maximum speed for 2 min and then left standing for 10 min. The homogenized mixture was then centrifuged at 3000 rpm for 5 min, followed by decantation of chloroform and methanol extract. This step was repeated thrice to extract out more lipids, and subsequently, the lipid extract was lyophilized and kept at 4 °C until use for further experiment. The PBL extract (20 mg ml−1 in n-dodecane, i.e., volume to be 500 μl) was filled in a 1 cm dialysis bag after sealing one end; the filter membrane was previously moistened in Milli-Q. Then, both the ends of the dialysis bag were sealed and kept for 24 h. 100 μl of all test samples were placed in the donor chamber above the filter membrane coated with PBLE (20 mg ml−1). The simulated cerebrospinal fluid (CSF: 119 mM NaCl, 2.5 mM KCl, 1.3 mM MgSO4, 2.5 mM CaCl2, 26.2 Mm NaHCO3, 1 mM NaH2PO4, and 11 mM glucose) was used as a receptor fluid. The formulations were allowed to diffuse from the donor chamber through the filter membrane to the receiver chamber filled, and the system was left undisturbed for 18 h, following which the concentration of TMZ in the receiver chamber was analysed by UV-Visible spectroscopy (PerkinElmer Lambda 25, PerkinElmer, USA) at 329 nm after appropriate dilutions.
Next, we investigated the stability of formulations (lyophilized 1 mL sample) as per the guidelines given by International Conference on Harmonization (ICH) under different storage conditions, i.e., 25 °C ± 2 °C, 65% relative humidity (RH), and 4 °C ± 2 °C. At specific time intervals, samples were withdrawn and resuspended with Milli-Q water followed by evaluation for appearance, particle size, polydispersity index, surface charge, and drug content analysis by high-performance liquid chromatography (HPLC).
Next, we performed the dialysis bag method to study the in vitro controlled release kinetics of targeted and non-targeted TMZ loaded nanovesicles using dialysis membrane-110 (12000–14000 of molecular weight cut-off, HiMedia) at 37 °C temperature at 100 rpm in simulated nasal fluid (SNF) [NaCl (0.87%), CaCl2. 2H2O (0.088%), KCl (0.31%), and BSA (0.636%)]46 and CSF (composition described previously)47 in type-II USP dissolution apparatus under sufficiently good sink conditions. At pre-determined time intervals, 1 ml aliquots from the release medium were withdrawn, and the same volume was replenished with a fresh medium. The concentration of TMZ present in withdrawn aliquots was measured after suitable dilutions using a UV-visible spectrophotometer at 329 nm.
4.5. In vitro dose–response and cellular uptake studies
The human U87MG glioblastoma multiforme cell line (over-expressing multi-drug resistance-1 {MDR-1}, Tf receptors)48 was selected for in vitro cellular studies. The cells were incubated under saturated humid environments at 37 °C with 5% CO2. Every alternate day medium changed for U87MG cells with subculture/plating at 75–80% confluency. The biocompatibility study was performed as per the previously reported method on normal cells, i.e., fibroblasts.49 Furthermore, we assessed different nanovesicle formulations’ anti-proliferative potential by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay by plating 5 × 104 cell per 200 μL per well in a complete medium followed by incubation in a 5% CO2 incubator to adhere for 24 h. After 24 h, the exhausted medium was replenished with a fresh medium followed by incubation for 72 h in the media containing graded concentrations of different formulations to be evaluated. Percent cell viability was measured using a tetrazolium-based MTT assay. The only medium-treated cells were used as the control. The IC50 values for all the test samples were evaluated using Prism 5.0 GraphPad software. The results were presented as mean ± SEM for three replicate measurements.Subsequently, for time-dependent U87MG cells cellular uptake of different formulations, 5 × 105cells per mL U87MG cells were seeded in a 24-well-plate for 48 h in a CO2 incubator. After 48 h, the exhausted medium was replenished with complete fresh medium and treated with 100 μL of rhodamine-6G (50 μg mL−1) labeled LNs-40 and LNs-Tf-40 and incubated for different time intervals at 37 °C. Similarly, concentration-dependent U87MG cells cellular uptake of different nanovesicles was carried out by incubating cells with dye-labeled nanovesicles for 1 h at 37 °C. At the end of the incubation period, cells were washed with Hank's balanced salt solution (HBSS) three–four times under ice-cold conditions followed by cell digestion. Time/concentration-dependent cellular uptake was quantified by mean fluorescence intensity measurement, using Flow cytometry analysis wherein untreated/unstained cells served as the negative control and results presented as mean ± SEM, (n = 3).
4.6. Intranasal brain transport and biodistribution of nanovesicles
Similarly, for the brain uptake study, the brain tissue was isolated at each time point and homogenized in phosphate buffered saline (PBS) acidified to obtain 10% brain homogenate and preserved at −80 °C until further studies. We extracted TMZ from plasma and tissue homogenates using ice-cold acetone followed by 2 min vortexing and centrifugation at 4000 rpm at 4 °C for 15 min. Subsequently, the supernatant was evaporated to dryness at 60 °C using a nitrogen gas evaporator. The left-over residue was dispersed in 100 μL mobile phase, i.e., 0.1% formic acid in water and acetonitrile (14:86 v/v), and pumped at 0.8 mL min−1 of flow rate. Drug quantification was performed by HPLC analysis using the C18 Cosmosil column (250 × 4.6 mm, 5 μm) at 329 nm after bio-analytical method validation and optimization to estimate free TMZ in the brain homogenates and plasma samples.
Furthermore, we performed the pharmacokinetic analysis to measure the concentrations of TMZ in brain homogenates and plasma at pre-determined times by applying the non-compartmental model. The maximum concentration (Cmax) and time required to reach maximum concentration (tmax) upon oral/intranasal administration of TMZ in plasma and brain, the elimination rate constant (Kel), half-life (t1/2), and area under the curve (AUC) were determined using Phoneix Winnonlin 6.3 version software, (USA).
Next, we investigated the nasal safety/toxicity potential of intranasal formulations by isolating the nasal tissues at 24 h post-treatment. Histopathological analysis was performed as reported by the previous method with minor modification.50 Concisely, nasal tissue was subjected for decalcification for 48–72 h followed by embedding and sectioning in paraffin blocks using a microtome (Nikon Fx-35A, Japan). The sections were subjected to H and E staining and images acquired under the optical microscope.
4.7. In vivo bio-distribution of 99mTc-labeled intranasal nanovesicles
For brain uptake and biodistribution analysis, we performed radio-labeling as per the previously reported method51 with slight modification. Briefly, saline control, non-targeted, targeted nanovesicles, and free drugs were radio-labeled with technetium by direct tagging using a reducing agent like the aqueous solution of stannous chloride (SnCl2). 1 mg mL−1 of SnCl2 solution (0.02 mL) was mixed with 1 mL of test formulation followed by pH adjustment at 6.8 using sodium bicarbonate buffer (50 mM) followed by filtration of the mixture through a 0.22 μm nylon-66 membrane filter. Subsequently, aqueous 99mTcO4 (2 mCi mL−1) was added to the filtered mixture and incubated for 15–20 min (factors that affect the maximum labeling amount, i.e., SnCl2, pH, and incubation time was pre-optimized). The radiolabeling efficiency of nanovesicles was evaluated by paper chromatography using the acetone mobile phase wherein paper acting as a stationary phase was cut over and below the mark of test formulation and region of the solvent front, respectively. The gamma counter (Triumph@, Gamma Medical Ideas, Northridge, USA) was used to check the radioactivity on the piece of paper to ensure that the test formulation had been labeled with radioactivity. The radiolabeled intranasal formulations (10 μL per nostril), oral/intranasal free TMZ as the positive control (dose 25 mg kg−1, 250 μL by oral gavage), and saline (negative control) were given to nude mice (male, 25–30 g) with partial isoflurane inhalation anaesthesia. Blood samples were drawn from retro-orbital plexus at different time intervals following administration to trace the plasma distribution, and retention profile of 99mTc labeled nanovesicles. The radioactivity of 99mTc labeled LNs-TMZ-Tf-40 and TLNs-TMZ-40 nanovesicles in blood and vital organs (heart, brain, lung, liver, kidney, and lungs) was quantified using a gamma counter. Results were expressed as the percentage of the dose administered that accumulated in each organ (% ID/cc).4.8. Therapeutic study in orthotopic GBM bearing NOD-SCID knockdown mice
To evaluate intranasal formulations’ anti-glioma efficacy, we established synergistic orthotopic GBM tumors in NOD-SCID mice, which showed a pathological correlation in humans and mice. Briefly, 7.5 × 106 cells in 6 μL sterile PBS were injected intracranially into mice at 2 mm right, 1 mm posterior to the bregma at a depth of 3 mm. We standardized the intracranial GBM bearing animal model by monitoring tumor development using a contrast-enhanced high-resolution micro-CT scan (PMOD Technologies Ltd, Zurich, Switzerland) and bioluminescence analysis. After two weeks, the animals showing average tumor sizes of approximately 5–10 mm3 were selected for further studies and subjected to further treatment. To optimize the dose, free TMZ solution was given at the dose equivalent to human dose, i.e., 75 mg m−2 day−1 was 25 mg kg−1 and 150 mg m−2 day−1 was 50 mg kg−1 of mouse body weight orally. Next, we randomized the tumor-bearing mice into six groups (n = 12) such as untreated control (negative control), intranasal LNs-TMZ-Tf-40, LNs-TMZ-40, LNs-Tf-40, LNs-40, and free TMZ solution (positive control) by oral and intranasal routes. All treatments were given once daily on day 16 and/or day 17 for five alternate days after tumor development, and mice were monitored for the progress of tumor development by constructing the 3D-tumor volume by OsiriX™ 7.5 version, software Geneva, Switzerland by micro-CT scan/bioluminescence analysis on pre-determined time points. Mice showing massive tumor burden with grade-4 symptoms were sacrificed, followed by brain and vital organ isolation, whereas survival analysis was carried out on remaining animals (n = 6). Mice were monitored for body weight and general health status, and neurological deficit scale scoring52 was used to score the clinical signs of GBM. For the histopathological study, poly-L-lysine-coated slides were used for formalin-fixed and paraffin-embedded tissue sections (4 μm). Additionally, uncoated slides were used for haematoxylin-eosin (H and E) staining of tissues section.4.9. Statistical analysis
Data were presented as mean ± SEM. GraphPad Prism 5.0 software was used to compare multiple-group and specific groups using one-way analysis of variance (ANOVA) and unpaired Student's t-test, respectively. Various pharmacokinetic factors were calculated using Basic software. P-value less than 0.05 indicated the statistically significant difference after normalizing all experimental animals’ dose and body weight.Author contributions
P.S. designed and performed all the experiments. B.M. contributed to animal studies. R.B., J.G., P.C., and S.D. conceived the idea and contributed to the design of the experiment. J.G. was involved in manuscript editing.Conflicts of interest
There are no conflicts to declare. The authors have filed patents related to this technology.Acknowledgements
We are thankful to the Lady Tata Memorial Trust (LTMT) for Ph.D. funding of P.S. The authors would like to thank IRCC and SAIF, IIT Bombay for research facilities.References
- H. Guerrero-Cazares, S. Y. Tzeng, N. P. Young, A. O. Abutaleb, A. Quiñones-Hinojosa and J. J. Green, ACS Nano, 2014, 8(5), 5141–5153, DOI:10.1021/nm501197v.
- H. Gao, Z. Yang, S. Cao, Y. Xiong, S. Zhang, Z. Pang and X. Jiang, Biomaterials, 2014, 35(7), 2374–2382, DOI:10.1016/j.biomaterials.2013.11.076.
- D. J. Brat, K. Aldape, H. Colman, D. Figrarella-Branger, G. N. Fuller, C. Giannini, E. C. Holland, R. B. Jenkins, B. Kleinschmidt-DeMasters, T. Komori, J. M. Kros, D. N. Louis, C. McLean, A. Perry, G. Reifenberger, C. Sarkar, R. Stupp, M. J. van den Bent, A. von Deimling and M. Weller, Acta Neuropathol., 2020, 139(3), 603–608, DOI:10.1007/s00401-020-02127-9.
- R. H. Bobo, D. W. Laske, A. Akbasak, P. F. Morrison, R. L. Dedrick and E. H. Oldfield, Proc. Natl. Acad. Sci. U. S. A., 1994, 91(6), 2076–2080, DOI:10.1073/pnas.91.6.2076.
- L. C. Hou, A. Veeravagu, A. R. Hsu and V. C. Tse, Neurosurg. Focus, 2006, 20(4), E5, DOI:10.3171/foc.2006.20.4.2.
- S. M. Chang, N. A. Butowski, P. K. Sneed and I. V. Garner, Neurosurg. Focus, 2006, 20(4), E4 Search PubMed.
- J. Perry, A. Chambers, K. Spithoff and N. Laperriere, Curr. Oncol., 2007, 14(5), 189–194, DOI:10.3747/co.2007.147.
- R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer and R. O. Mirimanoff, N. Engl. J. Med., 2005, 352(10), 987–996, DOI:10.1056/NEJMoa043330.
- C. Fang, K. Wang, Z. R. Stephen, Q. Mu, F. M. Kievit, D. T. Chiu, O. W. Press and M. Zhang, ACS Appl. Mater. Interfaces, 2015, 7(12), 6674–6682, DOI:10.1021/am5092165.
- M. Aryal, N. Vykhodtseva, Y. Z. Zhang and N. McDannold, J. Control. Release, 2015, 204, 60–69, DOI:10.1016/j.jconrel.2015.02.033.
- S. J. Madsen and H. Hirschberg, J. Biophotonics, 2010, 3(5–6), 356–367, DOI:10.1002/jbio.200900095.
- V. M. Pulgar, Front. Neurosci., 2019, 12, 1019, DOI:10.3389/fnins.2018.01019.
- S. Wang, Y. Meng, C. Li, M. Qian and R. Huang, Nanomaterials, 2015, 6(1), 3, DOI:10.3390/nano6010003.
- A. M. Mehta, A. M. Sonabend and J. N. Bruce, Neurotherapeutics, 2017, 14(2), 358–371, DOI:10.1007/s13311-017-0520-4.
- M. Touat, A. Idbaih, M. Sanson and K. L. Ligon, Ann. Oncol., 2017, 28(7), 1457–1472, DOI:10.1093/annonc/mdx106.
- P. G. Djupesland, J. C. Messina and R. A. Mahmoud, Ther. Delivery, 2014, 5(6), 709–733, DOI:10.4155/tde.14.41.
- K. Chung, I. Ullah, N. Kim, J. Lim, J. Shin, S. C. Lee, S. Jeon, S. H. Kim, P. P. Kumar and S. K. Lee, J. Drug Targeting, 2020, 28(6), 617–626, DOI:10.1080/1061186X.2019.1706095.
- F. Sabir, R. Ismail and I. Csoka, Drug Discovery Today, 2020, 25(1), 185–194, DOI:10.1016/j.drudis.2019.10.005.
- F. A. Bruinsmann, G. Richter Vaz, A. de Cristo Soares Alves, T. Aguirre, A. Raffin Pohlmann, S. Stanisçuaski Guterres and F. Sonvico, Molecules, 2019, 24(23), 4312, DOI:10.3390/molecules24234312.
- J. F. Hsu, S. M. Chu, C. C. Liao, C. J. Wang, Y. S. Wang, M. Y. Lai, H. C. Wang, H. R. Huang and M. H. Tsai, Cancers, 2021, 13(2), 195, DOI:10.3390/cancers13020195.
- J. S. Michael, B. S. Lee, M. Zhang and J. S. Yu, J. Transl. Int. Med., 2018, 6(3), 128–133, DOI:10.2478/jtim-2018-0025.
- P. S. Gaikwad and R. Banerjee, Ther. Delivery, 2018, 9(8), 571–592, DOI:10.4155/tde-2018-0022.
- M. Zhao, D. van Straten, M. Broekman, V. Préat and R. M. Schiffelers, Theranostics, 2020, 10(3), 1355–1372, DOI:10.7150/thno.38147.
- H. Strobel, T. Baisch, R. Fitzel, K. Schilberg, M. D. Siegelin, G. Karpel-Massler, K. M. Debatin and M. A. Westhoff, Biomedicines, 2019, 7(3), 69, DOI:10.3390/biomedicines7030069.
- R. H. Kang, J. E. Jang, E. Huh, S. J. Kang, D. R. Ahn, J. S. Kang, M. J. Sailor, S. G. Yeo, M. S. Oh, D. Kim and H. Y. Kim, Nanoscale Horiz., 2020, 5(8), 1213–1225, 10.1039/d0nh00077a.
- F. C. Lam, S. W. Morton and J. Wyckoff, et al. , Nat. Commun., 2018, 9, 1991, DOI:10.1038/s41467-018-04315-4.
- R. Manoharan, J. J. Baraga, R. P. Rava, R. R. Dasari, M. Fitzmaurice and M. S. Feld, Atherosclerosis, 1993, 103(2), 181–193, DOI:10.1016/0021-9150(93)90261-r.
- T. Sun, H. Wu, Y. Li, Y. Huang, L. Yao, X. Chen, X. Han, Y. Zhou and Z. Du, Oncotarget, 2017, 8(43), 74451–74465, DOI:10.18632/oncotarget.20165.
- A. T. Alex, A. Joseph, G. Shavi, J. V. Rao and N. Udupa, Drug Delivery, 2016, 23(7), 2144–2153, DOI:10.3109/10717544.2014.948643.
- T. K. Vyas, A. Shahiwala, S. Marathe and A. Misra, Curr. Drug Delivery, 2015, 2(2), 165–175, DOI:10.2174/1567201053586047.
- M. Agrawal, S. Saraf, S. Saraf, S. G. Antimisiaris, M. B. Chougule, S. A. Shoyele and A. Alexander, J. Control. Release, 2018, 281, 139–177, DOI:10.1016/j.jconrel.2018.05.011.
- O. Tsinman, K. Tsinman, N. Sun and A. Avdeef, Pharm. Res., 2011, 28(2), 337–363, DOI:10.1007/s11095-010-0280-x.
- J. Bicker, G. Alves, A. Fortuna, P. Soares-da-Silva and A. Falcao, Int. J. Pharm., 2016, 501(1–2), 102–111, DOI:10.1016/j.ijpharm.2016.01.074.
- T. P. Dorlo, T. A. Eggelte, P. J. de Vries and J. H. Beijnen, Analyst, 2012, 137(5), 1265–1274, 10.1039/c2an15641e.
- I. R. Gómez-Serranillos, J. Miñones, P. Dynarowicz-łatka Jr., J. Miñiones and E. Iribarnegaray, Langmuir, 2004, 20(3), 928–933, DOI:10.1021/la0303254.
- K. Nigam, A. Kaur, A. Tyagi, M. Nematullah, F. Khan, R. Gabrani and S. Dang, Drug Deliv. Transl. Res., 2019, 9(5), 879–890, DOI:10.1007/s13346-019-00622-5.
- S. Dixit, T. Novak, K. Miller, Y. Zhu, M. E. Kenney and A. M. Broome, Nanoscale, 2015, 7(5), 1782–1790, 10.1039/c4nr04853a.
- D. Ag-Seleci, V. Maurer, F. B. Barlas, J. C. Porsiel, B. Temel, E. Ceylan, S. Timur, F. Stahl, T. Scheper and G. Garnweitner, Int. J. Mol. Sci., 2021, 22(9), 4556, DOI:10.3390/ijms22094556.
- R. L. Shinde and P. V. Devarajan, Drug Delivery, 2017, 24(1), 152–161, DOI:10.1080/10717544.2016.1233593.
- L. Reyderman, P. Statkevich, C. M. Thonoor, J. Patrick, V. K. Batra and M. Wirth, Xenobiotica, 2004, 34(5), 487–500, DOI:10.1080/00498250410001685737.
- A. Calzolari, L. M. Larocca, S. Deaglio, V. Finisguerra, A. Boe, C. Raggi, L. Ricci-Vitani, F. Pierconti, F. Malavasi, R. De Maria, U. Testa and R. Pallini, Transl. Oncol., 2010, 3(2), 123–134, DOI:10.1593/tlo.09274.
- N. Raval, H. Jogi and P. Gondaliya, et al. , Sci. Rep., 2019, 9, 16047, DOI:10.1038/s41598-019-52390-4.
- Z. C. Soe, J. B. Kwon, R. K. Thapa, W. Ou, H. T. Nguyen, M. Gautam, K. T. Oh, H. G. Choi, S. K. Ku, C. S. Yong and J. O. Kim, Pharmaceutics, 2019, 11(2), 63, DOI:10.3390/pharmaceutics11020063.
- H. Ding, V. Sagar, M. Agudelo, S. Pilkka-Kanthikeel, V. S. Atluri, A. Raymond, T. Samikkannu and M. P. Nair, Nanotechnology, 2014, 25, 055101, DOI:10.1088/0957-4487/25/5/055101.
- S. Suri and R. Banerjee, J. Biomed. Mater. Res., Part A, 2006, 79(3), 650–664, DOI:10.1002/jbm.a.30917.
- P. S. Istvan, C. C. Tımea, F. Eva and S. Lajos, Bioact. Carbohydr. Diet. Fibre, 2013, 2, 157–163, DOI:10.1016/j.bcdf.2013.10.006.
- A. Rytter, T. Cronberg, F. Asztély, S. Nemali and T. Wieloch, J. Cereb. Blood Flow Metab., 2003, 23(1), 23–33, DOI:10.1097/01.WCB.0000034361.37277.1B.
- M. Rittierodt, T. Tschernig and K. Harada, Pathobiology, 2004, 71(3), 123–128, DOI:10.1159/000076466.
- S. Prijic, J. Scancar, R. Romih, M. Cemazar, V. B. Bregar, A. Znidarsic and G. Sersa, J. Membr. Biol., 2010, 236(1), 167–179, DOI:10.1007/s00232-010-9271-4.
- Z. Dong, H. Katsumi, T. Sakane and A. Yamamoto, Int. J. Pharm., 2010, 393(1–2), 244–252, DOI:10.1016/j.ijpharm.2010.04.021.
- A. Khan, S. S. Imam, M. Aqil, A. Ahad, Y. Sultana, A. Ali and K. Khan, Mol. Pharm., 2016, 13(11), 3773–3782, DOI:10.1021/acs.molpharmaceut.6b00586.
- J. Lee, S. H. Park and Y. Z. Kim, Brain Tumor Res. Treat., 2018, 6(1), 22–30, DOI:10.14791/btrt.2018.6.e1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr05460k |
| This journal is © The Royal Society of Chemistry 2022 |