
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Corallopyronin A: antimicrobial discovery to preclinical development
Anna K.
Krome
 abc,
Tim
Becker
ab,
Stefan
Kehraus
bd,
Andrea
Schiefer
bc,
Michael
Gütschow
e,
Lillibeth
Chaverra-Muñoz
f,
Stephan
Hüttel
f,
Rolf
Jansen
f,
Marc
Stadler
fg,
Alexandra
Ehrens
bc,
Domen
Pogorevc
gh,
Rolf
Müller
gh,
Marc P.
Hübner
bc,
Thomas
Hesterkamp
i,
Kenneth
Pfarr
bc,
Achim
Hoerauf
bc,
Karl G.
Wagner
*ab and
Gabriele M.
König
*bd
abc,
Tim
Becker
ab,
Stefan
Kehraus
bd,
Andrea
Schiefer
bc,
Michael
Gütschow
e,
Lillibeth
Chaverra-Muñoz
f,
Stephan
Hüttel
f,
Rolf
Jansen
f,
Marc
Stadler
fg,
Alexandra
Ehrens
bc,
Domen
Pogorevc
gh,
Rolf
Müller
gh,
Marc P.
Hübner
bc,
Thomas
Hesterkamp
i,
Kenneth
Pfarr
bc,
Achim
Hoerauf
bc,
Karl G.
Wagner
*ab and
Gabriele M.
König
*bd
aPharmaceutical Technology and Biopharmaceutics, University of Bonn, Germany. E-mail: karl.wagner@uni-bonn.de
bGerman Center for Infection Research (DZIF), Partner Site Bonn-Cologne, Germany
cInstitute for Medical Microbiology, Immunology and Parasitology, University Hospital Bonn, Germany
dInstitute for Pharmaceutical Biology, University of Bonn, Germany. E-mail: g.koenig@uni-bonn.de
ePharmaceutical & Medicinal Chemistry, University of Bonn, Germany
fDepartment of Microbial Drugs, Helmholtz Centre for Infection Research, Braunschweig, Germany
gGerman Center for Infection Research (DZIF), Partner Site Hannover-Braunschweig, Germany
hHelmholtz Institute for Pharmaceutical Research Saarland, Saarbrucken, Germany
iTranslational Project Management Office (TPMO), German Center for Infection Research (DZIF), Braunschweig, Germany
First published on 22nd June 2022
Abstract
Covering: August 1984 up to January 2022
Worldwide, increasing morbidity and mortality due to antibiotic-resistant microbial infections has been observed. Therefore, better prevention and control of infectious diseases, as well as appropriate use of approved antibacterial drugs are crucial. There is also an urgent need for the continuous development and supply of novel antibiotics. Thus, identifying new antibiotics and their further development is once again a priority of natural product research. The antibiotic corallopyronin A was discovered in the 1980s in the culture broth of the Myxobacterium Corallococcus coralloides and serves, in the context of this review, as a show case for the development of a naturally occurring antibiotic compound. The review demonstrates how a hard to obtain, barely water soluble and unstable compound such as corallopyronin A can be developed making use of sophisticated production and formulation approaches. Corallopyronin A is a bacterial DNA-dependent RNA polymerase inhibitor with a new target site and one of the few representatives of this class currently in preclinical development. Efficacy against Gram-positive and Gram-negative pathogens, e.g., Chlamydia trachomatis, Orientia tsutsugamushi, Staphylococcus aureus, and Wolbachia has been demonstrated. Due to its highly effective in vivo depletion of Wolbachia, which are essential endobacteria of most filarial nematode species, and its robust macrofilaricidal efficacy, corallopyronin A was selected as a preclinical candidate for the treatment of human filarial infections. This review highlights the discovery and production optimization approaches for corallopyronin A, as well as, recent preclinical efficacy results demonstrating a robust macrofilaricidal effect of the anti-Wolbachia candidate, and the solid formulation strategy which enhances the stability as well as the bioavailability of corallopyronin A.
1 Need for novel anti-infectives
Global health is threatened by infections with antibiotic resistant pathogens, which are on the rise. Antibiotics are a fundamental component in modern medicine, e.g., invasive surgeries can only be carried out safely if combined with effective antibiotic treatment options.1,2 However, anti-infectives are frequently overused, e.g., in food production. As a result, resistant infections are responsible for millions of deaths annually. In 2019, according to statistical models, 4.95 million deaths were associated with bacterial antimicrobial resistance. The same study attributed 1.27 million deaths globally directly to bacterial antimicrobial resistance.3 These alarming numbers led to the term “post-antibiotic era” in contrast to the “golden antibiotic era” of the previous century.4–6 Crucial factors against the emergence and transmission of resistant pathogens include better prevention and control of infectious diseases, as well as the sustainment of approved antibiotics via antimicrobial stewardship programs.7,8 Furthermore, novel antibiotics are urgently needed to treat infections caused by resistant pathogens or for infections for which drugs have not yet been developed, e.g., for many neglected tropical diseases.According to the World Health Organization (WHO), only 43 antibiotics and 27 non-traditional antibacterial agents were listed in the clinical pipeline in 2020.9 The majority are derivatives of well-established antibiotics and indeed only two of the eleven new antibiotics approved by the US Food and Drug Administration (FDA) and/or the European Medicines Agency (EMA) since July 2017 belong to new classes.9 Overall, the number and type of antibiotics in clinical development is insufficient to tackle the challenge of microbial resistance.10–15 In 2020, 292 antibacterial agents were in the preclinical stage.16–18 Of these preclinical candidates only five target RNA synthesis, a verified target for antibiotics. Included in the list is corallopyronin A (COR A),9 the focus of this review whose development is an example for a publicly funded project with the prospect for later private participation and joint development.19 This kind of cooperation has been trending in the last decades, because many major pharmaceutical companies, with few exceptions, have abandoned the field of antimicrobial research and development due to insufficient returns on their investments.19–21 Thus, publicly and privately funded research centres, universities, and university hospitals, as well as small- and medium-sized biopharmaceutical companies have entered the field of antimicrobial research. These undertakings are partly supported and maintained by established public and private grants22,23 and their impact can be seen in the number of antibiotics in the preclinical development.9,24
To guide and promote antimicrobial research and the consecutive development process of new antibiotics, the WHO has published a list of antibiotic-resistant priority pathogens, currently posing the greatest threat to global health.12,24 Furthermore, the WHO provided a list of 20 neglected tropical diseases, including many infectious diseases, for which novel drugs are highly needed.25 Two of them are onchocerciasis and lymphatic filariasis, which burden the public health system of affected countries. The therapy of these infectious diseases is currently limited to (semi-)annual mass drug administrations (MDA) of ivermectin or ivermectin combined with albendazole for onchocerciasis, and a triple therapy of ivermectin, albendazole, and diethylcarbamazine for lymphatic filariasis. However, diethylcarbamazine may cause serious adverse effects in patients suffering from onchocerciasis and life-threatening adverse events may occur in patients with the filarial nematode Loa loa following treatment with diethylcarbamazine or ivermectin. Therefore, the triple therapy is only recommended for regions not co-endemic for onchocerciasis and loiasis.26–29 In addition, the aforementioned therapies only temporally inhibit filarial embryogenesis and lead to the clearance of microfilariae, first stage larvae found in the blood or skin, and they lack a macrofilaricidal effect, i.e., killing the adult worms which can live for 5–15 years, continuously producing microfilariae.30–32 Novel modes of action and new drugs that provide a macrofilaricidal effect are, therefore, needed. The administration of doxycycline targeting Wolbachia, endosymbionts present in the majority of human pathogenic filariae, was shown to lead to permanent sterilization of the adult female filariae, as well as the favourable slow death of the adult worms over months.30,33,34 Doxycycline, a valuable treatment for individual anti-filarial therapy, is now recommended by the WHO against onchocerciasis and lymphatic filariasis.35 The lengthy therapy interval of 4–5 weeks and the contraindications for pregnant and breast-feeding women, as well as children below the age of 8 years led to the search for novel, faster acting drugs active against Wolbachia.34,36 COR A constitutes one of these novel agents currently in the preclinical phase with the aim to develop it as a treatment for human filarial infections (Fig. 1).34,37–42 The following sections highlight the discovery, biosynthesis, production, efficacy, and formulation strategies for COR A.
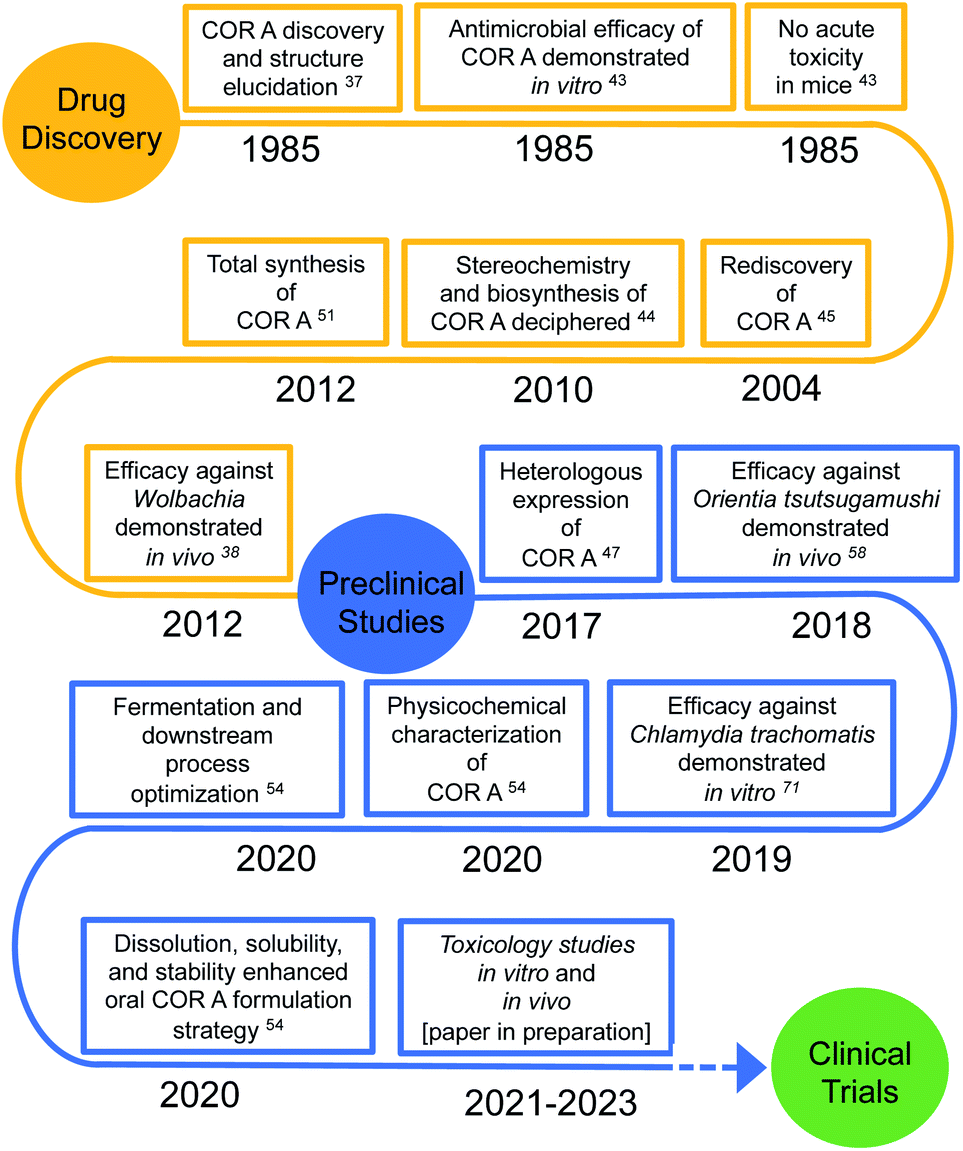 | ||
| Fig. 1 Chronological sequence of corallopyronin A (COR A) discovery and preclinical trials. | ||
2 Corallopyronin A
2.1 Drug discovery and structure elucidation
In a screening program for antibiotics from myxobacteria, COR A (Fig. 2A) was discovered by the Höfle and Reichenbach groups in the 1980s at the Helmholtz Centre for Infection Research (formerly named Gesellschaft für Biotechnologische Forschung (GbF), Braunschweig, Germany). Via liquid–liquid phase distribution and chromatography, the anti-infective COR A was isolated from the culture broth of the Corallococcus coralloides strain Cc127 (DSM 2550). In addition, the isomer corallopyronin A′ (COR A′) (Fig. 2B) and the cyclisation product corallopyronin C (COR C) (Fig. 2C) were obtained from the culture medium, both considered as isolation artefacts. Corallopyronin B (COR B) is a biosynthetically derived COR A derivative and only produced in very small quantities. To our knowledge, COR B has not been the focus of investigations, and is thus not discussed further in this review.37,43 The molecular formula of COR A was determined as C30H41NO7, leading to a molecular weight of 527.65 g mol−1. The chemical structures of COR A and its derivatives were determined via nuclear magnetic resonance spectroscopy (NMR), mass spectrometry (MS), and elemental analysis (EA).37 The configuration of the carbon–carbon double bonds, i.e., Δ,11,12 Δ,16,18 Δ,19,20 Δ,29,30 were already established in the first reports and annotated as trans configured, whereas the double bond Δ25,27 was determined as cis configured. The two chiral centers of COR A, C-7 and C-24, were found to be R configured, according to the Cahn–Ingold–Prelog system, by the group of König (Fig. 2A).44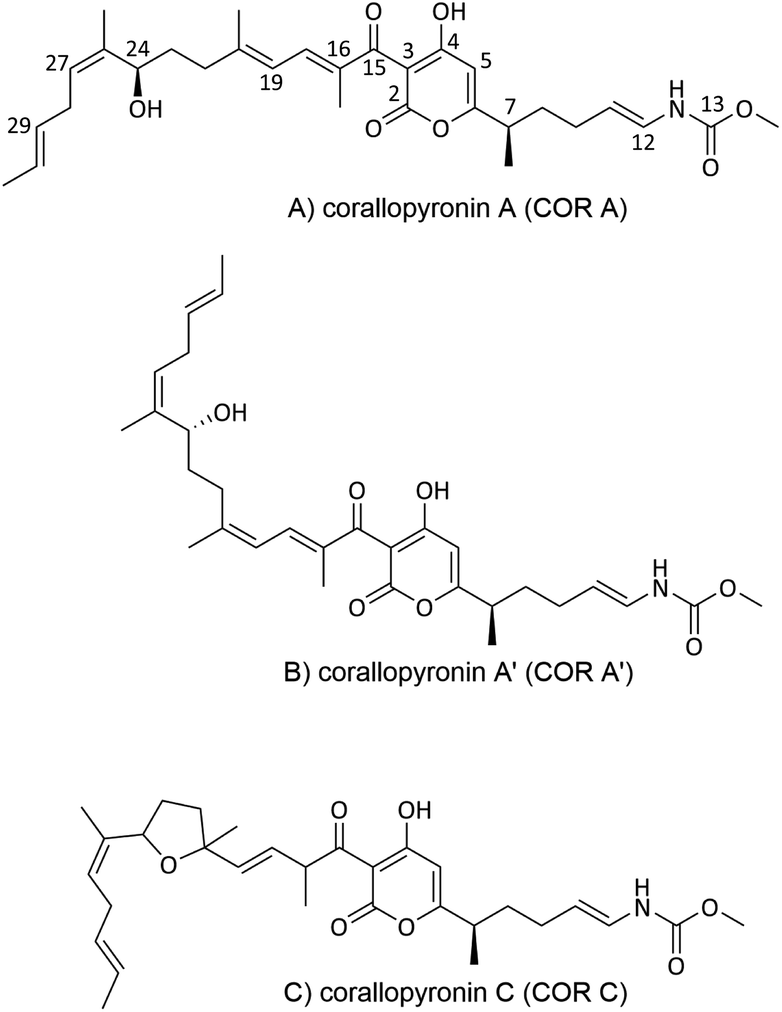 | ||
| Fig. 2 Chemical structures of corallopyronin A (COR A), and of its derivatives corallopyronin A′ (COR A′) and corallopyronin C (COR C). | ||
2.2 Biosynthesis
In 2000, the anti-infective COR A was rediscovered in the strain Corallococcus coralloides B035 by the group of König at the University of Bonn.45 This COR A producer strain was subsequently used by Erol et al. to describe the biosynthetic gene cluster of COR A.44 Later, attempts to establish a heterologous production platform by Sucipto et al. and Pogorevc et al. led to an even more detailed understanding of COR A biosynthesis.46,47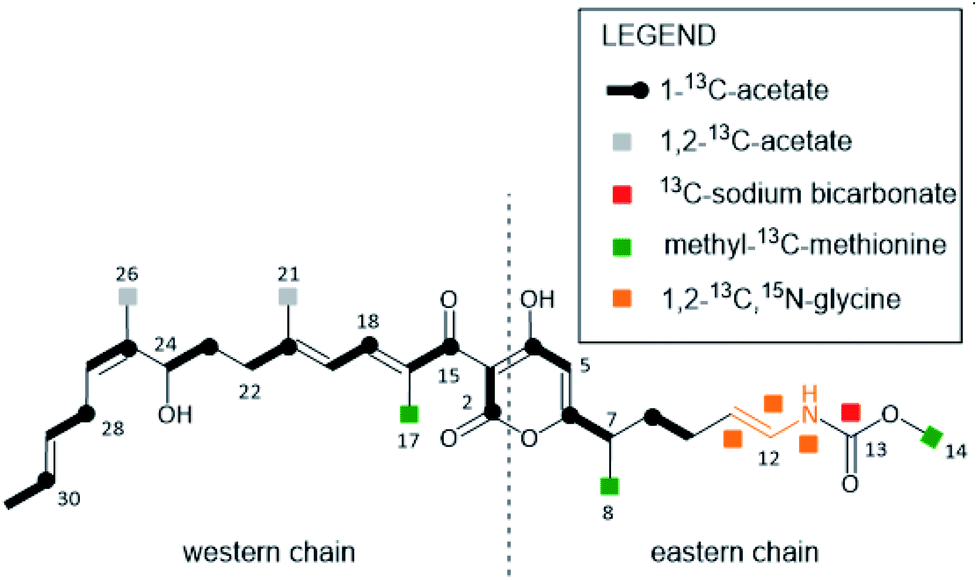 | ||
| Fig. 3 COR A building blocks deduced from feeding experiments with 13C- and 15N-labeled precursors. Figure adapted and modified from Erol et al.44 | ||
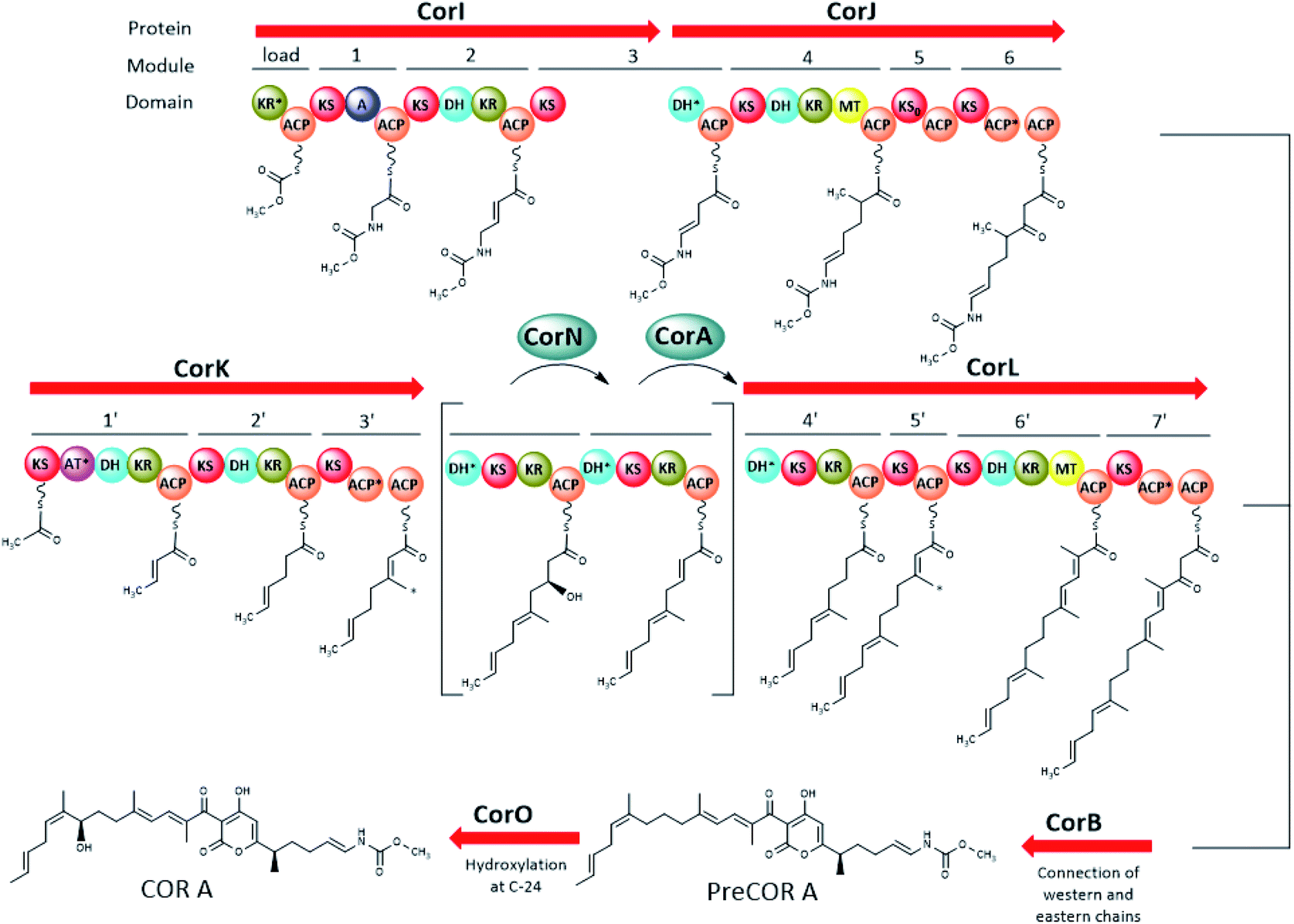 | ||
| Fig. 4 Major parts of the COR A biogenetic pathway including the enzymatic activity of CorB, CorI, CorJ, CorK, CorN, CorL, and CorO (domain abbreviations: acyl carrier protein (ACP), dehydratase domain (DH), ketoreductase domain (KR), ketosynthase domain (KS), inactive KS (KS0), methyltransferase domain (MT), adenylation domain (A)). The AT*, ACP*, and KR* domains are degraded. DH* in CorJ was proven to mediate a carbon–carbon double bond shift. DH* in CorL (module 6′) marks a putative “shift” domain. The two modules in brackets are an intermediate representation of modifications happening on module 4′ with CorN being a putative DH and CorA acting as ER. The black arrow indicates that the chains from modules 6 and 7′ are connected to form the pyrone ring. The asterisk (*) marks the carbons introduced by the β-branching cassette (CorC–G, not shown here). Figure adapted and modified from ref. 44 and 47. Not shown in this figure is the trans acting AT/KR CorA (in part) and the O-MT CorH. | ||
The biosynthesis of the eastern chain commences with carbonic acid, which becomes methylated by the O-methyltransferase CorH.48 Otherwise, the eastern chain is, in its major parts, generated by an assembly line encoded by two genes (CorI–CorJ) and includes PKS modules as well as one NRPS module. The latter is responsible for the incorporation of the amino acid building block glycine.46 Lohr et al. provided evidence that the domain CorJ DH* catalyses a carbon–carbon double-bond shift from the regular α, β position (i.e. C-10/C-11 in Fig. 3) in PKS reactions to the β, γ position (i.e. C-11/C-12 in Fig. 3) during COR A biosynthesis to finally yield the vinyl carbamate functionality.49
The PKS system necessary for the formation of the western chain is also encoded by two genes (CorK–L). Here, acetyl-CoA was identified as the starter unit, which subsequently is elongated with seven malonyl-CoA units. The assembly of the western chain includes the incorporation of the methyl groups C-21 and C-26, by a β-branching process, mediated in trans by enzymes encoded by the five genes CorC–CorG, whereas C-17 originates from the action of a MT domain found in CorL. The connection of the two chains, resulting in a pyrone ring is catalysed by the ketosynthase CorB.44 The CorB homologue MxnB was shown in vitro to catalyse a pyrone ring formation in the structurally related antibiotic myxopyronin.50 Modifying domains and enzymes encoded by the cor gene cluster were found to be responsible for a double-bond isomerization to result in Δ25,27 (putatively the N-terminal DH* domain of CorL) and hydroxylation at C-24 (CorO). From the metabolite profile of a CorN deletion mutant, CorN was assumed to be a putative dehydratase which may lead to dehydration of a C-23 hydroxylated intermediate in trans. The resulting double bond may be reduced by the trans ER activity of CorA.44,46,47
PreCOR A is an intermediate of COR A biosynthesis and its structure was first proposed by Erol et al. 2010 based on a putative biosynthetic model and mass spectrometry.44 However, isolation of this intermediate in 2010 revealed that the double bond position in the western chain was wrongly assigned, and corrected by Schäberle et al. in 2015 (Fig. 4).41
2.3 Production
Fermentation yields from C. coralloides Cc c127 originally reached 2 mg L−1 COR A.37 To enable further development, two approaches were investigated with the aim to enhance the production yield of COR A, (i) through chemical synthesis (Section 2.3.1) and (ii) using heterologous expression of the biosynthetic gene cluster (Section 2.3.2).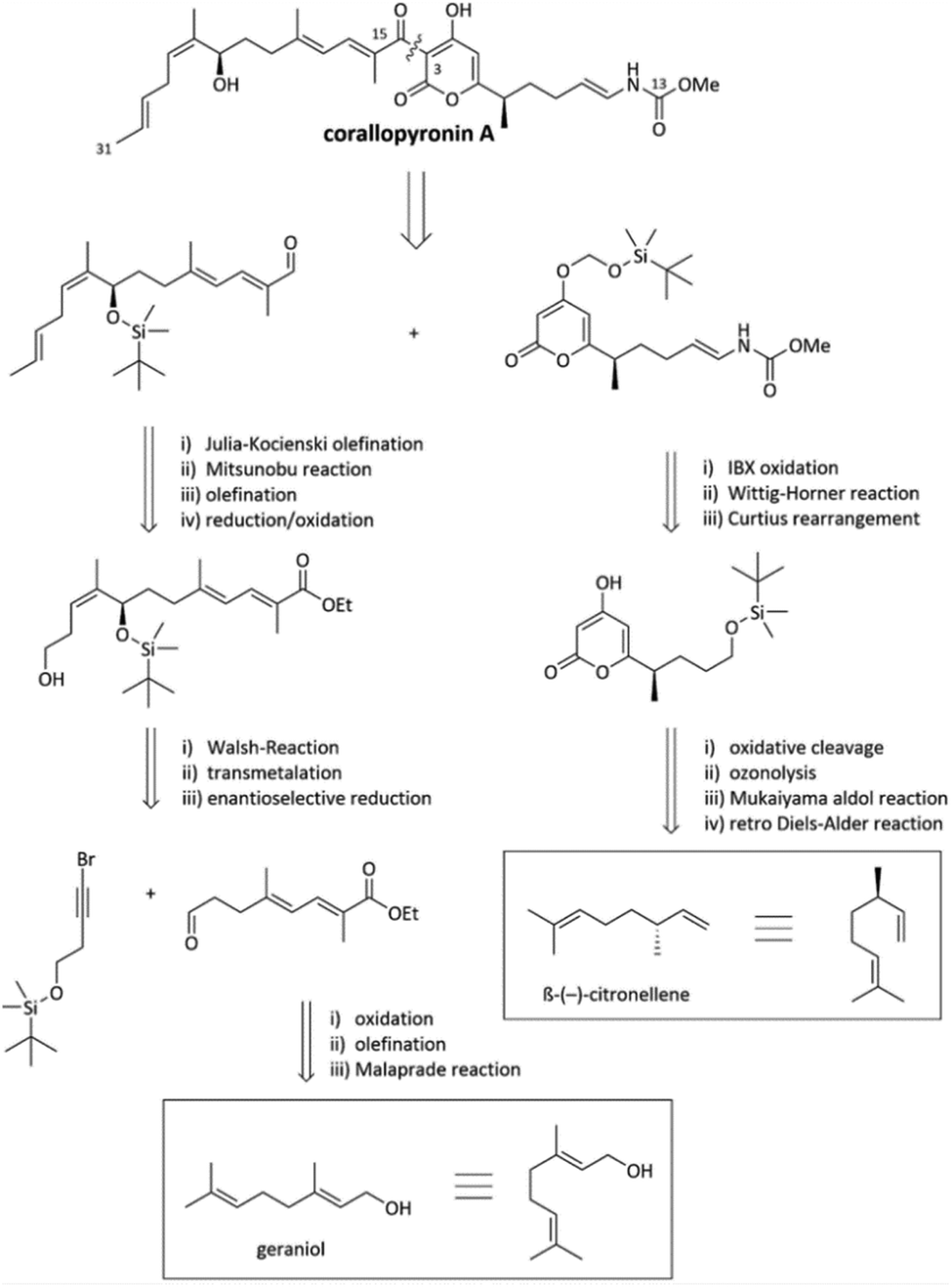 | ||
| Fig. 5 Excerpts from the retrosynthesis of COR A.51,53 | ||
The synthesis of the western chain was realized from the naturally occurring starting material geraniol after multiple chain elongation and modification reactions (Fig. 5). Regarding the synthesis of the eastern chain, the starting material β-(−)-citronellene (Fig. 5) was employed, which contains the desired configuration of the methyl group in position C-7. Synthetic steps include a retro-Diels–Alder reaction, a Mukaiyama aldol reaction, ozonolysis, Curtius rearrangement, and a Wittig–Horner reaction resulting in the eastern building block with the crucial pyrone and vinylogous carbamate unit.51 Recently, Sato and Chida published a second way to synthesize the western chain, consisting of the enantioselective allenylation of an aldehyde, hydroboration of the resulting 1,1-disubstituted allene and Migita–Kosugi–Stille coupling.52 The established synthesis enables the development of chemical variants of the anti-infective COR A with the potential to alter and optimize its pharmacological properties. However, due to the many steps and low overall yield of less than 1%,46 the presented synthesis does not represent a suitable method to produce larger amounts of COR A.
Functional experiments with a CorO deletion mutant led to a strain generating exclusively high levels of preCOR A (approx. 350 mg L−1). This demonstrated that CorO, a cytochrome P450 enzyme, is the enzyme responsible for the conversion of preCOR A to COR A in M. xanthus. Overexpression of CorO was done with the aim of converting all preCOR A to COR A, but did not result in stable conversion.47CorM was described as a type II TE domain not mandatory for COR A production itself, but important for high titres. Yet, CorM overexpression mutants decreased antibiotic production.47 Furthermore, an additional copy of the self-resistance protein CorP was co-expressed in M. xanthus DK1622:pDPO-Mxn116-Pvan-Tpase, but did not result in increased COR A titres.47
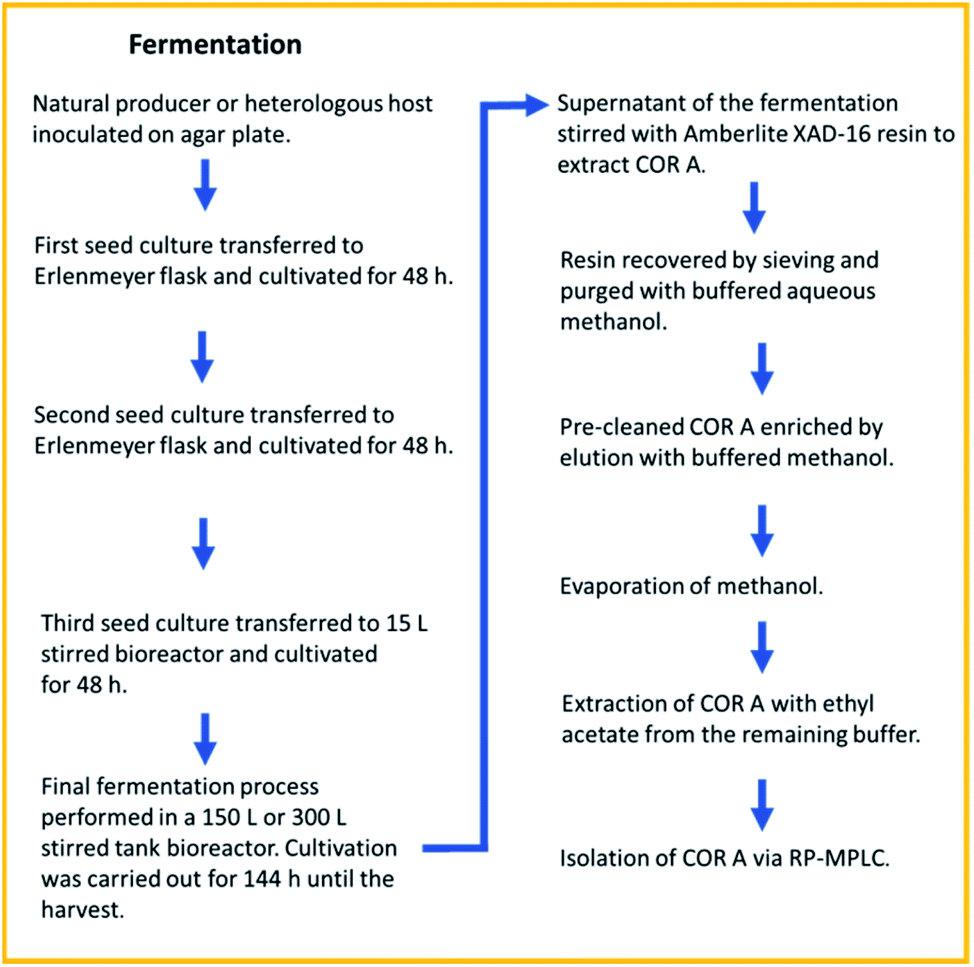 | ||
| Fig. 6 Fermentation and downstream process for COR A, resulting in a recovery rate of >80%. RP-MPLC (reversed-phase medium pressure liquid chromatography).54 | ||
The isolation of pure (90 to 99% purity) amorphous COR A (henceforth referred to as neat COR A in this review) was achieved by reversed-phase medium pressure liquid chromatography (MPLC).54 Due to the enhanced production yields of 100 mg L−1 and the achieved high purity of COR A, this process is currently used for providing material for the preclinical development. After three years of optimization, the fermentation and downstream processes are highly reproducible with a recovery rate of >80%. To our knowledge, this is the first time that the production of a preclinical candidate antibiotic using a heterologous production approach and scale-up in a pilot program has been done.
2.4 Quantification
For the novel biotechnologically produced active pharmaceutical ingredient (API) COR A, it is crucial to determine its precise and absolute quantity in each production batch, and also its concentration in samples generated during the development phase. In 2020, Krome et al. published the respective analytical methods.54 First, an absolute quantification method to analyse the COR A starting material, as it results from fermentations, was developed. The aim of these efforts was to establish COR A reference standard material. Quantitative 1H NMR spectroscopy using the internal standard dimethyl sulfone was selected as a suitable technique (Fig. 7).55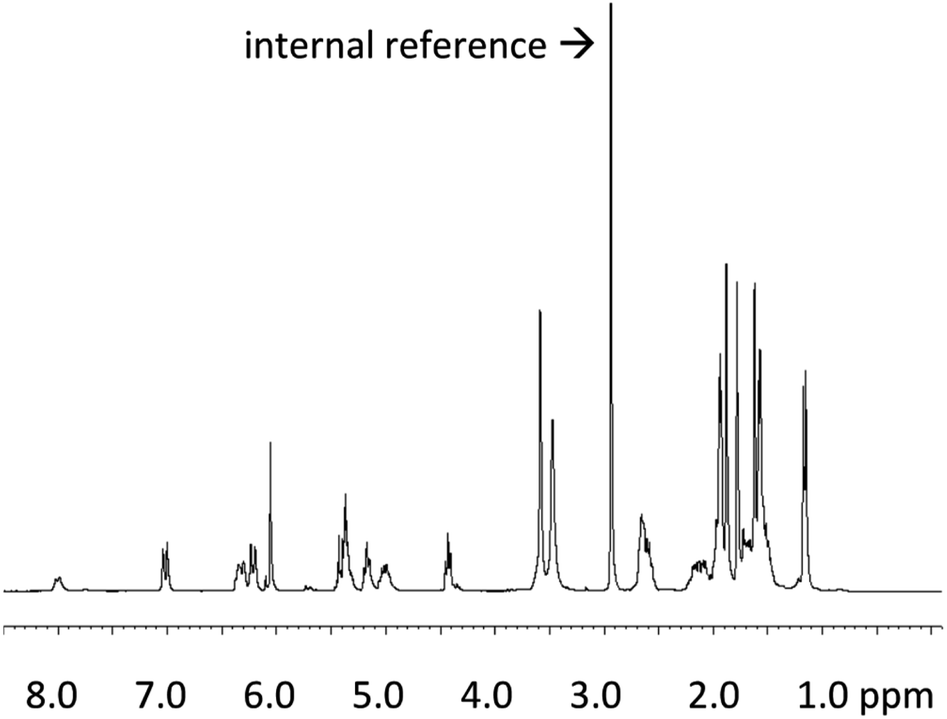 | ||
Fig. 7
1H NMR spectrum (300 MHz) of COR A produced by the strain Corallococcus coralloides B035 (COR A batch – content 99%) in acetonitrile-d3/D2O (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with the internal reference dimethyl sulfone.54 :1) with the internal reference dimethyl sulfone.54 | ||
For the quantitative calculation of the absolute COR A content, the singlet resonance at δ 6.06 (CH; 1H) of COR A as well as the singlet resonance of the internal reference (99.96% purity) at δ 2.99 (2 × CH3; 6H) were integrated. The COR A content was then calculated using a formula that compares the integrated proton NMR signal of COR A with that of the internal standard of known purity.54
Reversed-phase (RP) HPLC-DAD/MS was established to quantify the COR A content in samples assessing the stability of the API and to determine COR A concentrations in biological samples, e.g., plasma or organ homogenates.56 This method enabled the separation of COR A and the isomers COR A′ and COR C. The quantification of COR A was performed via an external calibration curve prepared with the COR A reference standard material quantified by the developed 1H NMR method.54
2.5 Mode of action
COR A was characterized to be a non-competitive inhibitor of the switch region of the bacterial DNA-dependent RNA polymerase (RNAP).57,58 Inhibitors of the bacterial RNAP are rare within antibiotics, but are regarded as promising novel anti-infectives. The most prominent representative of RNAP inhibitors is the potent antibiotic rifampicin and its congeners. Importantly, the binding site for COR A, i.e., the switch region of RNAP, is remote from that for rifampicin, which prevents the development of cross-resistance between these two APIs.57 In 2000 O'Neill et al. investigated the activity of several RNA polymerase inhibitors against rifampicin-resistant mutants of Staphylococcus aureus and found COR A to retain its activity towards these genotypes. These authors thus recommended the non-cytotoxic COR A for further development.59 The Wolbachia RNA polymerase COR A complex as shown in Fig. 8, visualizes the binding site of COR A. Out of all 292 anti-infectives currently in the preclinical phase, COR A is one of only five APIs that target bacterial RNA synthesis.9 COR A belongs to the α-pyrones, which are six-membered heterocyclic structures including a lactone moiety, representing a promising class of bioactive compounds.60–65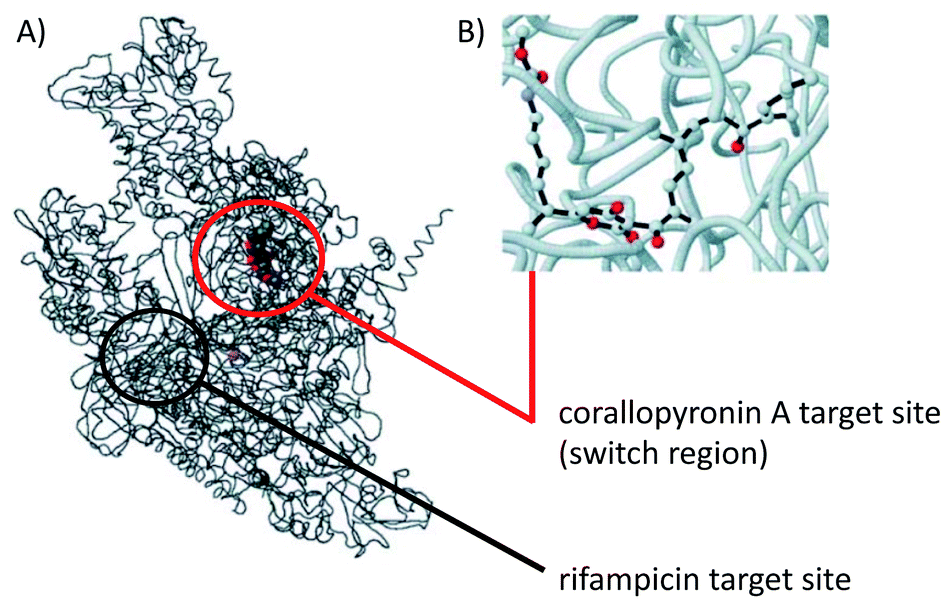 | ||
| Fig. 8 Model structure of the Wolbachia RNAP COR A complex. (A) Overall view, wRNAP (black), COR A (grey, red, blue), and magnesium ion in the active site (violet), including target sites for COR A and rifampicin. (B) Ball and stick model focused on COR A (oxygen in red, nitrogen in blue) bound to the wRNAP. Figure adapted from A. Schiefer, A. Schmitz, T. F. Schäberle, S. Specht, C. Lämmer, K. L. Johnston, D. G. Vassylyev, G. M. König, A. Hoerauf and K. Pfarr, J. Infect. Dis., 2012, 206, 249–257 by permission of Oxford University Press.38 Information about the target sites taken from Mukhopadhyay et al. 2008.57 | ||
Development of resistance towards COR A has been observed66 but ongoing studies in our laboratories in Bonn indicate that the rate is lower than that observed for rifampicin (Bierbaum, Hoerauf and Pfarr, data not yet published). For the structurally related compound myxopyronin, resistant mutants were also detected in S. aureus.67 The authors stated, that there is no cross-resistance between rifampicin and myxopyronin. Of major importance, was the finding that myxopyronin-resistant mutants had a lower level of resistance compared to rifampicin-resistant mutants. It was also found that the fitness costs for myxopyronin resistance are much higher than those for rifampicin-resistance, probably due to the vital function of the switch region of the RNAP which is the exclusive target of myxopyronin. The latter finding may be of clinical importance.67
At least against S. aureus, COR A should therefore be used in combination with other antibiotics like rifampicin to inhibit the formation of resistant mutants.
The biosynthetic gene cluster for COR A includes an open reading frame for the hypothetical protein CorP, which was found to be a self-resistance protein. However, Cor P does not confer resistance to COR A in E. coli, but to date in M. xanthus DK1622. The molecular basis of how Cor P leads to COR A resistance in M. xanthus is not yet elucidated and needs additional studies.47
2.6 Antimicrobial spectrum
COR A has been demonstrated to be active against Gram-positive and Gram-negative pathogens (Table 1). However, the activity of COR A was found mainly directed towards Gram-positive and intracellular pathogens inter alia Staphylococcus aureus, as shown in the course of its initial discovery by Irschik and Reichenbach in the 1980s.43 They also discovered that Escherichia coli, a Gram-negative bacterium, was susceptible to COR A if the gene encoding the outer membrane protein TolC, known for its efflux transmembrane transporter activity,68 was knocked out. Recently, the COR A congener myxopyronin B, also an inhibitor of RNA synthesis, was proposed as a lead structure for Clostridioides difficile infections.69| (A) Pathogen | Minimum inhibitory concentrationa (μg mL−1) | Source |
|---|---|---|
| a MIC = Lowest concentration of COR A, that prevented detectable growth. b Intracellular cell assay. c The antibiotic was applied on a 6 mm paper disc (5 μg per disc), the value represents the diameter of the inhibition zone, where bacterial colonies did not grow. d Mutant strain with increased permeability. | ||
| Bacillus cereus | 2 | 41 |
| Bacillus licheniformis DSM13 | 2 | 41 |
| Bacillus subtilis | 6.25 | 43 |
| Bacillus subtilis 168 | 16 | 41 |
| Bacillus megaterium | 0.39 | 43 |
| Chlamydia trachomatis Db | 0.5 | 71 |
| Chlamydia trachomatis L2b | 0.5 | 71 |
| Chlamydia muridarum | 0.5 | 71 |
| Chlamydia pneumoniae CWL029 | 1 | 71 |
| Corynebacterium mediolanum | 0.78 | 43 |
| Micrococcus luteus | 0.78 | 43 |
| Micrococcus luteus ATCC 4698 | 1 | 41 |
| Mycobacterium bovis | 16 | 38 |
| Orientia tsutsugamushi | 0.0078 | 58 |
| Rhizobium meliloti | 0.39 | 43 |
| Rickettsia typhi | 0.125 | 58 |
| Staphylococcus aureus | 0.097 | 43 |
| Staphylococcus aureus SG 511 | 0.5 | 41 |
| Staphylococcus simulans 22 | 1 | 41 |
| Wolbachia | 0.1 | 38 |
In the 2010s, the group of Hoerauf found COR A to be highly effective in vitro and in vivo against the Gram-negative intracellular Wolbachia spp. bacteria, and thus, selected this API as a preclinical candidate. The aim of this ongoing project is to develop COR A for the treatment of human filarial infections.30,38,40 Subsequently, the efficacy of COR A towards other intracellular Gram-negative bacteria was investigated. COR A is effective in vitro and in vivo against the human-pathogenic Orientia tsutsugamushi and hence, is a potential API to treat scrub typhus infections.58 The group of Rupp showed COR A to be in vitro effective against a third genus of Gram-negative intracellular pathogens, Chlamydia spp. including C. trachomatis D and L12, C. muridarium and C. pneumoniae.70,71 Due to limitations of the C. muridarium murine infection model, in vivo efficacy could not be clearly demonstrated. Therefore, this needs to be further investigated in other models to elucidate the full potential of COR A to treat genital chlamydial infections.71Table 1 lists all pathogens towards which COR A was found to be active, including gonococci (manuscript in preparation).
2.7 Physicochemical characterization
After COR A was selected as a preclinical candidate, its physicochemical characteristics were determined using a COR A batch of 93% purity. COR A is a vinylogous carboxylic acid and displayed a pKa value of 3.70. In accordance with its specific ionization properties, COR A showed high lipophilicity (logP = 5.42) and poor aqueous solubility (0.1 μg mL−1) at low pH values (pH 1.0–3.0). The ionization and subsequently the aqueous solubility of COR A increased at higher pH values (pH ≥ 4), while lipophilicity values, as reflected by the logD, decreased (Table 2).54
P) and distribution coefficient (logD) of neat COR A (COR A batch – content 93%) determined by a Pion's T3 apparatus54
| pH | Solubility (μg mL−1) | LogD |
|---|---|---|
| 1.0 | 0.11 | 5.42 → logP (neutral XH) |
| 2.0 | 0.11 | 5.41 |
| 3.0 | 0.14 | 5.31 |
| 4.0 | 0.40 | 4.85 |
| 5.0 | 2.98 | 3.97 |
| 6.0 | 28.86 | 3.00 |
| 6.5 | 91.13 | 2.52 |
| 7.0 | 288.00 | 2.09 |
| 7.4 | 723.20 | 1.81 |
| 8.0 | 2874.00 | 1.54 |
To estimate the permeability of the API COR A, a Caco-2 assay was performed, which predicted a high in vivo absorption for COR A in humans (2.0 × 10−5 cm s−1).53 However, neat COR A (COR A batch – content 99%) demonstrated poor dissolution and aqueous solubility in biphasic dissolution tests performed under simulated fasted human gut conditions (Fig. 9), using an in vivo predictive in vitro biphasic dissolution assay (BiPHa+, Fig. 9).54,72 Therefore, a dissolution and solubility enhanced formulation principle was required to achieve suitable bioavailability levels after oral administration.
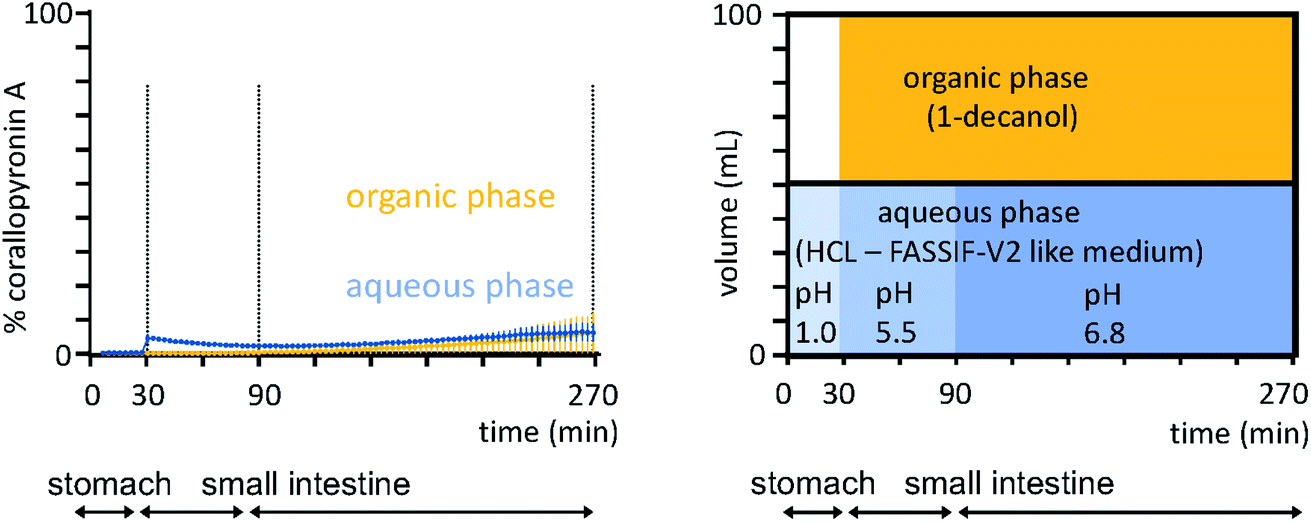 | ||
| Fig. 9 Biphasic dissolution media setup and profiles of the neat COR A performed under simulated fasted human gut conditions (n = 3, 100% ≙ 0.2 mg mL−1, COR A batch – content 99%).54 Concentration in organic phase predicts oral bioavailability. | ||
The solid-state of neat COR A was analysed by differential scanning calorimetry (DSC) and X-ray powder diffraction (XRPD). The DSC measurements demonstrated only one glass transition temperature (Tg) of 5 °C for neat COR A (COR A batch – content 99%). Since no melting point was found for the neat COR A sample during this experiment, COR A was determined to be purely amorphous, which was confirmed by XRPD analysis, where no Bragg-peaks were detected.53
Amorphous APIs, either as neat material or in solid formulations, tend to be chemically and physically unstable to thermal stresses above their Tg.73–76 Stability studies of the neat COR A were thus performed below and above its Tg. These stability tests were analysed using HPLC–DAD after the storage of the samples at different conditions (5 °C, 25 °C/60% relative humidity (RH), and 40 °C/75% RH) for 1, 2, 4, and 12 weeks. Stability of neat COR A inversely correlated with the storage temperature, i.e., higher temperatures led to lower COR A contents after several weeks of storage (Fig. 10).
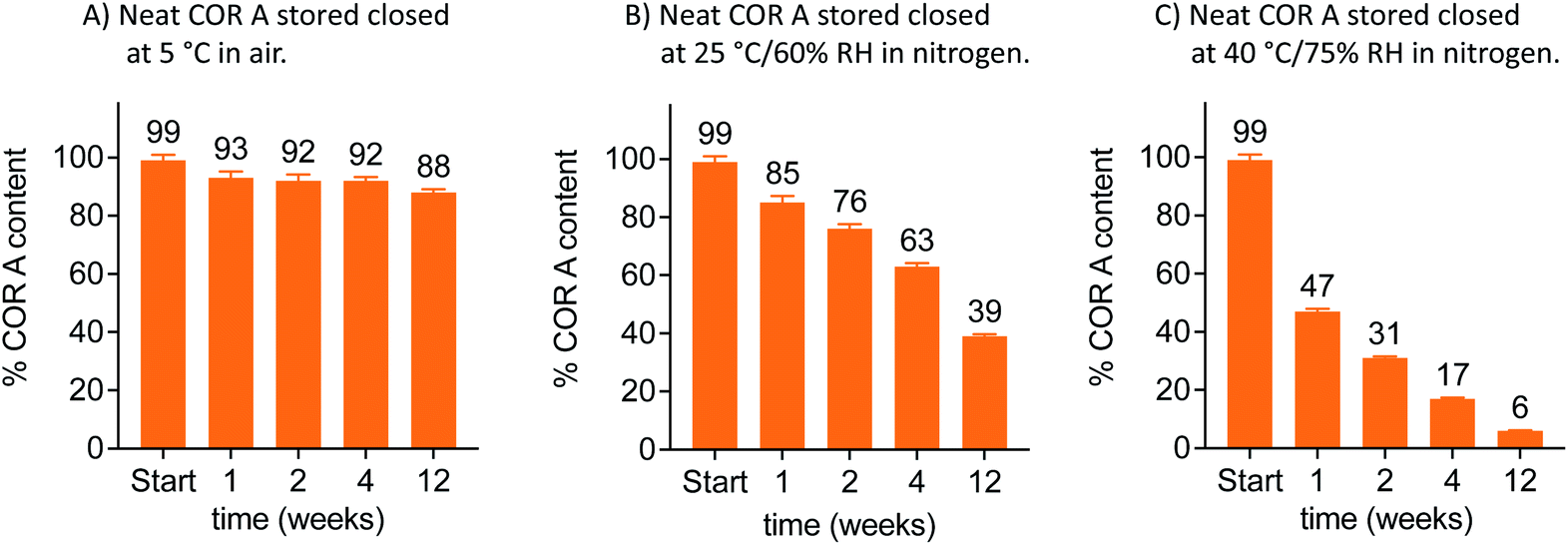 | ||
| Fig. 10 Stability analysis at 5, 25 and 40 °C via HPLC–DAD of neat COR A in closed twist-off glass vials (gas tight) and two different gas environments within the vials (air or nitrogen, n = 3, mean, SD, COR A batch – content 99%, RH = relative humidity).53,54 | ||
Whereas the COR A content rapidly decreased at temperatures ≥25 °C, there was a simultaneous increase of COR C.53 In addition, HPLC–DAD measurements demonstrated a higher instability of COR A in the presence of air (oxygen) when compared to storage under inert gases (Fig. 10). This is most likely due to autoxidation processes at the double bond system in COR A, which may cause the formation of hydroperoxides, which subsequently could decompose to aldehydes and ketones. The isomerization of COR A to COR A′ was not observed during the stability tests, but is reported to occur under acidic conditions.37
The above-mentioned physicochemical analysis of neat COR A clearly demonstrated the need for a stability enhanced formulation for this API to enable preclinical and clinical trials and to potentially introduce it into the market.54 In conclusion, the identified physicochemical COR A properties showing poor dissolution, poor solubility, poor stability, and good permeability, demand the development of COR A formulations with the ability to enhance dissolution, solubility, and stability.
2.8 Formulations
For intravenous application, a liquid solution (COR A > 20 mg mL−1) containing the following excipients was established: propylene glycol (20%), polyethylenglycol-15-hydroxystearate (20%), and phosphate buffered saline pH 7.4 (60%).53,54 For rodent efficacy studies with oral and intraperitoneal administration routes, a liquid formulation containing COR A and the excipient PEG 400 (50%) and phosphate buffered saline pH 7.4 (50%) was applied.30 A PEG 200 based liquid COR A formulation allowing an oral dose of 1000 mg kg−1 (100 mg mL−1) was developed for toxicity studies.53 All formulations exhibited sufficient in-use stability of COR A.53
Currently, the amorphous solid dispersion approach (ASD) represents the most promising solid oral formulation principle that has been used in preclinical PK and efficacy studies, and is intended to be applied in clinical trials and as a potential commercial formulation. Here, COR A is molecularly dispersed/dissolved in the amorphous polymer matrix povidone (PVP) or copovidone (PVP/VA) via spray drying out of an ethanolic solution.53,54 The resulting amorphous solid dispersion of the type of glass solutions demonstrated the required improvements of the neat COR A in respect to dissolution and solubility during the in vivo predictive in vitro biphasic dissolution tests (BiPHa+). The amount of COR A that partitioned into the organic phase, i.e., 1-decanol, is predictive for the amount of COR A that is passively absorbed into the human gut wall and subsequently, into the blood system. The results demonstrated a 5-fold increase of COR A that partitioned into the organic phase when embedded into the water-soluble polymer copovidone, and a 10-fold increase when embedded into the water-soluble polymer povidone (Fig. 11). The enhanced dissolution and solubility properties of COR A have important implications with regards to its pharmacokinetic properties and hence therapeutic efficacy after oral administration. COR A needs to dissolve in the gastrointestinal fluid in order to pass through the gastrointestinal membranes to reach the circulatory system, and ultimately its therapeutic target in sufficient quantities. These enhancements are related to the molecular dispersion of the lipophilic COR A molecules in the polymers povidone and copovidone, both of which exhibit good wettability and dissolution. Additionally, the fine ASD-particles resulting from the spray drying process favour fast dissolution due to an increased specific surface.53,54 Besides the dissolution and solubility enhancement of COR A, both ASD formulations also enhanced temperature stability of the API COR A (Fig. 12).
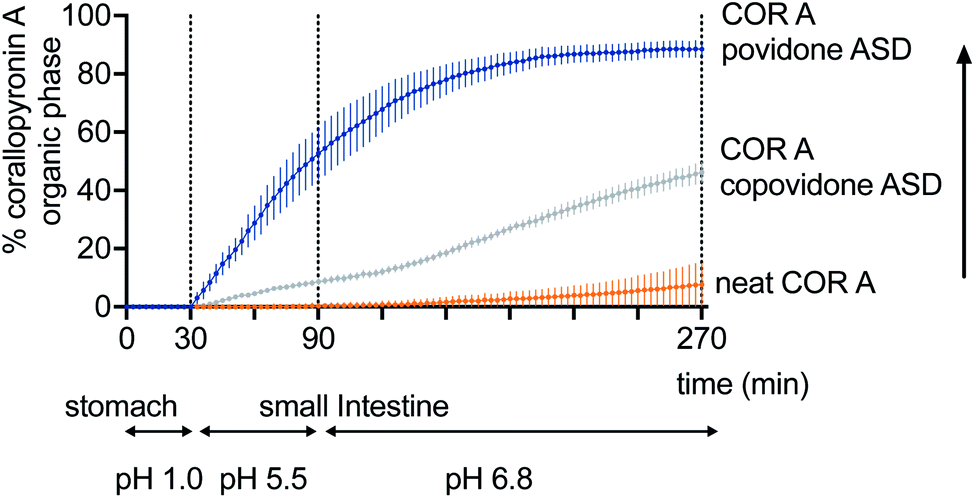 | ||
| Fig. 11 Increase of the fraction partitioning into the organic phase of the biphasic dissolution test: neat COR A < COR A-copovidone ASD formulation < COR A-povidone ASD formulation (COR A batch – content 99%).54 | ||
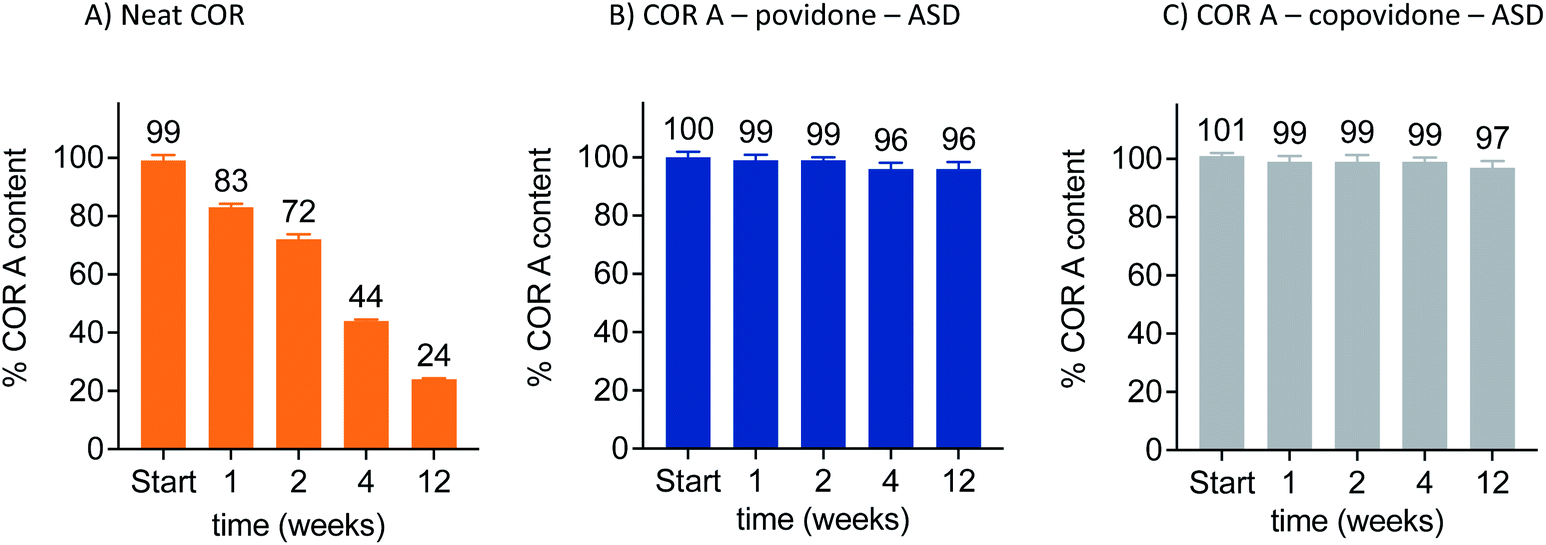 | ||
| Fig. 12 Increased stability of the COR A–ASD formulations compared to neat COR A when stored in closed twist-off glass vials under nitrogen at 30 °C/65% RH, determined via HPLC–DAD (COR A batch – content 99%).54 | ||
2.9 Pharmacokinetic analysis
Two of the aforementioned COR A formulations have been the subject of in vivo pharmacokinetic studies in BALB/c mice, i.e., the COR A ASD formulation comprising povidone for oral administration and the COR A intravenous liquid formulation (Fig. 13). Plasma profiles of COR A solution, intravenously applied, were used as a 100% reference for assessing the bioavailability of the solid ASD formulations. The pharmacokinetic parameters were calculated employing a non-compartmental and a two-compartmental approach. The in vivo plasma concentration–time profile of the oral (PO) administration of the spray-dried COR A–ASD formulation comprising povidone (administered suspended in PBS puffer, pH 7.4) demonstrated fast and high exposure (Cmax, Tmax, and AUC) (Fig. 13). These measurements confirmed the potential of this formulation principle to result in suitable COR A dissolution solubility, and permeability with an observed oral bioavailability of 59%.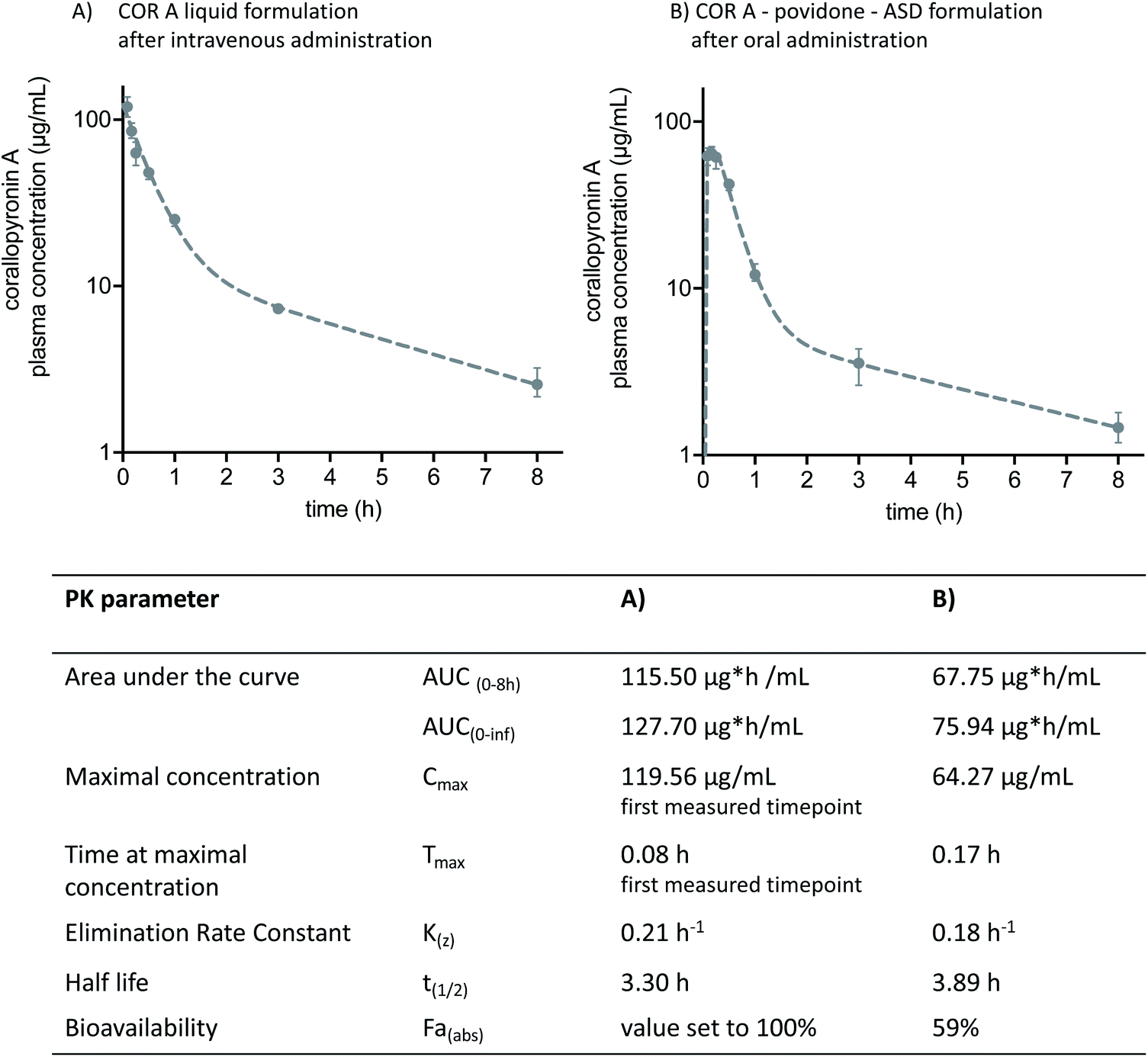 | ||
| Fig. 13 Two-compartment model pharmacokinetic log plots (median and interquartile range) and pharmacokinetic parameters of (A) intravenously administered COR A solution comprising propylene glycol, macrogol-15 hydroxy stearate, and PBS buffer, and (B) orally administered COR A–PVP–ASD suspension medium of PBS-buffer. The administered dose in both experiments was 36 mg kg−1 (COR A batch – content 99%), BALB/c mice (n = 4).53 | ||
Future studies are needed to investigate, whether the promising pharmacokinetics in mice (high Cmax and AUC values) achieved with the COR A-povidone–ASD formulation will translate into respective pharmacokinetics in other preclinical species and humans in comparison to the alternative ASD formulation based on a copovidone matrix. In case adjustments of the absorption kinetics (tmax and Cmax) would be beneficial for the efficacy against a pathogen like Wolbachia, the ASD-formulation principle allows for a multitude of combinations with various drug delivery technologies. Thus, tailor made release profiles of COR A will be available.
2.10 Pharmacodynamic analysis against Wolbachia
Wolbachia are essential obligate endosymbionts of most filariae that infect humans and rodents. They are found in all life-cycle stages and are needed for filarial embryogenesis,30,77–81 larval development,82,83 and adult worm survival.30,84–89 Therefore, these intracellular bacteria demonstrate a potential target to eliminate filariae and cure infections like lymphatic filariasis and onchocerciasis.90–93 The pharmacodynamic efficacy of COR A against Wolbachia was analysed using the Litomosoides sigmodontis rodent model.30,38,81 A larval model and a chronic infection setup were employed. In the larval model, clearance of >99.0% of the Wolbachia was found to be necessary to prevent the development of filaria into adult worms. In the chronic infection model, in which adult worm mate and reproduce, depletion of >99.0% of the Wolbachia was required to clear microfilariae from the blood stream and to kill adult filariae.30The L. sigmodontis larval model setup30,38,81 was used to quickly analyse the efficacy of different COR A formulations. The treatment started immediately after the infection with L. sigmodontis in BALB/c mice, simulating a prophylactic treatment. Good in vivo efficacy results were obtained if COR A (COR A batch – content 96%) was administered for 14 days intraperitoneally in a liquid formulation comprising PEG 400 and PBS (50/50) with a dose of 9 and 18 mg kg−1. The COR A treatment with the dose of 9 mg kg−1 cleared >99.0% of the Wolbachia. The treatment with a dose of 18 mg kg−1 cleared >99.9% (Fig. 14).30 Furthermore, the larval setup analysed the efficacy of the oral formulation comprising COR A (COR A batch – content 93%), propylene carbonate (PC), and mesoporous silica (Syloid® XDP 3050). This study showed that the treatment cleared >99.0% of Wolbachia and that it prevented the development of filariae into adult worms when administered orally for 14 days with a dose of 18 mg kg−1 once a day (QD, suspended in PBS pH 7.4 via gavage) (Fig. 14).30 The oral treatment with the COR A-povidone–ASD formulation (COR A batch – 99%) also applied in the larval setup cleared >99.9% of Wolbachia and prevented the development of filariae into adult worms when the COR A-povidone–ASD formulation was administered via gavage QD for 14 days with a dose of 36 mg kg−1 suspended in PBS.53
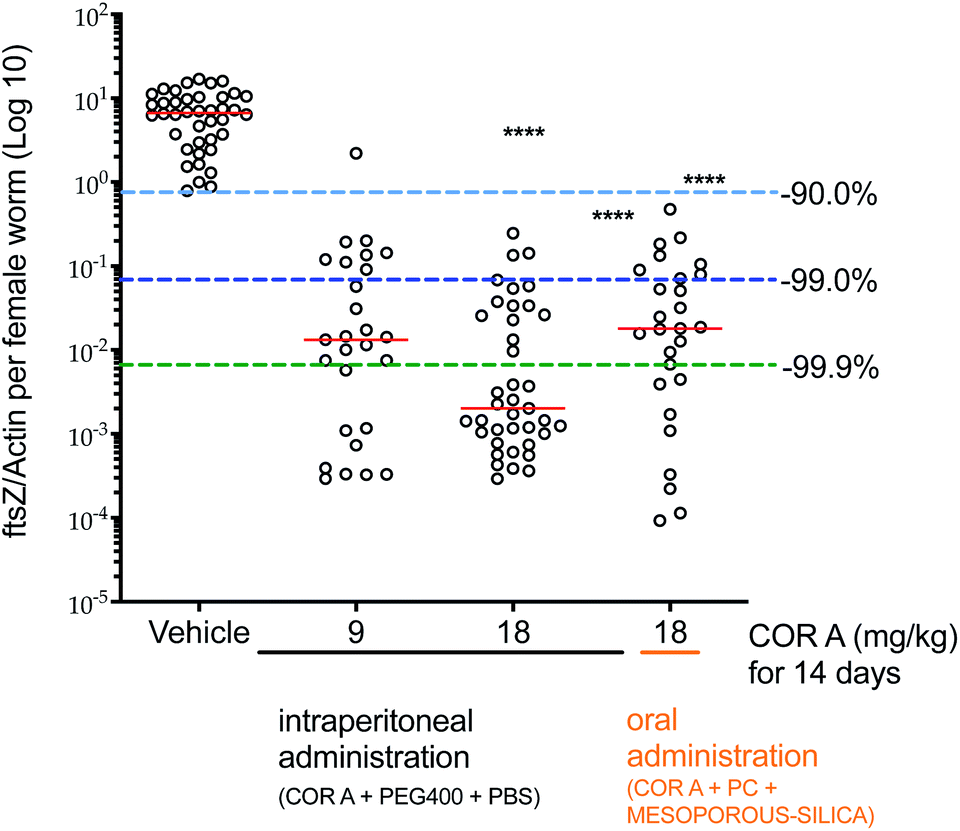 | ||
| Fig. 14 Pharmacodynamic effects of COR A against Wolbachia. Administration of COR A started on the day after the L. sigmodontis infection, BALB/c were treated with vehicle control or COR A formulations once a day at the indicated doses, treatment days and routes. Wolbachia ftsZ/filarial actin per female worm was calculated 35 days post treatment onset, n = 3–4 mice and n = 21–40 worms per group. Statistical analysis was applied using the Kruskal–Wallis test with the Dunn's multiple comparison post hoc test. ****P < 0.0001. Red lines indicate medians.30 | ||
In vivo pharmacodynamic studies using the L. sigmodontis chronic model were performed with the liquid COR A PEG 400/PBS (50/50) formulation (COR A batch – content 96%). The latter was administered intraperitoneally to Mongolian gerbils (Meriones unguiculatus) for 14 days. COR A depleted >99.9% of the Wolbachia, inhibited embryogenesis and cleared the blood-borne microfilariae when administered at a dose of 30 mg kg−1 twice a day. More importantly, COR A in this formulation resulted in the first published macrofilaricidal activity of a new anti-Wolbachia compound in this model.30
The efficacy results indicated that COR A, in a suitable formulation, is a promising anti-wolbachial drug candidate, which displayed better efficacy and reduced treatment time compared to doxycycline against filariasis in the L. sigmodontis rodent model.94 In addition, of all new anti-wolbachial compounds tested in a larger preclinical series of promising compounds, COR A was the only one showing clear and robust reduction of adult filarial worms.30
2.11 Toxicological analysis
A first indication that myxopyronin and COR A do not exert general toxicity can be retrieved from the literature, i.e. in 2009 Haebich and Nussbaum (Bayer Schering Pharma AG) reported low acute toxicity of myxopyronin A and COR A in mice (s.c., LD50 > 100 mg kg−1) in a review article.95 The toxicity data for myxopyronin A and B were originally published in 1983 by Irschik et al.96 ln 1985 an acute toxicological study was performed in mice with a dose level of 100 mg kg−1 COR A (s.c.) and no acute toxicology was observed.43 These studies however, did not provide experimental details on how toxicity was assessed. Our studies analysing the in vitro cytotoxicity of COR A towards cultured Hep G2 cells revealed no cytotoxic effect at a concentration of 0.2 μg mL−1 and only negligible cytotoxicity at 20 μg mL−1, whereas at 200 μg mL−1 COR A, liver cell viability was reduced by 89% in comparison to the DMSO control. To gain further insights into the possible cytotoxicity of COR A, a gene expression profile was generated using Hep G2 cells at a concentration of 15 μg mL−1 COR A. These experiments demonstrated only minimal effects of COR A treatment on the gene expression profile in comparison to DMSO.97 Additionally, during all our pharmacodynamic experiments, e.g. for up to 28 days and 35 mg kg−1,38 the rodents tolerated COR A well, with no indication for toxicology at the therapeutic dose levels. Also, in vitro studies assessing a potential cardiotoxicity analysed via the human ether-a-go-go (hERG) assay and the in vitro screening for genotoxicity via the micronucleus and AMES tests indicated no toxicity.98 Altogether, the early findings on low COR A toxicity43 together with our experience during numerous initial in vivo mouse experiments allowed us to judge the overall toxicity of COR A as minimal and led to the decision to move into translational studies with this compound.Relevant toxicological studies in a rodent and non-rodent species are currently being performed with rats and dogs. For both species, a 7 day repeated dose study (dose range finder study) will be performed, escalating COR A dose levels over a period of 7 days to reach a dose that elicits a toxicological effect. The results of the dose range finder study will be used as a starting point for a GLP-28 day repeated dose toxicology study. However, such studies demand large amounts of material, which will be accessible through production strategies outlined in Section 2.3.98
3 Concluding remarks and outlook
Novel anti-infectives are urgently needed to address the increased morbidity and mortality from infections with resistant pathogens and also those for which appropriate drugs are not available. This review describes the successful early preclinical development of COR A and serves as a showcase of how to proceed in similar cases. The DNA-dependent RNA polymerase inhibitor COR A was discovered in the early 1980s and was selected 30 years later as a preclinical candidate owing to its high potency against Wolbachia. Myxobacteria like C. coralloides are difficult to cultivate and thus to obtain sufficient amounts of API was an obstacle hard to overcome. The initial biopharmaceutical production via C. coralloides B035 was optimized and its biosynthesis elucidated, thus allowing for the subsequent development of an efficient heterologous production method. This resulted in a superior alternative to the described chemical synthesis. Moreover, the biopharmaceutical manufacturing approach offered a simplified downstream process resulting in highly purified COR A, which could not be achieved by crystallization after chemical synthesis due to the amorphous character of COR A. This lack of a rigid crystal lattice increased the molecular mobility of COR A in the amorphous solid state with pronounced isomerization during storage. Next to the poor stability of the neat drug substance, other physicochemical properties like dissolution and solubility were similarly poor. These issues could be addressed by embedding COR A molecularly dispersed in a polymeric matrix of either povidone or copovidone. The increased glass transition temperature of the resulting ASDs kinetically stabilized COR A and at the same time increased dissolution and solubility. The employed biphasic in vitro dissolution test was highly predictive for the in vivo PK and the obtained bioavailability of approximately 60% of the ASD formulation comprising COR A and the polymer povidone. Consequently, good efficacy against Wolbachia could be demonstrated in the L. sigmodontis larval rodent model. Further experiments also demonstrated good anti-Wolbachia efficacy of COR A against adult nematodes in the chronic L. sigmodontis model and other Gram-positive and intracellular Gram-negative pathogens.Current data on COR A, however are not sufficient yet to judge the future fate of this natural product as an antibiotic drug. The most critical point being the still missing detailed toxicological evaluation. Non-GLP adsorption, distribution, metabolism, elimination and toxicology studies are being completed and will be published soon. The non-GLP studies will be followed by GLP compliant studies. The next hurdle in the preclinical development of COR A is the dose range finder toxicology studies in rats and dogs. However, it is expected that the in vivo tolerability of COR A seen in mice and Mongolian gerbils treated daily with COR A for up to 28 days will be reflected in the formal toxicological studies. The pharmacokinetic evaluation of the developed solid COR A ASD formulations in rats and canines after oral administration is also to be carried out. The results of the canine studies will improve prediction accuracy regarding the pharmacokinetic profile in humans via the application of PBPK modelling. Thus, insightful information will be generated with respect to the intended oral dose of the developed COR A ASD formulations in humans. COR A is currently thoroughly studied in a preclinical package to hopefully demonstrate that it is safe and non-toxic. The results of preclinical studies – present and future – will be used to initiate a phase I trial to investigate COR A-formulations in humans, with the aim to establish an approved drug to cure infectious diseases. The review demonstrates how a hard to obtain, barely water soluble and unstable compound such as COR A can be developed making use of sophisticated production and formulation approaches.
4 Patents
The University of Bonn is the patent applicant for the following patents for the use of corallopyronin A for filarial infections: US 9168244 B2, US 9687470 B2, EP 2704708 B1. Inventors include A. Hoerauf, K. Pfarr, A. Schiefer, S. Kehraus and G. M. König. The Helmholtz Centre for Infectious Diseases GMBH is the patent applicant for the following patent PCT/EP2014/059677. Inventors include R. Müller.5 Author contributions
Conceptualization: A. K. Krome, G. M. König, K. G. Wagner. Data curation: A. K. Krome, S. Kehraus, G. M. König. Formal analysis: A. K. Krome, T. Becker, K. Pfarr, A. Schiefer. Funding acquisition: A. Hörauf, K. Pfarr, A. Schiefer, M. P. Hübner, K. G. Wagner, A. K. Krome, M. Stadler, R. Müller. Investigation: A. K. Krome, T. Becker, K. Pfarr, A. Schiefer, S. Kehaus. Methodology: A. K. Krome, T. Becker, S. Kehraus, R. Jansen, M. P. Hübner. Project administration: A. Hoerauf, A. Schiefer, T. Hesterkamp, K. Pfarr, M. P. Hübner. Resources: A. Hoerauf, M. P. Hübner, K. G. Wagner, G. M. König, M. Stadler, R. Müller. Software: A. K. Krome, T. Becker, K. Pfarr, A. Schiefer. Supervision: A. Hoerauf, M. P. Hübner, K. G. Wagner, G. M. König, M. Stadler. Validation: A. K. Krome, S. Kehraus, R. Jansen, M. Stadler. Visualization: A. K. Krome, G. M. König, S. Kehraus. Writing – original draft: A. K. Krome, G. M. König, K. G. Wagner. Writing – review and editing: A. K. Krome, G. M. König, S. Kehraus, K. G. Wagner, K. Pfarr, T. Becker, T. Hesterkamp, A. Hoerauf, M. P. Hübner, A. Schiefer, M. Gütschow, L. Chaverra-Muñoz, R. Jansen, M. Stadler, D. Pogorevc, R. Müller, S. Hüttel, A. Ehrens.6 Conflicts of interest
The authors declare no conflict of interest.7 Acknowledgments
The authors thank Tilman Aden, Helene Neufeld, and Marianne Koschel for their significant help and execution of the performed pharmacokinetic and pharmacodynamic studies in rodents. We thank Kerstin Schober, Silke Reinecke, and Steffen Bernecker for the production of high quality corallopyronin A via fermentation and the respective DSP process. The COR A project has received funding from the Deutsche Forschungsgemeinschaft grant number DFG PF 673/3-1 and KO 902/5-1, the German Center for Infection Research (https://www.DZIF.de) grant numbers TTU 09.807, 09.816, 09.914, the Federal Ministry of Education and Research (https://www.BMBF.de) grant number 16GW0229, and the European Regional Development Fund (https://www.EFRE.NRW.de) grant number EFRE-0400037.8 References
- R. Laxminarayan, A. Duse, C. Wattal, A. K. M. Zaidi, H. F. L. Wertheim, N. Sumpradit, E. Vlieghe, G. L. Hara, I. M. Gould, H. Goossens, C. Greko, A. D. So, M. Bigdeli, G. Tomson, W. Woodhouse, E. Ombaka, A. Q. Peralta, F. N. Qamar, F. Mir, S. Kariuki, Z. A. Bhutta, A. Coates, R. Bergstrom, G. D. Wright, E. D. Brown and O. Cars, Lancet Infect. Dis., 2013, 13, 1057–1098 CrossRef PubMed.
- A. Cassini, L. D. Högberg, D. Plachouras, A. Quattrocchi, A. Hoxha, G. S. Simonsen, M. Colomb-Cotinat, M. E. Kretzschmar, B. Devleesschauwer, M. Cecchini, D. A. Ouakrim, T. C. Oliveira, M. J. Struelens, C. Suetens, D. L. Monnet and Burden of AMR Collaborative Group, Lancet Infect. Dis., 2019, 19, 56–66 CrossRef.
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. K. Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. D. Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef CAS.
- A. Talebi Bezmin Abadi, A. A. Rizvanov, T. Haertlé and N. L. Blatt, J. Bionanosci., 2019, 9, 778–788 CrossRef.
- B. Ribeiro da Cunha, L. P. Fonseca and C. R. C. Calado, Antibiotics, 2019, 8, 45 CrossRef PubMed.
- E. Tacconelli and M. D. Pezzani, Lancet Infect. Dis., 2019, 19, 4–6 CrossRef PubMed.
- E. Charani and A. Holmes, Antibiotics, 2019, 8, 7 CrossRef.
- N. Fishman, Am. J. Infect. Control, 2006, 34, S55–S63 CrossRef PubMed.
- S. Paulin, M. Butler, H. Sati, R. Alm, L. Al-Sulaiman, P. Beyer, H. Getahun and S. Kotur Corliss, 2020 antibacterial agents in clinical and preclinical development: an overview and analysis, WHO, 2021 Search PubMed.
- M. S. Butler and D. L. Paterson, J. Antibiot., 2020, 73, 329–364 CrossRef CAS PubMed.
- M. A. Cooper and D. Shlaes, Nature, 2011, 472, 32 CrossRef CAS PubMed.
- S. Paulin and P. Beyer, 2019 Antibacterial agents in clinical development: an analysis of the antibacterial clinical development pipeline, WHO, 2019 Search PubMed.
- S. J. Projan and P. A. Bradford, Curr. Opin. Microbiol., 2007, 10, 441–446 CrossRef CAS PubMed.
- U. Theuretzbacher, S. Gottwalt, P. Beyer, M. Butler, L. Czaplewski, C. Lienhardt, L. Moja, M. Paul, S. Paulin, J. H. Rex, L. L. Silver, M. Spigelman, G. E. Thwaites, J.-P. Paccaud and S. Harbarth, Lancet Infect. Dis., 2019, 19, e40–e50 CrossRef CAS PubMed.
- U. Theuretzbacher, K. Bush, S. Harbarth, M. Paul, J. H. Rex, E. Tacconelli and G. E. Thwaites, Nat. Rev. Microbiol., 2020, 18, 286–298 CrossRef CAS PubMed.
- U. Theuretzbacher, M. Savic, C. Ardal and K. Outterson, Nat. Rev. Drug Discov., 2017, 16, 744–746 CrossRef CAS PubMed.
- U. Theuretzbacher, K. Outterson, A. Engel and A. Karlén, Nat. Rev. Microbiol., 2020, 18, 275–285 CrossRef PubMed.
- D. Hughes and A. Karlén, Upsala J. Med. Sci., 2014, 119, 162–169 CrossRef PubMed.
- B. Plackett, Nature, 2020, 586, S50–S52 CrossRef CAS.
- T. Hesterkamp, Curr. Top. Microbiol., 2016, 398, 447–474 CAS.
- D. M. Shlaes and P. A. Bradford, Pathog. Immun., 2018, 3, 19–43 CrossRef PubMed.
- T. F. Schäberle and I. M. Hack, Trends Microbiol., 2014, 22, 165–167 CrossRef PubMed.
- T. B. Nielsen, E. P. Brass, D. N. Gilbert, J. G. Bartlett and B. Spellberg, N. Engl. J. Med., 2019, 381, 503–505 CrossRef.
- P. Beyer and S. Paulin, Priority pathogens and the antibiotic pipeline: an update, Bull. World Health Organ., 2020, 98, 151 CrossRef PubMed.
- B. Abela-Ridder, G. Biswas, P. S. Mbabazi, M. Craven, A. Gerber, L. Hartenstein, J. Vlcek, M. N. Malecela, M. R. Polo, A. Tiendrebeogo, L. Castellanos, S. Nicholls, M. Jamsheed, Z. Lin, E. Gasimov, H. Atta and S. Warusavithana, Ending the neglect to attain the sustainable development goals: A road map for neglected tropical diseases 2021–2030, WHO, 2020 Search PubMed.
- F. Gobbi, E. Bottieau, O. Bouchaud, D. Buonfrate, F. Salvador, G. Rojo-Marcos, P. Rodari, J. Clerinx, B. Treviño, J. P. Herrera-Ávila, A. Neumayr, G. Calleri, A. Angheben, C. Rothe, L. Zammarchi, M. Guerriero and Z. Bisoffi, PLoS Negl. Trop. Dis., 2018, 12, e0006917 CrossRef CAS PubMed.
- K. Awadzi, Filaria J., 2003, 2, S6 CrossRef PubMed.
- H. Francis, K. Awadzi and E. A. Ottesen, Am. J. Trop. Med. Hyg., 1985, 34, 529–536 CrossRef CAS PubMed.
- B. O. L. Duke, B. Thylefors and A. Rougemont, Current views on the treatment of onchocerciasis with diethylcarbamazine citrate and suramin, WHO, 1981 Search PubMed.
- A. Schiefer, M. P. Hübner, A. K. Krome, C. Lämmer, A. Ehrens, T. Aden, M. Koschel, H. Neufeld, L. Chaverra-Muñoz, R. Jansen, S. Kehraus, G. M. König, D. Pogorevc, R. Müller, M. Stadler, S. Hüttel, T. Hesterkamp, K. Wagner, K. Pfarr and A. Hoerauf, PLoS Negl. Trop. Dis., 2020, 14, e0008930 CrossRef CAS PubMed.
- M. Gyapong, M. T. Ansari, L. Desir, A. Dorkenoo, D. Hollingsworth, J. Horton, A. Klion, N. Kshirsagar, D. McFarland, S. Njenga, R. Noordin, M. Parker, K. Ramaiah and R. Ramzy, Alternative mass drug administration regimes to eliminate lymphatic filariasis, WHO, 2017 Search PubMed.
- WHO team - control of neglected tropical diseases. WHO progress report on the elimination of human onchoceriasis 2016-2017, WHO, 2017.
- A. Y. Debrah, S. Specht, U. Klarmann-Schulz, L. Batsa, S. Mand, Y. Marfo-Debrekyei, R. Fimmers, B. Dubben, A. Kwarteng, M. Osei-Atweneboana, D. Boakye, A. Ricchiuto, M. Büttner, O. Adjei, C. D. Mackenzie and A. Hoerauf, Clin. Infect. Dis., 2015, 61, 517–526 CrossRef CAS PubMed.
- A. Hoerauf, L. Volkmann, C. Hamelmann, O. Adjei, I. B. Autenrieth, B. Fleischer and D. W. Büttner, Lancet, 2000, 355, 1242–1243 CrossRef CAS.
- WHO, Wkly. Epidemiol. Rec., 2019, 94, 415–419 Search PubMed.
- A. Ngwewondo, I. Scandale and S. Specht, Parasitol. Res., 2021, 120, 3939–3964 CrossRef PubMed.
- R. Jansen, G. Höfle, H. Irschik and H. Reichenbach, Liebigs Ann. Chem., 1985, 822–836 CrossRef CAS.
- A. Schiefer, A. Schmitz, T. F. Schäberle, S. Specht, C. Lämmer, K. L. Johnston, D. G. Vassylyev, G. M. König, A. Hoerauf and K. Pfarr, J. Infect. Dis., 2012, 206, 249–257 CrossRef CAS PubMed.
- A. Schmitz, S. Kehraus, T. F. Schäberle, E. Neu, C. Almeida, M. Roth and G. M. König, J. Nat. Prod., 2014, 77, 159–163 CrossRef CAS PubMed.
- T. F. Schäberle, A. Schiefer, A. Schmitz, G. M. König, A. Hoerauf and K. Pfarr, Int. J. Med. Microbiol., 2014, 304, 72–78 CrossRef PubMed.
- T. F. Schäberle, A. Schmitz, G. Zocher, A. Schiefer, S. Kehraus, E. Neu, M. Roth, D. G. Vassylyev, T. Stehle, G. Bierbaum, A. Hoerauf, K. Pfarr and G. M. König, J. Nat. Prod., 2015, 78, 2505–2509 CrossRef PubMed.
- K. Pfarr, A. Hoerauf, G. M. König, S. Specht, A. Schiefer, T. F. Schäberle, A. Schmitz and S. Kehraus, US Pat., US9168244B2, 2015 Search PubMed.
- H. Irschik, R. Jansen, G. Höfle, K. Gerth and H. Reichenbach, J. Antibiot., 1985, 38, 145–152 CrossRef CAS PubMed.
- Ö. Erol, T. F. Schäberle, A. Schmitz, S. Rachid, C. Gurgui, M. El Omari, F. Lohr, S. Kehraus, J. Piel, R. Müller and G. M. König, Chembiochem, 2010, 11, 1253–1265 CrossRef PubMed.
- D. Akkermann, Molekularbiologische und naturstoffchemische Untersuchungen ausgewählter Bakterienstämme, PhD thesis University of Bonn, 2004 Search PubMed.
- H. Sucipto, D. Pogorevc, E. Luxenburger, S. C. Wenzel and R. Müller, Metab. Eng., 2017, 44, 160–170 CrossRef CAS PubMed.
- D. Pogorevc, F. Panter, C. Schillinger, R. Jansen, S. C. Wenzel and R. Müller, Metab. Eng., 2019, 55, 201–211 CrossRef CAS PubMed.
- T. F. Schäberle, M. Mir Mohseni, F. Lohr, A. Schmitz and G. M. König, Antimicrob. Agents Chemother., 2014, 58, 950–956 CrossRef PubMed.
- F. Lohr, I. Jenniches, M. Frizler, M. J. Meehan, M. Sylvester, A. Schmitz, M. Gütschow, P. C. Dorrestein, G. M. König and T. F. Schäberle, Chem. Sci., 2013, 4, 4175–4180 RSC.
- H. Sucipto, J. H. Sahner, E. Prusov, S. C. Wenzel, R. W. Hartmann, J. Koehnke and R. Müller, Chem. Sci., 2015, 6, 5076–5085 RSC.
- A. Rentsch and M. Kalesse, Angew. Chem., Int. Ed., 2012, 51, 11381–11384 CrossRef CAS PubMed.
- Y. Nagashima, Y. Okada, T. Sato and N. Chida, Chem. Lett., 2019, 48, 1519–1521 CrossRef CAS.
- A. K. Krome, Dissolution, solubility, and stability enhanced formulation strategies for the novel anti-infective corallopyronin A, PhD thesis University of Bonn, 2021 Search PubMed.
- A. K. Krome, T. Becker, S. Kehraus, A. Schiefer, C. Steinebach, T. Aden, S. J. Frohberger, Á. L. Mármol, D. Kapote, R. Jansen, L. Chaverra-Muñoz, M. P. Hübner, K. Pfarr, T. Hesterkamp, M. Stadler, M. Gütschow, G. M. König, A. Hoerauf and K. G. Wagner, Pharmaceutics, 2020, 12, 1105 CrossRef CAS PubMed.
- S. Singh and R. Roy, Expert Opin. Drug Discov., 2016, 11, 695–706 CrossRef CAS PubMed.
- Y. V. Kazakevich and R. LoBrutto, HPLC for Pharmaceutical Scientists, John Wiley & Sons, 2007 Search PubMed.
- J. Mukhopadhyay, K. Das, S. Ismail, D. Koppstein, M. Jang, B. Hudson, S. Sarafianos, S. Tuske, J. Patel, R. Jansen, H. Irschik, E. Arnold and R. H. Ebright, Cell, 2008, 135, 295–307 CrossRef CAS PubMed.
- F. Kock, M. Hauptmann, A. Osterloh, T. F. Schäberle, S. Poppert, H. Frickmann, K.-D. Menzel, G. Peschel, K. Pfarr, A. Schiefer, G. M. König, A. Hoerauf, B. Fleischer and C. Keller, Antimicrob. Agents Chemother., 2018, 62(4), e01732–17 CrossRef CAS PubMed.
- A. O'Neill, B. Oliva, C. Storey, A. Hoyle, C. Fishwick and I. Chopra, Antimicrob. Agents Chemother., 2000, 44, 3163–3166 CrossRef PubMed.
- Z. S. Bhat, M. A. Rather, M. Maqbool, H. U. Lah, S. K. Yousuf and Z. Ahmad, Biomed. Pharmacother., 2017, 91, 265–277 CrossRef CAS PubMed.
- L. Fields, W. R. Craig, C. A. Huffine, C. F. Allen, L. M. Bouthillette, J. C. Chappell, J. T. Shumate and A. L. Wolfe, Bioorg. Med. Chem. Lett., 2020, 30, 127301 CrossRef CAS PubMed.
- H. Sucipto, S. C. Wenzel and R. Müller, Chembiochem, 2013, 14, 1581–1589 CrossRef CAS PubMed.
- J. S. Bauer, M. G. K. Ghequire, M. Nett, M. Josten, H.-G. Sahl, R. De Mot and H. Gross, Chembiochem, 2015, 16, 2491–2497 CrossRef CAS PubMed.
- M. Chu, R. Mierzwa, L. Xu, L. He, J. Terracciano, M. Patel, W. Zhao, T. A. Black and T.-M. Chan, J. Antibiot., 2002, 55, 215–218 CrossRef CAS PubMed.
- I. J. S. Fairlamb, L. R. Marrison, J. M. Dickinson, F.-J. Lu and J. P. Schmidt, Bioorg. Med. Chem., 2004, 12, 4285–4299 CrossRef CAS PubMed.
- K. Mariner, M. McPhillie, R. Trowbridge, C. Smith, A. J. O'Neill, C. W. G. Fishwick and I. Chopra, Antimicrob. Agents Chemother., 2011, 55, 2413–2416 CrossRef CAS PubMed.
- A. Srivastava, D. Degen, Y. W. Ebright and R. H. Ebright, Antimicrob. Agents Chemother., 2012, 56, 6250–6255 CrossRef CAS PubMed.
- A. Sharff, C. Fanutti, J. Shi, C. Calladine and B. Luisi, Eur. J. Biochem., 2001, 268, 5011–5026 CrossRef CAS PubMed.
- M. Brauer, J. Herrmann, D. Zühlke, R. Müller, K. Riedel and S. Sievers, Gut Pathog., 2022, 14, 4 CrossRef CAS PubMed.
- K. Shima, M. Wanker, R. J. Skilton, L. T. Cutcliffe, C. Schnee, T. A. Kohl, S. Niemann, J. Geijo, M. Klinger, P. Timms, T. Rattei, K. Sachse, I. N. Clarke and J. Rupp, mSphere, 2018, 3, e00412–e00418 CrossRef CAS PubMed.
- N. Loeper, S. Graspeuntner, S. Ledig, I. Kaufhold, F. Hoellen, A. Schiefer, B. Henrichfreise, K. Pfarr, A. Hoerauf, K. Shima and J. Rupp, Front. Microb., 2019, 10, 943 CrossRef PubMed.
- A. Denninger, U. Westedt, J. Rosenberg and K. G. Wagner, Pharmaceutics, 2020, 12, 237 CrossRef CAS PubMed.
- B. C. Hancock, S. L. Shamblin and G. Zografi, Pharm. Res., 1995, 12, 799–806 CrossRef CAS PubMed.
- D. Q. M. Craig, P. G. Royall, V. L. Kett and M. L. Hopton, Int. J. Pharm., 1999, 179, 179–207 CrossRef CAS PubMed.
- K. A. Graeser, J. E. Patterson, J. A. Zeitler, K. C. Gordon and T. Rades, Eur. J. Pharm. Sci., 2009, 37, 492–498 CrossRef CAS PubMed.
- S. Yoshioka and Y. Aso, J. Pharm. Sci., 2007, 96, 960–981 CrossRef CAS PubMed.
- M. J. Taylor, D. Voronin, K. L. Johnston and L. Ford, Cell. Microbiol., 2013, 15, 520–526 CrossRef CAS PubMed.
- U. Klarmann-Schulz, S. Specht, A. Y. Debrah, L. Batsa, N. K. Ayisi-Boateng, J. Osei-Mensah, Y. Mubarik, P. Konadu, A. Ricchiuto, R. Fimmers, S. Arriens, B. Dubben, L. Ford, M. Taylor and A. Hoerauf, PLoS Negl. Trop. Dis., 2017, 11, e0005156 CrossRef PubMed.
- S. Specht, S. Mand, Y. Marfo-Debrekyei, A. Y. Debrah, P. Konadu, O. Adjei, D. W. Büttner and A. Hoerauf, Parasitol. Res., 2008, 103, 1303–1309 CrossRef PubMed.
- A. Hoerauf, S. Mand, O. Adjei, B. Fleischer and D. W. Büttner, Lancet, 2001, 357, 1415–1416 CrossRef CAS.
- M. J. Taylor, T. W. von Geldern, L. Ford, M. P. Hübner, K. Marsh, K. L. Johnston, H. T. Sjoberg, S. Specht, N. Pionnier, H. E. Tyrer, R. H. Clare, D. A. N. Cook, E. Murphy, A. Steven, J. Archer, D. Bloemker, F. Lenz, M. Koschel, A. Ehrens, H. M. Metuge, V. C. Chunda, P. W. N. Chounna, A. J. Njouendou, F. F. Fombad, R. Carr, H. E. Morton, G. Aljayyoussi, A. Hoerauf, S. Wanji, D. J. Kempf, J. D. Turner and S. A. Ward, Sci. Transl. Med., 2019, 11, 483 Search PubMed.
- A. Albers, M. E. Esum, N. Tendongfor, P. Enyong, U. Klarmann, S. Wanji, A. Hoerauf and K. Pfarr, Parasites Vectors, 2012, 5, 12 CrossRef CAS PubMed.
- S. Arumugam, K. M. Pfarr and A. Hoerauf, Int. J. Parasitol., 2008, 38, 981–987 CrossRef PubMed.
- A. Y. Debrah, S. Mand, Y. Marfo-Debrekyei, L. Batsa, A. Albers, S. Specht, U. Klarmann, K. Pfarr, O. Adjei and A. Hoerauf, J. Parasitol. Res., 2011, 201617 Search PubMed.
- A. Hoerauf, S. Specht, M. Büttner, K. Pfarr, S. Mand, R. Fimmers, Y. Marfo-Debrekyei, P. Konadu, A. Y. Debrah, C. Bandi, N. Brattig, A. Albers, J. Larbi, L. Batsa, M. J. Taylor, O. Adjei and D. W. Büttner, Med. Microbiol. Immunol., 2008, 197, 295–311 CrossRef CAS PubMed.
- S. Mand, K. Pfarr, P. K. Sahoo, A. K. Satapathy, S. Specht, U. Klarmann, A. Y. Debrah, B. Ravindran and A. Hoerauf, Am. J. Trop. Med. Hyg., 2009, 81, 702–711 CrossRef CAS PubMed.
- M. J. Taylor, W. H. Makunde, H. F. McGarry, J. D. Turner, S. Mand and A. Hoerauf, Lancet, 2005, 365, 2116–2121 CrossRef CAS.
- J. D. Turner, N. Tendongfor, M. Esum, K. L. Johnston, R. S. Langley, L. Ford, B. Faragher, S. Specht, S. Mand, A. Hoerauf, P. Enyong, S. Wanji and M. J. Taylor, PLoS Negl. Trop. Dis., 2010, 4, e660 CrossRef PubMed.
- A. Y. Debrah, S. Mand, Y. Marfo-Debrekyei, L. Batsa, K. Pfarr, M. Buttner, O. Adjei, D. Buttner and A. Hoerauf, Trop. Med. Int. Health, 2007, 12, 1433–1441 CrossRef CAS PubMed.
- A. Hoerauf, O. Adjei and D. W. Büttner, Curr. Opin. Invest. Drugs, 2002, 3, 533–537 CAS.
- M. J. Taylor and A. Hoerauf, Parasitol. Today, 1999, 15, 437–442 CrossRef CAS PubMed.
- M. J. Taylor, A. Hoerauf and M. Bockarie, Lancet, 2010, 376, 1175–1185 CrossRef.
- M. Walker, S. Specht, T. S. Churcher, A. Hoerauf, M. J. Taylor and M.-G. Basáñez, Clin. Infect. Dis., 2015, 60, 1199–1207 CrossRef CAS PubMed.
- M. J. Taylor, A. Hoerauf and M. Bockarie, Lancet, 2010, 376, 1175–1185 CrossRef.
- D. Haebich and F. von Nussbaum, Angew. Chem., Int. Ed., 2009, 48, 3397–3400 CrossRef CAS PubMed.
- H. Irschik, K. Gerth, G. Höfle, W. Kohl and H. Reichenbach, J. Antibiot., 1983, 36, 1651–1658 CrossRef CAS PubMed.
- A. Schmitz, The antibiotic corallopyronin A and the corallorazines from the myxobaccterium Corallococcus coralloides B035, PhD thesis University of Bonn, 2013 Search PubMed.
- M. Miethke, M. Pieroni, T. Weber, M. Brönstrup, P. Hammann, L. Halby, P. B. Arimondo, P. Glaser, B. Aigle, H. B. Bode, R. Moreira, Y. Li, A. Luzhetskyy, M. H. Medema, J.-L. Pernodet, M. Stadler, J. R. Tormo, O. Genilloud, A. W. Truman, K. J. Weissman, E. Takano, S. Sabatini, E. Stegmann, H. Brötz-Oesterhelt, W. Wohlleben, M. Seemann, M. Empting, A. K. H. Hirsch, B. Loretz, C.-M. Lehr, A. Titz, J. Herrmann, T. Jaeger, S. Alt, T. Hesterkamp, M. Winterhalter, A. Schiefer, K. Pfarr, A. Hoerauf, H. Graz, M. Graz, M. Lindvall, S. Ramurthy, A. Karlén, M. van Dongen, H. Petkovic, A. Keller, F. Peyrane, S. Donadio, L. Fraisse, L. J. V. Piddock, I. H. Gilbert, H. E. Moser and R. Müller, Nat. Rev. Chem., 2021, 5, 726–749 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |