Structural diversity and biological relevance of benzenoid and atypical ansamycins and their congeners
Natalia
Skrzypczak
and
Piotr
Przybylski
*
Faculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznanskiego 8, 61-614 Poznan, Poland. E-mail: piotrp@amu.edu.pl
Received
18th January 2022
First published on 9th March 2022
Abstract
Covering: 2011 to 2021
The structural division of ansamycins, including those of atypical cores and different lengths of the ansa chains, is presented. Recently discovered benzenoid and atypical ansamycin scaffolds are presented in relation to their natural source and biosynthetic routes realized in bacteria as well as their muta and semisynthetic modifications influencing biological properties. To better understand the structure–activity relationships among benzenoid ansamycins structural aspects together with mechanisms of action regarding different targets in cells, are discussed. The most promising directions for structural optimizations of benzenoid ansamycins, characterized by predominant anticancer properties, were discussed in view of their potential medical and pharmaceutical applications. The bibliography of the review covers mainly years from 2011 to 2021.
Natalia Skrzypczak
Natalia Skrzypczak was born in Poznan in 1995. She obtained a bachelor's degree (2017) and a master's degree (2019) in chemistry at the Adam Mickiewicz University in Poznan, Poland. In 2019, she started her PhD studies under the supervision of Prof. Piotr Przybylski at the Faculty of Chemistry, AMU. She is a co-author of 4 publications. Her research interests include the novel approaches for transformation of ansamycins cores, based on modern synthetic methods (click chemistry).
Piotr Przybylski
Piotr Przybylski (MSc 2000; PhD – 2004; habilitation – 2011; full professor – 2019) was born in Poznan, Poland in 1975. He is a professor of organic chemistry at Department of Natural Products Chemistry (AMU in Poznan). He is the leader of the research team specialized in chemistry and medicinal chemistry of natural antibiotics, their derivatives and other biologically important molecules. His research interests are focused on modification and determination of SAR for natural products and their congeners, and with tautomerization, atropisomerization and zwitterionization processes. His current studies are related to cascade approaches enabling modification of lactone and lactam macrolides.
1. Introduction
Ansamycins, produced by different bacteria strains, can be distinguished from the other natural products by two characteristic structural portions – one is a rigid, usually aromatic “core” (red, Fig. 1) and the second is the so-called ansa bridge (black, Fig. 1), containing the lactam moiety (blue, Fig. 1), linked to the two nonadjacent positions of the core. These two structural moieties form a “molecular basket” of relatively high conformational lability, characteristic of chameleonic drugs.1–4 However, adherence to this definition has been inconsistent as per the literature. For example, kendomycin or rapamycin antibiotics have been described as “ansamycins” despite the absence of a lactam moiety in kendomycin and the absence of an aromatic “core” in rapamycin (Fig. 2).5 An analogous problem is related to the classification of rubradirin (Fig. 2) not including the characteristic lactam moiety but instead possessing the amine in its aliphatic chain. Hence, based on exclusively structural criteria, rubradirin should not be considered as classical ansamycin, although it shares a common biosynthetic pathway with ansamycins.6 Hence, to avoid any confusions concerning the classifications of ansamycins and taking into account recently strongly developed area of ansamycin mutasynthesis a more detailed description of these compounds is needed. We propose the use of the term natural “ansamycin” linked to the family of macrolactams, bearing relatively rigid central cores as benzenoid, naphthalenoid or atypical ones (e.g. alicyclic or heterocyclic ones), originated from AHBA or other-type small semisynthetic unsaturated precursors (mutasynthons), and produced in as a result of cooperation between polyketide (PKS type I) and amide synthases. This description of ansamycins is universal and comprises natural compounds having ansa chains with lactam groups, irrespective of whether they possess reduced or oxidized cores, as long as analogous biosynthetic pathways from the small precursor building the core are realized. Many published investigations have been devoted to structural studies, quantitative or qualitative detection, biological activity, biosynthesis and semisynthetic modifications of these natural products, and the results of these investigations have been very interesting, both from biological and chemical points of view.7–30 Newly discovered benzenoid ansamycin scaffolds and their modifications as potential attractive toward pharmaceutical and medical applications have stimulated detailed multidisciplinary studies that have aimed to determine the relationship between structure and useful biological effect. However, these studies are not trivial due to complexity of the structure and the different mechanisms of action of ansamycins. Informations about chemistry and biology of ansamycin antibiotics is still being updated. Funayama and Cordell, focused on providing an overview of the classification and biosynthesis of ansamycins (benzenoid and naphthalenoid) by the year 2000.27 Reviews concerning geldanamycin and ansamitocins, important members of benzenoid ansamycins, were earlier published by: Floss et al. (biosynthesis),23,31 Kirschning et al. (ansamycins and their modifications targeting Hsp90)32,33 and Moody et al. (geldanamycin anticancer properties)7 by the year 2013. Structural, biosynthetic, biological and analytical aspects of benzenoid ansamycins were also discussed in reviews from the years 1972 and 2010.15,23,24,27,29–31,34–36 Benzenoid and atypical ansamycins are being investigated at an increasing pace due to the new abilities to extract them from natural sources as well as due to the rapidly progressing modifications of these compounds via semisynthetic and mutasynthetic approaches or their combinations, in aim to optimize their biological properties. The review of the recent advances in the field of natural benzenoid and atypical ansamycins and their modifications is needed to highlight possible challenges concerning their biological applications.
Fig. 1 Exemplary structure of benzenoid ansamycin – ansamitocin P3 (AP3); visualized by Scigress (EU 3.1.8).
Fig. 2 Macrocyclic antibiotics which should not be named formally “ansamycins” because of the structural criterion – the lack at least one of characteristic structural motifs, i.e. the lactam bridge or the rigid and unsaturated core.
2. Divisions of benzenoid ansamycins by structure and source of origin
Funayama and Cordell divided ansamycins into two main groups regarding the type of the core and the length of the carbon ansa chain.27 Such an classification of ansamycins is in general reasonable but these two groups can be also more precisely divided further in view of the structure of the core, as shown in Fig. 3. In our structural division of ansamycins, the third group of atypical macrolactams appears as a consequence of developing mutasynthetic and combined approaches. Furthermore, in an addition to the earlier classification,27 we propose several subgroups of benzenoid ansamycins represented by a specific core type, namely: benzene, dibenzene, cyclohexa-2-enone, cyclohexa-2,5-dienone, benzoquinone or atypical (e.g. alicyclic or heterocyclic) and containing ansa chains of different lengths (Fig. 3). Such detailed structural division of ansamycins, despite labile oxidation state of the core, is reasonable as these natural products in quinol or quinoid forms have different chemical and biological features, i.e. physico–chemical parameters as lipophilicity or water solubility, binding modes to the target in cells or biological activity. To the best of our knowledge benzenoid ansamycins are produced solely by various bacteria strains, which can be associated with plants, terrestrial and marine organisms (Fig. 3). Titles of many publications often suggest plants as a source of ansamycins. Leistner et al. reported that higher plants usually do not have gene PKS type I machinery required to biosynthesis of benzenoid ansamycins.37 Furthermore, bioinformatic analysis also showed that PKS type I gene cluster is not present in plants, in contrast to PKS type III one, required for production of the other natural products of e.g. flavonoids group.38,39 Plants are able to perform only minor post-biosynthetic modifications of benzenoid ansamycin scaffolds by e.g. simple esterification as for ansamitocin P3.23,33
Fig. 3 Structural division of ansamycins regarding the type of the core (red) and the length of the ansa chain (top), and their source of origin (bottom). Bacteria strains, producing benzenoid ansamycins, associated with: plants are marked by green, mosses are brown, and those from water environment are blue.
3. Structure of the ansa bridge and atropisomerisation of benzenoid ansamycins having impact on their chemistry and biology
The ansamycin family of natural products is interesting in that their specific structural features have a great impact on the transport and molecular recognition processes with the target site of action, thereby influencing biological activity. In the structure of benzenoid ansamycins different numbers and configurations of the double bonds, conjugated with the lactam or to each other (Fig. 4), alter rigidity of the ansa chain changing the conformation flexibility needed to molecular recognition with the target in cells. Configurations of double bonds conjugated with the lactam moiety are (2E) and (4Z) in geldanamycin structure, whereas in the other benzenoid ansamycins such as ansamitocin P1 no analogous double bonds region at this portion of the ansa chain occurs. In the structure of ansamitocin P1 the conjugated double bond system is shifted away from the lactam moiety to the C(11) and C(13), where both double bonds display E-configuration. Ansatrienin A (mycotrienin I) has three mutually conjugated double bonds at the C(4), C(6) and C(8) positions, but they are not conjugated with the lactam moiety (Fig. 4). In case of herbimycins (Fig. 4), despite the fact that double bonds are in positions as those in geldanamycin structure, configurations of them are identical for herbimycin I (2E,4Z), but different for herbimycins J (2E,4E) and K (2Z,4Z), as evidenced by ECD and 1H–1H NOESY studies (Fig. 4).40 The most optimal configuration of double bonds in herbimycins from the anticancer activity viewpoint, seems to be (2E,4Z).40 The same number of the carbon atoms in the ansa chain, as for geldanamycin and herbimycins, possesses novel C15-benzenoid ansamycin DHQ5, where only one double bond is conjugated with the lactam.41 In contrast to DHQ5, in structure of C9-benzenoid microansamycin D (Fig. 4), the occurrence of double bonds, being conjugated to each other and to the C(3)-ketone group, contributed to increased rigidity of the ansa chain, reflected in the lack of essential antibacterial properties.42 Among all the above-mentioned examples of benzenoid ansamycins, only ansamitocins, of the mytansinoid group, have each N-methylated lactam moiety in the ansa bridge (Fig. 4).33 The basket-like structure of ansamycins enables chameleonic features of these natural products because relatively flexible ansa-bridge can adopt different conformations in response to the steric hindrances or environmental factors such as the nature of cell barriers. For ansamycin atropisomers the two generally possible different orientations of the ansa bridge relative to the rigid core are observed (Fig. 5). Hence, unique stereochemical properties, including atropisomerism (from Greek “a” meaning “not” and “tropos” meaning “turn”), were noted for benzenoid ansamycins. Experimental evidence has been provided for the occurrence of classical atropisomerism for benzenoid-type ansamycins as geldanamycin analogs, involving an extra cis–trans isomerization of the lactam (Fig. 5).43–48 Interestingly, geldanamycin and its C(17)-derivatives have been each detected in the two atropisomeric forms: with the trans-lactam in the ‘free’ form in solution and with the cis-lactam when bound to the molecular target (Fig. 5).32,46,48 The atropisomerization mechanism of this-type ansamycins is supported by the crystal structure of an intermediate form (Fig. 6), with the ansa chain placed between the two border locations of this chain, noted in (P,M) and (M,P) atropisomers.46 X-ray structure of this intermediate form between those of trans- and cis-lactam atropisomeric forms, is stabilized by an intramolecular H-bond O(11)–H⋯O(21), as shown in Fig. 6.46 Energy barrier of the interconversion between the two opposite atropisomeric forms of benzenoid-C15 ansamycins was reported as in the range from ∼16 to 37 kcal mol−1.48–50 The presence of an N-methylated lactam within the ansa chain of ansamitocins and maytansine implies, already in the ‘free’ form in solution or in solid, the formation of cis-lactam atropisomer, ready to be bound with the target (Fig. 1 and 4).51 In contrast to that, for geldanamycin the atropisomerization process is necessary for binding with the target because in the ‘free’ form in solution the trans-lactam atropisomeric form is predominant. In turn, the introduction of a bulky substituent at the C(19) of the quinone core of geldanamycin congeners generates steric hindrances with the oxygen of the lactam group and enforces conversion of the trans-lactam atropisomer into the cis-lactam one, in the ‘free’ form in solution (Fig. 7).45
Fig. 4 Different configuration of the double bonds within ansa bridge of benzenoid ansamycins.
Fig. 5 Atropisomers of geldanamycin implied by the two different arrangements of the ansa-bridge relative to the benzoquinone core; green and blue colour portions corresponds to P and M; visualized via Scigress (EU 3.1.8), Fujitsu.
Fig. 6 Superimposed arrangements of ansa bridge: in solution atropisomeric trans-lactam form (light blue), atropisomeric cis-lactam form bound with Hsp90 (orange), and the intermediate form, found in crystal, stabilized by an intramolecular H-bond O(11)H⋯OC(21) (violet; CCDC 1988071).46
Fig. 7 C(19)-functionalized analog of geldanamycin with 2-furyl moiety in a forced cis-lactam configuration and as atropisomer ready to bind with Hsp90 [visualised by Scigress (EU 3.1.8), Fujitsu; on the basis of X-ray data – CCDC 864025].45
The above-mentioned structural features have essential impact on ansamycins mechanism of action in cells. Despite molecular targets for benzenoid ansamycins being different, as e.g. tubulins52,53 or Hsp90 of bovine,54 yeast45,55 or leishmania56 (Fig. 8), the observed biological profile of these compounds is often similar, i.e. mainly the high anticancer potency with relatively high toxicity in normal cells are noted.
Fig. 8 Interactions between benzenoid-C15 ansamycins and their molecular targets in cells: (a) maytansine – β-tubulin PM060184 (PDB 4TV8);53 (b) premacbecin – Hsp90 from yeast (PDB 2VW5);55 (c) C(19)-phenyl analog of geldanamycin – Hsp90 from yeast (PDB 4ASG),45 visualized by Scigress (EU 3.1.8), Fujitsu. Assignment of colors is as following: intermolecular H-bonds stabilizing complexes (yellow dots), ansa chains of benzenoid ansamycins (orange), benzenoid cores (red), lactam groups (blue), amino acid codes taking part in formation of H-bonds (green); amino acid codes forming binding pockets (black).
Binding of maytansinoids to tubulins occurs via key H-bonds with the participation of the following moieties of the cis-lactam atropisomer: the lactam, C(7)–C(9) cyclic carbamate, and the hydroxyl at C(9), and the C(3)-tail, and following amino acid residues T180, V181, N102, K105, G100, and N101 of β-tubulin (PDB 4TV8; Fig. 8a).53 In turn, the hydrophobic stabilization of the maytansine–tubulin complex involves residues F404, W407, Y408, and V182 of β-tubulin. Thus, the presence of the N-methyl, chlorine and methoxy groups of the benzene core, and the C(4) epoxide group of the ansa chain, not involved in any interactions with the tubulin subunits, seems to be less important for maytansinoid affinity to the target. In contrast to tubulins, at binding with chaperones Hsp90 the substituents of the benzene or benzoquinone cores play an important role with formation of the complex. X-ray studies of a premacbecin complex with yeast Hsp90 indicated the involvement of the phenol group at C(18) of the core, the cis-lactam and carbamate in intermolecular H-bonding with residues D79, D40, F124 and indirectly with G123 (PDB 2VW5; Fig. 8b).55 Substitution of fluorine at C(18) of the benzene core improves stabilization of the complex with Hsp90, by an extra interaction with K98 (K112 in human orthologs).57 Geldanamycin, a bezoquinone-C15 ansamycin binds with ATP-binding pocket of Hsp90, of both human cancer and dopaminergic neural cells, in a manner slightly different to that discussed above for premacbecin of autolytimycins group (Fig. 8b).54 The presence of the two carbonyl groups in the benzoquinone core of geldanamycin does result in some new intermolecular interactions with K98/K112 at the expense of lost interaction with D40 of Hsp90, which is realized for premacbecin. Incorporation of an extra C(19) substituent into the benzoquinone core as in 19-Ph geldanamycin (Fig. 8c) does not alter markedly the binding mode, in respect of ansa chain and core, compared with geldanamycin.45 In turn, the replacement of the C(17)-methoxy group with an amine substituent for benzoquinone ansamycins was beneficial to an extra stabilization of the complex via formation of a strong H-bond C(17)N–H…−OOC(D39), as for e.g. 17-DMAP56 or 17-DMAG,43 irrespectively on the type of chaperone, i.e. human, bovine, yeast or leishmania. This short survey of binding modes of benzene or benzoquinone ansamycins with different targets reveals that one of atropisomers, containing the cis-lactam, is required to block the functionality of molecular targets, irrespectively of their types, i.e. tubulins or chaperones Hsp90. Functional groups attached to the benzenoid core are not always involved in stabilizing interactions with the target, whereas the ansa bridge groups take part in crucial interactions required for molecular recognition of benzenoid ansamycin, irrespectively on the target type, as suggested by the model of maytansine complex with β-tubulin.58
4. Biosynthesis of novel bioactive benzenoid ansamycins
Ansamycinscare synthesized in nature by various bacteria strains from AHBA, being a product of the modified shikimate acid pathway, and assembling of different basic building blocks of the ansa chain as acetate (C2), propionate (C3), butyryl (C4) or isobutyrylmalonyl (C6) units, irrespectively of the core type, as proved by studies with isotopically labeled precursors.9,20,59–62 The biosynthesis of benzenoid ansamycins is carried out by polyketide synthase of type I (PKS I) and by amide synthase catalyzing the macrolactamization. The post-PKS I tailoring of ansamycins scaffolds are based on the set of various simple reactions as e.g. oxidation, methylation, carbamoylation, elimination, which contributes to the structural variety of these natural products, especially in regard to the structure of the ansa chain.9,63
Natural benzene- and dibenzene-C9 ansamycins
Ansaetherone (1, Fig. 9) bearing fused benzene core with pyran motif, was isolated by the Hirota team from the Streptomyces sp. USF-4727 strain.64 The proposed biosynthetic pathway for this benzene-C9 ansamycin is realized from AHBA (m-C7N) assembled with three propionates (C3) and a one butanoate (C4) unit, and glucose as a precursor. It was found that 1 is intermediate in biosynthesis of tetrapetalones. Radical scavenging studies indicated activity of 1 in DPPH tests at μM level.64 Structural comparable to 1 are new diastereomeric cebulactams A1 (2, Fig. 9) and A2, biosynthesized by marine bacterium Saccharopolyspora cebuensis on the analogous biosynthetic pathway as 1, but not involving respective glycosyltransferase.65 These two natural, isomeric compounds have, different absolute configurations at C(4) stereogenic centers and different double bond configurations (E/Z) within their ansa chains, as indicated by 1H–1H NOESY and total synthesis.66,67 Unique benzene ansamycins, called juanlimycins A (3) and B (4), bearing the two benzene cores and the two C9 ansa chains, were isolated from Streptomyces sp. LC6, associated with leaves of Kandelia candel (Linn).68 Unusual biosynthetic pathway of these macrodilactams was initially performed by PKS I up to the fusion of AHBA part with tetraketide chain and then dimerization process of these intermediates is catalyzed by amide synthase (Fig. 10).68 The other transformations as hydroxylation at the benzene cores, condensation with benzoquinone, double dehydrations and the Mannich-type reaction, were postulated to be necessary to biosynthesis of juanlimycins (Fig. 10). Structures of 3 and 4 were evidenced by NMR and X-ray (for 3). Ansamycin 3, having two cis-lactams and decorated with the two identical anthranilate motifs, did show moderate inhibition of the secretion of SPI-1 effectors (Salmonella Pathogenicity Island).68
Fig. 9 Examples of benzenoid-C9 ansamycins: ansaetherone (1) and cebulactam A1 (2).
Fig. 10 Novel macrodilactams called juanlimycins A (3) and B (4) and their postulated biosyntheses based on Mannich-type reactions.
Natural benzene–C15 and benzoquinone–C15 ansamycins
Benzenoid ansamycins having C15 ansa chains (Fig. 3) are not only produced by bacteria strains associated with plants, but also by soil- or marine-derived bacteria. Recently, it was suggested that biosynthesis of the main scaffolds of maytansine and maytansinoids (Fig. 8), via proansamitocin stage, is realized mainly by cross-species, and the chlorination step is performed via cooperation of the plant Maytenus serrata with the endophytic bacterial community.69 The most widespread in nature are benzene-C15 ansamycins, as e.g. potent anticancer ansamitocins (Fig. 1 and 4).33 The 2-methoxymalonyl unit coupled with ACP (acyl carrier protein) has been identified as one of the biological precursors of ansamitocins P0–P5.70 Floss et al. found methyltransferase encoded by asm10, required to N-methylation in the biosynthesis of ansamitocins, to be dependent on the presence of SAM.71 Recently, the role of ACP linked to extender units in the biosynthetic pathway of ansamitocin P3 (Fig. 1) was studied by Zheng et al.72 They presented evidence for the preference of the AsmAT3 acyl transferase domain to catalyze the reaction involving exclusively (2S) but not (2R) diastereomer of methoxymalonyl-ACP (MOM-ACP) in biosynthesis of ansamitocin P3. Hence, in the biosynthetic pathway of ansamitocin P3, an extra enzyme, called epimerase, is required to utilize the (2R)-MOM-ACP or an alternatively (2R)-MOM-CoA and to make this process more efficient, despite AsmAT3 showing ∼300-fold preference for MOM-Asm14 over CoA-linked extender units at ansamitocins biosynthesis.72 An economical approach using a low-cost medium and A. pretiosum for producing ansamitocin P3 was reported by Hua et al.73 Natural maytansinoids are known tubulin polymerization inhibitors (Fig. 8a), binding at the site typical for vinblastine analogs, what contributes to their strong anticancer effects.52,53,74
A novel natural ansamycins, structurally close to maytansine or ansamitocins are the so-called ansacarbamitocins (5, Fig. 11), bearing glucosyl at the lactam and at least two carbamate units.75–78 These compounds, produced by A. pretiosum strain and of cis-lactam configuration, belong to a wider group called ansamitocinosids (6, Fig. 11). Ansacarbamitocins biosynthetic pathway includes double (5) or triple (6) carbamoylations within the main scaffold and the saccharide portion, where the activity of carbamoyltransferase asm21 is required.78 The Δ11, Δ13-diene system, shifted away from the lactam, is installed at processing of the nascent polyketide on module 3 of the asmPKS, where KS4 domain acts selectively toward rearranged intermediate tetraketide.79N-Glucosyl ansamitocinosides as 6, each containing an ester or carbamoyl tail at the C(3) position of the ansa chain, differ to each other with regards to the substituents at the C(4′) and C(6′) positions of the saccharide (Fig. 11). The incorporation of the saccharide unit into ansamitocinosides scaffold is enabled by N-demethylation with subsequent N-glycosylation of the lactam, performed by enzymes encoded by Asm10 and Asm25 genes, respectively.80 Recently, identification of the bacterial gene cluster asc for the biosynthesis of N-glucosyl maytansinoids in Amycolatopsis alba DSM 44262 and in A. pretiosum 31565 revealed an active participation of 3-O-methyl transferase and 3′-O-carbamyl-transferase enzymes in the biosynthetic pathway.81 Li et al. reported that C(9)-methoxy analog of 6 can be produced not only by A. pretiosum but also by the other Amycolatopsis alba DSM 44262 bacteria strain.82 Ansacarbomitocins exhibited modest antifungal activities against Septoria tritici, Erisiphe graminis, and Puccinia recondite.83
Fig. 11 Ansacarbomitocin 5, member of natural ansamitocinosides (6).
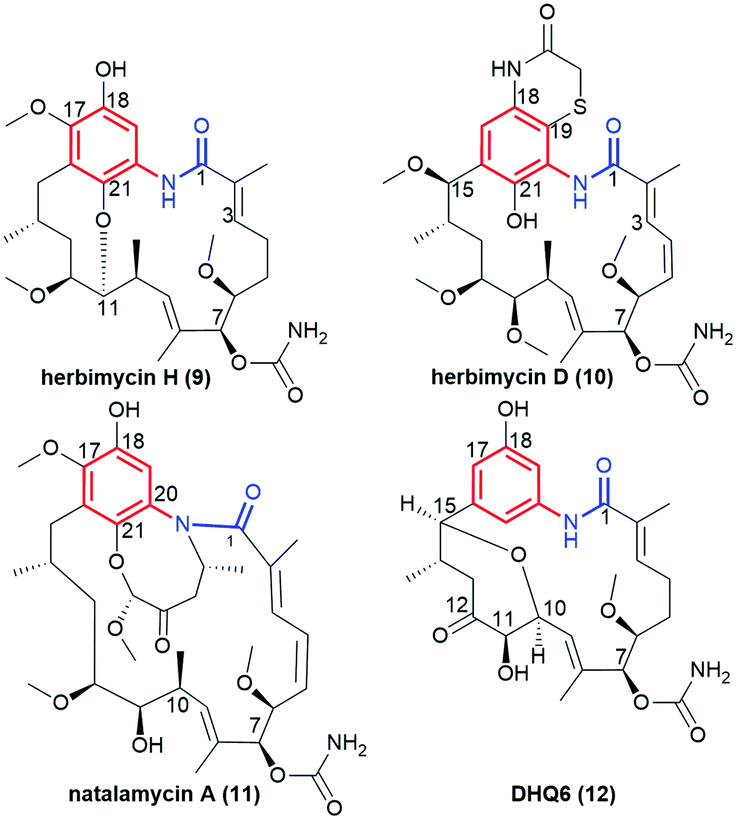
Another subgroup of natural benzene-C15 ansamycins, each lacking a chlorine atom at C(19), and often possessing an oxygen atom at C(18) or/and at C(21) are autolytimycin (7, Fig. 12),84 reblastatin (8, Fig. 12),84 herbimycins D, H, J, K (9, 10; Fig. 4 and 13),85,86 natalamycin (11, Fig. 13)87 and DHQ6 (12, Fig. 13). The presence of the oxygen at C(21) of the benzene core enables often an extra biosynthetic cyclizations, either between C(21) and C(11) of the ansa chain (see e.g. herbimycin 9) or between C(21) and the nitrogen of the lactam as for 11 (Fig. 13). The natural benzenoid-C15 ansamycins with the phenol at C(18) have rather rarely the ansa bridge incorporated into an extra cyclic system, as in DHQ5 (Fig. 4) and DHQ6 (12, Fig. 13), isolated from S. hygroscopicus subspec. duamyceticus JCM4427.41,88 Common biosynthetic pathways of 7 and 8 start from AHBA via seco-progeldanamycin to 7-hydroxy-7 intermediate, and next includes different post-PKS I tailoring (Fig. 12).84 In turn, the five-carbon membered aliphatic bridge, linking the core and the lactam moiety within 11 (Fig. 13), originates from 4-hydroxy-2-oxopentanoate unit (Fig. 14), being intermediate in 3-phenylpropionate catabolism.87 Herbimycin F, bearing 2-hydroxy and 4-keto substituents at the ansa chain, and herbimycin D (10, Fig. 13), called also heronamycin A, are secondary metabolites produced by Streptomyces sp. RM-7-15, isolated from soil near a coal fire in the Appalachian Mountains of Kentucky.85 Herbimycin D (10, Fig. 13) was also extracted alternatively from Australian marine-derived Streptomyces sp. (CMB-M0392) near Heron Island (Queensland) and characterized using X-ray and NMR methods.86 In contrast to 10, herbimycins D, H, J, K (Fig. 4 and 13) were isolated solely from the marine-derived Streptomyces sp. SCSGAA 0027 strain.40 In turn, natural ansamycin 11 was isolated from the termite-associated Streptomyces sp. M56 strain (South Africa).87 Thus, this-type ansamycins, being produced exclusively by bacteria strains, are widespread in the land and water environment. Structural analogs of benzoquinone-C15 herbimycins, intermediates in the biosynthesis of herbimycin A, possessing hydroxyls at C(11) and C(15) or at C(11) and C(4) as well as the C(4)–C(5) dihydro system, were isolated from Streptomyces sp. CPCC 200291.89 One of these herbimycins, having in the ansa chain a reduced double bond, was moderate anticancer active in cell line HCT116 (IC50 = ∼20 μM).
Fig. 12 An overview on biosynthetic pathways of autolytimycin (7) and reblastatin (8).
Fig. 13 Structural comparison of benzene-C15 ansamycins with rarely fused ansa chains and benzene cores, i.e. herbimycin H (9), herbimycin D (also called “heronamycin A”, 10), natalamycin A (11) and DHQ6 (12).
Fig. 14 Biosynthetic pathway yielding natalamycin A (11).
Polyketides 7 and 8 were very potent anticancer agents in MDA-MB-231, SKBR3, SKOV3 and PC3 cancer cells, with reduced toxic effects.90–93 Herbimycins J and K (Fig. 4) showed moderate anticancer effects whereas H and D (9 and 10, Fig. 13) exhibited neither any anticancer nor antifungal effects; despite affinity to Hsp90α at comparable level.40 Herbimycin 10, having higher conformational flexibility of the ansa chain showed, in addition, weak antibacterial potency.86 Interestingly, irrespectively on the type of the fused system in DHQ5 (Fig. 4) or DHQ6 (12, Fig. 13) the anticancer potency or inhibitory activity toward Hsp90 were not noted below IC50 = 20 μM.41 These results indicate clearly that flexibility of the ansa bridge, lost in most herbimycins, is required for attractive biological properties. Interestingly, the presence of a five-carbon bridge within 11 was beneficial for the antifungal activity against Pseudoxylaria X802 and Termitomyces T112 cultivar.87 Pseudoverticin B, a novel benzene-C15 ansamycin, was found in the extract of Streptomyces pseudoverticillus YN17707.94 This natural compound, containing 7-membered peroxide bridge joining C(17) and C(18) within the benzene core (analogous to that of 11), arrested the cell cycle of tsFT210 cells at the G0/G1 phase at MIC ∼10 μM. This result was better than that for geldanamycin (MIC ∼20 μM).
The most recognized natural benzoquinone-C15 ansamycin is geldanamycin (Fig. 4), produced by S. hygroscopicus.32 The modified 9 at C(4)–C(5), C(18) and C(21), showing Hsp90 inhibitory activity, was found to be produced by S. hygroscopicus DEM20745, collected from soil near Paraserinthes falactaria trees in Cangkringan, Indonesia.95 A unique 1,2-benzoquinone-C15 ansamycin – herbimycin E (13, Fig. 15) was isolated from the crude extract of Streptomyces sp. RM-7-15.85 Interestingly, the changed substitution pattern of the two carbonyl groups within the quinoid core of 13, does not alter the affinity to Hsp90, compared to geldanamycin. This result is explainable since neither C(18)-carbonyl nor C(19)-substituents are involved in stabilization of geldanamycin congeners at Hsp90 pocket (Fig. 8c).
Fig. 15 Structures of herbimycin E (13) and mccrearamycins B (14) and A (15).
Natural of atypical core–C15 ansamycins
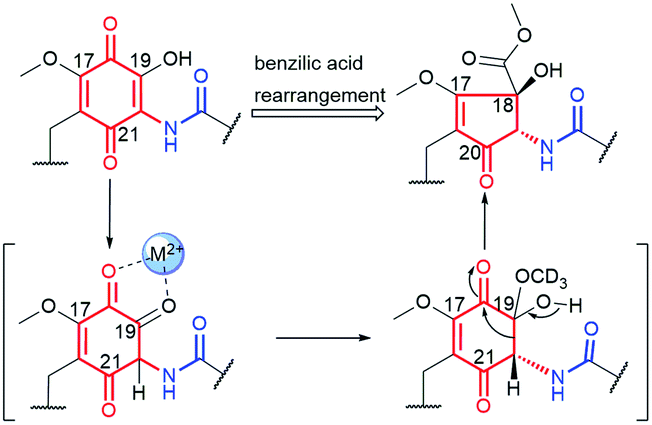
Mccrearamycins A and B (15 and 14, Fig. 15) of a unique cyclopentenone cores and typical C15 polyketide chain, as for geldanamycin, were isolated from coal-mine-derived Streptomyces sp. AD-23-14 (Eastern Kentucky).96 It was shown that the cyclopentenone cores of 14 and 15 can be formed in a result of metal M2+-mediated benzylic acid rearrangement via an ortho-quinone formation step (Fig. 16). This mechanism differs markedly from those of previously described biosynthetic pathways of benzenoid ansamycins. Compounds 14 and 15 showed moderate activity in A549 cancer cells and affinity toward Hsp90α, however, lower than did geldanamycin. In turn, mycothiol-containing compound 15 was found to be more anticancer active than 14. These biological studies together with the other ones concerning structurally simplified geldanamycin benzoquinone-C15 model,97 indicated that for the appearance of the anticancer potency of ansamycins, the suitably functionalized structure of the bridge is much more important than the structure of the core.
Fig. 16 Metal cation (M2+)-mediated benzylic acid rearrangement of 19-hydroxy geldanamycin leading to biosynthesis of atypical core of mccrearamycins 14 and 15.
Natural benzene–C17 ansamycins
Trienomycin-group of ansamycins, similarly as mycotrienin II (called also ansatrienin B) and cytotrienin A, have characteristic three double bond motif at C(4), C(6) and C(8) positions within the ansa bridge, distant from the lactam (Fig. 17). New members of benzene-C17 ansamycins named trienomycin I (16, Fig. 17) and H (17, Fig. 17), being natural derivatives of trienomycynol, were isolated from marine bacteria Ochrobactrum sp. OUCMDZ-2164.98 Their structures were elucidated using 2D NMR and ECD methods. Streptomyces cacaoi subsp. asoensis H2S5 associated with mosses was found also as the natural source of the other novel trienomycins bearing structurally different substituents at C(11) or C(13) (trienomycins 18-K, 19-J, 20-G, and cytotrienin A/21/, Fig. 17).99 Strecacansamycins A (23), B (24) and C (22) displayed in Fig. 18, were extracted from the same source as trienomycins 18–21, i.e. from S. cacaoi subsp. asoensis H2S5.100 Compounds 23 and 24 represent novel type benzenoid-C17 ansamycin scaffold, built on an atypical fused system of 7-, 6- and 5-membered rings (Fig. 18). Absolute configurations of stereogenic centers in strecacansamycins (22–24) were proved by ECD (verified by TD-DFT) and NMR methods. A bioinformatics-based antiSMASH analysis and feeding experiments with bacteria showed activity analogous hybrid PKS-NRPS gene cluster (nonribosomal peptide synthase) in biosyntheses of these novel scaffolds, similarly as for trienomycins or ansatrienins.100,101 The biosynthetic route to 23 and 24 is realized with including of D-Ala and a cyclohexyl moieties in the C(13) or at C(11) tails, oxidation of the p-diphenol moiety, and intramolecular Michael-type addition followed by diene–phenol-rearrangement giving a fused system of the three rings (Fig. 18). In the biosynthesis of 22, glucotransferase is used for the attachment of the L-glucose moiety at C(13) position.
Fig. 17 Novel trienomycins, having triene motif within the ansa chain, obtained from marine (16, 17) or mosses-associated (18–21) bacteria strains.
Fig. 18 Biosynthetic route to novel natural strecacansamycins A–C (22–24) involving alternatively activity of glucotransferase or FAD-dependent reductase.
Medium cytotoxic activity in A549 and K562 cancer cells was noted only for 17, in contrast to inactive 16.98 Generally, novel trienomycins showed even better anticancer effects than has doxorubicin in PC3 and HepG2 cells. Molecular recognition of the most potent 19 congeners at the binding site of inducible nitric oxide synthase (iNOS) explained their very high antineuroinflammatory potencies in BV-2 microglial cells.99 Compound 21, bearing C(11)-tail decorated with cyclopropyl ring, showed potent antiproliferative and proapoptotic activities, attributed to translation inhibition via interfering with eukaryotic elongation factor (eEF).102 This result is in line with the SAR of 23 and 24, where that containing C(13)-D-alanyl tail was less active (IC50 > 50 μM) than respective C(11)-functionalized ansamycin and doxorubicin. In turn, the attached 13-O-L-glucosyl moiety within 22, decreased anticancer activity relative to those of 23. The increased flexibility of the ansa chain in monoenomycins, obtained by the reduction of the two double bonds (Δ4, Δ6) within trienomycin scaffold, contributes to the enhanced anticancer activity in HeLa and MCF-7 cell lines (IC50 < 0.5 μM), relative to typical trienomycins.103 The result that the rigid structure of the ansa bridge and the presence of fused 5/6/7 tricyclic ring system in ansamycins 23 and 24 did not limit their anticancer potencies (IC50 = 0.61–7 μM) suggested clearly an alternative molecular mechanism of action for these compounds, relative to that based on the inhibition of Hsp90.
Another novel group of natural benzene-C17 ansamycins, containing bicyclic or tricyclic core systems are divergolides B, P, O′ and H (25, 26, 27 and 28, respectively; Fig. 19).40,104,105 Divergolides 25 and 28 have (3E)-double bond configuration, in contrast to the (3Z)-configuration found in 26 and 27, as evidenced by X-ray or NMR studies. Furthermore, structural studies indicated different absolute configurations for 26 and 27 than for 25 and 28 at C(6) (Fig. 19 and 20). These ansamycins are produced by the endophyte Streptomyces sp. of the mangrove tree B. gymnorrhiza using a biosynthetic pathway analogous to that later described for mutasynthetic benzene-C16 divergolides.104,105 The biosynthetic pathway for divergolides 25–28 requires an extra incorporation of one ethylmalonyl-CoA and one isobutyrylmalonyl-CoA units into the ansa chain (Fig. 20).104 In turn, the post-DivO stage with the formation of lactone motif in 25–28 involves, the Baeyer–Villiger oxidation by BVMO of the ansa chain, excluding the isobutyrylmalonyl tail at C(6) from the macrocyclic system, and subsequent bicyclization at the participation of the AHBA core (Fig. 20).105
Fig. 19 Novel natural benzene-C17 divergolides.
Fig. 20 Biosynthesis of divergolide H (28) with participation of monooxygenase BVMO and inclusion of an oxygen atom into the ansa bridge.
Divergolide B (25) revealed the best antibacterial potency of all studied at no notable cytotoxicity (IC50 > 10 μM).105 It should be noted here that the name “divergolide O” was used twice in the literature for the two totally different structures, one reported by Lu et al.106 for the benzene-C17 ansamycin 27 (here “divergolide O’ “), and the other by Qi et al.40 for the benzene-C16 ansamycin (“divergolide O”).
5. Mutasynthetic and genetic manipulation strategies opening access to novel functionalized benzenoid scaffolds
Structures of many natural products of promising biological properties are complex and often achieving their systematic modifications within the main pharmacophore portions solely by semisynthesis or total synthesis is challenging.33,107,108 Mutasynthetic approach appeared as a tool for obtaining medically relevant natural products on example of neomycin glyosidic antibiotic congeners, produced by mutant Streptomyces fradiae bacteria strain above 50 years ago.109 Since that time many natural products have been functionalized, in the way not available by a simple semi-synthetic approaches, not only among ansamycins but also among the other pharmaceutically relevant groups of natural products, e.g. lactone macrolides,110 nucleosides,111,112 peptides and macropeptides,113,114 aminocoumarines,115 anthracyclines,116 alkaloids,117 saccharides118 or terpenes.108 Mutasynthesis offers access to untypically-functionalized natural scaffolds, ready for further chemical transformations also in eukaryotic organisms as plants.119 Mutasynthesis (MBS, Fig. 21) is based on the manipulation of biosynthetic pathways via the feeding of mutated microorganisms with different unnatural precursors, called mutasynthons, prepared earlier by a simple semisynthetic transformations. In a mutasynthetic approach unnatural mutasynthons do not have to ‘compete’ with the natural precursors about access to key enzymes, because the latter are excluded from the early-stage of biosynthetic pathway by gene deletion, insertion or alteration, contrary to the other less efficient approach called precursor-directed biosynthesis (PDB, Fig. 21).18,108 In a PDB approach the unnatural precursor is added to the fermentation broth, natural and unnatural precursors are processed simultaneously by enzymes, and then different natural products are produced as a mixture, which should be separated. The biosynthetic pathways yielding secondary metabolites in nature are multistep and linear sequences of transformations of the main scaffold, with extra tailoring steps of peripheral groups. The inhibition or stimulation of biosynthetic transformations at different stages via gene manipulations, create opportunity to subject the structurally useful isolated intermediates to ‘external’ semi-synthetic modifications.120 Such mixed chem-bio or bio-chem approaches in different sequences enable the access to unique hybrid structures among ansamycins, which are unavailable or difficult to obtain exclusively by semi- or total-synthetic chemical transformations (Fig. 21).18 A new type hybrid scaffolds of natural products are also often built by hybrid-type microorganisms, i.e. recombinant strains by a combinatorial approach using engineered biosynthetic pathways.121,122 This approach, using hybrid wild-type strain, exploiting enzymes of the two independent biosynthetic pathways, can be aimed into atypical fusion of structural portions as e.g. the core and the ansa part belonging to ansamycins of different types (Fig. 21). Mixed approaches, where one organism (late-stage blocked mutant) produces intermediate for semisynthetic transformation, and next semisynthetic product is processed by the second microorganism (early-stage blocked mutant), contributes to the high structural diversity of benzenoid ansamycins, as reported by Taft et al.120 In turn, site-specific mutagenesis offers also the incorporation of atypical structural motifs into the scaffold by changing the molecular recognition specificity of enzymes taking part in biosynthesis, by alterations within the binding pocket or by subdomain swapping.122
Fig. 21 Exemplary differences between strategies of ansamycins functionalization via precursor-directed biosynthesis (PDB), mutasynthesis (MBS) and combinatorial approach, when referred to simple biosynthesis; a1 and a2 are enzymes of wild-type strain “a”; “b1” is enzyme system used by the other wild-type strain “b”; a1* and a2* are enzymes of mutant strain “a*”.
Recently, mutasynthesis, has been strongly developed towards linearly combined applications of microorganisms of selectively blocked gene clusters and semisynthetic methods, enabling multidirectional modifications of original ansamycin framework. Such an combined approach is highly desired to obtain atypical and systematic ansamycin scaffold functionalizations (Fig. 21, substituent X), enabling for further transformations (Fig. 21, substituent Y), and required to draw conclusions about biological importance of particular functional groups in biomolecules, i.e. comprehensive SAR studies. The mixed chem-bio concepts, with the pivotal role of mutasynthesis, were described by Kirschning et al.8,18,123
Benzene- and benzoquinone-C9 ansamycins obtained by genetic manipulations
Few ansamycin natural products having an C9 ansa chain and benzene or benzoquinone cores have been obtained so far. Novel natural products of this group, called aminoansamycins 29–33 (Fig. 22), were obtained by activation the cryptic ansamycins pentaketide gene cluster aas in Streptomyces sp. S35, isolated from soil (Kunming, Yunnan, China).124 The AasD hydroxylase takes part in the biosyntheses of 29–33, which is analogous to Nam7, active in the neoansamycins biosynthesis. Ansamycins 30–33 contain nonproteinogenic AHBA and anthranilate motifs attached to the C(9) position of their ansa chains (Fig. 22). Structures of new aminoansamycins, confirmed by ESI MS, 2D NMR and X-ray studies, revealed the presence of the trans-lactam group. The most active derivative, 30 decorating with an anthranilate motif, revealed quite potent antiproliferative activities (IC50s = 7–10 μM).124 The absence of the anthranilate or AHBA motifs at C(9) (29) decreased potency at least by 2-fold or even contributed to the loss of activity, similarly to the oxidation of the ansamycin core (32, 33).
Fig. 22 Biosynthetic pathway of aminoansamycins B, D, E, F and G (29–33) involving activity of AasD hydroxylase, similar to that taking part in biosynthesis of neoansamycins.
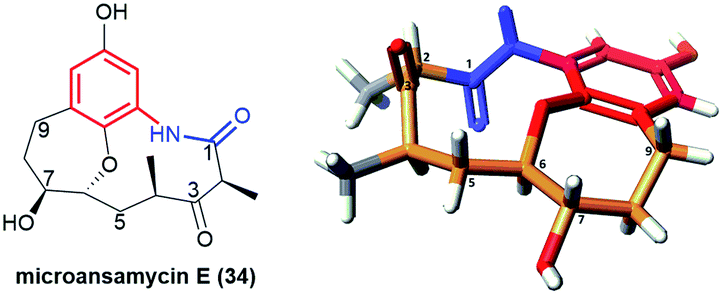
Pentaketides, named microansamycins E (34, Fig. 23) and F of the benzene-C9 subgroup, were obtained via overexpression of the specific regulator gene mas13, which was shown to activate the cryptic gene cluster in Micromonospora sp. HK160111mas13OE mutant strain.42 The biosynthetic pathway of these microansamycins occurs via step with the formation of the C(6)/C(7) epoxy intermediates. Unfortunately, these ansamycins, which have a “flattened” shape due to the presence of oxepane ring and high rigidity of the ansa chain, did not exhibit any antioxidant or antimicrobial activities.42
Fig. 23 Structure of microansamycin E (34) and its X-ray model (CCDC 1555915)42 visualized by Scigress (EU 3.1.8), Fujitsu.
Cyclohexa-2-enone-C9 and cyclohexa-2,5-dienone–C9 ansamycins obtained by genetic manipulations
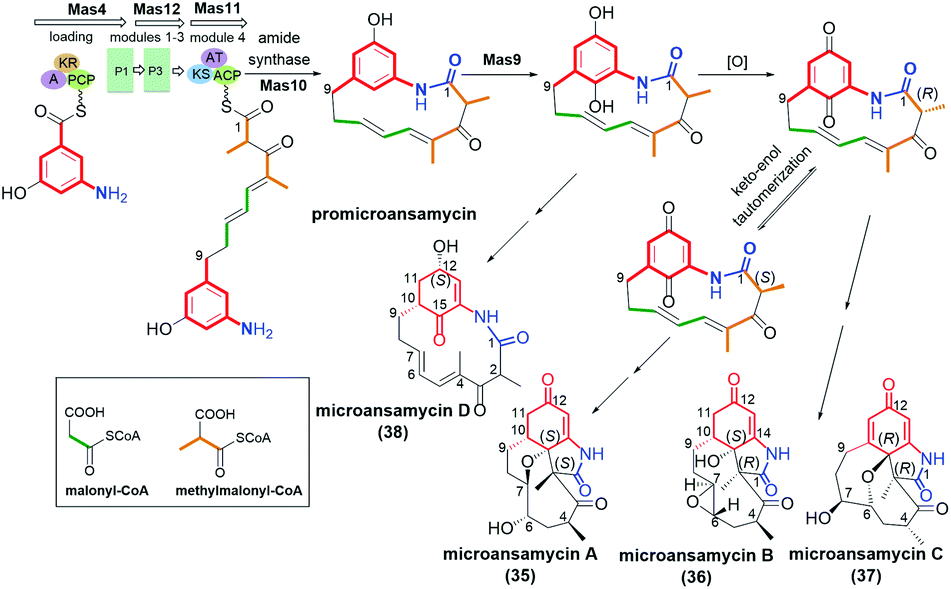
Ansamycins having partially saturated cores, i.e. microansamycins A–D (35–38, Fig. 24), are produced by Micromonospora sp. mutant strain after activation of mas cryptic gene cluster, similarly as for microansamycins E and F.42 Microansamycins 35–38 and those of the benzene-C9 group (34, Fig. 23), showed structural diversity, as indicated by X-ray and NMR studies of these secondary metabolites. High flexibility of promicroansamycin post-PKS modifications contributed to their some biosynthetic plasticity, as shown in Fig. 24. These modifications are based on tailoring the core substitution pattern, fusion of the core with the ansa bridge, and the inversion of configuration at C(2) via keto–enol equilibrium. Microansamycin D (38) of the highest flexibility of the ansa chain, exhibited moderate antioxidant activity.42
Fig. 24 Biosynthesis of microansamycins A–D (35–38) via activation of Mas cryptic gene cluster in Micromonospora sp. mutant strain.
Benzene-C15 and benzoquinone-C15 ansamycins obtained by genetic engineering and mutasynthesis
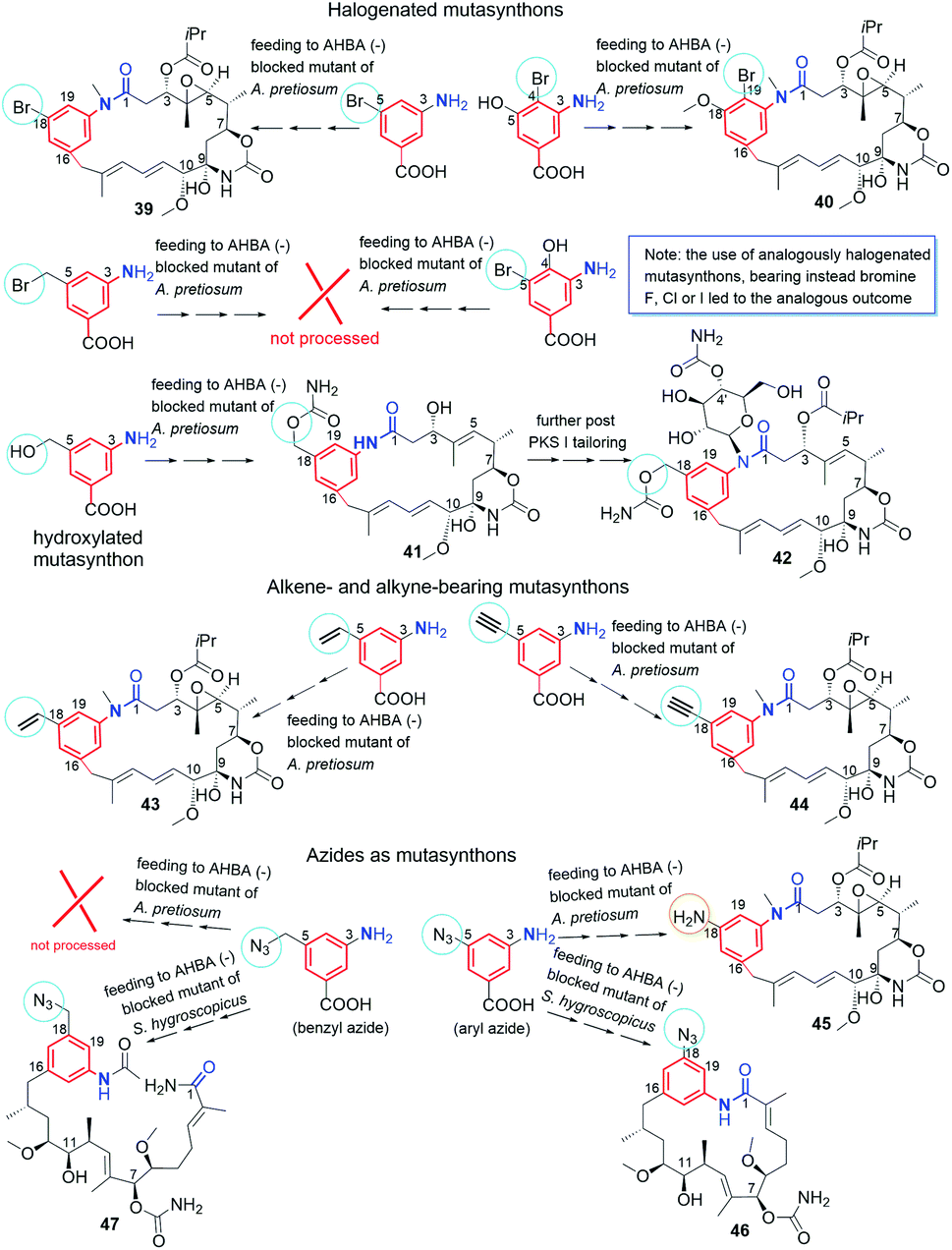
Supplementation of AHBA(−) blocked Actinosynnema pretiosum mutant strain with a number of structurally diverse aminobenzoic acid congeners (mutasynthons) yielded novel ansamycin derivatives (39–47, Fig. 25),125–128 bearing new functional groups as: alkyne, azide, vinyl, allyl, halogen, and hydroxyl-methylene at C(18) or C(19), convenient for further semisynthetic structural alterations, discussed further in the semisynthetic part. The presence of halogen at the core of the mutasynthon structure did not affect the ability to processing by PKS gene machinery into expected mutaproducts, although this process was dependent on the core substitution pattern (39, 40; Fig. 25).125,128,129 The efficiency of A. pretiosum gene machinery related to processing particular halogenated mutasynthons decreased with increasing size of the halogen F ∼ Cl > Br > I.125,128 Furthermore, the benzyl bromides or benzyl chlorides as well as mutasynthons with halogen at C(5) and phenol at C(4) in the AHBA-like core were not converted into respective ansamitocin-like derivatives (Fig. 25). The exchange of halogen and OH group between positions C(5) and C(4) within structure of mutasynthon, respectively, yielded successfully mutaproduct 40 (Fig. 25). In contrast to the above, supplementation A. pretiosum ΔAHBA mutant with benzyl alcohol mutasynthons contributed to successful formation of 41 and 42, as fermentation products. Participation of N-glucosyltransferase of A. pretiosum and UDP-glucose as a donor of glucosyl unit in post PKS I tailoring resulted in an extra attachment of the saccharide at the nitrogen of the lactam (42, Fig. 25). Structures of 41 and 42 suggested involvement of asm21 gene of A. pretiosum in multiple carbamoylations, not only performed at C(7)-hydroxyl of the ansa bridge, but also within the methylene-hydroxyl at C(18) of the core or at C(4′) of the saccharide (41, 42; Fig. 25).78 AHBA-like mutasynthons, showed in Fig. 25, were successfully processed by A. pretiosum or S. hygroscopicus ΔAHBA-blocked mutant strains, introducing different types of ansa bridges, i.e. those of ansamitocin-like or reblastatin-like (46, 47).127 In feeding experiments with ΔAHBA-blocked A. pretiosum and mutasynthon bearing aryl azide, an unusual enzymatic reduction of –N3 into –NH2 was noted (45, Fig. 25).127 When the aryl diamine mutasynthon was used, i.e. 3,5-diaminobenzoic acid, even no traces of respective anilino ansamitocin congener were detected as metabolite of A. pretiosum ΔAHBA-blocked strain. The explanation of this result was a high polarity of the aryl diamine mutasynthon, reducing ability to overcome the natural cell barriers and thereby decreasing accessibility of this substrate for the PKS I gene machinery. In turn, feeding of AHBA(−) S. hygroscopicus K390-61-1 with benzyl azides contributed to N-acylation of the core instead macrolactamization and yielded an open-ansa chain secondary metabolite (47, Fig. 25), formed as detoxification response.127 In contrast to this result, feeding of A. pretiosum ΔAHBA-blocked strain with benzyl azide resulted in a lack of processing of such mutasynthon (Fig. 25). Successful mutasynthetic attempts with assembling of the azide-decorated benzenoid core with the C15 ansa bridge of reblastatin (hybrids of ansamycins) prompted scientists to further explore of ansamycins functionalizations in the regard of their better matching to the molecular target in cells.
Fig. 25 Classical mutasynthesis with (−)AHBA blocked A. pretiosum and S. hygroscopicus strains and a set of selected, different mutasynthons yielding ansamitocin-like (39–45) and geldanamycin-like (46) scaffolds of high utility to further semisynthetic transformations (e.g. click as dipolar cycloaddition or Heck reaction of C–C coupling) as well as this of an open-chain structure (47), called seco-proreblastatinamide.
Novel fluorine-containing mutasynthons were successful precursors of C(17)-, C(18)-, C(19)- and C(21)-fluorinated reblastatin analogs of a very high binding affinity to Hsp90, where KDs(Hsp90) were in the range 0.5–57 nM (48–51, Fig. 26).57,130 The S. hygroscopicus ΔAHBA-blocked strain was effective in processing even multiple-fluorinated mutasynthons at different positions of the benzene ring, yielding natural products of ansa bridges as that in reblastatin (Fig. 26). Surprisingly, diamino (anilino)-mutasynthons, being earlier not accepted as substrates by A. pretiosum ΔAHBA blocked strain, were successfully transformed into reblastatin-like analogs with amine at C(18), when subjected mutasynthesis carried out by S. hygroscopicus K-390-61-1 ΔAHBA strain (52–56, Fig. 26).130 It should be noted that an extra phenol groups at C(17) or/and C(21) appeared in structures of these mutaproducts (53–55) in a result of enzymatic regioselective oxidations, performed by GdmL and GdmM, respectively. Furthermore, at this variant of mutasynthesis, the two-types non-quinone mutaproducts, the C(4)–C(5) saturated (52, 53) or C(4)–C(5) unsaturated (53, 55, 56) within the ansa chain, were isolated (Fig. 26). Kirschning team showed also the possibility of assembling alkyne/propargyl-containing core with the ansa bridge of reblastatin (57–60), using S. hygroscopicus K-390-61-1 ΔAHBA mutant strain (Fig. 26).130 It should be noted that alkyne/propargyl-containing mutaproducts were successfully processed by A. pretiosum ΔAHBA strain yielding ansamycins of ansamitocin-like bridges.129 Hybrid mutasynthetic strategy with the use of an early and late-stage blocked bacteria strains contributed also to obtaining of stereoisomeric ansamitocin-like mutaproducts 61–63 (Fig. 27).131,132 The first processing of AHBA by a late-stage blocked A. pretiosum HGF073, at chlorination and carbamoylation steps, gave proansamitocin as predominant product, and 10-epi-proansamitocin and 63 as minor products in a post-fermentation mixture. Proansamitocin after semisynthetic derivatization (reduction and esterification), was subjected processing by an early-stage blocked S. hygroscopicus yielding product 61 of the absolute configuration (9R,10R) and bearing 1,3-amide-enol and C(7)-carbamate. In turn, direct processing of 10-epi-proansamitocin by an early-stage blocked S. hygroscopicus led to formation of product 62 of absolute configuration (9S,10S) and containing C(7)/C(9) cyclic carbamate moiety.131 Formation of product 63 with blocked A. pretiosum HGF073 is a clear evidence of taking part in this process nonspecific epoxidase, which contributed directly to the formation of C(14)-hydroxyl and indirectly α,β,γ,δ-unsaturated ketone within the ansa bridge (Fig. 27).131 Furthermore, the mutasynthetic strategy with 3-amino-5-chlorobenzoic acid as mutasynthon and early-stage blocked (ΔAHBA) or late-stage blocked (Δasm19-acyltransferase) A. pretiosum mutant strains contributed also to the formation of unique C(7)-deoxygenated mutaproducts, being structural analogous to 61, despite lacking C(7) carbamoyl group.132 Formation of these deoxygenated ansamycins was postulated as a result of C(7)/C(8) elimination of water, followed by the two reductions of the intermediate enone in mutasynthetic pathway. Most of the above examples showed that the approach based on transformation sequence of the two different bacteria strains, that are blocked at different stages, with/or without further support of semisynthesis, can be a source of novel diastereoisomeric ansamycins, valuable for SAR studies. The efficient obtaining of benzene-C15 ansamycins, simultaneously modified with bulky substituents at C(17) and C(21), is difficult using solely mutasynthesis, with the exception of cases where fluorine- or diamine mutasynthons were used (Fig. 26). Hence, new hybrids having a geldanamycin-like substitution pattern of the core and ansamitocin-like ansa chain were obtained using analogous to the above-mentioned combined approach, but at different sequence of semisynthetic and mutasynthetic transformations (64, 65; Fig. 28).133 These hybrids with attached the two relatively bulky –OiPr substituents to the core, displayed atropisomerism (forms 65a and 65b, Fig. 28), due to the steric hindrance between the isopropoxy groups of the core and groups of the ansa chain. Because of the dynamic conformational changes, the NMR spectra of one of atropisomers lack the ansa chain proton and carbon atom signals, which is often phenomenon for benzenoid-C15 ansamycins and serious problem at their spectral characteristics. The crowded structure of the core also contributed to lack of inhibition the molecular target – chaperone Hsp90α, so the altered mechanism of action is probably realized in these cases.
Fig. 26 Classical mutasynthesis with (−)AHBA blocked S. hygroscopicus strain and mutasynthons bearing fluorine, amine and alkyne functional groups yielding reblastatin-like natural products (48–60) of high semi-synthetic utility (57–60) or attractive biological properties (48–56).
Fig. 27 Stereoisomeric mutasynthetic products 61–63, belonging to ansamitocins group, obtained using late-stage/Δasm12 (chlorination) and Δasm21 (carbamoylation)/and the early-stage ΔAHBA blocked mutants of S. hygroscopicus and A. pretiosum.
Fig. 28 Atropisomeric mutasynthetic products (a, b), belonging to ansamitocins group and obtained using the late-stage/Δasm12 (chlorination) and Δasm21 (carbamoylation)/and the early-stage ΔAHBA blocked mutants of S. hygroscopicus and A. pretiosum. On the right probable models of atropisomeric 65a and 65b ansamycins, both containing cis-lactam moiety due to the steric repulsions between groups of the ansa bridge and the core, visualized by Scigress (EU 3.1.8), Fujitsu.
The biological potency of mutaproducts 39–60 (Fig. 25–27), were studied in different cancer cell lines (Table 1). Despite these mutaproducts were good starting points to further structural semisynthetic optimizations, aimed toward better binding with targets in cells, the antiproliferative potencies some of them were at the level, even comparable to those of GDM and AP3. Compounds 39 and 40, containing bromine at C(19) of the core and ansa chains like ansamitocins, revealed comparable or even higher anticancer potencies than GDM and AP3. Toxicities of 39 and 40 were not investigated in normal cells so the selectivity of these derivatives is unknown. The introduction into the core or at the nitrogen of the lactam more bulky substituents decreased toxicity in normal cell lines at the expense of markedly limited anticancer activity (41, 42; Table 1). Surprisingly, assembling of C(18)-alkyne containing core with the ansamitocin ansa chain (44, Fig. 25) was a much more biologically beneficent (Table 1) than the assembling of the this-type core with the reblastatin-like ansa chain (57, Fig. 26). It should be noted that the replacement of acetylene with propargyl substituent at C(18) (59, 60; Fig. 26) or with azide (46, Fig. 25), decreased anticancer potency, irrespectively of the presence/absence of an extra phenol group at C(17) and the type of the ansa chain (Table 1). Mutaproduct 44, showing lower toxicity than AP3 in normal cells and the attractive anticancer potency in various cancer cell lines (IC50s = 0.12–1.3 nM), seemed to be convenient intermediate for further biological optimizations. Fusion of the C(18)-amine core with the ansamitocin ansa bridge (45, Fig. 25) also was an successful way for designing anticancer benzene-C15 congeners, active at nM level (Table 1).
Table 1The range of anticancer activity (IC50s [nM]) together with the IC50 values in selected PC-3 and SKOV-3 cancer cell lines of novel mutaproducts 39–60
Novel type of benzene-C15 ansamycins, called 5,10-seco-neoansamycins, bearing ansa chains mimicking those of naphthalenoid group, were obtained by genetic manipulations with Streptomyces sp. LZ355 (66–72, Fig. 29).134 Shen and co-workers designed a convenient pathway leading to benzenoid ansamycin scaffolds via disruption of nam7 hydroxylase, required to cyclization of the core belonging to naphthalenoid ansamycins, as e.g. neoansamycin A. Ansa chains of these seco-neoansamycins were different not only regarding the presence of α,β-unsaturated ketone (70) or 1,3-ketoenol (67, 68, 71) but also the presence/absence of an extra heterocyclic ring (69, 72), as evidenced by NMR and X-ray (Fig. 29).134 Newly obtained compounds showed moderate antiproliferative effects (IC50s = 20–30 μM).134 The most active ansamycin in SW480 cancer cell line (human colorectal adenocarcinoma), was 70 (IC50 = 9.5 μM) bearing the two α,β-unsaturated ketone motifs. In turn, the presence of glucose attached to the core contributed to complete inactivity of 68. Thus, the control of nam7 as a critical point of biosyntheses of naphthalenoid ansamycins can be successfully applied in future bioengineering novel benzenoid structures decorated with naphthalenoid-like ansa bridges of antiproliferative potency.
Fig. 29 Switching between biosynthetic pathways of naphthalenoid (neoansamycin A) and benzene-C15 (66–72) natural products with the use of genetic manipulation concerning activation or disruption of Nam7 hydroxylase.
Wu and co-workers reported that supplementing S. hygroscopicus N02Z-0421 with L-Met contributed to the formation of methanethiol followed by biosynthesis of sulfur-enriched ansamycins, i.e. 17,19-dimethylthioherbimycin A of moderate anticancer effect in HepG2 cells (IC50 ∼18 μM).135 In turn, the two new anticancer active C(4)-epi-hydroxy analogs of geldanamycin were produced by the S. hygroscopicus 17997 strain with activated cytochrome P450 oxidase.136 These two stereoisomeric new ansamycins showed comparable activity in HepG2 (IC50s ∼ 10 μM).
Mutasynthetic atypical core-C15 ansamycins
Great advance in the field of mutasynthetic designing of novel benzenoid ansamycin inhibitors of Hsp90 was idea to replace benzene/benzoquinone cores with the heterocyclic moieties (Fig. 30).137 Feeding of S. hygroscopicus mutant with heterocyclic mutasynthons, functionalized analogously to AHBA with carboxylic and amine groups, yielded ansamycin hybrids with structurally diverse heterocyclic cores (73–76, 78; Fig. 30) and alternatively open ansa bridge structures (77; Fig. 30).137 These experiments revealed, however, that not every AHBA-mimicking heterocycles can be processed by S. hygroscopicus ΔAHBA blocked mutant (Fig. 30). Heterocyclic mutasynthons were well-tolerated by PKS I gene machinery as long as they were not substituted with an relatively large groups as CH3, CF3, OCH3, OH, or Br (Fig. 30). Mutasynthon containing benzotriazole was processed up to the seco-derivative formation (77) and then its transformation was hampered at the stage of amide synthase, due to the nitrogen sp2 hybridization of amine. It was also demonstrated that the substitution pattern of the thiazole ring is crucial for the suitability of such blocks as substrates of PKS I. All heterocyclic-C15 ansamycins did not undergo any advanced post PKS I tailoring as e.g. oxidation of the core. Interestingly, thiophene-C15 ansamycin 73, obtained with a very good yield, revealed inhibitory activity toward different chaperones as: human Hsp90α and Hsp90β as well as bacterial X. campestris and H. pylori, at selectivity higher toward the latter ones than the former ones.137 Considering that the alterations of the core with heterocycles were beneficent for inhibition of bacterial Hsp90s, it seems that this new-type natural products of atypical cores can be considered as potential antibacterial agents, despite exhibiting simultaneously anticancer potency on μM level. These experiments also indicated that the ansa chain structure can be much more important than the structure of the core at molecular recognition with the target in cells.
Fig. 30 Attempts at obtaining novel atypical heterocyclic-C15 ansamycins (73–76, 78) via mutasynthetic approach with heterocyclic mutasynthons and polyketide 77.
Benzene–C16, benzene–C17 and benzoquinone–C17 ansamycins obtained by mutasynthesis and genetic manipulations
Benzene-C16 divergolide O (79, Fig. 31) and benzene-C17 divergolide P (80, Fig. 31), were produced by mutant Streptomyces sp. W112OE bacteria strain via an overexpression of div8.106 These natural products, belonging to the earlier discovered group of divergolides by Hertweck group,104,138 contain an oxygen atom as a part of the ansa chain and bicyclic spiroketal motif, but differ to each other with regards to configurations of C(3)C(4) and stereogenic centers at C(6) and C(15/16), as well as the length of the ansa chain and the structure of C(6)-substituent. Taking into regard the structural features needed for full functionality of ansamycins, the rigidity of ansa chains belonging to 79 and 80 strongly reduced their anticancer effects, in comparison to the active benzenoid ansamycins.40,106
Fig. 31 Benzene-C16 divergolide O (79) and benzene-C17 divergolide P (80) obtained via an overexpression of div8 in Streptomyces sp. W112OE bacteria.
Natural ansamycin with the lactam distant from the core by one carbon atom (81, Fig. 32), and containing ansa chain consisting of sixteen carbon atoms, was obtained by feeding of ΔAHBA-blocked S. hygroscopicus (K390-61-1) with 3-aminomethyl-5-hydroxybenzoic acid as mutasynthon.84 In turn, this mutasynthetic approach with 3-amino-5-hydroxymethylbenzoic acid mutasynthon unexpectedly yielded also macrolactones 82–85 (Fig. 32), structurally analogous to 81. Interestingly, 3-amino-5-hydroxymethylbenzoic acid mutasynthon and mutated A. pretiosum were employed also for obtaining classical ansamitocin-like derivatives 41 and 42 (Fig. 25), where no macrolactonization process was detected. It was concluded that in addition to formation of macrolactams, an unusual formation of the macrolactones at seco-proansamycin stage, is a result of the presence two possible conformers (rotation around C15–C16 bond), competing within the active site of amide synthase.84 Unfortunately, in contrast to the lactam-containing macrocycles, novel macrolactones 82–85 displayed low or no binding affinity to Hsp90 and lack of antiproliferative properties.84 This result underlined the importance of the lactam moiety of ansamycins at interactions with the target Hsp90. The 81–85 structural analogs, but containing C15 ansa chains and 1,2,3-triazole-cores, obtained with the help of click chemistry, showed neuronal biomarkers features.139
Fig. 32 Mutasynthetic benzene-C16 macrolactam (81) and benzene-C16 macrolactones (82–85).
A mutasynthetic approach with the mutant Streptomyces seoulensis IFB-A01 ΔCHC bacterial strain and cyclic mutasynthons yielded novel benzene-C17 dienomycins (86, Fig. 33), each containing fused 6-6-5 membered polycyclic system with the ansa chain, which were analogous to other isolated ansamycins, i.e. trienomycins (e.g.22, Fig. 18).140,141 The transformation of trienomycins into dienomycins at post-PKS I stage was evidenced experimentally as cascade consisting of: oxidation of the core at C(23) followed by [4 + 2] intramolecular Diels–Alder involving double bond of the ansa chain.140 Interestingly, the late-stage blocked mutant strain S. seoulensis IFB-A01 ΔCHC supplemented with less bulky mutasynthons, as e.g. three-, four- and five-membered cycloalkane carboxylic acids, activated an extra α,β-desaturase contributing to formation of an extra double bond in mutaproduct at the ring of C(11) tail (86, 89). Furthermore, a more bulky mutasynthons, containing 6- or 7-membered systems of carboxylic acids, were not processed by the mutant strain, irrespectively on the presence/absence of the heteroatom in the mutasynthon ring. Dienomycins of 86 type exhibited markedly lower anticancer potency than naturally biosynthesized trienomycins, but instead were good inhibitors of IL-6 production in RAW264.7 cells.140 Other peripheral groups modifications of ansatrienins, involving the incorporation of C(3)-hydroxyl/methoxyl groups at the expense of the lack of C(11) and C(13) substituents, were obtained via deletion of tailoring genes from the asc cluster of Streptomyces sp. XZQH13 mutant strains.142,143 Heterocyclization of the core benzene-C17 ansatrienins (87–89, Fig. 33) was also noted at mutasynthesis with Streptomyces sp. XZQH13 ΔastC strain and was found as a result of overexpression of LAL family regulator gene astG1.143,144 Unfortunately, most of this-type ansamycins were obtained in only small amounts, and therefore only partial structural characteristic was performed (HR-MS) without any biological assays.
Fig. 33 Mutasynthetic benzene-C17 dienomycin (86) and trienomycins-C17 of heterocyclized cores (87–90).
6. Semisynthetic modifications and biological properties of benzenoid ansamycins
Natural benzenoid ansamycins, especially congeners of geldanamycin (benzoquinone-C15) and ansamitocin-like (benzene-C15), despite their high and broad anticancer activities they often exhibit toxic effects in healthy cells, being the main obstacle in medical applications. Hence, many attempts at semisynthetic modifications of natural ansamycin scaffolds or their earlier-functionalized novel ‘unnatural’ natural scaffolds of mutaproducts, were aimed towards decreasing toxicity at conserved useful anticancer properties, in most cases.
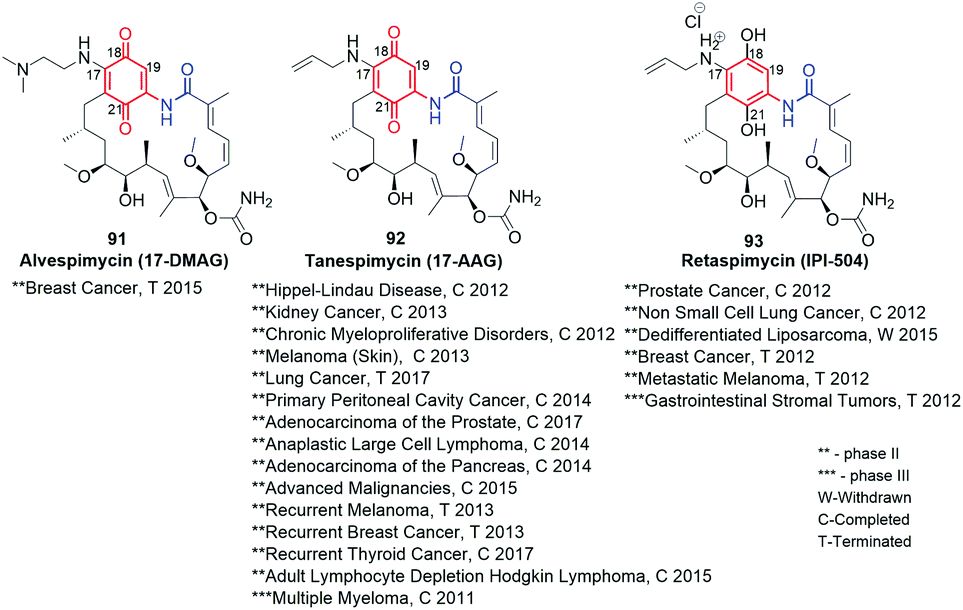
Despite the high anticancer activity of geldanamycin at submicromolar levels in SKBR3, PC3, U87, A549 and SKOV3,32,46 its application in therapy has been hindered (clinical trials terminated at phases I/II) due to toxic effects on healthy cells (hepatotoxicity) and low water solubility and bioavailability.145 The presence of a 1,4-benzoquinone core implicates possibility of 1,4-Michael conjugate addition/aromatization/oxidation cascade with incorporation of various nucleophiles at the C(17) or/and C(19) positions. This reactivity of geldanamycin towards glutathione, was used to explain the relatively high toxicity of this-group compounds.46,146,147 Semisynthetic C(17)-amine derivatives of geldanamycin, also in the reduced form of salt (91–93, Fig. 34), showed promising properties against various forms of cancer at reduced toxic effects, at different stages of clinical trials. In turn, earlier modifications of the ansa chain groups decreased in most cases the anticancer effects as a consequence of rigorous steric demands (tight binding, Fig. 8) at the ATP-binding pocket of chaperones Hsp90. Thus, the attractive anticancer properties of 91 (17-DMAG), 92 (17-AAG) and 93 (IPI-504) together with predominantly poor biological outcomes of structural alterations within the ansa bridge encouraged scientists to obtain and biological testing hundreds of new simple semisynthetic analogs of geldanamycin at C(17) or/and C(19), which were reviewed by Kirschning et al. up to 2013.32
Fig. 34 Clinically studied semisynthetic C(17)-geldanamycin derivatives with oxidized and the reduced the core.
Maytansinoids as ansamitocins (benzene-C15 ansamycins) showed even higher anticancer effects than geldanamycin derivatives but the toxicity in healthy cells was also a serious problem like for the latter. In contrast to geldanamycin derivatives, the modifications of maytansinoids within as well the core as the ansa bridge were better tolerated by their targets (tubulins, Fig. 8) without essential loss of biological activity. Hence, the semisynthetic modifications of these both structural parts of maytansinoids (natural, mutaproducts) were intensively studied up to 2008, as reported by Kirschning et al..33 Recent clinical studies of the most promising ansamitocins and their semisynthetic sulfur derivatives as their bioconjugates with antibodies has begun new era in designing of effective anticancer ansamycins (Fig. 35). Kadcyla (94), based on DM1 ansamitocin bioconjugate with has been approved by FDA against HER-2 positive breast cancer in 2013.148 Other bioconjugates of ansamitocins via expanded C(3)-substituent and bridged with Cys (95, Fig. 35) or Lys (96, Fig. 35) with antibody, are intensively studied in the last decade indicating usefulness of this drug-delivery strategy for anticancer therapy.149,150 In turn, mutasynthetic benzene-C15 reblastatin (Bioteca, UK; Fig. 26), being as potent inhibitor of Hsp90 as autolytimycin and geldanamycin, revealed very high anticancer activities.32 In order to avoid toxic effects such as those displayed by benzoquinone ansamycins, non-quinone reblastatin-like mutaproducts were excellent starting point for further semisynthetic transformations.130 Alternatively, the other strategy to limit the toxic effects was realized with the attachment to the benzoquinone core of ansamycin the C(17)- and/or C(19)-functional arms.
Fig. 35 Approved drug (94) and clinically studied semisynthetic C(3)-ansamitocin bioconjugates with antibodies (Y) (95; 96), where conjugation was obtained via participation of the amine of Lys or thiol of Cys of the antibody.
Semisynthetic modifications within the core of geldanamycin yielding new benzoquinone-C15
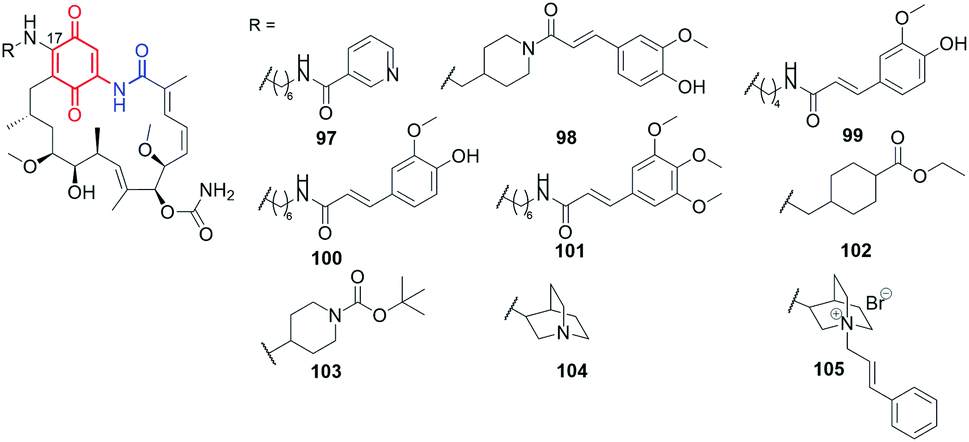
Using the lability of the C(17) methoxyl group at the core, new semisynthetic diamine analogs of geldanamycin, bearing at the C(17)-arms: aliphatic-heterocyclic (97, Fig. 36), heterocyclic-cinnamyl (98, Fig. 36), aliphatic-cinnamyl (99–101, Fig. 36)151 were obtained by Shen et al.151 These derivatives were tested against MDA-MB-231 cancer cells and showed either comparable or even higher anticancer activities with decreased hepatotoxicity compared to the clinically considered 92 (17-AAG) (Table 2). The substitution pattern of the cinnamoyl group as well as the length and basic nature of the diamine linker were crucial for the improved water solubility and the best anticancer effects (Table 2). A binding model of the 100-Hsp90 complex showed that the long C(17)-arm is directed toward a “new” region and is involved in π–π interaction with the amide group of Gln123 of Hsp90.152 The most active both in vitro and in vivo (mice xenograft model) 101 showed better inhibitory activity toward Hsp90α (Table 2) then did 92 (IC50(Hsp90) = 0.78 μM).151
Fig. 36 Semisynthetic geldanamycin derivatives bearing within the introduced C(17)-arms linkers of different length and cinnamoyl or heterocyclic end-motifs.
Table 2Anticancer and antiviral (HCV) activities and the toxicity (HDF) for geldanamycin (GDM), reblastatin and 101–108 C(17)-amine congeners
Compound
[μM]
GDM46,151,154
IC50(MDA-MB231) = 0.05
IC50(U87) = 0.81
IC50(SKBR3) = 0.87
IC50(A549) = 0.99
IC50(SKOV3) = 0.94
IC50(HDF) = 2.13
IC50(PC3) = 0.73
IC50(HCV) = 0.0048
Reblastatin46
IC50(SKBR3) = 0.6
IC50(PC3) = 0.018
IC50(SKOV3) = 0.125
101151
IC50(MDA-MB-231) = 0.19 ± 0.02
10246
IC50(SKBR3) = 0.59
IC50(U87) = 0.62
IC50(SKOV3) = 0.55
IC50(A549) = 0.59
IC50(PC3) = 0.67
IC50(HDF) = 2.15
10346
IC50(SKBR3) = 0.077
IC50(U87) = 0.084
IC50(SKOV3) = 0.083
IC50(A549) = 0.077
IC50(PC3) = 0.19
IC50(HDF) = 2.55
10449
IC50(MDA-MB231) = 0.14
IC50(SKOV3) = 1.16
IC50(MCF7) = 0.09
IC50(PC3) = 1.47
IC50(HeLa) = 1.06
IC50(U87) = 1.08
IC50(HepG2) = 1.25
IC50(A549) = 0.94
IC50(CCD39Lu) = 0.87
IC50(HDF) = 3.22
IC50(SKBR3) = 1.49
10549
IC50(MDA-MB231) = 4.66
IC50(SKBR3) = 2.02
IC50(MCF7) = 2.31
IC50(SKOV3) = 2.17
IC50(HeLa) = 8.14
IC50(PC3) = 2.05
IC50(HepG2) = >10
IC50(U87) = 2.26
IC50(CCD39Lu) = >10
IC50(A549) = 2.06
IC50(HDF) = 3.81
107154
IC50(HCV) = 0.00240
108155
IC50(MCF7) = 0.4 ± 0.05
IC50(GIST48) = 1.6
IC50(A549) = 0.1 ± 0.05
IC50(WI38) = 4.5
An analogous C(17) congeners were obtained by 1,4-Michael conjugate addition/aromatization/oxidation cascade (102–104; Fig. 36) and were found to be better inhibitors of Hsp90 (KDs 0.8–0.9 μM) than geldanamycin, reblastatin or 92.46,49 Postulated binding modes of these derivatives revealed key stabilizing interactions with K43 of Hsp90 (K58, in human ortholog). A very good affinity of 102–104 to the target in cells was in line with high activities against several cancer cell lines, even comparable or better when referred to standard compounds (Table 2). Compound 103 of lipophilicity clogP ∼3, aside from superior anticancer effects in SKOV-3 and A-549 cell lines, was also a more selective than GDM and 92, as indicated by high selectivity indices (SIs > 30).46 Basic derivatives as those bearing quinuclidine 104 showed also very good anticancer effects but at higher cytotoxicity in healthy cells. In turn, transformation of 104 into its salts, as e.g.105, increased water solubility, similarly as for 93 (Fig. 34), and decreased toxicity in normal cells at conserved anticancer effects.49 The use of 105 as mixtures with potentiators as polyethylenimine and doxorubicin enhanced anticancer activities at the expense of increased toxicity. Studies of these benzoquinone-C15 derivatives demonstrated lipophilicity on the level clogP = 2–3, which rather favors high potency, with a relatively increased toxic effects. In turn, well-balanced lipophilicity and water solubility contributed to conserved anticancer activity at limited toxicity in heathy cells. The exception was GDM analog with the methylene-18-crown-6 substituent at C(17), of low lipophilicity (clogP < 0), for which relatively high anticancer potency (IC50s = 0.66–1.09 μM) at markedly lower toxicity in normal cells was noted, compared to GDM or 92.46 This result was explained by the ability to form complexes by this ansamycin with Na+in vivo regulating the favorable balance between water solubility and lipophilicity.46
Many benzoquinone-C15 hybrids with biomolecules were obtained by semisynthetic modifications at C(17) to improve drug-delivery to the target site of action in cells (106–114, Fig. 37). In the aim to improve bioavailability, the prodrug of geldanamycin bearing β-galactose moiety at C(17) was obtained as the substrate of β-galactosidase, overexpressed in tumor cells (106, Fig. 37).153 Despite the known hepatotoxicity of GDM itself, after administration of 106, the tumor growth was suppressed at no significant damage of non-target organs in animals. In turn, the attachment of ester of phosphonic acid at C(17) of GDM core was reflected in attractive antiviral activity (anti-HCV) at extremely low IC50 = 2.4 nM (Table 2). This result was 2-fold better than that obtained for GDM.154 In contrast to that, the bi-substitution at C(17) and C(19) positions of the GDM core almost abolished anti-HCV potency. The other target of benzenoid ansamycins, i.e. telomerase, was discovered for triazole-bridged hybrids with acridine moiety at C(17), obtained using dipolar cycloaddition of CuAAC type (108, Fig. 37).155 In dependence on the length of the alkyl linker between triazole and the core, anticancer potencies for such hybrids were from good (IC50 ∼ 2 μM) to very good (IC50 ∼ 0.1 μM), in different cancer cells. The most important was, however, that the hybrid 108 revealed higher anticancer effects (Table 2), at lower toxicity in healthy cells (WI38), than 92.155
Fig. 37 Semisynthetic benzoquinone-C15 geldanamycin-like conjugates with biological relevant molecules or oligo- and polypeptides.
The synthesized GDM-biotin conjugate via oligoether-amide linkage at C(17) (109, Fig. 37), as excellent substrate binding with both isoforms of Hsp90 from human colorectal cancer cells (Hsp90α and Hsp90β), allowed to isolate of these proteins with high purities.156 An analogous oligoether linkage between GDM and apoptozole moieties was applied to designing effective Hsp70 and Hsp90 inhibitors (110, Fig. 37 and Table 3) of anti-leukemia potency higher than GDM.157 Semisynthetic construction of 110-type hybrids was based first on the attachment HOOC-oligoether amine at C(17) of GDM, and then the formation of the amide linkage with the apoptozole portion. Experimental studies showed that such type hybrids promote leukemia cell death through caspase-dependent apoptotic pathway.
Table 3IC50 values in cancer cell lines for geldanamycin (GDM), and its novel drug-delivery platforms as C(17)-amine congeners
Compound
[μM]
GDM157
IC50(HL60) = 2.30
IC50(Jurkat) = 4.8
IC50(KG1) = 3.20
IC50(K562) = 3.70
IC50(THP1) = 0.90
IC50(DU-145) = 0.03
IC50(U937) = 4.80
110157
IC50(HL60) = 0.73
IC50(U937) = 1.12
IC50(KG1) = 0.91
IC50(Jurkat) = 0.89
IC50(THP1) = 0.36
IC50(K562) = 0.63
111158
IC50(DU-145) = 1.2 ± 0.1
114159
IC50(HUVEC) = 33.2 ± 1.00
Recently, some greater attention has been paid toward semisynthetic formation of conjugates between GDM and oligo- or polypeptides (111–113, Fig. 37) as efficient drug-delivery platforms for anticancer therapy. Hybrid of GDM with hexadecapeptide (111, Fig. 37), degradable by cathepsin B, of improved water-solubility relative to GDM, showed good anticancer activities IC50s ∼ 1 μM (Table 3).158,159 In contrast to that, a more hydrophobic conjugates or peptide dendrimers with GDM revealed lower anticancer effects than 111. Structurally analogous to 111, pH-sensitive micelles based on GDM-levulinic acid-oligopeptide-PEG system (112, Fig. 37) were prepared with high drug loading efficiency (>30% of polymer) and releasing the identical active form as prodrug 106.160 In turn, drug-releasing system, containing conjugated aminohexyl-GDM as a portion of HPMA (oligopeptide)-copolymer bearing docetaxel (113, Fig. 37), induced expression of heat shock glucose regulating protein (GRP78) and showed time-dependent anticancer effects in human prostate cancer cells (DU145).161,162 Overall, the peptide conjugates with the functionalized C(17)-GDM analogs can be an effective strategy for directing the drug to the target site of action and allowing for its better accumulation in tumor tissues.
Regioselective introduction of various C(19) substituents into the GDM scaffold, at the conserved C(17)-methoxy group or amine parts of 91 or 92, was obtained using Suzuki–Miyaura or Stille coupling developed by Moody and co-workers (114–119; Fig. 38).163,164 Semisynthetic modifications of being considered clinically scaffolds of 91 (17-DMAG) and 92 (17-AAG) as well as GDM, by an incorporation of C(19)-alkyl, aryl or heterocyclic moieties, contributed to good anticancer effects (114–119; Fig. 38 and Table 4).45,165 Despite anticancer activity in breast cancer MDA468 cell line of these derivatives were comparable or lower than GDM or 92, the decreased toxicity in mouse and human hepatocytes and in HUEVEC or ARPE-19 human epithelial cell lines (Table 4), make them interesting group of anticancer drug candidates. Furthermore, this-type ansamycins, functionalized at C(19), showed inhibitory activity toward heat shock proteins of dopaminergic neural cells, demonstrating therapeutic potential against neurodegenerative diseases.45 Compound 115 was also more potent (at nM) in inducing Hsp70 and Hsp27 than 92 of higher anticancer potency (Table 4). The double substituted ansamycins at C(19) and C(17) (114–119), due to the steric hindrances between the newly attached C(19) substituent and the quinone or the lactam portions, occur in the cis-lactam configuration in “free” form in solution.45,163 The presence of this form, ready to be bound with Hsp90, explains high affinity of these ansamycins to the target (Fig. 8c). Similarly functionalized at the quinone core benzoquinone-C15 ansamycins, bearing an extra S–Me group at C(19), were obtained by Li et al. (120–122, Fig. 38).166 In general, these derivatives exhibited lower anticancer potency than GDM, with the exception of 122 (Table 4). Derivative 122 was the most potent (IC50s = 0.13–2.32 μM) of all studied ansamycins and GDM in tested cancer cell lines, at lower hepatotoxicity than GDM, in buffalo rat liver cell line (BRL) and measured as biomarker levels (aminotransferases ALT and AST) in the plasma samples. Unfortunately, symmetrical double substitution of the core at C(17) and C(19) with the tetrahydrofuran moieties (123, Fig. 38) contributed to markedly decreased antiviral (HCV) potency, when compared to promising derivative 107 and GDM (Table 4).154 Thus, the above examples showed that di-substitution of the benzoquinone-C15 ansamycins core at C(17) and C(19) is favorable for enhanced anticancer activity at reduced toxicity, but is not beneficent to antiviral properties. Catalytic approach to regio- and diastereoselective modifications of the cores and ansa bridges of benzoquinone ansamycins, via epoxidation of GDM using Asp-containing peptide/H2O2 or Asp-containing peptide/m-CPBA oxidation systems, have been proposed recently by Miller et al. (124–128, Fig. 38).167 Structures of new semisynthetic derivatives 124, 127 and 128 were determined by X-ray method and the oxidation reaction mechanisms were proposed with the help of experiments with model benzoquinones. The regio- and stereoselectivities of these epoxidations were attributed to formation H-bond between the peptide and carbamate group of the ansa chain in transition state. Compounds 125, 127 and 128 showed high affinity to the three isoforms of chaperones (Hsp90α, Hsp90β, Grp94) at KDs = 0.05–1.1 μM, whereas exclusively 125 showed comparable potency to GDM at nM level in SKBR-3.167
Fig. 38 Semisynthetic benzoquinone-C15 analogs of GDM, 91 (17-DMAG) and 92 (17-AAG) with an extra introduced substituents at C(19) of the core or with the reduced (123) or oxidized (124, 125) the core or oxygenated double bond within the ansa bridge (127, 128).
Table 4Anticancer and antiviral (HCV) potency together with toxicity (HUVEC) of double modified geldanamycin congeners
[μM]
GDM45,154,165,166
IC50(MDA468) = 0.06
IC50(BRL) = 2.41 × 10−3
IC50(HUVEC) = 0.041
IC50(HCV) = 0.00480
IC50(ARPE-19) = 0.10
9145,165,166
IC50(MDA468) = 10.5
IC50(ARPE-19) = 1.0
IC50(HUVEC) = 1.2
IC50(BRL) = 4.80 × 10−3
9245,165
IC50(TAMH) = 0.02
IC50(HUVEC) = 0.88
IC50(MDA468) = 0.61
IC50(ARPE-19) = 0.15
114a45
IC50(HUVEC) = 16.9
IC50(ARPE-19) > 20
114b45
IC50(HUVEC) > 20
IC50(ARPE-19) > 20
114c45,165
IC50(TAMH) = 4.61
IC50(ARPE-19) > 20
IC50(HUVEC) > 20
115a45,165
IC50(MDA468) = 7.45
IC50(ARPE-19) = 8.3
IC50(HUVEC) = 2.1
115b45,165
IC50(MDA468) = 88.37
IC50(ARPE-19) > 20
IC50(HUVEC) > 20
115c45,165
IC50(TAMH) = 8.00
IC50(HUVEC) = 17.7
IC50(MDA468) = 16.48
IC50(ARPE-19) = 8.2
122166
I C50(BRL) = 4.80 × 10−3
IC50(HeLa) = 1.42
IC50(HepG2) = 0.13
IC50(HCT116) = 1.11
IC50(MCF-7) = 2.32
123154
IC50(HCV) = 58.94
Modifications within the core and the ansa chain yielding new ansamitocin-like benzene-C15 derivatives
Novel semisynthetic triazole-branched, dendrimer-like ansamitocin hybrids (131, 132; Fig. 39) were obtained at good yields via a combined chem-bio strategy, employing products of PDB transformations, as well as their further semisynthetic modifications via alkyne–azide dipolar cycloaddition (CuAAC) with the use of CuBr/DIPEA/MeOH system.168 The alkyne-containing intermediates, as e.g.129 and 130 (Fig. 39), were prepared by feeding the mutant A. pretiosum strain with an acid-alkyne derivative and 3-amino-4-chlorobenzoic acid or 3-amino-4-bromobenzoic acid as biosynthetic precursors, analogously to the approach shown in Fig. 27. Among mutaproducts selected for further optimizations, 129 and 130 showed the best anticancer potencies and slightly decreased toxicities in HUVEC normal cells, when compared to those of AP3 (Table 5). Unfortunately, the formation of dendrimer-like structures of ansamitocins (131–134) was accompanied by reduced antiproliferative potency regarding AP3, but it was still good at nM level in most cases. The best antiproliferative potencies among dendrimer-like analogs were noted for 133 of the short aliphatic linker and the two triazole moieties (Table 5 and Fig. 39). Nevertheless, simple ‘click’ reaction product 135 showed markedly higher anticancer activity than the dendrimer-like 133 (Table 5). Hence, ansamycins functionalized with furan moiety (136, Fig. 40) were attractive toward the modern concepts regarding magnetically or thermocleavable controlled drug transport and release systems, based on nanoparticles (136a and 137a, Fig. 40).168–172 ‘Click’ strategy yielded thermocleavable conjugates between an anticancer ansamitocin-triazole-furan component and superparamagnetic nanostructured particles coated on a ferrite core/γ-Fe2O3 + Fe3O4/(136a, Fig. 40).170 These new biomaterials shown to be activated by conventional or external inductive heating via an oscillating electromagnetic field, initiating retro-Diels–Alder reaction, which released an active form of ansamycin in cancer cells.168,170 Such an approach was tested on mouse models and can be promising in cancer treatment via hyperthermia or chemotherapy. It should be added that 137, obtained from mutasynthetic 19-bromo AP3via Heck reaction and next immobilized on Fe3O4-nanoparticles (137a, Fig. 40), underwent efficient releasing under inductive heating within 30 min and exerted antiproliferative effects in several mammalian cancer cell lines.171 Without external heating source compound 137a did not exhibit any antiproliferative activity. The mutasynthetic 19-bromo AP3 was also used to semisynthetic construction of folate-AP3 conjugates via Heck reaction and oxidation into disulfide bridges (138, Fig. 41).126 Conjugate 138 showed good selectivity toward cancer cells with surface expressed folate receptors (FR+, Table 5).
Fig. 39 Semisynthetic structural optimization of C(3)-alkyne bearing ansamitocin-like derivatives 129 and 130via dipolar cycloaddition of CuAAC type.
Table 5Antiproliferative potency and toxicity (HUVEC) of AP3 and its novel ansamitocin-like derivatives and hybrids
[nM]
AP3168
IC50(L929) = 0.2
IC50(SKOV3) = 0.05
IC50(KB31) = 0.17
IC50(PC3) = 0.06
IC50(U937) = 0.01
IC50(HUVEC) = 0.02
IC50(A431) = 0.08
129168
IC50(L929) = 1.3
IC50(PC3) = 0.19
IC50(U937) = 0.03
IC50(MCF7) = 0.13
IC50(A431) = 0.16
IC50(HUVEC) = 0.1
IC50(SKOV3) = 0.1
130168
IC50(L929) = 0.42
IC50(PC-3) = 0.35
IC50(U937) = 0.02
IC50(MCF-7) = 0.06
IC50(A431) = 0.08
IC50(HUVEC) = 0.05
IC50(SKOV3) = 0.05
133168
IC50(L929) = 6.4
IC50(PC3) = 50
IC50(U937) = 8.7
IC50(MCF7) = 15.1
IC50(A431) = 12.4
IC50(HUVEC) = 3.8
IC50(SKOV3) = 4.1
135168
IC50(L929) = 4.9
IC50(SKOV3) = 1.6
IC50(KB31) = 0.41
IC50(PC3) = 2.2
IC50(A431) = 0.77
138126
IC50(KB31 – FR+) = 8
IC50(A549 – FR−) > 104
Fig. 40 Semisynthetic triazole ansamitocin-like derivatives 136 and 137, functionalized at the ansa chain and the core via dipolar cycloaddition of CuAAC type, used in combination with nanoparticles as drug-delivery platforms.
Fig. 41 The most active semisynthetic ansamitocin-like derivative 138, targeting at nM concentrations cancer cells with expressed foliate-receptors on the surface.
7. Conclusions
Our division of natural and semisynthetic ansamycins, based on the type of core and length of the ansa chain, has neatly demonstrated the high levels of structural diversity of benzenoid ansamycins (11 structural subgroups). We noted that many compounds despite lacking the lactam group in the ansa chain are also classified as ansamycins, what according to our strictly structural criteria of classification is a mistake. Hence, we propose to use the term “ansamycins” exclusively for natural compounds that are macrolactams, biosynthesized as a result of a cooperation of the polyketide synthase of type I with the amide synthase, and that contain a rigid core originating from AHBA, AHBA-like or synthetic mutasynthons.
Benzenoid ansamycins are biosynthesized by actinobacteria as A. pretiosum or by Streptomyces sp., being endophytic to various organisms such as: termites (benzene-C15 as natalamycin), higher plants (benzene-C15 as maytansine and its congeners; dibenzene-C9 juanlimycins), mosses (benzene-C17 as trienomycins). Natural ansamycins, such as cebulactams (benzene-C9), herbimycins (benzene-C15) and some of trienomycins (benzene-C17) were found as secondary metabolites of marine microorganisms. Biosynthetic plasticity of natural benzenoid ansamycins is well reflected in the structural diversity of their scaffolds found in nature, where often the ansa chain and the core are fused to each other by an extra cyclic/polycyclic systems and many alterations within the core and the ansa chain are performed on the post PKS I stages. Novel natural unique scaffolds contain e.g. cyclopentenone core within the so-called mccrearamycins or the two ansa chains and the two cores within dimeric juanlimycins. Fusion of the core and ansa chain parts or the presence of an extra unsaturated portions as results of biosynthetic pathways decrease structural flexibility of these ansamycins. Such a limited flexibility of the ansa chain, a key moiety in molecular recognition with the natural target, is often accompanied by decreased affinity toward original target site of action, as e.g. tubulins or chaperones (Hsp90α, Hsp90β, Grp94). Thus, due to the altered mechanism of action for some members of benzenoid ansamycins, instead expected anticancer properties, the other interesting activities as antiviral, antibacterial, antileishmanial or antioxidant, were noted. It is worth to mention that many natural ansamycin precursors consisting of open-chain structures have been found in bacteria extracts, and despite most of these precursors being relatively biologically inactive, further developing semisynthetic methods enabling their macrolactamizations could provide an interesting alternative way to search for novel drug candidates.
The use of genetically modified bacteria strains in mutasynthetic approach, opens access to novel and atypical-functionalized benzenoid ansamycin scaffolds, which are not available or difficult to obtain via solely multistep total synthetic or semisynthetic approaches. Mutasynthesis with the use of functionalized mutasynthons creates the possibility for installation of an additional carbon atom between the core and the lactam group as well as introducing a variety of different functional groups as azide, alkyne, alkene, halogen, –CH2–OH and –CH2–NH2 into the core or the ansa chain, useful for further semisynthetic modifications via amidation, esterification, SN2, Heck reaction or Huisgen dipolar cycloaddition of CuAAC type. Such wide-range of systematic functionalizations is very important to draw convincing SAR conclusions. Furthermore, it was showed that even the two atropisomeric forms of benzenoid ansamitocins, bearing sterically-crowded core, can be produced by mutasynthetic strategy with combination of an early- and late-stage blocked mutant strains. Mutasynthetic or genetic manipulations approaches to modifications of ansamycin structures enable replacement of benzenoid cores with the heterocyclic ones, and/or introduction of the lactone instead lactam. Moreover, this useful strategy offers also assembling of the core of one type-ansamycin with the ansa chain of the other-type ansamycin. Interestingly, some of novel mutaproducts among ansamitocin-like and geldanamycin-like revealed, without any semisynthetic modifications, anticancer properties comparable or even slightly higher (at nM level) than AP3, GDM or other active benzenoid ansamycins at lower toxic effects in normal cells. Some disadvantage of this approach is, sometimes low yield of the isolated mutaproduct, the greatest advantage is possibility of great structural alteration of benzenoid ansamycin scaffolds, enabling systematic SAR studies.
Semisynthetic modifications of benzenoid ansamycins are used separately or with combination with mutasynthesis as structural optimization step. The most explored semisynthetic modifications of benzenoid ansamycins are related to geldanamycin-like/mainly at the core – C(17) and C(19) positions/or ansamitocin-like compounds/mainly at C(3)/, due to the clinical trials of selected members of these both groups. Despite benzoquinone-C15 ansamycins are known from higher toxicity then benzene-C15 ones, the simple semisynthetic modifications of the former within the core [mainly at C(17) or C(19)] or conversion of them into bioconjugates allow to achieve some compromise between water solubility vs. lipophilicity and showed attractive anticancer effects. It should be noted that most of ansa bridge modifications within the benzoquinone-C15 ansamycins were relatively ineffective toward biological potency regulation. Recently, an interesting semisynthetic transformations of geldanamycin-like scaffolds, resulted in unusual epoxidation of the core and the ansa chain with the use of peptide-oxidation systems, yielded novel-type compounds binding with the three isoforms of chaperones. The semisynthetic approach has the limited range dictated by the reactivity of the quinone in the native ansamycin, and hence many type chemical functionalizations are unavailable when compared to the mutasynthetic approach. Other modern trend in semisynthetic transformations of ansamycins was formation of their functional hybrids, with biomolecules as e.g. peptides, saccharides, foliate, cinnamyl or phosphonate groups, serving as carriers of the active form of ansamycin into the cells. Many of ansamitocin-like mutaproducts, with the incorporated double or triple bonds, azide or halogens within the core or the ansa bridge, were subjected to semisynthetic optimizations via dipolar cycloadditions or Heck reactions linking them to biomolecules or to nanoparticles. Novel nanomaterials, containing ansamitocin-furan moiety link to nanoparticles, susceptible to Diels–Alder/retro-Diels–Alder reversible click reactions, were sensitive to electromagnetic field or standard heating in vivo or in model systems. Successful tests of these novel drug-delivery platforms, at releasing the active form of the anticancer agent at the target site of action, revealed their pharmaceutical potential.
Unfortunately, in some cases the same structure of ansamycin is being called two different names (e.g., ansatrienin B and mycotrienin II; herbimycin D and heronamycin A) or in other cases the two different structures were described by the same name (e.g., “divergolide O” and “divergolide O′” that we proposed). Another problem in the area of ansamycins involves overuse of terms such as “reblastatin-like”, “maytansine-like” and “geldanamycin-like”, in particular for reported compounds structurally much closer to groups totally different than those suggested by these names. Some reports have shown poor structural characterizations (e.g., exclusively on the basis of HR MS) due to the small amount of isolated ansamycin. Such characterization is worrying since many modern spectroscopic methods enabling spectral measurements, even from small amount of sample, are available today. For e.g. C(19) substituted geldanamycin-like ansamycins the lacking of resonance signals in NMR spectra and difficulties at structural characteristics were noted, similarly as for e.g. mutasynthetic and atropisomeric benzenoid ansamycins. This problematic spectral feature results from dynamic conformation equilibria concerning the mutual arrangement of the ansa bridge relative to the benzenoid core in solution.
8. Conflicts of interest
There are no conflicts to declare.
9. Acknowledgements
The authors are grateful for financial support from the Polish National Science Centre (NCN)—OPUS 13 project no. UMO-2017/25/B/ST5/00291.
10. Notes and references
A. Whitty, M. Zhong, L. Viarengo, D. Beglov, D. R. Hall and S. Vajda, Drug Discovery Today, 2016, 21, 712–717 CrossRefCASPubMed.
M. Rossi Sebastiano, B. C. Doak, M. Backlund, V. Poongavanam, B. Over, G. Ermondi, G. Caron, P. Matsson and J. Kihlberg, J. Med. Chem., 2018, 61, 4189–4202 CrossRefCASPubMed.
E. Danelius, V. Poongavanam, S. Peintner, L. Wieske, M. Erdelyi and J. Kihlberg, Chem.–Eur. J., 2020, 26, 5231–5244 CrossRefCASPubMed.
V. Poongavanam, Y. Atilaw, S. Ye, L. H. E. Wieske, M. Erdelyi, G. Ermondi, G. Caron and J. Kihlberg, J. Pharm. Sci., 2021, 110, 301–313 CrossRefCASPubMed.
H. B. Bode and A. Zeeck, J. Chem. Soc., Perkin Trans. 1, 2000, 323–328 RSC.
H. Hoeksema, S. A. Mizsak, L. Baczynskyj and L. Pschigoda, J. Am. Chem. Soc., 1982, 104, 5173–5181 CrossRefCAS.
R. R. A. Kitson and C. J. Moody, J. Org. Chem., 2013, 78, 5117–5141 CrossRefCASPubMed.
A. Kirschning and F. Hahn, Angew. Chem., Int. Ed., 2012, 51, 4012–4022 CrossRefCASPubMed.
Q. Kang, Y. Shen and L. Bai, Nat. Prod. Rep., 2012, 29, 243–263 RSC.
P. W. Piper and S. H. Millson, Open Biol. J., 2012, 2, 120138 CrossRefPubMed.
S. K. Gill and G. A. Garcia, Tuberculosis, 2011, 91, 361–369 CrossRefCASPubMed.
P. A. Aristoff, G. A. Garcia, P. D. Kirchhoff and H. D. Hollis Showalter, Tuberculosis, 2010, 90, 94–118 CrossRefCASPubMed.
M. X. Ho, B. P. Hudson, K. Das, E. Arnold and R. H. Ebright, Curr. Opin. Struct. Biol., 2009, 19, 715–723 CrossRefCASPubMed.
T.-W. Yu, L. Bai, D. Clade, D. Hoffmann, S. Toelzer, K. Q. Trinh, J. Xu, S. J. Moss, E. Leistner and H. G. Floss, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7968–7973 CrossRefCASPubMed.
S. Wings, H. Müller, G. Berg, M. Lamshöft and E. Leistner, Phytochemistry, 2013, 91, 158–164 CrossRefCASPubMed.
J.-K. Weng and J. P. Noel, in Methods in Enzymology, ed. D. A. Hopwood, Academic Press, 2012, vol. 515, pp. 317–335 Search PubMed.
X.-H. Nong, Z.-C. Tu and S.-H. Qi, Bioorg. Med. Chem. Lett., 2020, 30, 127168 CrossRefCASPubMed.
S. S. Hong, X. F. Cai, B. Y. Hwang, H. S. Lee, B.-N. Su, Y.-S. Hong and D. Lee, Tetrahedron Lett., 2010, 51, 351–353 CrossRefCAS.
J. Wang, W. Li, H. Wang and C. Lu, Org. Lett., 2018, 20, 1058–1061 CrossRefCASPubMed.
J. M. Jez, J. C.-H. Chen, G. Rastelli, R. M. Stroud and D. V. Santi, Chem. Biol., 2003, 10, 361–368 CrossRefCASPubMed.
C. J. Martin, S. Gaisser, I. R. Challis, I. Carletti, B. Wilkinson, M. Gregory, C. Prodromou, S. M. Roe, L. H. Pearl, S. M. Boyd and M.-Q. Zhang, J. Med. Chem., 2008, 51, 2853–2857 CrossRefCASPubMed.
R. R. A. Kitson, C.-H. Chang, R. Xiong, H. E. L. Williams, A. L. Davis, W. Lewis, D. L. Dehn, D. Siegel, S. M. Roe, C. Prodromou, D. Ross and C. J. Moody, Nat. Chem., 2013, 5, 307–314 CrossRefCASPubMed.
N. Skrzypczak, K. Pyta, P. Ruszkowski, M. Gdaniec, F. Bartl and P. Przybylski, Eur. J. Med. Chem., 2020, 202, 112624 CrossRefCASPubMed.
H. Onodera, M. Kaneko, Y. Takahashi, Y. Uochi, J. Funahashi, T. Nakashima, S. Soga, M. Suzuki, S. Ikeda, Y. Yamashita, E. S. Rahayu, Y. Kanda and M. Ichimura, Bioorg. Med. Chem. Lett., 2008, 18, 1588–1591 CrossRefCASPubMed.
P. Thepchatri, T. Eliseo, D. O. Cicero, D. Myles and J. P. Snyder, J. Am. Chem. Soc., 2007, 129, 3127–3134 CrossRefCASPubMed.
N. Skrzypczak, K. Pyta, P. Ruszkowski, P. Mikołajczak, M. Kucińska, M. Murias, M. Gdaniec, F. Bartl and P. Przybylski, J. Enzyme Inhib. Med. Chem., 2021, 36, 1898–1904 CrossRefCASPubMed.
Y.-S. Lee, M. G. Marcu and L. Neckers, Chem. Biol., 2004, 11, 991–998 CrossRefCASPubMed.
S. M. Kupchan, Y. Komoda, W. A. Court, G. J. Thomas, R. M. Smith, A. Karim, C. J. Gilmore, R. C. Haltiwanger and R. F. Bryan, J. Am. Chem. Soc., 1972, 94, 1354–1356 CrossRefCASPubMed.
J. M. Lambert and R. V. J. Chari, J. Med. Chem., 2014, 57, 6949–6964 CrossRefCASPubMed.
A. E. Prota, K. Bargsten, J. F. Diaz, M. Marsh, C. Cuevas, M. Liniger, C. Neuhaus, J. M. Andreu, K.-H. Altmann and M. O. Steinmetz, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13817–13821 CrossRefCASPubMed.
C. E. Stebbins, A. A. Russo, C. Schneider, N. Rosen, F. U. Hartl and N. P. Pavletich, Cell, 1997, 89, 239–250 CrossRefCASPubMed.
M.-Q. Zhang, S. Gaisser, M. Nur-E-Alam, L. S. Sheehan, W. A. Vousden, N. Gaitatzis, G. Peck, N. J. Coates, S. J. Moss, M. Radzom, T. A. Foster, R. M. Sheridan, M. A. Gregory, S. M. Roe, C. Prodromou, L. Pearl, S. M. Boyd, B. Wilkinson and C. J. Martin, J. Med. Chem., 2008, 51, 5494–5497 CrossRefCASPubMed.
A. K. Wernimont, W. Tempel, Y. H. Lin, A. Hutchinson, F. MacKenzie, A. Fairlamb, D. Cossar, Y. Zhao, M. Schapira, C. H. Arrowsmith, A. M. Edwards, C. Bountra, J. Weigelt, M. a. J. Ferguson, R. Hui, J. C. Pizarro and T. Hills, DOI:10.2210/pdb3q5j/pdb, To Be Published.
J. Hermane, I. Bułyszko, S. Eichner, F. Sasse, W. Collisi, A. Poso, E. Schax, J.-G. Walter, T. Scheper, K. Kock, C. Herrmann, P. Aliuos, G. Reuter, C. Zeilinger and A. Kirschning, ChemBioChem, 2015, 16, 302–311 CrossRefCASPubMed.
L. Li, L. Wang, Q.-D. You and X.-L. Xu, J. Med. Chem., 2020, 63, 1798–1822 CrossRefCASPubMed.
K. Watanabe, M. A. Rude, C. T. Walsh and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9774–9778 CrossRefCASPubMed.
M. G. Anderson, D. Monypenny, R. W. Rickards and J. M. Rothschild, J. Chem. Soc., Chem. Commun., 1989, 311–313 RSC.
C. G. Kim, A. Kirschning, P. Bergon, Y. Ahn, J. J. Wang, M. Shibuya and H. G. Floss, J. Am. Chem. Soc., 1992, 114, 4941–4943 CrossRefCAS.
C.-G. Kim, A. Kirschning, P. Bergon, P. Zhou, E. Su, B. Sauerbrei, S. Ning, Y. Ahn, M. Breuer, E. Leistner and H. G. Floss, J. Am. Chem. Soc., 1996, 118, 7486–7491 CrossRefCAS.
C. T. Walsh, ACS Infect. Dis., 2018, 4, 1283–1299 CrossRefCASPubMed.
T. Komoda, K. Akasaka and A. Hirota, Biosci., Biotechnol., Biochem., 2008, 72, 2392–2397 CrossRefCASPubMed.
S. M. Pimentel-Elardo, T. A. M. Gulder, U. Hentschel and G. Bringmann, Tetrahedron Lett., 2008, 49, 6889–6892 CrossRefCAS.
S. Yang, Y. Xi, J.-H. Chen and Z. Yang, Org. Chem. Front., 2014, 1, 91 RSC.
H. Pellissier, Beilstein J. Org. Chem., 2018, 14, 325–344 CrossRefCASPubMed.
J. Zhang, Z. Qian, X. Wu, Y. Ding, J. Li, C. Lu and Y. Shen, Org. Lett., 2014, 16, 2752–2755 CrossRefCASPubMed.
P. Kusari, S. Kusari, D. Eckelmann, S. Zühlke, O. Kayser and M. Spiteller, RSC Adv., 2016, 6, 10011–10016 RSC.
M. Zhao, Y. Fan, L. Wei, F. Hu and Q. Hua, Appl. Biochem. Biotechnol., 2017, 181, 1167–1178 CrossRefCASPubMed.
Y. Wu, Q. Kang, G. Shang, P. Spiteller, B. Carroll, T.-W. Yu, W. Su, L. Bai and H. G. Floss, ChemBioChem, 2011, 12, 1759–1766 CrossRefCASPubMed.
F. Zhang, H. Ji, I. Ali, Z. Deng, L. Bai and J. Zheng, ChemBioChem, 2020, 21, 1309–1314 CrossRefCASPubMed.
T. Liu, L. Bessembayeva, J. Chen, L.-J. Wei and Q. Hua, Bioresour. Bioprocess., 2019, 6, 1 CrossRef.
M. O. Steinmetz and A. E. Prota, Trends Cell Biol., 2018, 28, 776–792 CrossRefCASPubMed.
C. Lu, L. Bai and Y. Shen, J. Antibiot., 2004, 57, 348–350 CrossRefCASPubMed.
J. Ma, P.-J. Zhao and Y.-M. Shen, Arch Pharm. Res., 2007, 30, 670–673 CrossRefCASPubMed.
J. Ma, Y.-M. Shen, Y. Zeng and P.-J. Zhao, HCA, 2012, 95, 1630–1636 CrossRefCAS.
Y. Li, P. Zhao, Q. Kang, J. Ma, L. Bai and Z. Deng, Chem. Biol., 2011, 18, 1571–1580 CrossRefCASPubMed.
F. Taft, M. Brünjes, T. Knobloch, H. G. Floss and A. Kirschning, J. Am. Chem. Soc., 2009, 131, 3812–3813 CrossRefCASPubMed.
P. Zhao, L. Bai, J. Ma, Y. Zeng, L. Li, Y. Zhang, C. Lu, H. Dai, Z. Wu, Y. Li, X. Wu, G. Chen, X. Hao, Y. Shen, Z. Deng and H. G. Floss, Chem. Biol., 2008, 15, 863–874 CrossRefCASPubMed.
X. Li, X. Wu and Y. Shen, Org. Lett., 2019, 21, 5823–5826 CrossRefCASPubMed.
X.-M. Li, X.-M. Li and C.-H. Lu, J. Asian Nat. Prod. Res., 2017, 19, 946–953 CrossRefCASPubMed.
C. E. Snipes, D. O. Duebelbeis, M. Olson, D. R. Hahn, W. H. Dent, J. R. Gilbert, T. L. Werk, G. E. Davis, R. Lee-Lu and P. R. Graupner, J. Nat. Prod., 2007, 70, 1578–1581 CrossRefCASPubMed.
S. Eichner, T. Eichner, H. G. Floss, J. Fohrer, E. Hofer, F. Sasse, C. Zeilinger and A. Kirschning, J. Am. Chem. Soc., 2012, 134, 1673–1679 CrossRefCASPubMed.
K. A. Shaaban, X. Wang, S. I. Elshahawi, L. V. Ponomareva, M. Sunkara, G. C. Copley, J. C. Hower, A. J. Morris, M. K. Kharel and J. S. Thorson, J. Nat. Prod., 2013, 76, 1619–1626 CrossRefCASPubMed.
R. Raju, A. M. Piggott, Z. Khalil, P. V. Bernhardt and R. J. Capon, Tetrahedron Lett., 2012, 53, 1063–1065 CrossRefCAS.
K. H. Kim, T. R. Ramadhar, C. Beemelmanns, S. Cao, M. Poulsen, C. R. Currie and J. Clardy, Chem. Sci., 2014, 5, 4333–4338 RSC.
D. Lee, K. Lee, X. F. Cai, N. T. Dat, S. K. Boovanahalli, M. Lee, J. C. Shin, W. Kim, J. K. Jeong, J. S. Lee, C.-H. Lee, J.-H. Lee, Y.-S. Hong and J. J. Lee, ChemBioChem, 2006, 7, 246–248 CrossRefCASPubMed.
W. Zhao, B. Jiang, L. Wu, Y. Nan, J. Cui, L. Yu, Y. Wei, J. Li and G. Shan, J. Antibiot., 2015, 68, 476–480 CrossRefCASPubMed.
E. Schax, J.-G. Walter, H. Märzhäuser, F. Stahl, T. Scheper, D. A. Agard, S. Eichner, A. Kirschning and C. Zeilinger, J. Biotechnol., 2014, 180, 1–9 CrossRefCASPubMed.
Q. Zhao, C.-Z. Wu, J. K. Lee, S.-R. Zhao, H.-M. Li, Q. Huo, T. Ma, J. Zhang, Y.-S. Hong and H. Liu, J. Microbiol. Biotechnol., 2014, 24, 914–920 CrossRefCASPubMed.
S. Eichner, H. G. Floss, F. Sasse and A. Kirschning, ChemBioChem, 2009, 10, 1801–1805 CrossRefCASPubMed.
Z. Hu, Y. Liu, Z.-Q. Tian, W. Ma, C. M. Starks, R. Regentin, P. Licari, D. C. Myles and C. Richard Hutchinson, J. Antibiot., 2004, 421–428 CrossRefCASPubMed.
W.-X. Li, B. Han and C.-B. Cui, J. Asian Nat. Prod. Res., 2016, 18, 705–710 CrossRefCASPubMed.
A. Baksh, B. Kepplinger, H. A. Isah, M. R. Probert, W. Clegg, C. Wills, M. Goodfellow, J. Errington, N. Allenby and M. J. Hall, Nat. Prod. Res., 2017, 31, 1895–1900 CrossRefCASPubMed.
X. Wang, Y. Zhang, L. V. Ponomareva, Q. Qiu, R. Woodcock, S. I. Elshahawi, X. Chen, Z. Zhou, B. E. Hatcher, J. C. Hower, C.-G. Zhan, S. Parkin, M. K. Kharel, S. R. Voss, K. A. Shaaban and J. S. Thorson, Angew. Chem., Int. Ed., 2017, 56, 2994–2998 CrossRefCASPubMed.
M. M. Madathil, O. M. Khdour, J. Jaruvangsanti and S. M. Hecht, ACS Med. Chem. Lett., 2013, 4, 953–957 CrossRefCASPubMed.
Y. Fan, C. Wang, L. Wang, A. Chairoungdua, P. Piyachaturawat, P. Fu and W. Zhu, Mar. Drugs, 2018, 16, 282 CrossRefPubMed.
D. Tang, L.-L. Liu, Q.-R. He, W. Yan, D. Li and J.-M. Gao, J. Nat. Prod., 2018, 81, 1984–1991 CrossRefCASPubMed.
L.-L. Liu, Z.-F. Chen, Y. Liu, D. Tang, H.-H. Gao, Q. Zhang and J.-M. Gao, Org. Chem. Front., 2020, 4008–4018 RSC.
G. Shi, N. Shi, Y. Li, W. Chen, J. Deng, C. Liu, J. Zhu, H. Wang and Y. Shen, ACS Chem. Biol., 2016, 11, 876–881 CrossRefCASPubMed.
L. Lindqvist, F. Robert, W. Merrick, H. Kakeya, C. Fraser, H. Osada and J. Pelletier, RNA, 2010, 16, 2404–2413 CrossRefCASPubMed.
G. E. L. Brandt and B. S. J. Blagg, ACS Med. Chem. Lett., 2011, 2, 735–740 CrossRefCASPubMed.
Z. Xu, M. Baunach, L. Ding, H. Peng, J. Franke and C. Hertweck, ChemBioChem, 2014, 15, 1274–1279 CrossRefCASPubMed.
L. Ding, A. Maier, H.-H. Fiebig, H. Görls, W.-H. Lin, G. Peschel and C. Hertweck, Angew. Chem., 2011, 123, 1668–1672 CrossRef.
G. Zhao, S. Li, Z. Guo, M. Sun and C. Lu, RSC Adv., 2015, 5, 98209–98214 RSC.
K. J. Weissman, Trends Biotechnol., 2007, 25, 139–142 CrossRefCASPubMed.
W. T. Shier, K. L. Rinehart and D. Gottlieb, Proc. Natl. Acad. Sci. U.S.A., 1969, 63, 198–204 CrossRefCASPubMed.
S. R. Park, Y. J. Yoo, Y.-H. Ban and Y. J. Yoon, J. Antibiot., 2010, 63, 434–441 CrossRefCASPubMed.
M. Serpi, V. Ferrari and F. Pertusati, J. Med. Chem., 2016, 59, 10343–10382 CrossRefCASPubMed.
G. Niu, Z. Li, P. Huang and H. Tan, J. Antibiot., 2019, 72, 906–912 CrossRefCASPubMed.
M. R. Levengood, P. J. Knerr, T. J. Oman and W. A. van der Donk, J. Am. Chem. Soc., 2009, 131, 12024–12025 CrossRefCASPubMed.
R. Zhe and Z. Wenjun, Curr. Top. Med. Chem., 2016, 16, 1755–1762 CrossRefPubMed.
C. Anderle, S. Hennig, B. Kammerer, S.-M. Li, L. Wessjohann, B. Gust and L. Heide, Chem. Biol., 2007, 14, 955–967 CrossRefCASPubMed.
U. Gräfe, K. Dornberger, C. Wagner and K. Eckardt, Biotechnol. Adv., 1989, 7, 215–IN2 CrossRef.
S. Panjikar, J. Stoeckigt, S. O'Connor and H. Warzecha, Nat. Prod. Rep., 2012, 29, 1176–1200 RSC.
C. Feng, H. Ling, D. Du, J. Zhang, G. Niu and H. Tan, Microb. Cell Factories, 2014, 13, 59 CrossRefPubMed.
W. Runguphan, J. J. Maresh and S. E. O'Connor, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 13673–13678 CrossRefCASPubMed.
F. Taft, S. Eichner, T. Knobloch, K. Harmrolfs, J. Hermane and A. Kirschning, Synlett, 2012, 23, 1416–1426 CrossRefCAS.
M. S. A. Khan and M. M. Altaf, in New and Future Developments in Microbial Biotechnology and Bioengineering, ed. V. K. Gupta and A. Pandey, Elsevier, Amsterdam, 2019, pp. 131–139 Search PubMed.
J. J. Hug, D. Krug and R. Müller, Nat. Rev. Chem., 2020, 4, 172–193 CrossRefCAS.
F. Wesemann, A. Heutling, P. Wienecke and A. Kirschning, ChemBioChem, 2020, 21, 2927–2930 CrossRefCASPubMed.
F. Wei, Z. Wang, C. Lu, Y. Li, J. Zhu, H. Wang and Y. Shen, Org. Lett., 2019, 21, 7818–7822 CrossRefCASPubMed.
T. Knobloch, K. Harmrolfs, F. Taft, B. Thomaszewski, F. Sasse and A. Kirschning, ChemBioChem, 2011, 12, 540–547 CrossRefCASPubMed.
F. Taft, K. Harmrolfs, I. Nickeleit, A. Heutling, M. Kiene, N. Malek, F. Sasse and A. Kirschning, Chem.–Eur. J., 2012, 18, 880–886 CrossRefCASPubMed.
L. Mancuso, G. Jürjens, J. Hermane, K. Harmrolfs, S. Eichner, J. Fohrer, W. Collisi, F. Sasse and A. Kirschning, Org. Lett., 2013, 15, 4442–4445 CrossRefCASPubMed.
F. Taft, M. Brünjes, H. G. Floss, N. Czempinski, S. Grond, F. Sasse and A. Kirschning, ChemBioChem, 2008, 9, 1057–1060 CrossRefCASPubMed.
K. Harmrolfs, L. Mancuso, B. Drung, F. Sasse and A. Kirschning, Beilstein J. Org. Chem., 2014, 10, 535–543 CrossRefPubMed.
S. Mohammadi-Ostad-Kalayeh, F. Stahl, T. Scheper, K. Kock, C. Herrmann, F. A. Heleno Batista, J. C. Borges, F. Sasse, S. Eichner, J. Ongouta, C. Zeilinger and A. Kirschning, ChemBioChem, 2018, 19, 562–574 CrossRefCASPubMed.
S. Eichner, T. Knobloch, H. G. Floss, J. Fohrer, K. Harmrolfs, J. Hermane, A. Schulz, F. Sasse, P. Spiteller, F. Taft and A. Kirschning, Angew. Chem., Int. Ed., 2012, 51, 752–757 CrossRefCASPubMed.
T. Knobloch, G. Dräger, W. Collisi, F. Sasse and A. Kirschning, Beilstein J. Org. Chem., 2012, 8, 861–869 CrossRefCASPubMed.
G. Jürjens and A. Kirschning, Org. Lett., 2014, 16, 3000–3003 CrossRefPubMed.
J. Zhang, S. Li, X. Wu, Z. Guo, C. Lu and Y. Shen, Org. Lett., 2017, 19, 2442–2445 CrossRefCASPubMed.
S. Li, J. Cui, X. Lu, Z. Zheng, X. Liu, S. Ni, Y. Wang and L. Wu, J. Antibiot., 2013, 66, 499–503 CrossRefCASPubMed.
T. Li, S. Ni, C. Jia, H. Wang, G. Sun, L. Wu, M. Gan, G. Shan, W. He, L. Lin, H. Zhou and Y. Wang, J. Nat. Prod., 2012, 75, 1480–1484 CrossRefCASPubMed.
J. Hermane, S. Eichner, L. Mancuso, B. Schröder, F. Sasse, C. Zeilinger and A. Kirschning, Org. Biomol. Chem., 2019, 17, 5269–5278 RSC.
L. Ding, A. Maier, H.-H. Fiebig, H. Görls, W.-H. Lin, G. Peschel and C. Hertweck, Angew. Chem., Int. Ed., 2011, 50, 1630–1634 CrossRefCASPubMed.
S. Jogula, A. R. Soorneedi, J. Gaddam, S. Chamakuri, G. S. Deora, R. K. Indarapu, M. K. Ramgopal, S. Dravida and P. Arya, Eur. J. Med. Chem., 2017, 135, 110–116 CrossRefCASPubMed.
Y. N. Song, R. H. Jiao, W. J. Zhang, G. Y. Zhao, H. Dou, R. Jiang, A. H. Zhang, Y. Y. Hou, S. F. Bi, H. M. Ge and R. X. Tan, Org. Lett., 2015, 17, 556–559 CrossRefCASPubMed.
Y.-N. Song, W.-J. Zhang, S.-F. Bi, R.-H. Jiao, R.-X. Tan and H.-M. Ge, J. Antibiot., 2015, 68, 757–759 CrossRefCASPubMed.
J. Wang, X. Li, C. Lu and Y. Shen, ChemBioChem, 2018, 19, 256–262 CrossRefCASPubMed.
X. Li, J. Zhu, G. Shi, M. Sun, Z. Guo, H. Wang, C. Lu and Y. Shen, RSC Adv., 2016, 6, 88571–88579 RSC.
C. Xie, J.-J. Deng and H.-X. Wang, Curr. Microbiol., 2015, 70, 859–864 CrossRefCASPubMed.
T. Kim, G. Keum and A. N. Pae, Expert Opin. Ther. Pat., 2013, 23, 919–943 CrossRefCASPubMed.
W. Guo, P. Reigan, D. Siegel and D. Ross, Drug Metab. Dispos., 2008, 36, 2050–2057 CrossRefCASPubMed.
R. L. Cysyk, R. J. Parker, J. J. Barchi, P. S. Steeg, N. R. Hartman and J. M. Strong, Chem. Res. Toxicol., 2006, 19, 376–381 Search PubMed.
N. Joubert, A. Beck, C. Dumontet and C. Denevault-Sabourin, Pharmaceuticals, 2020, 13, 245 Search PubMed.
J. R. Junutula, K. M. Flagella, R. A. Graham, K. L. Parsons, E. Ha, H. Raab, S. Bhakta, T. Nguyen, D. L. Dugger, G. Li, E. Mai, G. D. Lewis Phillips, H. Hiraragi, R. N. Fuji, J. Tibbitts, R. Vandlen, S. D. Spencer, R. H. Scheller, P. Polakis and M. X. Sliwkowski, Clin. Cancer Res., 2010, 16, 4769–4778 CrossRefCASPubMed.
J. M. Lambert and C. Q. Morris, Adv. Ther., 2017, 34, 1015–1035 CrossRefCASPubMed.
Z. Li, L. Jia, J. Wang, X. Wu, H. Hao, Y. Wu, H. Xu, Z. Wang, G. Shi, C. Lu and Y. Shen, Eur. J. Med. Chem., 2014, 87, 346–363 CrossRefCASPubMed.
Z. Li, L. Jia, H. Tang, Y. Shen and C. Shen, RSC Adv., 2019, 9, 42509–42515 RSC.
K. H. Carruthers, G. Metzger, M. J. During, A. Muravlev, C. Wang and E. Kocak, Cancer Gene Ther., 2014, 21, 434–440 CrossRefCASPubMed.
G. Shan, Z. Peng, Y. Li, D. Li, Y. Li, S. Meng, L. Gao, J. Jiang and Z. Li, J. Antibiot., 2011, 64, 177–182 CrossRefCASPubMed.
S. Roe, M. Gunaratnam, C. Spiteri, P. Sharma, R. D. Alharthy, S. Neidle and J. E. Moses, Org. Biomol. Chem., 2015, 13, 8500–8504 RSC.
R. E. Connor, L. J. Marnett and D. C. Liebler, Chem. Res. Toxicol., 2011, 24, 1275–1282 Search PubMed.
S.-H. Park, W.-J. Kim, H. Li, W. Seo, S.-H. Park, H. Kim, S. C. Shin, E. R. P. Zuiderweg, E. E. Kim, T. Sim, N.-K. Kim and I. Shin, Sci. Rep., 2017, 7, 3537 Search PubMed.
V. Kolhatkar, J. Suárez and R. Kolhatkar, Bioorg. Med. Chem. Lett., 2015, 25, 3744–3747 CrossRefCASPubMed.
K. Greish, A. Ray, H. Bauer, N. Larson, A. Malugin, D. Pike, M. Haider and H. Ghandehari, J. Controlled Release, 2011, 151, 263–270 CrossRefCASPubMed.
Y. Bae, A. W. G. Alani, N. C. Rockich, T. S. Z. C. Lai and G. S. Kwon, Pharm. Res., 2010, 27, 2421–2432 CrossRefCASPubMed.
N. Larson, S. Roberts, A. Ray, B. Buckway, D. L. Cheney and H. Ghandehari, Macromol. Biosci., 2014, 14, 1735–1747 CrossRefCASPubMed.
N. Larson, A. Gormley, N. Frazier and H. Ghandehari, J. Controlled Release, 2013, 170, 41–50 CrossRefCASPubMed.
R. R. A. Kitson and C. J. Moody, Tetrahedron, 2021, 82, 131927 CrossRefCAS.
R. R. A. Kitson and C. J. Moody, Chem. Commun., 2013, 49, 8441 RSC.
C.-H. Chang, D. A. Drechsel, R. R. A. Kitson, D. Siegel, Q. You, D. S. Backos, C. Ju, C. J. Moody and D. Ross, Mol. Pharmacol., 2014, 85, 849–857 CrossRefPubMed.
Y. Li, J. Chen, J. Shen, J. Cui, L. Wu, Z. Wang and Z. Li, Cancer Chemother. Pharmacol., 2015, 75, 773–782 CrossRefCASPubMed.
M. J. Hilton, C. M. Brackett, B. Q. Mercado, B. S. J. Blagg and S. J. Miller, ACS Cent. Sci., 2020, 6, 426–435 CrossRefCASPubMed.
L. Mancuso, T. Knobloch, J. Buchholz, J. Hartwig, L. Möller, K. Seidel, W. Collisi, F. Sasse and A. Kirschning, Chem.–Eur. J., 2014, 20, 17541–17551 CrossRefCASPubMed.
M. D. Norris, K. Seidel and A. Kirschning, Adv. Ther., 2019, 2, 1800092 CrossRef.
K. Seidel, A. Balakrishnan, C. Alexiou, C. Janko, R.-M. Komoll, L.-L. Wang, A. Kirschning and M. Ott, Chem.–Eur. J., 2017, 23, 12326–12337 CrossRefCASPubMed.
L.-L. Wang, A. Balakrishnan, N.-C. Bigall, D. Candito, J. F. Miethe, K. Seidel, Y. Xie, M. Ott and A. Kirschning, Chem.–Eur. J., 2017, 23, 2265–2270 CrossRefCASPubMed.
S. Ullah, K. Seidel, S. Türkkan, D. P. Warwas, T. Dubich, M. Rohde, H. Hauser, P. Behrens, A. Kirschning, M. Köster and D. Wirth, J. Controlled Release, 2019, 294, 327–336 CrossRefCASPubMed.

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*








![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(21) (violet; CCDC 1988071).46
C(21) (violet; CCDC 1988071).46


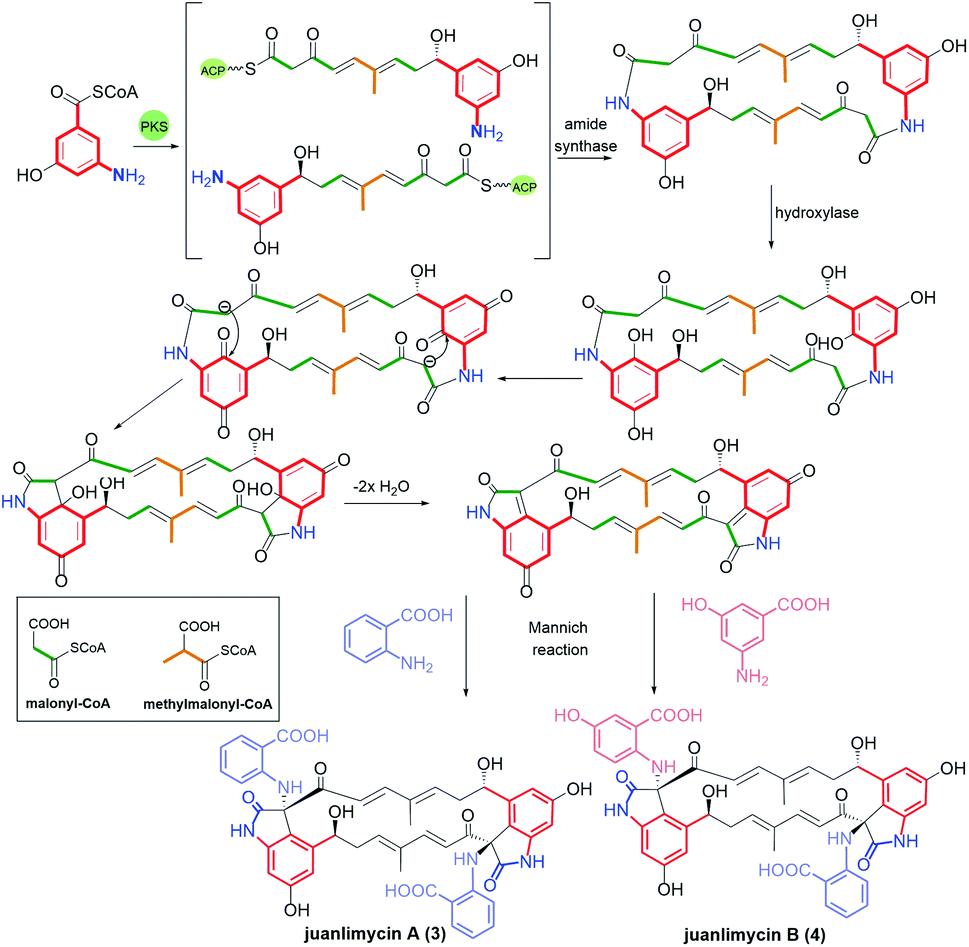
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 565 revealed an active participation of 3-O-methyl transferase and 3′-O-carbamyl-transferase enzymes in the biosynthetic pathway.81 Li et al. reported that C(9)-methoxy analog of 6 can be produced not only by A. pretiosum but also by the other Amycolatopsis alba DSM 44262 bacteria strain.82 Ansacarbomitocins exhibited modest antifungal activities against Septoria tritici, Erisiphe graminis, and Puccinia recondite.83
565 revealed an active participation of 3-O-methyl transferase and 3′-O-carbamyl-transferase enzymes in the biosynthetic pathway.81 Li et al. reported that C(9)-methoxy analog of 6 can be produced not only by A. pretiosum but also by the other Amycolatopsis alba DSM 44262 bacteria strain.82 Ansacarbomitocins exhibited modest antifungal activities against Septoria tritici, Erisiphe graminis, and Puccinia recondite.83