Modifications, biological origin and antibacterial activity of naphthalenoid ansamycins
Natalia
Skrzypczak
and
Piotr
Przybylski
*
Faculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznanskiego 8, 61-614 Poznan, Poland. E-mail: piotrp@amu.edu.pl
Received
14th January 2022
First published on 4th March 2022
Abstract
Covering: 2011 to 2021
Structural division of natural naphthalenoid ansamycins, regarding the type of the core and length of the ansa chain, and their biosynthetic pathways in microorganisms are discussed. The great biosynthetic plasticity of natural naphthalenoid ansamycins is reflected in their structural variety due to the alterations within ansa bridge or naphthalenoid core portions. A comparison between the biological potency of natural and semisynthetic naphthalenoid ansamycins was performed and discussed in relation to the molecular targets in cells. The antibacterial potency of naphthalenoid ansamycins seems to be dependent on the ansa chain length and conformational flexibility – the higher flexibility of the ansa chain the better biological outcome is noted.
Natalia Skrzypczak
Natalia Skrzypczak was born in Poznan in 1995. She obtained a bachelor's degree (2017) and a master's degree (2019) in chemistry at the Adam Mickiewicz University in Poznan, Poland. In 2019, she started her PhD studies under the supervision of Prof. Piotr Przybylski at the Faculty of Chemistry, AMU. She is a co-author of 4 publications. Her research interests include the novel approaches for transformation of ansamycins cores, based on modern synthetic methods (click chemistry).
Piotr Przybylski
Piotr Przybylski (M. Sc. 2000; PhD – 2004; habilitation – 2011; full professor – 2019) was born in Poznan, Poland in 1975. He is a professor of organic chemistry at Department of Natural Products Chemistry (AMU in Poznan). He is the leader of the research team specialized in chemistry and medicinal chemistry of natural antibiotics, their derivatives and other biologically important molecules. His research interests are focused on modification and determination of SAR for natural products and their congeners, and with tautomerization, atropisomerization and zwitterionization processes. His current studies are related to cascade approaches enabling modification of lactone and lactam macrolides.
1. Introduction
The term “ansamycin” was first suggested by Prelog and Oppolzer, to describe compounds being closely related to those discovered by Lüttringhaus (ansa compounds), and derives from the name for the characteristic ansa chain structural motif (ansa in Latin means “handle”).1,2 The discovery of the first naphthalenoid ansamycin antibiotics called rifamycins, was made by Professor Piero Sensi and his research group from Milan (Lepetit Research Laboratories).3–5 The basket-like structure of ansamycins (Fig. 1 and 2), consisting usually the aromatic core and flexible ansa bridge, has been shown to display some structural flexibility influencing biological properties, characteristic for chameleonic-type drugs.6–8 Taking into account recent achievements with mutasynthesis and isolation of novel natural scaffolds among macrolactams from bacteria strains, the term “ansamycins” can be used in a broader sense. Natural ansamycins can be described as a family of macrolactams, bearing relatively rigid central cores as benzenoid, naphthalenoid or atypical ones as e.g. alicyclic or heterocyclic ones, originated from AHBA or other-type small semisynthetic precursors (mutasynthons), and produced in as a result of cooperation between polyketide (PKS type I) and amide synthases (Fig. 2). The important role of naphthalenoid rifamycin-type antibiotics in medical therapy as antibacterial agents, especially against M. tuberculosis, encouraged us to report about their recently discovered natural sources, biogenesis, modifications, and structure–activity relationships.
Fig. 1 Structural analogy between the basket and 3-formylrifamycin SV, a member of naphthalenoid ansamycins.
Fig. 2 Structural division of naphthalenoid ansamycins regarding the core type and the length of the ansa chain (top) and natural sources of these ansamycins (bottom): soil-derived bacteria (black); marine bacteria (blue), bacteria associated with the higher plants (green).
2. Structural division of naphthalenoid ansamycins and their natural sources
Ansamycins can be roughly divided into three main groups called benzenoid, naphthalenoid, and atypical ones and further based on the length of the ansa chain. Funayama and Cordell divided ansamycins into two main subgroups and highlighted common biosynthetic paths of these natural products.9
The structural classification of ansamycins can be performed in a more detailed manner e.g. taking into account oxidized/reduced structure of the rigid core or the regioisomers of the quinone/iminoquinone moieties, as shown in Fig. 2. Thus, in an addition to the Funayama and Cordell classification, we propose several groups of naphthalenoid ansamycins, which can be divided into those having naphthalene, naphthalene-1,3(2H,4H)-dione, naphthalen-1(2H)-one, 2,3-dihydronaphthalene-1,4-dione, 3,4-dihydronaphthalen-1(2H)-one, 1,4-naphthoquinone, 1,2/1,4-iminonaphthoquinone or naphthalen-1(4H)-one cores and lactam ansa bridges of different length (Fig. 2). Some of biosynthetic, biological and analytical aspects of naphthalenoid ansamycins have been discussed in earlier reviews published between the years 1972 and 2011.9–23 Since that time many biological relevant natural naphthalenoid ansamycin scaffolds have been isolated from bacteria or semisynthetically modified, revealing differences in their modes of action. Isolation of novel naphthalenoid ansamycins of unique structure, in terms of the new core type or the length and structure of the ansa bridge, indicated a great structural diversity among these natural products and shed new light on alternative biosynthetic pathways realized in microorganisms. Comparison of structural division between benzenoid and naphthalenoid ansamycins indicates a greater structural variety of the latter than the former ones. Irrespectively on the type of the core or the length of the ansa bridge, the main natural source evidenced for naphthalenoid ansamycins are bacteria strains (Fig. 2; bottom), similarly as for the benzenoid ansamycins, which has been demonstrated. Different bacteria strains producing naphthalenoid ansamycins can be, however, derived from soil (Streptomyces sp.; Amycolatopsis sp.; Micromonospora sp.), water (Streptomyces sp.; Salinispora sp.; Micromonospora sp.) as well as from higher plants as e.g. trees or shrubs Manglietia hookeri (Streptomyces sp. CS).
3. Comparison between biosynthetic pathways of naphthalenoid and benzenoid ansamycins
Ansamycins are synthesized in nature via the polyketide pathway from AHBA, being a product of the modified shikimate pathway, and acetate with propionate units, irrespective of the type of the rigid core, as evidenced by experiments with isotopically labeled biosynthetic precursors (Fig. 3).12,24–27 The major difference between the biosyntheses of representative naphthalenoid (e.g. rifamycin B) and benzenoid (e.g. geldanamycin) ansamycins concerns the formation of the ansa chain (length and substitution) and the central core (type), as shown in Fig. 3. Studies with isotopically labeled biosynthetic units e.g. malonyl, methylmalonyl, glycolate, and methyl (methionine), required for producing geldanamycin and rifamycin B, clearly revealed key differences in biosynthetic pathways between these ansamycins. The three malonyl and eight methylmalonyl units are incorporated into the rifamycin B scaffold, where seven of methylmalonyl units are inserted into the ansa bridge portion and the one is required for cyclization of the naphthalenic core (orange, Fig. 3). In contrast to that, only four methylmalonyl, two glycolate and one malonyl units are inserted into the molecular “handle” of the geldanamycin basket by PKS I (Fig. 3).28 At the early stage of rifamycin B biosynthesis realized by bacteria, the closing of the naphthalene core via an intramolecular conjugate addition of Michael-type takes place (Fig. 3). Post-PKS I tailoring of rifamycin B and geldanamycin is based on cyclization catalyzed by amide synthase with the formation of rifamycin W or progeldanamycin, respectively. Rifamycin B post-PKS I transformations, apart from the lactam formation and methylation via SAM, involve acetylation of hydroxyl at C(11), attachment of 3-phosphoglyceric or 3-phosphohydroxypyruvate to the phenol group at the C(19), oxygen insertion between vinyl and naphthalene portions and the C1 unit lost at the C(14) position (asterisk, Fig. 3). In turn, post-PKS I modifications of progeldanamycin are concerned with oxidation of C(4)–C(5) positions, three O-methylations with SAM, and carbamoylation at C(7). Another difference between the above biosynthetic pathways is monooxygenation at C(17) and oxidation of p-phenol moiety into the quinone for geldanamycin, and the reduction of the quinone to the phenol form for rifamycin B. It should be noted that quinone or quinol forms of the above ansamycins are important for better molecular recognition with their different targets in cells. The absolute configurations of contiguous stereogenic centers in the polyketide chains of naphthalenoid and benzenoid ansamycins, can be predicted using a model developed by Funayama et al., analogous to the Celmer model referred to macrolactones (Fig. 4).29,30 As it can be deduced from Fig. 4, rifamycin B and streptovaricin C belonging to naphthalenoid ansamycins are biosynthesized using a common amide-head biosynthetic pathway; with the exception of the C(6) and C(7) positions, the absolute configurations of the stereogenic centers along the ansa chain of rifamycin B appear to be identical to those along the ansa chain of streptovaricin C. Most of the ansamycins, having seventeen carbon atoms in the polyketide chain, show the same structural sequence along the chain, i.e., PAPPPPPPAP, where P and A are propionate (methylmalonyl) and acetate (malonyl) units, respectively. Kanglemycins are the exception to this rule because they have an extra C1 unit.31,32 In contrast to the naphthalenoid-C17 ansamycins, those having a longer “handle” of the molecular basket, as e.g., actamycin with C23-ansa chain, show a different structural sequence along the bridge: AAAPAPAAPPPAP.
Fig. 3 Biosynthetic pathways of the two representatives of ansamycins: geldanamycin (benzenoid – benzoquinone-C15) and rifamycin B (naphthalenoid – naphthalene-C17).
Fig. 4 Model of expected stereochemistry within the ansa chains of naphthalenoid ansamycins according to Funayama et al. or Celmer et al.; p-propionate (orange) and A-acetate (green) units.
4. Structural aspects of naphthalenoid ansamycins influencing their physico-chemical and biological properties
Naphthalenoid ansamycins possess a different number of double bonds within the ansa bridge (Fig. 5). The number and position of double bonds along the ansa chain together with intramolecular interactions, as e.g. H-bonds, have an impact on the possibility to adopt various conformations of the ansa “handle” of naphthalenoid ansamycins in response to environmental factors such as the nature of cell barriers or steric demands of the molecular target. Due to the existence of the two structural portions, a rigid naphthalenoid core and the flexible-ansa chain bearing stereogenic centers, the relative arrangement of them contributes to atropisomerism (from Greek “a” meaning “not” and “tropos” meaning “turn”). This special case of stereoisomerism, dependent on the altered arrangement of the whole ansa bridge relative to the plane of the rigid core for naphthalenoid ansamycins, was also observed for the other medicinal relevant biomolecules as benzenoid ansamycins, alkaloids, purines or diaryl or aryl-heterocyclic drugs, where both axial and planar chiralities were well evidenced.33–36 Excellent, experimentally indicated examples of stereoisomerism (atropisomerism) among naphthalenoid ansamycins, induced by plane or axis, are streptovaricins (Fig. 6). Single crystal X-ray studies showed that one of the boronic esters of streptovaricin C (Fig. 5) has an opposite helicity of the ansa bridge to that present in most natural streptovaricins and other naphthalenoid ansamycins (Fig. 6).37,38 Furthermore, it was evidenced that for various rifamycins (including rifabutin, rifapentine, rifaximin, and rifampicin) the conformation of the ansa bridge and its arrangement relative to the central core are dependent on the phase (solid/liquid) or polarity and nature (protic/aprotic) of the solvent used.39–42 The conformation flexibility of the ansa chain for ansamycins, e.g. rifamycins, is not always concerned with atropisomerism, however, is crucial for chameleonic properties of this-type macrolactams, reflected in a facile adaptation to the changing cell environment during transport of the drug to the target site.43 Recently, it was demonstrated that interactions of naphthalenoid ansamycins (rifamycins) with water molecules lead to the formation of a “closed”-type structure (Fig. 7), with polar groups hidden toward the core, which increases the lipophilicity and enable diffusion of these antibiotics in bacterial cell membranes.44 However, a more hydrophilic rifamycin structure in solution, with polar groups exposed to the solvent, i.e., in the so-called “open” structure (Fig. 7), is necessary for the rifamycins to target bacterial RNA polymerases.45–47 Hence, a smooth conversion between these two-type structures with a displacement of the whole ansa bridge (Fig. 7), i.e. chameleonic nature, is highly important to the presence of attractive biological activity of naphthalenoid ansamycins.44
Fig. 5 Different configuration of double bonds within ansa bridge for exemplary naphthalenoid ansamycins.
Fig. 6 Newman projection of the axial stereoisomerism of streptovaricin C derivatives, implied by the two different orientations of the ansa bridge (bottom) relative to the aromatic core, visualized on molecular models (top) via Scigress (EU 3.1.8), Fujitsu. Models of streptovaricins for DFT optimizations were assumed using X-ray structure of p-bromobenzeneboronate ester of streptovaricin C triacetate.37
Fig. 7 Key conformers for a total functionality of rifamycins: an “open” ansa bridge conformer of lower lipophilicity and crucial for binding with bacterial RNA polymerases (top), and “closed” ansa-bridge conformer of higher lipophilicity needed to overcome natural cell barriers, visualized by Scigress (EU 3.1.8), Fujitsu.44
Another structural factor of naphthalenoid ansamycins is their zwitterionization as a result of the presence of both acidic and basic groups in scaffolds, which affects lipophilicity and water solubility in the cell environment. This phenomenon was the best researched for naphthalenoid rifamycins, where the proton of the phenol group was transferred to the most basic centre in the molecule (Fig. 7 and 8).44,48–50 It was indicated that despite unfavorable low water-solubility of the non-ionic form of rifamycins, the optimization of this physico-chemical parameter is realized by the proton transfer yielding zwitterions, at the active participation of the protic solvent. In turn, aprotic solvents favoured non-ionic form of rifamycins, better adapted to lipophilic cell barrier environment. This another chameleonic feature of rifamycins such as rifampicin or its various amine congeners was evidenced not only via 1D and 2D NMR and FT-IR spectroscopic studies in solutions but also was proved in crystal state where different solvents were applied.44,48,51,52 X-ray and CP-MAS NMR solid state studies showed that crystallization of the rifampicin from protic solvents yielded zwitterionic form (water-soluble) of the antibiotic whereas crystallization from aprotic solvents afforded the non-ionic form (lipophilic) of this antibiotic.42,49,53 The other example of chameleonic ansamycin is rifaximin, where the zwitterion formation was accompanied by the equilibrium between more lipophilic (closed) and more hydrophilic (open) conformers, supported by water or other protic solvent molecules (Fig. 7).44 Hence, the non-ionic structure of rifapentine, the antibiotic analogous to the above-mentioned rifamycins, in the solvate with methanol molecules54 seems to be incorrect. The problem of ansamycin zwitterionization, influencing the molecular recognition of the antibiotic with the target in cells (bacterial RNA polymerases – RNAPs, Fig. 9) and lipophilic permeability efficiency, is often neglected in the literature.50,55,56 In the work of Campbell et al., the structure of the non-ionic form of rifampicin (incorrect non-ionic pentahydrate crystal form – reinvestigated later as zwitterionic51) was used to build the model of interactions between this ansamycin and bacterial RNAPs (Fig. 9a).57 Considering the presence of ansamycin zwitterionic form in water and close proximity of the protonated basic piperazine substituent at C(3) of rifampicin or its congeners and the negatively charged carboxylate of a highly conserved E445 of RNAPs (distance 3.4 Å), alternative binding models of rifamycins with RNAPs were proposed (Fig. 9b).51,53,58–60 In one of these alternative binding patterns concerning rifampicin, the protonated piperazine arm at a slightly altered conformation takes part in a very strong H-bond interaction (rifampicin)N+–H…ŌOC-(E445RNAP) of the salt-type character (H-bond donor …acceptor ∼2.6 Å; angle ∼170°).53 It should be mentioned that new models for rifampicin and its amine congeners explained the structure–activity relationships at changing C(3)-substituent of congeners having comparable lipophilicity and water solubility.
Fig. 8 Regioselective zwitterionization of rifampicin and its amine derivatives containing different-types of basic “centers”.
Fig. 9 Binding modes of: (a) rifampicin to RNAP of Thermus aquaticus (PDB 1I6V);57 (b) rifampicin to RNAP of Thermus aquaticus53 (alternative model built on the basis of MOG-PM6 calculations with RNAP structure – PDB 1I6V); (c) rifabutin to RNAP of Thermus thermophilus (PDB 2A6E);61 (d) kanglemycin A to RNAP of Mycobacterium tuberculosis (Mtb) mutant S447 → L447 (PDB 6DCF), resitant to rifampicin;32 visualized by Scigress (EU 3.1.8, Fujitsu). Interatomic distances are marked by dots or dashed lines; key amino acids taking part in intermolecular H-bonds are marked by yellow (dark green codes); amino acids building the pocket and involved in hydrophobic interactions are marked by yellow – dark blue codes; the core of ansamycin was marked by the red and the lactam by a dark blue; in the structure of RNAP from Thermus thermophilus (c) the σ-finger region is marked by light blue whereas in the structure of RNAP mutant from Mtb (d) the mutated amino acid is marked by pink.
Bacterial RNAPs, as well-known targets in cells of naphthalenoid ansamycins, have identical key amino acids at analogous positions of the sequence, irrespectively on the bacteria strain (Fig. 9). Naphthalenoid ansamycin – rifabutin possesses spiro-piperidyl basic C(3)–C(4) arm, which is directed toward σ-finger of RNAPs (Fig. 9c),61 at nearly conserved interactions with β-fork loop, when compared to rifampicin. This model of rifabutin interactions, however, did not consider that C(3)–C(4) spiro-piperidyl arm possesses a basic nitrogen atom directed toward σ-finger of RNAPs, rich in acidic amino acids, as e.g. D514 or E515 (Fig. 9c). Following the above informations concerning binding of rifampicin with RNAPs, it was suggested that the C(3)–C(4) arm in the protonated form (in vivo), can be involved in H-bonding with D514 of the σ-finger.44 In contrast to the above rifamycins varying at the core substitution, kanglemycins are examples of naphthalenoid ansamycins modified with K-acid and K-sugar at C(20) and C(27) positions of the ansa bridge, respectively (Fig. 9d). Due to this structure of kanglemycins, their location at RNAPs pocket is different than those of rifampicin or rifabutin antibiotics, taking into account the distance to Mg2+ and Q426/510/390 (Fig. 9d).32 The substitution of naphthalenoid-C17 ansamycins at the ansa bridge with bulky substituents often results in hindered binding with bacterial RNAPs (one of the resistance mechanisms) and the markedly decreased or lack of antibacterial potency. Kanglemycins are, however, antibacterially potent, even against M. tuberculosis strains resistant to rifampicin, inter allia due to extra interactions of K-acid and K-sugar with R604 and R164 of RNAPs, respectively. It should be noted that in this altered binding model of kanglemycins, similarly to that of rifabutin one, there are lost interaction with amino acid at 447 positions of the sequence and weakened interactions between C(25) acetyl with the NH group of F430/514/394, when compared to rifampicin models (Fig. 9). Due to the altered importance of the interactions with amino acids of RNAPs, the binding pattern for kanglemycins can be good starting point at designing alternative antibiotics to rifampicin against multiresistant bacteria strains.
Recent NMR, X-ray, surface plasmon resonance (SPR), and site-directed mutagenesis studies on the affinity of rifamycins to viral targets have revealed the biological secret of rifampicin-like ansamycins bearing C(3)-arm, i.e. their binding ability with proteins of poxvirus, as e.g. viral trimeric protein D13, in a highly conserved region – F-ring at the central channel.62 It is a surprising result since known antibacterial antibiotics are usually not involved in inhibiting viral membranes assembly processes. The activity of rifampicin, which forms complexes with mutants of D13 (KDs = 19–285 μM), hinders molecular recognition between viral protein D13 and A17 and prevents virion formation. Hence, this alternative mechanism of action for rifamycins can be used for designing novel-type antiviral agents, based on the structure of naphthalene ansamycins.
The chameleonic nature of naphthalenoid ansamycins is important not only for transportation and binding with the target but can be essential from a bacteria resistance viewpoint, via increased/decreased affinity to efflux pump systems.44,63,64 The zwitterionization phenomenon for naphthalenoid ansamycins as rifamycins have also impact on their polymorphism in the crystal state, which is a very important problem discussed at potential pharmaceutical applications of this group of macrolactams in the treatment of bacterial infections (including tuberculosis).52,65–69
Thus, the chameleonic features of naphthalenoid ansamycins, concerned with zwitterionic/non-ionic structures and different conformations (sometimes related to stereoisomerism), should be taken into regard at their discussions focused on: the transport, binding with the target (SAR discussions), and bacteria resistance mechanisms.
5. Biosynthesis of naphthalenoid ansamycins
5.1. 1,4-Naphthoquinone-C23 ansamycins
The naphthoquinone-C23 ansamycin subgroup, of the longest ansa bridge among ansamycins, is relatively structurally homogeneous, and its members differ from each other mainly with regards to the groups present in their ansa chains and at the C(3) position of the core. Novel naphthomycins L (1), M (2) and N (3) were found to be secondary metabolites of Streptomyces sp. CS, associated with the tree/shrub tissues of Manglietia hookeri from China (Fig. 10).70 These natural products, in contrast to the earlier identified naphthomycins, have amine- or sulfur-containing substituents instead of chlorine (naphthomycin A), hydrogen (naphthomycin E) or hydroxyl (naphthomycin D) at C(3) (Fig. 10). Ansamycin 3 is a dimeric-like scaffold bridged by one 2-aminoethanethiol unit. The basic biological precursors of these naphthomycins are: AHBA, malonyl or methylmalonyl units whereas the sulfur source in biosyntheses of 2 and 3 is unknown. Structures of 1–3 were elucidated from HR ESI MS and 1D and 2D NMR, and several unknown genes were found in their biosynthetic clusters.70 Among these naphthomycins, only compound 1, bearing amine group at C(3), similarly as naphthomycin A, showed weak antifungal activities against several phytopathogenic fungi Fusarium, Verticillium, and Phyricularia at the lack of anticancer effects.70 Natural naphthoquinone-C23 naphthomycins O (4), P(5), Q (6), were obtained as secondary metabolites of mutant Streptomyces sp. CS/asm21-4 strain, or of non-mutant strain Streptomyces sp., associated with Chinese medicinal plant Maytenus hookeri, producing also other naphthomycins (Fig. 11).71 These new naphthomycins 4–6 differ from each other by a number of hydroxyls and double bonds, including their stereochemistries within the ansa chain, and different substituents at C(3) of the naphthoquinone core. Different structures of 4–6, including altered stereochemistry at C(26), C(29) and C(30) as well as the fused/non-fused core with the ansa chain were indicated by 1D and 2D NMR. Compounds 4–6 were quite potent against E. coli, S. typhimurium, S. aureus (MICs = 25 μg mL−1) and weak active against fungi as M. grisea and F. verticillioides (MICs = 200 μg mL−1).71 Structural diversity of naphthomycins can be helpful in the future at their biological optimizations, especially against Gram-negative bacteria strains, although, the mechanism of their activity is still not revealed.
Fig. 10 Structures of new naphthomycins L (1), M (2) and N (3) from Streptomyces sp. CS.
Fig. 11 Structures of new naphthomycins O (4), P (5) and Q (6) obtained from recombinatorial mutants of Streptomyces sp. CS/asm21-4.
5.2. Naphthalene-C17 ansamycins
Natural naphthalene-C17 ansamycins, called rifamorpholines A–E (7–11, Fig. 12), are produced by filamentous actinobacteria Amycolatopsis sp. Hca4 being associated with locust Locusta migratoria (Jiangsu Province, China).72 These natural rifamycins possess an extra fused ansa chain with the phenol group of the naphthalene core, as evidenced by X-ray studies of 7.72 Rifamorpholines 8–11, apart from the fused core with the ansa chain, possess ketone at C(18) and rare cyclopropane structural motif at C(19)–C(20). The natural production of rifamorpholines is based on the use of the gene cluster 19 having nearly the same organization (in particular a lack of orf32c and orf37c) as that producing rifamycins in Amycolatopsis mediterranei S699.72 The formation of demethyl desacetyl rifamycin S and transformation of the lactam via 1,6-cyclization yielding imino-quinone core were postulated as intermediate steps at biosyntheses of 7–11 (Fig. 13). Next, the transformations of the iminoquinone intermediate are realized in two directions: one involving recovering the lactam and epimerization at C(21) (7), and the second recovering the lactam, formation of α,β-unsaturated ketone at C(18) instead of the C(17)–C(18) double bond, followed by epimerization at C(21) and insertion of a one carbon atom at the ansa bridge (8–11).72 The mechanism of cyclopropyl formation within the ansa bridge has not been presented up to now. As the result of the above transformations, all rifamorpholines gained C(21S) absolute configuration, as proved by 2D NMR and X-ray studies. Rifamorpholines 7–11 were tested against several bacteria strains, and 8 was found to be the most active of all ansamycins against M. luteus at MIC = 0.5 μM. Importantly, the antibacterial activity of the most active 8 was identical to that of rifampicin against M. luteus and S. aureus strains, however, the mechanism of action of 8 was not discussed.72
Fig. 12 Structures of new rifamorpholines 7–11.
Fig. 13 Biosynthesis of the naphthalene-C17 ansamycins – rifamorpholines containing the naphthalene core fused with the ansa bridge (7–11) and a unique cyclopropyl moiety (8–11).
5.3. Naphthalene-1(4H)-one-C17 ansamycins
Ansavaricin D (12, Fig. 14), a new streptovaricin analog lacking the characteristic naphthodioxine motif, has recently been isolated from Streptomyces sp. S012, collected from a soil sample (Zhongshan Botanical Garden, Nanjing, China).73 Its structure was determined using NMR and HR-MS methods. Unfortunately, at a concentration below 100 μM, 12 did not inhibit the secretion of SPI-1 effectors of Salmonella enterica serovar Typhimurium. In turn, novel kanglemycins A (13), V1 (14) and V2 (15) were obtained by metagenomic survey of kng gene cluster and the use of soil-derived microbiome as well Amycolatopsis mediterranei var. kanglensis.32 Kanglemycin V2 (15, Fig. 14), possessing methylenedioxy moiety bridging C(4) and C(11), β-O-3,4-O,O′-methylene digitoxose deoxysugar at C(27), and gem-dimethylsuccinic acid portion (K-acid) attached at C(20), showed an attractive inhibitory activity, toward a rifampicin-resistant RNAP mutant (S447 → L447).32 Kanglemycins A and V1 (13, 14, Fig. 14), lacking methylene-dioxy bridge, are also natural congeners of rifamycins, possessing C(11) ketone or hydroxyl groups, and produced by Amycolatopsis mediterranei var. kanglensis.32 These natural compounds, shown to inhibit growth of the transcript at one step earlier than that shown for rifampicin. Binding affinity of 13 to RNAPs of rifampicin-resistant mutants (Fig. 9d), e.g. E. coli, was higher than rifampicin and the best for RNAPβD516V (IC50 (13) = 39 μg mL; IC50 (rifampicin) = 320 μg mL−1). Hence, the greatest advantage of 13 was, the first of all, its potency against rifampicin-resistant M. tuberculosis strain and also against several other pathogens as: S. pyogenes (MIC = 0.031 μg mL−1), B. subtilis (MIC = 1 μg mL−1), S. aureus MRSA (MIC = 1 μg mL−1). Different binding models of kanglemycins and rifampicin contributed to slightly lower activities of the former ones than rifampicin against most of tested strains, as e.g. standard M. tuberculosis strains (H37Rv; 1192/015).74 The explanation of this result is probably lost interactions between the ansamycin and important amino acids of RNAP (as e.g. S447), stabilizing mainly the core at the RNAP pocket, in consequence of the presence of bulky digitoxose deoxysugar portion attached to C(27).
Fig. 14 Structure of new naphthalene-1(4H)-one-C17 ansamycins: ansavaricin D (12) and kanglemycins: A (13), V1 (14), V2 (15).
5.4. 1,4-Naphthoquinone-C17 and 4-iminonaphthalen-1(4H)-one-C17 ansamycins
Chaxamycins A–D (16–19, Fig. 15), isolated from the Streptomyces sp. C34 strain collected from the soil of Chaxa Lagoon (Atacama Desert, northern Chile), were structurally characterized by 1D and 2D NMR methods.75,76 X-ray structure of 16 showed collective intramolecular H-bond system between hydroxyls at C(21), C(23) and C(25), H-bonded phenol at C(8) with the quinone oxygen at C(1) as well as the presence of trans-lactam moiety.75 The identical stereochemistry of the ansa chains of 16–19 was also independently confirmed by the total synthesis of the C(16)–C(28) portion.77 Compound 19, being closely structural related to classical rifamycins, showed attractive potency against S. aureus MRSA (MICs = 0.06–0.25 μg mL−1) and E. coli (MIC = 1.21 μg mL−1), at almost no inhibitory activity toward the ATP-ase function of human Hsp90.75 Furthermore, studies involving docking and inhibition tests confirmed that Hsp90α is not molecular target for chaxamycin 19, as its mechanism of action is probably closer to that realized for classical rifamycins binding to RNAPs (Fig. 9). Despite potency of 19 was weaker against S. aureus than rifampicin, in contrast to the latter it showed attractive potency toward Gram-negative strain.75
Fig. 15 Structure of new naphthoquinone-C17 ansamycins called chaxamycins A (16), B (17), C (18) and D (19), isolated from soil-derived Streptomyces sp. C34 strain.
Novel protostreptovaricins I–V (20–24, Fig. 16) were obtained independently from mutant strain Nocardia mediterranei N813 and S. pectabilis CCTCC M2017417, collected from a soil sample (Wuhan University campus, China).78 These ansamycins, being structural analogous to 16–18, differ from each other by the substituents at the ansa bridge at C(22) and C(26) as well as the substituent at C(6) of the core, as indicated by MS and NMR. The biosynthetic route of protostreptovaricins formation shares a common intermediate (prostreptovaricin) with the pathway yielding streptovaricins (Fig. 16). AntiSMASH analysis showed activity of cytochromes P450s, especially at hydroxylation processes being post-PKS tailoring stage of protostreptovaricins and streptovaricins. Notably visible role of P450 is at the oxidation of the methyl at C(25) into methylene hydroxyl group, which subsequently undergoes oxidation to the carboxylic group taking part in lactonization with the hydroxyl at C(22), as for streptovaricin F (25, Fig. 16).78 The precursor of C(3) methyl unit is SAM, similarly as C(12) carbon atom of dioxamethylene bridge of streptovaricins, whereby the formation of this bridge is catalyzed by type-I O-methyl transferase (StvM1).79 The presence of hydroxyl group at C(26) is essential for enhanced antibacterial potencies of protostreptovaricins against S. aureus strains (standard and MRSA, MICs = 16 μg mL−1), which was even better than those of some obtained streptovaricins.78 However, the most potent of all studied new ansamycin derivatives was streptovaricin F (25, Fig. 16; MIC = 0.125 μg mL−1), containing a tertiary hydroxyl group in the ansa chain at the analogous site [at C(29)] as that of the most active protostreptovaricin III (22), as well as one extra tertiary hydroxyl at C(21).78
Fig. 16 Common main biosynthetic route, via prostreptovaricin intermediate, leading to natural protostreptovaricins I–V (20–24) and streptovaricins with the key role of cytochrome P450s at post-PKS I tailoring of the structure (StvP1-P5, highlighted by colors).
Novel natural aminorifamycins 26 and 27 (Fig. 17), possessing an amine group at C(3), were isolated from cultures of Micromonospora sp. RJA4480.80 Stereochemistry of aminorifamycins was determined by X-ray structure of 26 as well as by 1D and 2D NMR studies both of them. Sporalactams A and B (28 and 29, Fig. 17), containing the core fused with the ansa bridge by C(3)–O–C(16) ether moiety and cyclopropane motif within the ansa bridge, were found also to be secondary metabolites of Micronospora sp.80 Sporalactams 28 and 29 showed very good antibacterial potency, especially against Gram-negative E. coli (MIC90s = 0.4–1.8 μM) and Gram-positive M. tuberculosis (MIC90s = 0.8–0.06 μM). However, ansamycins 26 and 27, bearing C(3)-amine substituent, showed the most attractive potency of all against S. aureus MRSA, E. coli and M. tuberculosis (standard and in macrophages) in the range far beyond expectations (MIC90s = 0.0001–0.7 μM), making them a very attractive starting point to drug design against hard to control pathogens. Such a biological outcome clearly shows that for very attractive potency relatively high flexibility of the ansa bridge is desired.80
Fig. 17 Structure of new naphthoquinone-C17 ansamycins called aminorifamycins (26, 27) and sporalactams A (28) and B (29), isolated from Northestern-Pacific marine-sediments bacteria of Micromonospora sp.
New 11,12-seco-rifamycin W analogs 30–32 (Fig. 18), originating from common biosynthetic precursor – rifamycin W, were found in an agar fermentation extract of Amycolatopsis mediterranei S699.81 These naphthoquinone-C17 ansamycins possess included oxygen heterocycles as oxirane, tetrahydrofuran or tetrahydropyran into the ansa bridge. Such type structures of 30–32 were indicated by HR-MS and 1D and 2D NMR. It should be mentioned that 30 and 32 possess (13Z)-configuration, in contrast to 31 of (13E)-configuration (Fig. 18). Biosynthesis of 31, containing both the lactam and the lactone portions within the ansa bridge, first requires glycosylation with L-rhamnose saccharide at C(6) and next the Baeyer–Villiger oxidation (C27–C28 oxidative cleavage) and an intramolecular transesterification with one of hydroxyls of the ansa bridge. Biosyntheses of 30 and 32 occur directly from rifamycin W, however, detailed mechanisms were not displayed. Ansamycins of greater heterocyclic ring motifs decorating their ansa chains, as 31 and 32, exhibited strong inhibitory activities toward DNA topoisomerase (Topo I) and Topo IIα at 50 μM, in contrast to weak effects noted for the other earlier-obtained 11,12-seco-rifamycins.81 In turn, the limited flexibility of the ansa bridges belonging to 30–32 contributed to the lack of typical for rifamycins antibacterial properties. A much higher plasticity in the production of structurally diverse rifamycin-like natural products was presented via deleting the rifT gene from Amycolatopsis mediterranei S699.82 This approach enabled to obtaining of 8-deoxy ansamycins as e.g.33 or 34 (Fig. 18), bearing tetrahydrofuran moieties at different positions of the ansa bridge. Compound 33, lacking C(8) phenol and possessing tetrahydrofuran ring next to the lactone, in contrast to 30–32 and 34, showed moderate antibacterial potency against S. aureus (MIC = 10 μg mL−1), and was inactive in cancer cells KG1.82
Fig. 18 Structure of new natural naphthoquinone-C17 ansamycins (30; 31 named rifamycinoside B, 33), derivatives of 11,12-seco-rifamycin W, as well as 8-deoxy derivatives obtained from gentic modified A. mediterranei ΔrifT strain (33, 34), all bearing oxygen heterocycles incorporated into the ansa bridge.
5.5. Naphthalene-1,3(2H,4H)-dione-C17 and 3,4-dihydronaphthalen-1(2H)-one-C17 ansamycins
Naphthalenoid ansamycins containing the saturated and deoxygenated quinone core are rather rare in nature. This group ansamycins includes ansavaricins C (35, Fig. 19) and E (36, Fig. 19), produced by Streptomyces sp. S012.73 Structures of these novel secondary metabolites, possessing identical 12E and 14Z configurations and decorating with tetrahydropyran and furan rings (35) or the two tetrahydropyran rings (36), were indicated by 1D and 2D NMR and HR-MS in negative and positive ion detection modes. The absolute configurations of C(7) and C(26) stereogenic centers in 35 were not proposed. The double fusion of the core at the C(23) and C(25) positions of the ansa chain was apparently responsible for the greater inhibitory activities observed for 36 than for 35 toward the secretion of SPI-1 effectors of Salmonella enterica serovar Typhimurium. Inhibitory activity of 36 at concentrations below 100 μM in an anti-T3SS assay can be useful for its applications against resistant pathogens.73 Ansavaricin J, showing nearly identical structure as 36, i.e. enriched by one phenol group at C(8) of the core, was isolated from the genetically modified Streptomyces spectabilis CCTCC M2017417 strain (ΔstvP5 cytochrome P450 gene deletion) producing also streptovaricins.83 This compound obtained by genetic manipulation, which structure was proved by 1D and 2D NMR and HR-MS, showed only limited antibacterial effect toward S. aureus MRSA strains. In turn, much more biological attractive were ansavaricins F–I, named as “polyketides of ansamycin class”.84 This description is, however, not precise because each of these ansavaricins F–I possesses cleavaged the ansa chain and, hence these polyketides can be only precursors or metabolites of ansamycins (called ansavaricins). Open-ansa chain ansavaricins were potent inhibitors toward human DNA topoisomerases (Topo I and Topo IIα) and one of them, i.e. ansavaricin H exhibited selectively cytotoxicity toward HeLa and MDA-MB-453 cancer cells at IC50s < 50 μM.84 It is a surprising result that for ansavaricins belonging to classical ansamycins with the ansa bridge the biological potency was lower or none, in contrast to their open-ansa chain congeners.
Fig. 19 Structure of new natural naphthalene-1,3(2H,4H)-dione-C17 (35, ansavaricin C) and dihydronaphthalen-1(2H)-one-C17 (36, ansavaricin E) ansamycins, isolated from botanical garden soil-derived (Nanjing, China) Streptomyces sp. S012 bacteria strain.
5.6. Naphthalenoid ansamycins of C15 ansa chains
The naphthoquinone-C15 ansamycins, similarly to other ansamycins, are solely secondary metabolites produced by bacteria. The most number and structurally diverse group of ansamycins with C15 ansa chains (Fig. 2) are recently-discovered ansaeomycins (exemplary 37–40; Fig. 20).85 These ansaeomycins were obtained through the expression of the asm gene cluster (cryptic red-ox gene) in a mutant of Streptomyces seoulensis. The activation of berberine bridge enzyme (BBE), i.e. oxidase AsmF, dictated the formation of natural ansamycins bearing C(8) phenol (37, 38, Fig. 21) whereas deletion of the AsmF gene contributed to a great structural variety of ansamycins structures, lacking C(8) phenol group (examples 39–41, Fig. 21).85 The key steps in the natural production of structural diverse ansaeomycins are isomerization (double bond shifts) and conformational change as well as Diels–Alder reactions of the triple unsaturated moiety near the lactam (Fig. 21). Formation of polyketide 41 of cleaved the ansa bridge was postulated as a simple seco-dehydration process. All novel ansaeomycins were characterized by HR-MS, 1D and 2D NMR in detail. Among all ansaeomycins obtained, only those containing C(8) phenol (37 and 38) showed moderate anticancer effects in cell line K562. Surprisingly, antibacterial screening of novel ansaeomycin scaffolds indicated potency of solely an open chain polyketide 41 toward S. aureus at MIC = 64 μM.85 These results revealed that the polyketides with the cleaved ansa chain could be also an interesting starting point toward antibacterial potency optimization and that the limited flexibility of the ansa bridge abolishes the antibacterial properties of ansaeomycins.
Fig. 20 Structure of new natural naphthalenoid-C15 ansaeomycins (37–40) and polyketide 41 with the cleavaged the ansa bridge structure.
Fig. 21 Biosynthetic pathway of natural ansaeomycins (37–40) and polyketide 41 with the cleavaged the ansa bridge structure.
Natural product 42 (rifamycininoside A, Fig. 22), belonging to naphthoquinone-C15 ansamycins, was isolated from Amycolatopsis mediterranei S699.81 It was postulated that 42 is formed on the common rifamycin biosynthetic pathway but with an extra oxidative decarboxylation and typical Baeyer–Villiger oxidation with C(25)–C(26) bond cleavage.81 The pattern of the cleavage within the ansa bridge, however, differs from those known contributing to formation divergolides R and S or hygrocins I and J,86,87 since Baeyer–Villiger oxidation is followed by rearrangement as shown in Fig. 23. As a result of this rearrangement an untypical propan-2-one motif, instead of usually occurring acetate, is being inserted at the ansa bridge structure.81 Subsequent glycosylation at C(6), followed by the rearrangement, yielded 42. It is worth noting, that the other type an intramolecular rearrangement with the nucleophilic attack of C(22) hydroxyl on the C(25) carbon (Fig. 23) yielded other ansamycins with tetrahydrofuran moiety, structurally analogous to those of longer C17 ansa chains (see e.g.31, Fig. 18). Compound 42, showed good inhibitory activities toward the topoisomerases Topo I and Topo IIα.81 The novel, untypical 8-deoxy rifamycin of the conserved 11,12-seco-rifamycin W scaffold (43, Fig. 22), classified to naphthoquinone-C15 ansamycins, has been obtained using the Amycolatopsis mediterranei S699 ΔrifT mutant strain.82 The structure of 43, is lacking one phenol group and the saccharide at the core as well as 13Z configuration is present, in contrast to 42. The presence of the propan-2-one motif in structures of 42 and 43 were proved using 1D and 2D NMR methods. Biological studies of 43 revealed moderate antibacterial activity against Staphylococcus aureus ATCC 25923 at MIC = 20 μg mL−1.82 The limited potency of 43 can be a result of the lack of O(8)H phenol group in the core, important at binding with bacterial RNAPs (interaction with S411, Fig. 9), however, the binding pose of this compound at RNAPs has not been revealed yet.
Fig. 22 Structure of new natural naphthoquinone-C15 ansamycins 42 (ansmitocinoside A) containing the lactone incorporated into the ansa bridge and L-rhamnose at the core and 43 with the unique propan-2-one motif attached at C(25) of the ansa chain.
Fig. 23 Biosynthetic rearrangements after Baeyer–Villiger oxidations: on the top, leading to exclusion of the propan-2-one moiety from the ansa chain backbone, as for ansamycin 42 (ansmitocinoside A); on the bottom, leading to formation of tetrahydrofuran moiety, present also in ansamycins of longer C17 ansa chains (e.g. product 31).
5.7. Naphthaleonoid-C13 ansamycins
Natural occurring ansamycins, consisting of an oxidized/reduced naphthalene moiety and bearing relatively short C13 ansa chain linked at several sites to the core are called ansalactams A (44), B (45), and D (46) (Fig. 24).88–90 Ansalactam 44 was extracted from marine sediment bacterium Streptomyces sp. CHN-189 and its stereochemistry was determined by 2D NMR and X-ray analysis of its reduced congener containing hydroxyls at C(1) and C(4).88 Biosynthetic isotopic studies of ansalactams indicated their main precursors as: AHBA, one malonyl, six methylmalonyl and one isobutyrylmalonyl units (Fig. 25).88,89 The inclusion of the isobutyrylmalonyl unit into the ansalactam scaffolds is enabled by activity of β-ketoacyl-ACP-synthase, dehydrogenase and crotonyl-CoA carboxylase producing isobutyrylmalonyl-CoA in Streptomyces sp. CHN-189. The biosynthetic plasticity of 44–46 is well reflected in the ability of 44 to be converted into 45 or 46via an extra cyclization involving the ansa chain and the core.89 In addition to 45 and 46, another natural secondary metabolite, an ansalactam C (47, Fig. 24) with the cleavaged ansa chain, is also produced by marine sediment bacterium Streptomyces sp. CHN-189.90 This metabolite obtained probably due to the Baeyer–Villiger type reaction from 44 can be further used in macrolactonization due to the possible approaching phenol by the carboxylic group. The above ansalactams 44–47 showed no anticancer effects but most of them were weak active against methicillin-resistant S. aureus at MICs = 31 μg mL−1 (45, 46) and MIC = 62 μg mL−1 (47). Thus, decreased flexibility of the ansa bridge or its cleavage contributed to limited antibacterial effects when compared to those of the most active rifamycins.
Fig. 24 Natural ansalactams A (44), B (45) and D (46), having C13 ansa chain fused with the core as well as ansalactam C (47) containing cleavaged the ansa bridge.
Fig. 25 The biosynthetic route leading to ansalactams A (44), B (45) and D (46), having C13 ansa chain fused with the core and ansalactam C (47) with the cleavaged ansa bridge.
Another small subgroup of naphthoquinone C13-ansa chain ansamycins includes those lacking the C(8) phenol and containing the lactone within the ansa chain and dihydro- or tetrahydro-2H-azepin-2-one ring next to the core (48–51, Fig. 26). Divergolide C (48), found in endophytic Streptomyces sp. associated with the mangrove tree Bruguiera gymnorrhiza,91,92 with its recently revised (15R) configuration, possesses structure identical to that of differently named “olimycin B”, isolated from the deep-sea bacterium Streptomyces olivaceus.93 Bioinformatic analysis indicated that Baeyer–Villiger monooxygenase (DivO) contributes to the presence of the lactone moiety in the ansa chain whereas an alternative exclusion of C(17) tail from the main backbone of the macrocyclic system of divergolides is also a result of the activity of P450-dependent oxidoreductase DivQ. Thus, these enzymes are essential at shaping the naphthalenoid ansamycin scaffolds of great structural variety concerning different ansa chain lengths, isolated from Streptomyces sp. W112 and bacterial endophytes (Fig. 27).87,94,95 Divergolide J (49, Fig. 26), bearing a partial saturated azepinone ring, has been obtained from the same source and on the similar biosynthetic pathway as those of 48. In turn, olimycin A (50, Fig. 26), isolated from Streptomyces olivaceus SCSIO T05, has the absolute configuration (15R), similarly as 48 and 49, and contains, in contrast to them, the saturated C(17) tail.93,96 Interestingly, a biosynthetic insertion of an O atom into the scaffold of olimycin A is realized by luciferase-type monooxygenase OvmM, an analogous to those producing divergolides (e.g.49), and acting before amide synthase OvmN.96 Inactivation of OvmM lead to production of an open-chain olimycins C and D.96 Structural close to the above ansamycins 48–50, are hygrocins A and B (52, 51; Fig. 26), isolated from the Streptomyces hygroscopicus ATCC25293 and Streptomyces sp. LZ35.97 Structure of hygrocin B (51), of the absolute configuration (15S), which is opposite relative to those of 48–50, was evidenced by 1D and 2D NMR. The biosynthetic pathway of 51 and 52 was based on the formation of the common intermediate prohygrocin and requires the presence of the redox genes hgc2 and hgc4 of the hgc cluster.97 The mutant strains ΔhgcL4, ΔhgcL2 and Δhgc5 and Δhgc11 were shown to be able to produce structurally diverse compounds 51–53 (Fig. 28 and 29). Classes I, II and O Baeyer–Villiger monooxygenases (BVMOs) play a key role in the incorporation of the lactone in the natural scaffolds, whereas oxygenation of prohygrocin is realized by luciferase-like homologue, namely monooxygenase Hgc3 (Fig. 28), of functionality similar to that producing olimycins (48 and 50).97 Compound 49 (Fig. 26) was shown to be inactive toward fungi and bacteria.98 Hygrocins 51 and 52 (Fig. 26) exhibit moderate potency against Aspergillus fumigatus, whereas the latter was also active toward S. pneumoniae.99 This result showed clearly that antibacterial potency is related to the not disturbed flexibility of the ansa bridge. In contrast to hygrocins 51 and 52, hygrocin E (53, Fig. 29) bearing fused core at C(1) with the ansa chain, were tested toward anticancer potency in MDA-MB-231 and PC3 cell lines. These tests did not show, however, the activity up to IC50 = 10 μM.97
Fig. 26 Naphthoquinone-C13 ansamycins, the so-called: divergolide C (48; also named-olimycin B), divergolide J (49), as well as olimycin A (50) and structurally related to them hygrocins B (51) and A (52).
Fig. 27 Biosynthesis of divergolide C (48, olimycin B) with the pivotal role of monooxygenase DivO and oxidoreductase DivQ.
Fig. 28 Biosynthesis of naphthoquinone-C13 hygrocins A (52), B (51) and their structural analog 53 (hygrocin E) of untypically fused the ansa chain with the core at C(1).
Fig. 29 Diastereomeric naphthalene-1(4H)-one-C13 hygrocins E (53), C (54), D (55), H (56), containing identical C(17)-methyl substituent within the ansa chain.
Diastereomeric ansamycins of naphthalene-1(4H)-one-C13 group and decorated with 5-membered lactam moiety, similar as 53, are hygrocins C (54), D (55) and H (56), produced by gdmAI-disrupted Streptomyces sp. LZ35.86,100 These hygrocins are structural different regarding the absolute configuration of the stereogenic center at C(13) and the double bond C(14)C(15), influencing the ansa chain conformation rigidity (Fig. 29).86,100 Structures of 54–56 were elucidated by HR-MS, 1D and 2D NMR as well as X-ray studies (structure of 54).86,100 It should be mentioned that hygrocin H (56), possesses an extra double bond at C(1), as result of dehydration process, in contrast to 53–55. Hygrocins 54–56 showed a quite high cytotoxic potency in MDA-MB-231, PC3 and HeLa cancer cells (IC50s = 0.5–5.0 μM), unlike to 53.86,100 This result suggested the impact of E/Z configuration at C(14) on anticancer activity, whereby the mechanism of this activity has not been revealed yet. This SAR among hygrocins is concerned probably with the fact that E-configuration limits flexibility of the ansa bridge.
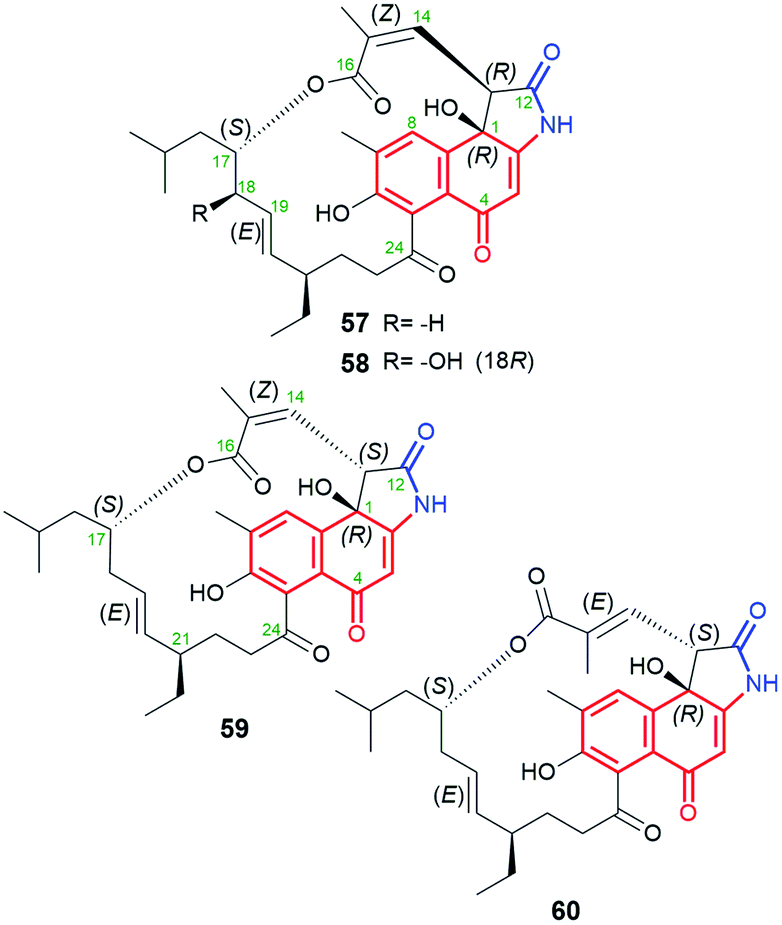
A very similar in structure to the above hygrocins, are divergolides T (57), U (58), V (59) and W (60, Fig. 30), containing also C13-ansa chains and isolated from marine-derived microorganisms Streptomyces sp. KFD18.101 Each of these compounds has an isobutyl group attached at C(17), whereas 59 and 60 have an inverted absolute configuration at C(13) relative to that of 57 and 58. Structures of 57–60 were confirmed by HR-MS, 1D and 2D NMR of them as well as by X-ray of 57. Interestingly, only 57 and 59 having (14Z) configuration, showed good potency in K562, A549, SGC-7901, and HeLa cancer cell lines, at IC50s = 2.8–20.9 μM, and apoptosis-inducing activity.101 This result is in agreement with that obtained for the above-discussed hygrocins 53–56, where only those of the absolute configuration (14Z), i.e. of more conformational labile the ansa bridge, showed notable anticancer potency.101
Fig. 30 Naphthalene-1(4H)-one-C13 stereoisomeric divergolides T (57), U (58), V (59) and W (60), containing identical C(17)-isobutyl substituent within the ansa chain, being produced by marine-derived microorganisms, i.e. Streptomyces sp. KFD18.
5.8. Naphthalenoid-C12 ansamycins
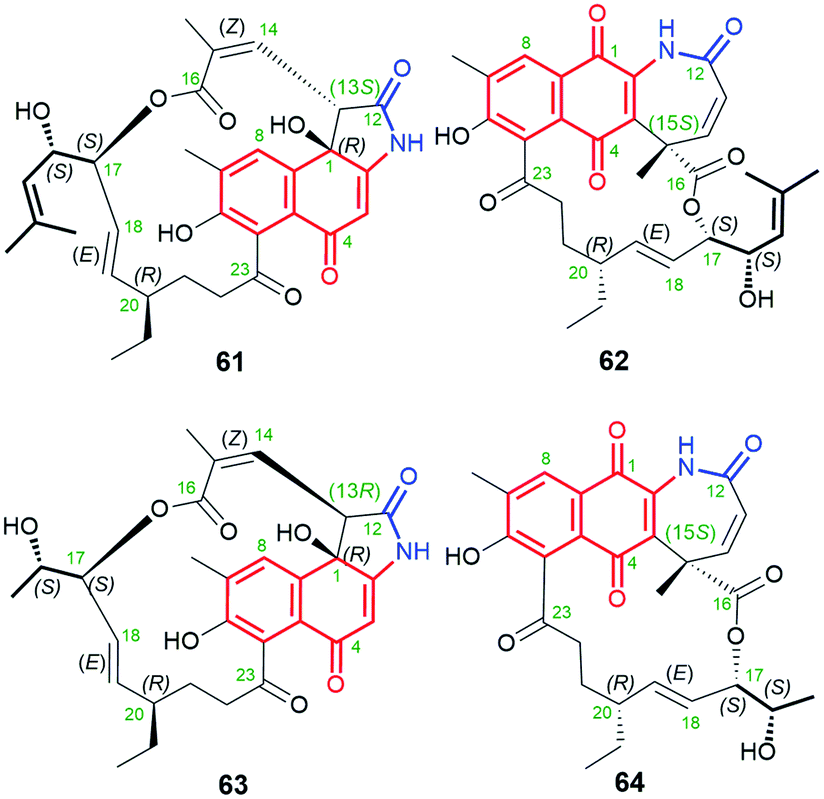
Polyketides, containing naphthalenoid core and C12 ansa chain, are shown in Fig. 31. Divergolide D (61, Fig. 31)91 and hygrocin F (63, Fig. 31)99 have common naphthalen-1(4H)-one core, whereas divergolide I (62, Fig. 31)98 and hygrocin G (64, Fig. 31)99 belong to the naphthoquinone-C12 ansamycins (Fig. 2). All of them have the lactone incorporated into the ansa bridge at C(16), enzymatically formed by a monooxygenases of BVMO-type. The stereochemistry of divergolide 62 has been revised from R into S at C(15), as evidenced by its total synthesis, 1D and 2D NMR, and comparison between X-ray structures and CD spectra of these ansamycins.93,102 It is worth to underlining that, apart from the above structural differences, compounds 61 and 63 also differ in the presence of opposite configurations at C(13). These hygrocins and divergolides possessing 2-propanol or 3-methylbut-2-en-1-ol C(17)-tails of identical stereochemistries were obtained by: the large-scale fermentation of Streptomyces sp. HKI0576 and HKI0595 associated with the mangrove tree Bruguiera gymnorrhiza (61, 62),91 overexpression of div8 in Streptomyces sp. W112 (61)87 and isolation from gdmAI-disrupted Streptomyces sp. LZ35 (62, 63 and 64).100 The biosynthetic pathway of these naphthalenoid-C12 ansamycins, despite suspected to be analogous to those of other hygrocins or divergolides of naphthalenoid-C13 group, was not however revealed or discussed. Compounds 63 and 61 (IC50s ranging from 1 to 2 μM) showed high anticancer activities in MDA-MB-231, PC3, as well as LXFA629L, PANC-1, RXF486L and Saos-2 cancer cells.91,100 Neither ansamycin 62 nor 64 was found to exhibit essential antifungal or anticancer potencies. Among tested divergolides with C12 and C13 chains (divergolides I–N), divergolide I (62) of shorter C12 ansa chain showed the strongest antibacterial activity (the best GIZs > 13 mm) against B. subtilis, S. aureus (MRSA), E. faecalis (vancomycin-resistant) and Mycobacterium vaccae.98 Thus, the profile of biological potency of C12 hygrocins and divergolides seems to be dependent on the type of the core, i.e. 1,4-naphthoquinone – antibacterial and naphthalen-1(4H)-one – anticancer.
Fig. 31 Members of naphthalenoid-C12 ansamycins group: divergolides D (61) and I (62) as well as hygrocins F (63) and G (64) of naphthalen-1(4H)-one or 1,4-naphthoquinone cores.
5.9. Naphthalenoid ansamycins of the shortest C11 ansa chains
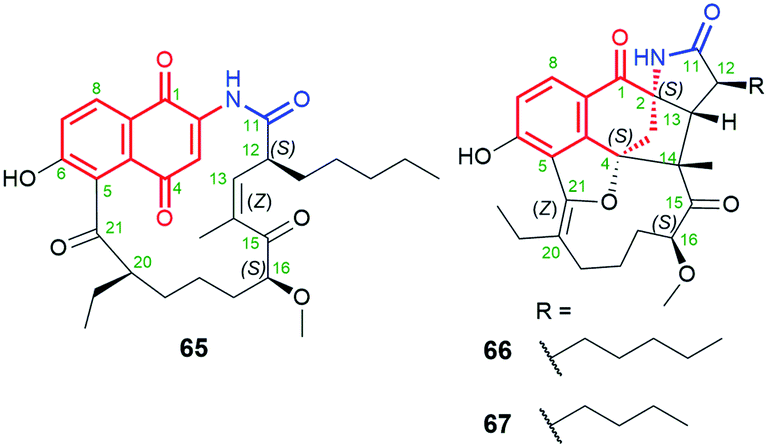
The naphthoquinone core is a predominant central motif in natural and the rarest ansamycin scaffolds of the shortest C11 ansa chains of all known naphthalenoid ansamycins. Neoansamycin A (65, Fig. 32) is a type of natural 1,4-naphthoquinone-C11 ansamycin, whereas neoansamycins B and C (66 and 67, respectively; Fig. 32), formed from 65, belong to 3,4-dihydronaphthalen-1(2H)-one-C11 ansamycins (Fig. 33).103,104 The X-ray structure of 66 provided evidence for stereochemistry of this-type of novel neoansamycins.104 These natural products were obtained through the activation of a cryptic ansamycin gene cluster in Streptomyces sp. LZ35.104 Experiments with a mutant strain Streptomyces sp. LZ35, revealed that nam7 hydroxylase is the key enzyme in switching between biosynthetic routes leading to the alternative formation of benzenoid ansamycins or neoansamycins in Streptomyces SR201nam1OEΔnam7.103,104 The long alkyl chains at C(12) in neoansamycins A–C structures suggested that PKS module 7 is able to incorporate carbon skeleton not only of n-butylmalonyl-CoA but also n-pentylmalonyl-CoA unit (Fig. 33). Antiproliferative tests indicated clearly that potency of 66, bearing the ansa chain of almost no flexibility, is lower than that of 65 (IC50 (SW480) = 6.7 μM). Surprisingly, the antibacterial screening of ansamycins revealed that both 65 and 66 showed potent activities towards S. aureus ATCC 25923 at low MICs = 3.125 μg mL−1.104 This result suggested that the antibacterial and anticancer mechanisms of these compounds are totally different.
Fig. 32 Structures of neoansamycins A–C (65–67), produced by nam7-disrupted Streptomyces sp. LZ35 SR201nam1OE strain.
Fig. 33 Biosynthesis of neoansamycins A and B (65 and 66) by nam7-disrupted Streptomyces sp. LZ35 SR201nam1OE strain.
6. Semisynthetic transformations of naphthalenoid ansamycins and their biological potency
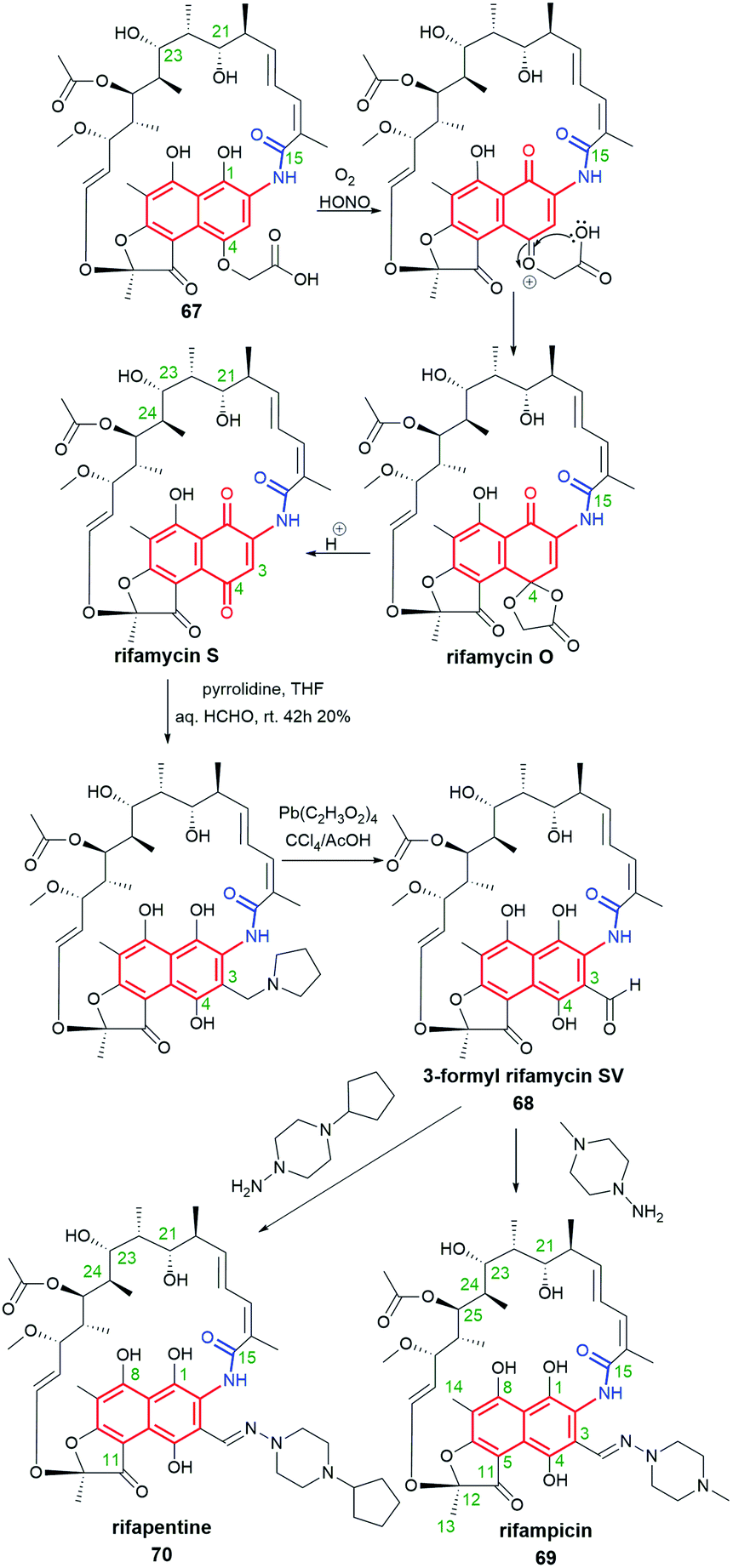
Despite many natural-occurring naphthalenoid ansamycins revealed important antibacterial or other type activities, among them are also those possessing relatively weak biological activities, whose semisynthetic modifications help to improve their biological relevance. Here an excellent example is natural rifamycin B (67, Fig. 34), of weak antibacterial potency, which subjected to semisynthetic structural alterations via structure of 3-formyl rifamycin SV (68), yielded one of the best known naphthalene-C17 semisynthetic antitubercular antibiotic – rifampicin (in US called also rifampin, 69, Fig. 34).3,105 The other reason why semisynthetic transformations are desired among the naphthalenoid rifamycins is concerned with the resistance of bacterial strains, including multidrug-resistance (MDR) to semisynthetic antibiotics as 69 or its congeners.10,106 Resistance of different bacteria to naphthalenoid-C17 rifamycins is realized via e.g. mutations of single amino acid residues of the target, ribosylation or glucosylation of hydroxyl groups of ansamycin, modification/hydrolysis of the C(3) arm of ansamycin, or via activation of the efflux pump systems in bacterial cells.107–109 In turn, modifications of rifamycin structures and/or the target performed by bacteria prevent stable binding of these ansamycins to the allosteric site of RNAPs, limits the permeability of the antibiotic through the natural cell barriers, or enhance affinity to efflux pump systems thereby decreasing the degree of accumulation of the antibiotic in bacterial cells.55,110 Recently it has been shown, that the loss of 69 antibacterial activity is a result of N-hydroxylation performed by flavoproteins such as rifampicin monooxygenase (RIFMO), co-working with NADPH, as evidenced by spectroscopic monitoring.111 Furthermore, the discovered novel metabolites of 69, being a result of the ansa bridge cleavage performed by monooxygenases (ROXs) from Streptomyces venezuelae, confirmed that structural alterations within naphthalenoid ansamycin scaffold are ‘double-edged swords’, not only useful for human at designing novel antibiotics but also for bacteria as a defense response against newly developed antibiotics.112,113 Thus, in view of this still-emerging problem of bacteria resistance, in order to achieve chemical stability, improved binding mode with the target, and better balanced physico-chemical properties, semisynthetic modifications of naphthalenoid ansamycins have been widely performed, especially among natural naphthalene-C17, 1,4-naphthoquinone-C17 and 4-iminonaphthalene-1(4H)-one-C17 scaffolds.
Fig. 34 Semisynthesis of potent antibiotics, rifampicin (69) and rifapentine (70), from natural rifamycin B (67) via intermediate 3-formylrifamycin SV (68), developed by Sensi et al.
6.1. Naphthalene-C17 ansamycins
It should be underscored here that only semisynthetic modifications of natural rifamycins have played a major role in pharmaceutical applications. A combined synthetic and genetic manipulation strategy with A. mediterranei S699 (AT6 mutant), targeting the ansa bridge, yielded rifamycin S and 69, both lacking methyl at C(24).114 These new C(24)-desmethyl derivatives showed improved activity against rifampicin-resistant M. tuberculosis, containing RNAPs with single amino acid mutations H526 → T526 and S531 → L531 (Table 1), compared to 69 (MICs > 50 μg mL−1). A such biological outcome suggested that the loss of C(24)-methyl, increasing the ansa bridge flexibility, and the presence of C(3)-arm of rifamycin-like derivatives, contribute to enhanced antitubercular potency against resistant strains.
Table 1Antibacterial potency of semisynthetic naphthalene-C17 ansamycins
3-Formyl-rifamycin SV (68, Fig. 34), an intermediate in synthesis of rifampicin, has been found to be an excellent starting point for >300 semisynthetic modifications.50 Other well-known and used in therapy semisynthetic analogs of 69 include: rifapentine (70, Fig. 34), a drug active against Gram-positive bacteria, and rifaximin (Fig. 7), active mainly against Gram-negative bacteria and used for the treatment of irritable bowel syndrome and hepatic encephalopathy.115–117
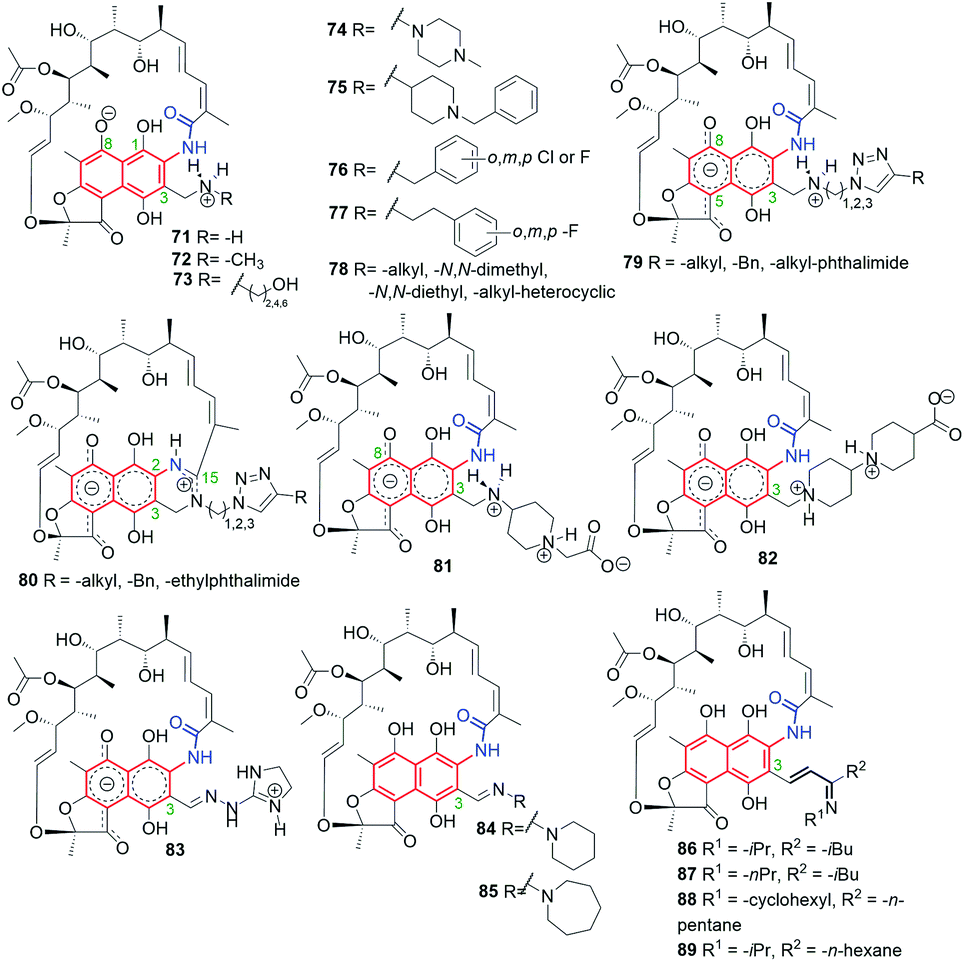
Comparison of X-ray structures of 69 in protic and aprotic solvents showed zwitterionic and non-ionic forms, respectively.48 Semisyntheses of amine products 71–78, via reductive alkylation of 68, gave zwitterions with the positively charged substituent at C(3) (Fig. 35).48,53 An analogous result to the above one was obtained for the crown-ether analogs (Fig. 8), where the protonation site was dependent on the basicity of the C(3) arm and their antibacterial activities were improved as a result of complexing them with small organic molecules, as e.g. histamine, (MICS. aureus = 0.125 μg mL; MICS. epidermidis = 0.125 μg mL−1), in the form of host–guest supramolecular complexes.49,118 Comparison between antibacterial potency of 69 with those of its amine analogs 71–78, revealed that a rigid and basic structure of the C(3) arm is important for high antibacterial properties.48,53 Derivatives, structurally similar to zwitterionic 71–78, obtained by Brucoli et al., were screened toward antibacterial potency against 69-resistant M. tuberculosis clinical isolates and “hypervirulent” HN-878 strains.119 Simple amine derivative 68 with benzylamine showed potency (MIC90 = 0.02 μM) comparable with 69 against HN-878 strain. In turn, other simple amine derivative of 68 with tryptamine exhibited better potency (MIC90 = 0.3 μM) than 69 against M. tuberculosis resistant strain (RpoBS522L). Furthermore, in this work an alternative binding region at RNAPs to that of 69 was proposed.119
Fig. 35 Semisynthetic analogs of 69, obtained from 68, and bearing different substituents at C(3) arm.
The most energy favorable binding models for 69 and its reduced hydrazine analog 74, containing also C(3)-piperazine arm, were postulated taking into account an extra interaction between the zwitterionic form of the antibiotic and a highly conserved E445 of RNAPs (Fig. 9).48,53 Such docking models were in line with the highest antibacterial potencies of these rifamycins among all studied. Furthermore, this result was also in agreement with attractive antimycobacterial activities of 75 (Fig. 35) and its hydrochloride salt. The biological result was explainable since C(3) arms of 75 and 75xHCl are able to interact with E445 of RNAPs, which was reflected in comparable potencies against M. bovis and M. tuberculosis H37Rv strains with those of 69 (Table 1).58
New C(3)-amine-triazole analogs of 79-type (Fig. 35) were not so antibacterial active as 75 and its salt (Table 1). Ansamycins of 79-type underwent an unexpected intramolecular cyclization between the lactam of the ansa chain and the C(3) arm, catalyzed by Cu2+, yielding zwitterionic amidine derivatives of 80-type. Novel cyclized and non-cyclized compounds, containing phthalimide moiety at C(3), showed comparable or improved antibacterial activity toward M. bovis, relative to 69.58 Recently, the assumption about a crucial interaction between the rifamycin C(3) arm and E445 (of RNAPs) led to obtain series of amine and hydrazone congeners of 81–83 types, with the attached basic and rigid substituents at C(3).60 Compounds 81 and 82 showed polar, double zwitterionic structures in solution, as indicated by NMR studies (Fig. 35).60 Amine congener 82 and hydrazone congener 83, displaying a good balance between solubility and lipophilicity and revealing favorable binding mode with RNAPs, showed markedly higher antibacterial potencies against S. aureus MRSA and MLSB than did the reference antibiotic 69 (Table 1).60 Interestingly, non-ionic and a more lipophilic hydrazone congeners 84 and 85 (Fig. 35), showed 16-fold better antibacterial potency against E. faecalis strain than 69, at still good antibacterial potencies toward resistant S. aureus. It should be also added that 82 and 83 were also more potent than 69 against E. faecalis (by 8-fold).60 Thus, the presence at C(3) rigid and basic substituents and achieving of well-balanced lipophilicity vs. water solubility for rifampicin-like derivatives provided high antibacterial potency not only against mycobacteria but also against other resistant pathogens. Taking into account the resistant mechanisms realized by bacteria, very potent and double zwitterionic derivative 82, bearing non-hydrolyzable C(3)-arm, seems to be very attractive toward future medicinal applications.
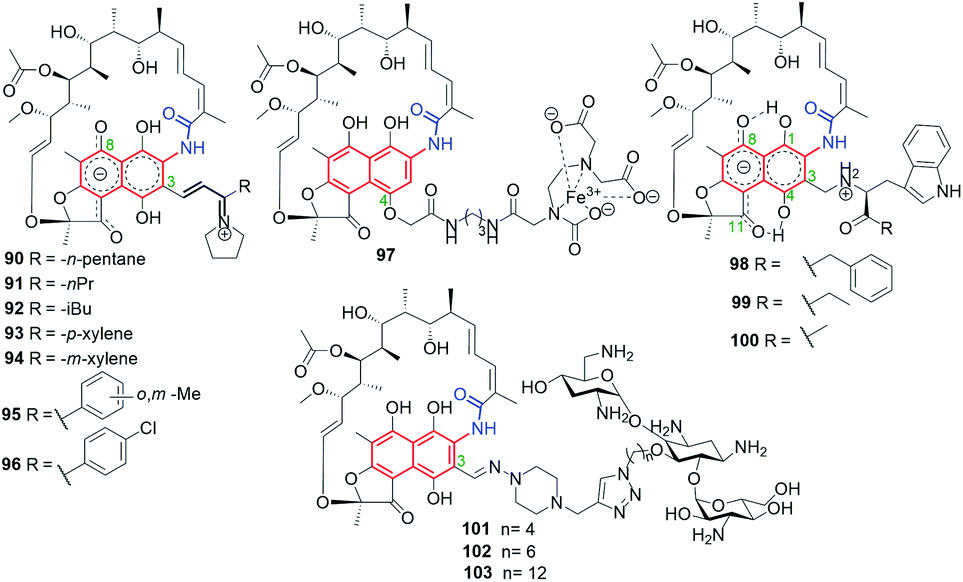
Bujnowski, Synoradzki and co-workers synthesized non-ionic C(3)-imine unsaturated rifamycin derivatives 86–89 (Fig. 35) by deploying a new protocol involving imine isomerization.121 This method has been extended toward being used for reactions with pyrrolidine and yielded dipolar products 90–96 (Fig. 36).122 Derivatives 86–92, due to their chemical instability in water, cannot be good drug candidates, in contrast to derivatives 93–96, which have shown good antimycobacterial activities, although not so improved, if compared to 69 or rifabutin.122
Fig. 36 Semisynthetic iminium analogs of 69 (90–96), and hybrid connections of rifamycins with EDTA (97), L-amino acid esters (98–100) and tobramycin (101–103).
Hybrid connections of rifamycin scaffold have been also obtained via initial functionalization not only the C(3) but also the C(4) position (Fig. 36).59,120,123 The conjugate 97, bearing a strongly complexing EDTA component, was proposed as an example of an “aggressive” agent, breaking polypeptide and polynucleotide chains near the areas of their binding to RNAP through the Fenton reaction.123 Semisynthetic hybrids with L-amino acid esters, which selected examples were shown in Fig. 36 (98–100), revealed structure–activity relationship (SAR) in dependence of the bulkiness of the substituent at the ester portion.59 The most potent of these compounds, derivative 100, with a small substituent at the ester moiety of L-Trp, showed very good antibacterial potency, on the level comparable to that of 69 against S. aureus, S. epidermidis and MRSA strains (Table 1).59 This result seems to be in line with that obtained for similar pharmacophore, i.e. tryptamine, attached at C(3) of rifamycin scaffold, which revealed also potent antibacterial activity.119 The molecular docking suggested that the loss of high antibacterial potency of rifamycin hybrids with L-amino acid esters in case of their bulky ester substituents is a result of steric hindrance with A447 of the RNAP binding pocket.
Click chemistry strategy was applied to obtain heterodimeric 101–103 rifamycin–tobramycin conjugates linked via triazole bridges (Fig. 36).120 This type conjugates, in dependence on the length of the linker, were effective against Gram-negative Pseudomonas aeruginosa efflux-deficient mutants, resistant to doxycycline and chloramphenicol. Furthermore, one of this-type semisynthetic conjugates showed improved potency relative to tobramycin against S. pneumoniae strain (Table 1), in case when the linker was the shortest (101). Thus, rifamycin-tobramycin hybrids can be useful for overcome the resistance of a Gram-negative P. aeruginosa to chloramphenicol and doxycycline in vitro and in vivo.
6.2. 1,4-Naphthoquinone-C17 and 4-iminonaphthalen-1(4H)-one-C17 ansamycins
Semisynthetic C(25)-carbamate derivatives 104–106 (Fig. 37), decorated at C(3) a morpholine moiety, belong to 1,4-naphthoquinone-C17 ansamycins.124 These ansamycins were shown to be even much more efficient than 69 against rapidly-growing mycobacteria such as M. abscessus, M. chelonae, M. fortuitum and M. smegmatis (Table 2). The most potent antimycobacterial potency showed piperidine derivative 106, however, the explanation of this result, in view of the known binding modes of rifamycins with RNAPs (Fig. 9) and taking into regard bacteria resistance mechanisms via attachment of bulky groups to the ansa bridge, is difficult.
Fig. 37 Semisynthetic analogs of 69, inspired by rifabutin (107) and rifalazil (108) structures.
Table 2Antibacterial potency of 4-iminonaphthalen-1(4H)-one-C17 ansamycins
Rifabutin (107, Fig. 9 and 37) is capable of inhibiting bacterial RNAPs and is derived from natural rifamycin S, for which further natural transformations, especifically those yielding rifamycin O, are mediated by transketolase Rif15 and the cytochrome P450 enzyme Rif16.127–130 High antibiotic activities of clinically-used 107, against both Gram-positive and Gram-negative bacteria, created opportunity to produce alternatives relative to 69 and encouraged further exploration of semisynthetic transformations of iminoquinone ansamycins, assigned to the 4-iminonaphthalen-1(4H)-one-C17 group (Fig. 2).109,130–132 Rifalazil (108) is an excellent example of a such semisynthetic ansamycin showing bactericidal potency against M. tuberculosis, Chlamydia trachomatis, Clostridioides difficile or Helicobacter pylori as stand alone antibiotic or with combination with the other antibiotics as e.g. vancomycin.10,133 Despite 108 helps to overcome resistance to other antibiotics, its toxic effects limited the clinical applications. Hence, novel semisynthetic benzoxazinorifamycins 109–111 (Fig. 37), being analogs of rifalazil and displaying high levels of affinity to RNAPs of wild-type and 69-resistant mutants of M. tuberculosis, were obtained from rifamycin S.46,125 Compound 109 showed good metabolic stability. Structures of complexes between 109 or 110 and RNAPs indicated important interaction of C(3)–C(4) tails with a σ-finger of RNAPs (Fig. 9c), disturbing in this way the template DNA position in RNAP channels.44,46 Furthermore, derivatives 109–111 showed comparable abilities at inhibiting transcription relative to those of 69 or 108 in mutant E. coli and M. tuberculosis strains.46 Antibacterial assays of 109–111 in aerobic (MABA) and anaerobic (LORA) conditions against M. tuberculosis H37Rv showed that the most potent is compound 109, even more active than 69 but lower active than 108 (Table 2). The most potent 109 inhibited the transcription process in vitro, catalysed by a wild-type M. tuberculosis RNAPs, at comparable IC50s ∼ 0.01 μM to those of 108.125 Furthermore, RNAPs of the mutants M. tuberculosis (D435 → V and S450 → L) were also better inhibited by 109 than by 108. Semisynthetic derivatives of kanglemycin A (13, Fig. 14) described as 112–115 in Fig. 37, with modified end-group of C(3)/C(4) benzoxazino tails or at the K-acid portion at C(20) (the so-called Kang-amides), showed attractive potencies against wild-type and 69-resistant S. aureus strains (Table 2).126 These studies indicated the presence of K-acid and the type of substituent at piperazine ring to be important for activity against 69-resistant bacteria, whereas the introduced benzoxazino moieties were shown to improve in vivo efficacy.126 The best antibacterial potencies against mutant S. aureus strains (H481 → Y) were noted for 112 and 113 ansamycins, which showed markedly improved properties relative to those of 13 and 108 (Table 2).
7. Conclusions
Our proposed division of natural and semisynthetic ansamycins (Fig. 2, top) based on the type of core and length of the ansa chain has neatly demonstrated the high levels of structural diversity of naphthalenoid ansamycins (23 structural subgroups). We also propose to use the term “ansamycins” as a family of macrolactams, bearing relatively rigid central cores as benzenoid, naphthalenoid, or atypical ones (e.g. heterocyclic, alicyclic), originated from AHBA or other-type small semisynthetic precursors (mutasynthons), and produced in a result of cooperation between polyketide (type I) and amide synthases. Due to the improved methods of extraction and genetic manipulations novel natural naphthalenoid ansamycin-like scaffolds, containing fused cores with heterocycles are available. Biosynthetic plasticity of naphthalenoid ansamycins is well reflected in the structural diversity of many naphthalenoid ansamycin congeners as divergolides, hygrocins, olimycins or 11,12-seco-rifamycin W analogs, bearing not only the lactam but also the lactone within the ansa bridge structure. Furthermore, there are also known examples of atypical fusion of the ansa chain with the core at C(1), as e.g. for hygrocin E or oligocyclic cores fused with the ansa chain, as noted for ansawaricin E and ansaeomycins. Among natural naphthalenoid ansamycins the shortest ansa chain is C11 (neoansamycins A–C) whereas the longest is C23 (naphthomycins L–Q).
The antibacterial potency of natural naphthalenoid ansamycins is moderate in most cases so the semisynthetic modifications of natural scaffolds are often required. One of exceptions are aminoansamycins, belonging to napththoquinone-C17 group, which antibacterial potencies are extremely high (even MICs = 0.0001 μM). It is interesting to note that mutasynthesis, often-used tool at optimization of anticancer activity of benzenoid ansamycins, has not been employed widely up to now in aim to modify the natural scaffolds of naphthalenoid ansamycins. Many natural ansamycin precursors containing cleavaged ansa chain have been found in bacterial extracts as e.g. ansavaricins F–I, and despite most of these precursors being relatively biologically inactive or of moderate potency, their macrolactamization and peripheral group modifications could provide alternatives of enhanced biological properties. The semisynthetic approach to modify naphthalenoid ansamycin natural scaffolds with the use of known ‘starting points’ as rifamycin SV, 3-formyl rifamycin SV, rifamycin S or kanglemycin A is predominant, in contrast to benzenoid ansamycins, where mutasynthesis is the main source of their structural diversity. It is noted also that some naphthalenoid ansamycins are called by different names for the identical structure as e.g., divergolide C and olimycin B.
Overview on the biological properties of naphthalenoid ansamycins shows that these compounds (especially semisynthetic derivatives) are predominantly powerful antibacterial agents, especially those of C17 ansa chains, against both Gram-positive and Gram-negative bacteria, although notable other-type potency as anticancer was also found for e.g. hygrocins 53–56. The review revealed the following dependence that with increasing length of the ansa chain within the naphthalenoid ansamycins, the change in biological potency profile from the anticancer into the antibacterial occur. Furthermore, the profile of biological activity of structurally close C12 hygrocins and divergolides, i.e. antibacterial or anticancer, seems to be dependent also on the type of the core. However, crucial for antibacterial potency of naphthalenoid ansamycins is conformational flexible ansa-bridge enabling adaptation of the molecule conformation (regulation of polarity/lipophilicity) to the outer environment during transportation to the target and enabling the molecular recognition of the ansamycin with the target. The best recognized molecular targets of naphthalenoid ansamycins are bacterial RNA-polymerases (RNAPs), which inhibition is related to antibacterial effects. Thus, target-directed designing of novel naphthalenoid ansamycin scaffolds, is reasonable way to obtain powerful antibacterials, even better than the clinically-used antibiotics as rifampicin (69), as demonstrated on semisynthetic C(3)-amine analogs, bearing rigid and basic amine arms, enabling zwitterionization. Interestingly, rifampicin-like derivatives possess the ability to interact with viral proteins, which are important at assembling viral membranes, therefore these compounds can be considered at designing novel antiviral agents.
8. Conflicts of interest
There are no conflicts to declare.
9. Acknowledgements
The authors are grateful for financial support from the Polish National Science Centre (NCN)-OPUS 13 project no. UMO-2017/25/B/ST5/00291.
10. Notes and references
V. Prelog and W. Oppolzer, Helv. Chim. Acta, 1973, 56, 2279–2287 CrossRefCASPubMed.
A. Lüttringhaus and H. Gralheer, Justus Liebigs Ann. Chem., 1942, 550, 67–98 CrossRef.
P. Sensi, A. M. Greco and R. Ballotta, Antibiot. Annu., 1959, 7, 262–270 Search PubMed.
P. Sensi, M. T. Timbal and G. Maffh, Experientia, 1960, 16, 412 CrossRefCASPubMed.
S. Bala, R. Khanna, M. Dadhwal, S. R. Prabagaran, S. Shivaji, J. Cullum and R. Lal, Int. J. Syst. Evol. Microbiol., 2004, 54, 1145–1149 CrossRefCASPubMed.
A. Whitty, M. Zhong, L. Viarengo, D. Beglov, D. R. Hall and S. Vajda, Drug Discovery Today, 2016, 21, 712–717 CrossRefCASPubMed.
M. Rossi Sebastiano, B. C. Doak, M. Backlund, V. Poongavanam, B. Over, G. Ermondi, G. Caron, P. Matsson and J. Kihlberg, J. Med. Chem., 2018, 61, 4189–4202 CrossRefCASPubMed.
V. Poongavanam, Y. Atilaw, S. Ye, L. H. E. Wieske, M. Erdelyi, G. Ermondi, G. Caron and J. Kihlberg, J. Pharm. Sci., 2021, 110, 301–313 CrossRefCASPubMed.
S. Funayama and G. A. Cordell, in Studies in Natural Products Chemistry, Elsevier, 2000, vol. 23, pp. 51–106 Search PubMed.
P. A. Aristoff, G. A. Garcia, P. D. Kirchhoff and H. D. Hollis Showalter, Tuberculosis, 2010, 90, 94–118 CrossRefCASPubMed.
J.-X. Zhang, X.-Y. Wang and K.-H. Tang, Chin. J. Antibiot., 2009, 34, 588–592 CAS.
H. G. Floss and T.-W. Yu, Chem. Rev., 2005, 105, 621–632 CrossRefCASPubMed.
H. B. Rode, D. M. Lade, R. Grée, P. S. Mainkar and S. Chandrasekhar, Org. Biomol. Chem., 2019, 17, 5428–5459 RSC.
Q. Kang, Y. Shen and L. Bai, Nat. Prod. Rep., 2012, 29, 243–263 RSC.
K. Watanabe, M. A. Rude, C. T. Walsh and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9774–9778 CrossRefCASPubMed.
C. G. Kim, A. Kirschning, P. Bergon, Y. Ahn, J. J. Wang, M. Shibuya and H. G. Floss, J. Am. Chem. Soc., 1992, 114, 4941–4943 CrossRefCAS.
C.-G. Kim, A. Kirschning, P. Bergon, P. Zhou, E. Su, B. Sauerbrei, S. Ning, Y. Ahn, M. Breuer, E. Leistner and H. G. Floss, J. Am. Chem. Soc., 1996, 118, 7486–7491 CrossRefCAS.
Biosynthesis, ed. J. W. Corcoran, Springer, Berlin, Heidelberg, 1981 Search PubMed.
S. Funayama, A. Nakagawa and S. Omura, Sendai, 1986, 28, 73–78 Search PubMed.
W. D. Celmer, Ann. N. Y. Acad. Sci., 1986, 471, 299–303 CrossRefCAS.
B. J. Rawlings, Nat. Prod. Rep., 2001, 18, 231–281 RSC.
J. Peek, M. Lilic, D. Montiel, A. Milshteyn, I. Woodworth, J. B. Biggins, M. A. Ternei, P. Y. Calle, M. Danziger, T. Warrier, K. Saito, N. Braffman, A. Fay, M. S. Glickman, S. A. Darst, E. A. Campbell and S. F. Brady, Nat. Commun., 2018, 9, 4147 CrossRefPubMed.
S. T. Toenjes and J. L. Gustafson, Future Med. Chem., 2018, 10, 409–422 CrossRefCASPubMed.
N. Skrzypczak, K. Pyta, P. Ruszkowski, M. Gdaniec, F. Bartl and P. Przybylski, Eur. J. Med. Chem., 2020, 202, 112624 CrossRefCASPubMed.
J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, Angew. Chem., Int. Ed., 2009, 48, 6398–6401 CrossRefCASPubMed.
S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505–513 CrossRefCASPubMed.
A. H. J. Wang, I. C. Paul, K. L. Rinehart and F. J. Antosz, J. Am. Chem. Soc., 1971, 93, 6275–6276 CrossRefCASPubMed.
K. L. Rinehart, W. M. Knoll, K. Kakinuma, F. J. Antosz, I. C. Paul, A. H. J. Wang, F. Reusser, L. H. Li and W. C. Krueger, J. Am. Chem. Soc., 1975, 97, 196–198 CrossRefCASPubMed.
L. Santos, M. A. Medeiros, S. Santos, M. C. Costa, R. Tavares and M. J. M. Curto, J. Mol. Struct., 2001, 563–564, 61–78 CrossRefCAS.
E. Rubio, I. Merino, A.-B. García, M.-P. Cabal, C. Ribas and M. Bayod-Jasanada, Magn. Reson. Chem., 2005, 43, 269–282 CrossRefCASPubMed.
L. Santos, F. Fant, M. A. Medeiros, F. a. M. Borremans, M. C. Costa and M. J. M. Curto, Magn. Reson. Chem., 2000, 38, 937–945 CrossRefCAS.
P. Przybylski, K. Pyta, K. Klich, W. Schilf and B. Kamieński, Magn. Reson. Chem., 2014, 52, 10–21 CrossRefCASPubMed.
E. Danelius, V. Poongavanam, S. Peintner, L. Wieske, M. Erdelyi and J. Kihlberg, Chem.–Eur. J., 2020, 26, 5231–5244 CrossRefCASPubMed.
K. Pyta, A. Janas, N. Skrzypczak, W. Schilf, B. Wicher, M. Gdaniec, F. Bartl and P. Przybylski, ACS Infect. Dis., 2019, 5, 1754–1763 CrossRefCASPubMed.
A. Bacchi and G. Pelizzi, J. Comput.-Aided Mol. Des., 1999, 13, 385–396 CrossRefCASPubMed.
V. Molodtsov, I. N. Nawarathne, N. T. Scharf, P. D. Kirchhoff, H. D. H. Showalter, G. A. Garcia and K. S. Murakami, J. Med. Chem., 2013, 56, 4758–4763 CrossRefCASPubMed.
V. Molodtsov, N. T. Scharf, M. A. Stefan, G. A. Garcia and K. S. Murakami, Mol. Microbiol., 2017, 103, 1034–1045 CrossRefCASPubMed.
K. Pyta, P. Przybylski, B. Wicher, M. Gdaniec and J. Stefańska, Org. Biomol. Chem., 2012, 10, 2385 RSC.
K. Pyta, P. Przybylski and F. Bartl, ChemPhysChem, 2015, 16, 938–942 CrossRefCASPubMed.
K. Bujnowski, L. Synoradzki, R. C. Darłak, T. A. Zevaco and E. Dinjus, RSC Adv., 2016, 6, 114758–114772 RSC.
B. Wicher, K. Pyta, P. Przybylski, E. Tykarska and M. Gdaniec, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, o209–o212 CrossRefCASPubMed.
B. Wicher, K. Pyta, P. Przybylski and M. Gdaniec, Cryst. Growth Des., 2018, 18, 742–754 CrossRefCAS.
K. Pyta, P. Przybylski, K. Klich and J. Stefańska, Org. Biomol. Chem., 2012, 10, 8283 RSC.
K. Zhou, J. Li and D. S. Zheng, J. Mol. Struct., 2010, 983, 27–31 CrossRefCAS.
M. R. Naylor, A. M. Ly, M. J. Handford, D. P. Ramos, C. R. Pye, A. Furukawa, V. G. Klein, R. P. Noland, Q. Edmondson, A. C. Turmon, W. M. Hewitt, J. Schwochert, C. E. Townsend, C. N. Kelly, M.-J. Blanco and R. S. Lokey, J. Med. Chem., 2018, 61, 11169–11182 CrossRefCASPubMed.
E. A. Campbell, O. Pavlova, N. Zenkin, F. Leon, H. Irschik, R. Jansen, K. Severinov and S. A. Darst, EMBO J., 2005, 24, 674–682 CrossRefCASPubMed.
E. A. Campbell, N. Korzheva, A. Mustaev, K. Murakami, S. Nair, A. Goldfarb and S. A. Darst, Cell, 2001, 104, 901–912 CrossRefCASPubMed.
K. Pyta, K. Klich, J. Domagalska and P. Przybylski, Eur. J. Med. Chem., 2014, 84, 651–676 CrossRefCASPubMed.
D. Czerwonka, J. Domagalska, K. Pyta, M. M. Kubicka, P. Pecyna, M. Gajecka and P. Przybylski, Eur. J. Med. Chem., 2016, 116, 216–221 CrossRefCASPubMed.
K. Pyta, A. Janas, M. Szukowska, P. Pecyna, M. Jaworska, M. Gajecka, F. Bartl and P. Przybylski, Eur. J. Med. Chem., 2019, 167, 96–104 CrossRefCASPubMed.
I. Artsimovitch, M. N. Vassylyeva, D. Svetlov, V. Svetlov, A. Perederina, N. Igarashi, N. Matsugaki, S. Wakatsuki, T. H. Tahirov and D. G. Vassylyev, Cell, 2005, 122, 351–363 CrossRefCASPubMed.
D. Garriga, S. Headey, C. Accurso, M. Gunzburg, M. Scanlon and F. Coulibaly, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8424–8429 CrossRefCASPubMed.
G. Li, J. Zhang, Q. Guo, J. Wei, Y. Jiang, X. Zhao, L.-L. Zhao, Z. Liu, J. Lu and K. Wan, J. Antibiot., 2015, 68, 431–435 CrossRefCASPubMed.
D. S. Raj, D. Kumar Kesavan, N. Muthusamy and S. Umamaheswari, Mater. Today: Proc., 2021, 45, 2976–2981 CAS.
G. Pelizza, M. Nebuloni, P. Ferrari and G. Gallo, Il Farmaco; Edizione Scientifica's, 1977, 32, 471–481 CAS.
G. C. Viscomi, M. Campana, M. Barbanti, F. Grepioni, M. Polito, D. Confortini, G. Rosini, P. Righi, V. Cannata and D. Braga, CrystEngComm, 2008, 10, 1074–1081 RSC.
M. M. de Villiers, M. R. Caira, J. Li, S. J. Strydom, S. A. Bourne and W. Liebenberg, Mol. Pharmaceutics, 2011, 8, 877–888 CrossRefCASPubMed.
C. Blandizzi, G. C. Viscomi and C. Scarpignato, Drug Des., Dev. Ther., 2014, 9, 1–11 Search PubMed.
L. de Pinho Pessoa Nogueira, Y. S. de Oliveira, J. d. C. Fonseca, W. S. Costa, F. N. Raffin, J. Ellena and A. P. Ayala, J. Mol. Struct., 2018, 1155, 260–266 CrossRef.
Y.-H. Yang, X.-L. Fu, L.-Q. Li, Y. Zeng, C.-Y. Li, Y.-N. He and P.-J. Zhao, J. Nat. Prod., 2012, 75, 1409–1413 CrossRefCASPubMed.
Y.-H. Yang, D.-S. Yang, G.-H. Li, R. Liu, X.-W. Huang, K.-Q. Zhang and P.-J. Zhao, Fitoterapia, 2018, 130, 17–25 CrossRefCASPubMed.
Y. S. Xiao, B. Zhang, M. Zhang, Z. K. Guo, X. Z. Deng, J. Shi, W. Li, R. H. Jiao, R. X. Tan and H. M. Ge, Org. Biomol. Chem., 2017, 15, 3909–3916 RSC.
Z. Zhang, J. Zhang, R. Song, Z. Guo, H. Wang, J. Zhu, C. Lu and Y. Shen, RSC Adv., 2017, 7, 5684–5693 RSC.
H. Mosaei, V. Molodtsov, B. Kepplinger, J. Harbottle, C. W. Moon, R. E. Jeeves, L. Ceccaroni, Y. Shin, S. Morton-Laing, E. C. L. Marrs, C. Wills, W. Clegg, Y. Yuzenkova, J. D. Perry, J. Bacon, J. Errington, N. E. E. Allenby, M. J. Hall, K. S. Murakami and N. Zenkin, Mol. Cell, 2018, 72, 263–274 CrossRefCASPubMed.
M. E. Rateb, W. E. Houssen, M. Arnold, M. H. Abdelrahman, H. Deng, W. T. A. Harrison, C. K. Okoro, J. A. Asenjo, B. A. Andrews, G. Ferguson, A. T. Bull, M. Goodfellow, R. Ebel and M. Jaspars, J. Nat. Prod., 2011, 74, 1491–1499 CrossRefCASPubMed.
J. F. Castro, V. Razmilic, J. P. Gomez-Escribano, B. Andrews, J. Asenjo and M. Bibb, Antonie van Leeuwenhoek, 2018, 111, 1433–1448 CrossRefPubMed.
M. Chen and W. R. Roush, J. Org. Chem., 2013, 78, 3–8 CrossRefCASPubMed.
Y. Liu, X. Chen, Z. Li, W. Xu, W. Tao, J. Wu, J. Yang, Z. Deng and Y. Sun, ACS Chem. Biol., 2017, 12, 2589–2597 CrossRefCASPubMed.
G. Sun, C. Hu, Q. Mei, M. Luo, X. Chen, Z. Li, Y. Liu, Z. Deng, Z. Zhang and Y. Sun, Nat. Commun., 2020, 11, 4501 CrossRefCASPubMed.
D. E. Williams, D. S. Dalisay, J. Chen, E. A. Polishchuck, B. O. Patrick, G. Narula, M. Ko, Y. Av-Gay, H. Li, N. Magarvey and R. J. Andersen, Org. Lett., 2017, 19, 766–769 CrossRefCASPubMed.
Y. Shi, J. Zhang, X. Tian, X. Wu, T. Li, C. Lu and Y. Shen, Org. Lett., 2019, 21, 900–903 CrossRefCASPubMed.
F. Ye, Y. Shi, S. Zhao, Z. Li, H. Wang, C. Lu and Y. Shen, Biomolecules, 2020, 10, 1265 CrossRefCASPubMed.
Z. Zhang, X. Wu, R. Song, J. Zhang, H. Wang, J. Zhu, C. Lu and Y. Shen, RSC Adv., 2017, 7, 14857–14867 RSC.
S. H. Liu, Y. Y. Wei, Y. N. Xing, Y. Chen, W. Wang, K. B. Wang, Y. Liang, R. H. Jiao, B. Zhang and H. M. Ge, Org. Lett., 2021, 23, 3724–3728 CrossRefCASPubMed.
S. Li, C. Lu, J. Ou, J. Deng and Y. Shen, RSC Adv., 2015, 5, 83843–83846 RSC.
G. Zhao, S. Li, Z. Guo, M. Sun and C. Lu, RSC Adv., 2015, 5, 98209–98214 RSC.
M. C. Wilson, S.-J. Nam, T. A. M. Gulder, C. A. Kauffman, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2011, 133, 1971–1977 CrossRefCASPubMed.
A. Lechner, M. C. Wilson, Y. H. Ban, J. Hwang, Y. J. Yoon and B. S. Moore, ACS Synth. Biol., 2013, 2, 379–383 CrossRefCASPubMed.
T. C. Le, I. Yang, Y. J. Yoon, S.-J. Nam and W. Fenical, Org. Lett., 2016, 18, 2256–2259 CrossRefCASPubMed.
L. Ding, A. Maier, H.-H. Fiebig, H. Görls, W.-H. Lin, G. Peschel and C. Hertweck, Angew. Chem., Int. Ed., 2011, 50, 1630–1634 CrossRefCASPubMed.
Z. Xu, L. Ding and C. Hertweck, Angew. Chem., Int. Ed., 2011, 50, 4667–4670 CrossRefCASPubMed.
C. Sun, C. Zhang, X. Qin, X. Wei, Q. Liu, Q. Li and J. Ju, Tetrahedron, 2018, 74, 199–203 CrossRefCAS.
Z. Xu, M. Baunach, L. Ding, H. Peng, J. Franke and C. Hertweck, ChemBioChem, 2014, 15, 1274–1279 CrossRefCASPubMed.
M. Pai, M. A. Behr, D. Dowdy, K. Dheda, M. Divangahi, C. C. Boehme, A. Ginsberg, S. Swaminathan, M. Spigelman, H. Getahun, D. Menzies and M. Raviglione, Nat. Rev. Dis. Primers, 2016, 2, 1–23 Search PubMed.
A. Tupin, M. Gualtieri, F. Roquet-Banères, Z. Morichaud, K. Brodolin and J.-P. Leonetti, Int. J. Antimicrob. Agents, 2010, 35, 519–523 CrossRefCASPubMed.
P. Spanogiannopoulos, M. Thaker, K. Koteva, N. Waglechner and G. D. Wright, Antimicrob. Agents Chemother., 2012, 56, 5061–5069 CrossRefCASPubMed.
R. Figueiredo, D. F. Ramos, C. Moiteiro, M. A. Medeiros, M. J. Marcelo Curto, J. Cardoso de Menezes, R. H. Pando, P. E. A. Silva and M. do C. Costa, Eur. J. Med. Chem., 2012, 47, 186–193 CrossRefCASPubMed.
J. Sarathy, V. Dartois, T. Dick and M. Gengenbacher, Antimicrob. Agents Chemother., 2013, 57, 1648–1653 CrossRefCASPubMed.
L.-K. Liu, H. Abdelwahab, J. S. Martin Del Campo, R. Mehra-Chaudhary, P. Sobrado and J. J. Tanner, J. Biol. Chem., 2016, 291, 21553–21562 CrossRefCASPubMed.
K. Koteva, G. Cox, J. K. Kelso, M. D. Surette, H. L. Zubyk, L. Ejim, P. Stogios, A. Savchenko, D. Sørensen and G. D. Wright, Cell Chem. Biol., 2018, 25, 403–412 CrossRefCASPubMed.
Y. Hoshino, S. Fujii, H. Shinonaga, K. Arai, F. Saito, T. Fukai, H. Satoh, Y. Miyazaki and J. Ishikawa, J. Antibiot., 2010, 63, 23–28 CrossRefCASPubMed.
A. Nigam, K. H. Almabruk, A. Saxena, J. Yang, U. Mukherjee, H. Kaur, P. Kohli, R. Kumari, P. Singh, L. N. Zakharov, Y. Singh, T. Mahmud and R. Lal, J. Biol. Chem., 2014, 289, 21142–21152 CrossRefCASPubMed.
H. L. Koo and H. L. DuPont, Curr. Opin. Gastroenterol., 2010, 26, 17–25 CrossRefCASPubMed.
F. Calanni, C. Renzulli, M. Barbanti and G. C. Viscomi, J. Antibiot., 2014, 67, 667–670 CrossRefCASPubMed.
O. Alfarisi, W. A. Alghamdi, M. H. Al-Shaer, K. E. Dooley and C. A. Peloquin, Expert Rev. Clin. Pharmacol., 2017, 10, 1027–1036 CrossRefCASPubMed.
P. Przybylski, K. Pyta, D. Czerwonka, M. M. Kubicka and M. Gajecka, Bioorg. Med. Chem. Lett., 2015, 25, 3903–3909 CrossRefCASPubMed.
M. Zloh, M. Gupta, T. Parish and F. Brucoli, Eur. J. Med. Chem., 2021, 225, 113734 CrossRefCASPubMed.
T. Idowu, G. Arthur, G. G. Zhanel and F. Schweizer, Eur. J. Med. Chem., 2019, 174, 16–32 CrossRefCASPubMed.
K. Bujnowski, L. Synoradzki, T. Zevaco and E. Dinjus, Tetrahedron, 2012, 68, 5925–5934 CrossRefCAS.
K. Bujnowski, L. Synoradzki, T. A. Zevaco, E. Dinjus, E. Augustynowicz-Kopeć and A. Napiórkowska, Tetrahedron, 2015, 71, 158–169 CrossRefCAS.
M. Kozlov, E. Nudler, V. Nikiforov and A. Mustaev, Bioconjugate Chem., 2013, 24, 443–447 CrossRefCASPubMed.
K. D. Combrink, A. R. Ramos, S. Spring, S. Schmidl, K. Elizondo, P. Morin, B. De Jesus and F. P. Maurer, Bioorg. Med. Chem. Lett., 2019, 29, 2112–2115 CrossRefCASPubMed.
S. K. Gill, H. Xu, P. D. Kirchhoff, T. Cierpicki, A. J. Turbiak, B. Wan, N. Zhang, K.-W. Peng, S. G. Franzblau, G. A. Garcia and H. D. H. Showalter, J. Med. Chem., 2012, 55, 3814–3826 CrossRefCASPubMed.
J. Peek, J. Xu, H. Wang, S. Suryavanshi, M. Zimmerman, R. Russo, S. Park, D. S. Perlin and S. F. Brady, ACS Infect. Dis., 2020, 6, 2431–2440 CrossRefCASPubMed.
F. Qi, C. Lei, F. Li, X. Zhang, J. Wang, W. Zhang, Z. Fan, W. Li, G.-L. Tang, Y. Xiao, G. Zhao and S. Li, Nat. Commun., 2018, 9, 2342 CrossRefPubMed.
A. Mejía, D. Luna, F. J. Fernández, J. Barrios-González, L. H. Gutierrez, A. G. Reyes, A. E. Absalón and S. Kelly, 3 Biotech, 2018, 8, 456 CrossRefPubMed.
S. A. Löw, B. M. Nestl, M. J. Weissenborn, F. Zepeck and B. Hauer, Org. Process Res. Dev., 2015, 19, 1544–1547 CrossRef.
T. Dick, Front. Microbiol., 2020, 11, 1–2 CrossRefPubMed.
D. J. Horne, C. Spitters and M. Narita, Int. J. Tuberc. Lung Dis., 2011, 15, 1485 CrossRefCASPubMed.
M. J. Maslow and C. Portal-Celhay, in Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases, ed. J. E. Bennett, R. Dolin, M. J. Blaser and W. B. Saunders, Philadelphia, 8th edn, 2015, pp. 339–349 Search PubMed.

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*


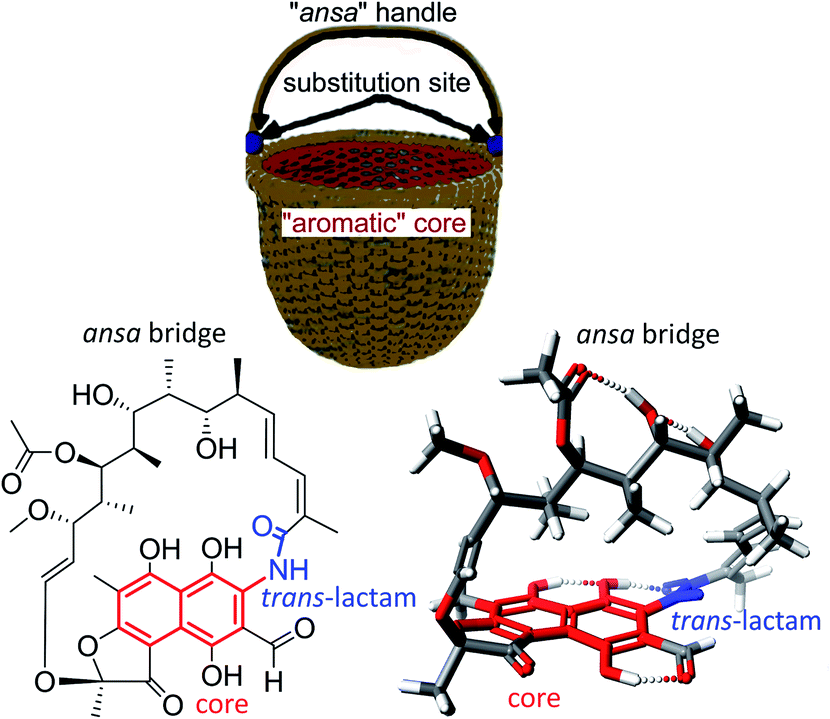
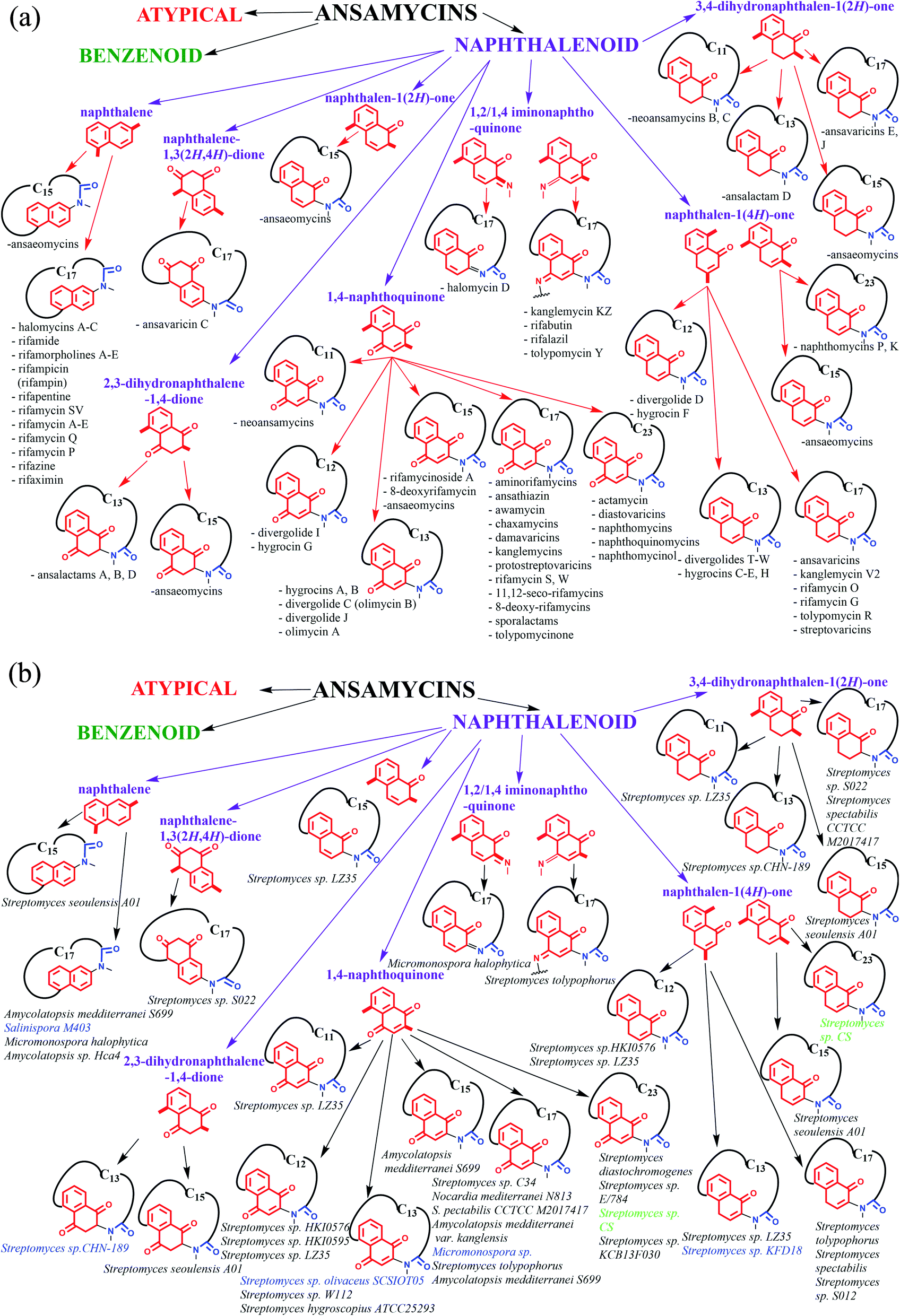
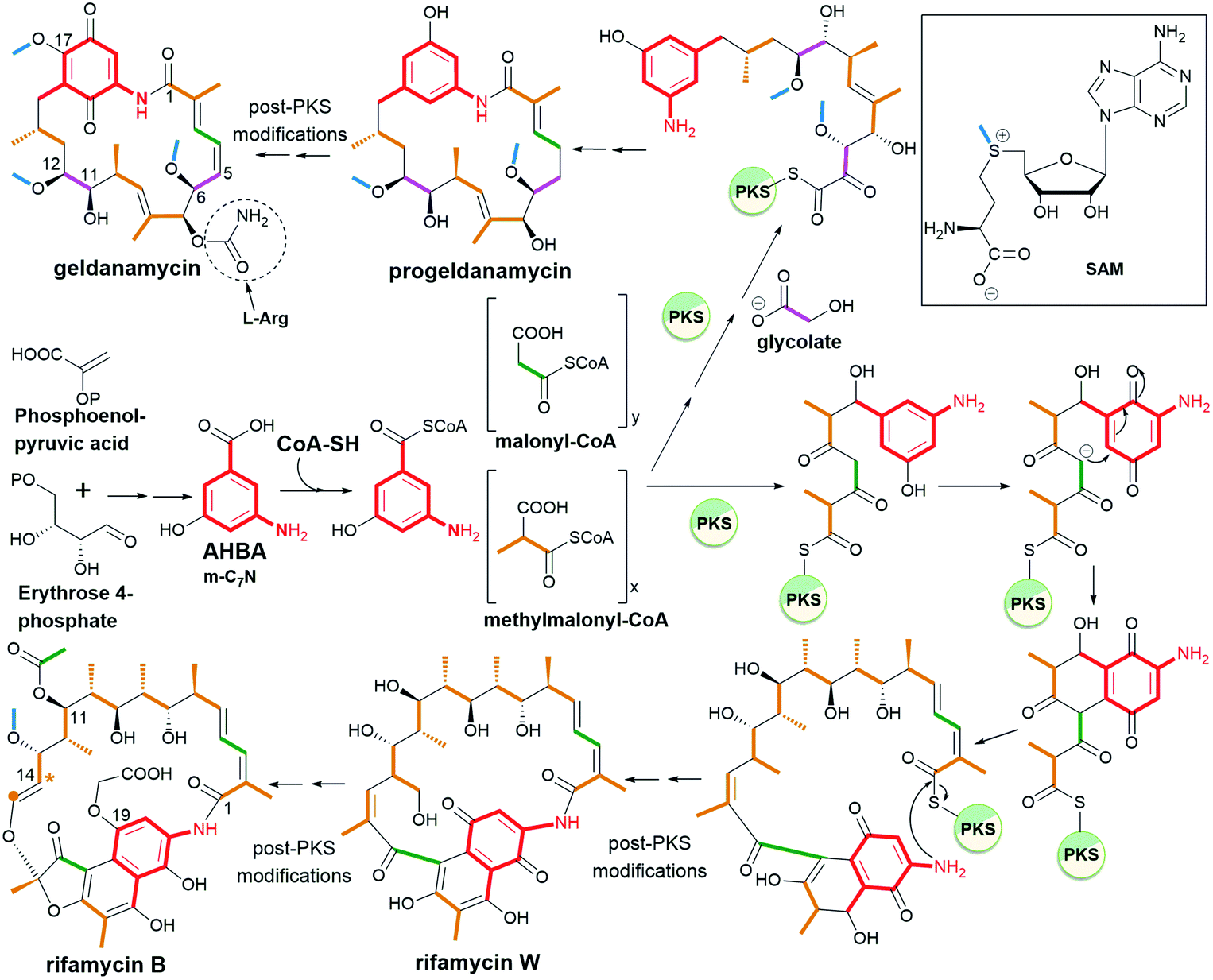
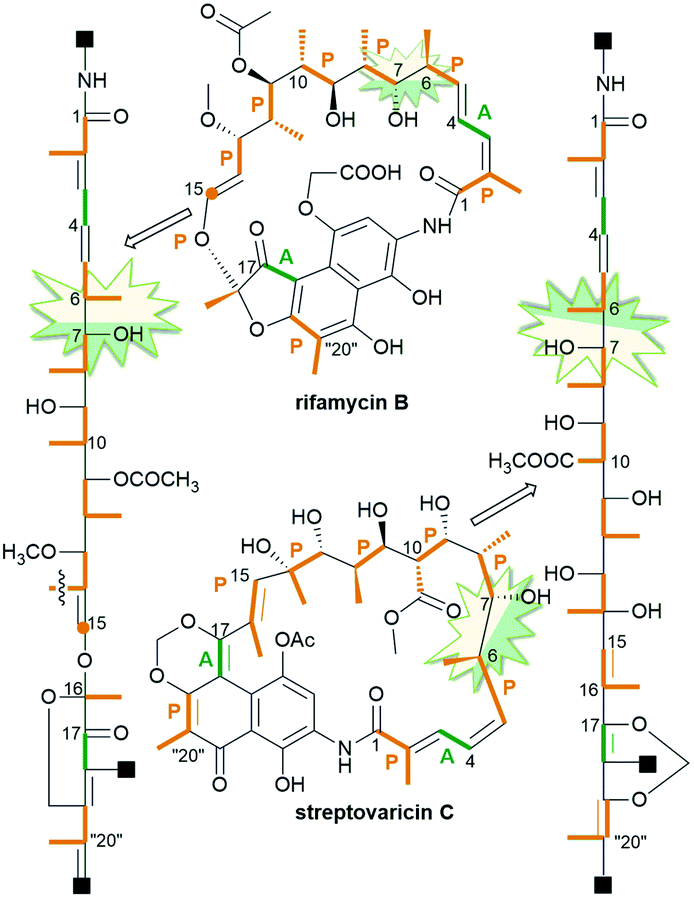
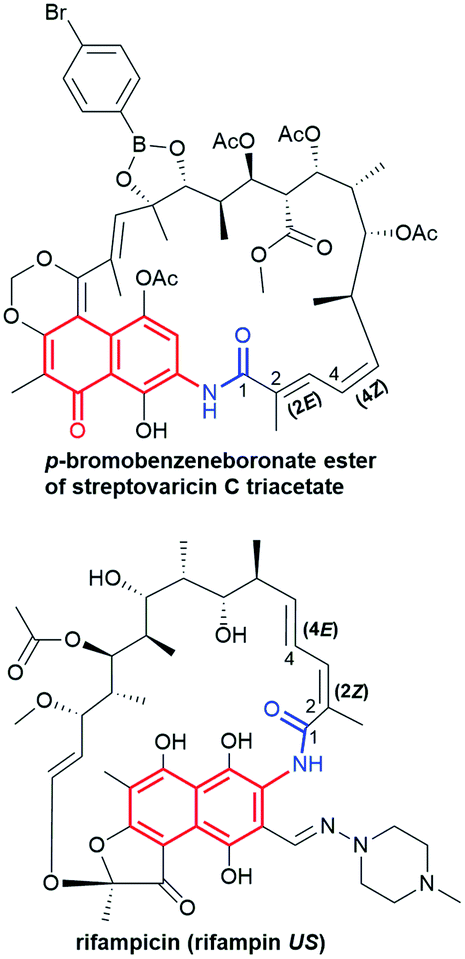
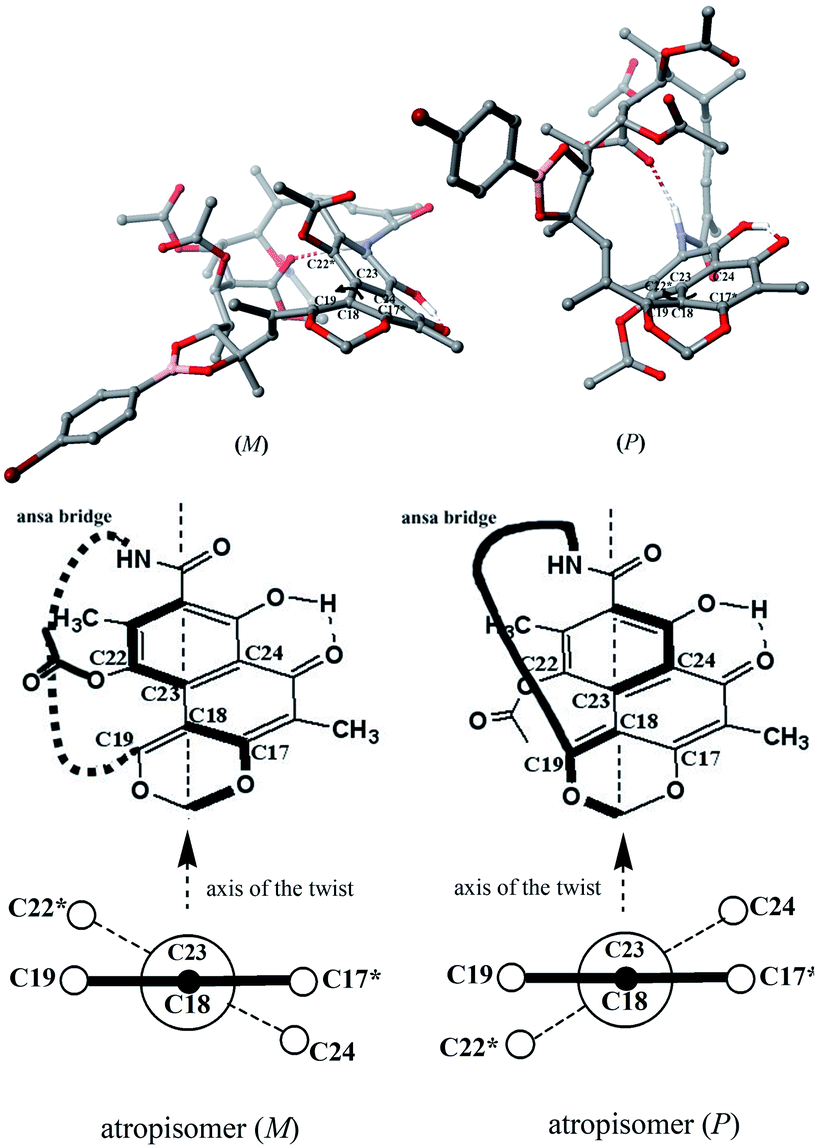

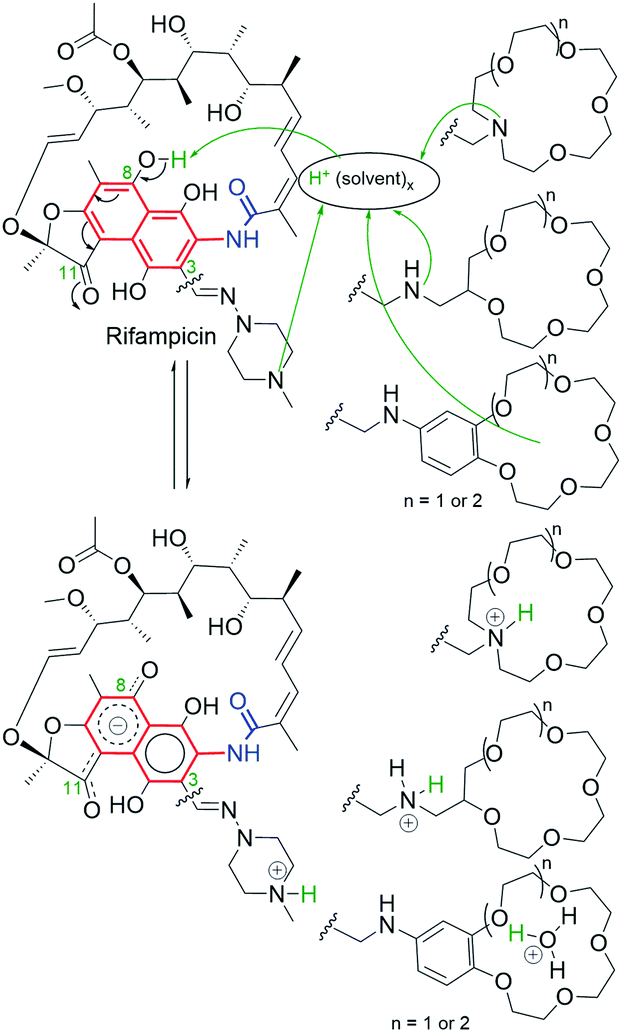
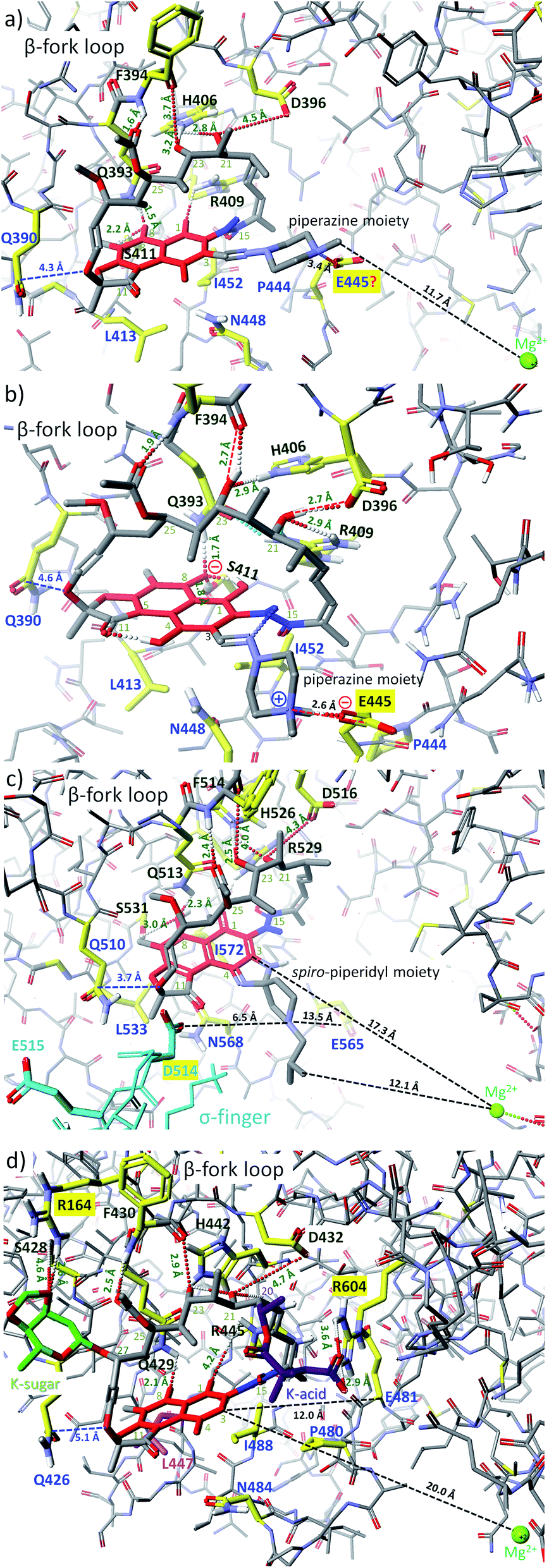
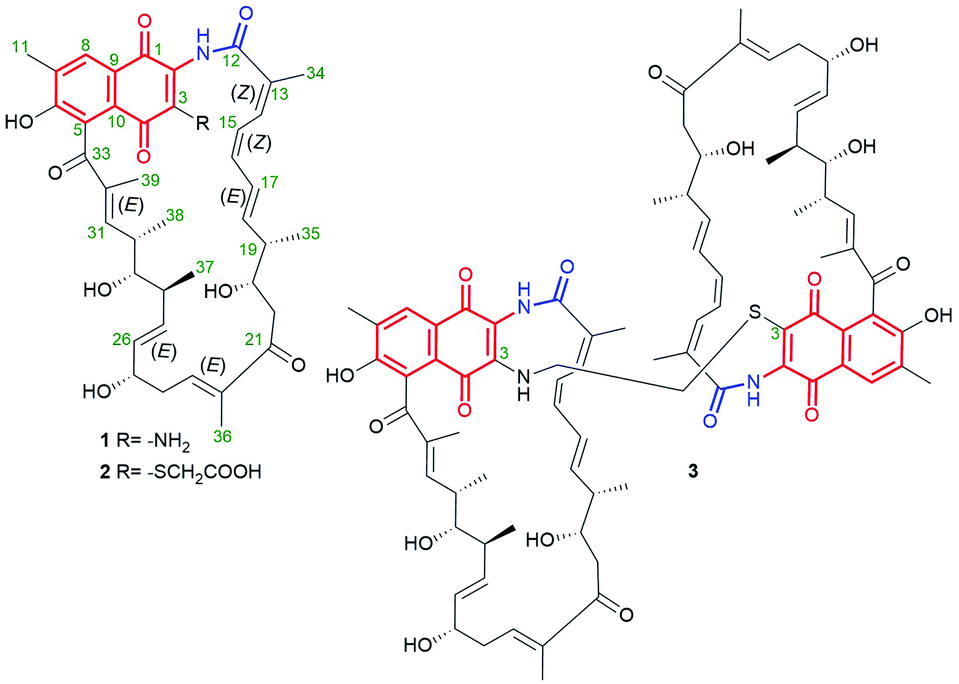
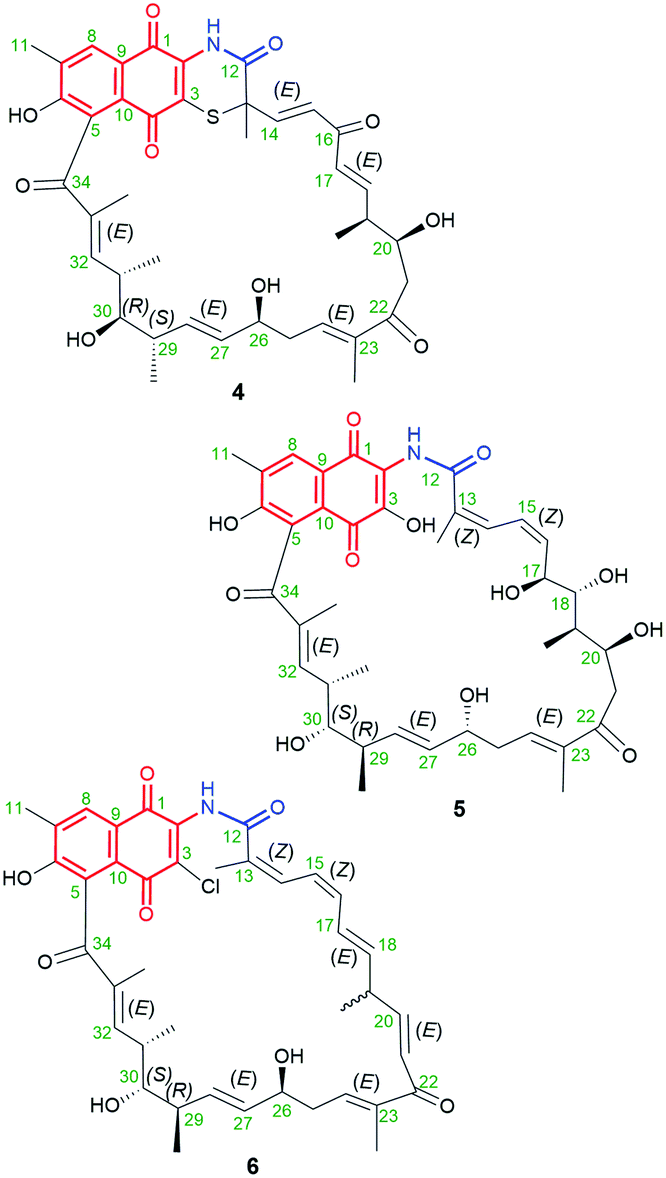
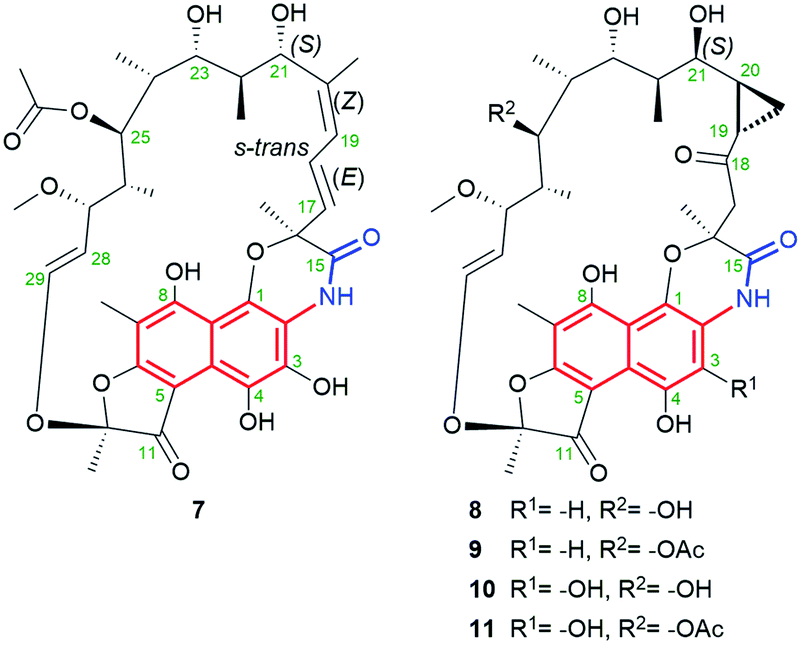
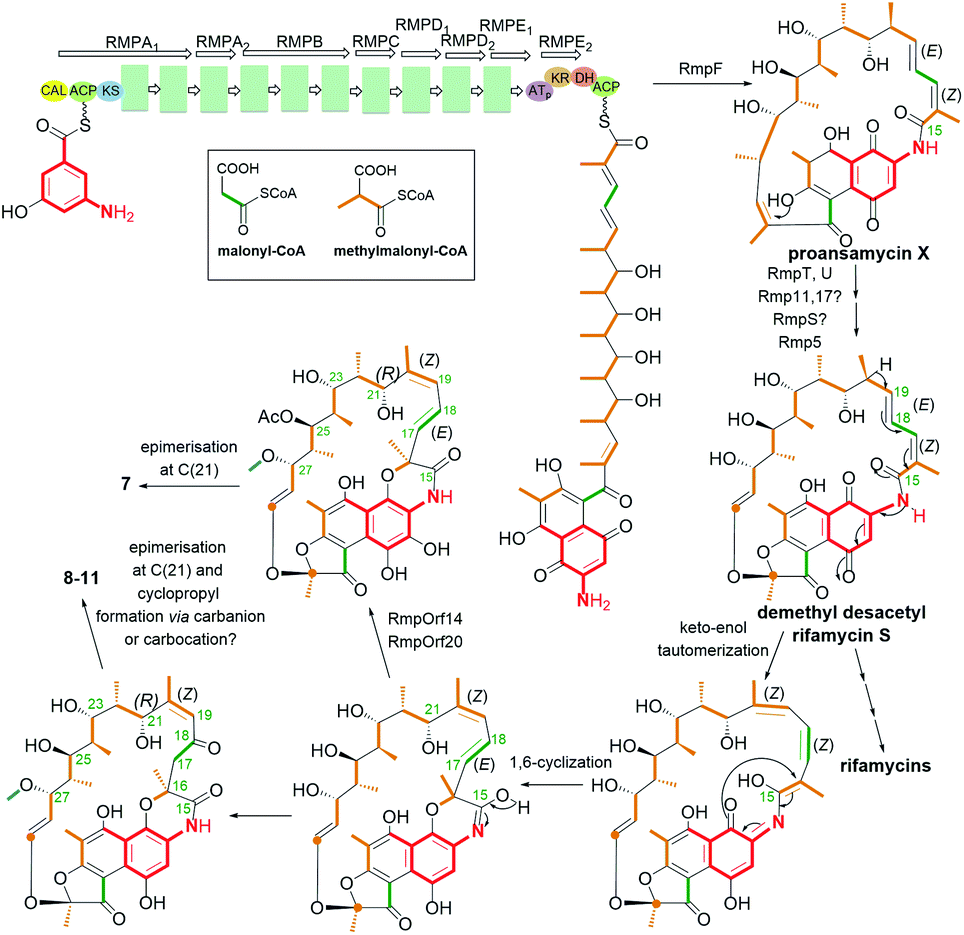
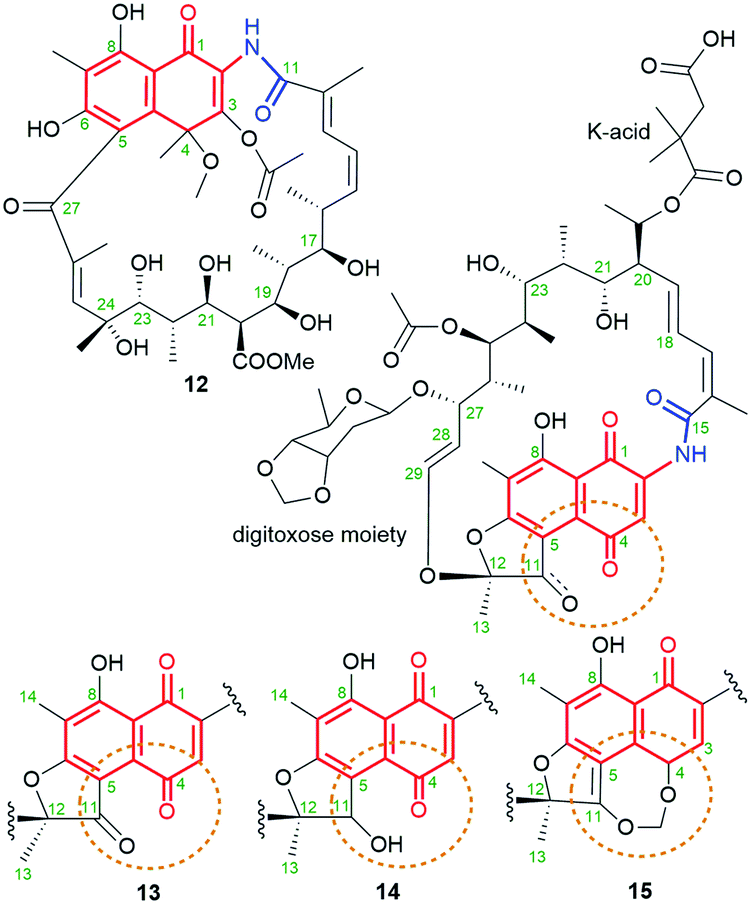
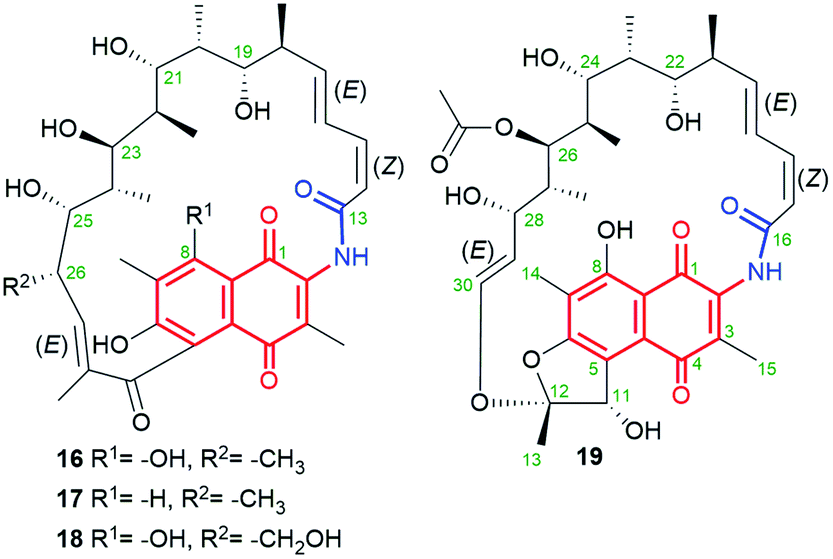

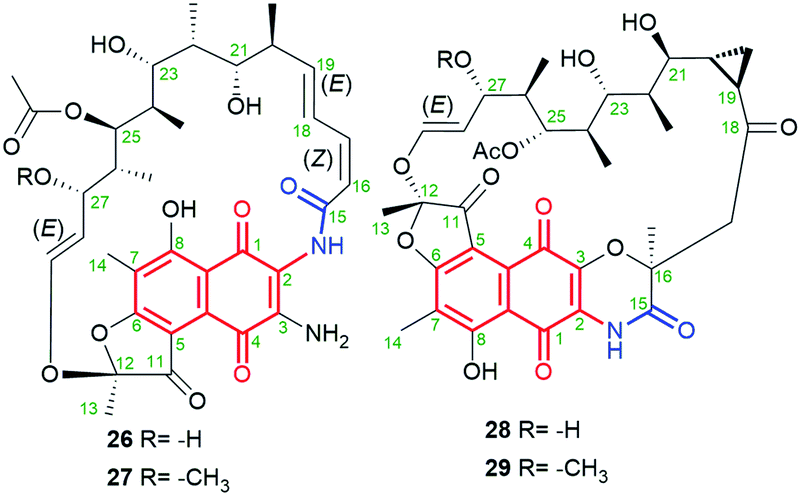
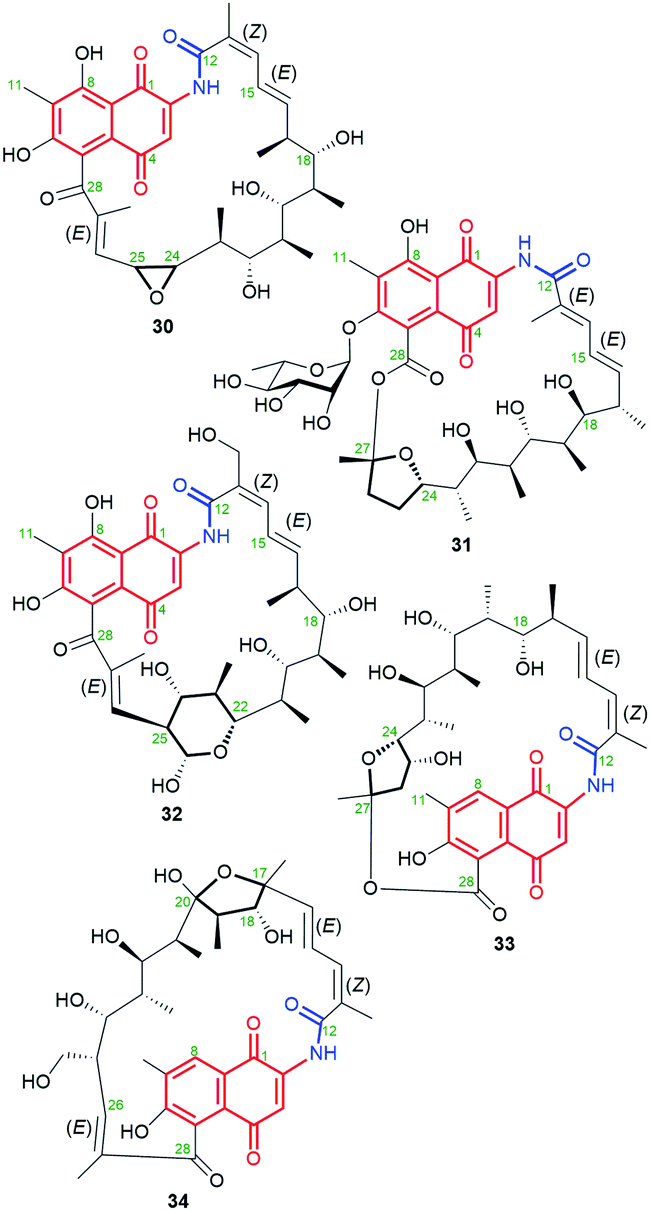

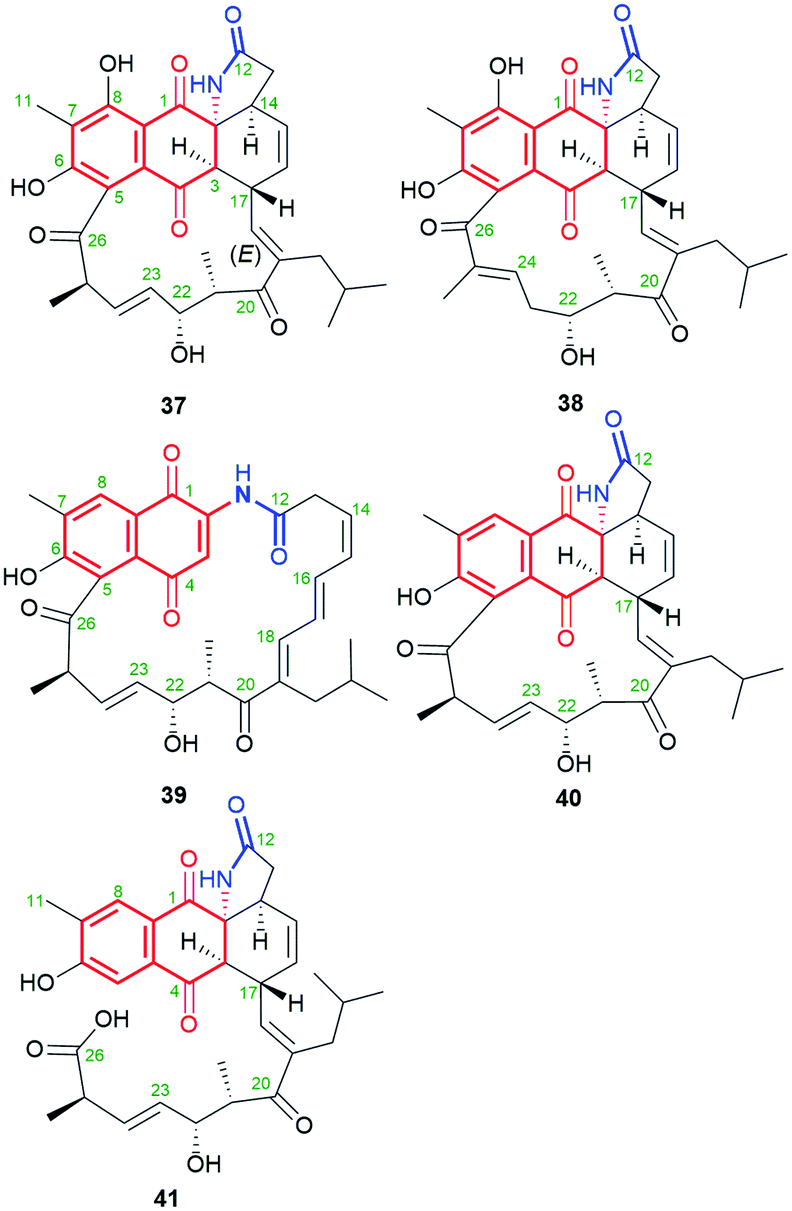
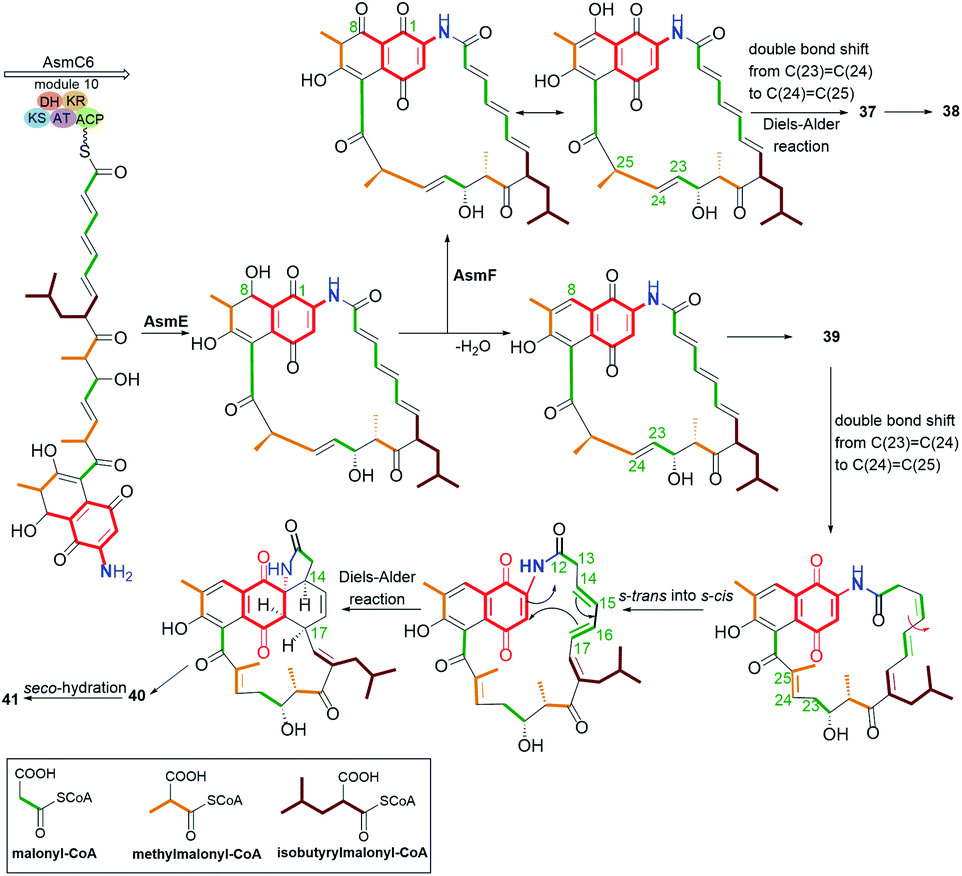
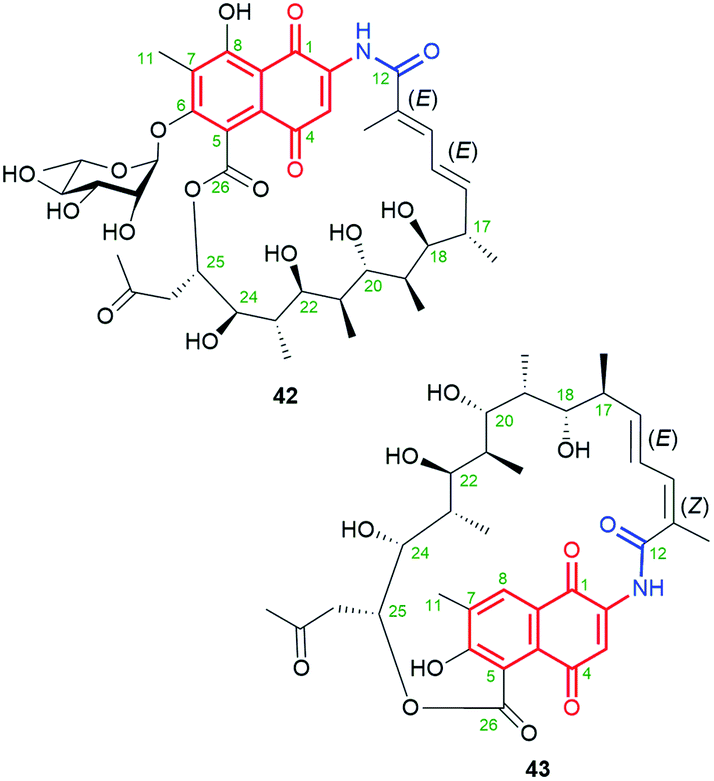
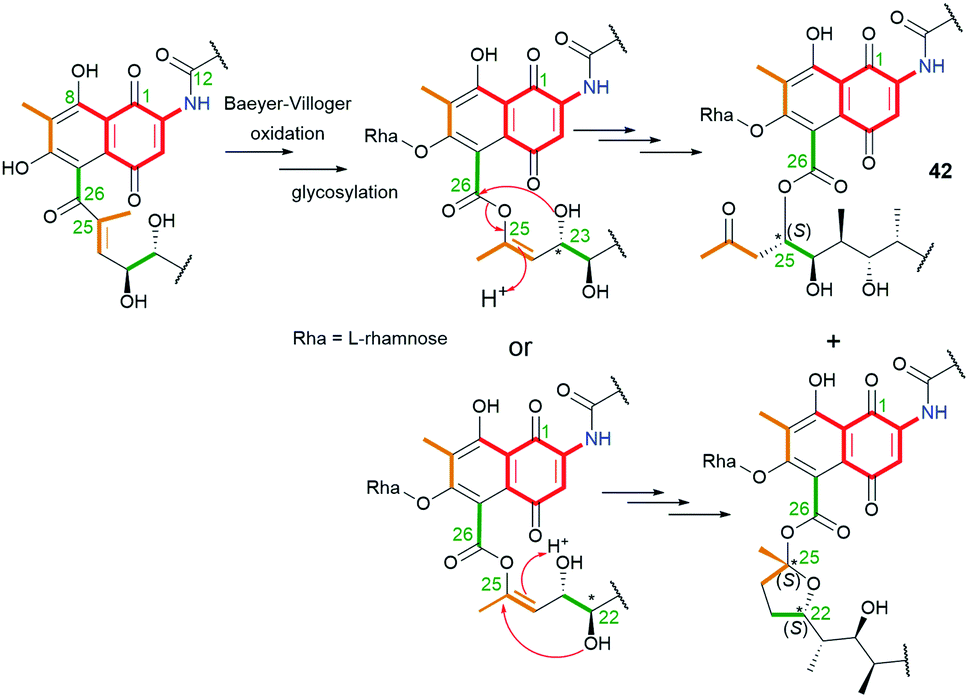
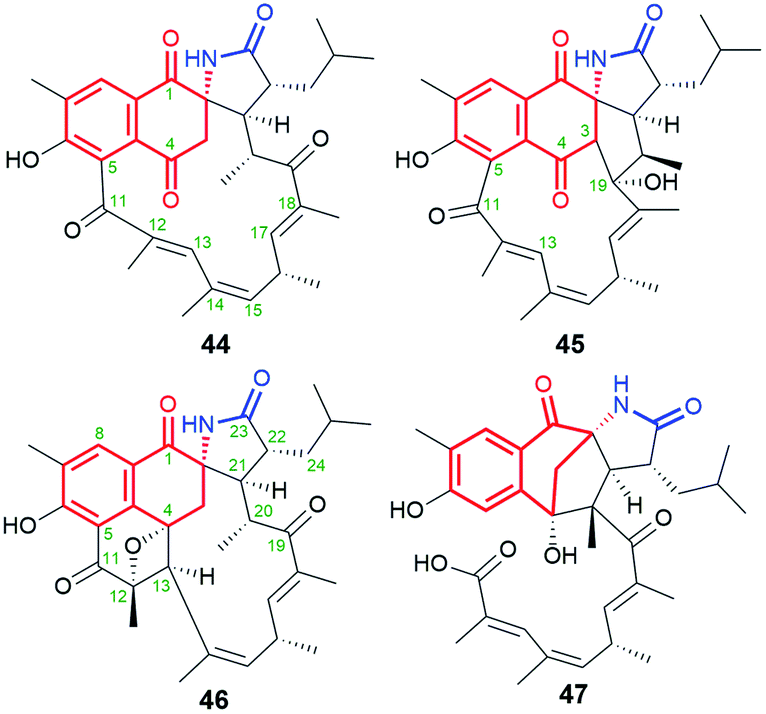
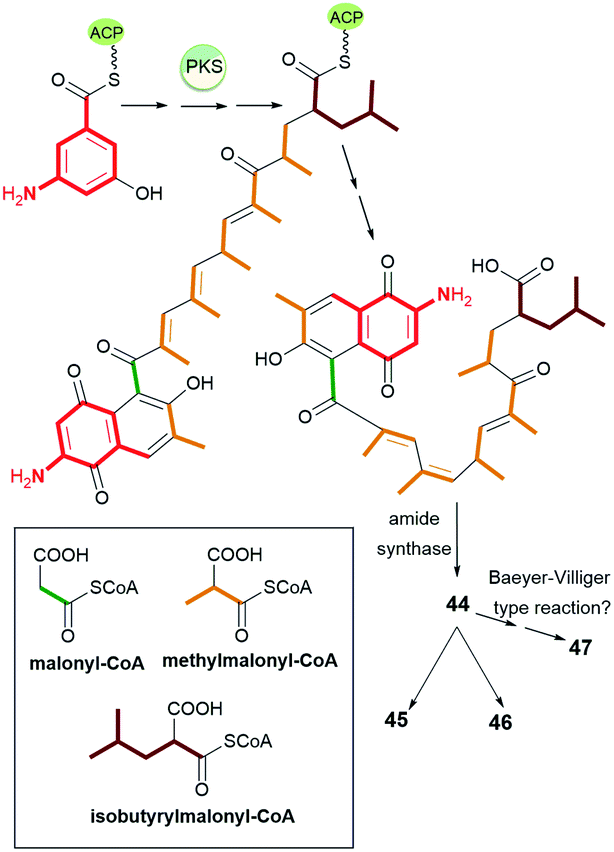
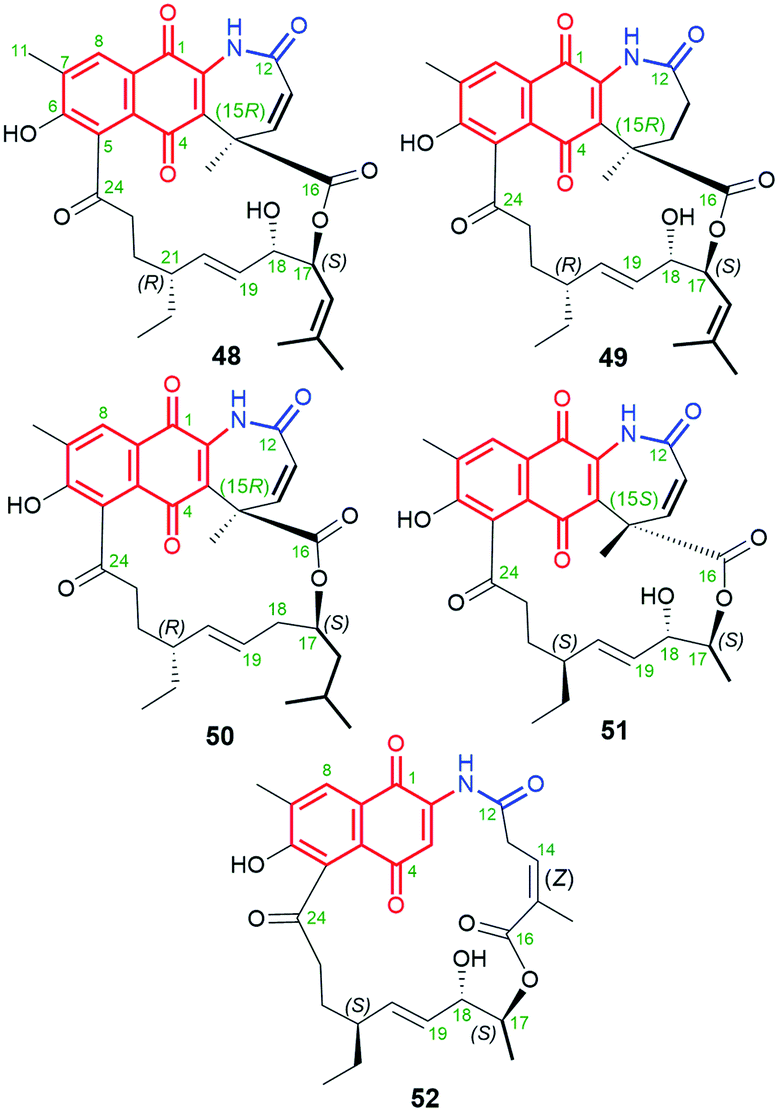
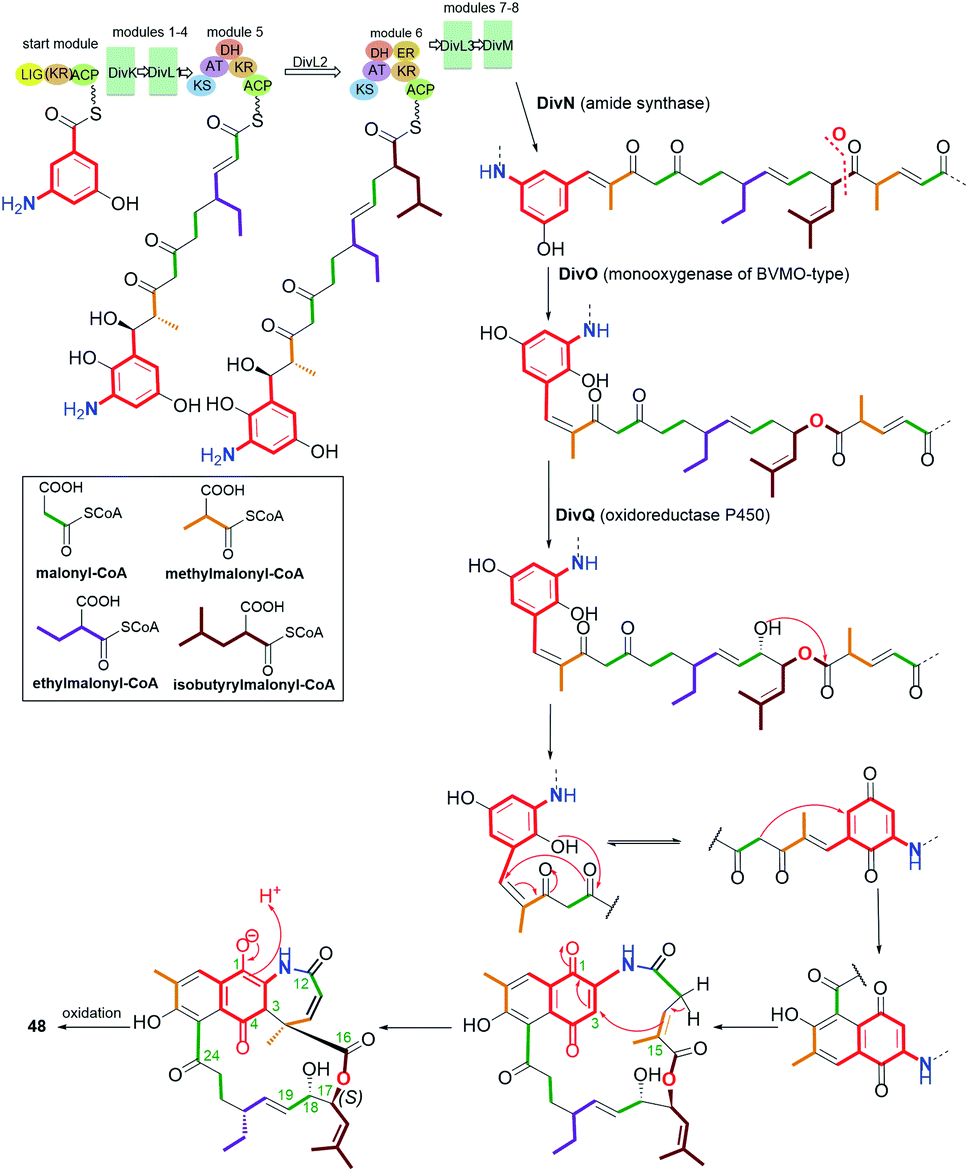
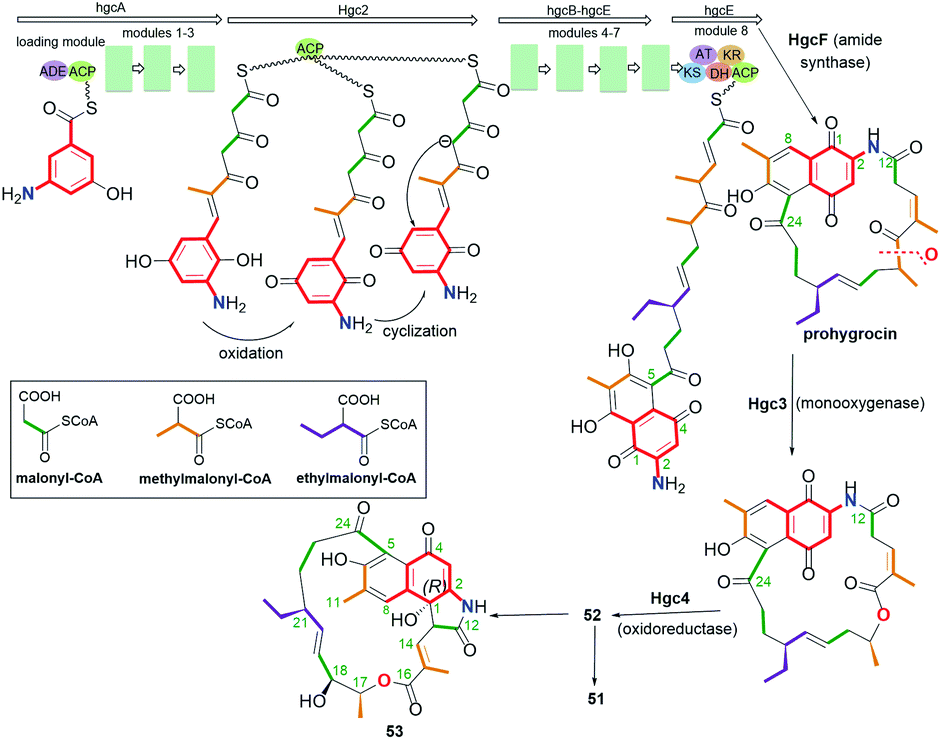
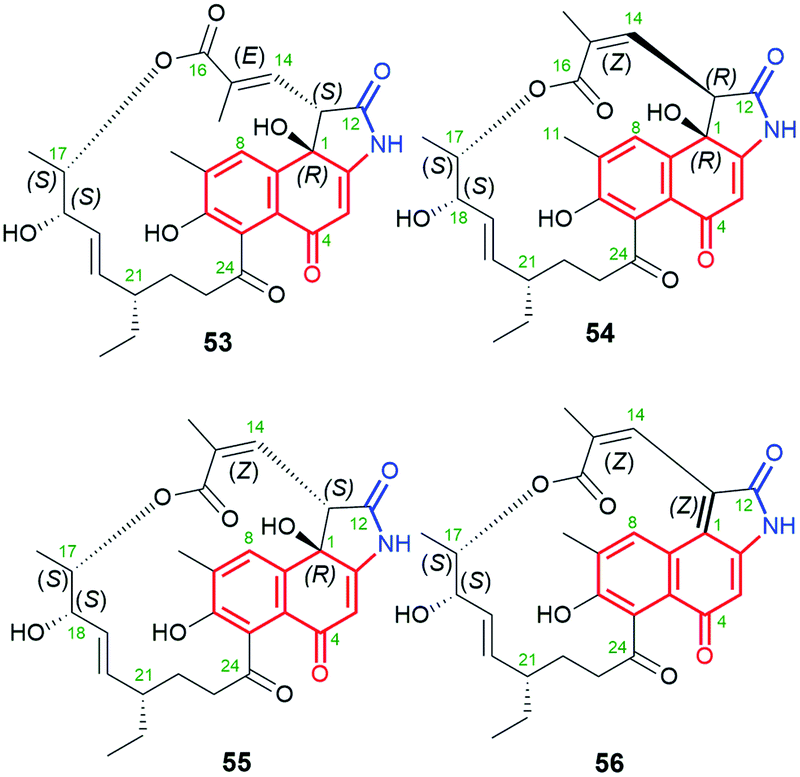
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(15), influencing the ansa chain conformation rigidity (Fig. 29).86,100 Structures of 54–56 were elucidated by HR-MS, 1D and 2D NMR as well as X-ray studies (structure of 54).86,100 It should be mentioned that hygrocin H (56), possesses an extra double bond at C(1), as result of dehydration process, in contrast to 53–55. Hygrocins 54–56 showed a quite high cytotoxic potency in MDA-MB-231, PC3 and HeLa cancer cells (IC50s = 0.5–5.0 μM), unlike to 53.86,100 This result suggested the impact of E/Z configuration at C(14) on anticancer activity, whereby the mechanism of this activity has not been revealed yet. This SAR among hygrocins is concerned probably with the fact that E-configuration limits flexibility of the ansa bridge.
C(15), influencing the ansa chain conformation rigidity (Fig. 29).86,100 Structures of 54–56 were elucidated by HR-MS, 1D and 2D NMR as well as X-ray studies (structure of 54).86,100 It should be mentioned that hygrocin H (56), possesses an extra double bond at C(1), as result of dehydration process, in contrast to 53–55. Hygrocins 54–56 showed a quite high cytotoxic potency in MDA-MB-231, PC3 and HeLa cancer cells (IC50s = 0.5–5.0 μM), unlike to 53.86,100 This result suggested the impact of E/Z configuration at C(14) on anticancer activity, whereby the mechanism of this activity has not been revealed yet. This SAR among hygrocins is concerned probably with the fact that E-configuration limits flexibility of the ansa bridge.