
Cheminformatic analysis of natural product-based drugs and chemical probes†
Samantha
Stone
a,
David J.
Newman
 b,
Steven L.
Colletti
c and
Derek S.
Tan
*ad
b,
Steven L.
Colletti
c and
Derek S.
Tan
*ad
aChemical Biology Program, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, New York 10021, USA. E-mail: tand@mskcc.org
bNIH Special Volunteer, Wayne, PA 19087, USA
cZymergen, Inc., 430 E 29th St, New York 10016, USA
dTri-Institutional Research Program, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, New York 10021, USA
First published on 3rd August 2021
Abstract
Covering: 1981 to 2019
Natural products continue to play a major role in drug discovery, with half of new chemical entities based structurally on a natural product. Herein, we report a cheminformatic analysis of the structural and physicochemical properties of natural product-based drugs in comparison to top-selling brand-name synthetic drugs, and a selection of chemical probes recently discovered from diversity-oriented synthesis libraries. In this analysis, natural product-based drugs covered a broad range of chemical space based on size, polarity, and three-dimensional structure. Natural product-based structures were also more prevalent in top-selling drugs of 2018 compared to 2006. Further, the drugs clustered well according to biosynthetic origins, but less so based on therapeutic classes. Macrocycles occupied distinctive and relatively underpopulated regions of chemical space, while chemical probes largely overlapped with synthetic drugs. This analysis highlights the continued opportunities to leverage natural products and their pharmacophores in modern drug discovery.
 Samantha Stone | Samantha Stone received her BA in Chemistry from Vassar College. She joined Memorial Sloan Kettering Cancer Center as a post-baccalaureate researcher with Derek Tan. She is now pursuing an MD-PhD at the University of Utah School of Medicine. |
 David J. Newman | David J. Newman, DPhil retired as Chief, Natural Products Branch, NCI in 2015, after 24 years at NCI (10 as Chief), preceded by 25 years in pharma. He was responsible for collection of microbes and marine invertebrates, and later plants following the retirement of Gordon Cragg from the Chief position, under the NCI's long-standing program for bioactive agents from Nature. With Gordon, the NCI Open Repository was established in 1993 for any researcher to use. He was trained in the U.K. as an analytical, organic, then microbial chemist. He still publishes extensively, frequently with Gordon in spite of “both being nominally retired”. |
 Steven L. Colletti | Steven L. Colletti, PhD has led drug discovery R&D for nearly 30 years across pharma and biotech. He is currently Senior Vice President of R&D at Zymergen. Prior to acquisition by Zymergen, as CSO he led the portfolio at Lodo Therapeutics in natural products drug discovery for oncology and infectious diseases. With 25 years of experience and leadership at Merck & Co., he contributed to programs in multiple therapeutic areas with diverse modalities, resulting in over a dozen preclinical candidates advanced toward clinical development. Beginning his career as a medicinal chemist, he is an inventor and author of 130 patents and publications. |
 Derek S. Tan | Derek S. Tan, PhD was born and raised in Rochester, New York. His parents, both chemists, discouraged him from going into chemistry and so, naturally, he became a chemist! He received his BS from Stanford University, working with Dale Drueckhammer, and PhD from Harvard University, working with Stuart Schreiber. He pursued postdoctoral training with Samuel Danishefsky at the Sloan Kettering Institute, where he joined the faculty in 2002. He is currently Member, Chair, and Tri-Institutional Professor in the SKI Chemical Biology Program, incumbent of the Eugene W. Kettering Chair, and Director of the Tri-Institutional PhD Program in Chemical Biology. |
1 Introduction
Natural products have played a major role in drug discovery, with over half of all small-molecule drugs based structurally on a natural product.1,2 This impact is even higher for certain indications, such as bacterial infections (71%) and cancer (65%). In contrast, most modern drug discovery efforts have turned away from the classical approach of screening natural product extracts to alternatives such as structure-based drug design and screening of synthetic libraries.3–5 Nonetheless, natural products remain invaluable starting points for drug discovery, both directly as lead compounds and indirectly as inspiration for synthetic analogues.6–8 Natural products provide structures that have implicitly evolved to bind to proteins and other biological targets, and are rich in stereochemistry and three-dimensional structure, features that have been correlated with increased binding specificity,9,10 decreased preclinical toxicity,11 and improved progression through clinical trials.12 Along these lines, there is increasing interest in exploring molecules that have “beyond-rule-of-5” properties, which is the case for many natural products.13–15 Further, natural products have provided means to address challenging targets,16 as in recent examples of Keap1-Nrf2 interaction inhibitors and K-Ras ligands.17,18We have previously carried out cheminformatic analyses that compare the structural features of drugs that are based on natural product structures to other drugs of purely synthetic origins.2,19 These studies have shown that drugs based on natural products tend to exhibit lower hydrophobicity and greater stereochemical content compared to their purely synthetic counterparts. Moreover, they suggest that the structural features found in natural products may be incorporated into synthetic drugs to target pharmaceutically-relevant chemical space and to increase the structural diversity available for drug discovery. Indeed, while natural product-based drugs continue to represent approximately half of all new chemical entity drug approvals (Fig. 1a), an increasing proportion have been generated by de novo synthesis based on natural product pharmacophores, particularly in the last 10 years (Fig. 1b).
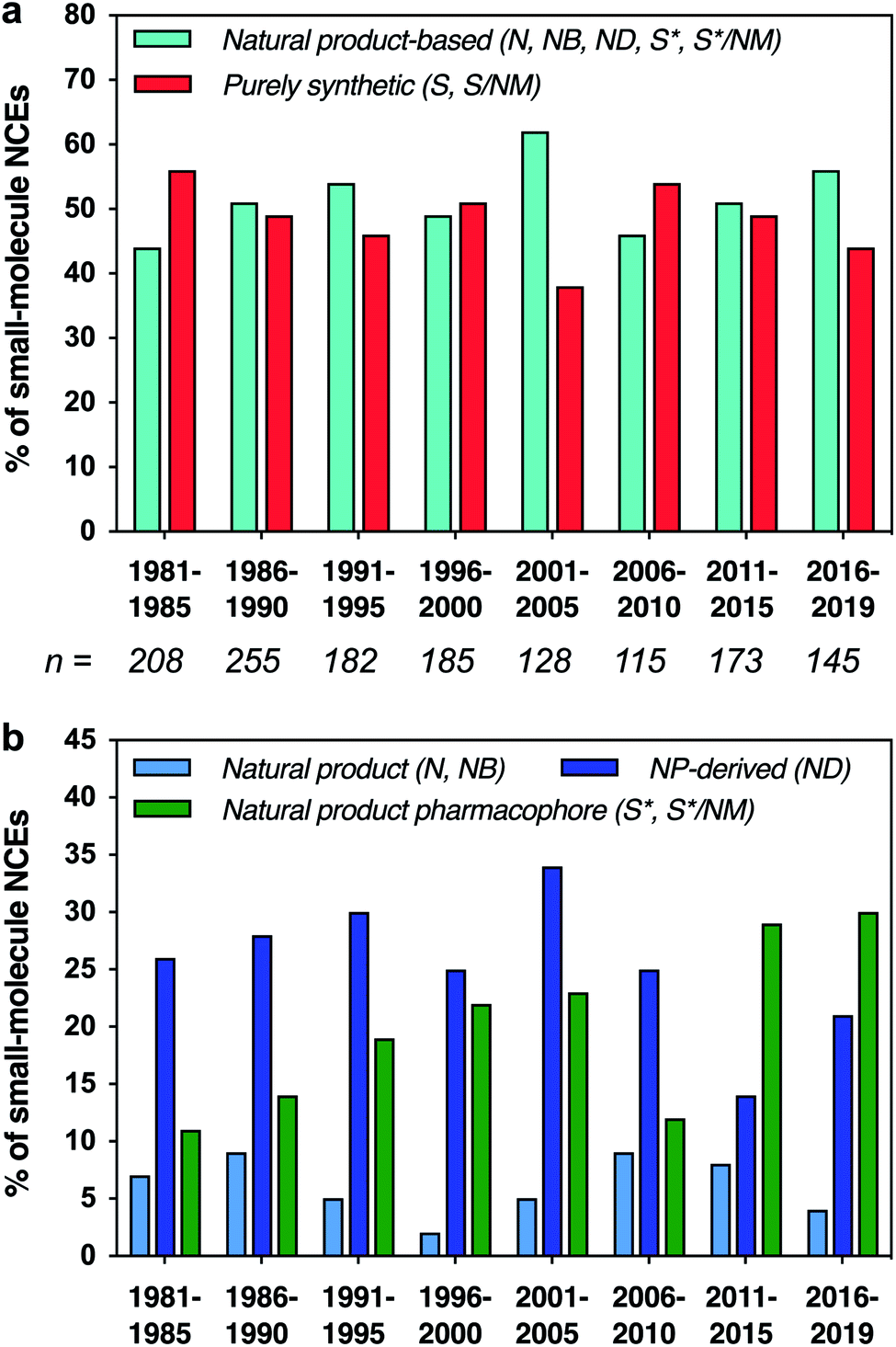 | ||
| Fig. 1 (a) Fractions of small-molecule new chemical entity (NCE) drug approvals based structurally on natural products vs. purely synthetic in origin. Total number (n) of small-molecule NCEs listed below each time period. (b) Fractions of NCEs that were unmodified natural products, defined natural product botanicals, natural product derivatives, or synthetic but based structurally on a natural product; purely synthetic drugs not shown. Data binned in 5 year periods, except final 4 year period. Categories as defined by Newman and Cragg: N = unaltered natural product; NB = botanical drug (defined mixture); ND = natural product derivative; S = synthetic drug; S* = synthetic drug (natural product pharmacophore); /NM = mimic of natural product.1 | ||
Herein, we present an updated and expanded analysis of the structural and physicochemical properties of natural product-based drugs approved between 1981–2019,1 in comparison to the top 40 best-selling brand-name drugs in 2006 and 2018,20 and a collection of chemical probes discovered recently from diversity-oriented synthesis (DOS) libraries.21 Further, we have classified these drugs by biosynthetic origin, macrocyclic structure, and therapeutic class to explore further the relationships between chemical space, chemotype, and drug function. Notably, we found a striking increase in the proportion of top-selling drugs that are based on natural products in 2018 compared to 2006. Principal component analysis (PCA) further showed that these top-selling drugs also occupy a larger range of chemical space in 2018 compared to 2006, and that the complete set of natural product-based drugs accesses an even more vast area. Interestingly, most of the recently discovered DOS probes overlap with purely synthetic drugs in the PCA plot. Moreover, the natural product-based drugs tend to cluster according to biosynthetic origins, and less so based on therapeutic classes. Taken together, these results indicate that there are rich opportunities for further exploitation of natural product-based structures in drug and probe discovery.
2 Results
2.1 Drug and probe compound datasets
To assess the structural features of natural product-based drugs in comparison to other compound classes, we assembled a dataset of 521 unique chemical structures (Table 1 and ESI Dataset†). The natural product-based drugs were compiled from the natural product (N) and natural product-derived (ND) categories in Newman and Cragg's latest compilation of new drug approvals between 1981–2019,1 building upon an earlier analysis by our groups.2 The list includes antibody–drug conjugates, for which we analyzed the small-molecule fragment separately. Large carbohydrates were analyzed as representative tetrasaccharide fragments, many of which were identical for the purposes of this analysis. For combination therapies, each component molecule was assessed and classified individually, with several assignments of older molecules discovered prior to 1981 made de novo as necessary. Molecules appearing in more than one combination were identified by generic name and dereplicated.| Category | Compounds | MW | HBD | HBA | ALOGPs | Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D D |
Rot | tPSA | Fsp3 | RngAr | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Unique | ||||||||||
|
a ALOGPs = calculated 1-octanol/water partition coefficient, Fsp3 = fraction sp3-hybridized carbons, HBA = hydrogen-bond acceptors, HBD = hydrogen-bond donors, LogD = calculated 1-octanol/water distribution coefficient (pH 7.4), MW = molecular weight, Rot = rotatable bonds, RngAr = aromatic rings, tPSA = topological polar surface area.
b In 2018, 28 out of the top 40 small-molecule drug products contained at least one natural product-based component.
c In 2006, 14 out of the top 40 small-molecule drug products contained at least one natural product-based component.
d Parameter averages for all compounds excluding (525 total, 471 unique) vs. only (51 total, 50 unique) ribosomal peptides are as follows: MW 501 vs. 2,288, HBD 3.1 vs. 31.4, HBA 7.4 vs. 35.6, ALOGPs 2.21 vs. 0.05, LogD 0.00 vs. −18.65, Rot 8.2 vs. 64.6, tPSA 135 vs. 926, Fsp3 0.58 vs. 0.54, RngAr 1.2 vs. 3.8.
|
|||||||||||
| Natural product drugs (N) | 88 | 77 | 611 | 5.9 | 10.1 | 1.96 | −1.40 | 11.0 | 196 | 0.71 | 0.7 |
| Natural product-derived drugs (ND) | 379 | 344 | 757 | 7.0 | 11.5 | 1.82 | −3.00 | 16.2 | 250 | 0.59 | 1.4 |
| Top 40 drugs in 2018: N, ND, S*, S*/NM (2018-N) | 37 | 34b | 473 | 2.4 | 6.0 | 2.11 | 1.78 | 7.8 | 111 | 0.50 | 1.9 |
| Top 40 drugs in 2018: S, S/SM (2018-S) | 17 | 15 | 444 | 1.9 | 5.1 | 2.83 | 2.49 | 6.5 | 95 | 0.33 | 2.7 |
| Top 40 drugs in 2018: all | 54 | 49 | 464 | 2.2 | 5.8 | 2.33 | 2.00 | 7.4 | 106 | 0.45 | 2.2 |
| Top 40 drugs in 2006: N, ND, S*, S*/NM (2006-N) | 15 | 14c | 367 | 2.4 | 5.0 | 2.08 | 0.40 | 7.6 | 90 | 0.54 | 1.6 |
| Top 40 drugs in 2006: S, S/SM (2006-S) | 30 | 27 | 355 | 1.1 | 3.9 | 3.15 | 2.37 | 5.4 | 61 | 0.33 | 2.3 |
| Top 40 drugs in 2006: all | 45 | 41 | 359 | 1.5 | 4.3 | 2.78 | 1.70 | 6.1 | 70 | 0.40 | 2.0 |
| Diversity-oriented synthesis probes (DOS) | 10 | 10 | 552 | 1.1 | 4.7 | 4.08 | 3.90 | 4.9 | 85 | 0.38 | 2.8 |
| All compoundsd | 576 | 521 | 673 | 5.8 | 10.1 | 2.01 | −1.79 | 13.6 | 211 | 0.58 | 1.5 |
For comparison, we also analyzed the top 40 best-selling, brand-name, small-molecule drugs as illustrated previously by Njarðarson and coworkers.20 Biologics such as monoclonal antibodies and other proteins were excluded from this analysis. Our previous cheminformatic analyses have used the top 40 small-molecule drugs from 2006.19,22–26 To assess changes in this collection over time, we also evaluated the top 40 small-molecule drugs from 2018. Both of these collections included drugs based on natural products (2006-N, 2018-N: N and ND classes also included in the complete natural product-based drug datasets above were dereplicated appropriately; S* and S*/NM classes were unique to these datasets) as well as others from purely synthetic origins (2006-S, 2018-S: S and S/NM classes). The presence of several multicomponent combination therapies resulted in greater than 40 unique chemical structures for each group. Strikingly, the fraction of natural product-based drugs increased dramatically in the 2018 collection (34 out of 49 unique structures = 69%, 28 out of 40 drug products = 70%) compared to the 2006 collection (14 out of 41 unique structures = 34%, 14 out of 40 drug products = 35%) (ESI Table S1†). Notably, the majority of these natural product-based compounds were produced by de novo synthesis (S*, S*/NM) in both the 2018 (23 out of 34 unique structures) and 2006 (10 out of 14 unique structures) collections, demonstrating the utility of natural product-based pharmacophores in modern drug discovery.
Finally, to expand the analysis further, we included a collection of 10 chemical probes discovered from DOS libraries and reviewed recently by Gerry and Schreiber.21 This collection represents a wide range of biological activities and is comprised mainly of polycyclic structures that might intuitively be viewed as resembling natural products more so than purely synthetic drugs (ESI Fig. S1†).
2.2 Structural and physicochemical properties of compound datasets
To calculate structural and physicochemical parameters, chemical structures were expressed as SMILES codes, obtained from our earlier analysis,2 from the PubChem database, or by manual conversion within ChemDraw. Compounds were then evaluated using our established set of 20 structural and physicochemical parameters that we have used in previous cheminformatic analyses (see ESI† for complete details).2,19,22 These parameters were selected because they are chemically interpretable and are readily accessed using free or widely-available tools.These included the Lipinski ‘rule of 5’ parameters for oral bioavailability,27 molecular weight (MW), hydrogen-bond donors (HBD), hydrogen-bond acceptors (HBA), and 1-octanol/water partition coefficient (ALOGPs), the last calculated using the method reported by Tetko (the capital P designates a logP calculation).28,29 The related parameter 1-octanol/water distribution coefficient at pH 7.4 (LogD) was also calculated using Instant JChem (ChemAxon). As solubility is often a limiting factor for synthetic drugs, we also calculated aqueous solubility (ALOGpS), using the Tetko algorithm (the capital S designates a logS calculation).29,30 Of note, the Tetko algorithm was unable to process 14 very large ribosomal peptides; to enable inclusion of these structures in the analysis, we used the average ALOGPs and ALOGpS values from the next three largest peptides as placeholders. We also evaluated the corresponding Instant JChem calculations, but observed extremely large values (ChemAxon LogP < −15; LogS > 60) and deemed these unreliable. It should be noted that Instant JChem also returned very large LogD values (< −28) for these 14 peptides, which were included in the analysis but should be viewed with caution.
We further included Veber's ‘rule of 2’ parameters for oral bioavailability,31 rotatable bonds (rot), and topological polar surface area (tPSA). To normalize for molecular size, we also calculated the van der Waals surface area (VWSA) and relative polar surface area (rPSA).
Natural product drugs tend to have fewer nitrogen atoms (N) and more oxygen atoms (O) than synthetic drugs,32 and, thus, counts of these atoms were included as well.
The fraction of sp3-hybridized carbons (Fsp3) and number of stereocenters (Stereo) were included as indirect indicators of molecular complexity and three-dimensional structure. Both parameters have been correlated with increased binding selectivity.9 Stereocenters have also been identified as a distinguishing feature of natural products compared to synthetic drugs32 and correlated with improved progression through clinical trials.12 To normalize the latter for molecular size, we also calculated stereochemical density as the number of stereocenters divided by the number of heavy atoms (Stereo/HA). Previous analyses from our group have described stereochemical density as Stereo/MW,2,19,22 but the number of heavy atoms was readily calculated herein using Instant JChem and provides a more relevant divisor with respect to molecular structure.
Parameters describing ring count (Rings), largest ring size (RngLg), ring systems (RngSys), and rings per ring system (RRSys) were included because some of these features distinguish natural products from synthetic drugs.32 Aromatic rings (RngAr) were also included as these have been correlated with increased preclinical toxicity and attrition rates in drug candidate progression.11
Parameters were calculated using Instant JChem and Tetko's Virtual Computational Chemistry Laboratory (http://www.vcclab.org).28–30 Average values and standard deviations of each structural and physicochemical descriptor, grouped by drug category, were calculated (Table 1 and ESI Table S2,† and Fig. 2 and ESI Fig. S2†). These calculations were performed both including and excluding ribosomal peptides, as these compounds proved to be outliers due to their high molecular weight and polarity (ESI Table S3†).
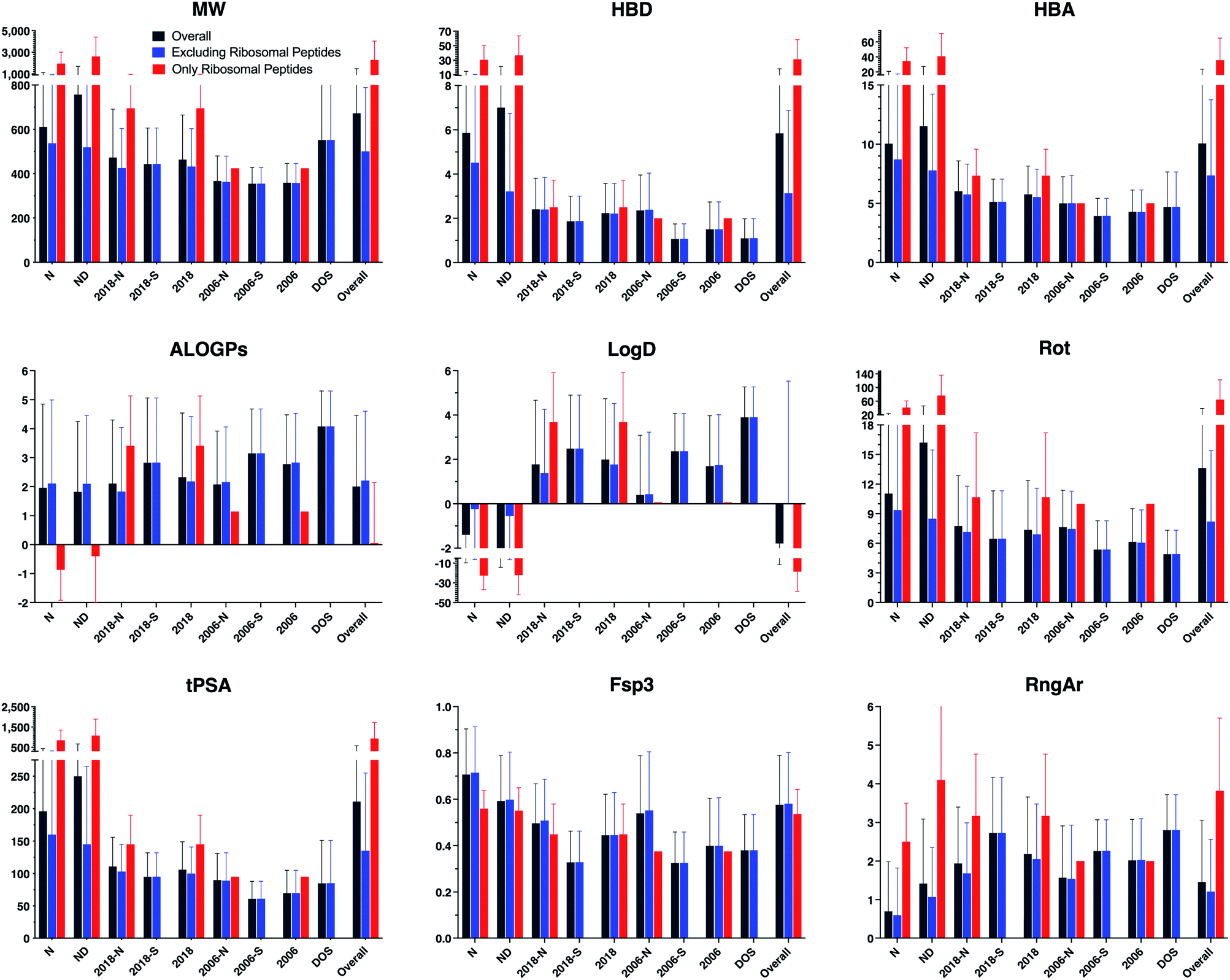 | ||
| Fig. 2 Bar graphs of selected structural and physicochemical properties of natural product drugs (N), natural product-derived drugs (ND), top 40 brand-name drugs from 2006 and 2018 from natural product (-N) and purely synthetic (-S) origins, and recently discovered chemical probes from diversity-oriented synthesis libraries (DOS). See ESI Fig. S2† for complete data. | ||
The natural product-based drugs (N, ND) had higher average values compared to purely synthetic drugs (2006-S, 2018-S) for multiple parameters, including molecular weight (MW), hydrogen-bonding groups (HBD, HBA), rotatable bonds (Rot), polar surface area (tPSA), total surface area (VWSA), oxygen counts (O), and stereocenters (Stereo). These trends were also observed when ribosomal peptides were excluded from the analyses, although the differences were somewhat smaller. In parameters normalized for molecular size, the natural products also had higher sp3 content (Fsp3) and stereochemical density (Stereo/HA) than the synthetic drugs, but more comparable relative polar surface area (rPSA). Conversely, the natural product-based drugs tended to have lower hydrophobicity (ALOGPs, LogD) and fewer aromatic rings (RngAr) compared to the synthetic drugs. In contrast, the natural product-based and synthetic drugs in this analysis had similar aqueous solubility (ALOGpS) and nitrogen counts (N). With respect to ring parameters, the natural product-based and synthetic drugs had similar ring counts (Rings), but the former tended to have larger rings (RngLg), and fewer but more complex ring systems (RngSys, RRSys).
Notably, the diversity-oriented synthesis probes (DOS) had the highest hydrophobicity (ALOGPs, LogD) and ring parameters (Rings, RngLg, RngSys, RngAr) and lowest solubility (ALOGpS) and relative polar surface area (rPSA) of any of the compound datasets. The DOS probes were similar to the natural product-based drugs for parameters associated with molecular size (MW, VWSA) and stereochemical complexity (Stereo, Stereo/HA), but more like the synthetic drugs for parameters relevant to polarity (HBD, HBA, tPSA), flexibility (Rot), and sp3 content (Fsp3).
2.3 Principal component analysis of compound datasets
We next used principal component analysis (PCA) to visualize the distributions of the compound sets in chemical space (Fig. 3 and ESI Fig. S3†).22 PCA is a statistical method that allows the complete 20-dimensional dataset to be visualized on two or three unitless, orthogonal axes, each representing a linear combination of the original 20 parameters.33–35 The method minimizes information loss in this dimensionality reduction, and the first three principal components retained 77% of the variance in the total dataset, with >90% of the variance incorporated within the first six principal components (ESI Table S4†).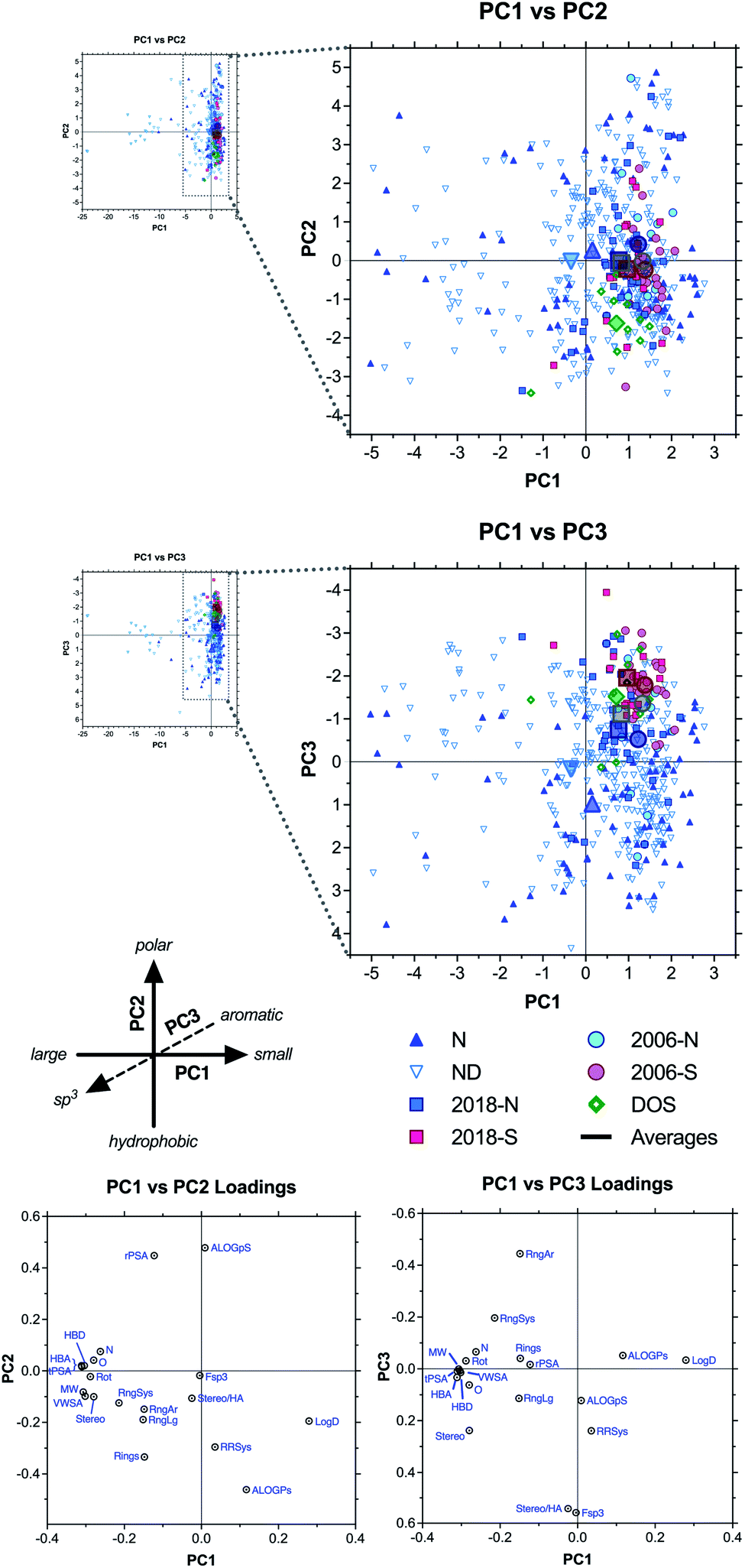 | ||
| Fig. 3 PCA and loading plots based on 20 structural and physicochemical parameters for natural product drugs (N), natural product-derived drugs (ND), top 40 brand name drugs in 2006 and 2018 derived from natural products (-N) or purely synthetic (-S), and recently discovered chemical probes from diversity-oriented synthesis libraries (DOS). See ESI† for complete data, including PC2 vs. PC3 plots (ESI Fig. S3†). | ||
Loading plots indicate that parameters associated with molecular size (HBA, tPSA, MW, HBD, VWSA; in order of magnitude) were highly correlated and had the strongest influence along the PC1 axis, reflected by the positioning of large drugs in the negative range (left) (Fig. 3, ESI Fig. S3, and ESI Table S5†). Polarity had a strong influence on PC2, with ALOGpS and rPSA correlating highly and providing the greatest contributions to variance in the positive direction (top), while ALOGPs conversely contributed to variance in the negative direction (bottom). In notable contrast, LogD contributed strongly to positive variance along PC1 but less so to negative variance along PC2, and this influence was seen in the positioning of several large but lipophilic compounds near the origin of PC1 (see below). This may be due to the high incidence of charged functionalities in large ribosomal peptide drugs, which impacts LogD but not ALOGPs (avg MW = 2,288, LogD = −18.65, ALOGPs = 0.05), resulting in an anticorrelation between size and LogD along PC1. However, this may also be an artifact of aberrantly large LogD values calculated by Instant JChem for large ribosomal peptides. Three-dimensional structure had a pronounced impact on PC3 positioning, with RngAr contributing to negative variance while Fsp3 and Stereo/HA being highly correlated and corresponding to positive variance.
Results of our PCA analysis indicate that natural product and natural product-derived drugs (blue) occupy a much greater range of chemical space compared to the purely synthetic drugs (red) in this analysis, consistent with previous studies.2,19,32 The natural product-based drugs extend deep into the negative range (left) of PC1 (Fig. 3), consistent with the larger size of these compounds observed in the parameter averages above (Table 1 and Fig. 2). The extreme outliers (−5 to −25) were all ribosomal peptides, such as the anticoagulant desirudin (Revasc; MW = 6963) and the diabetes drug lixisenatide (Lyxumia; MW = 4859), with carbohydrates such as heparan sulfate (Orgaran; representative tetramer MW = 1233) also extending deep into the negative range (to −8) (ESI Fig. S4†). Further analysis was facilitated by zooming in on a smaller region of the plot (Fig. 3 expansions).
The natural product-based drugs also extend further into the positive range (top) of PC2 compared to the synthetic drugs (Fig. 3), consistent with the presence of highly polar compounds such as the anticancer agent aminolevulinic acid (Levulan; ALOGPs = −2.85) and the hypolipidemic drug meglutol (Lipoglutaran; ALOGPs = −0.88) (ESI Fig. S4†). Finally, along PC3, the synthetic drugs clustered in the negative range, consistent with the high aromatic ring content of compounds such as the anticancer drug nilotinib (Tasigna; RngAr = 5), while the natural product-based drugs extended far into the positive range, based on their high three-dimensional character exemplified by compounds such as the cyclodextrin anesthesia antidote sugammadex (Bridion; Fsp3 = 0.89) and the halichondrin analogue anticancer drug eribulin (Halaven; Fsp3 = 0.88).
Among the top 40 brand-name drug sets (Fig. 3 and ESI Fig. S5†), segregation of natural product-based (-N) and purely synthetic (-S) molecules revealed PC3 as the primary axis of differentiation (blue vs. red), with the synthetic drugs all clustering in the negative range, again consistent with higher aromatic ring content (Table 1 and Fig. 2). The natural product-based drugs also spanned a wider range of positions along PC2 compared to the synthetic drugs, consistent with a wider range of polarities exemplified by the highly polar antidiabetic metformin (component of Janumet) (positive; ALOGPs = −1.83) and the highly hydrophobic antiviral pibrentasvir (component of Mavyret) (negative; ALOGPs = +5.97). However, the averages for the natural product and synthetic subsets remained comparable, particularly for the 2018 collection (squares). Interestingly, we noted a negative shift in the 2018 vs. 2006 averages along PC1 (squares vs. circles), indicative of a trend toward larger molecules within both the natural product-based and purely synthetic subsets, exemplified by molecules such as the natural product-derived anticancer drug everolimus (Afinitor/Certican; MW = 958) and the synthetic antiviral velpatasvir (component of Epclusa; MW = 883).
Interestingly, the DOS probes (green) clustered similarly to the synthetic drugs along PC1 and PC3, but were differentiated along PC2, where they extended deeper into the negative range (Fig. 3), consistent with the highly hydrophobic character observed in the parameter averages above (Table 1 and Fig. 2). Examples include the ENT1 (equilibrative nucleoside transporter 1) inhibitor rapadocin (ALOGPs = 5.87) and the Max transcription factor homodimer stabilizer KI-MS2-008 (ALOGPs = 4.71) (ESI Fig. S4†).
Thus, the PCA plots indicated that, as a class, natural product-based drugs exhibit higher structural diversity, larger molecular size, increased range of polarities, and more three-dimensional character compared to top-selling synthetic drugs. Recently identified probes from diversity-oriented synthesis collections shared many features with synthetic drugs, with the exception of increased hydrophobicity. These results contrast somewhat with the divergent characteristics of this small collection in the one-dimensional analyses of average parameter values above.
2.4 Classification by biosynthetic origin
We next investigated the locations of specific structural classes within the PCA analysis. The natural product-based drugs (including the 2018-N and 2006-N subsets of the top 40 drug collections) were assigned to one of eight major biosynthetic classes: alkaloid, carbohydrate, fatty acid, nucleoside, peptide, polyketide, terpenoid, or other (see ESI Dataset†). For molecules that belong to more than one class, designations were based on the largest fragment. Prevalent subcategories were also assigned (e.g., ribosomal vs. non-ribosomal peptides). Average parameter values were calculated for each category (ESI Table S6†) and the PCA data were revisualized with coding based on biosynthetic origin (Fig. 4 and ESI Fig. S6†).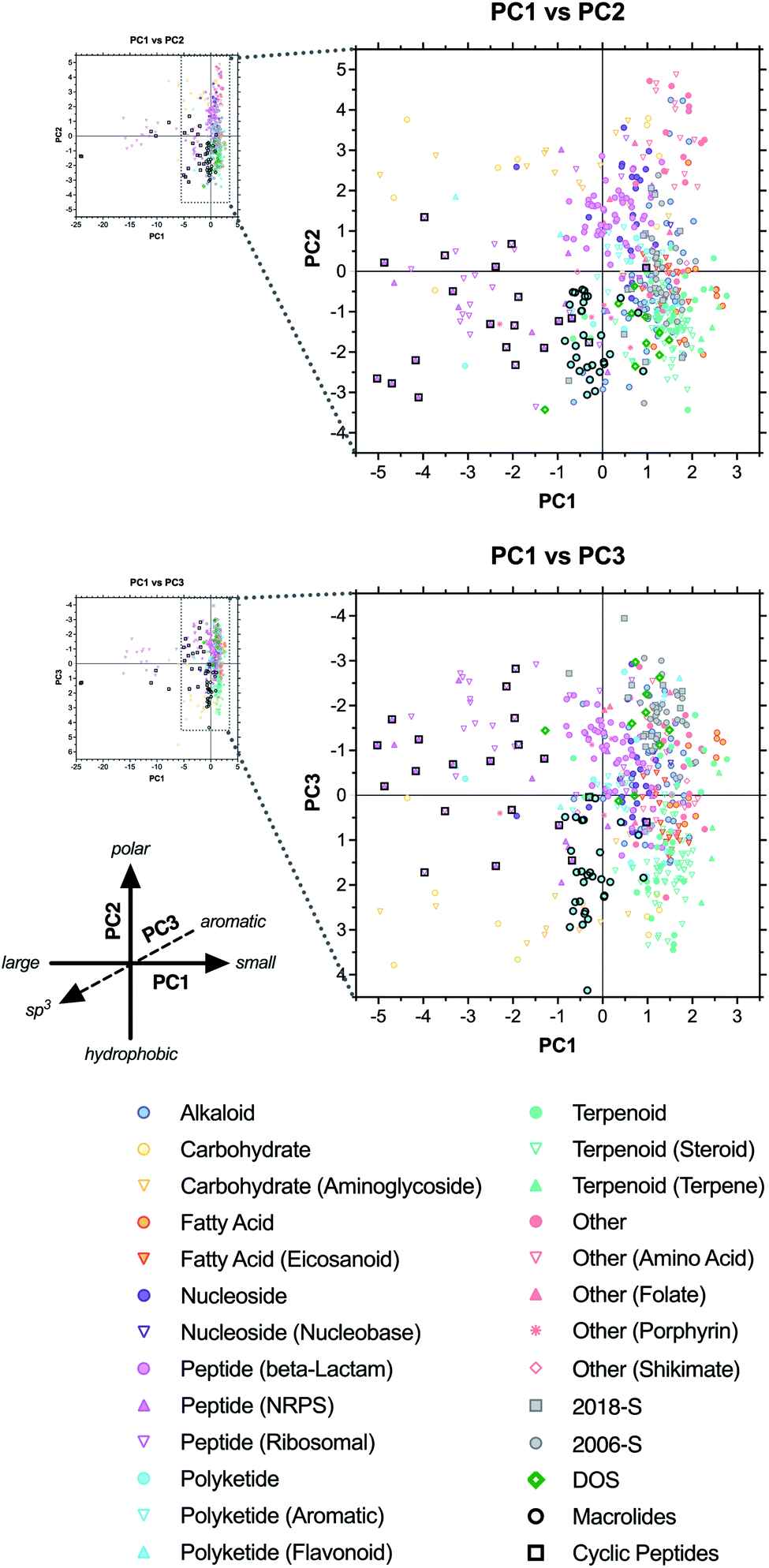 | ||
| Fig. 4 PCA plots of drugs coded by biosynthetic origin (colored markers) and macrocyclic structure (black outlines). All natural product-based drugs (N, ND, S*, S*/NM) are shown, in addition to purely synthetic drugs present in the top 40 collections (S, S/NM; gray markers), as well as chemical probes identified from diversity-oriented synthesis libraries (DOS; bold green diamonds). | ||
We observed that many of the biosynthetic classes segregated into distinct regions of the plots, consistent with a recent related analysis.36 Alkaloids (blue circles) were positioned in the positive range (right) along PC1, consistent with relatively small molecular sizes (avg MW = 402), spanned both the extreme positive and negative ranges of PC2, consistent with the range of polarities between metformin (component of Janumet; ALOGPs = −1.83) and dinalbuphine (Naldebain; ALOGPs = 5.34), and trended toward the negative range along PC3, consistent with the presence of at least one aromatic ring in almost all of these compounds (avg RngAr = 1.8) (ESI Fig. S7 and Table S6†).
In contrast, carbohydrates (yellow circles and triangles) extended across a wide span of PC1, ranging in size from oseltamivir (Tamiflu; MW = 312) to heparan sulfate (Orgaran; MW = 1233) (ESI Fig. S7†). These compounds clustered in the positive region (top) of PC2, and in the positive region of PC3, consistent with their high polarity (avg ALOGPs = 0.83) and rich stereochemical and three-dimensional features (avg Fsp3 = 0.73; avg Stereo/HA = 0.26) (ESI Table S6†).
Fatty acid derivatives (orange circles and triangles) clustered in the lower right of the PC1 vs. PC2 plot. As these compounds have moderate molecular weights (avg MW = 485), the positioning on PC1 may be impacted by the competing influence of their high LogD (3.05) (ESI Table S6†). Negative positioning along PC2 is consistent with their high hydrophobic character (avg ALOGPs = 4.59), with the antiobesity drug orlistat (Xenical; ALOGPs = 7.61) near the negative end of the range (ESI Fig. S7†).
Nucleosides (purple circles and triangles) clustered in the positive range (top) along PC2, indicating high polarity (avg ALOGPs = 0.19) but fell near the origin of PC1 and PC3, reflecting their relatively small molecular size (avg MW = 399) and a combination of aromatic character of the bases and three-dimensional character of the sugars (avg RngAr = 1.9; Fsp3 = 0.54) (ESI Table S6†). Exemplars include adenosine (Adenocard; MW = 267, ALOGPs = −1.21, RngAr = 2, Fsp3 = 0.5) and emtricitabine (component of Genvoya and Truvada; MW = 247, ALOGPs = −1.41, RngAr = 1, Fsp3 = 0.5) (ESI Fig. S7†).
The β-lactam subset of peptides (pink circles) also clustered similarly to the nucleosides, consistent with analogous structural influences (avg MW = 487, ALOGPs = 0.07, RngAr = 1.4, Fsp3 = 0.44), as in the cephalosporin antibiotic cefminox (Meicelin; MW = 518, ALOGPs = −1.26, RngAr = 1, Fsp3 = 0.56) (ESI Fig. S7 and Table S6†).
Peptides derived from both ribosomal (pink open triangles) and NRPS (pink closed triangles) pathways represent the largest structures in this analysis (avg MW = 2288, 1059, respectively), and this shifts them far into the negative range (left) of the PC1 axis, with ribosomal peptides such as desirudin (Revasc, MW = 6963) and lepirudin (Refludan, MW = 6979) occupying extreme outlier positions (ESI Fig. S7 and Table S6†). Both subsets occupied a wide range of positive and negative positions along PC2 and PC3, indicative of the competing characteristics of the peptide backbone and various side-chain functionalities (avg ALOGPs = 0.05, 2.68; RngAr = 3.8, 2.1; Fsp3 = 0.54, 0.62, respectively). Examples of this range of properties are seen in the polar, sp3-rich immunostimulant glycopin (Likopid/Licopid, ALOGPs = −2.23, Fsp3 = 0.74) and the hydrophobic, aromatic-rich antibiotic dalbavancin (Dalvance, ALOGPs = 3.58, RngAr = 7).
Polyketides (cyan circles and triangles) clustered near the origin along the PC1 axis, but in the negative range along PC2 and the positive range along PC3, indicating a combination of hydrophobic (avg ALOGPs = 3.12) and three-dimensional character (Fsp3 = 0.64) (ESI Table S6†). These characteristics are exemplified by sirolimus/rapamycin (Rapamune; ALOGPs = 4.85, Fsp3 = 0.75), whose high molecular weight (914) may be counteracted by its high LogD (7.45) in placing it near the origin of PC1 (ESI Fig. S7†). Interestingly, the subset of 14 aromatic polyketides (cyan open triangles) clustered closer to the origin along PC2 and PC3, indicative of relatively lower hydrophobicity (avg ALOGPs = 2.06) and higher aromaticity (RngAr = 2.1, Fsp3 = 0.46). This is seen in the positioning of the anticancer drug epirubicin (Farmorubicin; ALOGPs = 1.41, RngAr = 2, Fsp3 = 0.44).
Terpenoids (green circles and triangles) clustered tightly in the lower right quadrant of the PC1 vs. PC2 plot, and in the positive region along PC3, consistent with relatively low molecular weight, high hydrophobicity, and high three-dimensional character. Examples include nausea drug dronabinol/THC (Marinol; MW = 314, ALOGPs = 7.29, Fsp3 = 0.62) and the analgesic alloaromadendrene (component of Acheflan; MW = 204, ALOGPs = 3.70, Fsp3 = 0.87) (ESI Fig. S7†). Notably, this class overlapped substantially with the synthetic drugs in the PC1 vs. PC2 plot, but was well-differentiated along PC3, indicative of the differences in the sp3 and stereochemical vs. aromatic ring content between these groups.
The other category (peach markers) is comprised of drugs that do not fall into any of the other seven designations, such as amino acids, folates, porphyrins, and shikimates. Generally, these tended to be positioned in the upper right quadrant of the PC1 vs. PC2 plot, as well as in the negative range along PC3, indicating small sizes, high polarities, and low sp3 content. Examples include the multiple sclerosis drug dimethylfumarate (Tecfidera; MW = 144, ALOGPs = 0.45, Fsp3 = 0.33) and the analgesic acetominophen (component of Apadaz; MW = 151, ALOGPs = 0.51, Fsp3 = 0.12) (ESI Fig. S7†).
2.5 Classification by macrocyclic structure
Next, we investigated the positioning of macrocyclic drugs in the PCA plots. Macrocycles are an underexploited drug class that present large pharmacophores and can address challenging biological targets such as protein–protein interactions.17,37–42 These compounds often violate rule-based metrics applied to synthetic drugs, and are likely to lie in distinct regions of chemical space. Thus, we annotated polyketide-derived macrolides (black circles) and cyclic peptides (black squares) in the PCA analysis (Fig. 4 and ESI Table S7†). Both classes occupied regions of chemical space that were distinct from the purely synthetic drugs. The cyclic peptides localized primarily to the lower-left quadrant of the PC1 vs. PC2 plot, while spanning a range along the PC3 axis, consistent with relatively large, hydrophobic molecules having a variety of three-dimensional character. Examples of this include ribosomal peptide analogues pasireotide (Signifor; MW = 1047, LogD = −1.00, ALOGPs = 3.03, RngAr = 6, Fsp3 = 0.33) and plecanatide (Trulance; MW = 1682, LogD = −22.8, ALOGPs = −1.41, RngAr = 0, Fsp3 = 0.68), and the NRPS-derived antibiotics teicoplanin (Targocid; MW = 1880, LogD = −2.74, ALOGPs = 1.74, RngAr = 7, Fsp3 = 0.42) and daptomycin (Cubicin; MW = 1621, LogD = −22.1, ALOGPs = 0.93, RngAr = 3, Fsp3 = 0.53) (ESI Fig. S7†).
In contrast, the polyketide-derived macrolides clustered more tightly near the origin along PC1 and in the negative range along PC2 and the positive range along PC3, indicating trends toward comparatively smaller size (and concurrently higher LogD) and more sp3 content. Examples include the anticancer agent eribulin (Halaven; MW = 730, LogD = 0.20, ALOGPs = 1.26, RngAr = 0, Fsp3 = 0.88) and the antibiotic fidaxomicin (Dificid; MW = 1058; LogD = 7.10, ALOGPs = 5.59; RngAr = 1, Fsp3 = 0.63) (ESI Fig. S7†).
Interestingly, of the two DOS probes (green diamonds) with macrocyclic structures, rapadocin (36-membered ring) was positioned near the macrolides in the PC1 vs. PC2 plot (slightly below and to the left), consistent with its large size (MW = 1238) and high hydrophobicity (ALOGPs = 5.87), but in a distinct negative region along PC3, owing to its high aromatic ring content (RngAr = 4) (ESI Fig. S7†). Meanwhile, H3B-8800 (12-membered ring) fell on the other side of the macrolide cluster in the PC1 vs. PC2 plot (slightly above and toward the right end of the group), and near the origin along PC3 (at the top end of the group). This may be attributed to its relatively smaller size (MW = 556) and intermediate hydrophobicity (ALOGPs = 3.32) and sp3 content (Fsp3 = 0.58).
2.6 Classification by therapeutic class
Next, to assess the relationship between drug structure and function, compounds were grouped according to therapeutic indication in 12 major classes: anticancer, antiinflammatory, antimicrobial, antiulcer, antiviral, cardiovascular, central and peripheral nervous system (CNS & PNS), hormone therapy, immunomodulatory, metabolic disorders, respiratory, and other (see ESI Dataset†). In most cases, class assignments were made by grouping together relevant therapeutic indications reported previously by Newman and Cragg.1 However, in some cases (e.g., antiallergic indication), drugs were assigned individually to the most relevant class herein. In contrast to the analysis by biosynthetic classes above, drugs clustered less tightly according to therapeutic class, reflecting the structural diversity of some of the classes (Fig. 5 and ESI Table S8†).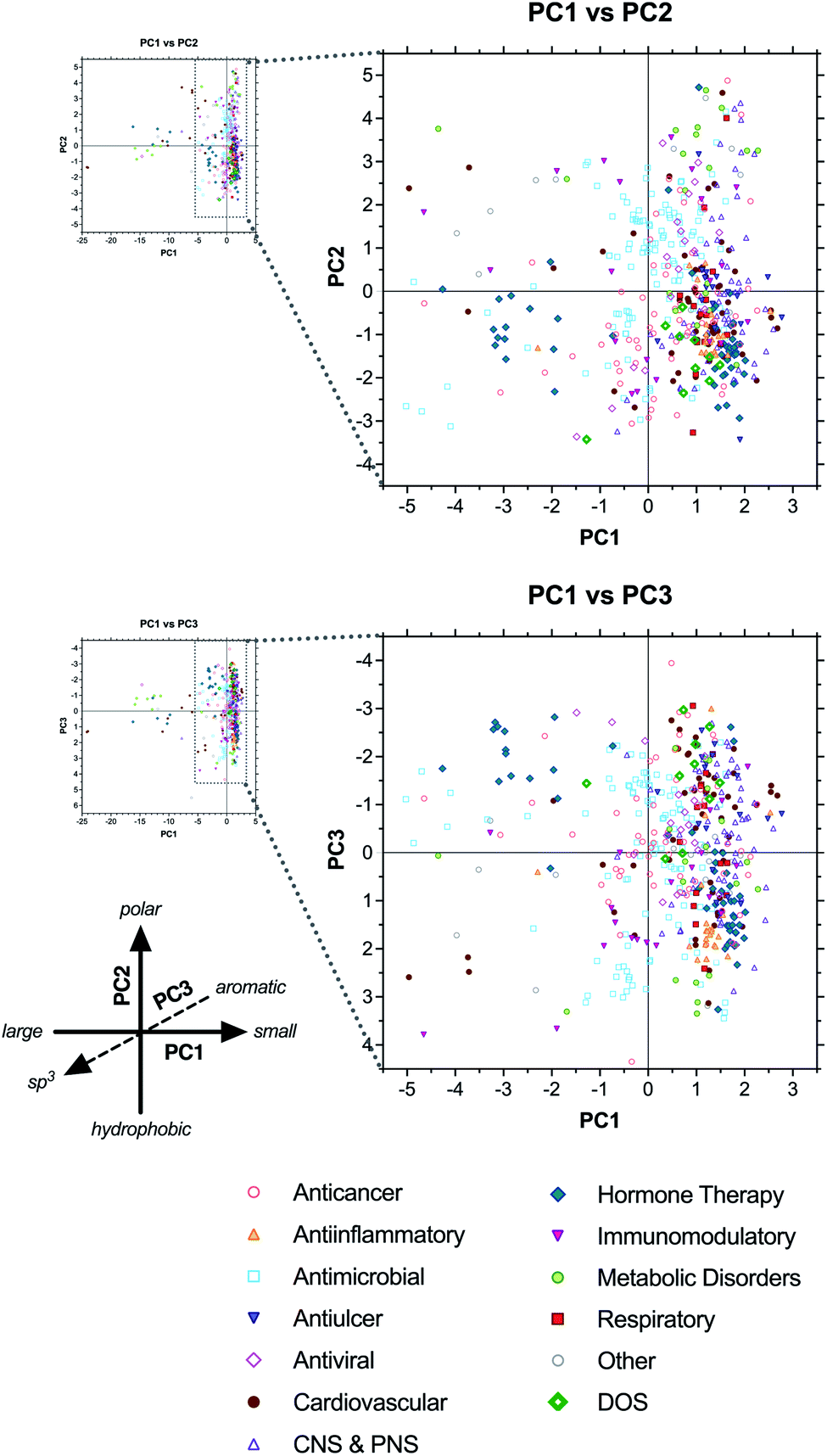 | ||
| Fig. 5 PCA plots of drugs coded by therapeutic class. All natural product-based drugs (N, ND, S*, S*/NM) are shown, in addition to purely synthetic drugs present in the Top 40 collections (S, S/NM), as well as chemical probes identified from diversity-oriented synthesis libraries (DOS; bold green diamonds). | ||
The anticancer (red open circles) and antimicrobial (cyan open squares) classes had the greatest numbers of compounds in the analysis (63 and 115 unique structures, respectively) and spanned the widest ranges of chemical space, consistent with the diversity of biological targets addressed by these drugs. For example, the antimicrobials included β-lactam inhibitors of penicillin binding protein and β-lactamase, glycopeptides that bind D-ala–D-ala motifs in cell wall biosynthesis intermediates, aminoglycosides that disrupt translational proofreading in the 30S ribosome subunit, and macrolides that block tRNA translocation in the 50S ribosome (ESI Fig. S8 and Table S9†). All of these classes occupied distinct regions of the PCA plots corresponding to their structures.
There were also cases of biological targets for which multiple structural solutions are seen in drugs. For example, topoisomerase II is targeted both by the anthracyclines, which are aromatic polyketides bearing aminoglycoside substituents, and etoposide phosphate (Etopophos), which is derived from a distinct lignan biosynthetic pathway (ESI Fig. S9 and Table S10†). Further, microtubule-stabilizing anticancer drugs include both the terpenoid-derived paclitaxel (Taxol) and docetaxel (Taxotere), and the polyketide-derived macrocycle ixabepilone (Ixempra), all of which bind the same site on tubulin. In these cases, distinct structures that shared the same target fell in overlapping or nearby regions of the PCA plots, and were not as dramatically separated as compounds having different targets. This suggests that there may be some correlation between individual biological targets and the structural properties of drugs that bind them, even if the specific structures are formally distinct.
The antiinflammatory category (orange triangles) exhibited the tightest clustering of all drug classes, falling primarily in the lower right quadrant of both the PC1 vs. PC2 plot and the PC1 vs. PC3 plot (Fig. 5). The majority of these compounds are structurally similar corticosteroids, with relatively low molecular size, high hydrophobicity, and high sp3vs. aromatic content (avg MW = 485, ALOGPs = 3.20, Fsp3 = 0.67, RngAr = 0.3) compared to the complete collection (avg MW = 673, ALOGPs = 2.01, Fsp3 = 0.58, RngAr = 1.5) (ESI Table S8†).
Antivirals (pink open diamonds) clustered near the origin along PC1, consistent with intermediate size (avg MW = 515 excluding enfuvirtide (Fuzeon) ribosomal peptide outlier), but spanned a wide range along PC2, indicating diverse polarities (ALOGPs −2.29 to +5.97). This class includes nucleosides, aminoglycosides, and peptidomimetics, accounting for this structural diversity (ESI Fig. S10 and Table S11†). Notably, the aminoglycosides clustered with the nucleosides in the PC1 vs. PC2 plot, but differentiated along PC3 based on sp3vs. aromatic character, with the former in the positive range (avg Fsp3 = 0.68, RngAr = 0) and the latter in the negative range (avg Fsp3 = 0.53, RngAr = 1.88).
Antiulcer drugs (blue inverted triangles) also fell at the positive (right) end of PC1, straddling the origin along PC2, and spanning a wide range along PC3 (Fig. 5 and ESI Table S12†). Most of these structures are polyisoprenoids and eicosanoids, having relatively low molecular weight, intermediate polarity, and moderate to high sp3 content (avg MW = 389, ALOGPs = 3.42, Fsp3 = 0.47, RngAr = 1.7) (ESI Table S8†). However, this class also includes several synthetic proton pump inhibitors that have high aromatic content (e.g., esomeprazole [Nexium], lansoprazole [Prevacid], pantoparazole [Protonix], rabeprazole [Aciphex]; RngAr = 3), explaining the diversity of positions along PC3.
Cardiovascular drugs (brown circles) were highly divergent, consistent with their wide range of targets, and included compounds falling in all four quadrants of both the PC1 vs. PC2 and PC1 vs. PC3 plots. Notably, this category included the heparin and hirudin families of anticoagulants, as well as atrial natriuretic peptide analogues, extending to the extreme negative (left) end of the PC1 axis owing to their large sizes (ESI Fig. S11 and Table S13†). In contrast, the statin family of polyketide-based drugs clustered in the positive range along PC1 based on their smaller structures. Notably, their positions along PC3 differentiated the natural products (lovastatin [Mevacor], simvastatin [Zocor], pravastatin [Mevalotin]) and their natural product-derived analogues (atorvastatin [Lipitor], rosuvastatin [Crestor]) based on sp3vs. aromatic content (avg Fsp3 = 0.75 vs. 0.34; RngAr = 0 vs. 3), demonstrating that drugs with the same molecular target may fall in somewhat distinct regions of chemical space.
CNS & PNS drugs (purple open triangles) were found exclusively in the positive range (right) of PC1, reflective of the typically small chemotypes associated with neurological targets, but spanned broad ranges along both PC2 and PC3 (Fig. 5). This broad category includes both CNS and PNS drugs with a variety of targets and mechanisms of action, consistent with this structural diversity.
Hormone therapies (green diamonds) segregated into three distinct clusters along PC1, corresponding to large peptides (e.g., growth hormone-releasing hormone and parathyroid hormone analogues), oligopeptides of 8–12 residues (e.g., gonadotropin-releasing hormone and somatostatin analogues), and smaller molecules (e.g., steroid contraceptives, eicosanoid abortifactents, and vitamin D analogues) (ESI Fig. S12†). The latter two clusters generally fell in the negative range along PC2, consistent with their hydrophobic character, but diverged along PC3, indicating contrasting levels of sp3vs. aromatic content.
Immunomodulatory drugs (inverted pink triangles) did not cluster tightly along any axis, indicative of their wide range of structures and targets. Even the subcategory of immunosuppressants included the large macrolide FKBP ligands tacrolimus (FK-506, Prograf), pimecrolimus (Elidel), sirolimus (rapamycin, Rapamune), and everolimus (Afinitor); the macrocyclic peptide cyclosporine (Sandimmune); the nucleoside mizoribine (Bredinin); the spermidine derivative gusperimus (Spanidin); and the terpenoids mycophenolate (Myfotic) and mycophenolate mofetil (CellCept) (ESI Fig. S13 and Table S14†).
Drugs used to treat metabolic disorders (lime circles) segregated into three main clusters, one in the extreme negative region (left) of PC1, another in the upper right quadrant of the PC1 vs. PC2 plot, and a third in the lower right quadrant. The first cluster included large antidiabetic peptides (e.g., glucagon and glucagon-like peptide mimetics), the second included a wide variety of small metabolite analogues (e.g., metformin, mlglastat, betaine), and the third was comprised mainly of synthetic antidiabetics (e.g., DPP4 inhibitors and PPAR-γ agonists) (see ESI Fig. S14 and Table S15†). Notably, the metabolite analogues fell in the extreme positive range along PC2, consistent with their highly polar structures, and generally in the positive range along PC3, indicative of their high sp3 content. In contrast, the synthetic compounds fell in the negative region of PC3, based on their high aromatic content.
Finally, respiratory drugs (red squares) segregated into two clusters in the upper and lower right quadrants of the PC1 vs. PC2 plot, indicative of relatively smaller, hydrophobic molecules (avg MW = 450, ALOGPs = 2.80) (ESI Table S8†). This class spanned a wide range along the PC3 axis, consistent with a variety of balances between sp3 and aromatic content (Fsp3 = 0.19 to 0.95; RngAr = 0 to 4). Examples used to treat asthma include the steroids budesonide (Pulmicort, Symbicort) and ciclesonide (Alvesco), the aromatic polyketide amlexanox (Solfa), the alkaloid salmeterol xinafoate (Advair), and the synthetic montelukast (Singulair) (ESI Fig. S15 and Table S16†).
3 Discussion
In this study, we used structural and physicochemical property analysis and principal component analysis (PCA) to investigate differences between natural product-based drugs, top-selling brand name drugs, and chemical probes discovered recently from DOS libraries. To explore temporal trends, we also compared the top 40 brand-name drugs in 2018 and in 2006. As a class, natural product-based drugs exhibit greater structural diversity, larger molecular size, an increased range of polarities, and more three-dimensional character compared to the purely synthetic drugs included in this analysis, although it must be noted that the latter were limited to a small set present in top-selling drugs. The DOS probes exhibited intermediate or drug-like characteristics in most cases, with the exception of tending to be highly hydrophobic. Although this may be perceived to impact their developability as therapeutics, it should be noted that they remain within the range of chemical space occupied by some natural product-based drugs.Moreover, we noted a significant increase in the number of natural product-based drugs and drug components in 2018 compared to 2006. Structurally, this was manifested in the property bar graphs and PCA plots by a slight shift on average toward larger and beyond-rule-of-5 molecules. This may reflect the increasing interest in investigating a wider range of structures, and natural products in particular, in drug discovery. The increased prevalence of these molecules in top-selling brand name drugs suggests that such efforts have been successful and may continue in the future. However, it should be noted that these molecules still generally cluster with purely synthetic drugs and do not extend out into the extreme regions of chemical space accessed by other natural product-based drugs.
Visualization of the PCA analyses encoded by biosynthetic origin revealed that drugs tend to cluster accordingly, with large peptide and carbohydrate molecules accessing remote regions of chemical space that are otherwise inaccessible to the other biosynthetic classes, and to purely synthetic drugs. Cyclic peptides were distributed across this region of chemical space, while macrolides clustered more tightly with other polyketide drugs having intermediate molecular size. Notably, the DOS probe rapadocin43 is also macrocyclic and fell in this general region of chemical space, indicating its accessibility via synthesis. Other DOS probes with polycyclic structures overlapped with regions populated by alkaloid and terpenoid natural product-based drugs. We note that rapadocin is at the size limit of natural polyketides, but well outside the normal range of the top-selling drug sets. This supports the idea that novel synthetic pathways can provide access to broader regions of chemical space that may be of clinical utility.
Sorting the dataset according to therapeutic class revealed that drugs generally did not cluster strictly according to therapeutic class, but rather by structural class, as would be expected given that the PCA is based on structural parameters. Some subsets of drugs did cluster according to molecular targets, although examples of divergent chemical solutions to individual targets were also evident. Perhaps the only exception to these trends were in the CNS & PNS and respiratory drugs, representing 26 and 8 subcategories, respectively, as originally reported by Newman and Cragg.1 In these cases, the pharmacological requirements for distribution in these tissue compartments may explain this apparent clustering.
4 Conclusions
Natural product-based drugs continue to play a major role in modern drug discovery. The prevalence of natural product-based molecules in top-selling brand name drugs has increased in 2018 compared to 2006, suggesting a resurgence of interest in this chemical diversity to address the increasing landscape of challenging therapeutic targets. Notably, the majority of these structures continue to be produced by de novo synthesis, rather than being derived directly from natural products. This may explain why they remain generally clustered near purely synthetic drugs, and do not reach the broader regions of chemical space accessed by larger natural products and derivatives. New synthetic strategies to access such larger molecules are being developed.43,44 Moreover, recent innovations in genome mining and activation of silent biosynthetic gene clusters are providing direct access to novel natural products.6,45,46 Thus, it will be of interest in the future to assess whether this increased access to novel and diverse natural product-based molecules translates to their increased prevalence in new drugs.5 Author contributions
S. S. carried out the experiments. D. J. N. provided data on drug origins. S. L. C. contributed to experimental design and analysis. S. S. and D. S. T. analyzed the experiments and wrote the manuscript with input from all coauthors.6 Conflicts of interest
S. L. C., as an employee of Lodo Therapeutics, facilitated partial financial support for this work. Lodo Therapeutics was subsequently acquired by Zymergen, Inc.7 Acknowledgements
We thank Prof. Jon T. Njarðarson (University of Arizona) and Prof. Igor V. Tetko (Helmholtz Zentrum München) for helpful discussions. Instant JChem was generously provided by ChemAxon. Financial support from Lodo Therapeutics (now Zymergen, Inc.) and the National Institutes of Health (R01 AI136795 to D. S. T. and CCSG P30 CA008748 to C. B. Thompson) is gratefully acknowledged.8 Notes and references
- D. J. Newman and G. M. Cragg, Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- C. F. Stratton, D. J. Newman and D. S. Tan, Cheminformatic comparison of approved drugs from natural product versus synthetic origins, Bioorg. Med. Chem. Lett., 2015, 25, 4802–4807 CrossRef CAS PubMed.
- R. L. M. van Montfort and P. Workman, Structure-based drug design: Aiming for a perfect fit, Essays Biochem., 2017, 61, 431–437 CrossRef.
- M. Song and G. T. Hwang, DNA-encoded library screening as core platform technology in drug discovery: Its synthetic method development and applications in DEL synthesis, J. Med. Chem., 2020, 63, 6578–6599 CrossRef CAS PubMed.
- L. H. Jones and M. E. Bunnage, Applications of chemogenomic library screening in drug discovery, Nat. Rev. Drug Discovery, 2017, 16, 285–296 CrossRef CAS PubMed.
- A. G. Atanasov, S. B. Zotchev and V. M. Dirsch, The International Natural Product Sciences Taskforce; Supuran, C. T. “Natural products in drug discovery: Advances and opportunities, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- B. Hong, T. Luo and X. Lei, Late-stage diversification of natural products, ACS Cent. Sci., 2020, 6, 622–635 CrossRef CAS PubMed.
- E. K. Davison and M. A. Brimble, Natural product derived privileged scaffolds in drug discovery, Curr. Opin. Chem. Biol., 2019, 52, 1–8 CrossRef CAS PubMed.
- P. A. Clemons, N. E. Bodycombe, H. A. Carrinski, J. A. Wilson, A. F. Shamji, B. K. Wagner, A. N. Koehler and S. L. Schreiber, Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 18787–18792 CrossRef CAS PubMed.
- P. A. Clemons, J. A. Wilson, V. Dancik, S. Muller, H. A. Carrinski, B. K. Wagner, A. N. Koehler and S. L. Schreiber, Quantifying structure and performance diversity for sets of small molecules comprising small-molecule screening collections, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 6817–6822 CrossRef CAS.
- T. J. Ritchie and S. J. F. MacDonald, “The impact of aromatic ring count on compound developability: Are too many aromatic rings a liability in drug design?” Drug Discov, Today, 2009, 14, 1011–1020 CAS.
- F. Lovering, J. Bikker and C. Humblet, Escape from flatland: Increasing saturation as an approach to improving clinical success, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS.
- B. C. Doak, J. Zheng, D. Dobritzsch and J. Kihlberg, How beyond rule of 5 drugs and clinical candidates bind to their targets, J. Med. Chem., 2016, 59, 2312–2327 CrossRef CAS PubMed.
- D. A. DeGoey, H.-J. Chen, P. B. Cox and M. D. Wendt, Beyond the rule of 5: Lessons learned from AbbVie's drugs and compound collection, J. Med. Chem., 2018, 61, 2636–2651 CrossRef CAS PubMed.
- M. D. Shultz, Two decades under the influence of the rule of five and the changing properties of approved oral drugs, J. Med. Chem., 2019, 62, 1701–1714 CrossRef CAS PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, The re-emergence of natural products for drug discovery in the genomics era, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed.
- F. Begnini, V. Poongavanam, B. Over, M. Castaldo, S. Geschwindner, P. Johansson, M. Tyagi, C. Tyrchan, L. Wissler, P. Sjoe, S. Schiesser and J. Kihlberg, Mining natural products for macrocycles to drug difficult targets, J. Med. Chem., 2021, 64, 1054–1072 CrossRef CAS PubMed.
- A. Bergner, X. Cockcroft, G. Fischer, A. Gollner, W. Hela, R. Kousek, A. Mantoulidis, L. J. Martin, M. Mayer, B. Muellauer, G. Siszler, B. Wolkerstorfer, D. Kessler and D. B. McConnell, KRAS binders hidden in nature, Chem. –Eur. J., 2019, 25, 12037–12041 CrossRef CAS.
- R. A. Bauer, J. M. Wurst and D. S. Tan, “Expanding the range of ‘druggable’ targets with natural product-based libraries: An academic perspective, Curr. Opin. Chem. Biol., 2010, 14, 308–314 CrossRef CAS PubMed.
- N. A. McGrath, M. Brichacek and J. T. Njarðarson, A graphical journey of innovative organic architectures that have improved our lives, J. Chem. Educ., 2010, 87, 1348–1349, see also: http://cbc.arizona.edu/njardarson/group/top-pharmaceuticals-poster Search PubMed.
- C. J. Gerry and S. L. Schreiber, Recent achievements and current trajectories of diversity-oriented synthesis, Curr. Opin. Chem. Biol., 2020, 56, 1–9 CrossRef CAS.
- T. A. Wenderski, C. F. Stratton, R. A. Bauer, F. Kopp and D. S. Tan, Principal component analysis as a tool for library design: A case study investigating natural products, brand-name drugs, natural product-like libraries, and drug-like libraries, Methods Mol. Biol., 2015, 1263, 225–242 CrossRef CAS.
- R. A. Bauer, T. A. Wenderski and D. S. Tan, Biomimetic diversity-oriented synthesis of benzannulated medium rings via ring expansion, Nat. Chem. Biol., 2013, 9, 21–29 CrossRef CAS PubMed.
- F. Kopp, C. F. Stratton, L. B. Akella and D. S. Tan, A diversity-oriented synthesis approach to macrocycles via oxidative ring expansion, Nat. Chem. Biol., 2012, 8, 358–365 CrossRef CAS.
- G. Moura-Letts, C. M. DiBlasi, R. A. Bauer and D. S. Tan, Solid-phase synthesis and chemical space analysis of a 190-membered alkaloid/terpenoid-like library, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 6745–6750 CrossRef CAS.
- R. A. Bauer, C. M. DiBlasi and D. S. Tan, The tert-butylsulfinamide lynchpin in transition-metal-mediated multiscaffold library synthesis, Org. Lett., 2010, 12, 2084–2087 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev., 1997, 23, 3–25 CrossRef CAS.
- I. V. Tetko, V. Y. Tanchuk and A. E. P. Villa, Prediction of n-octanol/water partition coefficients from PHYSPROP database using artificial neural networks and E-state indices, J. Chem. Inf. Comput. Sci., 2001, 41, 1407–1421 CrossRef CAS.
- I. V. Tetko, V. Y. Tanchuk, T. N. Kasheva and A. E. P. Villa, Internet software for the calculation of the lipophilicity and aqueous solubility of chemical compounds, J. Chem. Inf. Comput. Sci., 2001, 41, 246–252 CrossRef CAS PubMed.
- I. V. Tetko, V. Y. Tanchuk, T. N. Kasheva and A. E. P. Villa, Estimation of aqueous solubility of chemical compounds using E-state indices, J. Chem. Inf. Comput. Sci., 2001, 41, 1488–1493 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- M. Feher and J. M. Schmidt, Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry, J. Chem. Inf. Comput. Sci., 2003, 43, 218–227 CrossRef CAS PubMed.
- I. T. Joliffe and B. J. Morgan, Principal component analysis and exploratory factor analysis, Stat Methods Med Res, 1992, 1, 69–95 CrossRef CAS PubMed.
- I. T. Joliffe Principal Component Analysis; Springer: New York, New York, 2002 Search PubMed.
- J. E. A. Jackson. User's Guide to Principal Components; Wiley: Hoboken, New Jersey, 2003 Search PubMed.
- P. Ertl and T. Schuhmann, Cheminformatics analysis of natural product scaffolds: Comparison of scaffolds produced by animals, plants, fungi and bacteria, Mol. Inf., 2020, 39, e2000017 CrossRef.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, The exploration of macrocycles for drug discovery - An underexploited structural class, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- E. Marsault and M. L. Peterson, Macrocycles are great cycles: Applications, opportunities, and challenges of synthetic macrocycles in drug discovery, J. Med. Chem., 2011, 54, 1961–2004 CrossRef CAS PubMed.
- A. K. Yudin, Macrocycles: Lessons from the distant past, recent developments, and future directions, Chem. Sci., 2015, 6, 30–49 RSC.
- M. R. Naylor, A. T. Bockus, M.-J. Blanco and R. S. Lokey, Cyclic peptide natural products chart the frontier of oral bioavailability in the pursuit of undruggable targets, Curr. Opin. Chem. Biol., 2017, 38, 141–147 CrossRef CAS PubMed.
- A. Zorzi, K. Deyle and C. Heinis, Cyclic peptide therapeutics: Past, present and future, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS.
- M. D. Cummings and S. Sekharan, Structure-based macrocycle design in small-molecule drug discovery and simple metrics to identify opportunities for macrocyclization of small-molecule ligands, J. Med. Chem., 2019, 62, 6843–6853 CrossRef CAS.
- Z. Guo, S. Y. Hong, J. Wang, S. Rehan, W. Liu, H. Peng, M. Das, W. Li, S. Bhat, B. Peiffer, B. R. Ullman, C.-M. Tse, Z. Tarmakova, C. Schiene-Fischer, G. Fischer, I. Coe, V. O. Paavilainen, Z. Sun and J. O. Liu, Rapamycin-inspired macrocycles with new target specificity, Nat. Chem., 2019, 11, 254–263 CrossRef CAS.
- Y. Huang, M. M. Wiedmann and H. Suga, RNA display methods for the discovery of bioactive macrocycles, Chem. Rev., 2019, 119, 10360–10391 CrossRef CAS.
- G. D. Hannigan, D. Prihoda, A. Palicka, J. Soukup, O. Klempir, L. Rampula, J. Durcak, M. Wurst, J. Kotowski, D. Chang, R. Wang, G. Piizzi, G. Temesi, D. J. Hazuda, C. H. Woelk and D. A. Bitton, A deep learning genome-mining strategy for biosynthetic gene cluster prediction, Nucleic Acids Res., 2019, 47, e110 CrossRef CAS.
- K. Kingwell, An audience with Rick Brown, Nat. Rev. Drug Discovery, 2019, 18, 10–11 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1np00039j |
| This journal is © The Royal Society of Chemistry 2022 |