
Marine paralytic shellfish toxins: chemical properties, mode of action, newer analogues, and structure–toxicity relationship†
Joana F.
Leal
 and
Maria L. S.
Cristiano
*
and
Maria L. S.
Cristiano
*
Centre of Marine Sciences (CCMAR), Department of Chemistry and Pharmacy, Faculty of Science and Technology, University of Algarve, Campus de Gambelas, 8005-139 Faro, Portugal. E-mail: mcristi@ualg.pt
First published on 30th June 2021
Abstract
Up to the end of 2020
Every year, the appearance of marine biotoxins causes enormous socio-economic damage worldwide. Among the major groups of biotoxins, paralytic shellfish toxins, comprising saxitoxin and its analogues (STXs), are the ones that cause the most severe effects on humans, including death. However, the knowledge that currently exists on their chemistry, properties and mode of toxicological action is disperse and partially outdated. This review intends to systematically compile the dispersed information, updating and complementing it. With this purpose, it addresses several aspects related to the molecular structure of these toxins. Special focus is given to the bioconversion reactions that may occur in the different organisms (dinoflagellates, bivalves, and humans) and the possible mediators involved. A critical review of the most recently discovered analogues, the M-series toxins, is presented. Finally, a deep discussion about the relationship between the molecular structure (e.g., effect of the substituting groups and the net charge of the molecules) and the toxic activity of these molecules is performed, proposing the concept of “toxicological traffic light” based on the toxicity equivalency factors (TEFs).
 Joana F. Leal | Joana F. Leal completed her PhD in Chemistry in 2017 at the University of Aveiro, in Portugal. She concluded her Master in Analytical Chemistry and Quality in 2012 and her Degree in Biochemistry in 2010, both at the University of Aveiro. Currently, she is a researcher in Centre of Marine Sciences of Algarve (Portugal). She published several articles in scientific international journals as first author and participated in many international and national conferences. Her main areas of interest are Analytical and Environmental Chemistry and Organic Chemistry. Her research interests are currently focused on marine biotoxins, aquatic remediation and aquaculture. |
 Maria L. S. Cristiano | Maria L. S. Cristiano is full professor of Chemistry at the Department of Chemistry and Pharmacy, Faculty of Sciences and Technology, University of Algarve (FCT/UAlg). Present duties: dean of FCT/UAlg; director of the PhD course in Chemistry; PI of the R&D group Organic Reactivity and Medicinal Chemistry (OrgMedChem) of Centre of Marine Sciences (CCMar/UAlg). Memberships: IUPAC subcommittee “structural and mechanistic chemistry”; Portuguese Chemical Society; Royal Society of Chemistry (CCHEM). Research interests: Organic Reactivity; Heterocyclic Chemistry; anti-infectives. Research in the OrgMedChem group combines tools and approaches of Physical Organic and Medicinal Chemistry to investigate structure, reactivity, and properties of molecules with potential applications in human health and aquaculture. |
1. Introduction
Marine biotoxins or phycotoxins originate from natural phenomena, such as the harmful algae blooms (HABs), which are characterized by a massive growth of phytoplankton in marine ecosystems, able to produce toxic metabolites. The number of HABs episodes appear to be increasing, mainly potentiated by environmental and climatic conditions, and eutrophication.1 These natural toxins are produced by unicellular microalgae,2 bio-accumulate in aquatic species, like bivalves, xanthid crabs and pufferfish, through the food chain, and may be potentially toxic for humans, depending on the levels ingested.Among the most affected aquatic species, bivalves stand out for their growing interest and consequent increase of their production and consumption.3 Data for the period between 2010 and 2015 refer a global production of marine bivalves for human consumption higher than 15 million tonnes per year (average), representing a direct economic impact of more than 23 billion US dollars per year, an amount further increased if indirect contributions (e.g., preparation, manufacturing, transportation) are considered.4 The contamination of bivalves is a matter of great social and economic concern. Its effects impact on human health, on fishing and aquaculture (e.g., through catch restrictions), on monitoring and management (e.g., sampling and analysis of water and bivalves), as well as on tourism and recreational activities.5 It is complex to estimate the global costs associated with the contamination episodes by marine toxins, since they may be influenced by several factors.6 However, the studies available reporting global cost estimates point to many millions of dollars per year.7–9
Marine toxins may be classified according to three criteria: solubility (two groups), toxic effects (five groups) and chemical structure (eight groups).10–12 The classification of the most common marine toxins in temperate areas is depicted in Fig. 1, where known interrelationships between the categories are also evidenced. Marine toxins may be divided in five groups, according to the nature of their toxic effects in humans: paralytic shellfish poisoning (PSP); amnesic shellfish poisoning (ASP); neurotoxic shellfish poisoning (NSP); diarrheic shellfish poisoning (DSP); and azaspiracid poisoning (AZP). Regarding the chemical structure, yessotoxins (YTXs) are a distinct group that some authors12 have associated with the DSP group. However, the European food safety authority refers that YTXs do not induce diarrhoea nor other adverse effects in humans,13 so we considered more appropriate to dissociate YTXs from the DSP group. Similarly, cyclic imines (CIs) have not been associated to poisoning events in humans, and CIs regulatory limits in shellfish are also not established.14 Thus, in Fig. 1 the YTX and CI groups are dissociated from specific groups based on toxic effects. According to the Harmful Algal Events Dataset (HAEDAT), data by syndrome from 1980 to 2015 revealed that DSP and PSP feature the highest number of episodes, 1321 (43.1%) and 1090 (35.5%) respectively, followed by ASP with 329 episodes (10.7%).15
 | ||
| Fig. 1 Marine toxins and their classification, based on solubility, toxic effects, and chemical structure. | ||
Among the groups of marine biotoxins with the highest representation of toxic episodes we will focus on those responsible for PSP, because they are the ones that may lead to the most serious health consequences in humans, including death. The possible symptoms in humans,16–19 resulting from ingestion of significant amounts of these toxins, depicted in Fig. 2, depend on the doses ingested. The poisoning effects occur quickly, the first symptoms appearing within thirty minutes after ingestion, and if proper measures are not taken intoxicated individuals may die within few hours (1–5 hours post-poisoning). As an approved antidote or therapy for STXs poisoning is not yet known, only clinical measures are taken to try speed up detoxification and prevent more serious consequences. At an early stage, the use of activated charcoal to remove unabsorbed toxins may be considered. Artificial respiration, vomiting, cleaning of gastric contents (using a naso-gastric catheter), overhydration, as well as hemodialysis are some of the treatments that have been indicated.19,20 Despite the negative consequences associated with STXs, it should be mentioned their great therapeutic potential, namely as anaesthetics.21–23
 | ||
| Fig. 2 Possible symptoms resulting of STXs ingestion, organized according to their severity. | ||
The information disclosed in the literature regarding the chemistry, properties and toxicological action of STXs is disperse, partially outdated and often unrelated. In our view, a revision that systematically compiles, updates and correlates the dispersed information is both useful and timely, as it will facilitate further research in this area, encouraging the development of strategies to circumvent problems associated to their toxicity. Furthermore, to the best of our knowledge, this manuscript presents the first critical revision on the latest analogues, M-toxins, and highlights the recent findings about biotransformation in different organisms (dinoflagellates, bivalves, humans). Thus, this article is organized in four major parts: the first part focus on the main physicochemical characteristics of STXs, including their structure and chemical reactivity, which impact in their properties; the second part presents a biochemical perspective about the synthesis of STXs, as well as their mode of action; the third part displays the bioconversions between the STX analogues within the organisms, providing an in-depth discussion about the bio(chemical) reactions involved (also considering mediators) and their interaction with the different substituent groups; the fourth part provides an in depth analysis on existing evidence concerning the relationships between molecular structure and toxicity, among STX analogues.
2. Physicochemical characterization of STXs
Saxitoxin (STX) heads a long list of more than 50 toxins24 able to cause paralytic shellfish poisoning (PSP), and was the first molecule to be characterized from this group. As a whole, this set of toxins is known as paralytic shellfish toxins (PSTs), saxitoxin and its analogues (STXs), or simply as the saxitoxins (STXs)-group, since all members of the group share a common core structure,25,26 as shown in Fig. 3. Table 1 presents the various substituents along the STXs-group, also distributed by subgroups according to their characteristics. The categorization is based on the substituent group in R4 and the configurations α/β of the stereoisomers at C11 are also identified.25,27–31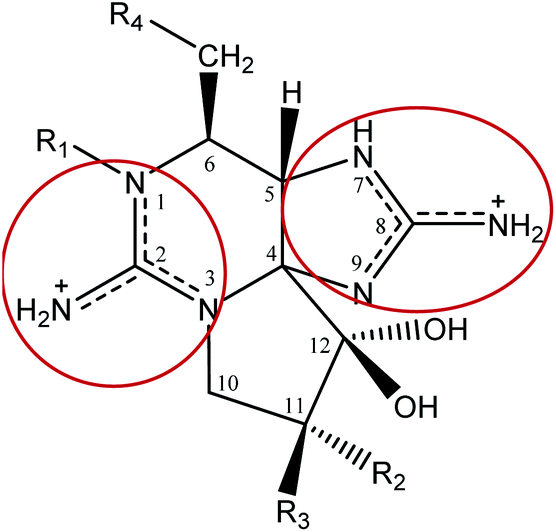 | ||
| Fig. 3 Representation of the molecular structure of STXs, with positive charge fully delocalized over the pyrimidine and imidazole groups (resonance structure representation shown inside the red circles). | ||
| R1 | R2 | R3 | R4 | Toxin | Molecular weighte | STX-subgroup |
|---|---|---|---|---|---|---|
| a Often referenced as 11-α,β-hydroxy STX. b Open ring at C4. c Additional structural characterization studies should be conducted. d Proposed in this work. e Note: the molecular weights presented correspond to uncharged species. | ||||||
| –H | –H | –H |

|
STX | 299 | Carbamate |
| –OH | –H | –H | NEO | 315 | ||
| –OH | –OSO3− | –H | GTX1 (α) | 411 | ||
| –H | –OSO3− | –H | GTX2 (α) | 395 | ||
| –H | –H | –OSO3− | GTX3 (β) | 395 | ||
| –OH | –H | –OSO3− | GTX4 (β) | 411 | ||
| –H | –OH | –H | M2αa | 315 | ||
| –H | –H | –OH | M2βa | 315 | ||
| –H | –OH | –OH | M4 | 331 | ||
| –H | –OH | –OH | M6b,c | 333d | ||
| –OH | –OH | –H | M8α | 331 | ||
| –OH | –H | –OH | M8β | 331 | ||
| –OH | –OH | –OH | M10 | 347 | ||
| –OH | –OH | –OH | M12b,c | 349d | ||
| –H | –H | –H |
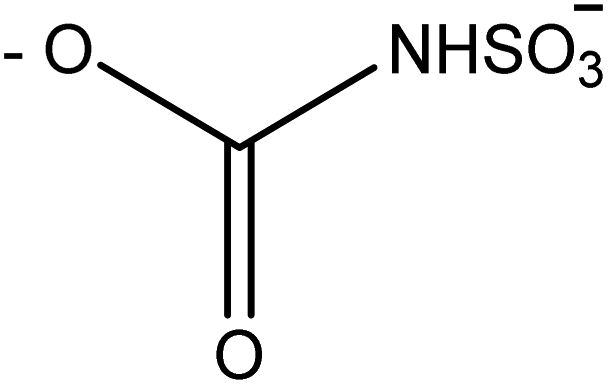
|
GTX5 or B1 | 379 | N-Sulfocarbamoyl |
| –OH | –H | –H | GTX6 or B2 | 395 | ||
| –H | –OSO3− | –H | C1 (α) | 475 | ||
| –H | –H | –OSO3− | C2 (β) | 475 | ||
| –OH | –OSO3− | –H | C3 (α) | 491 | ||
| –OH | –H | –OSO3− | C4 (β) | 491 | ||
| –H | –OH | –H | M1α | 395 | ||
| –H | –H | –OH | M1β | 395 | ||
| –H | –OH | –OH | M3 | 411 | ||
| –H | –OH | –OH | M5b,c | 413 | ||
| –OH | –OH | –H | M7α | 411 | ||
| –OH | –H | –OH | M7β | 411 | ||
| –OH | –OH | –OH | M9 | 427 | ||
| –OH | –OH | –OH | M11b,c | 429d | ||
| –H | –H | –H | –OH | dcSTX | 256 | Decarbamoyl |
| –OH | –H | –H | dcNEO | 272 | ||
| –OH | –OSO3− | –H | dcGTX1 (α) | 368 | ||
| –H | –OSO3− | –H | dcGTX2 (α) | 352 | ||
| –H | –H | –OSO3− | dcGTX3 (β) | 352 | ||
| –OH | –H | –OSO3− | dcGTX4 (β) | 368 | ||
| –H | –H | –H |
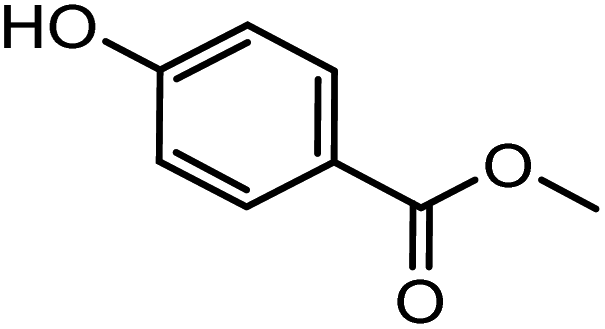
|
GC3 | 376 | Benzoate (para-hydroxy-) |
| –OH | –H | –H | GC6c | 392 | ||
| –OH | –OSO3− | –H | GC4 (α)c | 488 | ||
| –H | –OSO3− | –H | GC1 (α) | 472 | ||
| –H | –H | –OSO3− | GC2 (β) | 472 | ||
| –OH | –H | –OSO3− | GC5 (β)c | 488 | ||
| –H | –H | –H | –H | doSTX | 240d | Deoxydecarbamoyl |
| –OH | –OSO3− | –H | doGTX1 | 352d | ||
| –H | –OSO3− | –H | doGTX2 | 336d | ||
Concerning the structure characterization and spectral properties, the pioneering and most relevant studies of STX were performed by Casselman et al. (1960)32 (infrared – IR), Schantz et al. (1961, 1975)33,34 (ultraviolet-visible – UV-vis, IR, X-ray), Wong et al. (1971),35 Rogers and Rapoport (1980),36 and Shimizu et al. (1981)37 (nuclear magnetic resonance – NMR). Results disclosed indicate that STX does not absorb above 220 nm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 and has a specific rotation of +130 ± 5°.38 More recent studies39–41 (mass spectrometry – MS, NMR) have corroborated the chemical structure of STX and unveiled the structures for known derivatives. Currently, the focus is on identifying and characterizing novel derivatives or analogues.
33 and has a specific rotation of +130 ± 5°.38 More recent studies39–41 (mass spectrometry – MS, NMR) have corroborated the chemical structure of STX and unveiled the structures for known derivatives. Currently, the focus is on identifying and characterizing novel derivatives or analogues.
Structurally, STX belongs to the tetrahydro-purine alkaloid chemical class, comprising a 3,4-propinoperhydropurine tricyclic system42 and two guanidine groups that render it into a highly polar and hydrophilic molecule. STX is soluble in water and methanol, less soluble in ethanol and glacial acetic acid, and insoluble in non-polar solvents.33 The guanidine groups, shown in Fig. 3, are associated to two proton dissociations at pKa 8.1–8.4 and 11.3–11.6, assigned to the guanidinium moieties centred at C8 (imidazole guanidine) and C2 (pyrimidine guanidine), respectively.26,43–45 The first pKa range is abnormally low for a guanidinium deprotonation and the first studies had proposed proton dissociation from a hydroxyl group.35 However, potentiometric titrations and studies of STX in solution at different pH values, using 13C-NMR and 1H-NMR,36,37 as well as single crystal X-ray diffraction studies34 corroborated the attribution of the low pKa to the imidazole guanidine group. Following X-ray diffraction studies the authors reported C–N bonds of both guanidines to be equal in length within the experimental error (1.32 ± 0.01 Å), except for bond C(8)–N(9), whose length is 1.36 ± 0.01 Å.34 In turn, NMR studies36,37 revealed the existence of different STX molecular forms in solution, at different pH values. The low pKa assigned to the imidazole group may suggest that the three nitrogen atoms are not fully hybridized, this is, C(8) full resonance is not stabilized.46 The three equilibrium species proposed for STX in solution are shown in Fig. 4. It is reasonable to assume that the hydrated STX (monocation) is structurally favoured, due to the electron-withdrawing guanidinium groups which relieve the double-charge of the neighbouring carbons. After deprotonation, the keto form (at C12) appears as the dominant species.
 | ||
| Fig. 4 Molecular forms of STX in solution. | ||
The elucidation of the STX molecular formula and structure was difficult to achieve, mostly due to its physicochemical properties. STX is an amorphous hygroscopic compound, non-volatile and unable to crystalize in the form of chloride, acetate or sulphate salt.35,43 STX is stable in acidic media but extremely unstable under alkaline conditions, decomposing in the presence of oxygen with formation of a fluorescent degradation product ((i), in Fig. 5).33,46–48 Maximum molar extinction coefficients reported by different authors were at 235 nm and 333 nm,33 and at 236 nm and 324 nm.48 Regarding the STX derivatives, there are exceptions to the stability at low pH, namely within the N-sulfocarbamoyl subgroup. At high temperatures and acidic conditions (pH < 4), STX derivatives belonging to this subgroup may suffer hydrolysis, converting into the correspondent carbamate toxins.49
 | ||
| Fig. 5 Schematic representation of the degradation of STX to fluorescent purine derivatives. | ||
As pointed out, STXs are strongly affected by pH variations in solution, this phenomenon impacting on their toxicity and mode of action, as will be discussed in detail below. The relative ratio of each STX species at different pH is presented in Table 2 (adapted from Shimizu et al. (1981)37). Also, for some STX derivatives, the predominant net charge under acidic (acetic acid 0.1 M) and physiological conditions (around pH 7.4) is presented in Table 3 (adapted from Hall et al. (1990)26). From these data, it is noteworthy that despite the similarities in structure of toxins C1–C4 (N-sulfocarbamoyl toxins) only C3 and C4 are proposed to bear one negative charge under physiological conditions. By analogy with NEO,26,45 this may be explained by the presence of the hydroxyl group at N1 (R1 substituent) that presents an additional and lower pKa (probably also involving the guanidium group at C2). This would imply that, at physiological pH, C3 and C4 toxins would have a negative charge balance (two positive and three negative), while C1 and C2 toxins would have a neutral charge balance (two positive and two negative). The same principle may explain the neutral charge (two positive and two negative) of the GTX1/4 and the positive charge (two positive and one negative) of the GTX2/3, under physiological conditions.
| Saxitoxin species (charge) | pH | ||
|---|---|---|---|
| 7.7 | 8.4 | 9.8 | |
| Keto form (+1) | 1 | 24 | 36 |
| Hydrated (+1) | 23 | 37 | 62 |
| Hydrated (+2) | 76 | 39 | 2 |
| Under acidic conditions | Under physiological conditions | |||||||
|---|---|---|---|---|---|---|---|---|
| Charge | +2 | +1 | 0 | −1 | +2 | +1 | 0 | −1 |
| B1 | B2 | |||||||
| STX-group toxins | GTX2 | C1 | NEO | GTX1 | C3 | |||
| STX | GTX3 | C2 | STX | B1 | GTX4 | C4 | ||
| NEO | B2 | C3 | GTX2 | C1 | ||||
| GTX1 | C4 | GTX3 | C2 | |||||
| GTX4 | ||||||||
In addition to the strong influence of pH on the structure and stability of saxitoxins, temperature also significantly affects their stability. Several studies have demonstrated the stability of saxitoxins solutions in acid medium (pH 3–4), at low temperatures (−35 °C), for about 7 months50 or for 12 months.51 In the specific case of STX, literature reports indicate that its stability is maintained at least for 24 months, at −20 °C and −80 °C.52 However, the same authors observed a small decrease of STX stability, after 12 months at 4 °C. Also, they reported a lower stability of NEO, comparatively to STX, at −80 °C, −20 °C and 4 °C. The stability of STXs is significantly reduced at room temperature (25 °C) and was shown to depend on the structure of the analogue.50,51 At higher temperatures (>90 °C), the stability of each analogue drastically decreases to hours or even a few minutes53,54 and the differences in stability between analogues appear to be accentuated upon increasing the temperature. For example, while performing experiments at 110 °C and 120 °C, for 180 minutes, some authors53 found that GTX1/4 is more thermolabile than GTX2/3, reporting degradation rates (first-order reactions) about three times higher for GTX1/4. In short, high temperatures combined with high pH values lead to greater instability and faster decomposition of toxins. It is important to keep in mind that the pH and temperature variations potentiate interconversions between the various analogues, which could lead to an increase or a decrease in overall toxicity (a detailed discussion of this aspect is provided below).
Regarding the photochemistry, the lack of absorption evidenced by STX above 220 nm33 suggests photostability to medium- and long-wave UV light, and visible light. However, there is a study referring an irreversible decrease in the total number of high-affinity [3H]STX binding sites in rat synaptosomes, after exposure to UV radiation.55 Furthermore, a more recent study56 reported the gradual decomposition of dcSTX under UV radiation, using TiO2-based photocatalysts, which encourages further studies on the photochemistry of STXs aimed at a deeper knowledge about their photostability.
3. Biochemical perspective: synthesis and mode of action
Most of the STXs considered in this work occur naturally in the environment and are mainly produced by eukaryotes marine dinoflagellates belonging to genera Alexandrium (e.g., A. minutum, A. tamarense, A. catenella, A. fraterculus, A. fundyense, A. cohorticula), Gymnodinium (G. catenatum) and Pyrodinium (P. bahamense), which mostly occur in tropical and temperate climate zones, but also by cyanobacteria (prokaryotes), such as Anabaena, Cylindrospermopsis, Aphanizomenon, Planktothrix and Lyngbya.57,58 Although STXs may be produced by different organisms (marine eukaryotes or freshwater prokaryotes), studies59,60 comparing the blue-green alga Aphanizomenon flos-aquae with Gonyaulax toxins (G. catenella, G. tamarensis, G. excavata – currently corresponding to A. catenella and A. tamarense) revealed that their biosynthesis appears to be similar. In fact, a more recent study corroborates the involvement of similar genes in the STX synthesis, in cyanobacteria and dinoflagellates.61 This work will focus on STX analogues produced from marine dinoflagellates, which encompass a more representative part.Several publications report on the biosynthesis of STX and some of its analogues62–68 and the differences in the biosynthetic pathways proposed by the respective authors may be essentially ascribed to the evolution of knowledge. The complementarity of the genetic and enzymatic information provided by the most recent studies with the structural information has allowed a deeper understanding of the biosynthetic pathway of STX and analogues. Also, omics technologies have played a major role in the evolution of knowledge concerning the biosynthesis of STXs due to availability of methodologies to extract and evaluate biomolecules. Ideally, an interdisciplinary approach should be privileged, considering the different omics technologies (genomic, proteomic, transcriptomic, metabolomic), combined with multivariate data analysis. This will allow a greater complementarity of information and the identification of any flaws. In this context, some authors69 have even proposed a universal protocol that combines the different omics. On the other hand, the reductionist approach (one-to-one analysis/technology) should be abandoned since it does not take into account other biological factors that might affect the results, leading to misinterpretations. More detailed information on the omics perspective applied to STX biosynthesis may be found in a recent review by Akbar et al. (2020).70 Apart from those differences, the involvement of key players identified in Fig. 6 appears to be consensual, namely the gene stxA, which is composed by four catalytic domains (SxtA1 to SxtA4), arginine, acetyl-CoA, S-adenosylmethionine (SAM) and carbamoyl-phosphate (CP). Initially, the SxtA4 domain (aminotransferase, class II) mediates a Claisen-type condensation between propionyl-ACP (acyl carrier protein) and arginine. The biosynthetic pathway proceeds with the involvement of other Sxt genes, like SxtG, StxBC, SxtS and SxtU, and the occurrence of several chemical reactions, such as hydroxylation, epoxidation, and cyclization, culminating in the STX biosynthesis.
 | ||
| Fig. 6 Key players on STX biosynthetic pathway. SxtA: gene; Arg: arginine; SAM: S-adenosylmethionine; acetyl-CoA: acetyl-coenzyme A; CP: carbamoyl-phosphate. | ||
The biosynthesis of the STXs may be direct (as with STX), indirect, or may occur in both ways (e.g., decarbamoyl toxins42). In the indirect biosynthesis STX analogues are not biosynthesized by cyanobacteria or dinoflagellates, arising from bioconversion/biotransformation of other toxins directly biosynthesized by them, probably through processes mediated by enzymes71 associated with the STX biosynthetic pathway.
The laboratory synthesis of STX was firstly reported by Tanino et al. (1977).72 These authors achieved the total synthesis of d, l – saxitoxin (corresponds to (+), (−) – saxitoxin), through a complex synthetic route, with the yields over the different steps ranging from 30 to 75%. Since then, several other studies concerning the synthesis of saxitoxin have been published.73–76 Furthermore, some authors25,42,77 have reported synthetic routes for interconversion among the STX analogues, some of which extracted from biological matrices. The acid hydrolysis of STX and GTXs into decarbamoyl toxins is an example of the reactions that may occur.42
It has been proposed that STX acts through the reversible blockade of the voltage-gated sodium channels on excitable membranes. STX was shown to selectively bind to the receptor site 1 on the α sub-unit, located in a water-filled region formed by four P-loops,78 in a 1:1 ratio79 and with high affinity for STX (Kd ≈ 2 × 10−9 M).80 As the toxin molecule is too large to fully penetrate in the pore of Na+ channels, it obstructs the passage of ions through the membranes of the nerve cells81 thereby compromising the propagation of neuronal impulses in peripheral nerves and skeletal muscles (Fig. 7).
 | ||
| Fig. 7 Schematic representation of the normal Na+ flux through the pore of the sodium channel (left) and the sodium channel blocked by STX, avoiding the Na+ flux (right); adapted from Duran-Riveroll and Cembella.81 | ||
The mode of action of STX analogues is similar to that of the parent compound,82 but the binding affinity to the Na+ channels was shown to vary along the STX family, according to the structure and polarity of each member. Structural effects, ascribed to steric hindrance, are particularly notorious with the larger functional groups of some analogues, such as sulphate or benzoate. In turn, the net charge of molecules was hypothesized as another determinant difference in behaviour along the STX family. A low binding affinity to the Na+ channels may be associated with negative charges of the toxin species (e.g., N-sulfocarbamoyl C-toxins), which could imply a compensatory reduction of positive charges to balance the net charge.81,83 The variations in binding affinity translate into higher or lower toxicities to the living organism, suggesting that the increase of binding affinity and the consequent blockage of ion passage potentiates a greater toxicity. In addition to the effect caused by the presence/absence of the substituent groups, which will be discussed in depth in the final part of this article, the stereochemistry of the C11 substituent groups also affects the binding affinity to the receptor. For example, Strichartz (1984)84 pointed to a greater binding affinity (higher toxicity) when the –OSO3− group is in the β-position, compared to the corresponding α-isomer. The electrostatic effect of the sulfate group in the β-position stabilizes the cationic form of carbonyl carbon (at C12), the main reactive intermediate for the nucleophilic attack involved in the formation of a hemiketal bond.84
Concerning the specific mode of action of STXs, and identification of the groups implicated in toxicological effects, several studies44,84 have pointed to the strong participation of the guanidinium groups in binding to the Na+ channel, mainly the imidazole-guanidinium group in its cationic form, which are the toxophores involved in Na+ channel blockage. The hydroxyl groups at C12 (Fig. 4) were also shown to play an important role on the binding affinity.44,84 Thus, the guanidinium groups seem to be able to act as a cationic substitute of the sodium ion, electrostatically interacting with the anionic charges82 of amino acid residues located on the receptor site 1 of the Na+ channel. Despite the various studies on this topic, the chemical interactions whereby this binding happens are poorly understood, and different theories82,85,86 have been proposed. The first was presented by Hille (1975)85 who proposed a plugging model and advocated STX binding to the Na+ channel through formation of hydrogen bonds and an electrostatic interaction (ionic bond) between the guanidium group and an anionic site (carboxylate group) inside the channel. Later, Kao and Walker (1982)82 postulated that the electrostatic attraction between the active guanidium group and the fixed anionic charges occurs outside the cavity, but around the orifice of the Na+ channel, obstructing it totally or partially. In turn, Shimizu (1982)86 proposed a three-point interaction model with the flat surface of the excitable membrane, which contemplates an ion pairing (ionic bond) of the guanidium group with an anionic site and two hydrogen bonds with hydroxyl groups from C12. Another theory84 suggests the formation of a weak covalent bond with nucleophilic groups on Na+ channel, but considering a reversible blockade, the formation of covalent bonds seems unlikely.
More recent studies have confirmed the interaction of STXs with Na+ channels87 and unravelled their interactions with potassium (K+)88 and calcium (Ca2+)89 channels, showing that the molecular mode of action of STXs is different for each channel. In the case of K+ channels, specifically the human ether-á-go-go (hERG) K+ channels, a mechanism based on the channel voltage–inactivation relationship was proposed.88 According to this model, STXs block the ion conduction pore, like in the Na+ channel, and alter the energetics of channel gating through the shift of both the voltage-inactivation and voltage–activation processes. This means that the STXs induce a slower channel opening under strong depolarization, and a faster channel closure upon repolarization. As the hERG K+ channels are responsible for several electrical events, namely in myocardial repolarization, their inhibition may cause fatal cardiac arrhythmias. Furthermore, unlike with Na+ channels, four or more toxin molecules may bind to the K+ channels.88 Although the mode of action of STX in the Ca2+ channels is still poorly understood, evidence points to some resemblance to the STX action mode in the Na+ channels. Such similarity was ascribed to the analogy in structural organization between the α1 subunit of the Ca2+ channel and the α subunits of the Na+ channel, and to the identical amino acid sequence (about 25%) in the transmembrane regions.90 Some authors89 reported a reversible partial blockade of L-type Ca2+ channels, and a reduction in the Ca2+ current in ventricular myocytes in the presence of STX. However, voltage dependence in the blockade of L-type Ca2+ channel was not observed in their studies. Although STX interaction with different channels is possible, Na+ channel binding appears to be the interaction causing the most relevant and worrying toxicological effects of this group of toxins. The different binding affinities of the toxin to each of the different channels corroborating their relative toxicological impact. Binding affinities for Na+ channels range from 10−10 to 10−8 and between 10−6 and 10−5 for K+ and Ca2+ channels.88,89,91
When comparing the action modes in organisms, a question arises relating to the different toxicity of these molecules in humans and bivalves. Why is STX so toxic to humans and seemingly harmless to bivalves? From the literature, two main hypotheses are formulated to justify this difference. One of them considers that many species of bivalves have nerves and muscles working mainly through the voltage gated Ca2+ channels.92 As previously explained, STX has a lower binding affinity to the Ca2+ channels than to the Na+ channels, which means that higher doses would be needed in bivalves to cause the same level of toxicity. Nevertheless, among bivalves, some species are more resistant than others to the STX action,93,94 often presenting a complete isolation mechanism that works by closure of the protection valves when the external environment is unfavourable (e.g.: northern quahog or hard clam Mercenaria mercenaria in the presence of A. tamarense95). The other hypothesis, mainly focused on the binding affinity to the Na+ channels, is associated with a mutation of an amino acid residue, causing a decrease of the affinity at the STX-binding site in the Na+ channel.96 These authors demonstrated that clams Mya arenaria from regions often exposed to highly toxic algae, known as “red tides”, are more resistant (may accumulate STXs at higher concentrations) than those hardly ever exposed to them, showing a 1000-fold decrease in the STX binding affinity to the Na+ channel. This mechanism of natural selection, which has been mainly attributed to mutations in the physiological target, has been observed in several aquatic species, namely in bivalves97,98 and crustaceans.99,100
4. Bioconversion of STX toxins in the body
As previously mentioned, poisoning of humans by STXs occurs through the food chain, mainly by consumption of bivalves contaminated with these biotoxins. In turn, bivalves are natural filters of substances suspended in water, including biotoxins. The filtration/absorption capacity of the bivalves varies substantially between species101,102 and with their stage of development (age).103 It is affected by the species of dinoflagellates, which also varies with geographic location and season, and by the density of toxic dinoflagellate cells in the water column.101,104 Within the body, absorption may occur differentially in different organs and tissues (different absorption rates). The greatest absorption seems to occur in the digestive part (e.g., gastric content, digestive gland, viscera), followed by distribution to the remaining organs and tissues over time.18,19,94,102After absorption, various bioconversions among STXs may occur in the different organisms,94,101,105–108 as evidenced by the differences in toxin composition reported following several experiments that compared the toxic profiles in phytoplankton and bivalves,27,30,107,109–114 and in humans.17,115 The main chemical transformations proposed in those studies are summarised in Tables 4–6, which compile at least one bibliographic example of each reaction observed in dinoflagellates, bivalves, and humans, respectively. Several aspects may be discussed, namely the type of reactions and the intrinsic (e.g., substituents and substitution patterns) or external (e.g., method of analysis) factors that may influence them, as well as the biochemical targets (e.g., enzymes) involved and their specificity. The conversion reactions proposed for the most recently discovered STX analogues, the M-series toxins, will also be discussed. Note that the direction of the reactions shown in this table is not illustrative of their (ir)reversibility. In these specific cases, it is only intended to denote the reaction proposed by the respective authors of the study. In Fig. 12 we present a summary scheme where the reversibility of the reactions from the (bio)chemical point of view is considered.
| Bioconversion reactions (metabolic transformations), in dinoflagellates | Observations |
|---|---|
|
GTX2/3 → GTX1/4 conversion proposed in A. tamarense extracts106 and in G. catenatum120 |
| GTX5 → GTX6 and C1/2 → C3/4 conversions also suggested in G. catenatum120 | |
|
GTX2/3 → C1/2, STX → GTX5 and M2 → GTX2/3 conversions proposed in cultures of G. catenatum120,121 |
| GTX2/3 → C1/2 also suggested in cultures of A. tamarense122 | |
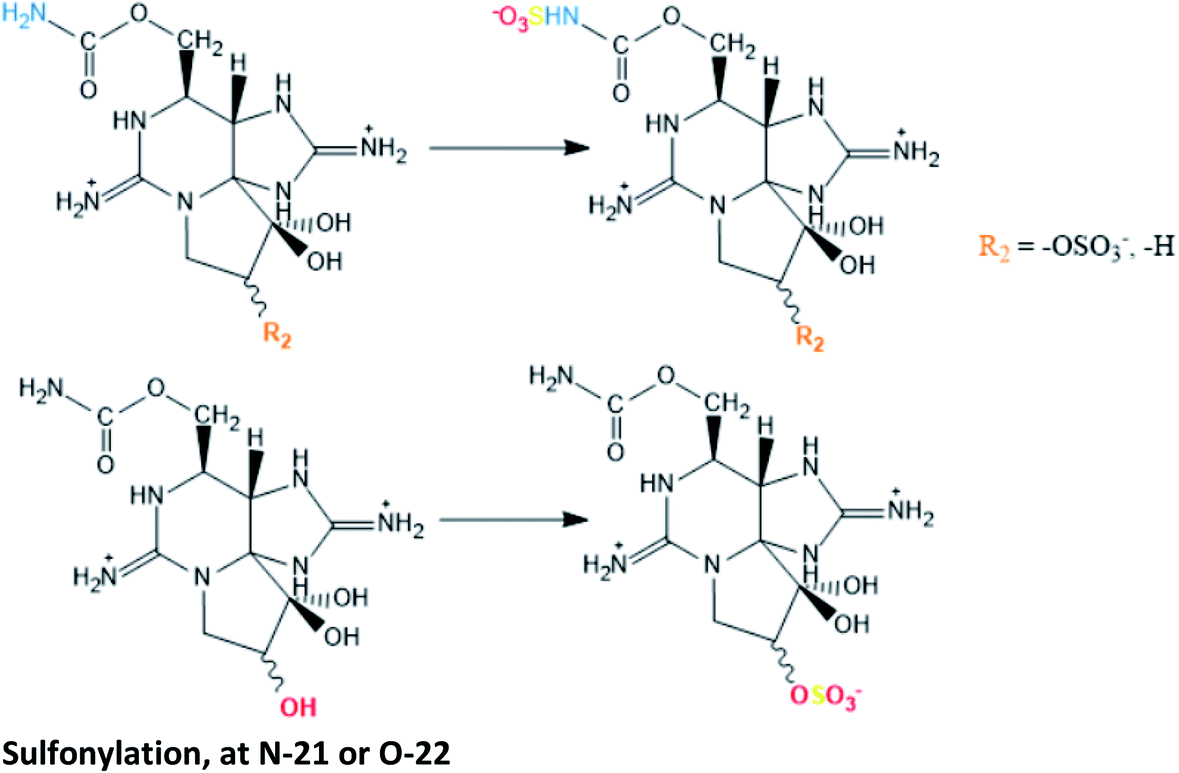
| Specific sulfotransferase (ST) enzymes have been reported to be responsible for sulfonations at N-21 (N-ST) and at O-22 (O-ST), both using the sulphate group from 3′-phosphoadenosine 5′-phophosulfate (PAPS) as source120,121 | |

|
STX → M2 conversion hinted in cultures of G. catenatum120,121 |
|
Reactions found in a G. catenatum strain27 |
4.1. Type of reactions and factors that influence them
As shown in Tables 4–6, the main chemical processes that lead to bioconversion among the STX family, in organisms, are reductive cleavage and elimination, hydrolysis, epimerization, oxidation, sulfurylation, glucuronidation, desulfurylation and hydroxylation. In turn, those reactions involve mostly the R4 (Fig. 3) substituent group (hydrolysis, sulfurylation), the –OH group at N1 (reductive elimination, oxidation), the substituent group at C11 (reductive cleavage, epimerization, desulfurylation, hydroxylation), and the –OH group at C12 (glucuronidation). Most of the reactions were suggested from studies carried out on bivalves (Table 5), comparing the toxins detected in these organisms with those detected in the dinoflagellates that served as food. Nevertheless, some common reactions (reductive cleavage, hydrolysis, oxidation, sulfurylation) and glucuronidation were also described using human samples (Table 6). More detailed reactions may be found in the ESI.†| Bioconversion reactions (metabolic transformations), in bivalves | Observations |
|---|---|

|
Described in different parts of the scallops Placopecten magellanicus;123 in digestive glands of scallop Patinopecten yessoensis contaminated with A. tamarense;124 in butter clams fed with A. catenella;125 in mussel Perna viridis contaminated with A. minutum (GTX1/4 → GTX2/3)126 |
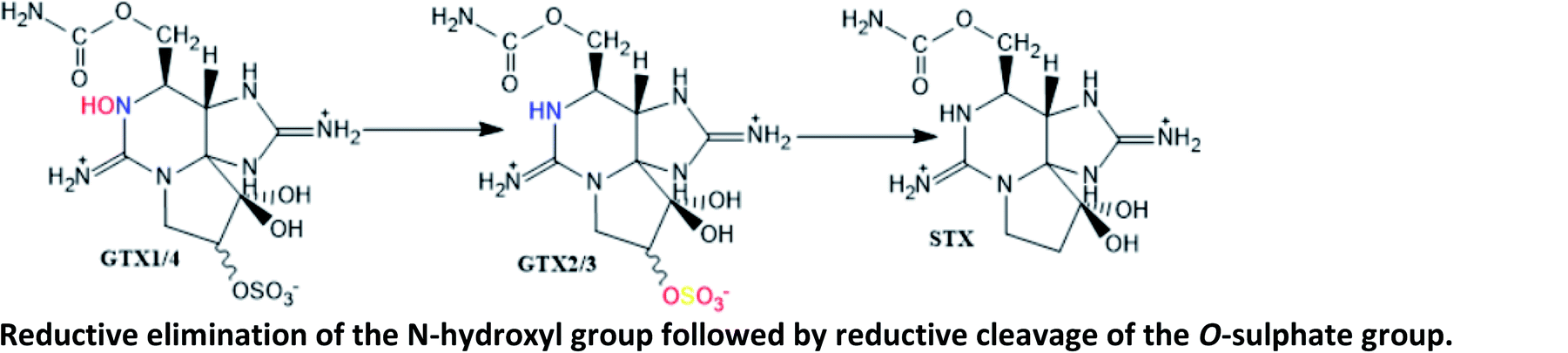
|
GTX2/3 → STX conversion was reported as involving bacteria isolated from intestines of a mussel Mytilus edulis.127 It was first proposed in other marine species (crabs and snails), involving the bacteria Vibrio and Pseudomonas spp. isolated from their viscera (incubation ≥ 20 h)128 |
| Reduction of the O-sulphate group at C11 from GTX1/4 and GTX2/3 to NEO and STX, respectively, is thought to be mediated by glutathione124 | |

|
C1/2 → GTX5 reported in digestive gland of mussels P. viridis fed with A. tamarense, but not in scallops Chlamys nobilis.110 As previously, this may be mediated by natural reductants, like glutathione |
| C3/4 → GTX6 reaction is also expected106 | |
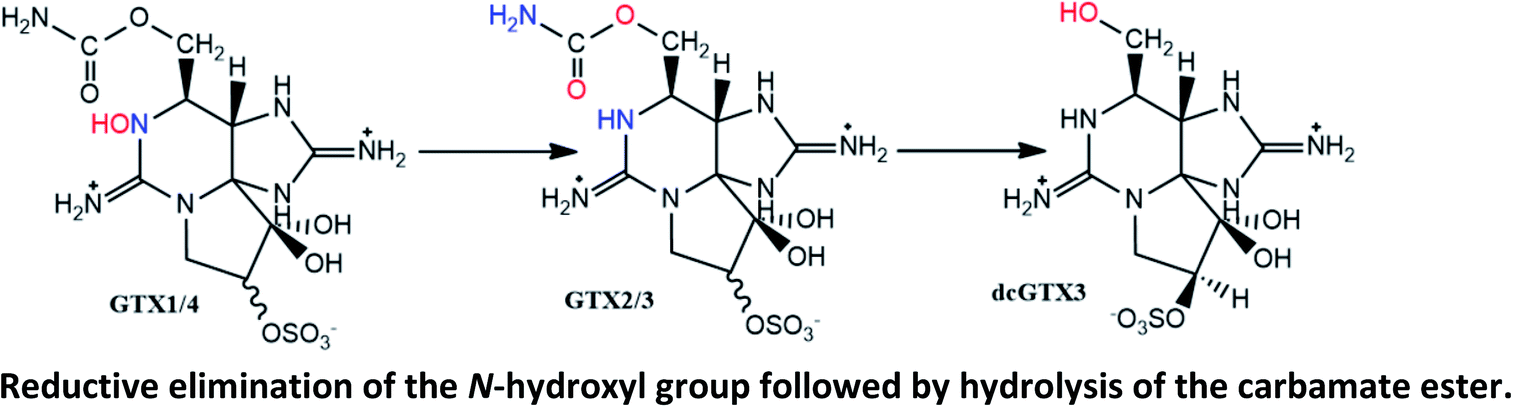
|
Described in mussels M. edulis (after 24 h).129 The increase of GTX2/3 was also observed in lesser extension in cockles Cerastoderma edule and Razorfish Ensis arcuatus, but only after 144 h |
| Not observed in queen scallops Chlamys opercularis (after 144 h) | |

|
Observed (except GC-toxins and M-toxins) in littleneck clam Protothaca staminea, using purified toxins from Gonyaulax sp.;71 from A. excavatum and A. minutum;130 incubated with a toxin mixture131 |
| Not detected in butter clams Saxidomus giganteus and mussels M. edulis, contaminated with Gonyaulax sp.;71 or in clams M. arenaria fed with Alexandrium spp.101 and incubated with a toxin mixture131 | |
| C1/2 → dcGTX2/3 reaction was also described in adductor muscle of scallops C. nobilis (higher extension) and in digestive gland of mussels P. viridis (lesser extension) fed with toxic A. tamarense.110 Additionally, GTX2/3 → dcGTX2/3 conversion was observed after 24 h in mussels M. edulis (dcGTX3 prevailed over dcGTX2), but not observed in queen scallops C. opercularis, after 144 h (ref. 129) | |
| These reactions appear to be promoted by the enzymatic activity, namely by hydrolytic enzymes, the carbamoylases71,111,130 | |
| M3 → dcM4 and M5 → dcM6 conversions reported in mussels M. edulis fed with toxic A. fundyense culture30 | |
| GC3 → dcSTX and GC1/2 → dcGTX2/3 conversions described in blue mussels (M. galloprovincialis), common cockles (C. edule), clams (R. decussata, V. pullastra, S. plana), offshore clams (D. trunculus, S. solida, C. gallina) and oysters (Crassostrea japonica) contaminated with G. catenatum.117 The carbamoylase activity exclusively targeted at benzoate STXs is proposed to be involved in these hydrolysis reactions of the carbamate ester | |

|
Suggested (except dcGTX3 → dcGTX2) in mussels M. edulis and Modiolus modiolus, oysters Crassostrea gigas, cockles Cardium edule, clams Arctica islandica and Mactra stultorum, sword razor Ensis ensis, scallops Pecten maximus contaminated with A. fundyense;30,107 and in oyster C. gigas (except GTX4 → GTX1) fed with A. minutum113 |
| C4 → C3 and dcGTX4 → dcGTX1 conversions are also expected106 | |

|
C1/2 → GTX2/3 and C3 → GTX1 reported in short-necked clams Tapes (Amygdala) japonica, mussels M. edulis and oysters C. gigas contaminated with two strains of A. tamarense isolated from toxic planktons109 |
| C1/2 → GTX2/3 reported in the northern quahog (clam) M. mercenaria exposed in laboratory to cultured isolates of Alexandrium (A. tamarense and A. fundyense),105 and in digestive glands of mussels P. viridis fed with A. tamarense110 | |
| GTX5 → STX documented in blue mussels132 | |
| Hydrolysis of the N-sulfa group was proposed by Oshima (1995),106 who also suggested GTX6 → NEO conversion | |
| M3 → M4 and M5 → M6 conversions suggested in mussels M. edulis fed with toxic A. fundyense culture.30 M5 → M6 also proposed based on STX analogues identified in clams Saxidomus purpuratus and scallops P. yessoensis133 | |

|
Bioconversions proposed in mussels M. galloprovincialis contaminated from A. tamarense strains29 |
| Also suggested in scallops Chlamys farreri and mussels M. galloprovincialis exposed to A. pacificum,114 although the authors did not detect M4 or M10 toxins | |
| C1/2 → M1 → M3 and GTX1/4 → M8 → M10 conversions suggested based on STX analogues identified in clams S. purpuratus and scallops P. yessoensis133 | |
| C1/2 → M1 → M3 and C3/4 → M7 → M9 described in mussels M. edulis fed with toxic A. fundyense culture30 | |
| Hydroxylation of M1 into M3 (second step of reaction) was suggested in several bivalve species (mussels M. galloprovinciallis, cookles C. edule, clams Ruditapes decussatus and Donax trunculus, and razor clams Ensis spp.) contaminated with G. catenatum.112 However, this author proposed that M1 is formed from GTX5 (instead from C1/2), also by hydroxylation at C11 | |
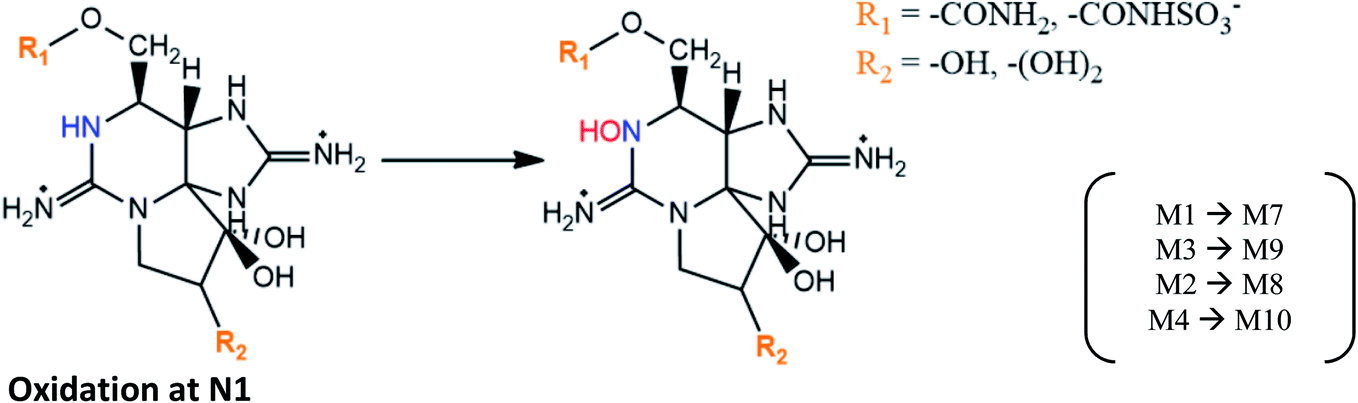
|
M1 → M7 and M3 → M9 proposed in mussels M. galloprovincialis contaminated from A. tamarense strains,29 and in mussels M. edulis fed with toxic A. fundyense culture30 |
| M2 → M8 and M4 → M10 suggested based on STX analogues identified in clams S. purpuratus and scallops P. yessoensis133 | |
| M10 formation was also proposed to occur from NEO in mussels M. galloprovincialis (an intermediate product may be formed)50 | |

|
Reaction suggested in mussels M. edulis fed with toxic A. fundyense culture30 |
| Bioconversion reactions (metabolic transformations), in humans | Observations |
|---|---|

|
Proposed to occur in gut samples134 |
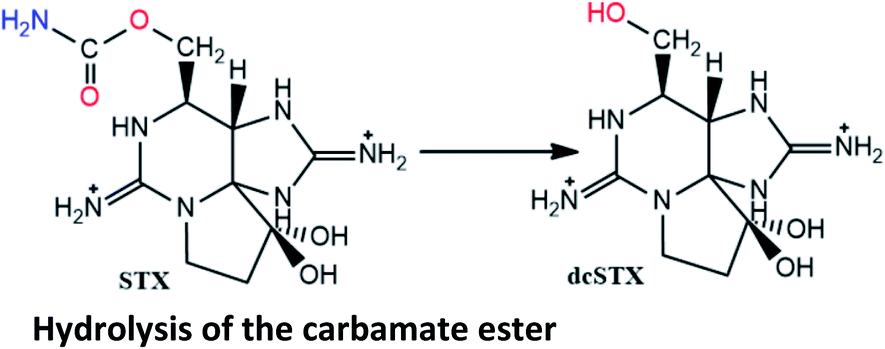
|
Reported in post-mortem analysis of human tissues (liver, kidney, lung)18 and human urine134 |
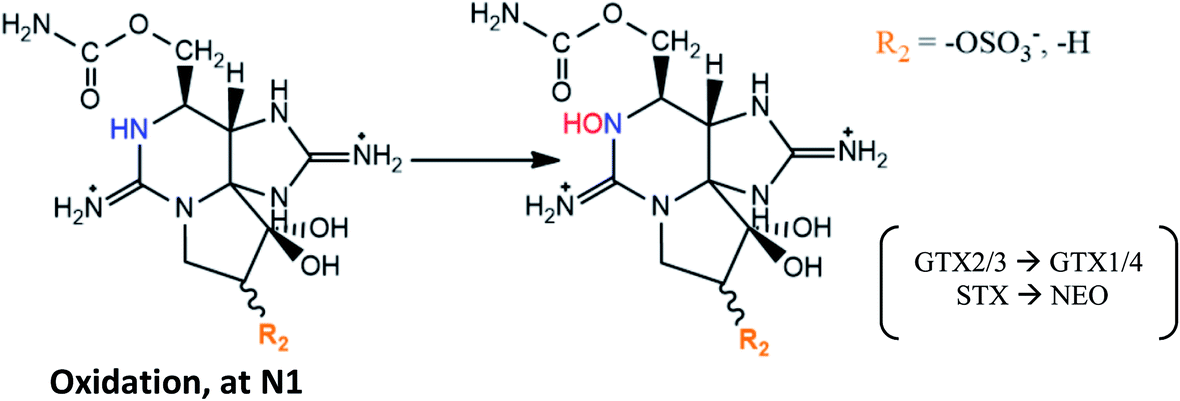
|
Described in post-mortem fluid and tissue samples18,134–136 |
| These reactions in humans occurred in presence of nicotinamide adenine dinucleotide phosphate oxidase (NADPH)135,136 | |

|
Proposed based on a larger proportion of C1 (and lower proportion of GTX2) in human serum than in cooked mussels17 |

|
Enzymatic transformations proposed in human liver microsomes after incubation with purified STXs135,136 |
| The reactions occurred in the presence of uridine 5′-diphospho-glucuronic acid (UDPGA), which transferred the glucuronosyl group to the toxin |
Based on this bibliographic survey, among the marine dinoflagellates, A. tamarense and G. catenatum appear to be the most studied, while the most used bivalves in the experiments are clams and mussels, which may be justified for different reasons. Clams represent the world's largest production of marine bivalves (38% of the total),4 which means greater sample availability, and higher economic impact. Mussels are known as biological indicators or sentinel organisms, since they usually accumulate toxins earlier, present high toxin uptake rate and tolerate the ingestion of higher amounts of toxins than other bivalves.94,116 The smaller number of studies in humans is related to the smaller availability of samples, since most analyzes are performed after the death of poisoned individuals.
When these studies are performed and bioconversion reactions are proposed it is important to keep in mind other factors unrelated to the chemical properties of the toxins and the metabolic capacities of the organisms. In fact, some steps of the sample preparation and analysis processes may influence the interpretation of the results, as has been reported by several authors.111,117–119 For example, heating the extracts in hydrochloric acid (in official mouse bioassay method) may convert N-sulfocarbamoyl toxins into the corresponding carbamate toxins.119 Similarly, the strong alkaline conditions associated to the derivatization step (in AOAC Official Method 2005.06, precolumn method) may lead to hydrolysis, contributing to formation of decarbamoyl analogues.117
Concerning the bioconversions that occur in organisms, it is possible to distinguish between those that are driven by chemical stability (e.g., epimerization), which are more related to the properties of each molecule (toxin), and those that are mediated by other substances associated with the metabolism of each species. Regarding the epimerization, several authors reported an interesting, yet unexplained, observation: β epimers appear to predominate in dinoflagellates, while α epimers are more commonly observed in bivalves.109,118,119 These observations suggest that β epimers are the first biosynthesis products in dinoflagellates.137 The proportion of α epimers increases gradually in bivalves during toxin uptake, evolving to the thermodynamic equilibrium, where a commonly accepted α:β ratio of about 3:1 is reached.106,119 Thus, the epimer ratio may be used as an indicator for the time of exposure to toxins, providing an estimate of when they began to accumulate in bivalves.110,138 This conversion seems to occur spontaneously through keto–enol tautomerism, but some contribution of the handling and storage of the samples should not be neglected since this equilibrium is affected by temperature and pH119 (the formation of α epimers is favoured at higher temperatures and pH.138
4.2. Mediators involved in the reactions and their specificity
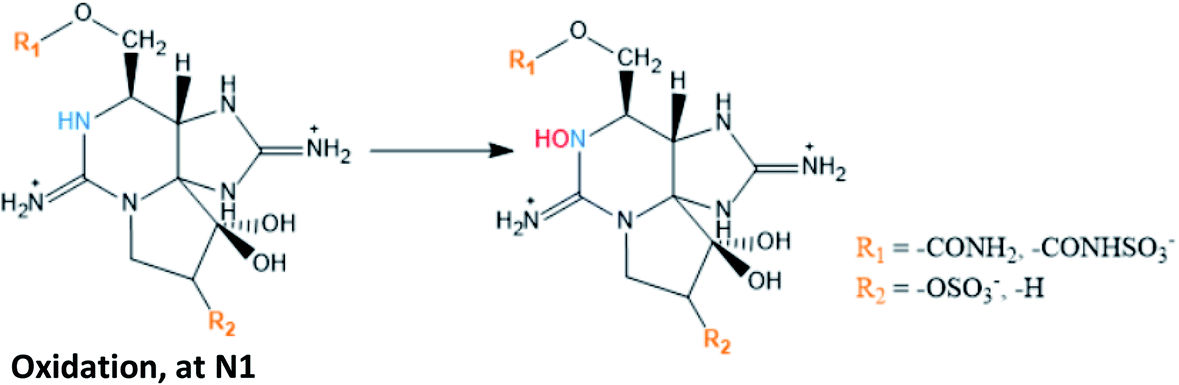
Among the bioconversion reactions involving other substances, those that are mediated by natural reducing agents, like glutathione (involved in reductive cleavage of the O-sulfate at C11), are distinguished from those that are mediated by enzymes, such as carbamoylases, sulfotransferases, UDPGT (uridine 5′-diphospho-glucuronosyltransferase) and NADPH (nicotinamide adenine dinucleotide phosphate) oxidase, the latter being involved in oxidation at N1 (substituent group R1). Beyond these, bio-transformations promoted by bacterial isolates from different bivalves have already been suggested, namely the reductive cleavage of the O-sulfate group at C11127 and the reductive elimination followed by hydrolysis of the carbamate ester.129 This work will focus mainly on the role of enzymes in the reactions since they are the most studied mediators.
Carbamoylases are hydrolytic enzymes commonly associated to the hydrolysis of the carbamate ester from N-sulfocarbamoyl, carbamate and benzoate toxins, often producing decarbamoyl toxins.71,111,117,130 Most studies that suggest the formation of decarbamoyl toxins with involvement of carbamoylases used clams as study organism, which led some authors138 to conclude that this type of reactions is uncommon among bivalves and may only occur in clams. However, other studies using different bivalve's species,110,117,129 as well as in humans,18 also demonstrated the occurrence of some of these reactions. Focusing on the enzymatic activity, it may be substrate-specific and/or species-specific. Regarding the substrate specificity, studies using the clams P. staminea revealed that the bioconversion into decarbamoyl toxins from N-sulfocarbamoyl toxins is faster than from carbamate toxins.71,130,131 The main difference between these two groups of toxins is the presence of a N-sulfate moiety in the R4 substituent of N-sulfocarbamoyl toxins, which is a more polar group. The proximity of this group with the site where hydrolysis occurs may facilitate the interaction of the substrate with the basic residues of the catalytic site, favouring the reaction.130 Furthermore, according to other authors139 who purified a carbamoylase from clam Mactra chinensis, its activity decreases with the presence of substituent groups at C11. They observed higher consumption rates for STX, NEO, GTX5 and GTX6, which have in common the absence of an O-sulfate group at C11, than for GTX1-4 and C1/2, bearing a O-sulfate group at C11, attributing this difference to a higher steric hindrance and negative charge of the sulfate group. These observations are partially in agreement with the results from previous studies130 insofar as, among the carbamate toxins, NEO and STX presented the fastest hydrolysis rates. Note that, unlike the authors previously mentioned, Lin et al. (2004)139 did not distinguish the greater or lesser specificity of the enzyme in the substrate depending on the presence or absence of a sulfate group in the R4 substituent group. Instead, they made that distinction based on the presence or absence of the sulfate group at C11. Despite the differences between studies, the results consistently show that the presence of the sulfate group and its position in the molecule (substrate) affects the specificity of the carbamoylases to the substrate. In addition to the substituent group effects, the stereochemistry of the O-sulfate group at C11 also seems to affect the substrate specificity. The β configuration favours a faster hydrolysis, comparatively to α configuration,129–131,139 which is probably related to steric hindrance interfering in the approach/interaction between the substrate and the catalytic site. These reactions are expected to occur in other organisms that rely on this enzyme. However, it is important to bear in mind that the enzyme specificity also varies between species, which means that the extent of the reactions may be greater or lesser in each of them. For example, comparing two species of the same type of bivalve (clams) fed during 3–6 hours with Alexandrium spp., S. solidissima was able to convert C1/2 into dcGTX2/3, while M. arenaria was not.101
The enzyme sulfocarbamoylase(I) proceeds similarly to other carbamoylases, but is more specific, only hydrolysing N-sulfocarbamoyl toxins into their decarbamoyl derivatives. This enzyme also showed more affinity for β than for the corresponding α epimers.140
Other enzymes commonly involved in bioconversion reactions are the sulfotransferases (ST). Studies using extracts of dinoflagellates showed that sulfotransferases appear to mediate specifically the sulfonylation of the R4 substituent group (N-ST), converting some carbamate toxins into N-sulfocarbamoyl toxins, and the sulfonylation of the C11 substituent group (O-ST), transforming M2 toxins into GTX2/3 toxins.120,121 In both cases, the enzymes act by transferring a sulfate group from 3′-phosphoadenosine 5′-phosphosulfate (PAPS), either to N-21 in the carbamoyl group (N-ST) or to O-22 in the C11 substituent group (O-ST). Among the substrates tested (STX, GTX2/3, GTX1/4, NEO, dcSTX, dcGTX2/3 and M2), the N-ST purified from G. catenatum was only specific to convert STX and GTX2/3 into GTX5 and C1/C2, respectively.120 The absence of specificity for dcSTX and dcGTX2/3 is expected since these toxins do not have a N-21 site for binding. In turn, NEO and GTX1/4 differ from STX and GTX2/3 in the –OH present at the N1 site, suggesting that this group may decrease the N-ST affinity. A similar study was carried out with N-ST from A. tamarense, confirming its specificity to convert GTX2/3 into C1/2, but revealing its inability to produce GTX5 from STX.122 Although it is tempting to attribute this difference to the different types of dinoflagellates under study (Gymnodinium spp. vs. Alexandrium spp.), the ability to convert GTX2/3 and STX into C1/2 and GTX5, respectively, has already been demonstrated using N-ST from A. catenella,141 which suggests once again that the activity of the enzyme is species-specific. Noteworthy, a sulfotransferase has also been implicated in the O-desulfation (from –OSO3− to –OH) at C11.29
Glucuronidation occurs by the transfer of glucuronic acid from UDPGA (uridine 5′-diphospho-glucuronic acid) to a substrate functional group, in this case the –OH at C12 of STXs. This reaction is catalysed by UGT (uridine 5′-diphospho-glucuronosyltransferase) and was proposed from experiments using human samples. The authors incubated UDPGA with STXs, obtaining the corresponding glucuronised forms.135,136 The conjugates may not be detected by fluorescence since this conjugation does not allow the oxidation reaction necessary for fluorescence detection. However, a β-glucuronidase capable of deconjugating the glucuronide and release the toxin may be used to enable analysis with fluorescence detection. The glucuronidation reactions followed a typical Michaelis–Menten kinetics and the obtained values for Vmax were shown to decrease in the following order: Gluc-STX > Gluc-NEO > Gluc-GTX2/3.136 Although the values are not significantly different, based on the standard deviations presented, the trend suggests that the glucuronidation may be harder in the STXs analogues containing substituent groups at C11, in this case, GTX2/3. This is a plausible hypothesis insofar as larger substituent groups at C11 impose a greater steric hindrance to C12 binding.
Fig. 8 shows a summary scheme of the main bioconversion reactions identified in this bibliographical survey, as well as the groups involved and their possible mediators.
 | ||
| Fig. 8 Summary scheme of main bioconversion reactions, groups involved and possible mediators. | ||
4.3. The latest STXs analogues
New STXs analogues have been proposed more recently, the M-series toxins. They belong mainly to the carbamate or N-sulfocarbamoyl sub-groups and differ from the others by the presence of one or two –OH group(s) at C11 (instead of –H or –OSO3−). Until now, 12 analogues (M1–M12) were identified in several bivalves,29,30,40,112,114,133 apart from the dcM (decarbamoylated) toxins that have also been detected.30 Their late discovery, comparatively to other STXs analogues, is related to their chemical properties and the methods commonly used for analysis. Most of the studies on STXs use liquid chromatography coupled with fluorescence detection (in accordance with the guidelines and the official method in force, AOAC Official Method 2005.06), but these compounds exhibit low fluorescence and thus require mass spectrometry detection.40,112 There are still few studies about M-toxins and a certain controversy around some aspects persists, namely regarding the organisms where they are detected and their structure. Some authors29,40 found them only in bivalves, but their presence was also detected in dinoflagellates.114 In what concerns to their structure, Dell'Aversano et al. (2008)40 proposed the structure of four (M1–M4) of the five (M1–M5) compounds detected, based on MS/MS spectra and NMR (1D and 2D) measurements. Vale (2010)112 detected some of these toxins in several bivalves contaminated with G. catenatum and suggested that M1 may be a hydroxylation product of GTX5 at C11 (reaction XLV, Table S1, in the S.I.),† and that M3 results from M1 by hydroxylation at C11 too. On the other hand, Ding et al. (2017)29 have proposed another precursor for M1, from C1/2 through an enzymatically mediated O-desulfonylation at C11 (reaction XXXII, S.I.†). Like the author112 previously mentioned, these authors29 also found that the analogues GTX5 and C1/2 were among the most abundant in the respective cultures of dinoflagellates, and one of the species under study is coincident (M. galloprovincialis), although coming from different geographical locations (Portugal vs. China). However, unlike Vale (2010),112 Ding et al. (2017)29 used bivalves contaminated from A. tamarense, which may explain the possible differences in precursor for M1. Other authors who corroborate the formation of M1 from C1/2 used A. fundyense30 and A. pacificum114 cultures, reinforcing the idea that the difference in precursors could be attributed to the different species (Gymnodinium spp. vs. Alexandrium spp.). Interestingly, the studies that pointed different precursors for M1 reported a coincident trend. In both situations, the authors referred a high abundance of GTX6 and C3/4 (potential precursors for M7 in the different species). However, M1 (from GTX5 or C1/2) was produced in greater abundance, suggesting that the presence of –OH group at N1 (in GTX6 and C3/4) may difficult the conversion to hydroxylated derivates at C11.112,114 A similar behaviour was noted by Qui et al. (2018)114 for the conversion of GTX2/3 (faster) and GTX1/4 (slower) into M2 and M8, respectively.Another apparent contradiction in the literature concerns the precursor for M5. Quilliam et al. (2017)30 revealed the structure of M5 based on MS/MS analysis and NMR measurements (1H, 13C). According to these authors, M5 derives from M1, following the opening of the ring at C4 (reaction XL, S.I.†) and may further originate M6 and dcM6 through N-desulfonylation (hydrolysis of the N-sulfo group, reaction XLII, S.I.†) and decarbamoylation (hydrolysis of the carbamate ester, reaction XLIV, S.I.†), respectively. However, Qui et al. (2018)114 mentioned that “M5 is formed from ring opening of M3, at the C-4 site”, citing Quilliam et al. (2017),30 who clearly referred that “M5 was not formed from M3”. So, this conclusion by Qui et al. (2018)114 unambiguously results from a misinterpretation of the data presented by Quilliam et al. (2017).30 Apart from this, Ding et al. (2017)29 proposed the conversion of M4 into M6 through ring opening at C4 (Fig. 9), unlike Quilliam et al. (2017)30 who advocate that M6 comes from M5 through hydrolysis of the N-sulfo group in the side chain, in agreement with other authors.133 Both studies29,30 were carried out using Alexandrium spp. (A. tamarense vs. A. fundyense) and mussels (M. galloprovinicallis vs. M. edulis). Quilliam et al. (2017)30 corroborated the M5 structure using NMR, in addition to the hydrophilic interaction liquid chromatography – tandem mass spectrometry (HILIC-MS/MS) analysis also performed by Ding et al. (2017).29 Their proposals could both be correct and chemically plausible. The difference could be justified by the greater or lesser kinetics of one reaction (ring opening at C4) in relation to the other (hydrolysis of the N-sulfogroup in the side chain). However, Ding et al. (2017)29 seem to have erroneously interpreted the MS spectra, since they assumed the m/z = 396.1 for the [M + H]+ ion related to M5. Although Quilliam et al. (2017)30 initially also assumed this m/z value, they later found that the true value is m/z = 414.1, which corresponds to a very weak signal in the MS spectrum. This apparent difficulty in detecting the true molecular ion of M5 is in line with previous studies done by other authors40 who, despite having reported the same [M + H]+ values for M1 and M5, assumed that the mass spectra of M5 and M1 were very different, suggesting a significant structural change between these analogues. Furthermore, the [M + H]+ values presented for M4 (332.1) and M6 (316.1) by Ding et al. (2017)29 are not coherent with the conversion pathway (via open-ring at C4) proposed by them, since in this situation a difference of only two mass units would be expected. Therefore, the conversion of M5 to M6 (N-desulfonylation) appears to be the most substantiated hypothesis. Assuming this, the value for the [M + H]+ ion related to M6 could be 334 instead of 316, as proposed by some authors.29,133 Taking into account all the questions and doubts related to these analogues (M-toxins), it is of great importance that more studies are developed in order to better elucidate and confirm the molecular structures, as well as to understand the conversions that may occur between them.
 | ||
| Fig. 9 Proposed paths to form M6 from M4 and from M5. | ||
Finally, in a summary about the bioconversion of STXs, it is important to draw attention to the fact that most of the studies presented here are based on in vitro experiments and considering one or more specific variables. It is thus essential to carry out more studies that consider a set of factors (e.g., enzymes, bacteria, substrates), better simulating what happens in vivo, which will eventually provide deeper knowledge about the competitions, inhibitions and/or inductions among the different chemical and metabolic processes. It is known that the reaction kinetics vary between species,71,110,114,129,131,142 which may be explained, among other factors, by the metabolic characteristics of each species. Additionally, the reactions are strongly affected by physical parameters such as temperature and pH.50–52 Thus, it is extremely difficult to compare reaction kinetics between studies because experimental conditions vary widely. Within the same study, some authors have tried to establish kinetic relationships between the different reactions.111,115,120,130,136,142 Most of their conclusions have already been integrated into this discussion.
Some of these bioconversions favour the excretion of toxins by the body, namely those that produce more hydrophilic molecules (e.g., glucuronidation, in humans).135,136 In fact, urine is the major route of toxin excretion in humans,17,115 despite some excretion also occurring by faeces.18 Regarding the excretion of toxins in bivalves, there appear to be two stages of clearance. The first phase occurs during the first day and is characterized by a rapid excretion, mainly of unassimilated toxins, while the second phase corresponds to a slower excretion of toxins assimilated and incorporated in the organs and tissues.93,110 In general, it is possible to categorize bivalves in two groups according to their detoxification/clearance rates. Rapid detoxifiers are those whose toxin elimination rates vary between 6 and 17% per day, which means that they may take a few weeks (less than 10) to reach regulatory levels (80 μg STXeq 100 g−1). Among the main species that fall into this group are, for example, the clam M. arenaria, the mussel M. edulis and the oyster Crassostrea iridescens. On the other hand, slow detoxifiers are those with elimination rates equal to or less than 1% per day. These organisms may take months or years to reach the regulatory limits. The clams S. giganteus and S. solidissima and the scallop Placopecten magellanicus are some examples of slow detoxifiers. More detailed information on these data may be found in literature.138
5. Structure–toxicity relationships
As has been discussed throughout this work, there are several factors that affect the concentration and distribution of biotoxins in the organism. A summary of these factors is presented in Fig. 10, highlighting the experimental conditions, the physicochemical factors directly related to the molecular structure and consequent binding affinity to the receptor channels, and the biological, metabolic, and environmental factors associated with the species. All these factors are interconnected and may affect the results and the toxicological profile of the analysed samples, at a specific time. | ||
| Fig. 10 Factors affecting the toxicological profile in the analyzed samples. | ||
Several studies report different levels of toxicity in different situations, from studies on a case-by-case basis. However, it is instrumental to understand the general trend of toxicity relationship between the molecules of this group of biotoxins. One of the best ways to establish this relationship is through the toxicity equivalency factor (TEF), defined as “the toxicity ratio of a compound from a chemical group that shares the same mode of action of a reference compound in the same group”, being the toxicity of the congener expressed as “a fraction of the toxicity of the reference compound in terms of potency, which is a pharmacological parameter that defines the amount of compound required for a certain effect”.143 When the compounds may cause death in humans, as is the case of this group of toxins, the most appropriate potency parameter is the median lethal dose (LD50), which refers to the dose that kills half the population of animals tested. The toxicity studies carried out by oral administration are more relevant and appropriate than those by intraperitoneal (i.p.) injection since the oral ingestion (through feeding) is the natural way of exposure. Nevertheless, both methodologies to determine LD50 present limitations and may originate unrealistic estimates. These specific issues, as well as the criteria that should be adopted to correctly derivatize TEFs, are properly discussed in a technical paper co-signed by FAO (Food and Agriculture Organization of the United Nations) and WHO (World Health Organization)143 and in another publication by the same authors.144 TEFs have often been estimated based on toxicity information obtained from the mouse bioassay (MBA),133,145,146 which provides the total toxicity of the various congeners existing in a sample. However, the MBA method is not considered a toxicological parameter and its results do not correlate with the acute toxicity.143 Nonetheless, TEFs estimated by Oshima (1995) (as cited in ref. 143) from the relative specific activity in MBA are still used as reference. Since then, several toxicity experiments have been performed considering i.p. injection147 and oral administration (by gavage and/or feeding)87,146,147 and, given the results obtained from these parameters, some revisions were proposed by the European and other international official entities. The last revision, made by a group of experts coordinated by FAO and WHO, considered the following criteria, by order of relevance: data from human cases (outbreaks) > oral LD50/toxicity data in animals > i.p. LD50/toxicity data in animals > MBA > in vitro data.143 The recommended TEFs are shown in Table 7. For future discussion, the last proposal made by FAO/WHO will be considered, not only because it is the most recent but mainly because it takes into account more realistic and representative criteria.
| Toxin | By MBAa | LD50 by i.p. injectionb | LD50 by oral administrationc | EFSAd proposal | FAO/WHOe proposal |
|---|---|---|---|---|---|
| a Estimated from the specific activity in MU per μmole determined by MBA (as cited in ref. 143). b From LD50 by i.p. injection.143,147 c From LD50 by oral administration (feeding).87,146 d Proposed by the scientific panel on Contaminants in the Food Chain (CONTAM Panel)10 based on acute i.p. toxicity in mice. e Proposed by the expert group coordinated by the FAO and WHO.143 Note: (mouse unit) is defined as the amount of toxin to kill a male mouse weighing 20 g after 15 min one MU.148 | |||||
| STX | 1 | 1.00 | 1 | 1.0 | 1.0 |
| NEO | 0.92 | 3.12 | 2.54 | 1.0 | 2.0 |
| GTX1 | 0.99 | 1.90 | 0.93 | 1.0 | 1.0 |
| GTX4 | 0.73 | 0.7 | 0.7 | ||
| GTX2 | 0.36 | 0.76 | 0.57 | 0.4 | 0.4 |
| GTX3 | 0.64 | 0.6 | 0.6 | ||
| GTX5 (B1) | 0.064 | 0.222 | 0.064 | 0.1 | 0.1 |
| GTX6 (B2) | — | 0.122 | <0.017 | 0.1 | 0.05 |
| C1 | 0.006 | 0.043 | — | 0.01 | |
| C2 | 0.096 | 0.1 | 0.1 | ||
| C3 | 0.013 | — | 0.01 | ||
| C4 | 0.058 | 0.1 | 0.1 | ||
| dcSTX | 0.51 | 0.79 | 0.37 | 1.0 | 0.5 |
| dcNEO | — | 0.058 | 0.22 | 0.4 | 0.2 |
| dcGTX1 | — | — | — | ||
| dcGTX2 | 0.15 | 0.11 | 0.2 | 0.2 | |
| dcGTX3 | 0.38 | 0.4 | 0.4 | ||
| dcGTX4 | — | — | — | ||
| M2 | — | 0.3 | — | ||
Based on Table 7 and Fig. 11, several conclusions may be drawn. The first of these is the clear evidence that, among the main groups of toxins, those belonging to the carbamate group (excluding M-toxins) exhibit the greatest toxicity, followed by those of the decarbamoyl and N-sulfocarbamoyl groups (excluding M-toxins). This agrees with conclusions reached by several authors71,130,131 and it is probably related to the substituent groups, once the main difference among them is the R4 group: –OCONH2 for carbamate, –OCONHSO3− for N-sulfocarbamoyl and –OH for decarbamoyl groups. Indeed, the N-sulfocarbamoyl toxins present the largest functional group, which might imply a higher steric hindrance comparatively to the other STX-groups and, consequently, a lower binding affinity to the receptor channels. Based solely on this criterion, one would expect a lower toxicity for the STX-carbamate group than for the STX-decarbamoyl, but that is not the case. It appears that the polarity of the molecule must also be considered, and decarbamoyl toxins are more polar than carbamate toxins. Among the STX-carbamate group, NEO is the most toxic, twice more than STX, the reference compound.
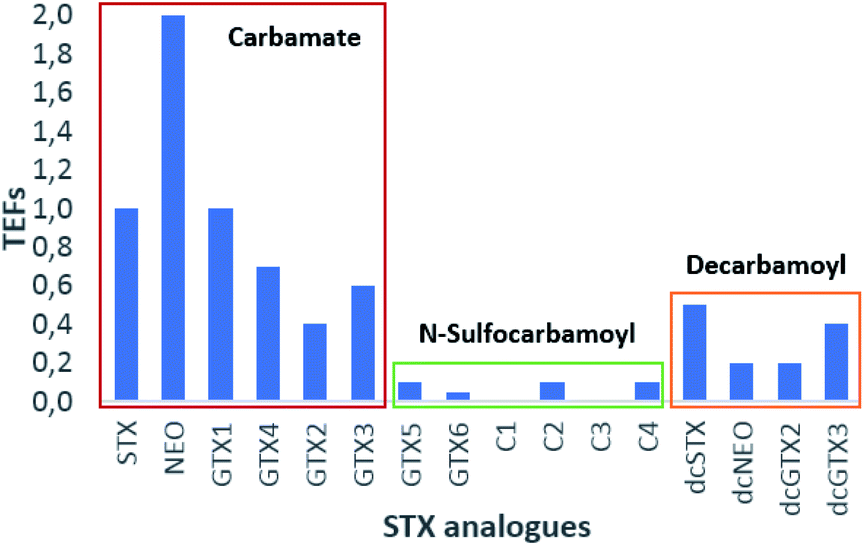 | ||
| Fig. 11 Graphic representation of TEFs proposed by FAO/WHO.143 | ||
The only feature that distinguishes these two analogues is the presence of –OH at N1 (R1 substituent group), suggesting that this group may favours the binding affinity to the channel receptor, thereby increasing the toxicity. Its presence on the molecule has been associated with a change in N1 reactivity and with the distribution of positive electrical charge on the guanidium group. According to some authors,149 a positive charge in N1 may benefit the dissociation of the hydroxyl proton, causing an uncommonly low pKa value of 6.75. Still in this group of toxins (STX-carbamate), there is another trend that should be highlighted and that distinguishes STX and NEO from GTX1-4.
Unlike STX and NEO, GTX1-4 bear –OSO3− groups at C11 that contribute to the decrease in toxicity, which may be due to the following reasons: (a) electronic repulsions between the sulfate group and the negatively charged amino acids in the receptor channel; (b) shift of the gem–diol–ketone equilibrium; (c) change in the charge distribution caused by the addition of a negative charge, possibly changing the pKa of the guanidium groups.78,84 Although the toxicities of GTX1-4 are generally lower than that of STX, GTX1/4 are more toxic than GTX2/3. In keeping with the justification proposed for the difference in toxicity between STX and NEO, the higher toxicities of GTX1/4 are attributed to the presence of OH at N1.78 Stereochemistry at C11 also appears to influence toxicity. From the data presented, GTX2 and dcGTX2 (α epimers) exhibit lower toxicity than GTX3 and dcGTX3 (β epimers), suggesting a lower binding affinity of the α epimers. However, this trend is reversed for GTX1/4, where GTX1 (α epimer) presents higher toxicity than GTX4 (β epimer). Although this hypothesis is not yet proven, it is possible that, once again, the presence of –OH in GTX1/4 affects the charge distribution and the intra- and inter-molecular interactions, thus affecting the apparent stereochemical effect at C11.
Focusing now on decarbamoyl toxins, an inverse toxicity trend between dcSTX and dcNEO is observed, comparatively to STX and NEO. This may suggest that the presence of the –OH group at N1 has a greater effect in increasing toxicity when R4 = –OCONH2 (carbamate STX analogues) than when R4 = –OH (decarbamoyl STX analogues), promoting different types of interactions in each of the cases. Concerning the N-sulfocarbamoyl toxins, since TEFs are so low and the errors associated with their determination are not shown, their comparison and discussion is not significant.
With respect to the M-series toxins, belonging to both the carbamate group (M2, M4, M6, M8, M10, M12) and the N-sulfocarbamoyl group (M1, M3, M5, M7, M9, M11), TEFs have not been disclosed so far, probably because these toxins were more recently discovered and there are fewer conclusive studies on their toxicities. Even so, from the information gathered so far it may be postulated that they exhibit low toxicity, as corroborated by the only TEF proposed for this series of toxins (for M2, in Table 7). Since this value is assigned to an M-toxin belonging to the carbamate group, it may be expected that M-toxins belonging to the N-sulfocarbamoyl group exhibit even lower toxicities. The low toxicity values for M1–M5 analogues were also corroborated by some authors133 following data from experiments using liquid chromatography (LC) coupled with fluorescence detection (unable to detect these analogues) and MBA.
TEFs for toxins of the benzoate group are also not known. Due the low toxicity attributed to the N-sulfocarbamoyl toxins, ascribed to the large functional group in R4, low toxicities for that group of toxins could also be expected. Nevertheless, the 4-hydroxyphenyl group in the side chain of these molecules impacts on hydrophobicity and reactivity, which may translate into differences on toxicological properties vis a vis other STX analogues.27 A study performed with the rat brain Na+ channel revealed a strong binding of GC1-3 to this channel,150 the authors confirming that these analogues are only slightly less potent than STX. Other authors151 found considerable proportions of GC toxins in toxin profiles from different G. catenatum cultures, drawing attention to the possibility of underestimating the toxicity of these toxins when the methods of preparation and analysis are not appropriate. It is worth remembering that the lack or low fluorescence of these toxins prevents their detection by the official chromatographic method in force. In addition, due their hydrophobicity, they can be lost while using reverse phase SPE cartridges to clean and prepare the sample.
Taking into account the TEFs proposed by FAO/WHO (2016)143 and the information collected about the bioconversion reactions that occur in the different organisms (Tables 4–6), the present work proposes the concept of a “toxicological traffic light” defined by four colours, from the most toxic to the least toxic: purple > red > yellow > green. Toxins for which TEFs are not yet established are identified in grey. The combination of all the information is shown in Fig. 12 and 13. In these figures, the possible reversibility of reactions is predicted. Out of these schemes there is still a set of theoretically expected reactions but their occurrence, in vitro or in living organisms, has not been reported to the best of our knowledge. Epimerization reactions are also not shown in this figure (due the scarcity of space), but they always occur at C11. In the cases of the C1/C2, C3/C4 and dcGTX2/3 epimeric pairs, in which the TEFs are classified in different levels of toxicity (different colours), the colour that appears in the scheme is the one that corresponds to a higher toxicity classification.
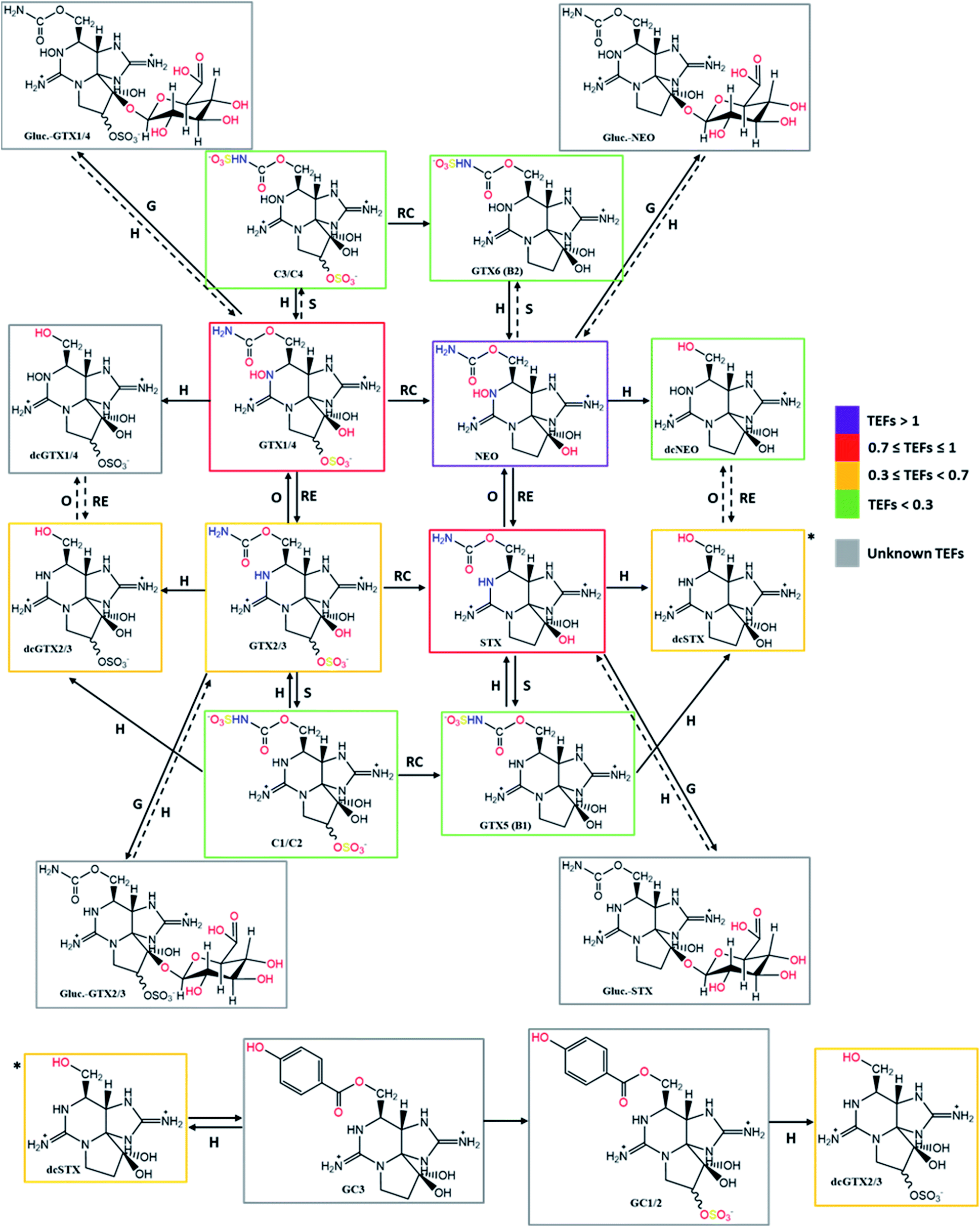 | ||
| Fig. 12 Summary scheme of the bioconversions among the different analogues of STXs categorized by colours according to the proposed toxicity scale based on TEFs. Solid arrows represent the bioconversions proposed by several authors, as described in Tables 4–6. All these bioconversions were reported to occur in organisms (dinoflagellates, bivalves, and humans). Dashed arrows correspond to chemical reactions proposed in this work, based on typical reactions already presented, assuming their reversibility. Type of reactions: RC – reductive cleavage, RE – reductive elimination, H – hydrolysis, O – oxidation, S – sulfurylation, D – desulfurylation, G – glucuronidation, Hx – hydroxylation. | ||
 | ||
| Fig. 13 Continued – summary scheme of the bioconversions among the different analogues of STXs categorized by colours according to the proposed toxicity scale based on TEFs. Solid arrows represent the bioconversions proposed by several authors, as described in Tables 4–6. All these bioconversions were reported to occur in organisms (dinoflagellates, bivalves, and humans). Dashed arrows correspond to chemical reactions proposed in this work, based on typical reactions already presented, assuming their reversibility. Type of reactions: RC – reductive cleavage, RE – reductive elimination, H – hydrolysis, O – oxidation, S – sulfurylation, D – desulfurylation, G – glucuronidation, Hx – hydroxylation. | ||
6. Conclusions
This work summarizes and discusses a set of complex and important aspects related to STXs or paralytic shellfish toxins (PSTs). Among the main groups of marine toxins capable of causing effects in humans, this set of toxins is likely the most dangerous, due to the severity of symptoms (may even induce human death in few hours). Human poisoning resulting from contaminated food consumption, in this specific case the bivalves, as well as the prohibitions to their capture, when biotoxins are identified in advance in the water column, represents great socio-economic losses. Although there are many research articles and some reviews on this topic, much remains to be discovered about STXs, so deeper investigations are urgently needed. During the preparation of this review several problems were encountered, namely a notorious difficulty in accessing some older articles, the frequent absence of properly substantiated explanations from a chemical point of view or the apparent contradictory information found in different articles. In view of this, our work aimed to compile and update the information about this group of toxins with respect to their physical–chemical properties, biosynthesis, mode of action, bioconversions that occur inside organisms and the relationship between structure and toxicity of the molecules.STX is an amorphous hygroscopic compound, non-volatile, unable to crystalize in the form of chloride, acetate, or sulfate salts, rendering its identification and characterization difficult. Some of the first studies date back to the 1960s and since then they have been complemented with information gathered using more robust and sensitive techniques. This work presents 43 STX analogues with origin (direct and/or indirect) in marine dinoflagellate eukaryotes, but there are already more than 50 analogues identified in the literature, also counting with those produced by freshwater prokaryotes. The biosynthesis of STX is a complex process involving, in its initial phase, the following key players: the gene stxA (SxtA1 to SxtA4), the amino acid arginine, acetyl-CoA, S-adenosylmethionine (SAM) and carbamoyl-phosphate (CP). Additionally, several reactions are involved, such as Claisen-type condensation, hydroxylation, epoxidation, and cyclization.
The molecular structure of STXs is highly susceptible to changes in pH and such changes strongly affect their binding affinity to the receptor channels, either by the balance of liquid charges or by their attractive/repulsive effect. Although interactions of STX with Ca2+ and K+ receptor channels have been described, the main route of interaction of these molecules is with Na+ receptor channels. In addition to the net charge (low binding affinities have been associated with negative charges), the different substituent groups of each analogue significantly influence the binding to the receptor channels. For example, the presence of large functional groups in the side chain of the molecules has been related to a lower binding affinity, in contrast to the presence of the –OH group at N1 which seems to potentiate the toxicity compared to other molecules containing only –H at N1. The presence of substituent groups in carbon 11 and their stereochemistry also contribute to the greater or lesser binding affinity.
The absorption, transformation, and excretion of toxins by or in the organisms is heavily dependent on a large set of factors, including the metabolic specificities of each species and the molecular structure of the toxins. This work provides a survey of 45 bioconversion reactions (available as S.I.†) in organisms (dinoflagellates, bivalves, and humans) reported by different authors, highlighting the higher number of reactions identified in bivalves, compared to dinoflagellates and humans. The main types of reaction are epimerization, hydrolysis, elimination and reductive cleavage, oxidation, sulfonylation, desulfonylation, hydroxylation and glucuronidation, some of these reactions being common to the different organisms. Many of these reactions are mediated by different players, namely bacteria, natural reducers such as glutathione, and various enzymes (e.g., carbamoylases, sulfotransferases, UDPGT and NADPH oxidase). All these bioconversions directly affect toxicity and one of the ways to establish the toxicity relationship among analogues is through the toxicity equivalency factors (TEFs). According to the most recently known update of these values, by FAO and WHO, the toxic character of the most-well known STX analogues varies as follows: NEO > STX ≈ GTX1 > GTX4 > GTX3 > dcSTX > GTX2 ≈ dcGTX3 > dcGTX2 ≈ dcNEO > GTX5 ≈ C2 ≈ C4 > GTX6 > C1 ≈ C3, which contradicts the older idea that NEO had similar or lower toxicity than STX (mainly based on MBA results). Although TEFs are not yet known for the most recently discovered toxins, namely those of the M-series, the few data collected so far seem to suggest a low toxicity among them. This work also concluded that the standardized HPLC methods with fluorescence detection are not adequate to specifically detect and quantify some of these toxins due to their low or residual fluorescence.
To sum up, our work proposes some explanations from a chemical perspective, highlight some recent findings about bioconversions and relate the various sub-topics about STXs. It significantly contributes for a better comprehension about STXs and identifies some challenges (e.g., adaptation of some analysis methods) and gaps of knowledge (e.g., toxicity of M-toxins and those from benzoate group), providing guidance on research needs and ways forward.
7. Future perspectives
The detection and discovery of the new STX analogues by using different techniques, such as mass spectrometry, highlights the need to adapt and improve standard chromatographic methods to a new reality. This will allow a deeper knowledge about the biotoxins produced directly by the organisms or from bioconversions and will contribute to the determination of their toxic potential. Given the complexity associated with the various bioconversions that may occur and their relationship with the increase or decrease in toxicity, and the controversies that seem to exist, mainly regarding the most recently discovered analogues, it is imperative to carry out further studies that contribute to a better understanding of what happens in each species, or at least in each organism. For example: why do some reactions seem to be so dependent on the species of dinoflagellate used (Gymnodinium spp. vs. Alexandrium spp.), while others are common to such different organisms (dinoflagellates, bivalves, humans)? Will it be possible to develop an analytical method sufficiently sensitive and robust to quantify the different analogues in the same sample, rather than combining analytical methods to identify and quantify specific subgroups of STX to obtain a complete toxicological profile? On the other hand, is it really important to develop such a method? Note that if analogues not detected in the reference method currently in force do not have relevant toxicity, then it may not be worth investing in methods improvement or development. However, if the toxicity is high (as seems to be the case for benzoate group toxins), the reference method should include its identification and quantification. To be able to decide about this, more toxicological studies are needed, with special focus on toxins whose TEFs are not yet established. The acquisition of this knowledge will greatly contribute to a better prevention and monitoring and, consequently, to the reduction of socio-economic impacts.Another aspect that must be considered in future studies is the issue of climate change and its consequences, namely the acidification of oceans and the increase in sea water temperature. There are works reporting some of these effects, not only on shell calcification processes, but also in terms of the physiological parameters, like pH.152–154 Remember that the action of this group of toxins is highly vulnerable to changes in pH and this may be reflected in greater or lesser toxicity. Also, the increase in the temperature of ocean waters affects the metabolism of bivalves and some authors,155 who carry out their studies with oysters, suggest a decrease in detoxification rates, which means that the toxins persist for longer periods in the organism. In the medium and long term, the consequences of climate change may alter what we currently know about PSTs, and we must be prepared for that.
Finally, despite the negative connotation associated with these toxins, they also have a great therapeutic potential, as previously introduced. As consequence of their reversible interaction with the receptor channels, at the neuronal level, they may be used as anaesthetics, namely STX and NEO.21–23 Some authors23 used rats and blocked the percutaneous sciatic nerve with different STXs. They observed that, for 60 minutes, the analgesic effect was achieved with median concentrations of 34 ± 2, 58 ± 3 and 268 ± 8 μmol L−1 of NEO, STX and dcSTX, respectively. These results are in keeping with the higher toxicity of NEO (lesser dose required) and lower toxicity of dcSTX (higher dose required for the same effect). More recent experiments, in humans, revealed the efficacy of a 50 μg-dose NEO injected in the subcutaneous plane, achieving a complete anaesthetic effect for 3 hours.21 The anesthetic effects achieved with these low levels of concentrations corroborate the great potential of these toxins for therapeutic purposes, so the investment in this area of research appears to be very promising.
8. Conflicts of interest
There are no conflicts to declare.9. Acknowledgements
This study received funds from FCT – Foundation for Science and Technology, through project UIDB/04326/2020, and from the project MAR-01.03.01-FEAMP-0049 co-financed by the Operational Program Mar 2020, Portugal 2020 and the European Union (EU), through the European Maritime Affairs and Fisheries Fund (EMFF). The authors gratefully acknowledge the financial support. The authors also thank the AQUA-CIBUS network for their help in discussion of some issues.10. References
- M. L. Wells, B. Karlson, A. Wulff, R. Kudela, C. Trick, V. Asnaghi, E. Berdalet, W. Cochlan, K. Davidson, M. De Rijcke, S. Dutkiewicz, G. Hallegraeff, K. J. Flynn, C. Legrand, H. Paerl, J. Silke, S. Suikkanen, P. Thompson and V. L. Trainer, Harmful Algae, 2020, 91, 101632 CrossRef.
- H. P. van Egmond, Anal. Bioanal. Chem., 2004, 378, 1152–1160 CrossRef CAS.
- FAO, The State of World Fisheries and Aquaculture 2020. Sustainability in action, Rome, 2020 Search PubMed.
- A. C. Smaal, J. G. Ferreira, J. Grant, J. K. Petersen and Ø. Strand, Goods and Services of Marine Bivalves, Springer, 2019, p. 598 Search PubMed.
- I. Sanseverino, D. Conduto, L. Pozzoli, S. Dobricic and T. Lettieri, Algal bloom and its economic impact, European Comission, 2016 Search PubMed.
- E. Berdalet, L. E. Fleming, R. Gowen, K. Davidson, P. Hess, L. C. Backer, S. K. Moore, P. Hoagland and H. Enevoldsen, J. Mar. Biol. Assoc. U. K., 2015, 62 Search PubMed.
- P. Hoagland and S. Scatasta, in Ecology of Harmful Algae, ed. E. Granéli and J. T. Turner, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 391–402 Search PubMed.
- K. Dyson and D. D. Huppert, Harmful Algae, 2010, 9, 264–271 CrossRef.
- S. Martino, F. Gianella and K. Davidson, Harmful Algae, 2020, 99, 101912 CrossRef CAS PubMed.
- EFSA, EFSA J., 2009, 1019, 1–76 Search PubMed.
- H. Daguer, R. B. Hoff, L. Molognoni, C. R. Kleemann and L. V. Felizardo, Curr. Opin. Food Sci., 2018, 24, 43–55 CrossRef.
- P. Visciano, M. Schirone, M. Berti, A. Milandri, R. Tofalo and G. Suzzi, Front. Microbiol., 2016, 7, 1051 CrossRef PubMed.
- EFSA, EFSA J., 2008, 907, 1–62 Search PubMed.
- CONTAM, EFSA J., 2010, 8, 1628 Search PubMed.
- HAEDAT, Harmful Algal Events Dataset (HAEDAT), 2020, http://haedat.iode.org/ Search PubMed.
- N. W. Lagos and D. Andrinolo, in Seafood and freshwater toxins: pharmacology, physiology, and detection, ed. L. M. Botana, Marcel Dekker,Inc., New York, 2000, pp. 203–215 Search PubMed.
- B. D. Gessner, P. Bell, G. J. Doucette, E. Moczydlowski, M. A. Poli, F. Van Dolah and S. Hall, Toxicon, 1997, 35, 711–722 CrossRef CAS PubMed.
- C. García, M. del Carmen Bravo, M. Lagos and N. Lagos, Toxicon, 2004, 43, 149–158 CrossRef PubMed.
- D. Montebruno, Med., Sci. Law, 1993, 33, 243–246 CrossRef CAS.
- C. Belin and B. Raffin, Les espèces phytoplanctoniques toxiques et nuisibles sur le littoral français de 1984 à 1995, résultats du REPHY (réseau de surveillance du phytoplancton et des phycotoxines) (Toxic and harmful phytoplankton species on the French coast from 1984 to 1995), 1998 Search PubMed.
- A. J. M. D. Rodriguez-Navarro, N. P. D. Lagos, M. M. D. Lagos, I. M. D. Braghetto, A. M. D. Csendes, J. M. D. Hamilton, C. M. D. Figueroa, D. M. S. Truan, C. M. S. Garcia, A. M. D. Rojas, V. M. S. Iglesias, L. M. D. Brunet and F. M. D. Alvarez, Anesthesiology, 2007, 106, 339–345 CrossRef CAS.
- H. J. Adams, M. R. Blair and B. Takman, Arch. Int. Pharmacodyn. Ther., 1976, 224, 275–282 CAS.
- D. S. Kohane, N. T. Lu, A. C. G. Gökgöl-Kline, C. B. Berde, M. Shubina, Y. Kuang, S. Hall and G. R. Strichartz, Reg. Anesth. Pain Med., 2000, 25, 52–59 CrossRef CAS.
- M. Wiese, P. M. D'Agostino, T. K. Mihali, M. C. Moffitt and B. A. Neilan, Mar. Drugs, 2010, 8, 2185–2211 CrossRef CAS PubMed.
- M. V. Laycock, P. Thibault, S. W. Ayer and J. A. Walter, Nat. Toxins, 1994, 2, 175–183 CrossRef CAS PubMed.
- S. Hall, G. Strichartz, E. Moczydlowski, A. Ravindran and P. B. Reichardt, in Marine Toxins – Origin, Structure, and Molecular Pharmacology, ed. S. Hall and G. Strichartz, American Chemical Society, 1990, vol. 418, pp. 29–65 Search PubMed.
- A. Negri, D. Stirling, M. Quilliam, S. Blackburn, C. Bolch, I. Burton, G. Eaglesham, K. Thomas, J. Walter and R. Willis, Chem. Res. Toxicol., 2003, 16, 1029–1033 Search PubMed.
- P. Vale, J. Chromatogr. A, 2008, 1190, 191–197 CrossRef CAS.
- L. Ding, J. Qiu and A. Li, J. Agric. Food Chem., 2017, 65, 5494–5502 CrossRef CAS PubMed.
- M. A. Quilliam, A. Li, N. Lewis, P. McCarron, K. Thomas and J. A. Walter, Mar. Fresh-Water Harmful Algae. Proc. 17th Int. Conf. Harmful Algae., 2017, pp. 118–121 Search PubMed.
- A. D. Turner, R. G. Hatfield, B. H. Maskrey, M. Algoet and J. F. Lawrence, TrAC, Trends Anal. Chem., 2019, 113, 124–139 CrossRef CAS.
- A. A. Casselman, R. Greenhalgh, H. H. Brownell and R. A. B. Bannard, Can. J. Chem., 1960, 38, 1277–1290 CrossRef CAS.
- E. J. Schantz, J. D. Mold, W. L. Howard, J. P. Bowden, D. W. Stanger, J. M. Lynch, O. P. Wintersteiner, J. D. Dutcher, D. R. Walters and B. Riegel, Can. J. Chem., 1961, 39, 2117–2123 CrossRef CAS.
- E. J. Schantz, V. E. Ghazarossian, H. K. Schnoes, F. M. Strong, J. P. Springer, J. O. Pezzanite and J. Clardy, J. Am. Chem. Soc., 1975, 97, 1238–1239 CrossRef CAS.
- J. L. Wong, R. Oesterlin and H. Rapoport, J. Am. Chem. Soc., 1971, 93, 7344–7345 CrossRef CAS PubMed.
- R. S. Rogers and H. Rapoport, J. Am. Chem. Soc., 1980, 102, 7335–7339 CrossRef CAS.
- Y. Shimizu, C.-P. Hsu and A. Genenah, J. Am. Chem. Soc., 1981, 103, 605–609 CrossRef CAS.
- E. J. Schantz, J. D. Mold, D. W. Stanger, J. Shavel, F. J. Riel, J. P. Bowden, J. M. Lynch, R. S. Wyler, B. Riegel and H. Sommer, J. Am. Chem. Soc., 1957, 79, 5230–5235 CrossRef CAS.
- L. Sleno, D. A. Volmer, B. Kovačević and Z. B. Maksić, J. Am. Soc. Mass Spectrom., 2004, 15, 462–477 CrossRef CAS.
- C. Dell'Aversano, J. A. Walter, I. W. Burton, D. J. Stirling, E. Fattorusso and M. A. Quilliam, J. Nat. Prod., 2008, 71, 1518–1523 CrossRef.
- C. Graves, K. Thomas, E. Bond, S. Crain and M. A. Quilliam, CRM-STX-f, a certified calibration solution reference material for Saxitoxin, National Research Council Canada, Halifax, 2011 Search PubMed.
- Y. Shimizu, in Seafood and freshwater toxins: pharmacology, physiology, and detection, ed. L. M. Botana, Marcel Dekker, Inc., New York, 2000, pp. 151–172 Search PubMed.
- G. B. Elyakov, V. A. Stonik and T. N. Makar'eva, Chem. Heterocycl. Compd., 1977, 13, 345–359 CrossRef.
- C. Y. Kao, Ann. N. Y. Acad. Sci., 1986, 479, 52–67 CrossRef CAS PubMed.
- Y. Shimizu, Ann. N. Y. Acad. Sci., 1986, 479, 24–31 CrossRef CAS.
- Y. Shimizu, in Fortschritte der Chemie organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Grisebach and G. W. Kirby, Springer Vienna, Vienna, 1984, pp. 235–264 Search PubMed.
- H. A. Bates and H. Rapoport, J. Agric. Food Chem., 1975, 23, 237–239 CrossRef CAS PubMed.
- J. L. Wong, M. S. Brown, K. Matsumoto, R. Oesterlin and H. Rapoport, J. Am. Chem. Soc., 1971, 93, 4633–4634 CrossRef CAS PubMed.
- H. P. Van Egmond, K. M. Jonker, M. Poelman, P. Scherpenisse, A. G. Stern, P. Wezenbeek, A. A. Bergwerff and H. J. Van den Top, Food Addit. Contam., 2004, 21, 331–340 CrossRef CAS PubMed.
- Y. Che, L. Ding, J. Qiu, Y. Ji and A. Li, J. Agric. Food Chem., 2020, 68, 1427–1435 CrossRef CAS PubMed.
- W. M. Indrasena and T. A. Gill, Food Chem., 2000, 71, 71–77 CrossRef CAS.
- A. Alfonso, M. C. Louzao, M. R. Vieytes and L. M. Botana, Toxicon, 1994, 32, 1593–1598 CrossRef CAS.
- Y. Nagashima, T. Noguchi, M. Tanaka and K. Hashimoto, J. Food Sci., 1991, 56, 1572–1575 CrossRef CAS.
- W. M. Indrasena and T. A. Gill, J. Food Sci., 2000, 65, 948–953 CrossRef CAS.
- J. B. Weigele and R. L. Barchi, J. Neurochem., 1980, 35, 430–435 CrossRef CAS.
- Y. Tominaga, T. Kubo and K. Hosoya, Catal. Commun., 2011, 12, 785–789 CrossRef CAS.
- P. F. Solter and V. R. Beasley, in Haschek and Rousseaux's Handbook of Toxicologic Pathology, Elsevier Inc., 2013, pp. 1155–1186 Search PubMed.
- Background document for development of WHO Guidelines for Drinking-water Quality and Guidelines for Safe Recreational Water Environments, Cyanobacterial toxins: saxitoxins, WHO, 2020 Search PubMed.
- M. Ikawa, K. Wegener, T. L. Foxall and J. J. Sasner, Toxicon, 1982, 20, 747–752 CrossRef CAS.
- E. Jackim and J. Gentile, Science, 1968, 162, 915–916 CrossRef CAS.
- J. D. Hackett, J. H. Wisecaver, M. L. Brosnahan, D. M. Kulis, D. M. Anderson, D. Bhattacharya, F. G. Plumley and D. L. Erdner, Mol. Biol. Evol., 2013, 30, 70–78 CrossRef CAS PubMed.
- Y. Shimizu, M. Norte, A. Hori, A. Genenah and M. Kobayashi, J. Am. Chem. Soc., 1984, 106, 6433–6434 CrossRef CAS.
- S. Gupta, M. Norte and Y. Shimizu, J. Chem. Soc., Chem. Commun., 1989, 1421–1424 RSC.
- R. Kellmann, T. K. Mihali, Y. J. Jeon, R. Pickford, F. Pomati and B. A. Neilan, Appl. Environ. Microbiol., 2008, 74, 4044–4053 CrossRef CAS.
- S. Tsuchiya, Y. Cho, K. Konoki, K. Nagasawa, Y. Oshima and M. Yotsu-Yamashita, Org. Biomol. Chem., 2014, 12, 3016–3020 RSC.
- S. Tsuchiya, Y. Cho, R. Yoshioka, K. Konoki, K. Nagasawa, Y. Oshima and M. Yotsu-Yamashita, Angew. Chem., Int. Ed., 2017, 56, 5327–5331 CrossRef CAS.
- S. W. Chun, M. E. Hinze, M. A. Skiba and A. R. H. Narayan, J. Am. Chem. Soc., 2018, 140, 2430–2433 CrossRef CAS PubMed.
- J. R. Chekan, T. R. Fallon and B. S. Moore, Curr. Opin. Chem. Biol., 2020, 59, 119–129 CrossRef CAS PubMed.
- L. Valledor, M. Escandón, M. Meijón, E. Nukarinen, M. J. Cañal and W. Weckwerth, Plant J., 2014, 79, 173–180 CrossRef CAS.
- M. Afiq Akbar, N. Yuziana, M. Yusof, N. I. Tahir, A. Ahmad, G. Usup, F. K. Sahrani and H. Bunawan, Mar. Drugs, 2020, 18, 24 Search PubMed.
- J. J. Sullivan, W. T. Iwaoka and J. Liston, Biochem. Biophys. Res. Commun., 1983, 114, 465–472 CrossRef CAS.
- H. Tanino, T. Nakata, T. Kaneko and Y. Kishi, J. Am. Chem. Soc., 1977, 99, 2818–2819 CrossRef CAS.
- P. A. Jacobi, M. J. Martinelli and S. Polanc, J. Am. Chem. Soc., 1984, 106, 5594–5598 CrossRef CAS.
- P. A. Jacobi, in Strategies and Tactics in Organic Synthesis ed. T. Lindberg, Academic Press, 1989, vol. 2, pp. 191–219 Search PubMed.
- J. J. Fleming and J. Du Bois, J. Am. Chem. Soc., 2006, 128, 3926–3927 CrossRef CAS.
- V. R. Bhonde and R. E. Looper, J. Am. Chem. Soc., 2011, 133, 20172–20174 CrossRef CAS PubMed.
- T. Akimoto, A. Masuda, M. Yotsu-Yamashita, T. Hirokawa and K. Nagasawa, Org. Biomol. Chem., 2013, 11, 6642–6649 RSC.
- G. Choudhary, L. Shang, X. Li and S. C. Dudley, Biophys. J., 2002, 83, 912–919 CrossRef CAS.
- R. P. Hartshorne and W. A. Catterall, J. Biol. Chem., 1984, 259, 1667–1675 CrossRef CAS.
- G. J. Doucette, M. M. Logan, J. S. Ramsdell and F. M. Van Dolah, Toxicon, 1997, 35, 625–636 CrossRef CAS PubMed.
- L. M. Duran-Riveroll and A. D. Cembella, Mar. Drugs, 2017, 15, 28 CrossRef.
- C. Y. Kao and S. E. Walker, J. Physiol., 1982, 323, 619–637 CrossRef CAS PubMed.
- L. M. Durán-Riveroll, A. D. Cembella, C. J. Band-Schmidt, J. J. Bustillos-Guzmán and J. Correa-Basurto, Toxins, 2016, 8, 129 CrossRef.
- G. Strichartz, J. Gen. Physiol., 1984, 84, 281–305 CrossRef CAS PubMed.
- B. Hille, Biophys. J., 1975, 15, 615–619 CrossRef CAS.
- Y. Shimizu, Pure Appl. Chem., 1982, 54, 1973 CAS.
- E. Alonso, A. Alfonso, M. R. Vieytes and L. M. Botana, Arch. Toxicol., 2016, 90, 479–488 CrossRef CAS PubMed.
- J. Wang, J. J. Salata and P. B. Bennett, J. Gen. Physiol., 2003, 121, 583 CrossRef CAS.
- Z. Su, M. Sheets, H. Ishida, F. Li and W. H. Barry, J. Pharmacol. Exp. Ther., 2004, 308, 324 CrossRef CAS.
- W. A. Catterall, S. Cestèle, V. Yarov-Yarovoy, F. H. Yu, K. Konoki and T. Scheuer, Toxicon, 2007, 49, 124–141 CrossRef CAS PubMed.
- G. P. Rossini and P. Hess, in Molecular, Clinical and Environmental Toxicology: Volume 2: Clinical Toxicology, ed. A. Luch, Birkhäuser Basel, Basel, 2010, pp. 65–122 Search PubMed.
- C. Y. Kao, in Algal Toxins in Seafood and Drinking Water, ed. I. A. N. R. Falconer, Academic Press, San Diego, 1993, pp. 75–86 Search PubMed.
- K. S. Tan and J. Ransangan, in Reviews of Environmental Contamination and Toxicology, ed. D. M. Whitacre, Springer International Publishing, Cham, 2015, vol. 235, pp. 1–25 Search PubMed.
- V. M. Bricelj, J. H. Lee, A. D. Cembella and D. M. Anderson, Mar. Ecol.: Prog. Ser., 1990, 63, 177–188 CrossRef CAS.
- S. E. Shumway and T. L. Cucci, Aquat. Toxicol., 1987, 10, 9–27 CrossRef.
- V. M. Bricelj, L. Connell, K. Konoki, S. P. MacQuarrie, T. Scheuer, W. A. Catterall and V. L. Trainer, Nature, 2005, 434, 763–767 CrossRef CAS.
- R. G. Kvitek and M. K. Beitler, Mar. Ecol.: Prog. Ser., 1991, 69, 47–54 CrossRef.
- S. P. MacQuarrie and V. M. Bricelj, Mar. Ecol.: Prog. Ser., 2008, 366, 59–74 CrossRef CAS.
- V. Roncalli, P. H. Lenz, M. C. Cieslak and D. K. Hartline, Sci. Rep., 2017, 7, 14201 CrossRef.
- S. P. Colin and H. G. Dam, Harmful Algae, 2002, 1, 113–125 CrossRef.
- V. M. Bricelj, A. D. Cembella, D. Laby, S. E. Shumway and T. L. Cucci, in Harmful and Toxic Algal Blooms, ed. T. Yasumoto, Y. Oshima and Y. Fukuyo, Intergovernmental Oceanographic Commission of UNESCO, 1996, pp. 405–408 Search PubMed.
- C. García, F. Pérez, C. Contreras, D. Figueroa, A. Barriga, A. López-Rivera, O. F. Araneda and H. R. Contreras, Mar. Ecol.: Prog. Ser., 2015, 32, 984–1002 Search PubMed.
- A. Ortiz, J. M. Navarro, G. Pizarro, P. A. Villanueva and C. J. Segura, Mar. Environ. Res., 2019, 144, 240–245 CrossRef CAS PubMed.
- J. Oyaneder Terrazas, H. R. Contreras and C. García, Toxins, 2017, 9, 190 CrossRef.
- V. M. Bricelj, J. H. Lee and A. D. Cembella, Mar. Ecol.: Prog. Ser., 1991, 74, 33–46 CrossRef CAS.
- Y. Oshima, in Harmful Marine Algal Blooms, ed. P. Lassus, G. Arzul, E. Erard-Le Denn, P. Gentien and C. Marcaillou-Le Baut, Lavoisier Publishing, 1995, pp. 475–480 Search PubMed.
- E. Jaime, G. Gerdts and B. Luckas, Harmful Algae, 2007, 6, 308–316 CrossRef CAS.
- A. D. Turner, A. M. Lewis, A. O'Neil and R. G. Hatfield, Toxicon, 2013, 65, 41–58 CrossRef CAS PubMed.
- M. Asakawa, K. Miyazawa, H. Takayama and T. Noguchi, Toxicon, 1995, 33, 691–697 CrossRef CAS PubMed.
- M. C. Choi, D. P. H. Hsieh, P. K. S. Lam and W. X. Wang, Mar. Biol., 2003, 143, 927–934 CrossRef CAS.
- M. L. Artigas, P. J. V. Vale, S. S. Gomes, M. J. Botelho, S. M. Rodrigues and A. Amorim, J. Chromatogr. A, 2007, 1160, 99–105 CrossRef CAS PubMed.
- P. Vale, Toxicon, 2010, 55, 162–165 CrossRef CAS.
- M. Guéguen, R. Baron, M. Bardouil, P. Truquet, H. Haberkorn, P. Lassus, L. Barillé and Z. Amzil, Ecol. Modell., 2011, 222, 3394–3402 CrossRef.
- J. Qiu, F. Meng, L. Ding, Y. Che, P. McCarron, D. G. Beach and A. Li, Aquat. Toxicol., 2018, 200, 233–240 CrossRef CAS PubMed.
- S. DeGrasse, V. Rivera, J. Roach, K. White, J. Callahan, D. Couture, K. Simone, T. Peredy and M. Poli, Deep Sea Res., Part II, 2014, 103, 368–375 CrossRef CAS.
- M. Samsur, Y. Yamaguchi, T. Sagara, T. Takatani, O. Arakawa and T. Noguchi, Toxicon, 2006, 48, 323–330 CrossRef CAS.
- P. Vale, J. Chromatogr. A, 2008, 1195, 85–93 CrossRef CAS.
- A. D. Cembella, S. E. Shumway and R. Larocque, J. Exp. Mar. Biol. Ecol., 1994, 180, 1–22 CrossRef CAS.
- B. Krock, C. G. Seguel and A. D. Cembella, Harmful Algae, 2007, 6, 734–744 CrossRef CAS.
- Y. Sako, T. Yoshida, A. Uchida, O. Arakawa, T. Noguchi and Y. Ishida, J. Phycol., 2001, 37, 1044–1051 CrossRef CAS.
- T. Yoshida, Y. Sako, A. Uchida, T. Kakutani, O. Arakawa, T. Noguchi and Y. Ishida, Fish. Sci., 2002, 68, 634–642 CrossRef CAS.
- D. Wang, S. Zhang and H. Hong, Chin. J. Oceanol. Limnol., 2007, 25, 227–234 CrossRef CAS.
- Y. Shimizu and M. Yoshioka, Science, 1981, 212, 547 CrossRef CAS PubMed.
- S. Sakamoto, S. Sato, T. Ogata and M. Kodama, Fish. Sci., 2000, 66, 136–141 CrossRef CAS.
- M. K. Beitler and J. Liston, in Toxic Marine Phytoplankton, ed. E. Graneli, B. Sundström, L. Edler and D. M. Anderson, Elsevier Science, New York, 1990, pp. 257–262 Search PubMed.
- J. K. Andres, A. T. Yñiguez, J. M. Maister, A. D. Turner, D. E. B. Olano, J. Mendoza and L. Salvador-Reyes, Toxins, 2019, 11, 468 CrossRef CAS.
- Y. Kotaki, Nippon Suisan Gakkaishi, 1989, 55, 1293 CrossRef.
- Y. Kotaki, Y. Oshima and T. Yasumoto, Nippon Suisan Gakkaishi, 1985, 51, 1009–1013 CrossRef CAS.
- E. A. Smith, F. Grant, C. M. Ferguson and S. Gallacher, Appl. Environ. Microbiol., 2001, 67, 2345–2353 CrossRef CAS PubMed.
- A. Buzy, P. Thibault and M. V. Laycock, J. Chromatogr. A, 1994, 688, 301–316 CrossRef CAS.
- M. D. Fast, A. D. Cembella and N. W. Ross, Harmful Algae, 2006, 5, 79–90 CrossRef CAS.
- J. J. Sullivan, Paralytic shellfish poisoning: analytical and biochemical investigation, University of Washington, 1982 Search PubMed.
- A. Li, J. Ma, J. Cao, Q. Wang, R. Yu, K. Thomas and M. A. Quilliam, Food Addit. Contam., Part A, 2012, 29, 1455–1464 CrossRef CAS.
- L. E. Llewellyn, M. J. Dodd, A. Robertson, G. Ericson, C. de Koning and A. P. Negri, Toxicon, 2002, 40, 1463–1469 CrossRef CAS.
- C. García, A. Rodriguez-Navarro, J. C. Díaz, R. Torres and N. Lagos, Toxicon, 2009, 53, 206–213 CrossRef.
- C. García, A. Barriga, J. C. Díaz, M. Lagos and N. Lagos, Toxicon, 2010, 55, 135–144 CrossRef.
- Y. Oshima, S. I. Blackburn and G. M. Hallegraeff, Mar. Biol., 1993, 116, 471–476 CrossRef CAS.
- V. M. Bricelj and S. E. Shumway, Rev. Fish. Sci., 1998, 6, 315–383 CrossRef CAS.
- H.-P. Lin, Y. Cho, H. Yashiro, T. Yamada and Y. Oshima, Toxicon, 2004, 44, 657–668 CrossRef CAS.
- Y. Cho, N. Ogawa, M. Takahashi, H.-P. Lin and Y. Oshima, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 1277–1285 CrossRef CAS PubMed.
- T. Yoshida, Y. Sako, T. Kakutani, A. Fujii, A. Uchida, Y. Ishida, O. Arakawaand and T. Noguchi, in Harmful Algae, ed. B. Reguera, J. Blanco, M. L. Fernandez and T. Wyatt, Xunta de Galicia and IOC of UNESCO, Vigo, 1998, pp. 366–369 Search PubMed.
- M. J. Botelho, F. Marques, R. Freitas, A. Pires, E. Pereira and C. Vale, Mar. Environ. Res., 2020, 154, 104839 CrossRef CAS.
- FAO/WHO, Toxicity equivalence factors for marine biotoxins associated with bivalve molluscs, 2016, p. 108 Search PubMed.
- L. M. Botana, P. Hess, R. Munday, A. Nathalie, S. L. DeGrasse, M. Feeley, T. Suzuki, M. van den Berg, V. Fattori, E. G. Gamarro, A. Tritscher, R. Nakagawa and I. Karunasagar, Trends Food Sci. Technol., 2017, 59, 15–24 CrossRef CAS.
- C. Vale, A. Alfonso, M. R. Vieytes, X. M. Romarís, F. Arévalo, A. M. Botana and L. M. Botana, Anal. Chem., 2008, 80, 1770–1776 CrossRef CAS PubMed.
- A. I. Selwood, C. Waugh, D. T. Harwood, L. L. Rhodes, J. Reeve, J. Sim and R. Munday, Toxins, 2017, 9, 73 CrossRef.
- R. Munday, K. Thomas, R. Gibbs, C. Murphy and M. A. Quilliam, Toxicon, 2013, 76, 77–83 CrossRef CAS.
- Y. Oshima, M. Machida, K. Sasaki, Y. Tamaoki and T. Yasumoto, Agric. Biol. Chem., 1984, 48, 1707–1711 CAS.
- J. L. Penzotti, G. Lipkind, H. A. Fozzard and S. C. Dudley Jr, Biophys. J., 2001, 80, 698–706 CrossRef CAS.
- L. Llewellyn, A. Negri and M. Quilliam, Toxicon, 2004, 43, 101–104 CrossRef CAS PubMed.
- A. P. Negri, C. J. S. Bolch, S. Geier, D. H. Green, T.-G. Park and S. I. Blackburn, Harmful Algae, 2007, 6, 774–780 CrossRef CAS.
- F. Gazeau, L. M. Parker, S. Comeau, J.-P. Gattuso, W. A. O'Connor, S. Martin, H.-O. Pörtner and P. M. Ross, Mar. Biol., 2013, 160, 2207–2245 CrossRef CAS.
- X. Zhao, W. Shi, Y. Han, S. Liu, C. Guo, W. Fu, X. Chai and G. Liu, Mar. Environ. Res., 2017, 125, 82–89 CrossRef CAS.
- C. Peng, X. G. Zhao, S. X. Liu, W. Shi, Y. Han, C. Guo, X. Peng, X. L. Chai and G. X. Liu, Mar. Ecol.: Prog. Ser., 2017, 575, 107–117 CrossRef CAS.
- H. Farrell, F. Seebacher, W. O'Connor, A. Zammit, D. T. Harwood and S. Murray, GCB Bioenergy, 2015, 21, 3402–3413 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1np00009h |
| This journal is © The Royal Society of Chemistry 2022 |