
Recent advances in the synthesis of ent-kaurane diterpenoids
Xiangbo
Zhao†
,
Bastien
Cacherat†
,
Qifei
Hu
and
Dawei
Ma
 *
*
State Key Laboratory of Bioorganic & Natural Products Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: madw@sioc.ac.cn
First published on 15th July 2021
Abstract
Covering: 2015 to 2020
The ent-kaurane diterpenoids are integral parts of tetracyclic natural products that are widely distributed in terrestrial plants. These compounds have been found to possess interesting bioactivities, ranging from antitumor, antifungal and antibacterial to anti-inflammatory activities. Structurally, the different tetracyclic moieties of ent-kauranes can be seen as the results of intramolecular cyclizations, oxidations, C–C bond cleavages, degradation, or rearrangements, starting from their parent skeleton. During the past decade, great efforts have been made to develop novel strategies for synthesizing these natural products. The purpose of this review is to describe the recent advances in the total synthesis of ent-kaurane diterpenoids covering the period from 2015 to date.
 Xiangbo Zhao | Xiangbo Zhao was born and raised in Xuzhou, China and obtained his BSc degree at Northwest University (Xi'an, China) in 2012. Then, he went to Shanghai Institute of Organic Chemistry (SIOC) to pursue his PhD degree in Professor Dawei Ma's group, working on the total synthesis of ent-kaurane diterpenoids and napelline alkaloids. After graduation in 2017, he continued his work in the same group for one year and then joined the Macmillan group as a post-doc at Princeton University in 2019, where he mainly focused on the development of new cross-coupling methods using metallaphotoredox catalysis. He is currently research associate in SIOC and his research interest includes total synthesis of natural products and photo-induced methodologies. |
 Bastien Cacherat | Bastien Cacherat studied at the École Nationale Supérieure de Chimie de Mulhouse from 2011 to 2014, earning a M.Sc. in sciences, technologies, health and research, together with a Diplôme d’Ingénieur in chemistry. As a doctoral student supported by a Max-Planck-Gesellschaft fellow-ship in the laboratory of Prof. Bill Morandi at the Max-Planck-Institut für Kohlenforschung, he then developed new strategies in organic synthesis, using either transition metals or Brønsted acids. Subsequently, he moved to Shanghai, China to undertake a postdoctoral fellowship with Prof. Dawei Ma at the Shanghai Institute of Organic Chemistry, where he mainly focused on the total synthesis of a marine alkaloid. He is currently research chemist at Spirochem AG, a CRO based in Basel, Switzerland. |
 Qifei Hu | Qifei Hu was born in Xi'an, China and graduated from Nankai University (Tianjin, China) in 2015. Then, he continued his education under the supervision of Prof. Dawei Ma at Shanghai Institute of Organic Chemistry (Shanghai, China), working on the total synthesis of rearranged ent-kaurane diterpenoids. In 2020, he received his PhD and joined Pharmaron Inc. as a R&D Senior Scientist in the same year. His research interest includes total synthesis of natural products with significant bioactivities and efficient methodologies in drug research and development. |
 Dawei Ma | Dawei Ma received his PhD in 1989 from Shanghai Institute of Organic Chemistry and did his postdoctoral studies at the University of Pittsburgh and Mayo Clinic. He returned to SIOC in 1994 and was appointed as research professor in 1995. His research interests currently focus on the development of new synthetic methodologies, the total synthesis of complex natural products, and their SAR and action mode studies, as well as the discovery of small modulators for target proteins and special biological processes. |
1 Introduction
In 1961, the simplest ent-kaurane diterpenoid, ent-kaurene (1), was detected for the first time in New Zealand in the leaf essential oil in Agathis, a member of Araucariaceae, a conifer family (Fig. 1).1 The plant being locally called kauri pine, this type of diterpene with negative optical rotation was consequently called ent-kaurene, “ent” meaning enantiomeric. | ||
| Fig. 1 ent-Kaurene skeleton. | ||
As a family composed of structurally diverse and biologically active natural products of terrestrial plants, ent-kaurane diterpenoids, with their 20 C-atoms, account to date for more than 1000 varieties of natural enantiomers that have been either identified or isolated, or both. Most of them were isolated from the plants of the Isodon genus,2 which have long been known in Asian traditional medicine for their curative properties, especially because of the bioactive diterpenoids that they contain. Moreover, not only do Isodon, as a group of flowering plants in the Labiatae (or Lamiaceae) family, feature numerous ent-kauranoids, but other families, for example Asteraceae (or Compositae), Annonaceae, Pteridaceae, Euphorbiaceae, as well as liverworts and microbial metabolites, are also a source of a plethora of ent-kauranoids.2c Sun and co-workers notably achieved the separation, the identification as well as biological activity testing of more than 300 diterpenoids from the Isodon species.2a,b In recent years, more and more studies have shown that most of these natural products showcase excellent properties, including but not limited to antibacterial, anticancer, antifungal, antitumor and antiviral biological activities. Concerning biosynthesis, it appears that the biosynthesis of ent-kaurene is well known.3a,b Indeed, geranylgeranyl pyrophosphate (GGPP, 2), the universal precursor of all plant diterpenes, is first converted to copalyl diphosphate (CPP) under the action of the enzyme named CPP synthase or CPS or sometimes known as kaurene synthase A. It is important to mention that two products, either ent-CPP or syn-CPP, can result from this transformation. It depends whether the enzyme is ent-CPP synthase or syn-CPP synthase, respectively. When using ent-CPP synthase, the desired ent-CPP intermediate is obtained and then transformed into ent-kaurene in a few steps under the action of ent-kaurene synthase (KS) or kaurene synthase B. The classic mechanism has also been described (Scheme 1).4d Starting from ent-CPP (3), the cyclization cascade generates the tricyclic intermediate PA (4) with a tertiary carbocation at C-8. The double bond at C-14 then intramolecularly attacks the carbocation at C-8, which results in the formation of the key tetracyclic beyeranyl-13-cation intermediate PB (5), with a secondary carbocation at C-13. A 1,2-alkyl migration then occurs, forming the ent-kaurenyl-16-cation intermediate PE (6) with a more stable tertiary carbocation. Alternatively, Tantillo's calculation work on the ent-kaurene biosynthetic pathway indicates that secondary carbocations can be avoided by combining two or more mechanistic steps into concerted processes that lead to tertiary carbocations.3c,d Therefore, the tertiary carbocation PE (6) is also expected to be formed from PA (4) via a concerted cyclization/alkyl shift process. Finally, ent-kaurene is generated through proton removal which quenches the tertiary carbocation, yielding the desired exocyclic olefin.
 | ||
| Scheme 1 Biosynthesis of ent-kaurene. | ||
For this parent ent-kaurene carboskeleton, oxidation, C–C bond cleavage, fragmentation or rearrangement within these diterpenoids are incredibly common and lead to a variety of additional structures. Based on structural features, ent-kaurane diterpenoids were classified into different sub-classes: the C-20 non-oxygenated ent-kauranoids (such as pharicin A, 7), the C-20 oxygenated ent-kauranoids (such as xerophilusin I, 8), the seco-ent-kauranoids (such as sculponeatin N, 12), nor- or rearranged-ent-kauranoids (such as maoecrystal V, 14), and grayananes (such as principinol D, 15) (Fig. 2).2ent-Kauranoids exhibit numerous frameworks and oxidation patterns, as well as a variety of bioactivities, and so these molecules have naturally attracted extensive attention from synthetic chemists and a tremendous amount of total syntheses were disclosed over the past decade.4–6 To the best of our knowledge, there have been several elegant reviews focusing on the total synthesis of these diterpenoids between 1962 (the first total synthesis of ent-kaurane diterpenoids by Ireland and co-workers)5a and 2014, published by the Thomson, Hong, Lei, and Schindler groups.4a–d Many of the classic methods that were invented for total synthesis of these natural products in this period, such as Mori's fragmentation of unique cyclopropane intermediate,5f Corey's cationic cyclization,5h,i and Yang's oxidative dearomatization/intramolecular Diels–Alder reaction,6g remain effective in this field and continue as fruitful resources to inspire total synthesis and method development to this day.4e
 | ||
| Fig. 2 Structures of related ent-kauranes. | ||
During the past seven years, rapid development of new synthetic strategies and methodologies, particularly unprecedented C–C bond formation reactions, has offered numerous new opportunities for the total synthesis of ent-kaurane and related diterpenoids. Dozens of unique synthetic strategies have been developed towards accessing the more recently isolated, highly oxidized, and unusual members of this family.7–11 In this review, we cover the recent total syntheses of the ent-kauranoids from 2015 to date.
2 Total syntheses of C-20 non-oxygenated ent-kauranoids
The C-20 non-oxygenated ent-kaurane diterpenoids are the most widely distributed products among ent-kauranes, accounting for about one third of the whole family. Their structural particularity is the presence of a C-20 methyl at C-10, whereas an H-atom is usually found at C-5 or C-9, two positions that are rarely oxidized. Most of the natural products that were isolated from fragrant plant genera contain several oxidized carbons, including C-15 that is usually connected to an O-atom.2a During past years, several elegant strategies have been developed to assemble these natural products, which are summarized below.2.1 Asymmetric total syntheses of (+)-lungshengenin D and (+)-1α,6α-diacetoxy-ent-kaura-9(11),16-dien-12,15-dione (Ma)
In 2017 the Ma group completed the first asymmetric total syntheses of two highly oxidized ent-kaurane diterpenoids, lungshengenin D and 1α,6α-diacetoxy-ent-kaura-9,16-dien-12,15-dione,7a in which a [3.2.1] bicyclic unit was designed as a common intermediate. This unit was established by using a Hoppe homoaldol reaction and a Lewis acid-mediated Mukaiyama–Michael-type cyclization as the key steps at the early stage, making this strategy remarkably convergent.As outlined in Scheme 2, the authors started the synthesis from enone 16, which was transformed into ester 17 in two steps. This ester was subjected to a Pd-catalyzed intramolecular allylation to create a quaternary carbon center in 82% yield and 87% ee.12 After reduction and isomerization of the chiral ketone 18, the resulting enone 19 was subsequently converted into [3.2.1] bicyclic enone 20 through a Pd-catalyzed cycloalkenylation. Ketone protection of 20, followed by silyl ether cleavage and Ley–Griffith oxidation delivered the designed aldehyde building block 21.
 | ||
| Scheme 2 The first asymmetric total synthesis of 1α,6α-diacetoxy-ent-kaura-9(11),16-dien-12,15-dione by the Ma group. | ||
In a parallel procedure, asymmetric reduction of α-methylcyclohexenone 22 with (R)-CBS gave allylic alcohol 23 in 90% yield and 99% ee. NaH-mediated condensation of 23 with N,N-diisopropylcarbamoyl chloride provided the desired cyclohexenyl carbamate 24. After condensation of 24 with the aldehyde 21 under typical Hoppe homoaldol reaction conditions,13 an unprecedented intramolecular Mukaiyama–Michael cyclization reaction was conducted under the action of boron trifluoride diethyl etherate, successfully yielding a diastereomeric mixture 26 after quenching the reaction with aqueous sodium bicarbonate. Next, hydrolysis of the ester group in 26 and subsequent oxidation furnished a ketone, which was treated with DBU to afford C-5 epimerization product 27.
Using 27 as a key intermediate, total synthesis of (+)-1α,6α-diacetoxy-ent-kaura-9(11),16-dien-12,15-dione was achieved first. The two carbonyl groups in 27 were subsequently reduced with L-selectride and DIBAL-H to produce diol 28 stereoselectively. Acetylation of 28 followed by ketal deprotection and oxidation state adjustment of the [3.2.1] bicyclic fragment delivered the target molecule. In order to convert the intermediate 27 into the pentacyclic ent-kaurane diterpenoid lungshengenin D, an additional hydroxyl group should be introduced to create the requisite ether linkage (Scheme 3). To this end, Ma and coworkers transformed two ketone groups in 27 into the disilyl enol ether, which was subjected to selective oxidation of less hindered A-ring with DDQ and then cleavage of the silyl ether and ketal protecting groups to deliver trione 30. Through this operation the more reactive C-2 position was blocked and now C-11 became the more reactive one. After installing a hydroxyl group at this position to form 31, intramolecular etherification was carried out under the assistance of HCl to afford pentacyclic intermediate 32. Next, deoxygenation of C-12 was achieved via a selective reduction of C-12 carbonyl group, mesylation of the resultant alcohol, and subsequent reduction with LAH. The resultant diol was oxidized with IBX to give 33, which was converted into lungshengenin D after 4 steps.
 | ||
| Scheme 3 The first asymmetric total synthesis of lungshengenin D by the Ma group. | ||
2.2 Asymmetric total syntheses of (−)-pharicin A, (+)-pharicinin B, (+)-7-O-acetylpseurata C and (+)-pseurata C (Ding)
In 2017, the Ding group developed a new strategy to forge the highly oxygenated bicyclo[3.2.1]octane core unit of ent-kaurane diterpenoids bearing a β-OH at C-14.7b This new strategy featured an oxidative dearomatization-induced [5+2] cycloaddition/pinacol-type 1,2-acyl migration cascade to construct the core structure. The implementation of this strategy with subsequent retro-aldol/aldol reaction and singlet oxygen ene reaction allowed for the first asymmetric total syntheses of the natural products pharicin A, pharicinin B, 7-O-acetylpseurata C and pseurata C in a rather convergent way.As depicted in Scheme 4, conversion of the Wieland–Miescher ketone 35 into the bicyclic ketone 36via the formation of corresponding chiral alcohol was performed in 6 steps. The resulting epoxide 36 underwent Eschenmoser–Tanabe cleavage to deliver the acetylene aldehyde 37 with the side chains at C-10 and C-5 positions. A couple of reaction sequences allowed the access to a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of vinylphenols at C-7, which was further transformed to tetracyclic diketone 42 upon treatment with PIFA. The ODI (oxidative dearomatization-induced) [5+2] cycloaddition first connected C-9 to C-8 and C-11 to C-12, and then a pinacol-type 1,2-acyl migration reaction occurred to cause C14–C12 cleavage and subsequent creation of a C14–C13 linkage. Finally, a retro-aldol/aldol process and a singlet oxygen reaction were employed to adjust the oxidation state,7b which allowed the total syntheses of several natural products, namely pharicin A, pharicinin B, 7-O-acetylpseurata C and pseurata C after a few steps.
:1 mixture of vinylphenols at C-7, which was further transformed to tetracyclic diketone 42 upon treatment with PIFA. The ODI (oxidative dearomatization-induced) [5+2] cycloaddition first connected C-9 to C-8 and C-11 to C-12, and then a pinacol-type 1,2-acyl migration reaction occurred to cause C14–C12 cleavage and subsequent creation of a C14–C13 linkage. Finally, a retro-aldol/aldol process and a singlet oxygen reaction were employed to adjust the oxidation state,7b which allowed the total syntheses of several natural products, namely pharicin A, pharicinin B, 7-O-acetylpseurata C and pseurata C after a few steps.
 | ||
| Scheme 4 The first asymmetric total syntheses of (−)-pharicin A, (+)-pharicinin B, (+)-7-O-acetylpseurata C and (+)-pseurata C by the Ding group. | ||
2.3 Total syntheses of (±)-11β-hydroxy-16-kaurene, (±)-11α-hydroxy-16-kaurene, (±)-liangshanin G and (±)-gesneroidin B (Ma)
In 2019, the Ma group reported the total syntheses of four kaurane diterpenoids by using a twisted and extremely reactive enone, 6-methylenebicyclo[3.2.1]oct-1-en-3-one (46) (Scheme 5), as a Diels–Alder reaction dienophile.7c The [3.2.1] bridgehead enone, prepared through bromide elimination, enabled the construction of the tricyclic or tetracyclic adducts through Diels–Alder cycloaddition at the early stage of a synthesis. | ||
| Scheme 5 The total syntheses of (±)-11β-hydroxy-16-kaurene, (±)-11α-hydroxy-16-kaurene, (±)-liangshanin G and (±)-gesneroidin B by the Ma group. | ||
The synthesis of the [3.2.1] bridgehead enone started with the preparation of bicyclic carboxylic acid 44 from methyl 3-methoxybenzoate 43 in 75% yield over three steps (Scheme 5).14 The carboxylic acid group was exchanged with a bromide through Barton halodecarboxylation with halothane as a bromide source. As proof of concept, the resulting bromide 45 underwent Cs2CO3-mediated bromide elimination to form the desired dienophile 46, which spontaneously underwent Diels–Alder reactions with some dienes, namely 2,3-dimethyl-1,3-butadiene, Danishefsky's diene, furan and a diene derived from 1-(cyclohex-1-en-1-yl)ethan-1-one, to provide the corresponding cycloadducts.7c However, under the same conditions the diene 47 did not give a satisfactory result. The authors then found that when using another inorganic base, K3PO4, instead, at 100 °C and in neat conditions, the desired product 48 could be formed in 48% yield as a 1:1 diastereomeric mixture. The slow addition of the dienophile to the diene/K3PO4 mixture appeared to be critical. Then, the ketone at C-11 was reduced with DIBAL-H to form alcohol 49 in 47% yield, together with its separable C-10 epimer. With the alcohol 49 in hand, four kaurane diterpenoids, namely (±)-11β-hydroxy-16-kaurene, (±)-11α-hydroxy-16-kaurene, (±)-liangshanin G and (±)-gesneroidin B, were rapidly prepared through simple oxidation state adjustments.
2.4 Total synthesis of (−)-glaucocalyxin A (Jia)
In 2020, the Jia group reported the asymmetric total synthesis of (−)-glaucocalyxin A, a highly oxidized ent-kaurane diterpenoid.7d The synthesis featured a Mn(III)-mediated radical cyclization to form a C-14 oxygenated bicyclo[3.2.1]octane ring at the early stage and an intramolecular Diels–Alder reaction to form the key tetracyclic core.The synthesis started with a highly enantioselective conjugate addition of Grignard reagent 50 onto the cyclic α,β-unsaturated ketone 51 followed by enolate ethoxycarbonylation with Mander's reagent (Scheme 6). The resulting ketoester 52 and its corresponding enol were alkylated with 3-bromo-1-trimethylsilyl-1-propyne (53) in a diastereoselective fashion to result in β-ketoester 54. The alkynyl ketone 55 was then prepared after selective C(sp3)–Si bond cleavage, followed by a Mn(III)-mediated oxidative cyclization to deliver C14-oxygenated bicycle 55 in 45% yield. Next, desilylation followed by reduction of the ketone and TBS protection of the resulting alcohol yielded ester 56. The ester was then converted to aldehyde 57 before a Yamamoto aldol reaction achieved the formation of 58 as major diastereoisomer together with the undesired epimer at C-7 (3:1 dr). After MOM protection of the resulting alcohol, the ester was successively converted to a Weinreb amide, to a ketone upon Grignard addition and finally to silyl enol ether 59. An intramolecular Diels–Alder reaction then delivered the tetracyclic compound 60 in excellent yield but under pretty harsh reaction conditions. The key tetracyclic core was obtained after the gem-dimethyl group at C-4 was installed upon addition of “anhydrous” TBAF and concentrated MeI, leading to the formation of ketone 61 in 86% yield. After installations of the desired alcohol at C-14 and ketone at C-15, the total synthesis of (−)-glaucocalyxin A was finally achieved.
 | ||
| Scheme 6 The total synthesis of (−)-glaucocalyxin A by the Jia group. | ||
2.5 Total synthesis of (−)-euphoranginol C and (−)-euphoranginone D (Lou)
In 2020, the Lou group completed the asymmetric total synthesis of (−)-euphoranginol C and (−)-euphoranginone D.7e The synthesis featured a notable De Mayo reaction to rapidly generate the bicyclic [3.2.1]octane moiety in the target molecules.The synthesis commenced with the preparation of polyenoid 62 over four steps, which was used in a BF3·Et2O-induced biomimetic cationic cyclization to afford halogenated decalin 63 (Scheme 7). Benzyl protection of the newly formed secondary alcohol in 63, followed by formylation and reduction, provided an allylic alcohol, which was further converted to allylic bromide 64 with PBr3. Next, treatment of 64 with the enolate of 65 generated coupling products 66 and epi-66, which both underwent the De Mayo reaction under irradiation with 254 nm light, triggering a smooth [2+2] photocycloaddition. ent-Kaurene-type 70 and ent-phyllocladene 71 were obtained from the intermediates 67 and 68 after acidic work-up, respectively. It is noteworthy that the treatment of intermediate 67 with BF3·2AcOH resulted in its C9 epimerization via an oxonium intermediate to give ent-kaurene-type 70. In parallel, the De Mayo reaction was attempted on substrate epi-66, finally affording C9-epi-kaurane-type 72 as a single isomer. With the desired intermediate 70 in hand, several transformations were carried out to accomplish the total synthesis. Hydrolysis of vinyl ether 70 with 3 N HCl and regioselective methenylation in the presence of phosphine salts produced olefin 73. Next, a radical reduction, acid-promoted esterification and removal of the benzyl group afforded euphorangiol C, which was transformed into euphoranginone D upon further oxidation.
 | ||
| Scheme 7 The total synthesis of (−)-euphoranginol C and (−)-euphoranginone D by the Lou group. | ||
2.6 Total synthesis of (±)-1α-hydroxykauran-12-one, (±)-12-oxo-9,11-dehydrokaurene and (±)-12α-hydroxy-9,11-dehydrokaurene (Lei)
In 2020, the Lei group developed a concise approach for assembling the core structure of ent-kaurane diterpenoids,7f which relied on a rhodium-catalyzed [3+2+1] cycloaddition to furnish the B ring and C ring in one step, as well as a palladium-mediated cycloalkenylation to construct the key [3.2.1] ring system. Based on this synthetic strategy they achieved the first total syntheses of (±)-1α-hydroxykauran-12-one, (±)-12-oxo-9,11-dehydrokaurene and (±)-12α-hydroxy-9,11-dehydrokaurene in a protecting-group-free manner.The synthesis started with commercially available enone 74 that underwent Michael addition and alkynylation in two steps to yield 77 (Scheme 8). After a brief optimization, the key Rh(I)-catalyzed [3+2+1] cycloaddition of 77 and CO occurred smoothly to deliver the desired 78 in 52% yield with a diastereomeric ratio of 5.8:1 at C8. It is noteworthy that two rings, three carbon–carbon bonds, and one quaternary stereogenic center were created in this step through a remarkable [3+2+1] cycloaddition.15 Next, Pd-mediated 5-endo oxidative cyclization of a 5-vinyl silyl enol ether generated from the tricyclic 78 produced 79 in 79% yield. The success in creating the requisite [3.2.1] ring in this step relied on increasing the equivalents of Pd(OAc)2 and conducting the reaction at a relatively low temperature. Next, regioselective Mukaiyama hydration of the diene 79 occurred at the less hindered part and the resultant tertiary alcohol was subjected to dehydration to provide a terminal alkene, which was reduced with NaBH4 leading to the formation of two isomers, 80 and 81. Due to the undesired C16 stereochemistry provided by hydrogenation of the terminal double bond in 81, a hydroxyl-directed reduction strategy was then studied to overcome this drawback. As a result, face-selective Luche reduction of 81 afforded C12-β-OH 82, which underwent hydroxyl-directed reduction with RANEY® Ni and H2 to give enone 83 with the desired C16 stereochemistry upon a MnO2-mediated oxidization. Reduction of 83 under Birch conditions enabled the first total synthesis of (±)-1α-hydroxykauran-12-one. In a parallel procedure, Barton–McCombie deoxygenation of the isomer 80 delivered (±)-12-oxo-9,11-dehydrokaurene. Luche reduction of 84 furnished C12-β-OH 85, which was treated with 2 N HCl at 50 °C to give a mixture of unreacted 85 and the desired (±)-12α-hydroxy-9,11-dehydrokaurene.
 | ||
| Scheme 8 The total synthesis of (±)-1α-hydroxykauran-12-one, (±)-12-oxo-9,11-dehydrokaurene and (±)-12α-hydroxy-9,11-dehydrokaurene by the Lei group. | ||
3 Total syntheses of C-20 oxygenated ent-kauranoids
In contrast to unoxidized C-20 ent-kaurane diterpenoids, these types of diterpenes usually feature either an oxidized methylene group, an aldehyde group or a carbonyl group at C-20. Consequently, they can form additional oxygen bridges, such as acetal, hemiketal, ketone, lactone or ether with carbons at the C-3, C-4, C-7, C-11 or C-14 position. Three papers about the total syntheses of this subgroup have been published during the past few years, which are discussed below.3.1 Total synthesis of (±)-maoecrystal P (Luo)
In 2018, the Luo group completed the racemic total synthesis of maoecrystal P, an ent-kaurene diterpenoid oxygenated at C-20.8a The synthesis featured a highly regioselective and diastereoselective intermolecular Diels–Alder cycloaddition for creating a tetracyclic system from an enone-aldehyde and a diene embedded in a substituted bicyclo[4.1.0] skeleton. Another key point in this synthesis was a SmI2-mediated carbonyl–alkene reductive coupling to install the key [3.2.1] ring system.As shown in Scheme 9, BF3·OEt2-mediated intermolecular Diels–Alder cycloaddition between the enone-aldehyde 86 and the diene 87 quickly produced tetracyclic compound 88. The subsequent reduction, acetyl protection and allylic oxidation afforded the desired enone 90, which underwent Pd-catalyzed hydrogenolysis of the cyclopropane ring to give a pair of diastereoisomers 91 at C-16. Decarboxylation of 91 and acetonide protection led to the formation of 92, which was converted into aldehyde 93 through two-step redox manipulations. Carbonyl–alkene reductive cyclization of 93 with the assistance of SmI2 proceeded smoothly to afford tetracyclic intermediate 94, which successively underwent DMP oxidation of the hydroxyl group at C-15 and introduction of a methylene group at C-16 to produce 96. Oxidation of the enol ether moiety in 96 followed by deprotection furnished triol 97. TBAF-mediated ketal formation of 97 provided acetonide 98, which was converted into maoecrystal P after 5 steps.
 | ||
| Scheme 9 The first total synthesis of maoecrystal P by the Luo group. | ||
3.2 Total syntheses of (±)-eriocalyxin B, (±)-neolaxiflorin L and (±)-xerophilusin I (Lee)
In 2018, the Lee group completed the racemic total syntheses of three diterpenoids, namely eriocalyxin B, neolaxiflorin L and xerophilusin I.8b These natural products are oxidized at C-20 and are named 7,20-epoxy-ent-kauranoids after forming the C-7,20 hemiketal bridge. The syntheses relied on an intramolecular Diels–Alder cycloaddition to install the AB ring system, and a cascade Lewis acid-mediated intramolecular Mukaiyama–Michael/carbocyclization reaction to set up the CD ring system. After construction of the requisite tetracyclic core, the authors planned that a redox relay would successively undergo (1) selective reduction of C-12 ketone; (2) α-oxygenation at C-6 thanks to the residual ketone at C-7; (3) C-7,20 hemiketal bridge formation through cis-AB to trans-AB ring equilibrium; (4) OH-directed allylic C–H oxidation at C-15 and H-bonding controlled reduction of the carbonyl at C-6; and finally (5) selective oxidation at C-1.The linear synthesis mainly started with an intermolecular Diels–Alder cycloaddition between the α-formylcyclohexenone 101 and the enone 100 after conversion of the latter to silyl enol ether (Scheme 10). With the AB ring system now formed, the subsequent reduction, diol protection and oxidation yielded the enone 104. After strong Lewis acids screening, it was found that dimethyl aluminum chloride, in the presence of lithium bromide, could be used for realizing the expected tandem Mukaiyama–Michael/carbocyclization reaction. With the desired tetracyclic core 105 in hand, the authors carried out C-12 deoxygenation through mesylate 106 and introducing a hydroxyl group at the C-6 position to afford ketone 107, whose free hydroxyl group was oxidized and then silyl ether cleavage was conducted to produce hemiketal 108. During this course the cis-AB ring had been equilibrated to the trans-AB ring. A Riley oxidation then introduced a hydroxyl group at C-15, LAH reduced the carbonyl group at C-6 and other oxidation state adjustments led to the formation of the three C-20 oxygenated diterpenoids eriocalyxin B, neolaxiflorin L and xerophilusin I.
 | ||
| Scheme 10 The first total syntheses of (±)-eriocalyxin B, (±)-neolaxiflorin L and (±)-xerophilusin I by the Lee group. | ||
3.3 Total synthesis of (−)-oridonin (Luo)
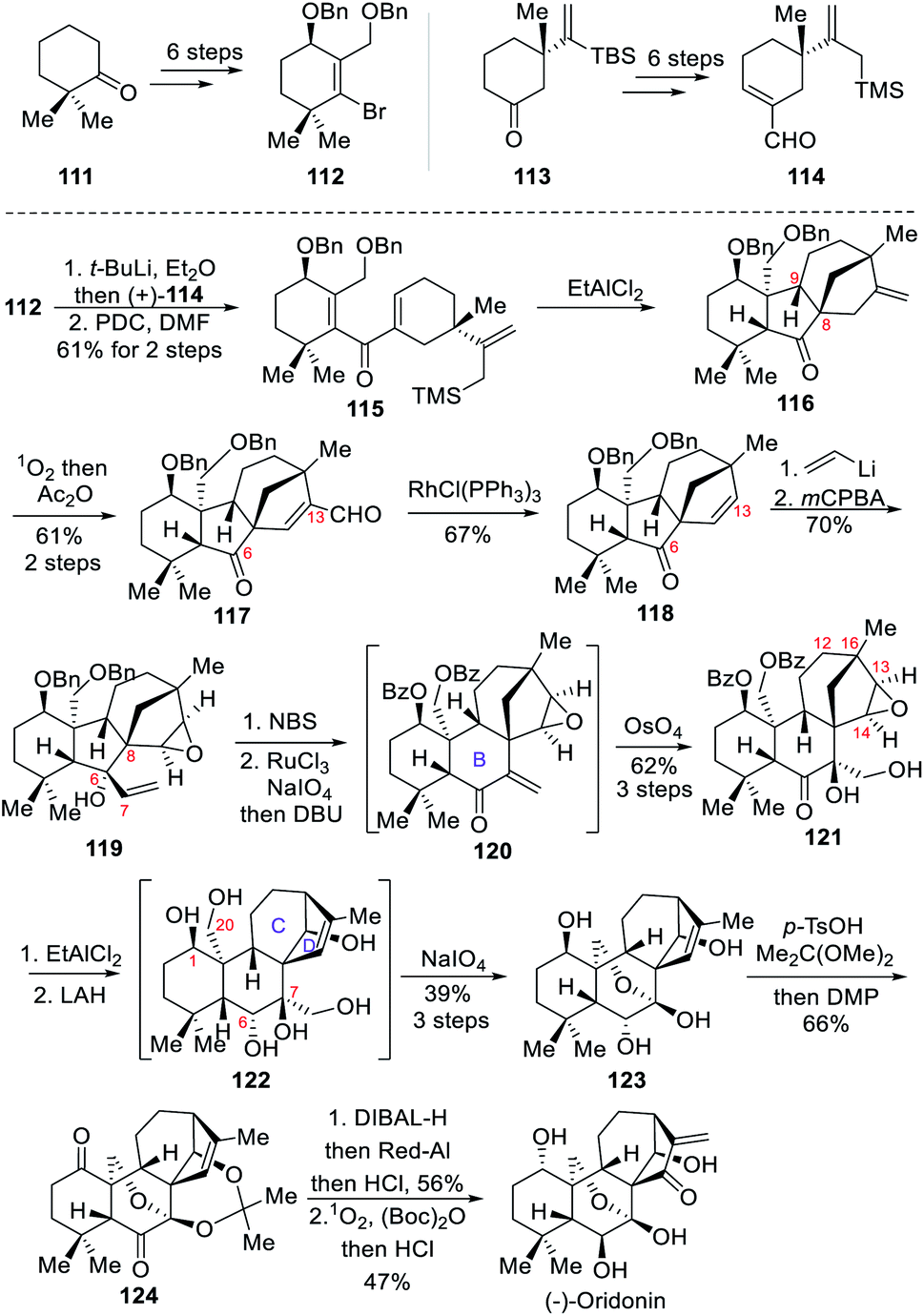
In 2019, the Luo group reported the asymmetric total synthesis of the highly oxidized (−)-oridonin.8c It features an interrupted Nazarov/Hosomi–Sakurai reaction cascade to build the key tetracyclic core and an important stereogenic center at C-8.The synthesis commenced with the preparation of two key building blocks, 112 and 114, over several simple transformations (Scheme 11). With these two fragments in hand, Li–Br exchange between 112 and t-BuLi yielded the corresponding vinyl lithium, and the addition onto the first fragment 114 afforded secondary alcohols, which were converted to ketone 115 upon addition of Cornforth reagent PDC. For the key interrupted Nazarov/Hosomi–Sakurai reaction, the authors discovered that excess EtAlCl2 was effective, and ring closing yielded the intermediate 116 as a major product, in which the stereogenic center at C-8 was established through anti-addition of the allyl silane to the hydrogen at C-9. Next, subjecting 116 to singlet oxygen ene reaction followed by acetate anhydride treatment provided aldehyde 117. A Rh(I)-catalyzed deformylation delivered the ketone 118, which underwent 1,2-addition to form an allylic alcohol, and then selective epoxidation of the C13–C14 alkene with mCPBA to form 119 in 70% overall yield. The desired B-ring was then formed through ring expansion via bromination of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond with NBS and subsequent semipinacol rearrangement. The resultant bromide was oxidized with RuCl3/NaIO4 to convert two benzyl ether moieties to the corresponding benzoyl esters, and then treated with DBU to eliminate HBr to give enone 120, which underwent Upjohn dihydroxylation to form diol 121. The desired C/D ring system was then implemented upon addition of EtAlCl2 which induced the key rearrangement. The product was then treated with excess LAH to reduce the ketone at C-6 and cleavage of the two benzoyl protecting groups to form the polyol 122. The hemiketal 123 was then formed after oxidative cleavage of the less sterically hindered vicinal diol with NaIO4. Now the stage was set for inversion of the C-1 and C-6 stereogenic centers. The authors envisioned that oxidation/reduction operation at these positions might give the desired results. Accordingly, acetonide protection of the 1,3-diol unit in 123 followed by DMP oxidation produced diketone 124. After some tries, they found that sequentially reducing C-1 ketone with DIBAL-H and C-6 ketone with Red-Al could gave the desired stereochemistry at both positions, and the resultant diol was subjected to acidic work-up and another singlet oxygen ene reaction, and treatment with Boc2O and HCl sequentially to furnish (−)-oridonin.
C double bond with NBS and subsequent semipinacol rearrangement. The resultant bromide was oxidized with RuCl3/NaIO4 to convert two benzyl ether moieties to the corresponding benzoyl esters, and then treated with DBU to eliminate HBr to give enone 120, which underwent Upjohn dihydroxylation to form diol 121. The desired C/D ring system was then implemented upon addition of EtAlCl2 which induced the key rearrangement. The product was then treated with excess LAH to reduce the ketone at C-6 and cleavage of the two benzoyl protecting groups to form the polyol 122. The hemiketal 123 was then formed after oxidative cleavage of the less sterically hindered vicinal diol with NaIO4. Now the stage was set for inversion of the C-1 and C-6 stereogenic centers. The authors envisioned that oxidation/reduction operation at these positions might give the desired results. Accordingly, acetonide protection of the 1,3-diol unit in 123 followed by DMP oxidation produced diketone 124. After some tries, they found that sequentially reducing C-1 ketone with DIBAL-H and C-6 ketone with Red-Al could gave the desired stereochemistry at both positions, and the resultant diol was subjected to acidic work-up and another singlet oxygen ene reaction, and treatment with Boc2O and HCl sequentially to furnish (−)-oridonin.
 | ||
| Scheme 11 The total synthesis of (−)-oridonin by the Luo group. | ||
4 Total syntheses of seco-ent-kauranoids
The seco-ent-kaurane diterpenoids can be generated through oxidative cleavage of the C–C bond of their biogenetic prototype and this phenomenon occurs naturally. This class of diterpenes are numerous, mainly dominated by 6,7-seco-ent-kaurane diterpenoids that can be divided into two types according to different structures: enmein-type and spirolactone-type. Some total syntheses toward this subgroup are listed below.4.1 Total synthesis of (±)-trichorabdal A and (±)-maoecrystal Z (Liang)
In 2018, the Liang group disclosed their total syntheses of trichorabdal A and maoecrystal Z.9a Both natural products were prepared from the same [3.2.1] bicyclic intermediate 135, whose oxidation at C-15 would lead to trichorabdal A whereas a retro-aldol/aldol tandem reaction would feature concomitant C6–C8 bond formation and C8–C15 bond cleavage to afford maoecrystal Z. Their common synthetic route featured several intramolecular radical cyclizations, including a cross-ring radical cyclization to forge the C9–C10 bond together with the spirocyclic quaternary carbon center at C-10; a Ueno–Stork radical cyclization to connect C-15 to C-8 and establish an all-carbon quaternary carbon center at C-8; and a reaction between cyclohexenone and terminal alkyne in 132 to forge the bicyclo[3.2.1]octane ring in diketone 133. The lactone ring in 135 was constructed by oxidative cleavage of the five-membered ring α-hydroxyketone.The synthetic route towards the key intermediate 135 started with compound 125 whose γ-position was added onto the aldehyde 126 upon n-BuLi deprotonation (Scheme 12). Once the two structures united together, in situ TBS deprotection with TBAF yielded the desired lactone 127 in 83% yield. The first radical cyclization was initiated with AIBN, and its combination with Bu3SnH afforded spirocyclic compound 128. Subjecting 128 to subsequent reduction and dearomatization and introducing two more carbons delivered the designed precursor 129 for key Ueno–Stork cyclization. In this particular case, the Ueno–Stork cyclization reaction appeared challenging, and numerous attempts led to either decomposition, deiodination or aromatization. The authors addressed this challenge through 1,4-addition of thiophenol to break the 1,6-enone system that was apparently preventing the formation of the C8–C15 bond. In such case the desired product 131 was obtained in 89% yield under standard conditions. After a few simple transformations, the desired terminal alkyne and cyclization precursor 132 was obtained. Another radical cyclization occurred under the action of AIBN and Bu3SnH to produce bicyclo[3.2.1]octane 133 with the connection of C-13 to C-16.16 Next, the stereogenic center at C-5 was fixed through redox reactions, and protecting group manipulations allowed the final formation of 134. After a hydroxyl group was introduced at C-20, at the α-position of a carbonyl group, an oxidative cleavage occurred under the action of aqueous orthoperiodic acid to form a lactol intermediate. Treatment of the lactol with LAH to remove the benzoate protecting group and subsequent oxidation of the resulting primary alcohol with PCC provided the key intermediate 135. Trichorabdal A was prepared in a few steps through introduction of an additional carbonyl group and TBS deprotection. Concerning the preparation of maoecrystal Z, the authors designed a reverse aldol/aldol reaction. To that end, the TBS group in the key intermediate 135 was first replaced with an acetate group to give 136. Next, the allylic alcohol resulting from Riley oxidation at C-15 underwent the expected reverse aldol/aldol reaction under basic conditions, furnishing the natural product maoecrystal Z.
 | ||
| Scheme 12 The total syntheses of (±)-trichorabdal A and (±)-maoecrystal Z by the Liang group. | ||
4.2 Asymmetric total syntheses of (−)-enmein, (−)-isodocarpin and (−)-sculponin R (Dong)
In 2018, the Dong group reported the total syntheses of (−)-enmein, (−)-isodocarpin and (−)-sculponin R,9b which were prepared from the same tetracyclic intermediate. At the early stage of the synthetic route, a Diels–Alder reaction was utilized to quickly construct the A/B ring system. Next, a one-pot acylation/alkylation/lactonization reaction was employed to construct both the C ring and the quaternary carbon center at C-8. Finally, a reductive alkenylation was used to achieve the construction of the D/E rings.The asymmetric synthesis started with a Diels–Alder reaction between the chiral diene 137 and the maleic anhydride 138 (Scheme 13). After aqueous hydrochloric acid quenching, the desired addition product 139 was obtained in 52% yield as a major isomer. The anhydride 139 was treated with LAH to selectively reduce more sterically hindered carbonyl group in the anhydride and the ketone to afford lactone 140.
 | ||
| Scheme 13 The asymmetric total syntheses of (−)-enmein, (−)-isodocarpin and (−)-sculponin R by the Dong group. | ||
Under the action of Li/NH3, the 1,4-cyclohexadiene was obtained through Birch reduction of 140, whose lactone was reduced to hemiacetal and the benzyl ether protecting group at C-1 underwent smooth cleavage. After acidic work-up, the 1,4-cyclohexadiene was converted to cyclohexenone, while the hemiacetal was transformed to the caged acetal, finally yielding caged compound 141. TMS protection at C-1 prior to hydrogenation of the α,β-unsaturated ketone 141 furnished the desired C-9 stereocenter. Newhouse's procedure was then applied, yielding the α,β-unsaturated ketone 142.17 The one-pot acylation/alkylation/lactonization reaction could now be performed to install the quaternary center at C-8. For that purpose, the β-ketoester was formed first, and 2,3-dibromopropene was then introduced at the alpha position. The C ring in the advanced key intermediate 144 was finally obtained after acidic work-up to remove the TMS group. In order to prepare sculponin R, LiHMDS/TBSCl treatment of the key intermediate 144 yielded the corresponding silyl enol ether, which was subsequently transformed into γ-hydroxyenone via epoxide formation. With the oxidation at C-11 fixed, compound 146 underwent reduction to allylic alcohol at C-14. The [3.2.1] bridge ring was then implemented through a radical cyclization reaction and a few simple transformations yielded the natural product (−)-sculponin R. Back to the key intermediate 144, its reduction with L-selectride generated a lithium enolate, which underwent a palladium-catalyzed alkenylation reaction under the action of Cs2CO3 and Pd(PPh3)4 to construct the [3.2.1] bicyclic ring in 145. From that point, simple transformations led to the total syntheses of both (−)-enmein and (−)-isodocarpin.
4.3 Asymmetric total syntheses of (+)-longirabdiol, (−)-longirabdolactone and (−)-effusin (Li)
In 2019, the Li group described their asymmetric total syntheses of (+)-longirabdiol, (−)-longirabdolactone and (−)-effusin through late-stage redox modification of a common key intermediate oxygenated at C-19.9c The key steps towards these target molecules included several free-radical-based reactions, namely a copper-catalyzed tandem decarboxylative radical cyclization/alkenylation reaction to build the cis-γ-lactone; an intramolecular Giese reaction to transform a carboxylic acid to a precursor for forming an unsaturated spirolactone; and a vinyl radical cyclization to install the required bicyclo[3.2.1]octane ring.The synthesis started with commercially available 2-cyclohexanonecarboxylate 148 that underwent a 5-step procedure to yield compound 149 in 87% ee and 35% overall yield (Scheme 14). The carboxylic acid in 149 was converted to the redox-active ester 151 in 3 steps and 85% overall yield. Next, the free-radical decarboxylative cyclization/olefination reaction was carried out to form the C5–C6 bond and introduce the side chain at C-10. The authors found that the reaction of 151 with β-bromostyrene proceeded best with copper(II) chloride and DavePhos as a catalyst system to yield the desired γ-lactone 152. The use of both 2-bromopropanoic acid as an additive and zinc powder proved essential to the reaction. After the styrene group in 152 underwent oxidative cleavage to form the desired carboxylic acid 153 in 46% yield, construction of the C-10 quaternary stereocenter continued with the esterification using Takeda's conditions.18 Although a mixture of diastereoisomers was obtained, LDA treatment followed by BOMCl addition yielded a single diastereomer through 1,3-diaxial interaction. Upon reduction of the lactone, TBS protection of the resulting diol, and removal of the t-butyl ester protecting group, acid 154 was obtained in 54% overall yield. The acid was then converted into a redox active ester and was then exposed to 2,2,2-trifluoroethyl acrylate through decarboxylative Giese reaction19 to produce the desired product 155, which was subjected to benzyl ether deprotection and subsequent oxidation with benzeneseleninic acid anhydride to give spirolactone 156. After that, the strategy of the Li group to build the C ring resembles that of the Thomson team.6f The 1,4-addition of the allyl cuprate species followed by the in situ reaction with allyl iodide introduced two side chains, and then an intramolecular RCM reaction occurred, yielding compound 157 in 65% overall yield. LDA was then used to deprotonate compound 157 prior to allylation with 2,3-dibromopropene which yielded compound 158 as single diastereomer. The bicyclo[3.2.1]octane ring was then formed through a 5-exo radical annulation and subsequent allylic oxidation and DMP oxidation at C-15 yielded the ketone 159. The natural product (+)-londirabdiol was finally obtained after global deprotection using LiBF4. Another DMP oxidation of (+)-londirabdiol would furnish (−)-longirabdolactone, whereas a regioselective acetylation of the C-19 hydroxyl group prior to DMP oxidation would furnish (−)-effusin.
 | ||
| Scheme 14 The asymmetric total syntheses of (+)-longirabdiol, (−)-longirabdolactone and (−)-effusin by the Li group. | ||
5 Total syntheses of nor- or rearranged-ent-kauranoids
With a unique skeleton structure, the classification of this group of the ent-kaurane diterpenoids is challenging. They are usually the results of either the formation of one or several new C–C bonds, or of one or several C–C bond rearrangements, bonds that are usually connected to the ent-kaurene framework. The degradation of one or several C-atoms can also lead to new compounds with different and interesting structures. Some synthetic efforts towards this class of natural products are introduced below.5.1 Total syntheses of (±)-jungermannenone B and (±)-jungermannenone C (Lei)
In 2015, the Lei group reported the scalable racemic total syntheses of the rearranged ent-kaurene jungermannenones B and C.10a The protecting-group-free synthesis featured an intramolecular electrophilic hydroarylation to construct the tricyclic system and an unprecedented regioselective 1,6-dienyne reductive cyclization reaction to close the rearranged bicyclo[3.2.1]octane bridged ring.The authors started the synthesis with inexpensive and readily available geraniol (160) that underwent substitution reaction to yield the aromatic diene 162 (Scheme 15), which in turn underwent a Ru(III)-catalyzed tandem cyclization followed by chromium trioxide oxidation to afford tricyclic ketone 164. Birch reduction of 164 followed by acid hydrolysis yielded dienone 167. A propargyl group was then selectively introduced, and an allylic alcohol was formed through Luche reduction of the enone to afford a precursor for the key radical cyclization. Upon n-Bu3SnH/AIBN treatment, the expected intramolecular 1,6-dienyne reductive radical cyclization proceeded smoothly to create the key bicyclo[3.2.1]octane bridged ring in tetracyclic intermediate 170. From that point, selective oxidative cleavage of exocyclic double bond provided the ketone 171 in 83% yield. Finally, the racemic syntheses of jungermannenones B and C were both completed after final installation of exo-enone by α-methylenation.
 | ||
| Scheme 15 The total synthesis of (±)-jungermannenone B and (±)-jungermannenone C by the Lei group. | ||
5.2 Asymmetric total synthesis of (−)-maoecrystal V (Baran)
The combination of striking structural features and exciting bioactivity exhibited by the ent-kaurane diterpenoid maoecrystal V has attracted extensive interest within the organic community since 2004.6g–k In 2016, the Baran group developed a new strategy for asymmetric total synthesis of (−)-maoecrystal V in 11 steps.10b In the synthesis, the chirality was introduced through an asymmetric 1,4-addition of an enone. A pinacol rearrangement together with a double bond isomerization was used to convert the [3.2.1] bridged ring to the required [2.2.2] bridged ring.Starting from cyclohexanone (172), an alkenyl fragment was smoothly introduced through asymmetric copper-catalyzed 1,4-Michael addition of an allyl silane using a TADDOL-derived phosphine-phosphite ligand L2 in toluene/MeTHF (Scheme 16). The resulting product 174 underwent α-acetoxylation after successive deprotonation with LiTMP, oxygen atom transfer with Davis' oxaziridine followed by acetylation to finally yield compound 175. An intramolecular Hosomi–Sakurai reaction catalyzed by EtAlCl2 was then performed, which successfully installed the [3.2.1] bridged ring of alcohol 176 through an anti-Baldwin cyclization. After successive methylation, hydrolysis and oxidation, the resulting ketone 177 underwent addition of the corresponding Grignard reagent of 178 that was generated after Mg/I exchange in the presence of turbo Grignard.20 After heating the resultant alcohol 179 under acidic conditions, both a pinacol rearrangement and olefin isomerization took place to produce diketone 180 in 45% isolated yield. The next challenging step was introducing a hydroxymethyl group regioselectively at C-10. With this aim in mind, the authors extended 180 to its sodium enolate form, added a LaCl3·2LiCl solution and quenched the resulting thermodynamically stable enolate with a formaldehyde gas to achieve the construction of the all-carbon quaternary carbon center and yielded 181. The regioselective reduction of the ketone at C-5 was made possible through protection of the carbonyl at C-8 by means of ketal formation. Subsequent treatment of the ketal 182 with LiBH4/Zn(OTf)2 reduced the carbonyl at C-5 to the desired hydroxyl with the required configuration. The resulting alcohol 183 underwent a few protecting group adjustments prior to the introduction of a cyano group at C-8. The δ-valerolactone 186 was then formed upon saponification, therefore installing the last ring of the pentacyclic core. After DMDO epoxidations, both the epoxides were opened upon InI3/MgI2 treatment to form 188 after DMP oxidation. In this case an iodohydrin resulted from one of the epoxides, whereas the other epoxide underwent 1,2-hydride shift, installing the desired stereochemistry at C-16. Finally, upon oxidation of 188 with oxone followed by the spontaneous elimination of the α-iodoso species, the 11-step total synthesis of maoecrystal V was successfully completed.
 | ||
| Scheme 16 The asymmetric total synthesis of (−)-maoecrystal V by the Baran group. | ||
5.3 Asymmetric total syntheses of (+)-jungermatrobrunin A, (−)-1α,6α-diacetoxyjungermannenone C and (+)-12-hydroxy-1α,6α-diacetoxy-ent-kaura-9(11),16-dien-15-one (Lei)
In 2019, the Lei team completed the asymmetric total synthesis of the rearranged ent-kaurene (+)-jungermatrobrunin A in 13 steps, along with the syntheses of (−)-1α,6α-diacetoxyjungermannenone C and (+)-12-hydroxy-1α,6α-diacetoxy-ent-kaura-9(11),16-dien-15-one.10c Interestingly, the natural product (+)-jungermatrobrunin A contains a scarcely observed peroxide bridge in the ent-kaurene diterpenoid family. The synthesis featured a 1,4-asymmetric addition catalyzed by a chiral Cu(I) complex to introduce the chirality and a radical-mediated reductive 1,6-dienyne cyclization to construct the bicyclo[3.2.1]octene ring. The key peroxide bridge of (+)-jungermatrobrunin A was implemented at the last step through a Schenck ene reaction upon exposure of the intermediate (−)-1α,6α-diacetoxyjungermannenone C to irradiation with a 24 W white LED. Interestingly, the authors observed that (−)-1α,6α-diacetoxyjungermannenone C is in equilibrium with (+)-12-hydroxy-1α,6α-diacetoxy-ent-kaura-9(11),16-dien-15-one upon UV irradiation or sunlight exposure of either one or the other.The synthesis started with the 1,4-asymmetric addition of styrene-derived 190 onto the α,β-unsaturated ketone 189 catalyzed by both a chiral copper(I) complex and Schwartz's reagent (Scheme 17).21 The desired chiral adduct 191 was obtained in 78% yield and 88% ee. Subsequent α-arylation under the action of a palladium(II)/NHC catalytic system completed the ring closure to afford 192 after simple methylation, whose benzylic carbon was then oxidized by chromium trioxide yielding the diketone 193. After Luche reduction, the resulting triol 194 underwent a Birch reduction followed by an acidic work-up to deliver 2,4-dienone 195. The propargyl side chain was stereoselectively introduced at C-13 after the TMS protection of the diol 195 was carried out. Next, Luche reduction of the new 2,4-dienone followed by MOM protection of the resultant alcohol afforded the key intermediate 196. The free radical reductive cyclization of 196 proceeded smoothly to install bicyclo[3.2.1]octane unit and lead to the formation of 197.10a A Lemieux–Johnson oxidation then delivered the ketone 198 upon oxidative cleavage, whose TMS protecting groups were exchanged for acetates to deliver the natural product (−)-1α,6α-diacetoxyjungermannenone C after a methylene group was introduced. The last task was exposure of this compound to 24 W white LED light to furnish (+)-jungermatrobrunin A through a Schenck ene reaction and singlet oxygen participation. The interconversion of the rearranged [3.2.1] bridged ring structure to the ent-kaurane [3.2.1] bridged ring structure was also noted upon UV irradiation or upon sunlight exposure of either one or the other. The synthesis of (+)-12-hydroxy-1α,6α-diacetoxy-ent-kaura-9(11),16-dien-15-one was therefore also completed.
 | ||
| Scheme 17 The asymmetric total syntheses of (+)-jungermatrobrunin A, (−)-1α,6α-diacetoxyjungermannenone C and (+)-12-hydroxy-1α,6α-diacetoxy-ent-kaura-9(11),16-dien-15-one by the Lei group. | ||
5.4 Asymmetric total syntheses of (+)-ent-kauradienone and (−)-jungermannenone C (Lei)
In 2019, the Lei team also completed the total syntheses of two other diterpenoids.10d The first one, (+)-ent-kauradienone, was prepared in 12 steps and its reduction and subsequent photoinduced skeletal rearrangement delivered a rearranged ent-kaurane diterpenoid, jungermannenone C.Based on their previous results,10a,c the authors embarked on the preparation of other natural products knowing that a skeletal rearrangement of the bicyclo[3.2.1]octene ring system of ent-kaurane diterpenoids can be induced at a late stage by the action of light. With jungermannenone C as main target molecule, the authors started the synthesis from 6-heptyn-1-ol (199) (Scheme 18). The formation of the (alkenyl)dimethylalane through carboalumination with zirconocene dichloride followed by iodination resulted in compound 200 that underwent Suzuki–Miyaura cross-coupling to form 202. Next, a Swern oxidation was performed, allowing for an enantioselective organo-SOMO cycloaddition using MacMillan's catalyst 203.22 The resulting chiral compound 204 was obtained in 72% yield and 85% ee. The subsequent C-4 methylation of 205, aldehyde deoxygenation, benzylic oxidation, Birch reduction and acidic hydrolysis delivered the desired 2,4-dienone 206. The dienyne 207 was then prepared through α-propargylation and a radical-mediated reductive cyclization to install the key bicyclo[3.2.1]octene ring. A Lemieux–Johnson oxidation followed by α-methylenation smoothly delivered the first compound, (+)-ent-kauradienone, in an excellent overall yield. The asymmetric total synthesis of jungermannenone C was completed after Luche reduction and a late-stage rearrangement of the ent-kaurane-type skeleton upon UV irradiation at 254 nm or sunlight exposure. These results in turn implied that photoinduced skeletal rearrangement may occur in the biosynthesis of ent-kaurane-type diterpenoids.
 | ||
| Scheme 18 The asymmetric total syntheses of (+)-ent-kauradienone and (+)-jungermannenone C by the Lei group. | ||
5.5 Total synthesis of (+)-farnesin (Gao)
In 2020, the Gao team completed the asymmetric total synthesis of farnesin, an acafamane-type diterpenoid.10e This natural product features a cis-fused tetracyclic structure (syn-syn-syn hydrofluorenol ABC ring) and an extra lactone A′ ring. Farnesin and, in a broader context, all acafamane-type diterpenoids are biosynthetically derived from ent-kaurene derivatives through B-ring cleavage followed by ring regeneration.23 To prepare the highly strained ABC ring system, a stereocontrolled excited-state Nazarov cyclization was implemented by the authors.The synthesis commenced with the preparation of 2,4-dienol 212 over three steps, which was used in an asymmetric Diels–Alder reaction with methyl acrylate 211 (Scheme 19). The resulting bicyclic compound 213 was obtained in 87% yield thanks to an optimized chiral ZnII/MgII bimetallic system with (R)-octahydrobinaphthol L3 as a ligand.24 After methylation at C-4 to afford 214, acetonide protecting group was removed prior to oxidative cleavage of the resulting diol to provide aldehyde 215.
 | ||
| Scheme 19 The total synthesis of (+)-farnesin by the Gao group. | ||
The synthesis of the second fragment started from diastereoselective alkylation of 217 with allyl bromide 216, and the resultant diene 218 was subjected to Ru-catalyzed RCM reaction to afford bromocyclohexene 219 in 75% yield. Reductive removal of the chiral auxiliary in 219 and oxidation generated an aldehyde, which was subjected to a NHC-catalyzed hydroxymethylation and subsequent TBS protection to produce 220. A Wittig olefination finally delivered the second fragment 221, which underwent an intermolecular addition onto the first fragment 215 after treatment with t-BuLi. After IBX oxidation of the resulting alcohol, dienone 222 was obtained in 67% yield over two steps. The key Nazarov reaction was then implemented since non-aromatic dicylic divinyl ketone can engage in cyclization under acidic conditions or by photolysis.25 The latter apparently allowed the formation of the desired product through disrotatory cyclization via an excited-state Nazarov reaction, which resulted in the formation of the desired highly strained tetracyclic intermediate 223 in 68% yield using flow chemistry. TBS deprotection followed by SmI2-mediated radical reaction then reduced the enone in 223. Although the ketone at C-7 was reduced in the meantime, it was recovered through DMP-oxidation, which ultimately delivered the aldehyde 224. The formation of the C8–C15 bond through NaOMe-mediated intramolecular aldol condensation achieved the construction of the bicyclo[3.2.1]octane unit. Oxidation of the resulting alcohol then delivered the methyl ether 225, which underwent demethylation followed by dehydration through sulfonate ester formation to afford the acafamane-type framework 226. The total synthesis of (+)-farnesin was fulfilled after oxidation state adjustments at C-6, C-7 and C-15 over 7 steps.
6 Total syntheses of grayananes
Grayananes are among a broader class of rearranged kaurane diterpenoids and their tetracyclic framework is formed through rearrangement of the kaurane 6,6-A/B ring system to the 5,7-ring system.26 They feature a central seven-membered ring in a characteristic [5,7,6,5]-tetracyclic carbon skeleton decorated with 7–10 stereogenic centers, as well as a dense arrangement of hydroxyl groups. Before 2019, only two total syntheses of these family members had been disclosed. One was reported by Matsumoto and co-workers more than forty years ago, in which a relay total synthesis (>39 steps) of grayanotoxin II based on a key biomimetic photo-santonin rearrangement was described.27a,b Another one was disclosed by Shirahama and co-workers in 1994, in which an enantioselective total synthesis of grayanotoxin III was accomplished in 38 steps.27c Very recently, three more synthetic studies on these family members were reported, which are briefly summarized below.6.1 Total synthesis of (+)-principinol D (Newhouse)
In 2019, the Newhouse group developed a convergent route towards the total synthesis of (+)-principinol D.11a This synthesis featured a 1,2-addition to couple the two designed fragments, a SmI2-mediated reductive cyclization to form the central seven-membered ring of the grayanane skeleton, and a Ni-catalyzed α-vinylation reaction to install the requisite bicyclo[3.2.1]octane ring.As outlined in Scheme 20, the synthesis started with a vicinal difunctionalization of 1-cyclohex-2-enone 227. The addition of a vinyl Grignard reagent onto the enone was performed using a catalytic amount of CuBr–DMS complex together with excess DMPU. The following ethoxycarbonylation with Mander's reagent resulted in the formation of the β-ketoester 228. After allylation with 2,3-dibromopropene, reduction of the ester using LiBH3NMe2 through enolate formation with the weak base Zn(TMP)2 yielded the primary alcohol 229. A TBS protection of the alcohol 229 was performed prior to the α-vinylation using a Ni(II) precatalyst and LiHMDS as base. The TBS group of the resulting bicyclo[3.2.1]octane ring was then cleaved under acidic conditions to afford 230. The ketone was then reduced with SmI2 in the presence of HMPA and PhSH, presumably acting as H-atom donor.28 The desired diol 231 was obtained in both high yield and diastereoselectivity, and the primary alcohol was converted into alkyl iodide through Appel reaction, whereas the secondary alcohol was MOM-protected to finally yield the first key fragment 232. The treatment of 232 with t-BuLi enabled addition onto the enantioenriched aldehyde 233, and the resulting secondary alcohol was MOM-protected to form the desired adduct 234 in 23% overall yield. TBS deprotection followed by DMP oxidation firstly formed an enone, and its terminal alkene then underwent Lemieux–Johnson oxidation to finally yield the aldehyde 235. The key seven-membered ring and therefore 236 was now formed in 63% yield upon SmI2/H2O treatment. With the aim of installing exocyclic olefin at C-10, the secondary alcohol was first oxidized to ketone using DMP and then treated with Me3SiCH2Li to form β-hydroxysilane 237. The tertiary alcohol at C-16 was firstly installed through Mukaiyama hydration. Then, the secondary alcohol at C-3 was formed upon reduction of the ketone with LiEt3BH at 65 °C. Upon heating, the β-hydroxysilane was eliminated to deliver the alkene at C-10 through Peterson olefination. Global MOM deprotection finally delivered (+)-principinol D in a total of 19 steps.
 | ||
| Scheme 20 The total synthesis of (+)-principinol D by the Newhouse group. | ||
6.2 Total syntheses of (±)-rhodomolleins XX and XXII (Ding)
In 2019, the Ding group reported the total syntheses of (±)-rhodomolleins XX and XXII, two highly oxidized grayanoids that were prepared in 23 and 22 steps, respectively.11b Notably, it features a Ti(III)-mediated reductive ring opening of a [2.2.1]-bicyclic ketoepoxide and a Dowd–Beckwith rearrangement to establish the bicyclo[3.2.1]octane ring system.The synthesis started with the 7-step preparation of β-ketoester 239 from 3-hydroxy-2-methoxybenzaldehyde 238 (Scheme 21). A Regitz diazo transfer delivered an α-diazo-β-ketoester, which then gave the cyclopropanation product 240 under the catalysis of a Cu(II) salen analogue. Treatment of 240 with TMSOTf and (TMSOCH2)2 resulted in protection of the C-2 ketone together with ring opening of the cyclopropane to deliver the styrene 241, which was subjected to selective epoxidation of the alkene and subsequent Pd-catalyzed hydrogenation to produce alcohol 242. The ethyl ester 242 was then converted to vinyl ketone 243 through subsequent β-keto phosphonate formation, followed by a HWE olefination. After the deprotection of silyl ether in 243 with TBAF, PIDA-mediated intramolecular ODI Diels–Alder cycloaddition smoothly occurred to afford desired tetracyclic product 244 in 70% yield. The SmI2-mediated reductive demethoxylation of 244 followed by selective epoxidation of the alkene with DMDO provided the exo-epoxide 245 as a single diastereomer in 83% yield over two steps. The key Ti(III)-mediated reductive epoxide opening then took place in a rather regioselective fashion thanks to the free alcohol at C-6. The resulting radical intermediate 246 then underwent a Dowd–Beckwith rearrangement reaction. In this case the subsequent selective cleavage of the C12–C16 bond was supposedly enabled by the titanoxy group at C-14, which might slow down the rate of radical quenching at C-13, resulting in the reversible formation of the cyclopropoxy radical 247 prior to the construction of the key bicyclo[3.2.1]octane ring in 248 through C13–C16 bond linkage.29
 | ||
| Scheme 21 The total synthesis of (±)-rhodomolleins XX and XXII by the Ding group. | ||
Moving on to the total syntheses of (±)-rhodomolleins XX and XXII, the acetylation of the two free hydroxyl groups in 248 and subsequent regioselective methylenation of the C-16 ketone produced enol 249. After treatment of 249 with PhSeCl and pyridine to afford enone 250 through α-selenylation and subsequent spontaneous elimination of the resultant selenide, regioselective Mukaiyama hydration was conducted to deliver triol 251. Both the hydroxyl groups at C-2 and C-14 were protected as TBS-ethers prior to Grignard addition to the C-10 ketone that delivered 252. Without further purification, the synthesis of (±)-rhodomollein XXII was achieved after acidic work-up in 77% yield over two steps. For the synthesis of (±)-rhodomollein XX, the intermediate 252 was converted into α-hydroxyketone 253 (d.r. > 20:1) through a MeReO3-catalyzed Rubottom oxidation. TBS group deprotection finally yielded (±)-rhodomollein XX in 64% yield over three steps.
6.3 Total synthesis of (−)-rhodomollanol A (Ding)
Rhodomollanol A is a highly oxidized hexacyclic diterpenoid that was isolated by Yao and co-workers from the leaves of Rhododendron molle G. in 2017.30 Unlike typical grayanoids, rhodomollanol A possesses an unusual [3,5,7,5,5,5] hexacyclic carbon framework, which features a 7-oxabicyclo[4.2.1]nonane core. Biosynthetically, this target molecule was proposed to be derived from grayananes via a set of biochemical transformations.31 In 2020, the Ding group reported the asymmetric total synthesis of (−)-rhodomollanol A.11c The key elements in their synthesis include an oxidative dearomatization-induced (5+2) cyclo-addition/pinacol-type 1,2-acyl migration cascade to construct the bicyclo[3.2.1]octane skeleton, a retro-Dieckmann fragmentation/vinylogous Dieckmann cyclization cascade to forge the bicyclo[3.3.0]octane ring, and a photo-Nazarov cyclization/intramolecular cycloetherification cascade to assemble the 7-oxabicyclo[4.2.1]nonane core structure of the natural product.The synthesis started with the 1,2-addition of the corresponding aryllithium reagent of 256 to the aldehyde 255 that was prepared in 7 steps from bicyclic enone 254 (Scheme 22). The resulting alcohols 257 underwent PDC oxidation and subsequent NaBH4 reduction to yield the desired epimer 258 at C-7. After THP protection of the alcohol 258, an ODI (5+2) cycloaddition/pinacol-type 1,2-acyl migration cascade was carried out to form tetracyclic intermediate 259 as a single diastereomer after THP deprotection with TFA. Treatment of 259 with TMSOTf and 1,2-bis(trimethylsiloxy)ethane allowed the formation of enone 260 through retro-Dieckmann fragmentation/vinylogous Dieckmann cyclization. With 260 in hand, the alcohol at C-7 was converted into xanthate ester, which was subjected to Barton–McCombie deoxygenation to furnish 261. DIBAL-H treatment of 261 to cleave the benzoyl protecting group and reduce the enone formed allylic alcohol 262, in which the hydroxyl group was then critical to direct the vanadyl acetylacetonate-catalyzed epoxidation through hydrogen bonding. The resulting β-epoxide 263 underwent (PhSeO)2O-mediated oxidation and Luche reduction to afford α-alcohol 265. As mentioned by the authors, TMS group protection of the alcohol was necessary to efficiently perform the subsequent photo-Nazarov cyclization/intramolecular ring-opening cycloetherification cascade reactions. As a result, the desired product 266 was ultimately formed in 56% yield.
 | ||
| Scheme 22 The total synthesis of (−)-rhodomollanol A by the Ding group. | ||
To continue the synthesis, ketone 267 was obtained through deprotonation of the enone 266 with dimsyl sodium32 and methylation with methyl iodide in extremely anhydrous solvent. The Δ (ref. 5 and 6) double bond in compound 267 underwent dihydroxylation with OsO4-pyridine complexes followed by hydrolysis of the osmate ester with NaHSO3. The resulting cis-diol was then efficiently protected as an acetonide 268 upon PPTs and 2-methoxypropene treatment. Inversion of the stereogenic center at C-1 was then performed through successive enone formation, conversion to hydrazine, stereoselective reduction and allylic diazene rearrangement to yield 269. The epoxy compound 269 was then reduced to alkene 270 upon Ti(III)-mediated reductive ring opening of the epoxide, Chugaev elimination, and acetal and acetonide deprotections. Finally, C-5 hydroxyl group-directed epoxidation through hydrogen bonding and reduction of the ketone at C-12 achieved the asymmetric total synthesis of rhodomollanol A.
7 Conclusion and outlook
As mentioned above, we have witnessed great progress in the total synthesis of ent-kaurane and related diterpenoids during the past seven years. Around 43 diverse target molecules have been synthesized during this period, which were demonstrated in 20 published articles. The total number of finished molecules is probably higher than what was done from 1962 to 2014. These achievements are highly reliant on using many concise retrosynthetic disconnections and innovative synthetic strategies, particularly those unprecedented C–C bond formation reactions such as novel radical-triggered cyclization, metal-mediated cascade cyclization, unusual Diels–Alder cycloaddition, and photoinduced skeletal rearrangement. These inventive ideas have been well demonstrated to overcome key challenges associated with the constitution of bicyclo[3.2.1]octane skeleton and its embedded ring system of ent-kaurane-type diterpenoids. It is interesting that some synthetic strategies are quite general to some extent for diversely synthesizing ent-kaurane and related diterpenoids, which are exemplified by the unified approach utilizing dearomatizative cycloaddition that the Ding group took to access three different kinds of target molecules. Other remarkable progress is that many of the reported synthetic routes to the same (or closely similar) target molecules have been regularly updated and simplified. The striking case is Baran's synthetic routes towards maoecrystal V only needing about 11 steps, which would have been unthinkable a decade ago. One can expect that in this field more recently isolated and highly unusual target molecules of this big family will be synthesized in the near future.Nevertheless, a large part of the already isolated and characterized ent-kauranoids still represent an unsolved challenge for the synthetic community, as they remain inaccessible by the reported strategies. Moreover, there is undoubtedly a plethora of ent-kauranoids that have been neither isolated nor characterized from their natural source. Consequently, new, more divergent and efficient strategies remain highly desirable, particularly unified strategies to efficiently access a subgroup of ent-kaurenes, which will help to close the gap between the number of ent-kaurenes isolated and those synthesized. Additionally, the biological activities of most isolated ent-kauranoids have not been systematically evaluated because sufficient material was not available. With the progress in total synthesis of ent-kauranoids, researchers should pay more attention to obtaining more material, particularly those with unique structural features, and testing the biological activities of the corresponding natural products. Such studies will be of benefit for detailed investigations of the mechanism of action and therapeutic potential of ent-kauranoids.
8 Conflicts of interest
There are no conflicts to declare.9 Acknowledgements
Financial support of this research from Chinese Academy of Sciences (supported by the Strategic Priority Research Program, grants XDB20020200 and QYZDJ-SSW-SLH029) and the National Natural Science Foundation of China (grants 21621002, 21831009 and 21991110) is acknowledged.10 References
- P. Quitt, E. Mosettig, R. C. Cambie, P. S. Rutledge and L. H. Briggs, J. Am. Chem. Soc., 1961, 83, 3720 CrossRef.
- (a) H.-D. Sun, S.-X. Huang and Q.-B. Han, Nat. Prod. Rep., 2006, 23, 673 RSC; (b) M. Liu, W.-G. Wang, H.-D. Sun and J.-X. Pu, Nat. Prod. Rep., 2017, 34, 1090 RSC; (c) P. A. García, A. B. Oliveira and R. Batista, Molecules, 2007, 12, 455 CrossRef PubMed.
- (a) P. M. Dewick, Medicinal Natural Products, A Biosynthetic Approach, Wiley, 2008, p. 17 Search PubMed; (b) J. MacMillan and M. H. Beale, in Diterpene Biosynthesis in Comprehensive Natural Product Chemistry, ed. D. E. Cane, Pergamon, 2000 Search PubMed; (c) Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2010, 132, 5375 CrossRef CAS PubMed; (d) D. J. Tantillo, Chem. Soc. Rev., 2010, 39, 2847 RSC.
- For the reviews of synthesis of ent-kauranoids, see: (a) K. E. Lazarski, B. J. Moritz and R. J. Thomson, Angew. Chem., Int. Ed., 2014, 53, 10588 CrossRef CAS; (b) L. Zhu, S.-H. Huang, J. Yu and R. Hong, Tetrahedron Lett., 2015, 56, 23 CrossRef CAS; (c) M.-J. Du and X.-G. Lei, Youji Huaxue, 2015, 35, 2447 CrossRef CAS; (d) P. S. Riehl, Y. C. Deporre, A. M. Armaly, E. J. Groso and C. S. Schindler, Tetrahedron, 2015, 71, 6629 CrossRef CAS; (e) W. Li, J.-J. Wang and D.-W. Ma, Progress in Chemistry, 2019, 31, 1460 Search PubMed.
- For selected synthesis of intact ent-kauranoids between 1962 and 2014, see: (a) R. A. Bell, R. E. Ireland and R. A. Partyka, J. Org. Chem., 1962, 27, 3741 CrossRef CAS; (b) S. Masamune, J. Am. Chem. Soc., 1964, 86, 288 CrossRef CAS; (c) R. E. Ireland and L. M. Mander, Tetrahedron Lett., 1965, 6, 2627 CrossRef; (d) R. B. Turner, K. H. Ganshirt, P. E. Shaw and J. D. Tauber, J. Am. Chem. Soc., 1966, 88, 1776 CrossRef CAS; (e) R. F. Church, R. E. Ireland and J. A. Marshall, J. Org. Chem., 1966, 31, 2526 CrossRef CAS PubMed; (f) K. Mori, Y. Nakahara and M. Matsui, Tetrahedron, 1972, 28, 3217 CrossRef CAS; (g) F. E. Ziegler and J. A. Kloek, Tetrahedron, 1977, 33, 373 CrossRef CAS; (h) E. J. Corey, G. Wess, Y. B. Xiang and A. K. Singh, J. Am. Chem. Soc., 1987, 109, 4717 CrossRef CAS; (i) E. J. Corey and K. Liu, J. Am. Chem. Soc., 1997, 119, 9929 CrossRef CAS; (j) E. C. Cherney, J. C. Green and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 9019 CrossRef CAS; (k) J. T. S. Yeoman, V. W. Mak and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 11764 CrossRef CAS; (l) L. Zhu, J. Luo and R. Hong, Org. Lett., 2014, 16, 2162 CrossRef CAS PubMed; (m) J. T. S. Yeoman, J. Y. Cha, V. W. Mak and S. E. Reisman, Tetrahedron, 2014, 70, 4070 CrossRef CAS.
- For selected syntheses of oxidatively cleaved or rearranged ent-kauranoids between 1962 and 2014, see: (a) E. Fujita, M. Shibuya, S. Nakamura, Y. Okada and T. Fujita, J. Chem. Soc., Perkin Trans. 1, 1974, 165 RSC; (b) L. A. Paquette, D. Backhaus, R. Braun, T. L. Underiner and K. Fuchs, J. Am. Chem. Soc., 1997, 119, 9662 CrossRef CAS; (c) J. Y. Cha, J. T. S. Yeoman and S. E. Reisman, J. Am. Chem. Soc., 2011, 133, 14964 CrossRef CAS; (d) J. T. S. Yeoman, V. W. Mak and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 11764 CrossRef CAS; (e) Z. Pan, C. Zheng, H. Wang, Y. Chen, Y. Li, B. Cheng and H. Zhai, Org. Lett., 2014, 16, 216 CrossRef CAS PubMed; (f) B. J. Moritz, D. J. Mack, L. Tong and R. J. Thomson, Angew. Chem., Int. Ed., 2014, 53, 2988 CrossRef CAS. Synthesis of maoecrystal V: (g) J. Gong, G. Lin, W. Sun, C.-C. Li and Z. Yang, J. Am. Chem. Soc., 2010, 132, 16745 CrossRef CAS; (h) F. Peng and S. J. Danishefsky, J. Am. Chem. Soc., 2012, 134, 18860 CrossRef CAS PubMed; (i) P. Lu, Z. Gu and A. Zakarian, J. Am. Chem. Soc., 2013, 135, 14552 CrossRef CAS PubMed; (j) P. Lu, A. Mailyan, Z. Gu, D. M. Guptill, H. Wang, H. M. L. Davies and A. Zakarian, J. Am. Chem. Soc., 2014, 136, 17738 CrossRef CAS; (k) C. Zheng, I. Dubovyk, K. E. Lazarski and R. J. Thomson, J. Am. Chem. Soc., 2014, 136, 17750 CrossRef CAS PubMed.
- (a) X. Zhao, W. Li, J. Wang and D. Ma, J. Am. Chem. Soc., 2017, 139, 2932 CrossRef CAS; (b) C. He, J.-L. Hu, Y.-B. Wu and H.-F. Ding, J. Am. Chem. Soc., 2017, 139, 6098 CrossRef CAS PubMed; (c) J. Wang and D. Ma, Angew. Chem., Int. Ed., 2019, 58, 15731 CrossRef CAS PubMed; (d) J. Guo, B. Li, W. Ma, M. Pitchakuntla and Y. Jia, Angew. Chem., Int. Ed., 2020, 59, 15195 CrossRef CAS; (e) Z. Xu, Y. Zong, Y. Qiao, J. Zhang, X. Liu, M. Zhu, Y. Xu, H. Zheng, L. Fang, X. Wang and H. Lou, Angew. Chem., Int. Ed., 2020, 59, 19919 CrossRef CAS PubMed; (f) J. Wang, B. Hong, D. Hu, Y. Kadonaga, R. Tang and X. Lei, J. Am. Chem. Soc., 2020, 142, 2238 CrossRef CAS PubMed.
- (a) F. Su, Y.-D. Lu, L.-R. Kong, J.-J. Liu and T.-P. Luo, Angew. Chem., Int. Ed., 2018, 57, 760 CrossRef CAS PubMed; (b) L.-Z. Zhu, W.-J. Ma, M.-X. Zhang, M. M.-L. Lee, W.-Y. Wong, B. D. Chan, Q.-Q. Yang, W.-T. Wong, W. C.-S. Tai and C.-S. Lee, Nat. Commun., 2018, 9, 1283 CrossRef PubMed; (c) L. Kong, F. Su, H. Yu, Z. Jiang, Y. Lu and T. Luo, J. Am. Chem. Soc., 2019, 141, 20048 CrossRef CAS PubMed.
- (a) Z. Lv, B. Chen, C. Zhang and G. Liang, Chem.–Eur. J., 2018, 24, 9773 CrossRef CAS PubMed; (b) S. Pan, S. Chen and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 6333 CrossRef CAS PubMed; (c) J.-P. Zhang, Z.-J. Li, J.-M. Zhuo, Y. Cui, T. Han and C. Li, J. Am. Chem. Soc., 2019, 141, 8372 CrossRef CAS.
- (a) W. Liu, H. Li, P.-J. Cai, Z. Wang, Z.-X. Yu and X. Lei, Angew. Chem., Int. Ed., 2016, 55, 3112 CrossRef CAS PubMed; (b) A. Cernijenko, R. Risgaard and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 9425 CrossRef CAS PubMed; (c) J. Wu, Y. Kadonaga, B. Hong, J. Wang and X. Lei, Angew. Chem., Int. Ed., 2019, 58, 10879 CrossRef CAS PubMed; (d) B. Hong, W. Liu, J. Wang, J. Wu, Y. Kadonaga, P.-J. Cai, H.-X. Lou, Z.-X. Yu, H. Li and X. Lei, Chem, 2019, 5, 1671 CrossRef CAS; (e) Y. Que, H. Shao, H. He and S. Gao, Angew. Chem., Int. Ed., 2020, 59, 7444 CrossRef CAS.
- (a) A. Turlik, Y.-F. Chen, A. C. Scruse and T. R. Newhouse, J. Am. Chem. Soc., 2019, 141, 8088 CrossRef CAS PubMed; (b) K. Yu, Z.-N. Yang, C.-H. Liu, S.-Q. Wu, X. Hong, X.-L. Zhao and H.-F. Ding, Angew. Chem., Int. Ed., 2019, 58, 8556 CrossRef CAS PubMed; (c) J. Gao, P. Rao, K. Xu, S. Wang, Y. Wu, C. He and H. Ding, J. Am. Chem. Soc., 2020, 142, 4592 CrossRef CAS PubMed.
- A. Y. Hong and B. M. Stoltz, Eur. J. Org. Chem., 2013, 2745 CrossRef CAS PubMed.
- (a) D. Hoppe and T. Hense, Angew. Chem., Int. Ed., 1997, 36, 2282 CrossRef CAS; (b) D. Hoppe, Synthesis, 2009, 2009, 43 CrossRef.
- S. Chow, C. Kreß, N. Albæk, C. Jessen and C. M. Williams, Org. Lett., 2011, 13, 5286 CrossRef CAS.
- L. Jiao, M. Lin, L.-G. Zuo and Z.-X. Yu, Org. Lett., 2010, 12, 2528 CrossRef CAS.
- (a) G. Stork and R. Mook Jr, J. Am. Chem. Soc., 1987, 109, 2829 CrossRef CAS; (b) M. Toyota, M. Yokota and M. Ihara, Tetrahedron Lett., 1999, 40, 1551 CrossRef CAS; (c) M. Toyota, M. Yokota and M. Ihara, J. Am. Chem. Soc., 2001, 123, 1856 CrossRef CAS PubMed; (d) M. Toyota, K. Matsuura, M. Yokota, M. Yonaga, H. Sugimoto and M. Ihara, Chem. Pharm. Bull., 2004, 52, 1153 CrossRef CAS PubMed.
- D. Huang, Y. Zhao and T. R. Newhouse, Org. Lett., 2018, 20, 684 CrossRef CAS PubMed.
- K. Takeda, A. Akiyama, H. Nakamura, S. Takizawa, Y. Mizuno, H. Takayanagi and Y. Harigaya, Synthesis, 1994, 10, 1063 CrossRef.
- T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. Novak, J. Z. Zhong, L. R. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 260 CrossRef CAS PubMed.
- H. Ren, A. Krasovskiy and P. Knochel, Org. Lett., 2004, 6, 4215 CrossRef CAS PubMed.
- R. M. Maksymowicz, P. M. C. Roth and S. P. Fletcher, Nat. Chem., 2012, 4, 649 CrossRef CAS PubMed.
- S. Rendler and D. W. C. Macmillan, J. Am. Chem. Soc., 2010, 132, 5027 CrossRef CAS.
- N. P. Sahu, K. Koike, S. Banerjee, B. Achari, Z. Jia and T. Nikaido, Tetrahedron Lett., 1997, 38, 8405 CrossRef CAS.
- (a) D. E. Ward and M. S. Souweha, Org. Lett., 2005, 7, 3533 CrossRef CAS PubMed; (b) J. Ishihara, S. Nakadachi, Y. Watanabe and S. Hatakeyama, J. Org. Chem., 2015, 80, 2037 CrossRef CAS.
- (a) S. Cai, Z. Xiao, Y. Shi and S. Gao, Chem.–Eur. J., 2014, 20, 8677 CrossRef CAS PubMed; (b) S. Gao, Q. Wang and C. Chen, J. Am. Chem. Soc., 2009, 131, 1410 CrossRef CAS PubMed; (c) F. Churruca, M. Fousteris, Y. Ishikawa, M. v. W. Rekowski, C. Hounsou, T. Surrey and A. Giannis, Org. Lett., 2010, 12, 2096 CrossRef CAS PubMed.
- (a) L. Wang and G. Qin, Nat. Prod. Res. Dev., 1997, 9, 82 CAS; (b) Y. Li, Y.-B. Liu and S.-S. Yu, Phytochem. Rev., 2013, 12, 305 CrossRef CAS; (c) C.-H. Li, J.-Y. Zhang, X.-Y. Zhang, S.-H. Li and J.-M. Gao, Eur. J. Med. Chem., 2019, 166, 400 CrossRef CAS.
- (a) N. Hamanaka and T. Matsumoto, Tetrahedron Lett., 1972, 13, 3087 CrossRef; (b) S. Gasa, N. Hamanaka, S. Matsunaga, T. Okuno, N. Takeda and T. Matsumoto, Tetrahedron Lett., 1976, 17, 553 CrossRef; (c) T. Kan, S. Hosokawa, S. Nara, M. Oikawa, S. Ito, F. Matsuda and H. Shirahama, J. Org. Chem., 1994, 59, 5532 CrossRef CAS.
- (a) J. M. Mayer, Annu. Rev. Phys. Chem., 2004, 55, 363 CrossRef CAS PubMed; (b) T. V. Chciuk, W. R. Anderson Jr and R. A. Flowers, J. Am. Chem. Soc., 2016, 138, 8738 CrossRef CAS PubMed; (c) S. S. Kolmar and J. M. Mayer, J. Am. Chem. Soc., 2017, 139, 10687 CrossRef CAS PubMed.
- P. Dowd and W. Zhang, Chem. Rev., 1993, 93, 2091 CrossRef CAS.
- J. Zhou, G. Zhan, H. Zhang, Q. Zhang, Y. Li, Y. Xue and G. Yao, Org. Lett., 2017, 19, 3935 CrossRef CAS PubMed.
- T. Masutani, M. Hamada, E. Kawano, J. Iwasa, Z. Kumazawa and H. Ueda, Agric. Biol. Chem., 1981, 45, 1281 CAS.
- G. Stork, D. Niu, A. Fujimoto, E. R. Koft, J. M. Balkovec, J. R. Tata and G. R. Dake, J. Am. Chem. Soc., 2001, 123, 3239 CrossRef CAS PubMed.
Footnote |
| † Both authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2022 |