
Recent advances in the biosynthesis strategies of nitrogen heterocyclic natural products
Bo
Gao†
 a,
Bo
Yang†
b,
Xudong
Feng
*a and
Chun
Li
*abc
a,
Bo
Yang†
b,
Xudong
Feng
*a and
Chun
Li
*abc
aKey Laboratory of Medical Molecule Science and Pharmaceutics Engineering, Ministry of Industry and Information Technology, Institute of Biochemical Engineering, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing, China. E-mail: xd.feng@bit.edu.cn; lichun@tsinghua.edu.cn
bSynBio Research Platform, Collaborative Innovation Center of Chemical Science and Engineering, Key Laboratory of Systems Bioengineering, Ministry of Education, School of Chemical Engineering and Technology, Tianjin University, Tianjin, China
cKey Laboratory for Industrial Biocatalysis, Ministry of Education, Department of Chemical Engineering, Tsinghua University, Beijing, China
First published on 10th August 2021
Abstract
Covering: 2015 to 2020
Nitrogen heterocyclic natural products (NHNPs) are primary or secondary metabolites containing nitrogen heterocyclic (N-heterocyclic) skeletons. Due to the existence of the N-heterocyclic structure, NHNPs exhibit various bioactivities such as anticancer and antibacterial, which makes them widely used in medicines, pesticides, and food additives. However, the low content of these NHNPs in native organisms severely restricts their commercial application. Although a variety of NHNPs have been produced through extraction or chemical synthesis strategies, these methods suffer from several problems. The development of biotechnology provides new options for the production of NHNPs. This review introduces the recent progress of two strategies for the biosynthesis of NHNPs: enzymatic biosynthesis and microbial cell factory. In the enzymatic biosynthesis part, the recent progress in the mining of enzymes that synthesize N-heterocyclic skeletons (e.g., pyrrole, piperidine, diketopiperazine, and isoquinoline), the engineering of tailoring enzymes, and enzyme cascades constructed to synthesize NHNPs are discussed. In the microbial cell factory part, with tropane alkaloids (TAs) and tetrahydroisoquinoline (THIQ) alkaloids as the representative compounds, the strategies of unraveling unknown natural biosynthesis pathways of NHNPs in plants are summarized, and various metabolic engineering strategies to enhance their production in microbes are introduced. Ultimately, future perspectives for accelerating the biosynthesis of NHNPs are discussed.
 Bo Gao | Bo Gao is a master's student at the Beijing Institute of Technology, mentored by Prof. Chun Li. He received his BS degree from the Beijing Institute of Technology in 2018. His research is focused on enzyme engineering for natural product biosynthesis. |
 Bo Yang | Bo Yang is currently a PhD student under the supervision of Prof. Chun Li at Tianjin University. Her research is focused on the efficient production of natural products in the microbial cell factory through metabolic engineering and synthetic biology. |
 Xudong Feng | Dr Xudong Feng is an assistant professor at the Beijing Institute of Technology. He obtained his PhD degree from the University of Auckland in 2014. His research interest is focused on the mining and engineering of key enzymes in natural products synthesis. He has published 50 peer-reviewed research papers in this field. |
 Chun Li | Dr Chun Li is a professor at the Tsinghua University and Distinguished Young Scholar of Natural Science Foundation of China. He received his PhD degree in Biochemical Engineering from the Tianjin University in 2001. He is a senior expert on synthetic biology, biocatalysis, and enzyme engineering, with more than 15 years of research experience. Since the past ten years, his research has been focused on the biosynthesis of plant natural products in microorganisms and the development of strategies and tools for increasing the productivity of plant natural products and the robustness of microbes. He has published 307 peer-reviewed research papers. |
1. Introduction
Nitrogen heterocyclic natural products (NHNPs) are primary or secondary metabolites containing nitrogen heterocyclic (N-heterocyclic) skeletons, which have a wide range of biological activities. Among them, the primary metabolites of NHNPs include purines, pyrimidines, three N-heterocyclic amino acids (tryptophan, proline and histidine), and some members of the vitamin B family. They play important roles in storing genetic information, supplying energy, and participating in various important metabolic pathways of organisms.1–3 The secondary metabolites of NHNPs include alkaloids, pseudoalkaloids, biogenic amine derivatives, and β-lactam antibiotics. Most alkaloids and pseudoalkaloids contain indole, isoquinoline, pyrrole, pyridine, or other nitrogen-containing heterocyclic skeletons, which belong to NHNPs.4–12 A few alkaloids and pseudoalkaloids without nitrogen atoms in the heterocyclic ring do not belong to NHNPs. N-heterocyclic alkaloids and pseudoalkaloids have been widely used for developing new medicines and pesticides due to their remarkable anti-tumor, anti-bacterial, anti-fungal, insect toxicity, and other pharmacological activities. For example, morphine is widely used as an anesthesia due to its analgesic effects;13 scopolamine is used for treating gastric ulcers due to its effect of relieving gastrointestinal cramps;14 camptothecin is used as a chemotherapy drug for leukemia and gastric cancer.15Nitrogen has a significant effect on the activity of the heterocyclic skeleton. On the one hand, nitrogen has a lone pair of electrons in the heterocyclic ring, which shows weak basicity. On the other hand, polar N–H bond exhibits weak acidity. These characteristics of the nitrogen element enable the N-heterocyclic skeleton to bind a variety of bioactive molecules, which improves the bioavailability of NHNPs and makes them exhibit excellent physiological activities. For example, five-membered heterocycles such as pyrrole and pyrazole usually form electron-rich compounds due to the lone pair of electrons of the nitrogen atom participating in the cyclic conjugated π bond and are prone to electrophilic reactions. The lone pair of electrons of six-membered heterocyclic nitrogen atoms such as pyridine and quinoline do not participate in conjugation and usually form electron-deficient compounds, and are prone to nucleophilic reactions. The combined action of the N-heterocyclic skeleton and the surrounding modification groups determine the biological activity of NHNPs. For example, indole is an electron-rich aromatic N-heterocycle. Many natural products containing indole rings are both weakly basic and weakly acidic, and can form π–π interactions with various active substances or receptors. Due to the unsaturated bond, the indole nucleus also has potential reducibility.16 A variety of natural products can be obtained by introducing substituents of different properties into the indole ring, which can exhibit various physiological activities such as anti-cancer, antibacterial, anti-oxidant, anti-viral, and anti-convulsant.17 Diketopiperazine is an N-heterocyclic compound formed by the dehydration condensation of two amino acids. The diketopiperazine skeleton contains two amide bonds; thus, there are two hydrogen bond acceptors and hydrogen bond donor sites, which allow them to bind to a variety of enzymes and acceptors. Adding modified groups to the diketopiperazine skeleton can form many natural products with higher receptor binding ability, inhibiting a variety of target proteins and produce a variety of pharmacological activities such as anticancer, anti-inflammatory, and anti-cardiovascular diseases.18
At present, more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 NHNPs have been reported.19 In the past two centuries, researchers have been continuously discovering new NHNPs and exploring their potential values, and remarkable progress has been achieved. However, the low native content of NHNPs severely restricts the industrial applications.5,10 The traditional strategy is to directly extract NHNPs from plants. Several high-value NHNPs such as vincristine and morphine, are still obtained through large-scale plant cultivation, which exhibit many disadvantages. Firstly, most plants have a long growth cycle; thus, they are easily affected by environmental factors, such as insects, pests, and climate. Secondly, the extraction of active ingredients from the plants not only causes environmental pollution but also faces complex separation and purification problems. For microbe-derived NHNPs, indistinct biosynthetic pathways have hindered their industrial production.
000 NHNPs have been reported.19 In the past two centuries, researchers have been continuously discovering new NHNPs and exploring their potential values, and remarkable progress has been achieved. However, the low native content of NHNPs severely restricts the industrial applications.5,10 The traditional strategy is to directly extract NHNPs from plants. Several high-value NHNPs such as vincristine and morphine, are still obtained through large-scale plant cultivation, which exhibit many disadvantages. Firstly, most plants have a long growth cycle; thus, they are easily affected by environmental factors, such as insects, pests, and climate. Secondly, the extraction of active ingredients from the plants not only causes environmental pollution but also faces complex separation and purification problems. For microbe-derived NHNPs, indistinct biosynthetic pathways have hindered their industrial production.
Researchers are committed to the artificial synthesis of NHNPs to overcome the shortcomings of the traditional methods. Chemical synthesis is currently the most common method of obtaining NHNPs since it can provide different synthetic routes, obtain multiple analogs, and screen the best products.20–24 However, for NHNPs with complicated structures (e.g., opioid alkaloids and terpenoid alkaloid), chemical synthesis has severe disadvantages, including multiple steps, low yield, by-product production, as well as poor regio- and stereo-specificity. In addition, similar to plant extraction, chemical synthesis can also cause serious environmental pollution.
For the development of biotechnology, biosynthetic strategies provide a promising option for obtaining NHNPs, especially those with complicated structures. The biosynthesis of NHNPs has the advantages of high efficiency, rigid specificity, and environment friendliness, which is attracting increasing attention from researchers. With the aid of sequencing technology, transcriptomics, and proteomics, researchers have elucidated the synthetic pathways of multiple NHNPs in native hosts.9,25 Generally, N-heterocyclic structures are enzymatically synthesized by condensing amines or imine precursors, which were then modified to form NHNPs after oxidation, reduction, methylation, amination, acetylation, or glycosylation. Initially, plant cell engineering was used to produce NHNPs. However, the complex metabolic network of the plant cells and the interaction among the metabolic pathways severely restrict the synthesis of target NHNPs.26 The emergence of DNA synthesis technology has provided a breakthrough in the biosynthesis of NHNPs. Researchers can conveniently reconstruct metabolic networks in easily-cultivable model microorganisms or use recombinant enzymes to synthesize NHNPs. Due to their high chemo-, regio-, and stereoselectivity toward natural structures, enzymatic biosynthesis and microbial cell factories are the most widely used biosynthetic strategies for obtaining NHNPs. Moreover, the rapid development of metabolic engineering, protein engineering, and synthetic biology have significantly improved the yield of NHNPs.8,27
This review introduces the progress of biosynthesis of NHNPs, mainly in the recent five years. Secondary metabolites containing N-heterocyclic structures were selected as the object of review. For the enzymatic biosynthesis part, the important enzymes for producing typical NHNP skeletons are classified as three major groups including polyketide synthase/non-ribosomal peptide synthase (pyrrole/piperidine), cyclodipeptide synthases (diketopiperazine), and Pictet–Spenglerase (isoquinoline/β-carboline). Their mechanism and mining strategies are summarized. Subsequently, post-modification enzymes add various functional groups to the above skeletons to form valuable NHNPs. The strategies developed for engineering post-modification enzymes to improve their stability, activity, and specificity are also elucidated. The construction of a multi-enzyme cascade for the biosynthesis of precursors of NHNPs, including isoquinoline, pyrrole, and piperidine derivatives, has also been introduced. Tropane alkaloids (TAs) and tetrahydroisoquinoline (THIQ) alkaloids are selected as the representative compounds in the section of NHNPs production in microbial cell factories. The recent strategies developed for the decryption of the biosynthesis pathway of NHNPs are summarized. Then, the construction and regulation strategy of the NHNPs biosynthesis in microbial cell factories are introduced. This content can provide guidance for researchers who are engaged in deciphering the unknown synthetic pathways of natural products and optimizing the synthetic efficiency of the cell factories. Finally, the challenges in the biosynthesis of NHNPs have been discussed.
2. Enzymatic biosynthesis of NHNPs
The biosynthesis of NHNPs depends on the participation of multiple enzymes. Enzymatic biosynthesis has great potential for NHNPs production, especially for those with complex structures. Enzymatic biosynthesis in vitro allows the precise control of reaction conditions such as temperature and pH, which enables the effective synthesis of NHNPs without being affected by the inherent metabolic network of the host. This section introduces the progress of enzymatic biosynthesis of NHNPs in vitro. First, the key enzymes are summarized for the synthesis of N-heterocyclic skeletons, which provide the basic structure for NHNPs. Subsequently, the tailoring enzymes for introducing the functional groups into the N-heterocyclic skeleton to provide activity toward NHNPs are discussed. Recently, dramatic progress has been made to improve the properties of tailoring enzymes by protein engineering; thus, this part is especially emphasized. The key enzymes for forming skeletons can be combined with different tailoring enzymes to construct an in vitro cascade reaction system in order to produce diverse NHNPs and their derivatives, and this content is introduced at the end of this section (Fig. 1). | ||
| Fig. 1 The schematic diagram of the enzymatic biosynthesis of NHNPs. | ||
2.1 Enzymatic biosynthesis of N-heterocyclic skeletons of NHNPs
The biosynthesis of NHNPs comprises two steps: the formation of N-heterocyclic skeletons and post-modification. In nature, the key enzymes for the synthesis of N-heterocyclic skeleton of secondary metabolites mainly include polyketide synthase/non-ribosomal peptide synthase (PKS/NRPS), cyclodipeptide synthase (CDPS), and Pictet–Spenglerase (PSase). These enzymes can synthesize common five-membered and six-membered N-heterocyclic skeletons, such as pyrrole, piperidine, and piperazine (Fig. 2). Then, these skeletons are decorated with various functional groups by post-modification enzymes, such as cytochrome P450, decarboxylase, methyltransferase, acyltransferase, and prenyltransferase to form NHNPs with diverse structures.28–30 The discovery of enzymes involved in NHNPs biosynthesis provides availability for industrial applications. Recently, great progress has been made in terms of mining the enzymes that can synthesize N-heterocyclic skeletons. Thus, we reviewed the latest progress in the mining and in vitro characterization of these enzymes in this part.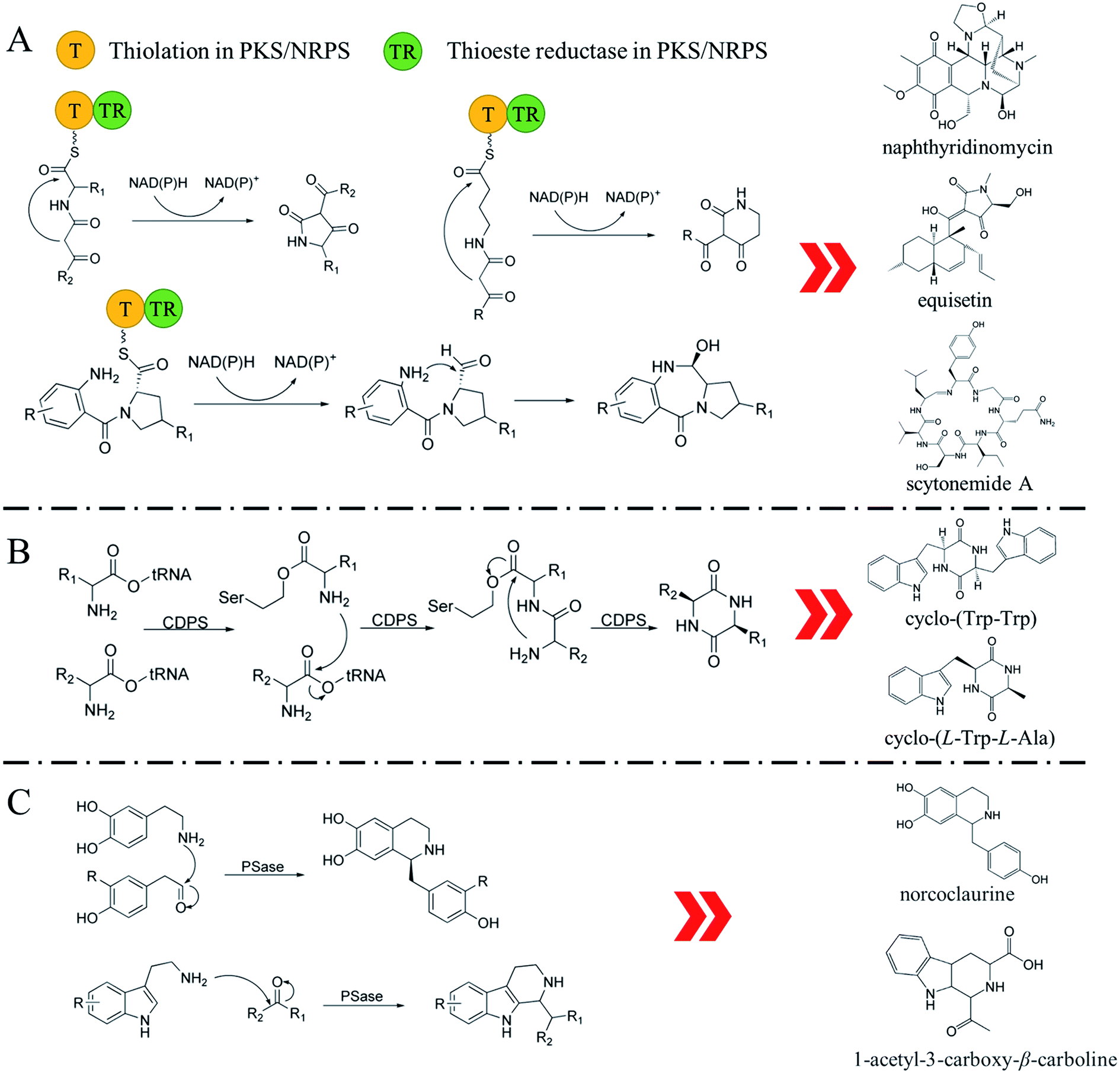 | ||
| Fig. 2 The reaction mechanism of key enzymes that synthesize N-heterocyclic skeletons. (A) PKS/NRPS. (B) CDPS. (C) PSase. | ||
 | ||
| Fig. 3 Typical N-heterocyclic skeletons synthesized by TR. | ||
 | ||
| Fig. 4 Typical N-heterocyclic skeletons synthesized by CDPS. (A) BcmA. (B) NozA and NcdA. | ||
 | ||
| Fig. 5 Typical N-heterocyclic skeletons synthesized by PSase. (A) McbB and NscB. (B) StnK2. | ||
 | ||
| Fig. 6 Typical N-heterocyclic skeletons synthesized by other key enzymes. (A) CnsA. (B) KtzT. (C) ω-Transaminase. (D) IRED. | ||
2.2 Tuning the tailing enzyme properties for NHNPs skeleton decoration by protein engineering
The multiple structures of NHNPs provide diverse bioactivities, which depend on the post-modification of the N-heterocyclic skeletons. However, post-modifications are usually the rate-limiting steps in NHNPs biosynthesis. To solve this problem, protein engineering strategies are widely used (Fig. 7). Enzymes with high activity, stability, substrate promiscuity, and selectivity can be obtained by mutating critical residues or fragments.55–57 Directed evolution and rational design strategies are widely used in the construction of engineered enzymes. The mutant library was generated by random or rational mutation, and then the mutants in the library were screened under specific pressure. Mutants with improved properties were utilized as the templates for the next round of mutations until the desired mutants were obtained.58,59 Protein engineering of post-modification enzymes for NHNP biosynthesis has made significant progress recently and this section mainly introduces the latest progress of this point. | ||
| Fig. 7 Construction of engineered enzymes to synthesize NHNPs. (A) Process of protein engineering. (B) Influence of protein engineering on the enzyme properties. | ||
000 clones. They screened sixteen mutants with increased activity in the library, which contained five amino acid residue mutations. Single-site mutations were made to these five residues, and two key residues S14 and K97 that affected the enzyme activity were determined. Saturated mutations were then performed on these two residues, and a double mutant S14P/K97A with 4.4-fold increased hydroxylation activity was obtained. However, random mutation requires the screening of a large number of mutants, which reduces the efficiency. Some residues on the substrate binding pocket directly affect the activity; thus, saturation mutation on these residues can obtain mutants with increased activity efficiently. Fan et al.63 performed saturation mutation on the substrate binding residue R224 of the cyclic dipeptide C4-prenyltransferase from Aspergillus fumigatus. R244 was proposed to bind to the side chain of substrate and has been verified to affect the activity. Thirteen mutants at R244 showed up to 76-fold higher turnover number toward seven cyclic dipeptides. Brockmeyer et al.64 mutated two residues, T82 and Y196, in the substrate binding pocket P1 of a CDPS from Nocardiopsis prasina. The mutant T82V/Y196F showed 8-fold increased activity in the synthesis of cyclo-(L-Tyr-L-Tyr) and 10-fold increased activity in the synthesis of cyclo-(L-Tyr–L-Phe).
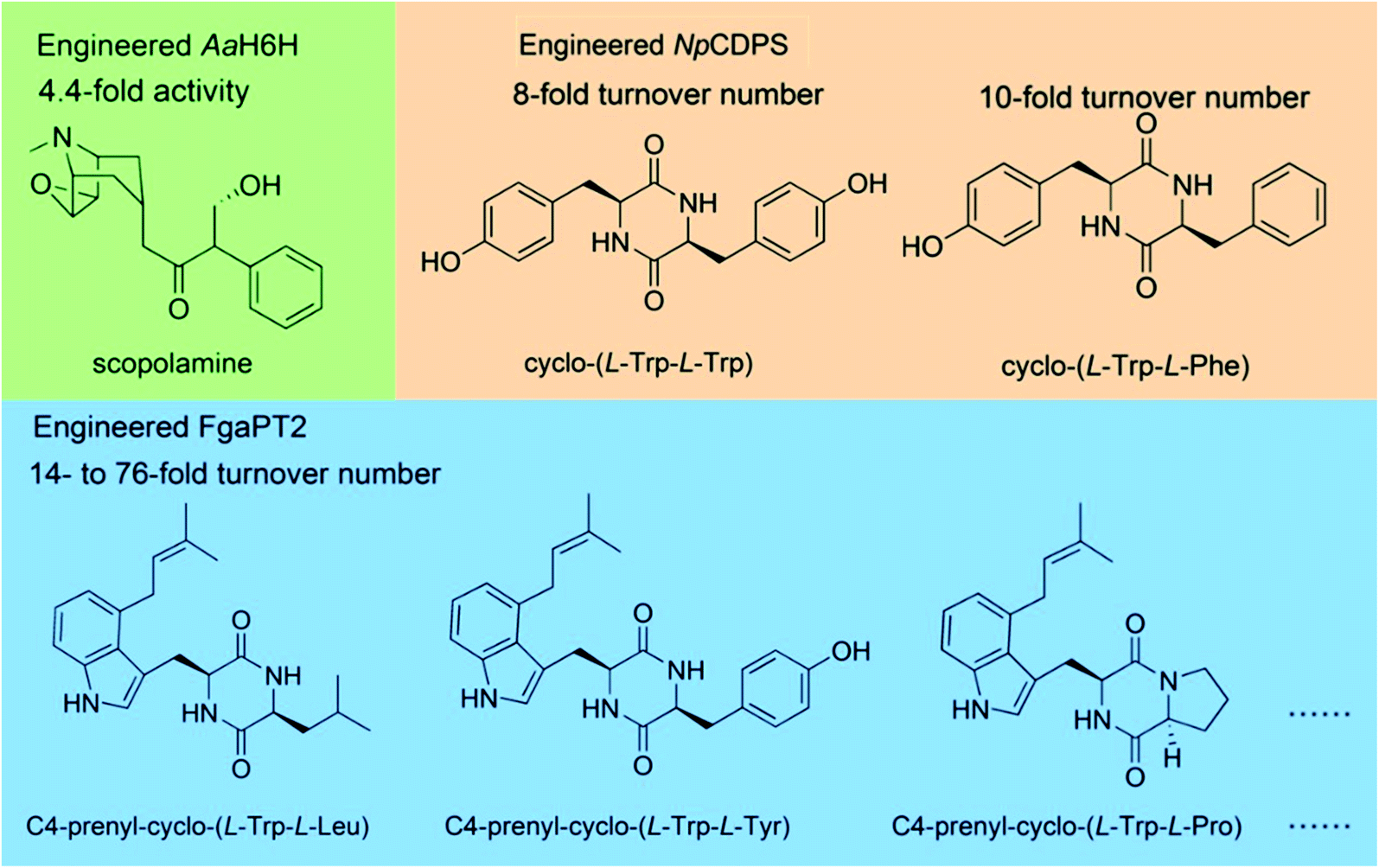 | ||
| Fig. 8 Engineering the enzyme activity to synthesize NHNPs and their derivatives. | ||
 | ||
| Fig. 9 Engineering the substrate promiscuity to synthesize NHNPs and their derivatives. (A) TrpB. (B) wi-WelO15. (C) S9OMT. (D) RebH. | ||
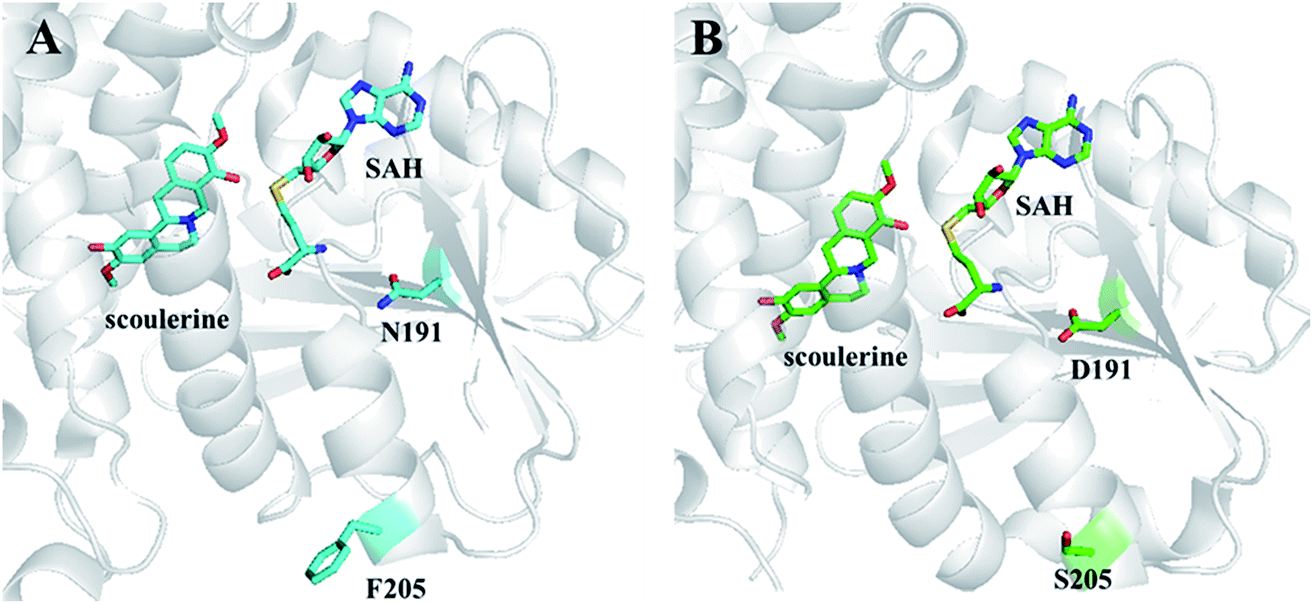 | ||
| Fig. 10 Crystal structures of S9OMT wt (A) and S9OMT DS (B). Substrates and mutated residues are indicated by sticks. The images were reproduced from ref. 71. | ||
The synthesis of NHNPs often involves the cooperation of multiple cascade enzymes, and the overall rate depends on the activity of the rate-limiting enzymes. Usually, rate-limiting enzymes with low activity reduce the supply of precursors, leading to the accumulation of intermediates and the reduction of the target NHNPs. Improving the substrate promiscuity can avoid the accumulation of the intermediate products to improve the overall rate. Halotryptamine is a common precursor for the biosynthesis of halogen-containing indole alkaloids. Tryptophan halogenase was introduced to form halotryptophan from L-Trp in plants, and then halotryptophan was transformed into halotryptamine by endogenous tryptophan decarboxylase (TDC). However, TDC showed low activity toward halotryptophan, resulting in reduced halotryptamine production. To solve this problem, Glenn et al.72 selected a 7-L-tryptophan halogenase (RebH) from actinomycetes and converted it into 7-tryptamine halogenase. Based on the crystal structure of RebH, they mutated several residues on the active site. Among them, RebH Y455W catalyzed the 7-chlorination of tryptamine instead of the natural substrate tryptophan with higher selectivity. The activity of RebH Y455W toward tryptophan was reduced by 10 times, while the activity toward tryptamine was increased by about 3 times. The change in the substrate specificity of RebH Y455W was interpreted as preventing tryptophan from binding to the active site (Fig. 9D). RebH Y455W was introduced to Catharanthus roseus to produce high value-added 12-chloro-19,20-dihydroakuammicine without accumulating 7-chlorotryptophan.
 | ||
| Fig. 11 Engineering the reaction specificity to synthesize NHNPs and their derivatives. (A) xcTDO. (B) CYP102A1. (C) DHAPPS. (D) Prenyltransferase. | ||
2.3 Multi-enzyme cascade strategy for the biosynthesis of NHNPs
Multi-enzyme cascade systems can convert commercially available raw materials into high-value NHNPs with ideal stereospecificity, which are difficult to achieve by chemical strategies. These multi-enzyme cascade systems usually contain three types of enzymes, namely, precursor synthesis enzymes, N-heterocycle synthesis enzymes, and post-modification enzymes. Multi-enzyme cascade systems can combine a variety of enzymes that cannot be combined by natural biosynthetic pathways. Multiple enzymatic reactions can be carried out simultaneously in the same reactor. By changing the recipe of enzymes and optimizing the reaction conditions, a large number of NHNPs have been efficiently synthesized. Recently, many researchers have combined norcoclaurine synthase (NCS), transaminase, and IRED with other precursor synthesis enzymes and post-modification enzymes to construct multi-enzyme cascades for the synthesis of NHNPs. | ||
| Fig. 12 Enzyme cascade constructed to synthesize isoquinoline compounds. (A), (B), and (C) Construction of the NCS-containing enzyme cascade to synthesize isoquinoline compounds. (D) Construction of the IRED-containing enzyme cascade to modify isoquinoline compounds. | ||
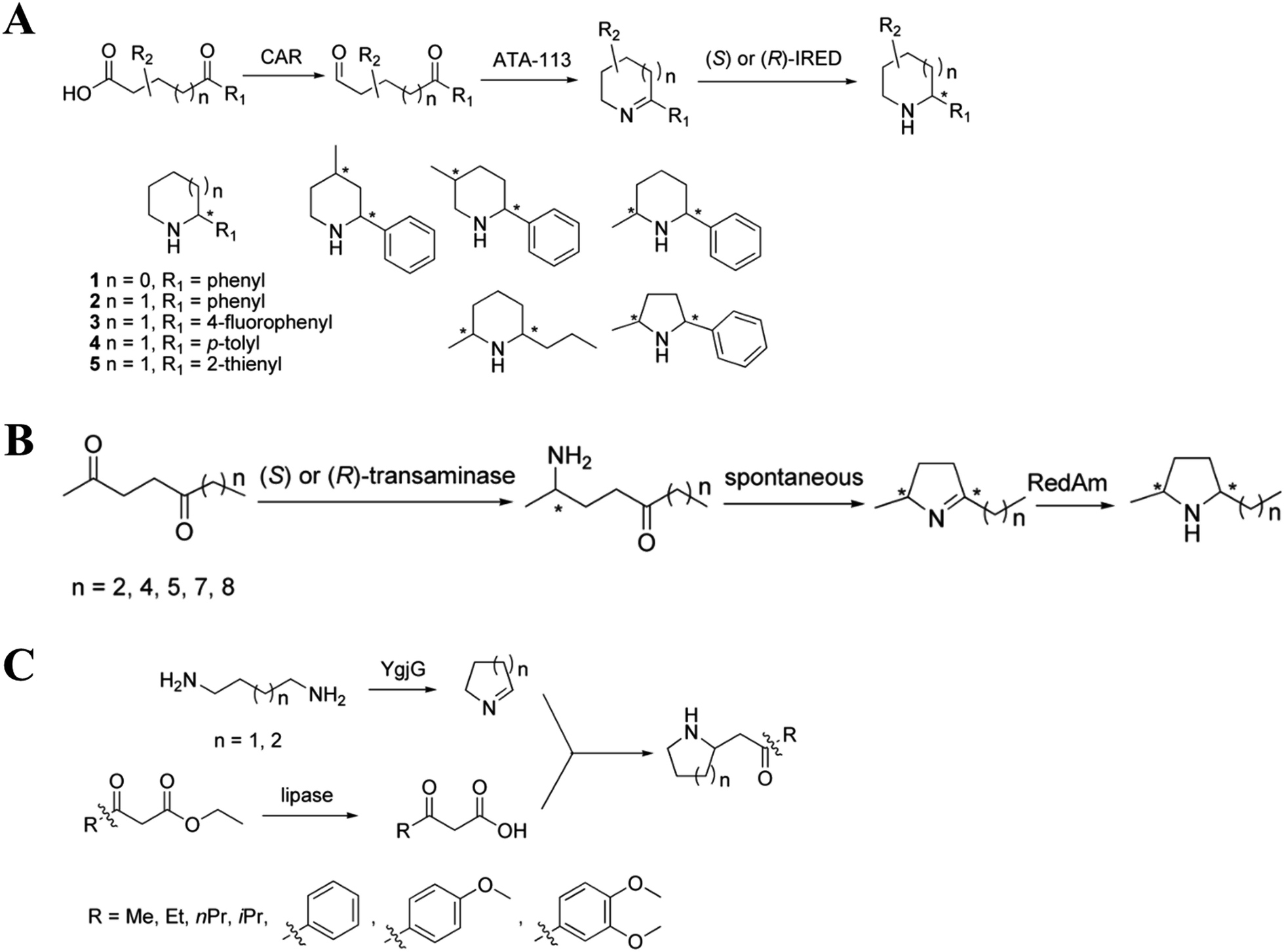 | ||
| Fig. 13 Enzyme cascade constructed with transaminase/IRED. (A) Construction of transaminase and IRED-containing enzyme cascade to synthesize piperidine compounds. (B) Construction of transaminase and RedAm-containing enzyme cascade to synthesize pyrrole compounds. (C) Construction of α,ω-transaminase-containing enzyme cascade to synthesize pyrrole and piperidine compounds. | ||
 | ||
| Fig. 14 Enzyme cascade constructed with other key enzymes. (A) Enzyme cascade constructed with strictosidine synthases. (B) Enzyme cascade constructed with prenyltransferases. (C) Enzyme cascade constructed with type III PKS. | ||
3. Production of NHNPs in microbial cell factory
There are some complex problems that remain to be solved in the process of enzymatic biosynthesis, for example, membrane proteins need to be attached to the membrane for displaying activity, and a number of enzymes rely on expensive cofactors. Microbial cell factories possess complete membrane structure and cofactor synthesis ability, which is an alternative or complementary strategy to synthesize high-value NHNPs.26 One of the dominating obstacles in the heterologous synthesis of NHNPs is the indistinct metabolic pathways and unknown enzymes required for the catalytic reaction. Based on the comprehensive analysis and in-depth mining of multiple omics, including genome, transcriptome, proteome, and metabolome, the natural synthesis pathways of large number of NHNPs, such as morphine, noscapine, and sanguinarine, become gradually unambiguous, which has paved the way for heterologous synthesis in microorganisms. E. coli and Saccharomyces cerevisiae are the most widely adopted heterologous chassis due to the short growth cycle, clear genetic background, and simple culture conditions.87 Various metabolic engineering strategies have been utilized to overcome the problem of inefficient production of NHNPs in microbes. Recently, researchers reconstructed the complete biosynthetic pathways in engineered strains by utilizing simple carbon and nitrogen sources for NHNPs production.8,9,88 Huang et al.89 summarized the entire research process of TAs biosynthesis in plants. The discovery process and related study of each enzyme in the pathway were also discussed. In contrast, this review mainly focused on the research progress of TAs and THIQ alkaloids in the past five years, which had made significant breakthroughs, and summarized the strategies of mining new enzymes and optimizing the production of NHNPs in microbial cell factories.3.1 Deciphering the biosynthesis pathway of NHNPs
The main challenge in the biosynthesis of NHNPs is that the natural synthesis pathways or the enzymes required for NHNPs synthesis have not been completely elucidated; thus, it is impractical to achieve biosynthesis in heterologous hosts.90 Focused on this scientific issue, many studies have been carried out. Several strategies are developed for pathway deciphering: (1) co-expression analysis based on the Pearson correlation coefficient of transcriptome data, coupled with sequence alignment and phylogenetic tree analysis, to identify new genes, (2) adjacent genes located in the same gene cluster, especially those with similar expression profiles, are likely to be related to the function and catalyze continuous reactions in the same pathway, (3) transient expression of candidates in Nicotiana benthamiana leaves and adding the substrate exogenously or harnessing the engineered microbe platform strains to verify the catalytic function of the target genes. | ||
| Fig. 15 The biosynthesis pathway of TAs. The dashed arrow represents uncharacterized reaction steps. AR, L-arginase; ODC, Ornithine decarboxylase; PMT, putrescine N-methyltransferase; DAO, diamine oxidase; PYKS, pyrrolidine ketide synthase; CYP82M3, tropinone synthase; TRI, tropinone reductase I; TRII, pseudotropine reductase II; AT4, aromatic amino acid aminotransferase; PPAR, phenylpyruvic acid reductase; UGT1, phenyllactate UDP-glycosyltransferase; LS, littorine synthase; CYP80F1, littorine mutase/monooxygenase; H6H, hyoscyamine 6β-hydroxylase. | ||
Tropinone is the simplest plant tropane in structure. The genes related to tropane biosynthesis in A. belladonna were preferentially expressed in the roots, and some candidates were obtained by dissecting the transcriptome of roots.90 Combining the strategy of gene silencing and functional verification, two pivotal enzymes, pyrrolidine ketide synthase (AbPYKS), and tropinone synthase (AbCYP82M3), which catalyzed the successive conversion of N-methylpyrrolinium to tropinone, were identified. So far, the biosynthesis pathway of tropinone has been elucidated, paving the way for producing tropinone and derivatives in heterogenous microbes. However, the catalytic mechanism of PYKS is still elusive. RNA-Seq was performed on the hairy roots (where TAs was accumulated) of A. acutangulus, Datura stramonium, and A. belladonna, and 11 putative type III polyketide synthase (PKS) candidates were identified through phylogenetic tree analysis and amino acid sequence alignment.93 By activity verification in vitro, AaPKS2, AbPKS3, and DsPKS1 were effective for the formation of 4-(1-methyl-2-pyrrolidinyl)-3-oxobutanoic acid from N-methylpyrrolinium. Among them, AbPKS3 was the previously reported AbPYKS.90 According to the crystal structure of AaPYKS (formerly AaPKS2), it was found that malonyl-CoA produced 3-oxo-glutaric acid under the catalysis of PYKS, and then 3-oxo-glutaric acid reacted with N-methylpyrrolinium spontaneously to generate 4-(1-methyl-2-pyrrolidinyl)-3-oxobutanoic acid. The newly discovered mechanism provided a reference for the synthesis of other plant alkaloids.
Subsequently, tropinone, the molecular skeleton of TAs, was catalyzed by tropine-forming reductase (PtTRI) and pseudotropine-forming reductase (PtTRII) to form tropine and pseudotropine, respectively, in Przewalskia tangutica.94 Compared with TRI from Brugmansia arborea,95Hyoscyamus niger,96 and Datura stramonium,95 PtTRI possessed the highest affinity for tropinone. The same function of StTRI and StTRII from Solanum tuberosum was proved.97 The reduction ability of TRII was stronger than that of TRI in potato leaves. One interesting phenomenon was that when TRI was disrupted or overexpressed, there was no obvious effect on the accumulation of calystegines, which are the products of the competitive pathway.
Multiple strategies including conserved domain analysis, tissue-specific expression profiles, and phylogenetic tree were adopted to parse the transcriptomes of A. belladonna, and three functional enzymes, namely, phenylpyruvic acid reductase (AbPPAR),98 phenyllactate UDP-glycosyltransferase (UGT1), and littorine synthase (LS),99 were identified, which characterized the biosynthetic pathway of littorine completely. AbPPAR was the first enzyme discovered in plants to reduce phenylpyruvic acid to phenyllactic acid.98 The suppression of AbPPAR in transgenic root cultures disrupted phenyllactic acid production. Tropine and phenyllactylglucose, the glycosylation product of UGT1 from phenyllactate, were condensed via LS to generate littorine.
The chemodiversity of alkaloid was diverse in Mandragora spp. Three common alkaloids, namely, hyoscyamine, anisodamine, and scopolamine, are downstream metabolites of littorine. Scopolamine has the highest market demand due to its fewer side effects and stronger pharmacological activities. For example, it was used as the sedative or in the treatment of motion sickness. The successive hydroxylation and epoxidation of hyoscyamine to anisodamine and scopolamine were achieved by the catalysis of hyoscyamine 6β-hydroxylase (H6H). The functional inactivation of H6H by the substitution of a catalytic residue Gly220 to Cys in M. officinarum led to the accumulation of hyoscyamine and the lack of anisodamine and scopolamine.100 The polymorphism of TAs content was conducive for understanding the catalytic mechanism of crucial enzymes and guiding further rational modification. In the hairy root cultures of Scopolia lurida, the accumulation of scopolamine and anisodamine were significantly improved by overexpressing H6H from Hyoscyamus niger (HnH6H) than that when introducing H6H from S. lurida (SlH6H).101 HnH6H may also be more suitable for scopolamine production in microbial cell factories.
 | ||
| Fig. 16 The biosynthesis pathway of THIQs. CYP76AD5, tyrosine hydroxylase; Aro8, aromatic aminotransferase I; Aro9, aromatic aminotransferase II; DODC, DOPA decarboxylase; Aro10, phenylpyruvate decarboxylase; NCS, norcoclaurine synthase; 6OMT, norcoclaurine 6-O-methyltransferase; CNMT, coclaurine N-methyltransferase; CYP80B3, (S)-N-methylcoclaurine 3′-hydroxylase isozyme; 4′-OMT, 3′-hydroxy-N-methylcoclaurine 4′-omethyltransferase; REPI, reticuline epimerase; SalSyn, salutaridine synthase; SalR, salutaridine reductase; SalAT, salutaridine 7-O-acetyltransferase; THS, thebaine synthase; CODM, codeine 3-O-demethylase; T6ODM, thebaine 6-O-demethylase; COR, codeinone reductase; NISO, neopinone isomerase. | ||
To promote the biosynthesis of thebaine derivatives, a new codeinone reductase (COR) isoform, COR-B, exhibited better catalytic performance than the previously characterized isoforms.104 The reversible conversion between codeine and codeinone, morphine and morphinone, and the irreversible synthesis of neopine or neomorphine from neopinone or neomorphinone, respectively, could be achieved by COR with the dependence on NADP(H). It was found that the expression level of PR10-3 was the highest in the proteome and transcriptome of three opium poppy chemotypes and COR-coupled enzymatic reaction identified PR10-3 as a neopinone isomerase (NISO) to catalyze the isomerization of neopinone and codeinone, neomorphinone and morphinone, which were considered as spontaneous reactions previously.105 The introduction of NISO into the in vitro system or engineering yeast containing T6ODM and COR-B promoted the biosynthesis of codeine and morphine from thebaine. Different from the spontaneous transformation, the recently identified THS103 and NISO105 significantly promoted the synthesis of products, which is beneficial to the heterologous production of thebaine derivatives.
In the synthesis branch of noscapine, four genes catalyzing the generation of noscapine from (S)-reticuline were located in a ten-gene cluster in the genome of opium poppy, and the transcription of the gene cluster was associated with the accumulation of noscapine.106 The catalytic functions of CYP82X1, CYP82X2, AT1, and CXE1, whose encoding genes resided in the same gene cluster, were characterized by utilizing the substrates formed in the previous steps, thus clarifying the biosynthetic routes of noscapine.107 Virus-induced gene silencing was further harnessed to confirm the roles of these four genes in opium poppy. Acetylation introduced by AT1 acted as a protective group to hinder hemiacetal generation, which was necessary for the oxidation of CYP82X1.
Biosynthetic gene cluster of noscapine and the genes responsible for converting (S)-reticuline to thebaine were located on chromosome 11 in the draft genome of opium poppy, which were co-expressed in stems.108 Similar results were also discovered in which the expression patterns of the genes distributed in close proximity on the chromosomes tended to be similar,109 and the function of genes in the same gene cluster might be related.106 Mining key enzymes based on gene clusters is an effective strategy to parse the unknown metabolic pathways.
The biosynthesis pathways of numerous bioactive molecules from medicinal plants are still a mystery. Until recently, the nearly complete synthetic pathway of phenethylisoquinoline colchicine, a fascinating molecule used to treat gout and anti-inflammatory diseases, has been deciphered, which provided an effective strategy for pathway discovery.110 Each candidate methyltransferase or cytochrome P450 identified from Gloriosa superba transcriptome was transiently expressed in N. benthamiana leaves individually, and soaked in chemically synthesized substrate. The leaf extracts were analyzed to verify the function of the enzyme. The mined enzymes were gradually superimposed for the next round of screening, and the de novo synthesis of N-formyldemecoline in N. benthamiana was realized by combining the upstream module of the 1-phenethylisoquinoline scaffold. The residual three steps from N-formyldemecoline to colchicine remain to be illuminated.
3.2 Heterologous biosynthesis of NHNPs in microbial cell factory
Various strategies were combined to promote the efficient synthesis of NHNPs in microbial cell factory. Taking TAs and BIAs as examples, the techniques employed to boost the production mainly include: (A) enriching precursors through the overexpression of upstream enzymes or rate-limiting enzymes, introducing heterologous pathways or blocking the competition pathways; (B) enhancing the enzyme activities including codon optimization of heterologous enzymes, introduction of more efficient enzymes, enhancement of cofactors pool, or truncation of plant P450 signal peptide to modify the function; (C) identifying and optimizing the metabolic bottlenecks to regulate intracellular metabolic flux distribution; (D) modular stepwise fermentation and integrating transporters to promote the transport of intermediates in the co-culture (Fig. 17, Table 1). | ||
| Fig. 17 The strategies for optimizing the biosynthesis of NHNPs in the microbial cell factory. (A) Enriching the supply of precursors. (B) Enhancing the enzyme activities. (C) Identifying and optimizing the metabolic bottlenecks. (D) Modular stepwise fermentation. | ||
| Species | Products | Structures | Engineering strategy | Titer | References |
|---|---|---|---|---|---|
| E. coli | N-Methylpyrrolinium |
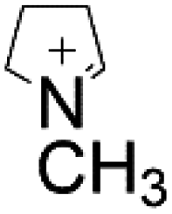
|
Screening enzymes with high activities | 3.02 mg L−1 | 111 |
| S. cerevisiae | N-Methylpyrrolinium |
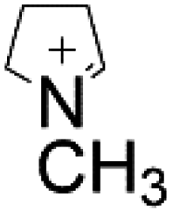
|
Screening enzymes with high activities, disruption of the competitive pathways, overexpression of the key genes | 17.82 mg L−1 | 111 |
| S. cerevisiae | Tropine |

|
Comparison of the catalytic performance of enzymes, enrichment of the precursor | 0.13 mg L−1 | 112 |
| Pseudotropine |

|
0.08 mg L−1 | |||
| S. cerevisiae | Putrescine |

|
Optimization of endogenous and exogenous pathways, disruption of the competitive pathways, recognition of the metabolic bottlenecks | 34 mg L−1 | 113 |
| Tropine |

|
5.9 mg L−1 | |||
| Cinnamoyltropine |

|
6.0 μg L−1 | |||
| S. cerevisiae | (S)-Norcoclaurine |

|
Optimization of the gRNAs and genomic loci, synthetic DNA landing pad (LP) system | 130 μg L−1 | 114 |
| S. cerevisiae | (S)-Reticuline |

|
Disruption of the side reactions, overexpression of the key enzymes, introduction of more efficient enzymes | 4.6 g L−1 | 115 |
| E. coli | Thebaine |
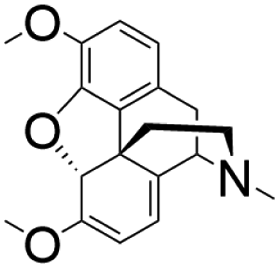
|
The N-terminus truncation of plant-derived P450s, selection of appropriate CPR, addition of heme precursor, stepwise fermentation | 2.1 mg L−1 | 116 |
| S. cerevisiae | Noscapine |

|
Optimization of N-terminal tags, promoters and culture temperatures, upregulation of the expression level of rate-limiting enzymes | 1.64 μM | 117 |
| S. cerevisiae | Noscapine |

|
Truncation of the N-terminal signal peptide, overexpression of the bottleneck enzymes, increasement of NADPH pool, optimization of fermentation conditions | 2.21 mg L−1 | 118 |
(S)-Norcoclaurine undergoes 4-step continuous methylation and hydroxylation to form (S)-reticuline, the key precursor of BIAs. Pyne et al.115 established a high-yield yeast platform with a titer of 4.6 g L−1 (S)-reticuline for producing natural or synthetic THIQ alkaloids. Several efforts have been made for the efficient production of (S)-reticuline. Five NADPH-dependent reductases and dehydrogenases were knocked out to prevent 4-HPAA from generating byproducts 4-hydroxyphenylacetic acid (4-HPAC) or tyrosol through oxidation or reduction reaction, respectively. However, there were still other unrecognized enzymes that catalyzed the aforementioned side reactions. Single gene knockout identified that Gre2 or Hfd1 could reduce or oxidize 4-HPAA to produce fused products. The simultaneous disruption of gre2 and hfd1 destroyed more than 80% of 4-HPAC and tyrosol synthesized intracellularly. The overexpression of enzymes in the upstream pathways or the introduction of more efficient enzymes, such as NCS from Coptis japonica (CjNCS) and CYP76AD5, provided sufficient precursors, 4-HPAA and dopamine, and further dramatically increased (S)-reticuline production. Utilizing the promiscuity of NCS, the condensation of endogenous amino acids or exogenous added amino acids with dopamine can realize the de novo synthesis of diverse non-canonical THIQs, enriching the structure of THIQs and broadening the application range of THIQ products.
Many types of BIAs were derived from (S)-reticuline, for instance, thebaine, noscapine, sanguinarine, and magnoflorine. During the biosynthesis of thebaine, which originated from L-tyrosine, E. coli with a high amino acid productivity was selected as the host. The N-terminus truncation of plant-derived P450s, including STORR and SalS, the selection of appropriate CPR, and the addition of the heme precursor, 5-aminolevulinic acid (5-ALA), were performed to improve the catalytic performance of P450s.116 Tyrosinase, responsible for transforming L-tyrosine to levodopa (L-DOPA), was able to degrade tetrahydropapaveroline (THP), which was the intermediate during thebaine production. The content of (R,S)-THP would influence the composition of R/S-reticuline, and the high concentration of (R,S)-THP reduced (R)-reticuline accumulation. In the process of thebaine production in E. coli, IPTG was found to possess an opposite effect on the synthesis of (R,S)-reticuline and thebaine. Given the interrelationship between the above pathways, the biosynthetic pathway of thebaine from glycerol was divided into four modules, with each module completed via different E. coli strains in a stepwise fermentation manner. Stepwise culture method divided the whole metabolic pathway into several parts and allowed each module to be optimized separately, increasing the thebaine titer to 2.1 mg L−1. The introduction of corresponding downstream pathways could also achieve the efficient synthesis of (S)-reticuline derivatives.
Noscapine, a potential anticancer medicine, is another typical BIAs. The identification of a ten-gene cluster in charge of noscapine biosynthesis in the opium poppy genome provided a pivotal foundation for the de novo synthesis in S. cerevisiae.107 The incorporation of heterogeneous pathways enabled the bioconversion of (S)-canadine to noscapine.117 Functional O-methyltransferases from plant sources were usually dimers. Consistent with the previous characterization results in vitro, monomer PsMT2 or PsMT3 was inactive when acting on narcotoline alone but their simultaneous expression might act as a heterodimer to methylate the 4′-O of narcotoline to form noscapine. The optimization of N-terminal tags, promoters, and culture temperatures, combined with the upregulating of the expression level of the rate-limiting enzymes, ultimately increased noscapine production to 1.64 μM from norlaudanosoline. In-depth studies were carried out sequentially to improve the yield. The introduction of 25 heterologous enzymes and 6 endogenous enzymes reconstituted the complete synthesis pathway of noscapine.118 In order to enhance the metabolic flux of the product, different modifications were implemented to different rate-limiting enzymes, such as truncating the N-terminal signal peptide of NCS, adopting codon-optimized tyrosine hydroxylase (TyrH) mutant, and overexpressing the bottleneck enzymes CYP82X2 and S9OMT. NADPH was the electron donor of plant cytochrome P450s, and increased the NADPH pool, contributing to enhanced catalytic efficiency. The overexpression of some endogenous genes for NADPH regeneration, either individually or in combination, significantly promoted noscapine production. The optimization of medium composition and fermentation conditions was a basic but effective strategy to boost the product synthesis.121 Under the optimal conditions, the yield of noscapine of up to 2.21 mg L−1 was achieved, which was increased by 300 times. Feeding tyrosine derivatives and utilizing the promiscuity of enzymes realized the biosynthesis of halogenated BIAs, which extended the application of the platform strains and offered an alternative approach for the synthesis of synthetic compounds.
For products with long and complicated metabolic pathways, modular co-culture engineering strategy has been widely utilized to alleviate the heavy burden of multi-enzyme expression on the host or to avoid inhibition or competition among the pathways.122 Efficient transport of intermediates is critical for a multistrain co-culture. The transportation of berberine in Coptis japonica was mediated by the ATP-binding cassette (ABC) protein family including CjMDR1 (ref. 123) and CjABCB2,124 and multiantimicrobial extrusion protein (MATE) family containing CjMATE1.125 However, the transporter of opiate alkaloids in opium poppy is still elusive. In the gene cluster responsible for thebaine synthesis, there was a putative purine uptake permease (PUP), a transporter with affinity for alkaloids, which was termed as BUP1 pseudogene.126 Eight additional BUPs (BUP2-BUP9) were identified in combination with transcriptome mining and sequence similarity analysis, among which, BUP1 localized on the plasma membrane and was verified to be capable of transporting a wide range of alkaloids in engineered yeast. In the three engineering yeast strains, the biosynthesis of codeine from DOPA was accomplished in a stepwise culture way, and the overexpression of BUP1 promoted the conversion significantly. The proposed transportation mechanism of BUP1 was to translocate the intermediates synthesized by the sieve element to the laticifer. In the stepwise fermentation mediated by multiple strains, introducing BUP1 would facilitate the transfer of the intermediates among different strains, thus promoting the synthesis of opiate alkaloids. There are plenty of transporters that remain to be parsed.
4. Conclusions and prospective
In summary, many biosynthetic pathways have been elucidated with the development of multi-omics and bioinformatics. A large number of new NHNPs are discovered annually, which are potential materials for manufacturing new food additives, pesticides, and drugs. Biosynthesis offers an alternative and efficient method for obtaining NHNPs, which can avoid large-scale planting, save land resources, and improve economic efficiency. Moreover, the biosynthesis of NHNPs from biomass resources can convert inorganic carbon sources such as CO2 into high-value products and promote carbon balance. Despite the breakthroughs that have been made in enzyme engineering and microbial cell factory, the yield is still low and far from meeting the market demand. First, compared with chemical methods, only a few types of nitrogen-heterocyclic skeletons can be obtained by enzymatic methods. Enzymes that synthesize morpholine, pyrazole, tri/tetrazole, tri/tetrazine, and other high-value N-heterocyclic skeletons are still rarely reported. Second, most of the reported enzymes are not stable enough to meet industrial needs, which prevents them from showing higher benefits than chemical methods. Thirdly, the synthesis pathways of many NHNPs have not been fully elucidated, and researchers cannot find alternative enzymes to synthesize the target products. Today, researchers are committed to employing multiple strategies to optimize the synthesis, for example, utilizing retro-biosynthesis algorithm to predict the synthesis route of compounds, metabolic engineering coupled with a metabolic network model to optimize the metabolic flux, and biosensors for the dynamic regulation of metabolic processes. The biosynthesis of NHNPs will be further combined with big data and artificial intelligence technologies to meet the needs of higher data analysis and information integration. By designing high-throughput screening and detection methods, more enzymatic synthesis platforms and microbial cell factories will be constructed to achieve the efficient biosynthesis of NHNPs.5. Conflicts of interest
There are no conflicts to declare.6. Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFA0901800), the Natural Science Foundation of China (No. 21736002, and 21878021), and the Beijing Nova Program of Science and Technology (No. Z191100001119099), Beijing Natural Science Foundation (No. M21010 and 2212017).7. References
- C. G. Acevedo-Rocha, L. S. Gronenberg, M. Mack, F. M. Commichau and H. J. Genee, Curr. Opin. Biotechnol., 2019, 56, 18–29 CrossRef CAS PubMed.
- T. Iyanagi, Biochim. Biophys. Acta Bioenerg., 2019, 1860, 233–258 CrossRef CAS.
- T. J. Titcomb and S. A. Tanumihardjo, Compr. Rev. Food Sci. Food Saf., 2019, 18, 1968–1984 CrossRef CAS.
- J. A. Joule, in Heterocyclic Chemistry In the 21st Century: A Tribute To Alan Katritzky, ed. E. F. V. Scriven and C. A. Ramsden, Elsevier Academic Press Inc, San Diego, 2016, vol. 119, pp. 81–106 Search PubMed.
- K. W. Wang and X. J. Gao, Mini Rev. Org. Chem., 2016, 13, 402–409 CrossRef CAS.
- S. S. Zhou and J. G. Jiang, Curr. Med. Chem., 2019, 26, 1833–1848 CrossRef CAS PubMed.
- V. Amirkia and M. Heinrich, Phytochem. Lett., 2014, 10, 48–53 Search PubMed.
- S. Schlager and B. Drager, Curr. Opin. Biotechnol., 2016, 37, 155–164 CrossRef PubMed.
- A. Diamond and I. Desgagne-Penix, Plant Biotechnol. J., 2016, 14, 1319–1328 CrossRef CAS.
- L. Chen, Q. Y. Zhang, M. Jia, Q. L. Ming, W. Yue, K. Rahman, L. P. Qin and T. Han, Crit. Rev. Microbiol., 2014, 42, 454–473 Search PubMed.
- K. Gademann and C. Portmann, Curr. Org. Chem., 2008, 12, 326–341 CrossRef CAS.
- S. Asamizu, Biosci. Biotechnol. Biochem., 2017, 81, 871–881 CrossRef CAS.
- C. Hartley, F. Moultrie, A. Hoskin, G. Green, V. Monk, J. L. Bell, A. R. King, M. Buckle, M. van der Vaart, D. Gursul, S. Goksan, E. Juszczak, J. E. Norman, R. Rogers, C. Patel, E. Adams and R. Slater, Lancet, 2018, 392, 2595–2605 CrossRef CAS.
- B. Muller-Krampe, M. Oberbaum, P. K. Dipl-Math and M. Weiser, Pediatr. Int., 2007, 49, 328–334 CrossRef.
- S. Gezici and N. Sekeroglu, Anticancer Agents Med. Chem., 2019, 19, 101–111 CrossRef CAS.
- T. V. Sravanthi and S. L. Manju, Eur. J. Pharm. Sci., 2016, 91, 1–10 CrossRef CAS.
- V. Sharma, P. Kumar and D. Pathak, J. Heterocycl. Chem., 2010, 47, 491–502 CAS.
- A. D. Borthwick, Chem. Rev., 2012, 112, 3641–3716 CrossRef CAS.
- Z. X. Qing, P. Yang, Q. Tang, P. Cheng, X. B. Liu, Y. J. Zheng, Y. S. Liu and J. G. Zeng, Curr. Org. Chem., 2017, 21, 1920–1934 CrossRef CAS.
- B. K. Hong, H. H. Li, J. B. Wu, J. Zhang and X. G. Lei, Angew. Chem., Int. Ed., 2015, 54, 1011–1015 CrossRef CAS PubMed.
- P. Q. Huang, S. Y. Huang, L. H. Gao, Z. Y. Mao, Z. Chang and A. E. Wang, Chem. Commun., 2015, 51, 4576–4578 RSC.
- Y. M. Huang, Y. Liu, C. W. Zheng, Q. W. Jin, L. Pan, R. M. Pan, J. Liu and G. Zhao, Chem.–Eur. J., 2016, 22, 18339–18342 CrossRef CAS PubMed.
- S. Y. Xu, J. Zhang, D. H. Ma, D. Y. Xu, X. G. Xie and X. G. She, Org. Lett., 2016, 18, 4682–4685 CrossRef CAS PubMed.
- Q. Zhang, F. M. Zhang, C. S. Zhang, S. Z. Liu, J. M. Tian, S. H. Wang, X. M. Zhang and Y. Q. Tu, J. Org. Chem., 2019, 84, 12664–12671 CrossRef CAS.
- N. Gerhards, L. Neubauer, P. Tudzynski and S. M. Li, Toxins, 2014, 6, 3281–3295 CrossRef CAS PubMed.
- C. Dziggel, H. Schafer and M. Wink, Biotechnol. J., 2017, 12, 1600145 CrossRef.
- S. Kishimoto, M. Sato, Y. Tsunematsu and K. Watanabe, Molecules, 2016, 21, 1078 CrossRef PubMed.
- T. Awakawa and I. Abe, Beilstein J. Org. Chem., 2019, 15, 1545–1551 CrossRef CAS PubMed.
- P. J. Facchini and J. S. Morris, Front. Plant Sci., 2019, 10, 1058 CrossRef.
- X. Liu, X. Zhu, H. Wang, T. Liu, J. Cheng and H. Jiang, Synth. Syst. Biotechnol., 2020, 5, 187–199 CrossRef.
- A. Miyanaga, F. Kudo and T. Eguchi, Nat. Prod. Rep., 2018, 35, 1185–1209 RSC.
- A. Nivina, K. P. Yuet, J. Hsu and C. Khosla, Chem. Rev., 2019, 119, 12524–12547 CrossRef CAS.
- C. Beck, J. F. G. Garzon and T. Weber, Biotechnol. Bioprocess Eng., 2020, 25, 886–894 CrossRef CAS.
- M. J. Jaremko, T. D. Davis, J. C. Corpuz and M. D. Burkart, Nat. Prod. Rep., 2020, 37, 355–379 RSC.
- P. D. Walker, A. N. M. Weir, C. L. Willis and M. P. Crump, Nat. Prod. Rep., 2021, 38, 723–756 RSC.
- A. Zipperer, M. C. Konnerth, C. Laux, A. Berscheid, D. Janek, C. Weidenmaier, M. Burian, N. A. Schilling, C. Slavetinsky, M. Marschal, M. Willmann, H. Kalbacher, B. Schittek, H. Brotz-Oesterhelt, S. Grond, A. Peschel and B. Krismer, Nature, 2016, 535, 511–516 CrossRef CAS.
- F. Hemmerling and F. Hahn, Beilstein J. Org. Chem., 2016, 12, 1512–1550 CrossRef CAS.
- M. W. Mullowney, R. A. McClure, M. T. Robey, N. L. Kelleher and R. J. Thomson, Nat. Prod. Rep., 2018, 35, 847–878 RSC.
- C. Gui, Q. L. Li, X. H. Mo, X. J. Qin, J. Y. Ma and J. H. Ju, Org. Lett., 2015, 17, 628–631 CrossRef CAS.
- P. Borgman, R. D. Lopez and A. L. Lane, Org. Biomol. Chem., 2019, 17, 2305–2314 RSC.
- M. Gondry, I. B. Jacques, R. Thai, M. Babin, N. Canu, J. Seguin, P. Belin, J. L. Pernodet and M. Moutiez, Front. Microbiol., 2018, 9, 46 CrossRef PubMed.
- S. Meng, W. Han, J. Zhao, X. H. Jian, H. X. Pan and G. L. Tang, Angew. Chem., Int. Ed., 2018, 57, 719–723 CrossRef CAS PubMed.
- E. D. James, B. Knuckley, N. Alqahtani, S. Porwal, J. S. Ban, J. A. Karty, R. Viswanathan and A. L. Lane, ACS Synth. Biol., 2016, 5, 547–553 CrossRef CAS.
- R. Roddan, J. M. Ward, N. H. Keep and H. C. Hailes, Curr. Opin. Chem. Biol., 2020, 55, 69–76 CrossRef CAS.
- E. Eger, A. Simon, M. Sharma, S. Yang, W. B. Breukelaar, G. Grogan, K. N. Houk and W. Kroutil, J. Am. Chem. Soc., 2020, 142, 792–800 CrossRef CAS.
- H. Lechner, P. Soriano, R. Poschner, H. C. Hailes, J. M. Ward and W. Kroutil, Biotechnol. J., 2018, 13, 1700542 CrossRef.
- T. Mori, S. Hoshino, S. Sahashi, T. Wakimoto, T. Matsui, H. Morita and I. Abe, Chem. Biol., 2015, 22, 898–906 CrossRef CAS.
- Q. Chen, S. F. Zhang and Y. C. Xie, J. Biotechnol., 2018, 281, 137–143 CrossRef CAS PubMed.
- X. Z. Wang, D. K. Kong, T. T. Huang, Z. X. Deng and S. J. Lin, Org. Biomol. Chem., 2018, 16, 9124–9128 RSC.
- K. L. Chen, C. Y. Lai, M. T. Pham, R. J. Chein, Y. Tang and H. C. Lin, Org. Lett., 2020, 22, 3302–3306 CrossRef CAS.
- Y. L. Du, H. Y. He, M. A. Higgins and K. S. Ryan, Nat. Chem. Biol., 2017, 13, 836–838 CrossRef CAS PubMed.
- R. C. Simon, B. Grischek, F. Zepeck, A. Steinreiber, F. Belaj and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 6713–6716 CrossRef CAS.
- N. Borlinghaus, S. Gergel and B. M. Nestl, ACS Catal., 2018, 8, 3727–3732 CrossRef CAS.
- Z. Xu, P. Yao, X. Sheng, J. Li, J. Li, S. Yu, J. Feng, Q. Wu and D. Zhu, ACS Catal., 2020, 10, 8780–8787 CrossRef CAS.
- H. Renata, Z. J. Wang and F. H. Arnold, Angew. Chem., Int. Ed., 2015, 54, 3351–3367 CrossRef CAS PubMed.
- P. Molina-Espeja, J. Vina-Gonzalez, B. J. Gomez-Fernandez, J. Martin-Diaz, E. Garcia-Ruiz and M. Alcalde, Biotechnol. Adv., 2016, 34, 754–767 CrossRef.
- F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS PubMed.
- C. A. Tracewell and F. H. Arnold, Curr. Opin. Chem. Biol., 2009, 13, 3–9 CrossRef CAS PubMed.
- R. E. Cobb, R. Chao and H. M. Zhao, Biotechnol. Adv., 2013, 59, 1432–1440 CAS.
- R. Parra-Cruz, P. L. Lau, H. S. Loh and A. Pordea, J. CO2 Util., 2020, 37, 1–8 CrossRef CAS.
- L. S. Xu, F. K. Han, Z. Dong and Z. J. Wei, Microorganisms, 2020, 8, 519 CrossRef CAS PubMed.
- Y. D. Cao, Y. C. He, H. Li, G. Y. Kai, J. H. Xu and H. L. Yu, J. Biotechnol., 2015, 211, 123–129 CrossRef CAS PubMed.
- A. Fan and S. M. Li, Appl. Microbiol. Biotechnol., 2016, 100, 5389–5399 CrossRef CAS.
- K. Brockmeyer and S. M. Li, J. Nat. Prod., 2017, 80, 2917–2922 CrossRef CAS PubMed.
- M. Herger, P. van Roye, D. K. Romney, S. Brinkmann-Chen, A. R. Buller and F. H. Arnold, J. Am. Chem. Soc., 2016, 138, 8388–8391 CrossRef CAS PubMed.
- J. Murciano-Calles, D. K. Romney, S. Brinkmann-Chen, A. R. Buller and F. H. Arnold, Angew. Chem., Int. Ed., 2016, 55, 11577–11581 CrossRef CAS PubMed.
- D. K. Romney, J. Murciano-Calles, J. E. Wehrmuller and F. H. Arnold, J. Am. Chem. Soc., 2017, 139, 10769–10776 CrossRef CAS.
- C. E. Boville, D. K. Romney, P. J. Almhjell, M. Sieben and F. H. Arnold, J. Org. Chem., 2018, 83, 7447–7452 CrossRef CAS PubMed.
- S. Duewel, L. Schmermund, T. Faber, K. Harms, V. Srinivasan, E. Meggers and S. Hoebenreich, ACS Catal., 2020, 10, 1272–1277 CrossRef CAS.
- A. Zqdlo-Dobrowolska, L. Hammerer, T. Paykov-Keller, K. Gruber and W. Kroutil, ACS Catal., 2020, 10, 1094–1101 CrossRef PubMed.
- T. R. Valentic, J. T. Payne and C. D. Smolke, ACS Catal., 2020, 10, 4497–4509 CrossRef CAS.
- W. S. Glenn, E. Nims and S. E. O'Connor, J. Am. Chem. Soc., 2011, 133, 19346–19349 CrossRef CAS.
- Y. X. Wei, C. Lu, S. S. Jiang, Y. Y. Zhang, Q. C. Li, W. J. Bai and X. Q. Wang, Angew. Chem., Int. Ed., 2020, 59, 3043–3047 CrossRef CAS PubMed.
- Y. S. Li and L. L. Wong, Angew. Chem., Int. Ed., 2019, 58, 9551–9555 CrossRef CAS PubMed.
- C. J. Vavricka, T. Yoshida, Y. Kuriya, S. Takahashi, T. Ogawa, F. Ono, K. Agari, H. Kiyota, J. Li, J. Ishii, K. Tsuge, H. Minami, M. Araki, T. Hasunuma and A. Kondo, Nat. Commun., 2019, 10, 2015 CrossRef PubMed.
- G. Liao, P. Mai, J. Fan, G. Zocher, T. Stehle and S. M. Li, Org. Lett., 2018, 20, 7201–7205 CrossRef CAS PubMed.
- B. R. Lichman, E. D. Lamming, T. Pesnot, J. M. Smith, H. C. Hailes and J. M. Ward, Green Chem., 2015, 17, 852–855 RSC.
- A. Bonamore, L. Calisti, A. Calcaterra, O. H. Ismail, M. Gargano, I. D'Acquarica, B. Botta, A. Boffi and A. Macone, Chemistryselect, 2016, 1, 1525–1528 CrossRef CAS.
- V. Erdmann, B. R. Lichman, J. X. Zhao, R. C. Simon, W. Kroutil, J. M. Ward, H. C. Hailes and D. Rother, Angew. Chem., Int. Ed., 2017, 56, 12503–12507 CrossRef CAS PubMed.
- L. Yang, J. M. Zhu, C. H. Sun, Z. X. Deng and X. D. Qu, Chem. Sci., 2020, 11, 364–371 RSC.
- S. P. France, S. Hussain, A. M. Hill, L. J. Hepworth, R. M. Howard, K. R. Mulholland, S. L. Flitsch and N. J. Turner, ACS Catal., 2016, 6, 3753–3759 CrossRef CAS.
- B. Z. Costa, J. L. Galman, I. Slabu, S. P. France, A. J. Marsaioli and N. J. Turner, ChemCatChem, 2018, 10, 4733–4738 CrossRef CAS.
- J. L. Galman, I. Slabu, F. Parmeggiani and N. J. Turner, Chem. Commun., 2018, 54, 11316–11319 RSC.
- E. M. Fischereder, D. Pressnitz and W. Kroutil, ACS Catal., 2016, 6, 23–30 CrossRef CAS.
- V. Wohlgemuth, F. Kindinger and S. M. Li, Appl. Microbiol. Biotechnol., 2018, 102, 2671–2681 CrossRef CAS.
- J. Wang, N. Ding, Y. Wu, X. P. Shi, B. W. Qi, X. Liu, X. H. Wang, J. Li, P. F. Tu and S. P. Shi, RSC Adv., 2020, 10, 23566–23572 RSC.
- A. Cravens, J. Payne and C. D. Smolke, Nat. Commun., 2019, 10, 3634 CrossRef PubMed.
- A. Sharma, D. Amin, A. Sankaranarayanan, R. Arora and A. K. Mathur, Biotechnol. Lett., 2020, 42, 11–23 CrossRef CAS.
- J.-P. Huang, Y.-J. Wang, T. Tian, L. Wang, Y. Yan and S.-X. Huang, Nat. Prod. Rep., 2021 Search PubMed.
- M. A. Bedewitz, A. D. Jones, J. C. D'Auria and C. S. Barry, Nat. Commun., 2018, 9, 5281 CrossRef CAS.
- L. Cui, F. Huang, D. Zhang, Y. Lin, P. Liao, J. Zong and G. Kai, Mol. Genet. Genomics, 2015, 290, 1367–1377 CrossRef CAS PubMed.
- T. Zhao, S. Li, J. Wang, Q. Zhou and Z. Liao, ACS Synth. Biol., 2020, 9, 437–448 CrossRef CAS PubMed.
- J. P. Huang, C. Fang, X. Ma, L. Wang, J. Yang, J. Luo, Y. Yan, Y. Zhang and S. X. Huang, Nat. Commun., 2019, 10, 4036 CrossRef PubMed.
- N. Wu, D. Jian, M. Xiang, M. Chen, X. Lan, Z. Liao and X. Liu, Biotechnol. Appl. Biochem., 2019, 66, 597–606 CrossRef CAS.
- W. Qiang, K. Xia, Q. Zhang, J. Zeng, Y. Huang, C. Yang, M. Chen, X. Liu, X. Lan and Z. Liao, Phytochemistry, 2016, 127, 12–22 CrossRef CAS.
- T. Hashimoto, K. Nakajima, G. Ongena and Y. Yamada, Plant Physiol., 1992, 100, 836–845 CrossRef CAS.
- N. Küster, S. Rosahl and B. Dräger, Planta, 2017, 245, 355–365 CrossRef PubMed.
- F. Qiu, C. Yang, L. Yuan, D. Xiang, X. Lan, M. Chen and Z. Liao, Org. Lett., 2018, 20, 7807–7810 CrossRef CAS.
- F. Qiu, J. Zeng, J. Wang, J.-P. Huang, W. Zhou, C. Yang, X. Lan, M. Chen, S.-X. Huang, G. Kai and Z. Liao, New Phytol., 2020, 225, 1906–1914 CrossRef CAS PubMed.
- D. Schlesinger, R. Davidovich Rikanati, S. Volis, A. Faigenboim, V. Vendramin, F. Cattonaro, M. Hooper, E. Oren, M. Taylor, Y. Sitrit, M. Inbar and E. Lewinsohn, Plant Sci., 2019, 283, 301–310 CrossRef CAS PubMed.
- X. Lan, J. Zeng, K. Liu, F. Zhang, G. Bai, M. Chen, Z. Liao and L. Huang, Biochem. Biophys. Res. Commun., 2018, 497, 25–31 CrossRef CAS PubMed.
- R. Lenz and M. H. Zenk, J. Biol. Chem., 1995, 270, 31091–31096 CrossRef CAS PubMed.
- X. Chen, J. M. Hagel, L. Chang, J. E. Tucker, S. A. Shiigi, Y. Yelpaala, H. Y. Chen, R. Estrada, J. Colbeck, M. Enquist-Newman, A. B. Ibáñez, G. Cottarel, G. M. Vidanes and P. J. Facchini, Nat. Chem. Biol., 2018, 14, 738–743 CrossRef CAS.
- M. Dastmalchi, L. Chang, M. A. Torres, K. K. S. Ng and P. J. Facchini, Plant J., 2018, 95, 631–647 CrossRef CAS.
- M. Dastmalchi, X. Chen, J. M. Hagel, L. Chang, R. Chen, S. Ramasamy, S. Yeaman and P. J. Facchini, Nat. Chem. Biol., 2019, 15, 384–390 CrossRef CAS.
- T. Winzer, V. Gazda, Z. He, F. Kaminski, M. Kern, T. R. Larson, Y. Li, F. Meade, R. Teodor, F. E. Vaistij, C. Walker, T. A. Bowser and I. A. Graham, Science, 2012, 336, 1704–1708 CrossRef CAS PubMed.
- T.-T. T. Dang, X. Chen and P. J. Facchini, Nat. Chem. Biol., 2015, 11, 104 CrossRef CAS.
- L. Guo, T. Winzer, X. Yang, Y. Li, Z. Ning, Z. He, R. Teodor, Y. Lu, T. A. Bowser, I. A. Graham and K. Ye, Science, 2018, 362, 343–347 CrossRef CAS.
- G. Kustatscher, P. Grabowski and J. Rappsilber, Mol. Syst. Biol., 2017, 13, 937 CrossRef PubMed.
- R. S. Nett, W. Lau and E. S. Sattely, Nature, 2020, 584, 148–153 CrossRef CAS.
- Y. Ping, X. Li, B. Xu, W. Wei, W. Wei, G. Kai, Z. Zhou and Y. Xiao, ACS Synth. Biol., 2019, 8, 257–263 CrossRef CAS PubMed.
- Y. Ping, X. Li, W. You, G. Li, M. Yang, W. Wei, Z. Zhou and Y. Xiao, ACS Synth. Biol., 2019, 8, 1257–1262 CrossRef CAS PubMed.
- P. Srinivasan and C. D. Smolke, Nat. Commun., 2019, 10, 3634 CrossRef.
- L. Bourgeois, M. E. Pyne and V. J. J. Martin, ACS Synth. Biol., 2018, 7, 2675–2685 CrossRef CAS PubMed.
- M. E. Pyne, K. Kevvai, P. S. Grewal, L. Narcross, B. Choi, L. Bourgeois, J. E. Dueber and V. J. J. Martin, Nat. Commun., 2020, 11, 3337 CrossRef CAS.
- A. Nakagawa, E. Matsumura, T. Koyanagi, T. Katayama, N. Kawano, K. Yoshimatsu, K. Yamamoto, H. Kumagai, F. Sato and H. Minami, Nat. Commun., 2016, 7, 10390 CrossRef CAS.
- Y. Li and C. D. Smolke, Nat. Commun., 2016, 7, 12137 CrossRef CAS.
- Y. Li, S. Li, K. Thodey, I. Trenchard, A. Cravens and C. D. Smolke, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, E3922–E3931 CrossRef CAS.
- C. Ronda, J. Maury, T. Jakociunas, S. A. B. Jacobsen, S. M. Germann, S. J. Harrison, I. Borodina, J. D. Keasling, M. K. Jensen and A. T. Nielsen, Microb. Cell Factories, 2015, 14, 97 CrossRef.
- J. Lian, M. HamediRad, S. Hu and H. Zhao, Nat. Commun., 2017, 8, 1688–1696 CrossRef.
- J. S. Morris, M. Dastmalchi, J. Li, L. Chang, X. Chen, J. M. Hagel and P. J. Facchini, in Synthetic Biology and Metabolic Engineering in Plants and Microbes, Pt A: Metabolism in Microbes, ed. S. E. O'connor, 2016, vol. 575, pp. 143–178 Search PubMed.
- S.-F. Yuan, X. Yi, T. G. Johnston and H. S. Alper, Microb. Cell Factories, 2020, 19, 143–154 CrossRef CAS.
- N. Shitan, I. Bazin, K. Dan, K. Obata, K. Kigawa, K. Ueda, F. Sato, C. Forestier and K. Yazaki, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 751–756 CrossRef CAS.
- N. Shitan, F. Dalmas, K. Dan, N. Kato, K. Ueda, F. Sato, C. Forestier and K. Yazaki, Phytochemistry, 2013, 91, 109–116 CrossRef CAS PubMed.
- K. Takanashi, Y. Yamada, T. Sasaki, Y. Yamamoto, F. Sato and K. Yazaki, Phytochemistry, 2017, 138, 76–82 CrossRef CAS PubMed.
- M. Dastmalchi, L. Chang, R. Chen, L. Yu, X. Chen, J. M. Hagel and P. J. Facchini, Plant Physiol., 2019, 181, 916–933 CrossRef CAS.
Footnote |
| † With equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2022 |