
Macroemulsion-mediated synthesis of fibrous ZnO microrods and their surface morphology contribution to the high photocatalytic degradation rate†
Didi Prasetyo
Benu
 abc,
Amelia
Andriani
bd,
Nadiatus
Silmi
ab,
Fry Voni
Steky
ab,
Fainan
Failamani
b,
Brian
Yuliarto
ef,
Rino Rakhmata
Mukti
bfg and
Veinardi
Suendo
*bf
abc,
Amelia
Andriani
bd,
Nadiatus
Silmi
ab,
Fry Voni
Steky
ab,
Fainan
Failamani
b,
Brian
Yuliarto
ef,
Rino Rakhmata
Mukti
bfg and
Veinardi
Suendo
*bf
aDoctoral Program of Chemistry, Faculty of Mathematics and Natural Sciences, Institut Teknologi Bandung, Bandung, 40132, Indonesia
bDivision of Inorganic and Physical Chemistry, Faculty of Mathematics and Natural Sciences, Institut Teknologi Bandung, Bandung, 40132, Indonesia. E-mail: vsuendo@chem.itb.ac.id
cDepartment of Chemistry, Universitas Timor, Kefamenanu, 85613, Indonesia
dDepartment of Pharmacy, Sekolah Tinggi Ilmu Kesehatan Adila, Bandar Lampung, 35144, Indonesia
eAdvanced Functional Materials (AFM) Laboratory, Engineering Physics Department, Institut Teknologi Bandung, 10 Ganesha Street, Bandung, 40132, Indonesia
fResearch Center for Nanosciences and Nanotechnology, Institut Teknologi Bandung, Bandung, 40132, Indonesia
gCenter for Catalysis and Reaction Engineering, Institut Teknologi Bandung, Bandung, 40132, Indonesia
First published on 22nd November 2022
Abstract
In this work, fibrous ZnO microrods were synthesized through a macroemulsion-mediated solvothermal method using two different precursors: zinc nitrate and zinc acetate. Initially, we optimized urea concentration using the zinc acetate precursor, producing homogeneous ZnO microrods. We then applied this optimum condition to synthesize ZnO using zinc nitrate. XRD and Raman spectroscopy analyses confirmed that both precursors resulted in ZnO with a wurtzite crystal structure. Raman spectroscopy also reveals the presence of the B1L silent mode in both samples, which is caused by crystal defects. Morphological observation using FESEM shows that both samples have rod shapes with different diameters and lengths but similar aspect ratios. Moreover, the detailed morphology indicates that the ZnO synthesized using zinc acetate has a more uniform fibrous morphology than the ZnO synthesized using zinc nitrate. The uniform fibrous morphology might be induced by the organic counterions, the acetate ion (CH3COO−), in the mixture during the synthesis process. The uniform fibrous ZnO microrod provides excellent properties, including a higher surface area, smaller bandgap energy, and higher total density of defects. The photocatalytic activity of the synthesized ZnO was determined through in situ observation using time-dependent photoluminescence spectroscopy measurements. The photocatalytic test shows that the uniform fibrous ZnO microrods have higher photocatalytic efficiency (73.82%) within 60 minutes of irradiation and a higher photodegradation rate (k = 0.1745 min−1). The recovery test confirmed that the ZnO_Ac_U2 photocatalyst also has higher stability. The higher photocatalytic activity might be due to the synergistic effect among the higher surface area, smaller bandgap energy, higher total density of defects, and uniform fibrous morphology. Moreover, the in situ measurement using time-dependent photoluminescence spectroscopy can collect data at decent intervals, resulting in a more reliable value of the rate constant.
1. Introduction
Wurtzite zinc oxide (ZnO) is an attractive semiconductor material due to its wide bandgap energy (3.37 eV) and large exciton binding energy (60 meV) at room temperature.1,2 ZnO has several advantages, including good thermal and chemical stability, environmental friendliness, low toxicity, and low production cost.3,4 These advanced characteristics make ZnO a promising material for many applications, including optoelectronic devices,5,6 batteries,7,8 sensors,9,10 catalysts,11,12 photocatalysts,13,14 and piezocatalysts.15–17ZnO crystal preferentially grows in the [002] direction, in which O2− and Zn2+ ions stack alternately, forming ZnO nano-/microrods.18,19 Therefore, forming ZnO nano-/microrods is relatively easier than other shapes. Currently, there are many reports about the preparation of ZnO nano-/microrods for various applications, including photocatalysts.20,21 As a photocatalyst in an aqueous system, the rod shape of ZnO has many advantages, such as being more vigorous while stirring22 and a higher surface-to-volume ratio.23 ZnO nano-/microrods with higher aspect ratios have a higher surface area, which facilitates higher adsorption of target molecules, leading to higher photocatalytic activity.24,25 The common strategy to increase the surface to volume ratio is reducing the crystallite size. However, it is hard to obtain very small ZnO nanorods due to the high surface energy of small nanorods. Moreover, small nanorods have low mechanical strength. Therefore, another strategy should be employed to increase the surface to volume ratio of ZnO nano-/microrods to go further than merely controlling the particle size and morphology. This strategy will penetrate to the level of modifying the morphology of the particles’ surfaces, i.e., surface roughening and texturization. Here, ZnO nano-/microrods with a rough surface can be considered as an alternative solution to increase the surface to volume ratio.
In recent years, ZnO nano-/microrods have been successfully prepared using different synthesis methods, including chemical vapor deposition,26,27 molecular beam epitaxy,28,29 electrodeposition,30,31 sputtering,32,33 and the hydrothermal/solvothermal method.10,34 However, recent works have produced ZnO with almost a smooth surface. Therefore, it is challenging to synthesize ZnO with a rough surface, leading to a higher surface-to-volume ratio. Many research groups also reported that the structural and textural properties, including size, shape, porosity, and types of defects, play vital roles in the functional properties of ZnO.35–37 Therefore, it is important to control the structural and textural properties to enhance the functional performance of ZnO materials for various applications, including photocatalysts.
Compared with other synthesis methods, the hydrothermal/solvothermal process provides a simple and convenient way to control ZnO nanostructures’ size, shape, and morphology with a relatively high yield.38 In our research group, we successfully prepared different materials (polyaniline,39,40 silica,41–44 alumina,45,46 and zinc oxide14) with a controllable morphology using the macroemulsion-mediated solvothermal method. Recently, the synthesis of ZnO within a macroemulsion resulted in holey ZnO nanosheets. In this work, we modified the previous synthesis method by changing the pH of the initial solution (pH = 11). We expected that the alkaline condition of the precursor solution would facilitate a different crystal growth behavior, resulting in particles with different shapes and morphologies.
In our previous work, urea plays an important role in forming ZnO particles.14 Therefore, optimizing the amount of urea resulting in the desired morphology of ZnO is essential. Apart from the pH, the counterion of the precursor also plays an important role in the crystal growth of ZnO, resulting in a different morphology.47–49 Particles surrounded by organic ions would provide slower crystal growth than those surrounded by inorganic ions.47 In this work, we used two precursors: zinc acetate, the hydrolytic counterion, and zinc nitrate, the non-hydrolytic counterion. The synthesis condition was based on the optimum amount of urea producing homogeneous ZnO microrods. Rather than nitrate, the acetate counterion participates in the hydrolysis reaction, resulting in a higher amount of OH−, which promotes the formation of the growth unit Zn(OH)42−.49 Since zinc acetate is less acidic than zinc nitrate, the acetate counterions would be able to maintain the change of pH during synthesis under alkaline conditions. Therefore, we expected that the properties of acetate counterions would facilitate the formation of a more homogeneous surface of ZnO particles.
In this work, we investigated the photocatalytic activity of synthesized ZnO by using in situ time-dependent photoluminescence spectroscopy, in which rhodamine B solution was used as an organic pollutant. The kinetics of photocatalytic degradation was obtained by measuring the photoluminescence intensity decay. Compared with other methods (e.g., absorption), the observation method using in situ time-dependent photoluminescence spectroscopy can collect data at decent intervals, resulting in a more reliable rate constant value.
2. Experimental section
2.1 Materials
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O), zinc acetate dihydrate (Zn(CH3COO)2·2H2O), cetyltrimethylammonium bromide (CTAB), n-butanol, toluene, urea, and NaOH were obtained from Merck. All materials are of analytical quality and used without purification.2.2 Synthesis of fibrous ZnO microrods
We synthesized the fibrous ZnO microrods using a macroemulsion system under solvothermal conditions. We used zinc acetate and zinc nitrate precursors. Using zinc acetate dihydrate, we optimized the amount of urea and then used the optimum amount of urea to synthesize ZnO using zinc nitrate. In a typical process, we prepared two types of solutions representing the aqueous and organic phases. First, the zinc precursor, CTAB, and urea were dissolved in deionized water to form the aqueous phase. After forming a homogeneous solution, 8 g of NaOH was introduced. Meanwhile, the organic phase was prepared by mixing toluene and n-butanol. Then, the aqueous phase was introduced dropwise into the organic phase under vigorous stirring until a stable emulsion was obtained. Next, the emulsion was transferred into a 50 mL Teflon-lined stainless-steel autoclave and heated in an electric oven at 120 °C for 24 h. The obtained powder was filtered and rinsed consecutively with deionized water, ethanol, and acetone. The powder was then dried in a drying oven and further calcined under the atmosphere at 400 °C for 4 h. The synthesized samples were denoted as ZnO_x_Uy, in which x refers to the precursor and y refers to the molar ratio of urea to the precursor. The detailed compositions of chemicals in each variation can be seen in Table 1.| Sample name | Composition | ||||||
|---|---|---|---|---|---|---|---|
| Zinc precursor (g) | Urea (g) | CTAB (g) | NaOH (g) | H2O (mL) | Toluene (mL) | n-Butanol (mL) | |
| ZnO_Ac_U0 | 1.4553 | 0 | 0.4957 | 8 | 11.87 | 17.17 | 1.21 |
| ZnO_Ac_U0.5 | 1.4457 | 0.1966 | 0.4925 | 8 | 11.80 | 17.06 | 1.20 |
| ZnO_Ac_U1 | 1.4363 | 0.3907 | 0.4893 | 8 | 11.72 | 16.95 | 1.19 |
| ZnO_Ac_U2 | 1.4179 | 0.7713 | 0.4830 | 8 | 11.57 | 16.73 | 1.18 |
| ZnO_Nit_U2 | 1.8900 | 0.7586 | 0.4750 | 8 | 11.38 | 16.45 | 1.16 |
2.3 Characterizations
The structure of the synthesized particles was characterized using Raman spectroscopy and powder X-ray diffraction. Raman spectra were collected using a Bruker Senterra Raman spectrometer equipped with a 50× Olympus long-working-distance MPlan semiapochromat objective lens. The samples were excited using a 532 nm Nd–YAG diode-pumped solid-state (DPSS) laser with a laser power of 20 mW, an integration time of 10 s, and the co-addition of 2 times. During the measurements, the Andor iDus DU420A CCD detector was in deep-cool mode at −80 °C. The diffractogram of synthesized samples was obtained using a Rigaku MiniFlex powder X-ray diffraction analyzer with Cu Kα radiation. The diffraction data were recorded at a speed of 10° min−1 with a step of 0.02°. The Rietveld refinement method was employed in Fullprof software (Version 7.40 – January 2021) to calculate the lattice parameters. In addition, the Bragg and Scherrer equations were used to calculate the synthesized samples’ interplanar spacing and crystallite size.The morphology of the synthesized particles was observed using a scanning electron microscope (SEM Hitachi SU3500) with an accelerating voltage of 10 kV. The detailed morphology and surface of ZnO microrods were observed using a field emission scanning electron microscope (FESEM Hitachi SU8000) with an accelerating voltage of 5 kV. We measured the diameter and length of particles from the SEM images and made the distribution curve. The N2 physisorption isotherms of synthesized particles were collected using a Belsorp Mini II instrument. The surface area and pore size distribution were calculated using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively. The optical properties of the synthesized samples were studied using the diffuse reflectance spectroscopy (DRS) technique on an integrating sphere equipped Evolution-220 UV-vis spectrometer. The bandgap energy of the synthesized samples was determined from their Tauc plots based on the Kubelka–Munk equation for direct bandgap. The photoluminescence (PL) measurement was employed to study the type of defects and the luminescence properties of the synthesized ZnO samples. The samples were excited using a UV LED with a peak center at 371 nm (see Fig. S1, ESI†). The measurement was performed at room temperature, and the PL spectra were recorded using an Ocean Optics USB4000 UV-vis spectrometer equipped with a 405 nm long-wave pass edge filter (Semrock EdgeBasic, BLP01-405R-25).
2.4 Photocatalytic activity test
We performed a time-dependent photodegradation study in this work by modifying the method reported in our previous study.14 In a typical process, 8 mg of the synthesized ZnO was mixed with 8 mL of rhodamine B solution (5 ppm). First, the mixture was stirred in the dark for 5 minutes. After that, the mixture was stirred for 60 minutes under irradiation with a UV LED with a peak center at 371 nm (see Fig. S1, ESI†). The fluorescence spectra of rhodamine B solution were collected using a StellarNet GREEN-Wave spectrometer. We calculated the evolution of photocatalytic efficiency using the following equation: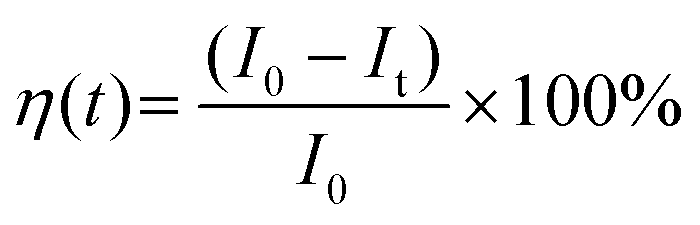 | (1) |
The first-order kinetic model was adapted to determine the kinetics of photodegradation, as described using the following equation:
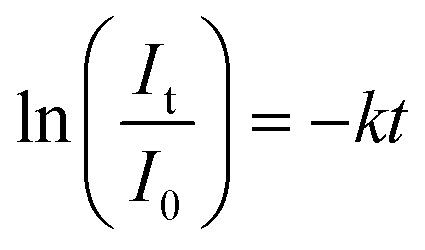 | (2) |
We also performed recovery tests to check the stability of the synthesized ZnO photocatalyst. Each ZnO photocatalyst was used for three cycles of photodegradation. After the first cycle, the powder of the ZnO photocatalyst was separated using centrifugation and rinsed with deionized water. The photocatalyst was then used for the next cycle.
3. Results and discussion
3.1 Optimization of the amount of urea
In our previous work, urea is essential in forming nanostructured ZnO and its properties.14 The urea hydrolysis is vital for the formation of zinc carbonate hydroxide and the subsequent ZnO crystal.50,51 Therefore, optimizing the amount of urea resulting in the desired morphology of ZnO is important. Since urea acts as a hydrolyzing and homogeneous precipitation agent, a certain ratio between urea and zinc precursor would produce a uniform morphology of synthesized ZnO. The pH of the mixture before and after solvothermal treatment can be seen in Table S1 (ESI†). We can see that the solvothermal treatment did not significantly change the pH of the mixture. In other words, the formation of ZnO from zinc acetate under alkaline conditions did not alter the pH.Fig. 1 exhibits the morphology, XRD pattern, and absorption spectra of the synthesized ZnO with different urea-to-zinc precursor molar ratios. Fig. 1a1 and a2 show the SEM images of the ZnO_Ac_U0 sample at different magnifications. The synthesis condition without urea produced particles with inhomogeneous shapes and sizes, consisting of microrods and aggregated nanoparticles. A similar morphology was also observed in the samples of ZnO_Ac_U0.5 (Fig. 1b1 and b2) and ZnO_Ac_U1 (Fig. 1c1 and c2). The uniform microrod particles were observed in the ZnO_Ac_U2 sample (Fig. 1d1 and d2). Therefore, we can say that the ZnO_Ac_U2 sample contains the optimum amount of urea producing uniform microrod particles.
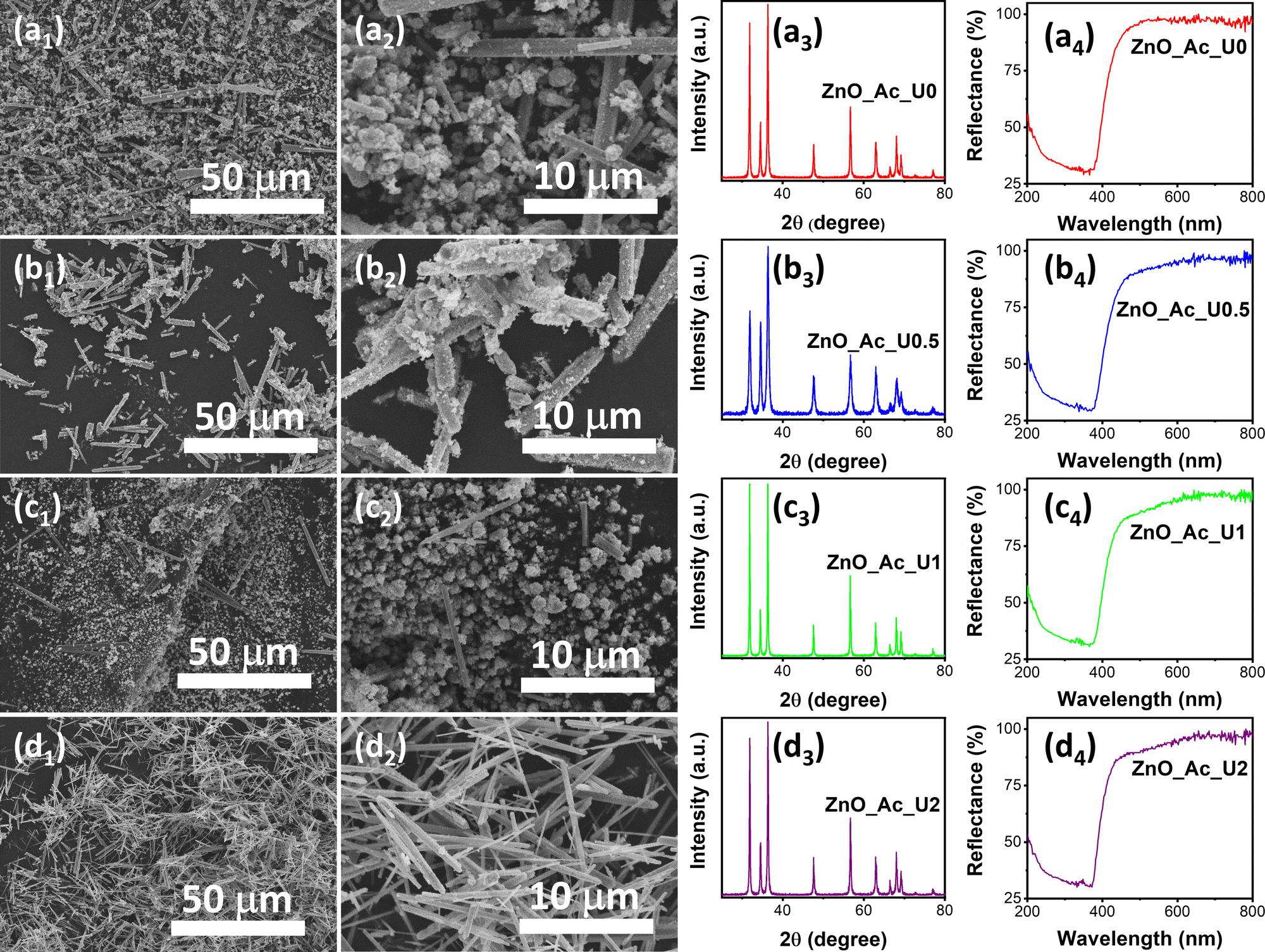 | ||
| Fig. 1 SEM images, XRD pattern, and absorption spectra of the synthesized ZnO with different urea to zinc precursor molar ratios; (a) ZnO_Ac_U0, (b) ZnO_Ac_U0.5, (c) ZnO_Ac_U1, and (d) ZnO_Ac_U2. | ||
We also characterized the synthesized samples using XRD and UV-vis DRS. Fig. 1a3, b3, c3, and d3 represent the XRD patterns of ZnO_Ac_U0, ZnO_Ac_U0.5, ZnO_Ac_U1, and ZnO_Ac_U2 samples, respectively. All samples have a similar diffraction pattern. The patterns match the wurtzite ZnO crystal (CIF 2300450).52 We did not observe any impurity peaks, indicating that all synthesized ZnO samples are of high purity. It is well known that wurtzite ZnO crystal has a semiconductor characteristic. The absorption spectra confirm the semiconductor characteristic in Fig. 1a4, b4, c4, and d4. We can see that all samples have an absorption edge at around 400 nm. The absorption is attributed to the transition of an electron from the valence band (VB) to the conduction band (CB).53
3.2 Structural properties
The structure of particles synthesized using different precursors was analyzed using powder X-ray diffraction and Raman spectroscopy. We get information about the crystalline phase and the crystalline parameters, including lattice parameters, interplanar spacing, and crystallite size, from the XRD measurements. Fig. 2a represents the XRD pattern of synthesized ZnO samples and the Rietveld refinement method calculation. Both samples’ diffraction peaks perfectly match the wurtzite ZnO crystal structure (space group symmetry P63mc, CIF 2300450).52 The crystal structure of wurtzite ZnO can be seen in Fig. 2b. We did not observe any impurities or peaks shifting, indicating that both synthesized samples are pure ZnO with a wurtzite crystal structure. However, we found errors in the peak intensity of ZnO samples compared with the reference, showing a different preferential growth of synthesized ZnO samples. Table 2 displays the synthesized ZnO samples’ lattice parameters, interplanar spacing, and crystallite size. We found similar lattice parameters and interplanar spacing values for both samples. Meanwhile, the crystallite sizes of both samples are pretty different. The crystallite sizes of ZnO_Nit_U2 are approximately 1.5 times higher than that of ZnO_Ac_U2, indicating the faster crystal growth of ZnO_Nit_U2. Particles surrounded by inorganic ions (NO3−) would have faster crystal growth than those surrounded by organic ions (CH3COO−).49 In more detail, the crystallite size ratio of ZnO_Nit_U2/ZnO_Ac_U2 is about 1.47, 1.58, and 1.41 in [10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0], [0002], and [101] directions, respectively. The similar crystallite size ratio in all directions indicates that both precursors have similar preferential crystal growth orientation.
0], [0002], and [101] directions, respectively. The similar crystallite size ratio in all directions indicates that both precursors have similar preferential crystal growth orientation.
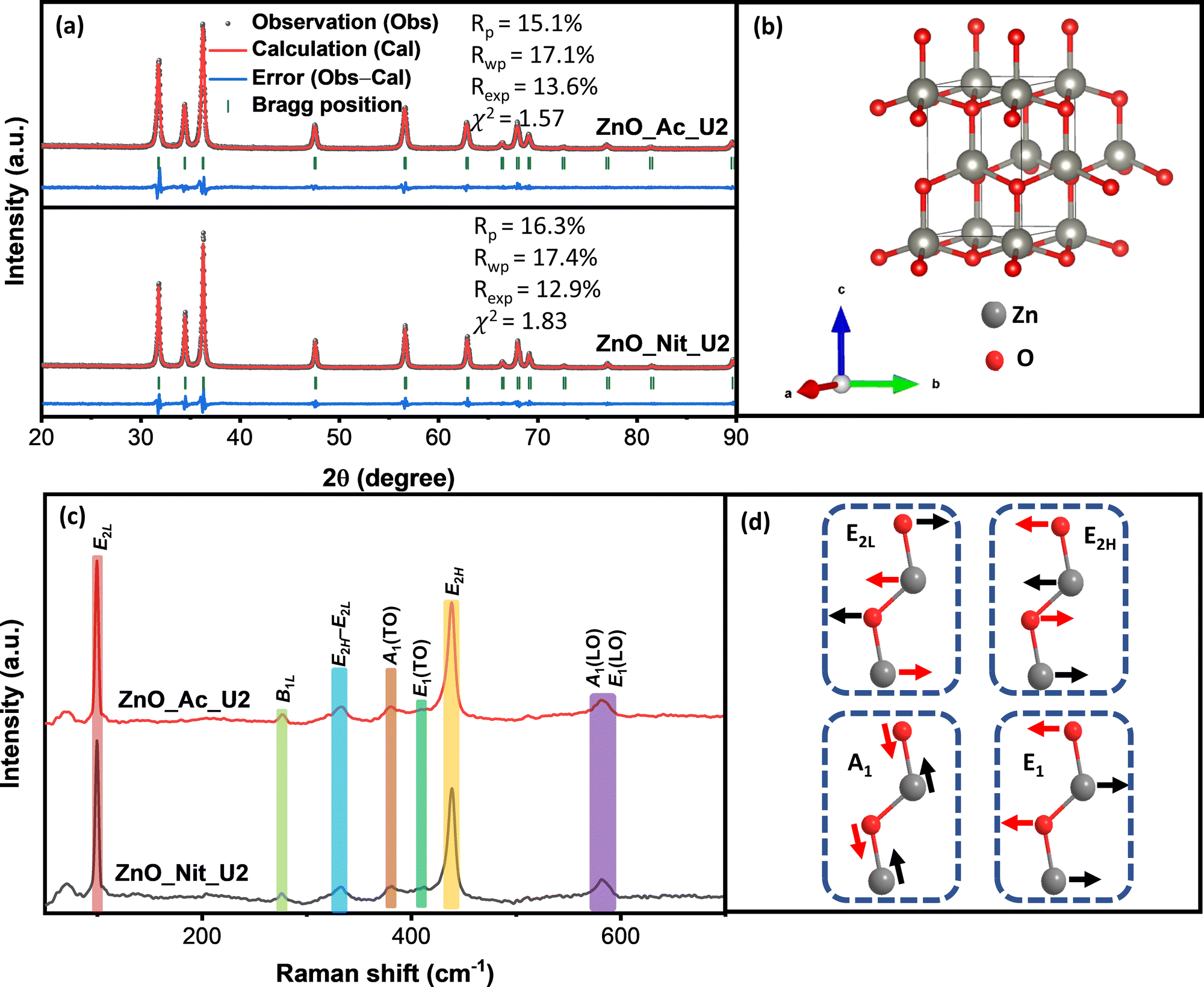 | ||
| Fig. 2 (a) XRD pattern of the synthesized ZnO particles and the calculation based on the Rietveld refinement method. (b) Crystal structure of wurtzite ZnO extracted from CIF 2300450. (c) Raman spectra of the synthesized ZnO samples. (d) Vectoral orientation of several vibrational modes, where the red arrows specify the dominant atom displacement. | ||
| Sample name | Lattice parameters (Å) | Interplanar spacing (Å) | Crystallite size (nm) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| a | b | c | (100) |
(0002) | (101) |
[100] |
[0002] | [101] |
|
| ZnO_Nit_U2 | 3.2495 | 3.2495 | 5.2956 | 2.8163 | 2.6045 | 2.4777 | 37.7315 | 40.4483 | 37.0053 |
| ZnO_Ac_U2 | 3.2521 | 3.2521 | 5.2122 | 2.8042 | 2.6001 | 2.4738 | 25.6723 | 25.9700 | 26.1648 |
In the wurtzite ZnO crystal structure, the optical phonons at the Γ point of the Brillouin zone have the irreducible representation:54–56
| Γopt = 1A1 + 2B1 + 1E1 + 2E2 | (3) |
In contrast with the E2 phonons, the A1 and E1 polar phonons are split into transverse optical (TO) and longitudinal optical (LO). The small peak at 380 cm−1 and weak shoulder at 410 cm−1 correspond to A1(TO), and E1(TO) modes, respectively. These peaks indicate that ZnO crystallites propagate in a direction other than the c-axis typical of ZnO particles. The intensity ratio between A1(TO) and E2H indicates the fraction of polar planes, {0002} and {100}, in the synthesized ZnO samples. We found that the intensity ratio between A1(TO) and E2H is about 0.16 for ZnO_Nit_U2 and 0.15 for ZnO_Ac_U2. The results indicate that both samples have almost the same fraction of polar planes, about 60%.60 This interpretation confirmed that both samples have a similar crystal growth orientation, consistent with the XRD analysis.
Moreover, the broad peak at 570–590 cm−1 represents the modes of A1(LO) and E1(LO). The high intensity of A1(LO) + E1(LO) modes indicates a high density of oxygen vacancies and interstitial zinc defects.59 The sharp peak at 333 cm−1 is attributed to multi-phonon E2H–E2L. The small peak at 276 cm−1 is the B1L mode, a silent mode. The appearance of the silent mode in the synthesized ZnO samples is due to the breakdown of the translation crystal symmetry caused by the presence of defects and impurities.61,62 Therefore, from the Raman analyses, we can say that the synthesized ZnO samples contain a high density of defects.
3.3 Morphology, size, and porosity
The detailed morphologies of the ZnO_Nit_U2 and ZnO_Ac_U2 were observed using FESEM (Fig. 3). Fig. 3a displays the FESEM image of ZnO_Nit_U2 at low magnification, exhibiting a rod-like shape of ZnO particles. At higher magnification (Fig. 3b), we observed two types of morphology: rod-like particles with a smooth and rough surface. The rough surface of the ZnO_Nit_U2 sample is clearly shown in Fig. 3c. The rod-like particles contain sub-unit nanocrystals at the particle's surfaces, resulting in a rough surface. Similar to the ZnO_Nit_U2 sample, ZnO_Ac_U2 particles also have a rod-like shape, as shown in Fig. 3d. However, the FESEM image at higher magnification (Fig. 3e) shows that all particles have rough surfaces. The detailed morphology of ZnO_Ac_U2 particles can be seen in Fig. 3f. It is observed that the particles have a fibrous morphology provided by the sub-unit crystals at the surface of the particles. The uniform fibrous morphology of the ZnO_Ac_U2 particles might be induced by the mixture's organic counterions (CH3COO−). The uniform fibrous morphology of the ZnO_Ac_U2 particles might also provide highly accessible active sites in catalytic applications, such as photocatalysts.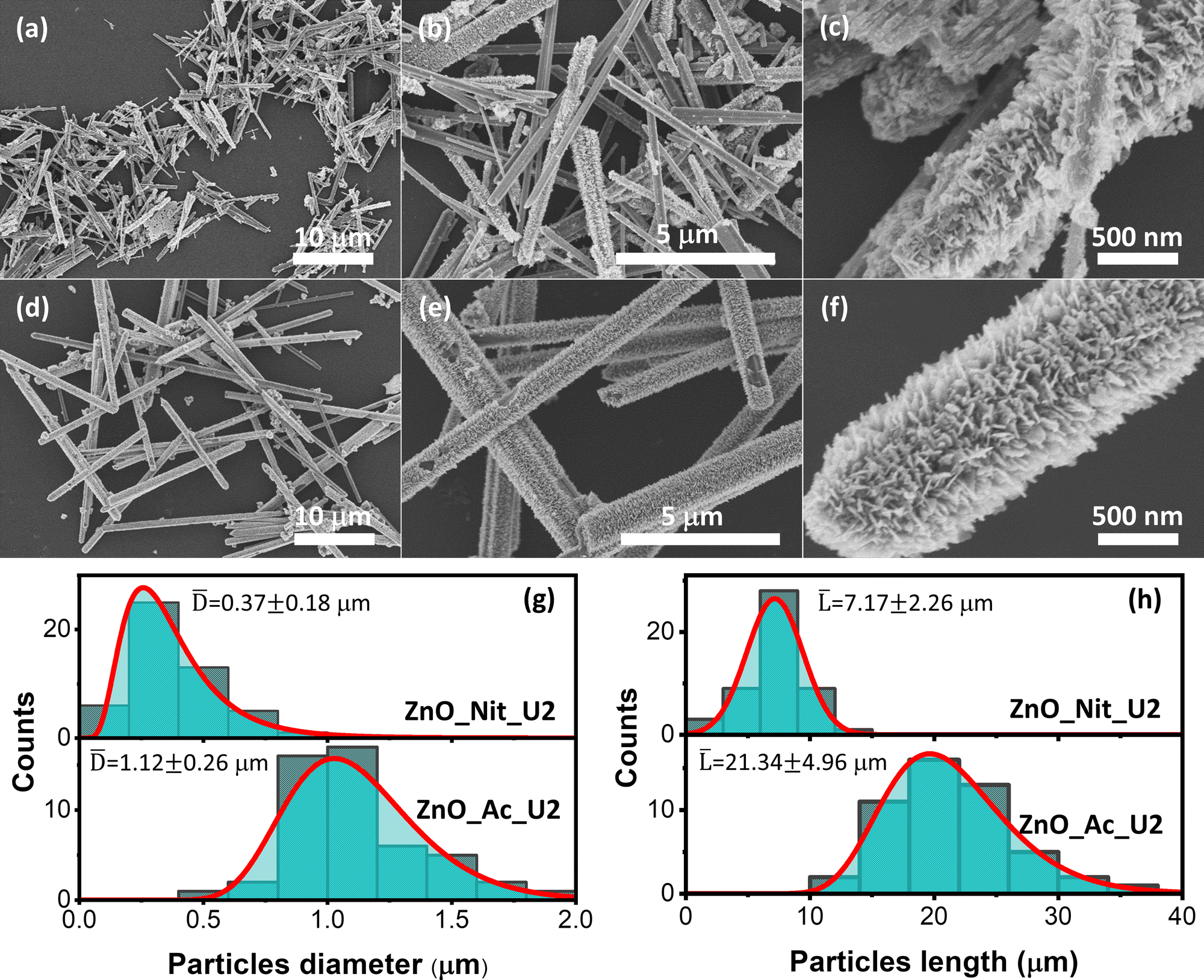 | ||
| Fig. 3 FESEM images of fibrous ZnO microrods synthesized using different precursors; (a–c) zinc nitrate and (d–f) zinc acetate. The particle size distribution of synthesized ZnO; (g) distribution of particle diameter, and (h) distribution of particle length. | ||
From the FESEM images, we also observed that both synthesized ZnO particles differ in diameter and length. Therefore, we calculated the diameter and length of both synthesized particles and determined the diameter and length distribution. Fig. 3g shows the particle diameter distribution of ZnO_Nit_U2 and ZnO_Ac_U2 samples. The diameter of ZnO_Nit_U2 particles is smaller than that of ZnO_Ac_U2. The average diameter of ZnO_Ac_U2 (1.12 ± 0.26 μm) is about three times higher than that of ZnO_Nit_U2 (0.37 ± 0.18 μm).
Moreover, in Fig. 3h, the length of ZnO_Ac_U2 (21.34 ± 4.96 μm) particles is also approximately three times higher than that of ZnO_Nit_U2 (7.17 ± 2.26 μm). Although the diameter and length of both samples are very different, the aspect ratio (length-to-diameter ratio) is similar. The aspect ratio is 19.38 and 19.05 for ZnO_Nit_U2 and ZnO_Ac_U2, respectively. The similar aspect ratio of both samples indicates that both precursors have similar preferential crystal growth orientation, and the results agree with XRD measurements. The large particles of ZnO_Ac_U2 can provide good mechanical stability when applied as a catalyst.
Understanding nanostructured materials’ surface area and porosity is essential to understanding their utility and quality.63 Specifically, a higher surface area of materials provides a higher number of available active sites, leading to high catalytic activity.64 Here, we provided the textural properties of the synthesized ZnO samples based on N2 physisorption measurements. Fig. 4 exhibits synthesized ZnO samples’ N2 physisorption isotherm and BJH pore size distribution. The N2 adsorption–desorption isotherms of both samples display the type III isotherms based on the IUPAC classification.65 The type III isotherm indicates the weak interaction of N2 with ZnO particles, in which the N2 molecules are clustered around the most favorable sites on the surface of nanoporous or microporous ZnO particles. The rough surface is probably the most favorable site based on the FESEM images (Fig. 3).
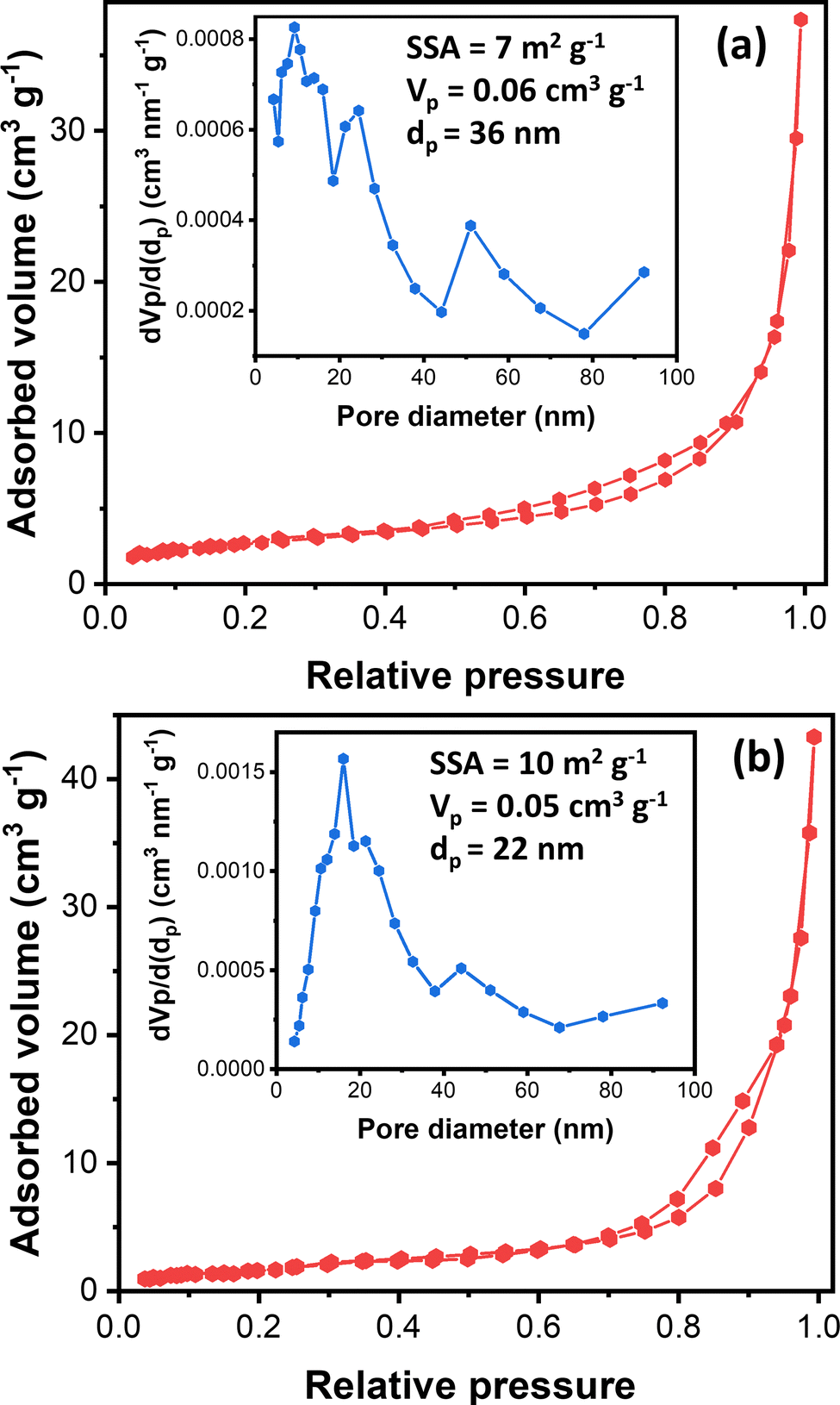 | ||
| Fig. 4 N2 physisorption isotherm of fibrous ZnO microrods synthesized using different precursors; (a) zinc nitrate and (b) zinc acetate. The inset exhibits the BJH pore distribution and textural parameters (SSA = specific surface area, Vp = total pore volume, and dp = average pore diameter) of the corresponding samples. | ||
Moreover, the isotherms resemble the type H3 hysteresis loop, possibly due to macropores not filling with pore condensate.65 Macropore presence agrees with the BJH pore size distribution (inset, Fig. 4). The BJH pore size distribution displayed that both samples have pore sizes in the mesopore and macropore range, with the most frequent pore diameters being 9.2 nm and 16.2 nm for ZnO_Nit_U2 and ZnO_Ac, respectively. However, the average pore diameter of ZnO_Nit_U2 (36 nm) is more significant than that of ZnO_Ac_U2 (22 nm). The total pore volume of ZnO_Nit_U2 (0.06 cm3 g−1) is also larger than that of ZnO_Ac_U2 (0.05 cm3 g−1). In contrast, the BET surface area of ZnO_Nit_U2 (7 m2 g−1) is smaller than that of ZnO_Ac_U2 (10 m2 g−1). The higher surface area of ZnO_Ac_U2 might be provided by the uniform fibrous morphology of ZnO_Nit_U2 particles (see Fig. 3d–f).
3.4 Optical properties
We employed UV-vis DRS to determine the electronic properties of synthesized ZnO. Fig. 5 exhibits the absorption spectra of both synthesized samples and their Tauc plots to determine the bandgap energy value. The absorption spectra in Fig. 5a show that both samples have an absorption edge at around 410 nm, indicating a semiconductor characteristic. The absorption band is attributed to the intrinsic bandgap absorption of wurtzite ZnO for the electron transition from the VB to the CB.66 Moreover, it is clear that the area beneath the absorption edge (Aedge) of the ZnO_Ac_U2 sample is larger than that of ZnO_Nit_U2 (inset, Fig. 5a). The larger value of Aedge indicates the higher total density of defects.14 Therefore, we can say that synthesized ZnO_Ac_U2 has a higher total defect density than ZnO_Nit_U2. The results agree with the surface area of ZnO_Ac_U2 particles, in which the higher surface area particles should have a higher total density of defects.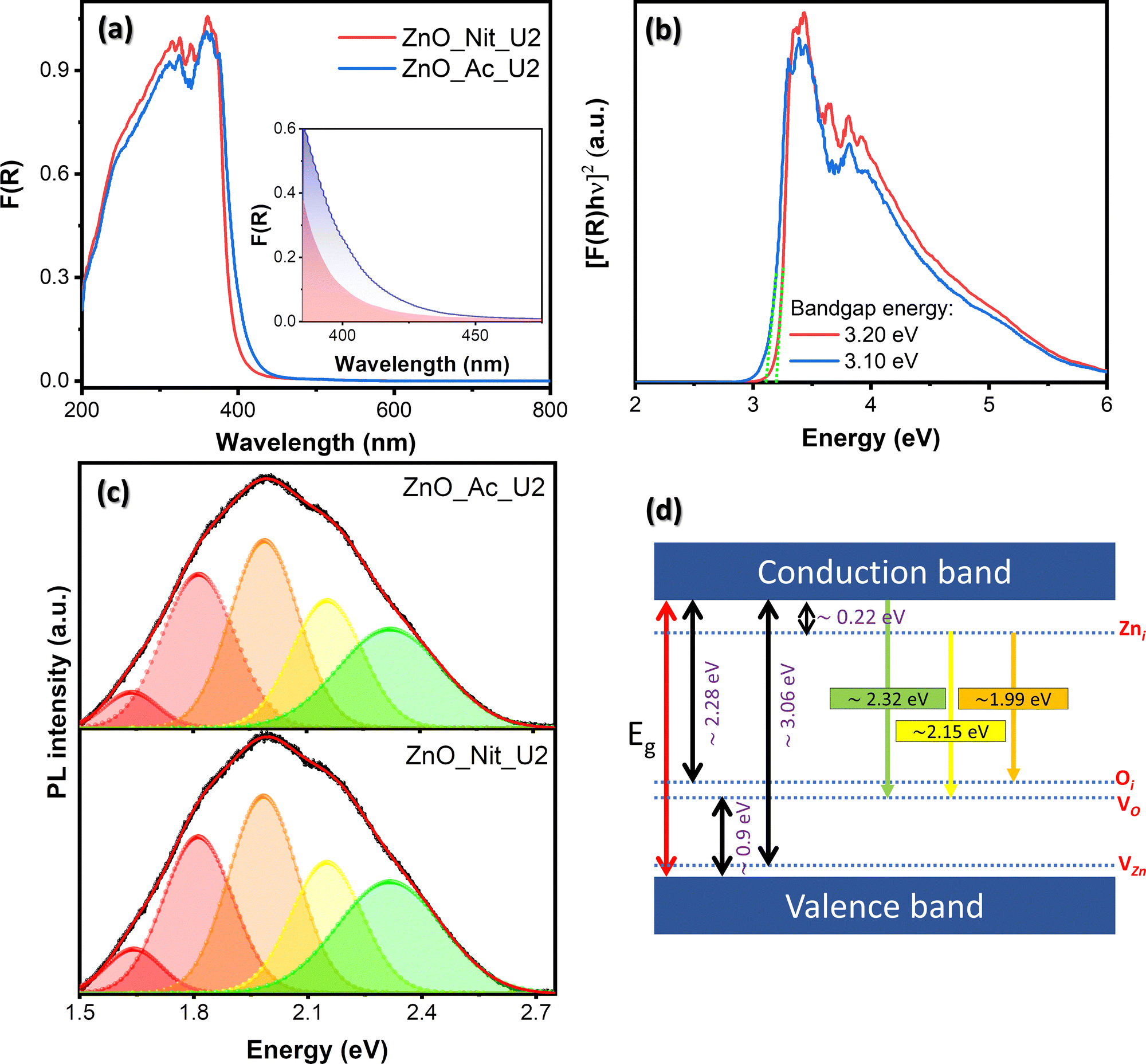 | ||
| Fig. 5 (a) Absorption spectra of the synthesized fibrous ZnO microrods, where the inset shows the area beneath the absorption edge (Aedge). (b) Tauc plots of the synthesized fibrous ZnO microrods. (c) Deconvoluted PL spectra of the synthesized fibrous ZnO microrods. (d) Schematic band diagram of emission provided by the defect levels. | ||
The bandgap energies were determined based on Tauc plots using the Kubelka–Munk equation for direct bandgap (Fig. 5b).67 From the Tauc plots, we obtained both samples’ direct band gap energy values, which are 3.20 eV and 3.10 eV for ZnO_Nit_U2 and ZnO_Ac_U2, respectively. The lower bandgap energy of ZnO_Ac_U2 might be provided by the uniform fibrous morphology of ZnO_Ac_U2 particles. In addition, the advanced characteristics of ZnO_Ac_U2, such as high surface area, the high total density of defects, and lower bandgap energy, might provide good photocatalytic activity.
Room temperature PL spectroscopy was employed to study the type of defects and the luminescence properties. Fig. 5c exhibits the deconvoluted PL spectra of the synthesized ZnO samples. The raw data can be seen in Fig. S2 (ESI†), and the deconvoluted peak properties can be seen in Table S2 (ESI†). Based on the deconvoluted PL spectra of both samples, we obtained at least five luminescence centers in the visible region. Visible emissions observed in the synthesized ZnO samples generally resulted from various intrinsic defects, such as zinc interstitials (Zni), oxygen vacancies (VO), zinc vacancies (VZn), and oxygen interstitials (Oi).68,69 Theoretically, the Zni, Oi, and VZn energy levels are around 0.22, 2.28, and 3.06 eV below the CB, respectively, while the VO level is 0.9 eV above the VB.68 The schematic band diagram of emission provided by the defect levels can be seen in Fig. 5d.
We observed peaks corresponding to green emission at 2.32 eV and yellow emission at 2.15 eV. These emissions in both samples are due to electron transition from the CB to the VO level and from the Zni level to the Vo level, respectively.68,70 The orange emission (1.99 eV) is attributed to the transition from the Zni to the Oi level.68 The red emission observed at 1.82 eV and 1.65 eV can be assigned as the surface defect of positively charged oxygen vacancies (VO+).71,72 Generally, the PL peak area of ZnO_Ac_U2 is higher than that of ZnO_Nit_U2, indicating a higher total density of defects in ZnO_Ac_U2 (see Table S2, ESI†). The results agree with the total density of defects obtained from the absorption spectra (inset, Fig. 5a). A higher area under the PL spectra indicates a higher surface defect. Here, the surface defect may serve as the carrier trap to prevent the electrons and holes from recombination.73
3.5 Photocatalytic performance
In this work, we adopted rhodamine B as a model pollutant representing the organic dye waste in assessing the photocatalytic performance of the synthesized ZnO samples. The experiment was performed by irradiating the rhodamine B solution and ZnO photocatalyst using a UV LED with the peak center at 371 nm. Fig. S1 (ESI†) exhibits the comparison of the absorption spectra of ZnO_Ac_U2 with the emission spectra of the UV LED. These spectra indicate that the UV LED can promote an electron from the valence band to the conduction band of ZnO. Therefore, the photocatalytic properties of ZnO could be activated by irradiation using a UV LED.The photocatalytic degradation of rhodamine B was performed in a microreactor containing only 8 mL of rhodamine B solution and 8 mg of ZnO photocatalyst. The photodegradation process was in situ observed using time-dependent photoluminescence spectroscopy. Fig. 6 represents the photocatalytic degradation results of rhodamine B using ZnO_Nit_U2 and ZnO_Ac_U2 photocatalysts. The photocatalytic efficiency and rate constants are summarized in Table 3. Fig. 6a and d exhibit the PL spectra evolution of rhodamine B in the presence of ZnO_Nit_U2 and ZnO_Ac_U2 photocatalysts, respectively. For both photocatalysts, it is clear that the PL intensities tend to decrease with the irradiation time. We then calculated the photocatalytic efficiency using eqn (1). The plots representing the change in photocatalytic efficiency with the irradiation time can be seen in Fig. 6b (ZnO_Nit_U2) and Fig. 6e (ZnO_Ac_U2), and the maximum photocatalytic efficiency after 60 minutes of irradiation can be seen in Table 3. We obtained the maximum photocatalytic efficiency within 60 minutes of irradiation, which is 65.55% and 73.82% when using ZnO_Nit_U2 and ZnO_Ac_U2 photocatalysts, respectively. The digital images (inset, Fig. 6b and e) indicate that the color of the rhodamine B solution after 60 minutes of photodegradation using ZnO_Ac_U2 is more transparent than that of the ZnO_Nit_U2. The higher photocatalytic activity of ZnO_Ac_U2 might be due to the synergistic effect among the higher surface area, smaller bandgap energy, higher total density of defects, and uniform fibrous morphology.
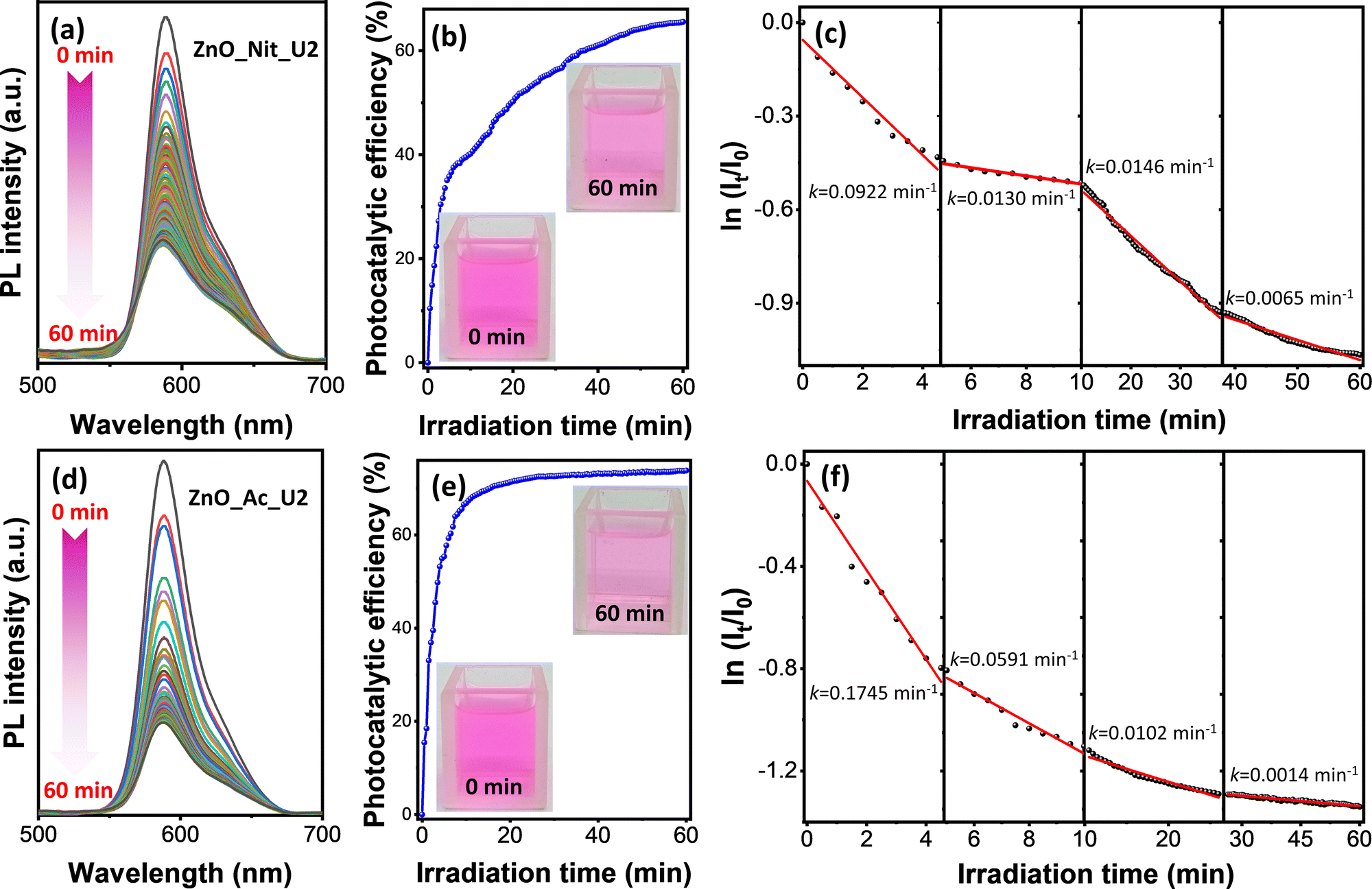 | ||
| Fig. 6 Photodegradation results of rhodamine B using ZnO_Nit_U2 (a–c) and ZnO_Ac_U2 (d–f) photocatalysts; (a and d) evolution of PL intensity during the photocatalytic degradation process, (b and e) photocatalytic efficiency plot with the corresponding digital images of rhodamine B solution at t = 0 min and 60 min, and (c and f) first-order kinetic plot. | ||
| Photocatalysts | Photocatalytic efficiency (%) | Rate constants, k (× 10−2 min−1) | |||
|---|---|---|---|---|---|
| k 1 | k 2 | k 3 | k 4 | ||
| ZnO_Nit_U2 | 65.55 | 9.22 | 1.30 | 1.46 | 0.65 |
| ZnO_Ac_U2 | 73.82 | 17.45 | 5.91 | 1.02 | 0.14 |
Photocatalytic degradation of rhodamine B is a multistep reaction with many intermediate compounds, as reported in previous articles.74–76 During the photodegradation process, rhodamine B would be degraded to form intermediate species and finally decomposed, forming CO2, H2O, NO3−, and NH4+.75 Since the photocatalytic degradation of rhodamine B is a multistep reaction, the photodegradation rate might have several rate constants. The rate constant value was obtained by separating the first-order kinetic plots into several linear plots. The first-order kinetic plots can be seen in Fig. 6c (ZnO_Nit_U2 photocatalyst) and Fig. 6f (ZnO_Ac_U2 photocatalyst), as well as the corresponding rate constants. The plot with an uncondensed x-axis can be seen in Fig. S3 (ESI†), and the detailed linear fitting can be seen in Fig. S4 (ESI†). From those plots, we obtained four linear plots representing four rate constants, in which the k1, k2, k3, and k4 correspond to steps 1, 2, 3, and 4, respectively. The four degradation steps are consistent with the previously reported work, in which four photocatalytic degradation processes include N-de-ethylation, chromophore cleavage, opening-ring, and mineralization.76 The obtained rate constants can be seen in Table 3. We found that the k1 and k2 when using the ZnO_Ac_U2 photocatalyst are much higher than those of ZnO_Nit_U2. The results indicate that the ZnO_Ac_U2 photocatalyst provided a higher initial photodegradation rate than ZnO_Nit_U2. In contrast, the k3 and k4 when using the ZnO_Ac_U2 photocatalyst are smaller than those of ZnO_Nit_U2. The smaller value of k3 and k4 in the ZnO_Ac_U2 photocatalyst might be due to the formation of numerous intermediate species from previous steps (steps 1 and 2).
Here, we would like to discuss the photodegradation kinetics of rhodamine B when using the ZnO_Ac_U2 photocatalyst. We adopted the photocatalytic degradation model reported by Li and co-workers to explain the physical meaning of the rate constants obtained in this work.77 The photocatalytic degradation rate for the surface decomposition of rhodamine B on ZnO (r) is represented by the following equation:
| r = kΘOHΘRhB | (4) |
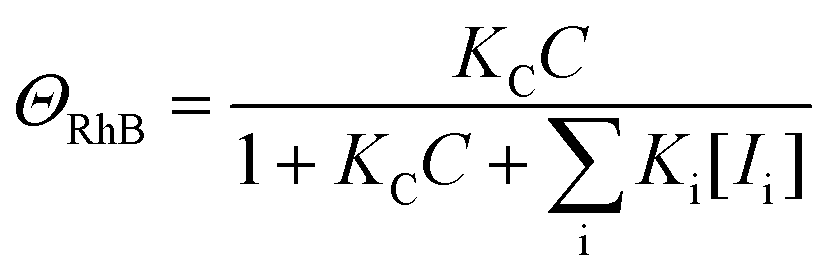 | (5) |
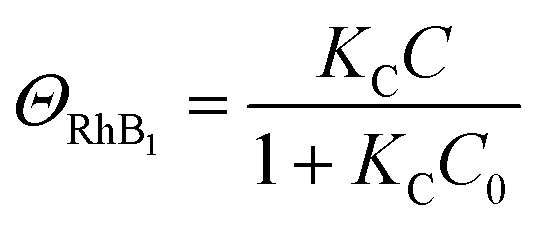 | (6) |
| k1 = kΘOH | (7) |
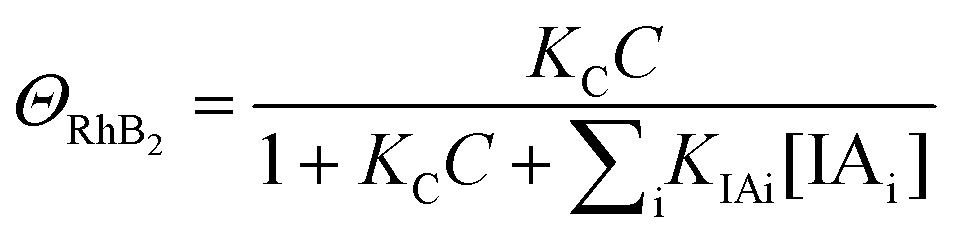 | (8) |
| k2 = k1ΘRhB2 | (9) |
Since ΘRhB2 < ΘRhB1, the value of k2 would be smaller than k1.
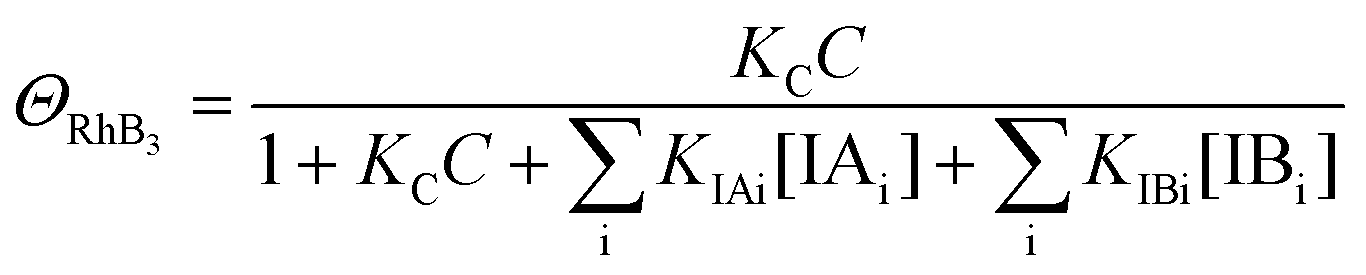 | (10) |
| k3 = k1ΘRhB3 | (11) |
Since ΘRhB3 < ΘRhB2 < ΘRhB1, the value of k3 < k2 < k1.
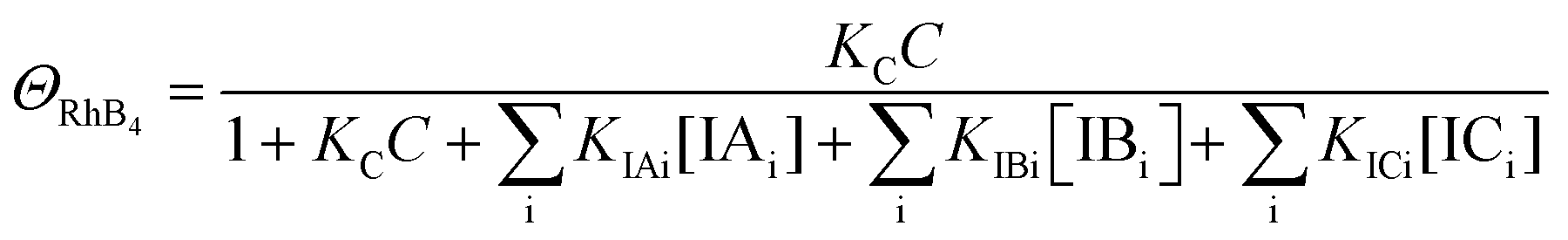 | (12) |
| k4 = k1ΘRhB4 | (13) |
Since ΘRhB4 < ΘRhB3ΘRhB2 < ΘRhB1, the value of k4 < k3 < k2 < k1.
For comparison, we also checked the decay of rhodamine B PL intensity without photocatalysts. The experimental data can be seen in Fig. S5 (ESI†). Without a ZnO photocatalyst, the PL intensity decay is very slow (see Fig. S5a, ESI†). The first-order kinetic plot in Fig. S5b (ESI†) shows that the initial rate constant is around 8.9 × 10−3 min−1. On the other hand, the rate constant with the ZnO_Ac_U2 photocatalyst is around twenty times higher than without a photocatalyst. This experimental fact indicates that the degradation of rhodamine B in the presence of a ZnO photocatalyst is dominated by the photocatalytic process rather than the photolysis process.
Moreover, to prove that the fibrous morphology plays a significant role in the photocatalytic activity, we also compared the photocatalytic performance of the fibrous ZnO microrods with synthesized ZnO nanorods with smooth surfaces. The morphology of synthesized ZnO nanorods and the photocatalytic performance can be seen in Fig. S6 (ESI†). In Fig. S6a (ESI†), we can see that the synthesized ZnO has a rod-like shape with a diameter of about 100–200 nm. The decay of rhodamine B PL intensity with irradiation time and the first-order kinetic plot can be seen in Fig. S6b and c (ESI†), respectively. We found that the photocatalytic degradation rate of the smooth ZnO nanorods is about 9.24 × 10−2 min−1. This value is similar to the rate constant from ZnO_Nit_U2 but smaller than that of ZnO_Ac_U2. The similar rate constant value of smooth ZnO nanorods to ZnO_Nit_U2 might be due to the smaller size of the smooth ZnO nanorods. The smooth ZnO nanorods gain surface area from the smaller particles, while the ZnO_Nit_U2 gains surface area from the partial surface roughness. Therefore, both samples have similar values of rate constants. However, the rate constant of ZnO_Ac_U2 is still twice higher than that of smooth ZnO nanorods.
We also performed recovery tests of both ZnO_Ac_U2 and ZnO_Nit_U2 photocatalysts for three cycles. The raw data of the recovery test can be seen in Fig. S7 (ESI†), while the photocatalytic performance obtained from the recovery test can be seen in Fig. 7. Fig. 7a and b shows the evolution of photocatalytic efficiency when using ZnO_Ac_U2 and ZnO_Nit_U2 photocatalysts, respectively. The evolution of photocatalytic efficiency exhibits a different pattern for different cycles. Moreover, we can see that both photocatalysts can reproduce the same maximum photocatalytic efficiency (η60) at the second cycle. However, at the third cycle, the η60 value decreased drastically, especially for the ZnO_Nit_U2 photocatalyst.
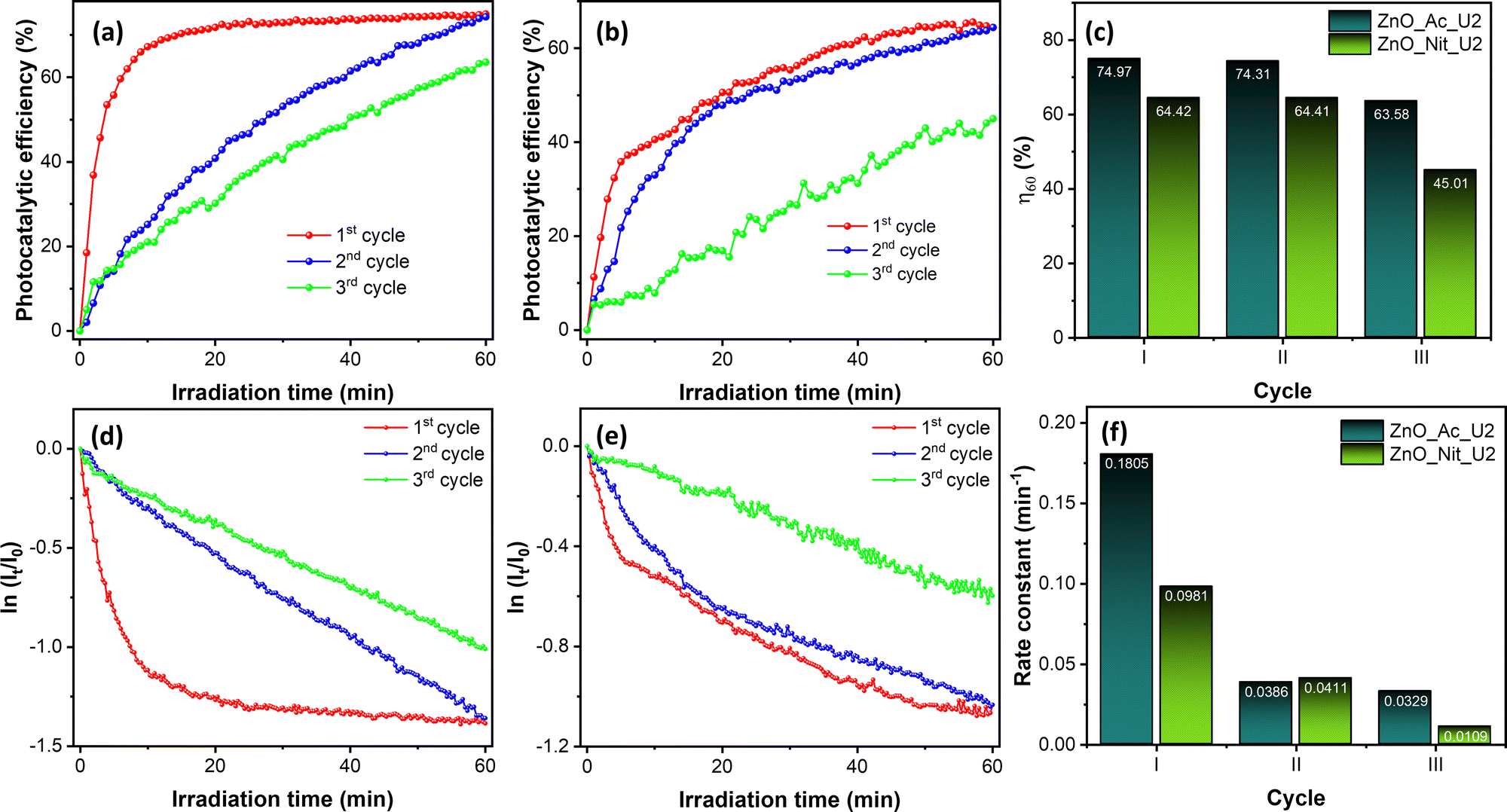 | ||
| Fig. 7 The results of the recovery test; (a) the evolution of photocatalytic efficiency of the ZnO_Ac_U2 photocatalyst and (b) ZnO_Nit_U2 photocatalyst for each cycle. (c) Photocatalytic efficiency after 60 minutes of irradiation. (d) The first-order kinetic plot of the ZnO_Ac_U2 photocatalyst and (e) ZnO_Nit_U2 photocatalyst for each cycle. (f) The initial rate constant obtained from the first-order kinetic plot in the range 1–5 minutes. | ||
The exact value of the η60 is plotted in Fig. 7c. The ZnO_Ac_U2 photocatalyst is more stable than the ZnO_Nit_U2 photocatalyst. In the third cycle, the η60 value of ZnO_Ac_U2 was 63.68%, while that of ZnO_Nit_U2 decreased drastically to 45.01%.
Fig. 7d and e show the first-order kinetic plot when using ZnO_Ac_U2 and ZnO_Nit_U2 photocatalysts, respectively. For both photocatalysts, it is clear that the initial rate is pretty different for each cycle. After the first cycle, the initial rate constant decreased dramatically. The exact value of the initial rate constant is plotted in Fig. 7f. For the ZnO_Ac_U2 photocatalyst, the initial rate constant decreased from 0.1805 min−1 (first cycle) to 0.0386 min−1 (second cycle) and then decreased to 0.0329 min−1 (third cycle). The rate constant did not change significantly from the second to third cycles. Meanwhile, for the ZnO_Nit_U2 photocatalyst, the initial rate constant decreased from 0.0981 min−1 (first cycle) to 0.0411 min−1 (second cycle) and then continued to decrease to 0.0109 min−1. The results about the value of the initial rate constant also indicate that the ZnO_Ac_U2 photocatalyst is more stable than the ZnO_Nit_U2 photocatalyst. The drastically decreased initial rate constant from the first to the second cycle might be due to the coverage of the ZnO surface by rhodamine B and intermediate molecules from the first cycle. Since rhodamine B is not completely degraded, it is suggested to rinse the photocatalyst several times before being used in the next cycle. This treatment might remove the rhodamine B and intermediate molecules from the surface of the photocatalyst.
We also compared the rate constant obtained in this work and the rate constant reported in previous articles. The comparison can be seen in Table 4. We can see that the rate constant obtained in this work is higher than almost all the last reported rate constant values. The rate constant of the ZnO_Ac_U2 photocatalyst is smaller than that of the holey ZnO nanosheet photocatalyst reported in our previous work.14 The high photodegradation rate of the ZnO_Ac_U2 photocatalyst might provide an alternative photocatalyst to degrade organic pollutants from wastewater.
| Photocatalysts | Catalyst loading (g mL−1) | Concentration of rhodamine B | Initial rate constant, k (min−1) | Ref. |
|---|---|---|---|---|
| Fibrous ZnO microrods (ZnO_Ac_U2) | 1 | 5 ppm | 0.1745 | This work |
| Holey ZnO nanosheets | 1 | 5 ppm | 0.2550 | 14 |
| Flower-like ZnO | 1 | 5 ppm | 0.0072 | 78 |
| ZnO nanoparticles | NA | 7 ppm | 0.1320 | 79 |
| ZnO nanoparticles | 0.0126 | 5 ppm | 0.0538 | 80 |
| Fluffy ZnO spheres | 0.2 | 10−5 M | 0.0332 | 81 |
| Rod-like ZnO nanoparticles | 0.5 | 10−5 M | 0.0633 | 82 |
| Rice-like ZnO nanoparticles | 0.5 | 10−5 M | 0.0432 | 82 |
| Disc-like ZnO nanoparticles | 0.5 | 10−5 M | 0.0245 | 82 |
Besides the high photocatalytic activity of ZnO_Ac_U2, we also would like to say that our in situ observation of photodegradation using time-dependent photoluminescence spectroscopy might provide more reliable rate constants. Compared with the standard method (i.e., absorption), this method can collect data at a decent interval. Therefore, we can separate the first-order kinetic plot into several linear plots, resulting in several rate constants. The rate constants reported in the previous articles are too small because they only have a few data over a long period. For example, Lai and co-workers only have 5 data for 50 minutes of irradiation.81 Therefore, they might only obtain the average rate constants in that time range by fitting the first-order kinetic plot. In contrast, our method can provide several meaningful rate constants at each step of photodegradation.
4. Conclusions
This work reports the synthesis of fibrous ZnO microrods using a macroemulsion-mediated solvothermal method under solvothermal conditions. Using the zinc acetate precursors, we optimize the amount of urea, producing homogeneous ZnO microrods. We found that the morphology of the ZnO_Ac_U2 sample is more uniform than other samples. We then applied this optimum condition to synthesize ZnO using zinc nitrate. Different types of precursors result in ZnO with different properties and photocatalytic activity. XRD and Raman spectroscopy analyses confirmed that both precursors resulted in ZnO with a wurtzite crystal structure. Morphological observation using FESEM shows that both samples have rod shapes with different diameters and lengths. ZnO_Nit_U2 is smaller and shorter than ZnO_Ac_U2, but ZnO_Nit_U2's aspect ratio (length to diameter ratio) is bigger than ZnO_Ac_U2. The detailed morphology shows that ZnO_Ac_U2 has a more uniform fibrous morphology than ZnO_Nit_U2. The uniform fibrous morphology of ZnO_Ac_U2 provides a higher surface area, smaller bandgap energy, and high total density of defects. The photocatalytic activity of the synthesized ZnO was determined through in situ observation using time-dependent photoluminescence spectroscopy measurements. The photocatalytic test shows that ZnO_Ac_U2 has a higher photocatalytic efficiency (73.82%) within 60 minutes of irradiation and a higher photodegradation rate (k = 0.1745 min−1). The higher photocatalytic activity of ZnO_Ac_U2 might be due to the synergistic effect among the higher surface area, smaller bandgap energy, higher total density of defects, and uniform fibrous morphology. The recovery test confirmed that the ZnO_Ac_U2 photocatalyst also has higher stability. The in situ measurement using time-dependent photoluminescence spectroscopy can collect data at decent intervals, resulting in a more reliable rate constant value. By separating the first-order kinetic plot into four linear plots, we obtained four values of rate constants. The values of rate constants correspond to four photodegradation steps, including N-de-ethylation, chromophore cleavage, opening-ring, and mineralization.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The research was financially supported by Penelitian, Pengabdian Masyarakat, dan Inovasi (PPMI) Institut Teknologi Bandung 2022. D. P. B. thanks the National Institute for Material Science (NIMS) for the support through the International Cooperative Graduate Program (ICGP). N. S. thanks The Ministry of Education, Culture, Research, and Technology of the Republic of Indonesia for the PMDSU (Program Magister menuju Doktor untuk Sarjana Unggul) scholarship. D. P. B. and V. S. thank Dr Yessi Permana and Ms Eunike K. Salduna for supporting the XRD measurements.References
- D. M. Bagnall, Y. F. Chen, Z. Zhu, T. Yao, M. Y. Shen and T. Goto, Appl. Phys. Lett., 1998, 73, 1038–1040 CrossRef CAS.
- S. Panigrahi, S. Sarkar and D. Basak, ACS Appl. Mater. Interfaces, 2012, 4, 2709–2716 CrossRef CAS.
- C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536–551 CrossRef CAS.
- M. Adeel, M. Saeed, I. Khan, M. Muneer and N. Akram, ACS Omega, 2021, 6, 1426–1435 CrossRef CAS PubMed.
- A. B. Djurišić, A. M. C. Ng and X. Y. Chen, Prog. Quantum Electron., 2010, 34, 191–259 CrossRef.
- J. A. Anta, E. Guillén and R. Tena-Zaera, J. Phys. Chem. C, 2012, 116, 11413–11425 CrossRef CAS.
- C. Guo, Q. Wang, J. He, C. Wu, K. Xie, Y. Liu, W. Zhang, H. Cheng, H. Hu and C. Wang, J. Phys. Chem. Lett., 2020, 11, 905–912 CrossRef CAS.
- Y. Yan, Y. Zhang, Y. Wu, Z. Wang, A. Mathur, H. Yang, P. Chen, S. Nair and N. Liu, ACS Appl. Energy Mater., 2018, 1, 6345–6351 CrossRef.
- N. L. W. Septiani, Y. V. Kaneti, B. Yuliarto, Nugraha, H. K. Dipojono, T. Takei, J. You and Y. Yamauchi, Sens. Actuators, B, 2018, 261, 241–251 CrossRef CAS.
- N. L. W. Septiani, A. G. Saputro, Y. V. Kaneti, A. L. Maulana, F. Fathurrahman, H. Lim, B. Yuliarto, Nugraha, H. K. Dipojono, D. Golberg and Y. Yamauchi, ACS Appl. Nano Mater., 2020, 3, 8982–8996 CrossRef CAS.
- L. Martínez-Suárez, N. Siemer, J. Frenzel and D. Marx, ACS Catal., 2015, 5, 4201–4218 CrossRef.
- S. R. Kelly, X. Shi, S. Back, L. Vallez, S. Y. Park, S. Siahrostami, X. Zheng and J. K. Nørskov, ACS Catal., 2019, 9, 4593–4599 CrossRef CAS.
- V. Sharma, A. Kumar, A. Kumar and V. Krishnan, Chemosphere, 2022, 287, 132119 CrossRef CAS.
- A. Andriani, D. P. Benu, V. Megantari, B. Yuliarto, R. R. Mukti, Y. Ide, S. Chowdhury, M. A. Amin, Y. V. Kaneti and V. Suendo, New J. Chem., 2022, 46, 9897–9908 RSC.
- X. Li, J. Wang, J. Zhang, C. Zhao, Y. Wu and Y. He, J. Colloid Interface Sci., 2022, 607, 412–422 CrossRef CAS PubMed.
- Y. Li, H. Chen, L. Wang, T. Wu, Y. Wu and Y. He, Ultrason. Sonochem., 2021, 78, 105754 CrossRef CAS PubMed.
- S. Zheng, X. Li, J. Zhang, J. Wang, C. Zhao, X. Hu, Y. Wu and Y. He, J. Environ. Sci., 2023, 125, 1–13 CrossRef.
- X. Zhang, J. Qin, Y. Xue, P. Yu, B. Zhang, L. Wang and R. Liu, Sci. Rep., 2015, 4, 4596 CrossRef.
- V. Kumar, R. Gupta and A. Bansal, ACS Appl. Nano Mater., 2021, 4, 6212–6222 CrossRef CAS.
- P. K. Aspoukeh, A. A. Barzinjy and S. M. Hamad, Int. Nano Lett., 2022, 12, 153–168 CrossRef CAS.
- W. Zhou, X. Zhang, D. Zhao, M. Gao and S. Xie, Sci. China: Phys., Mech. Astron., 2013, 56, 2243–2265 CrossRef CAS.
- A. Gupta, H. J. H. Clercx and F. Toschi, Eur. Phys. J. E: Soft Matter Biol. Phys., 2018, 41, 116 CrossRef CAS.
- L. K. Harris and J. A. Theriot, Trends Microbiol., 2018, 26, 815–832 CrossRef CAS PubMed.
- B. Witkowski, Acta Phys. Pol., A, 2018, 134, 1226–1246 CrossRef CAS.
- A. Das and R. G. Nair, J. Alloys Compd., 2020, 817, 153277 CrossRef CAS.
- S. Kumar, P. D. Sahare and S. Kumar, Mater. Res. Bull., 2018, 105, 237–245 CrossRef CAS.
- Q. Zhang and C. Li, Nanomaterials, 2019, 9, 1339 CrossRef CAS.
- M. Asghar, K. Mahmood, M. Y. Raja and M. A. Hasan, Adv. Mater. Res., 2013, 622–623, 919–924 Search PubMed.
- O. W. Kennedy, M. L. Coke, E. R. White, M. S. P. Shaffer and P. A. Warburton, Mater. Lett., 2018, 212, 51–53 CrossRef CAS.
- S. N. Sarangi, J. Phys. D: Appl. Phys., 2016, 49, 355103 CrossRef.
- A. C. Cruickshank, S. E. R. Tay, B. N. Illy, R. Da Campo, S. Schumann, T. S. Jones, S. Heutz, M. A. McLachlan, D. W. McComb, D. J. Riley and M. P. Ryan, Chem. Mater., 2011, 23, 3863–3870 CrossRef CAS.
- C. Baratto, RSC Adv., 2018, 8, 32038–32043 RSC.
- A. Thote, I. Jeon, H.-S. Lin, S. Manzhos, T. Nakagawa, D. Suh, J. Hwang, M. Kashiwagi, J. Shiomi, S. Maruyama, H. Daiguji and Y. Matsuo, ACS Appl. Electron. Mater., 2019, 1, 389–396 CrossRef CAS.
- T. R. Chetia, M. S. Ansari and M. Qureshi, ACS Appl. Mater. Interfaces, 2015, 7, 13266–13279 CrossRef CAS PubMed.
- D. Chandra, S. Mridha, D. Basak and A. Bhaumik, Chem. Commun., 2009, 2384–2386 RSC.
- S. Chatterjee, P. Bhanja, D. Ghosh, P. Kumar, S. Kanti Das, S. Dalapati and A. Bhaumik, ChemSusChem, 2021, 14, 408–416 CrossRef CAS.
- A. Kumar and V. Krishnan, Adv. Funct. Mater., 2021, 31, 2009807 CrossRef CAS.
- C. Wang, E. Wang, E. Shen, L. Gao, Z. Kang, C. Tian, C. Zhang and Y. Lan, Mater. Res. Bull., 2006, 41, 2298–2302 CrossRef CAS.
- A. N. Utami, M. Reza, D. P. Benu, A. I. Fatya, B. Yuliarto and V. Suendo, Polym.-Plast. Technol. Mater., 2020, 59, 1350–1358 CAS.
- M. Reza, A. N. Utami, A. N. Amalina, D. P. Benu, A. I. Fatya, M. K. Agusta, B. Yuliarto, Y. V. Kaneti, Y. Ide, Y. Yamauchi and V. Suendo, New J. Chem., 2021, 45, 5958–5970 RSC.
- E. Febriyanti, V. Suendo, R. R. Mukti, A. Prasetyo, A. F. Arifin, M. A. Akbar, S. Triwahyono, I. N. Marsih and Ismunandar, Langmuir, 2016, 32, 5802–5811 CrossRef CAS PubMed.
- V. Megantari, E. Febriyanti, D. P. Benu, M. Reza, F. V. Steky, B. Yuliarto, R. R. Mukti and V. Suendo, Polym.-Plast. Technol. Mater., 2020, 59, 1359–1369 CAS.
- N. Silmi, E. Febriyanti, A. Andriani, R. Arsyad, F. V. Steky, R. R. Mukti and V. Suendo, Mater. Chem. Phys., 2021, 265, 124492 CrossRef CAS.
- E. Febriyanti, N. Silmi, V. Suendo, R. R. Mukti, P. U. Vivitasari, D. R. Adhika, Y. Majima, Suprijadi and Ismunandar, Langmuir, 2022, 38, 1368–1379 CrossRef CAS.
- D. P. Benu, V. Suendo, R. R. Mukti, E. Febriyanti, F. V. Steky, D. R. Adhika, V. V. Tanuwijaya and A. B. Nugraha, Bull. Chem. React. Eng. Catal., 2019, 14, 542 CrossRef CAS.
- D. P. Benu, A. Hardian, R. R. Mukti, B. Yuliarto, N. Fukumitsu, Y. Ide, Y. Yamauchi, Y. V. Kaneti and V. Suendo, Microporous Mesoporous Mater., 2021, 111055 CrossRef CAS.
- M. Pudukudy and Z. Yaakob, Solid State Sci., 2014, 30, 78–88 CrossRef CAS.
- M. M. J. van Rijt, B. M. Oosterlaken, R. R. M. Joosten, L. E. A. Wijkhuijs, P. H. H. Bomans, H. Friedrich and G. de With, CrystEngComm, 2020, 22, 5854–5861 RSC.
- K. Sahu and A. K. Kar, Cryst. Growth Des., 2021, 21, 3656–3667 CrossRef CAS.
- Y. Miao, H. Zhang, S. Yuan, Z. Jiao and X. Zhu, J. Colloid Interface Sci., 2016, 462, 9–18 CrossRef CAS.
- Y.-C. Liang, C.-S. Hung and W.-C. Zhao, Nanomaterials, 2020, 10, 1352 CrossRef CAS.
- M. Schreyer, L. Guo, S. Thirunahari, F. Gao and M. Garland, J. Appl. Crystallogr., 2014, 47, 659–667 CrossRef CAS.
- Y. Shi, C. Zhu, L. Wang, C. Zhao, W. Li, K. K. Fung, T. Ma, A. Hagfeldt and N. Wang, Chem. Mater., 2013, 25, 1000–1012 CrossRef CAS.
- R. Jothilakshmi, V. Ramakrishnan, R. Thangavel, J. Kumar, A. Sarua and M. Kuball, J. Raman Spectrosc., 2009, 40, 556–561 CrossRef CAS.
- M. Šćepanović, M. Grujić-Brojčin, K. Vojisavljević, S. Bernik and T. Srećković, J. Raman Spectrosc., 2010, 41, 914–921 CrossRef.
- R. Cuscó, E. Alarcón-Lladó, J. Ibáñez, L. Artús, J. Jiménez, B. Wang and M. J. Callahan, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 165202 CrossRef.
- Y. Song, S. Zhang, C. Zhang, Y. Yang and K. Lv, Crystals, 2019, 9, 395 CrossRef CAS.
- V. Russo, M. Ghidelli, P. Gondoni, C. S. Casari and A. Li Bassi, J. Appl. Phys., 2014, 115, 073508 CrossRef.
- S. Marković, I. Stojković Simatović, S. Ahmetović, L. Veselinović, S. Stojadinović, V. Rac, S. D. Škapin, D. Bajuk Bogdanović, I. Janković Častvan and D. Uskoković, RSC Adv., 2019, 9, 17165–17178 RSC.
- J. Thyr, L. Österlund and T. Edvinsson, J. Raman Spectrosc., 2021, 52, 1395–1405 CrossRef CAS.
- H. Souissi, S. Jabri, A. Souissi, G. Amiri, P. Gemeiner, A. Lusson, P. Galtier, B. Dkhil, V. Sallet, M. Oueslati and A. Meftah, J. Appl. Phys., 2018, 123, 025705 CrossRef.
- F. J. Manjón, B. Marí, J. Serrano and A. H. Romero, J. Appl. Phys., 2005, 97, 053516 CrossRef.
- D. P. Benu, J. Earnshaw, A. Ashok, K. Tsuchiya, I. Saptiama, B. Yuliarto, V. Suendo, R. R. Mukti, N. Fukumitsu, K. Ariga, Y. V. Kaneti and Y. Yamauchi, Bull. Chem. Soc. Jpn., 2021, 94, 502–507 CrossRef CAS.
- F. V. Steky, V. Suendo, R. R. Mukti, D. P. Benu, M. Reza, D. R. Adhika, V. V. Tanuwijaya and A. B. Nugraha, Bull. Chem. React. Eng. Catal., 2019, 14, 513–520 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- Y. Shi, C. Zhu, L. Wang, C. Zhao, W. Li, K. K. Fung, T. Ma, A. Hagfeldt and N. Wang, Chem. Mater., 2013, 25, 1000–1012 CrossRef CAS.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef.
- C. H. Ahn, Y. Y. Kim, D. C. Kim, S. K. Mohanta and H. K. Cho, J. Appl. Phys., 2009, 105, 013502 CrossRef.
- K. H. Tam, C. K. Cheung, Y. H. Leung, A. B. Djurišić, C. C. Ling, C. D. Beling, S. Fung, W. M. Kwok, W. K. Chan, D. L. Phillips, L. Ding and W. K. Ge, J. Phys. Chem. B, 2006, 110, 20865–20871 CrossRef CAS PubMed.
- L. S. Vlasenko and G. D. Watkins, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 125210 CrossRef.
- H. Kaftelen, K. Ocakoglu, R. Thomann, S. Tu, S. Weber and E. Erdem, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 014113 CrossRef.
- H. H. Kim, Y. Lee, Y. J. Lee, J. Jeong, Y. Yi, C. Park, S.-Y. Yim, B. Angadi, K.-J. Ko, J.-W. Kang and W. K. Choi, ACS Photonics, 2020, 7, 723–734 CrossRef CAS.
- J. Xu, Y. Teng and F. Teng, Sci. Rep., 2016, 6, 32457 CrossRef CAS PubMed.
- Y. A. B. Neolaka, Z. S. Ngara, Y. Lawa, J. N. Naat, D. P. Benu, A. Chetouani, H. Elmsellem, H. Darmokoesoemo and H. Septya Kusuma, J. Environ. Chem. Eng., 2019, 7, 103482 CrossRef CAS.
- T. S. Natarajan, M. Thomas, K. Natarajan, H. C. Bajaj and R. J. Tayade, Chem. Eng. J., 2011, 169, 126–134 CrossRef CAS.
- Z. He, C. Sun, S. Yang, Y. Ding, H. He and Z. Wang, J. Hazard. Mater., 2009, 162, 1477–1486 CrossRef CAS PubMed.
- Y. Li, S. Sun, M. Ma, Y. Ouyang and W. Yan, Chem. Eng. J., 2008, 142, 147–155 CrossRef CAS.
- N. Rana, S. Chand and A. K. Gathania, J. Mater. Sci.: Mater. Electron., 2016, 27, 2504–2510 CrossRef CAS.
- J. Wang, J. Yang, X. Li, B. Feng, B. Wei, D. Wang, H. Zhai and H. Song, Powder Technol., 2015, 286, 269–275 CrossRef CAS.
- D. Blažeka, J. Car, N. Klobučar, A. Jurov, J. Zavašnik, A. Jagodar, E. Kovačević and N. Krstulović, Materials, 2020, 13, 4357 CrossRef.
- Y. Lai, M. Meng and Y. Yu, Appl. Catal., B, 2010, 100, 491–501 CrossRef CAS.
- S.-Y. Pung, W.-P. Lee and A. Aziz, Int. J. Inorg. Chem., 2012, 2012, 1–9 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj04862k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |